
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRemarkable elasticity and enzymatic degradation of bio-based poly(butylene adipate-co-furanoate): replacing terephthalate†
Hyeri
Kim‡
a,
Taeho
Kim‡
a,
Sejin
Choi
 a,
Hyeonyeol
Jeon
a,
Dongyeop X.
Oh
ab,
Jeyoung
Park
ab,
Youngho
Eom
*c,
Sung Yeon
Hwang
*ab and
Jun Mo
Koo
*a
a,
Hyeonyeol
Jeon
a,
Dongyeop X.
Oh
ab,
Jeyoung
Park
ab,
Youngho
Eom
*c,
Sung Yeon
Hwang
*ab and
Jun Mo
Koo
*a
aResearch Center for Bio-based Chemistry, Korea Research Institute of Chemical Technology (KRICT), Ulsan 44429, Republic of Korea. E-mail: crew75@krict.re.kr; jmkoo@krict.re.kr
bAdvanced Materials and Chemical Engineering, University of Science and Technology (UST), Daejeon 34113, Republic of Korea
cDepartment of Polymer Engineering Pukyong National University, Busan, 48513, Republic of Korea. E-mail: eomyh@pknu.ac.kr
First published on 5th August 2020
Abstract
Biodegradable polyesters, such as poly(butylene adipate-co-terephthalate) (PBAT), can trigger the hydrolysis of esters in a natural environment. However, biodegradability does not guarantee its eco-friendliness due to the concerns associated with terephthalic acid. Substitution by biomass-based counterparts no longer ensures “truly” eco-friendly biodegradable polyesters. In this study, we have used poly(butylene adipate-co-furanoate) (PBAF), which utilizes a biomass-derived substituent, furan-2,5-dicarboxylic acid (FDCA), and have proven that this is superior from the perspective of both mechanical performance and enzymatic degradation. Compared to its terephthalate counterpart, PBAF exhibits remarkable elasticity upon stretching: when released beyond 800% extension, PBAT almost malfunctioned, whereas PBAF exhibited instant elastic recovery. This results in PBAT and PBAF having a similar tensile strength in the range of 50–70 MPa at the ring fraction of 50 to 80 mol%. During the enzymatic degradation using lipase from Thermomyces lanuginosus, PBAF with a ring fraction of 50 mol% solely undergoes hydrolysis due to enzyme specificity. Ex situ spectroscopic analysis confirms that the participation of FDCA ester groups in the early stages of degradation triggered the successive hydrolytic reaction. Thus, understanding the structural characteristics of FDCA would open new opportunities in the field of sustainable plastic industries.
Introduction
Plastic, a petroleum product, was once regarded as an essential commodity, primarily because of its widespread applications and its convenience of use. In fact, the damage caused by petroleum products and their wastes was easily overlooked. Currently, however, the effects of cumulative plastic pollution on soil, atmosphere, and marine ecosystems have become extremely severe, affecting everyday lives, and are a legitimate threat to future generations.1–5 Consequently, efforts are being made globally to ban plastic disposal into the sea/landfills and restrict its incineration, while exploring an alternative that has the advantages of plastic. Understanding the importance of developing biodegradable and sustainable plastics has never been higher.6–13Commercially available biodegradable plastics, such as poly(lactic acid), have already made an impact on the plastic industries that changed the course of polymer developments. Such success has encouraged the world to explore new biomass based materials that can improve existing polymers. One of the representative examples of a biodegradable polymer that can have high potential for substituents is poly(butylene adipate-co-terephthalate) (PBAT), which is commercially known as Ecoflex®, Wango, and Eastar Bio.14 It is an aliphatic–aromatic copolyester whose crystallinity, mechanical properties, and biodegradability can be changed by controlling the composition ratio of its aliphatic and aromatic contents.15,16 Generally, a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio of the aromatic and aliphatic contents is maintained during the synthesis of PBAT, in order to have tear resistance, flexibility, and water resistance similar to those of low-density polyethylene.17,18 This is to ensure that the former can be used for making various products such as food packaging materials,19–21 mulching films,22–24 and biomedical materials.25–27
:50 ratio of the aromatic and aliphatic contents is maintained during the synthesis of PBAT, in order to have tear resistance, flexibility, and water resistance similar to those of low-density polyethylene.17,18 This is to ensure that the former can be used for making various products such as food packaging materials,19–21 mulching films,22–24 and biomedical materials.25–27
However, PBAT is a petroleum-based polyester that leaves behind non-consumable and toxic monomers after degradation, particularly terephthalic acid (TPA). TPA is a chemically synthesized monomer that is ranked in the top 50 most abundantly produced chemicals worldwide.28 It is one of the most commonly used monomers for producing polyesters such as poly(ethylene terephthalate) (PET) and poly(butylene terephthalate) (PBT). However, the synthesis of TPA produces byproducts that affect the environment and human health.29–33 Hence, effective substitutes for TPA are being explored to overcome its limitations.
Amongst the possible candidates, biomass derived furan-2,5-dicarboxylic acid (FDCA) is the most potential substitute due to the similarities in its structural, chemical, and physical properties to those of TPA.34–39 However, in contrast to the highly symmetric and planar structure of TPA, which has π–π stacking,40 the molecular packing in FDCA is lost due to an inherent angle of 129.4° between the carboxylates. Yet, it adopts a spiral motif and a diverse nonlinear architecture within a polymer chain and acquires attractive properties.41 To compare FDCA directly with TPA, it is important to understand the physical and chemical roles of FDCA within the polymer chain.
Numerous studies on the biodegradation behavior of FDCA-based polyesters have investigated the macroscopic transitions and function of counterparts and have discussed the consequential interpretations.42–46 However, a comparative and fundamental understanding of the structural role of FDCA and its effect on the biodegradation mechanism is crucial to validate the expansion of its applications.
To mitigate plastic pollution and to substitute TPA-based polyesters, we conducted a comparative study to reveal the structural role and benefits of using FDCA. We proposed a FDCA-based polyester, poly(butylene adipate-co-furanoate) (PBAF), whose synthetic protocol would be identical to that of PBAT. PBAF was expected to exhibit competing mechanical properties and superior degradability. Thermal studies of the two polyesters revealed the effects of structure and composition on the crystallization and melting behaviors. The analysis, in turn, aided in interpreting the unique degradation behavior in chemical and enzymatic media. The sequence of degradation and the transitions during the degradation were determined through two-dimensional correlation spectroscopy (2DCOS),47–49 in order to understand the contribution of the FDCA moiety in the enzymatic degradability of PBAF. The segmental motion of chains and kinetic parameters such as the activation energy were investigated using dynamic mechanical analysis (DMA) and the corresponding Arrhenius plot, respectively, to gain insights into the molecular affinity and thermodynamics of the polymer–enzyme system during degradation.
Results and discussion
Synthesis of poly(butylene adipate-co-furanoate) with remarkable mechanical elasticity
Two series of copolyesters, using petrochemical and bio-based monomers, were synthesized by a conventional two-step melt polymerization method, such that all the synthesized copolyesters had a similar molecular weight (Mn) of ±3000 g mol−1. The polymerization procedure and results are shown in Fig. 1a and Table S1.† In this synthesis, the dimethyl forms of TPA (dimethyl terephthalate, DMT) and FDCA (dimethyl-2,5-furandicarboxylate, DMFD) were used, while maintaining the molar ratio of methyl carboxylate to 1,4-butanediol (BD) at 1.6. The ester-exchange reaction occurred at two temperatures, 180 and 210 °C, owing to the reactivity difference of the methyl and hydroxyl leaving groups. Initially, the reaction was conducted at 180 °C to allow the complete reaction of DMT/DMFD with BD, ensuring the expected aromatic composition ratio and preventing severe discoloration. Finally, the temperature was increased to 210 °C to allow the reaction of the remaining adipic acid (AA). Despite the separate ester-exchange stages, the transesterification mechanism ensured that the copolyester is a random copolymer. The composition ratio of each copolyester was determined from the 1H NMR spectrum, in which the integrated ratio of the peaks (Fig. S1 and Table S1†) was calculated to show that the composition ratio matches with the feed ratio.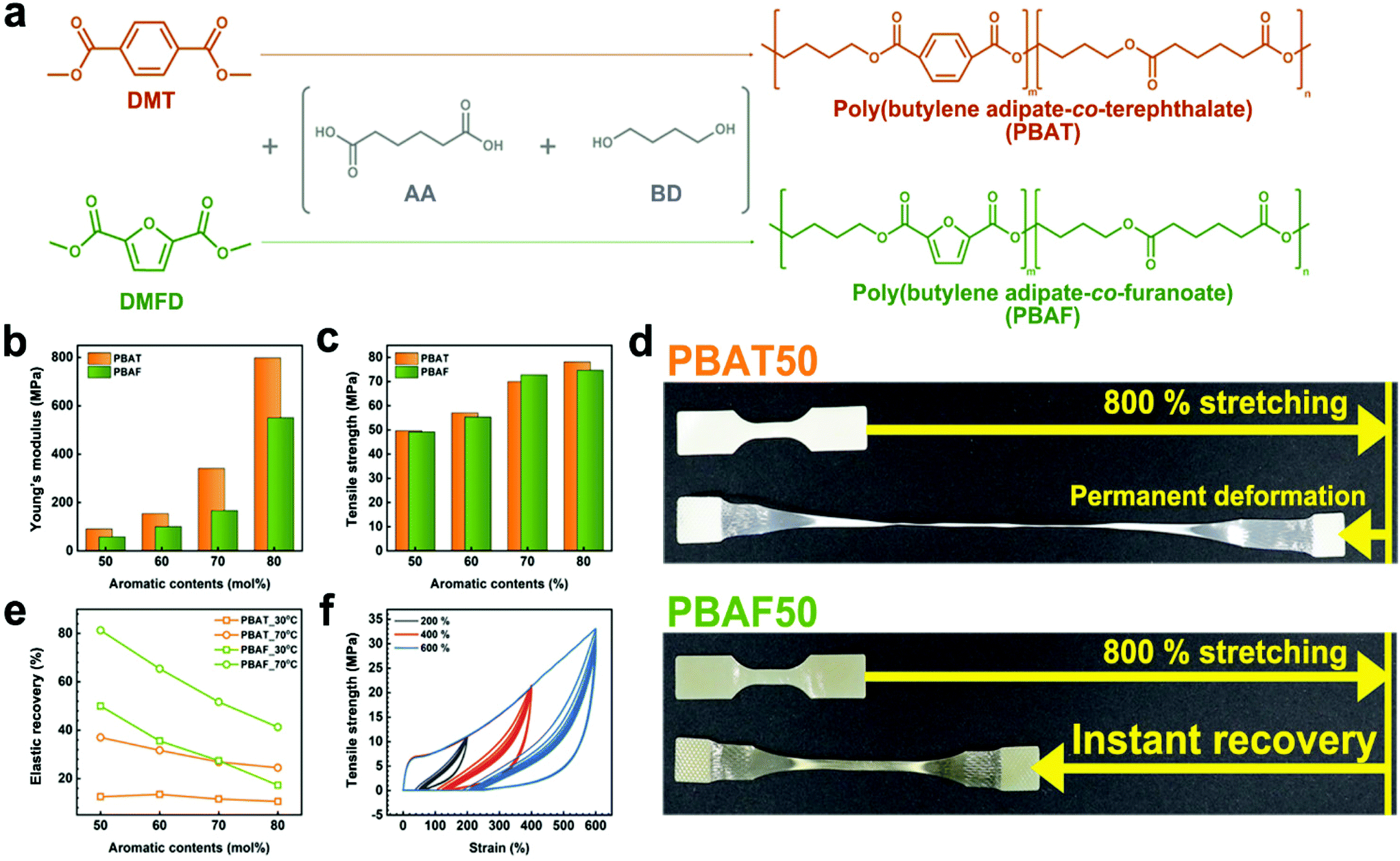 | ||
| Fig. 1 (a) Synthesis of terephthalate-based poly(butylene adipate-co-terephthalate) (PBAT) and furanoate-based poly(butylene adipate-co-furanoate) (PBAF). Comparative analysis of (b) Young's modulus and (c) tensile strength. (d) Visual comparison of the elastic recovery of PBAT50 and PBAF50. (d) Elastic recovery test of PBATs and PBATs (at 30 °C (square) and 70 °C (circle)) with different aromatic contents. (f) Hysteresis test on PBAF50 at 200%, 400%, and 600% strains. | ||
To support and establish that the sequence of copolyesters is random, sequence distribution was calculated (Table S2 and Fig. S2†). As can be seen in Table S2,† the randomness ‘R’ converges to the value of 1, confirming that the distribution obeys Bernoullian statistics. If R < 1, = 1, = 2, and = 0, it means that the polymer is a block copolymer, random copolymer, alternating copolymer, homopolymer, respectively.50,51 Therefore, the result confirms that synthesized copolyesters are completely random.
The poorer mechanical properties of PBAF as compared to PBAT are expected, considering that FDCA has an asymmetric, semi-rigid structure while TPA is symmetric (Fig. S3†). Although the low-strain property (i.e., Young's modulus) met the aforementioned expectation, surprisingly, the high-strain property (i.e., tensile strength) was comparable between the two polymers. Fig. 1b, c and S4 and Table S3† show that the tensile strengths of both the copolyesters increase with increasing aromatic content because a higher ring fraction imparts a stiffer chain structure. The tensile modulus of PBATs is almost 1.5 times greater than that of PBAFs (Fig. 1b). However, the ultimate tensile strength of PBAFs is comparable to that of PBATs and ranges from 49.2 to 74.7 MPa at a ring fraction of 50–80 mol%. This suggests that the strain-induced chain orientation and the lateral alignment compensate for the inferior chain regularity of PBAFs. Thus, the difference in the mechanical performances of the two copolyesters becomes narrow, and finally negligible, with increasing tensile strain. It is worth noting that the stiffening of the stretched PBAF chains alone cannot completely explain the high mechanical strength.
A huge difference in the elastic recovery is observed when the copolyesters are released after 800% stretching. While PBAT50 (aromatic content of 50 mol%) lost its mechanical performance due to an irreversible deformation, PBAF50 exhibited instant elastic recovery (Fig. 1d and Movie S1†). It is obvious that this remarkable elasticity amplifies the resistance of PBAF to uniaxial stretching at high strains, i.e., tensile strength. It is evident from Fig. 1e that the elastic recovery of PBATs at 30 °C over 12 h is as low as 10% and does not exceed 40% when the heating temperature was increased to 70 °C for the same duration. However, particularly for PBAF50, recoveries of 50% at 30 °C and 81% at 70 °C were observed; these are 4.0 and 2.2 times, respectively, greater than those of PBAT50. In the hysteresis tests, the highly elastic PBAF50 exhibited a slight drop in the ultimate tensile strength from 11.0 to 10.8 MPa (−1.1%), 21.2 to 20.3 MPa (−4.2%), and 33.1 to 28.6 MPa (−13.6%) after 10 cycles of 200%, 400%, and 600% strains, respectively (Fig. 1f). Such remarkable elasticity of PBAF50 seems to be derived from an entropy spring of the semi-rigid chain structure. As is known, stretched and oriented polymeric chains decrease the entropy which stimulates chains to recover their recoiled state upon unloading. Then, the symmetric and planar TPA moiety stabilizes the lateral packing of the aligned chains, disturbing the elastic recovery. On the other hand, the asymmetric FDCA moiety prefers recoiling to packing, resulting in reversible deformation. In addition, the immediate ring-flip of the furan ring accelerates the entropy recovery. Thus, from the long-term perspective, PBAFs have superior durability compared to PBATs.
The elasticity of polymers is determined not only from their chemical structure but also by the extent of entanglement with the neighboring chains. The entangled state can be traced using a rheological approach. Fig. 2 presents the rheological properties of molten PBAT50 and PBAF50 at 220 °C. It should be noted that PBAF50 has higher complex viscosity (η*) and storage modulus (G′) compared to PBAT50 over the entire frequency range observed (Fig. 2a). Generally, the rheological properties are predominantly affected by three factors: chemical structure of the chain (chain rigidity), size of the chain (molecular weight), and degree of entanglement (physical network). If the molecular weight difference of the comparative groups is negligible, the rheological parameters are commonly proportional to the chain rigidity. However, an opposite trend was observed in our case, indicating that PBAF50 possesses a stronger physical network compared to PBAT50 in the molten phase. Generally, the physical network is created by either a heterogeneous structure or a spatial entanglement of the chains.52 The former can be ascertained by the evaluation of yield stress (τy), which represents the minimum energy required to collapse the heterogeneous structure.53 It is clear from the Casson plots (Fig. 2b) that τy values of both the polymers are close to zero, signifying only a little heterogeneity in the molten state. Accordingly, the higher G′ of PBAF50 as compared to PBAT50 can be exclusively ascribed to a higher number of chain entanglement. It is conjectured that the asymmetric and distorted FDCA can inter-diffuse and entangle with the aliphatic segments more efficiently as compared to the rigid TPA. In addition, the relatively polar nature of oxygen-containing FDCA contributes further to the enhanced intermolecular interaction and strengthened chain entanglement.41 As a result, the recoverable elastic strain (SR), a quantitative indicator of the rheological elasticity, in PBAF50 is twice that in PBAT50 at various frequencies (Fig. 2c).54
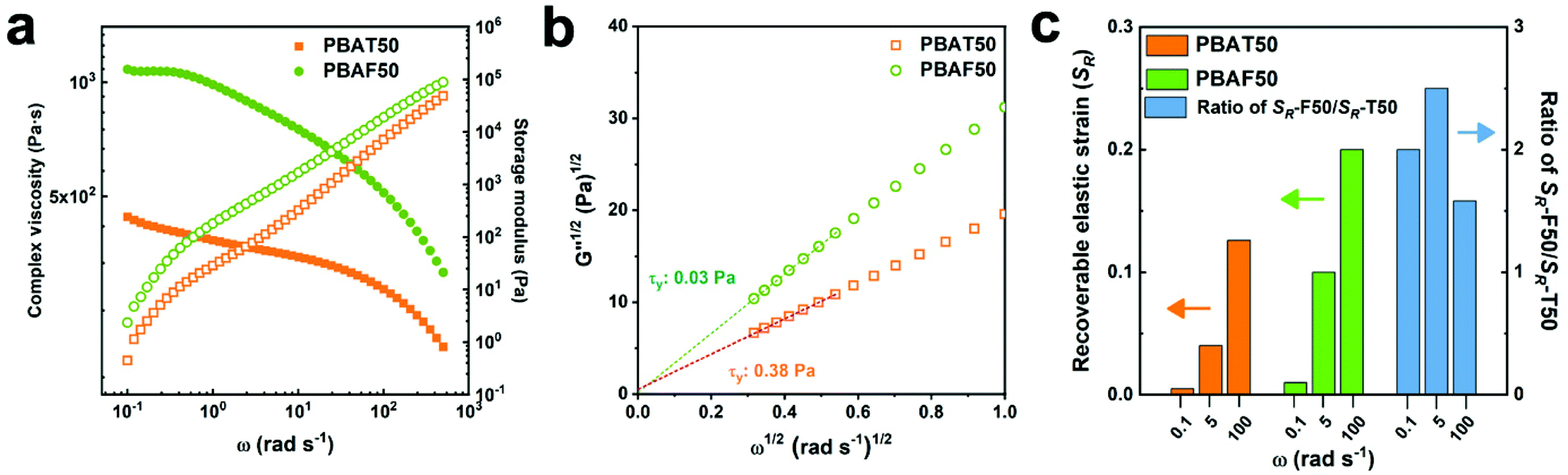 | ||
| Fig. 2 Rheological properties of PBAT50 and PBAF50 at 220 °C: (a) complex viscosity (η*) and storage modulus (G′) in the angular frequency range 0.05–500 rad s−1. (b) Casson plot for obtaining the yield stress (τy), and (c) recoverable elastic strains (SR) of PBAT50 and PBAF50 and their ratios (SR-F50/SR-T50). | ||
Structural and spectroscopic analysis of chemical and enzymatic degradation
The prominent effects of different aromatic structures can be better understood from the degradation in two different media and the weight loss as a result of the degradation (Fig. 3a and b). NaOH (1 M) at pH 13.4 was used as the chemical medium to accelerate the degradation. PBAF50 and PBAF70 were degraded by 60% and 49%, respectively, of their original weight within 6 days. These were 2 and 7 times, respectively, higher than the corresponding PBAT compositions (Fig. 3a). A similar trend in thickness reduction was also observed (Fig. S5†). | ||
| Fig. 3 (a) Chemical and (b) enzymatic degradation of PBATs and PBAFs at ring fractions of 50 and 70 mol% showing weight reduction, schematic of degradation, and SEM images of degraded samples. | ||
The faster degradation of PBAFs than PBATs originates from the inherent structural irregularity in the distorted and asymmetric FDCA. In contrast, the planar TPA with the benzene ring has a compact stacking of polymer chains, which severely impedes the degradation. Accordingly, two distinct tendencies can be observed in the chemical degradation: (1) high degradation rate of PBAFs compared to PBATs and (2) increased degradation rate at the lower ring fraction. Probably, these trends are strongly related to the penetration rate of hydroxyl anions (Fig. 3a).
During the enzymatic degradation using Thermomyces lanuginosus lipase (TLL), interestingly, PBAF50 underwent degradation without any trend, similar to the chemical degradation (Fig. 3b). In contrast to hydroxyl anions, TLL lipase is a highly selective catalyst owing to its size. TLL is a single chain protein consisting of 269 amino acids and has a molecular weight of 31700 g mol−1. It is spherical in shape, with an effective radius of ∼50 Å.55 Compared to hydroxyl anions (effective ionic radius of 0.1 Å),56 TLL is an eminently bulky complex with a strong tendency to form bimolecular aggregates. Therefore, the enzymatic hydrolysis is not simply triggered by a physical penetration of the enzymes but is triggered by the enzyme specificity that originates upon the precise docking of the substrate onto oriented active sites on the enzyme; the precise docking accelerates specific reactions. In this regard, PBAF50 is the only polymer that satisfies the criterion of enzyme specificity. The peculiar morphology of the degraded PBAF50 reveals that the TLL enzymes attacked only those regions of the polymer chain in which the structural density or chain conformation was favorable for coordination with the enzyme molecules (Fig. 3b).
The enzyme specificity of PBAF50 can be confirmed from its structural analysis (Fig. 4). In the first heating scan in differential scanning calorimetry (DSC) (Fig. 4a and b), both the copolyesters exhibit two melting endotherms (Tm1 and Tm2), which are representative of the crystalline structures of the aliphatic- and aromatic-rich phases, respectively. With an increase in the aromatic ring content, the peak position of Tm1 remains almost constant at about 55 °C whereas that of Tm2 increases drastically (i.e., strongly dependent on the aromatic content) for both the copolyesters, indicating a substantial contribution of the aromatic ring to the crystalline phase (Fig. 4a and b). Tm2 of PBAFs is about 50 °C lower than that of PBATs for the same aromatic contents, and, moreover, the dominance of the aromatic content is comparatively low. The heats of fusion at Tm1 and Tm2 (ΔHm1 and ΔHm2) for the two copolyesters were compared (Fig. 4c and Table S4†). While ΔHm2 exceeds ΔHm1 at all the compositions in PBATs, the trend of ΔHm1 and ΔHm2 at 50 and 60 mol% of the aromatic content in PBAFs is different (Fig. 4d). In particular, for PBAF50, the crystalline phase is predominantly composed of the aliphatic component, with higher ΔHm1. This structural difference between the two copolyesters can be determined from the different crystallizabilities of TPA and FDCA. As expected, the former promotes rapid crystallization due to the favorable π–π stacking of the benzene rings, while the latter undergoes a relatively slow crystallization due to the poor packing of distorted furan rings. Thus, low FDCA contents enable aliphatic chains to crystallize completely over a long time, depending on their molecular potential. It is because of this that an overwhelming ΔHm1 of PBAF50 is obtained. Moreover, the structure of PBAF50 with an evidently larger aliphatic crystalline phase supports the enzymatic selectivity.
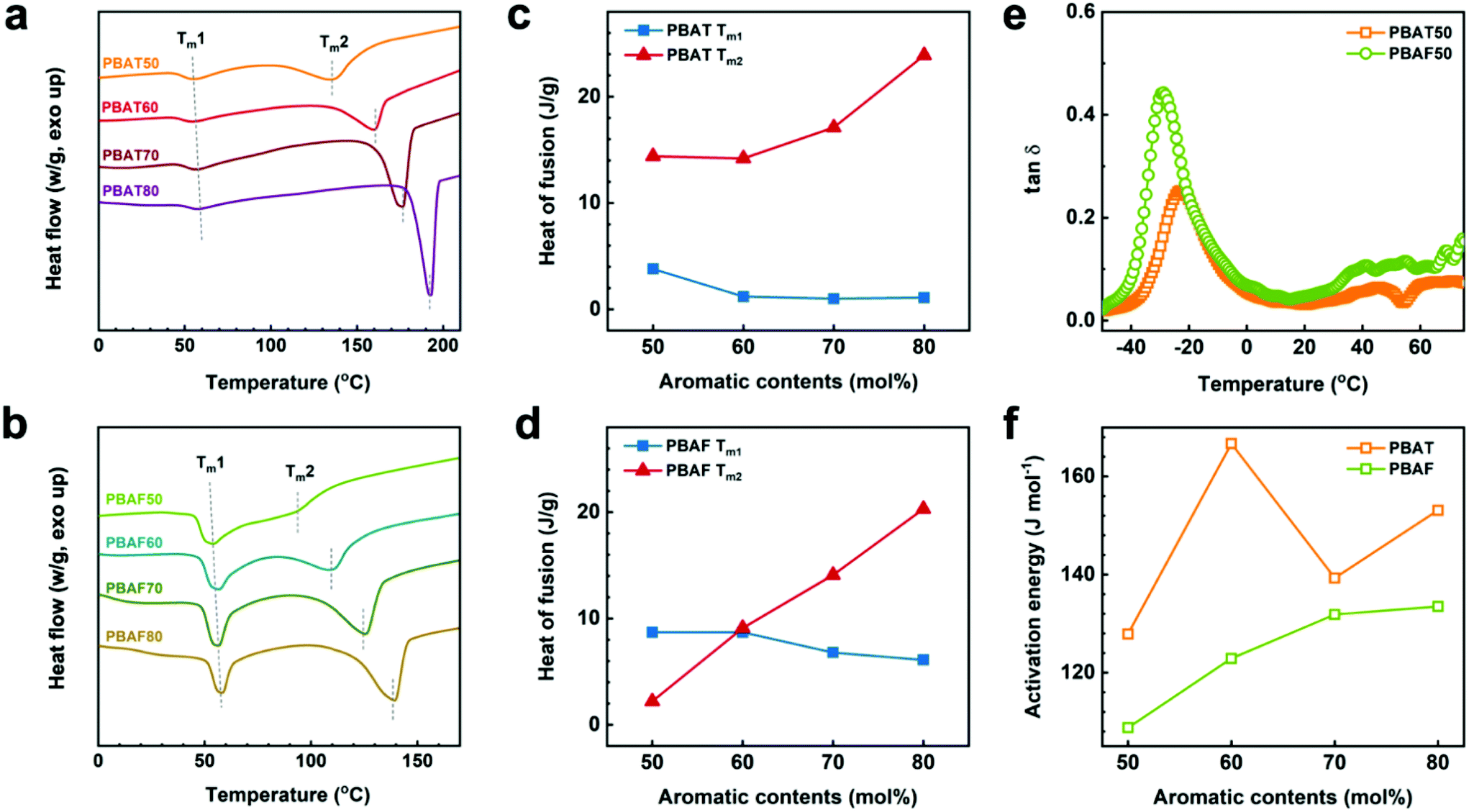 | ||
| Fig. 4 1st heating scan of (a) PBATs and (b) PBAFs. Heats of fusion of (c) PBATs and (d) PBAFs. (e) Loss factor (tanδ) curves of PBAT50 and PBAF50 at a frequency of 1 Hz. (f) Activation energies (Ea) of PBATs and PBAFs at various aromatic contents calculated using the Arrhenius equation. | ||
Apart from the crystalline phase-dependent microstructure, a conformational change of the individual chains through a segmental motion significantly influences the enzyme selectivity during the degradation. An easier conformational change of the polymer chains provides better coordination with the enzyme, thereby leading to the enzymatic selectivity. The segmental motion of the chain and the relative conformational change can be explained based on the DMA (Fig. 4e and f). The peak position of loss tangent (tanδ) indicates the glass transition temperature (Tg) of each polymer.57 Thus, the peak intensity (i.e., the tanδ value) of PBAF50 (0.44) is almost twice that of PBAT50 (0.25). Theoretically, a higher tanδ value corresponds to a higher Tg value and less solid-like characteristics of a polymer owing to the enhanced segmental motion of the chains.58 In this regard, it can be expected that the presence of asymmetric FDCA aids in active conformational changes during the hydrolysis. The fact that the favorable chain motion of PBAFs imparts enzyme specificity can be quantitatively confirmed from the activation energy (Ea) of the segmental motion. Fig. 4f and Fig. S6† show the Arrhenius plots for calculating the activation energies. The lower Ea values of PBAFs compared to PBATs over the entire 50–80 mol% ring fraction confirm that the activation of the segmental motion of the asymmetric FDCA-containing chains is more facile. Apart from the kinetic motion of the polymer chains, the furan ring also enhances the chemical affinity between the polar active sites of the enzymes and PBAF chains due to the existence of a polar oxygen atom. To evaluate the polar interactions, we calculated the theoretical solubility parameters of PBAF50 and PBAT50 using the Van Krevelen group contribution method (Fig. S7 and Table S5†).59 Such theoretical parameters offer an indirect evidence of the chemical affinity between the polymers and the enzymes possessing numerous polar functional groups. It is worth noting that the polar (δp) and hydrogen bonding (δh) terms of the individual solubility parameters and the resulting total solubility parameter noticeably increase upon replacing TPA with FDCA. Particularly, δh at a ring fraction of 50 mol% for PBAF50 (δh = 8.9) is 1.3 times that for PBAT50 (δh = 6.7). This is because of the enhanced polarity originating from the oxygen-containing furan ring. From the viewpoints of the kinetic motion and thermodynamic affinity of PBAF50, it is believed that the enzyme preferably draws the ester groups adjacent to the polar furan ring. Additionally, the segmental motion of PBAF, which has a low Ea, favors the conformational change and imparts the enzymatic specificity.
Using advanced Fourier transform infrared (FT-IR) spectroscopy, another crucial clue to the enzymatic selectivity of PBAF50 during its ex-situ enzymatic degradation was obtained—hydrolysis of the ester groups adjacent to the furan ring triggered the enzymatic degradation. Fig. 5a shows the FT-IR spectrum on each day of the enzymatic degradation of PBAF50; the FT-IR spectra during the chemical degradation of PBAF50 and PBAT50 are also shown in Fig. S8† for comparison. The FT-IR spectra of the chemically and enzymatically hydrolyzed specimens were collected at intervals of 24 and 12 h, respectively. No significant changes in the spectrum were observed during the chemical degradation, probably because the hydrolyzed chains rapidly diffuse into the KOH solution and rarely remain on the specimen surface; this was further confirmed from the morphology observed by scanning electron microscopy (SEM, Fig. 3a). In contrast to chemical degradation, significant changes in the spectrum were observed during the enzymatic degradation. The intensity of the carbonyl peak (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of PBAF50 at 1720 cm−1 drastically decreased with increasing time of degradation, along with the simultaneous appearance of a new carbonyl peak at around 1662 cm−1 from the hydrolyzed PBAF (Fig. 5a); the intensity of the new peak also increased with degradation time. Also, the peak increase in CC at 1574 cm−1 and the peak increase/shift in C–O–C at 1223 and 1024 cm−1 of the furan ring indicate that the ester groups of FDCA are prone to hydrolysis during the enzymatic degradation. To the best of our knowledge, in the field of biodegradable polymers, this is the first evidence of the participation of FDCA in enzymatic degradation.
O) of PBAF50 at 1720 cm−1 drastically decreased with increasing time of degradation, along with the simultaneous appearance of a new carbonyl peak at around 1662 cm−1 from the hydrolyzed PBAF (Fig. 5a); the intensity of the new peak also increased with degradation time. Also, the peak increase in CC at 1574 cm−1 and the peak increase/shift in C–O–C at 1223 and 1024 cm−1 of the furan ring indicate that the ester groups of FDCA are prone to hydrolysis during the enzymatic degradation. To the best of our knowledge, in the field of biodegradable polymers, this is the first evidence of the participation of FDCA in enzymatic degradation.
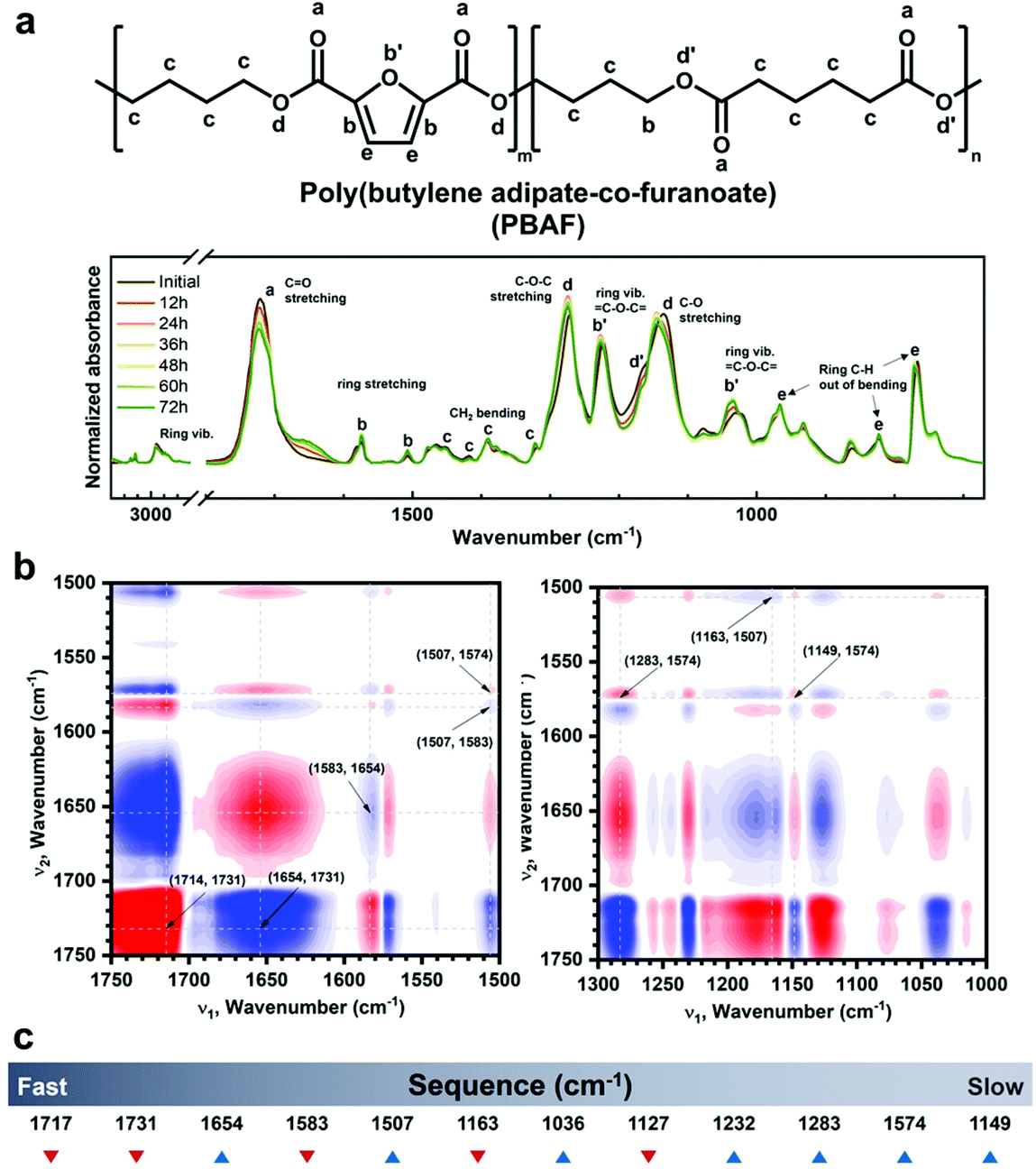 | ||
| Fig. 5 (a) One-dimensional FT-IR spectra of PBAF50 during enzymatic degradation. Two-dimensional correlation spectroscopy (2DCOS) analysis to confirm the degradation sites and to obtain sequential information on degradation. (b) Homogeneous (left) and heterogeneous (right) synchronous 2DCOS spectra. (c) Sequence of peak transition during degradation. | ||
To further investigate the degradation mechanism of PBAF50, the time-dependent sequential changes in the FT-IR peaks were clarified using advanced analytical tools such as 2DCOS. 2DCOS is one of the most powerful and versatile spectral analysis tools that can establish the sequence of spectral changes under a given condition (in our case, the degradation time is a perturbation factor for the spectral change).60 Synchronous and asynchronous 2DCOS obtained from the transient IR spectra of PBAF50 could identify the coincidental and sequential changes in the spectral intensities (Fig. 5b). With the exception of diagonally positive autopeaks (a result of the autocorrelation function of intensity variation) in synchronous 2DCOS, the positive cross peaks (red) imply that the intensities of two bands follow the same trend (either increasing or decreasing), whereas the negative cross peaks (blue) imply that the intensities of two bands follow the opposite trend (one is increasing while the other is decreasing). When designated cross-peaks from the synchronous spectrum match the sign of the asynchronous spectrum, the intensity change on the x-axis occurs before that on the y-axis and vice versa.
Fig. 5b shows the representative 2DCOS of PBAF50 during the enzymatic degradation; the remaining spectra are presented in Fig. S9.† The peak assignments for each matrix are summarized in Table S6.† The overall sequence of the peak transition (Fig. 5c) during the enzymatic degradation of PBAF50 is as follows: 1717↓ > 1731↓ > 1654↑ > 1583↓ > 1507↑ > 1163↓ > 1036↑ > 1127↓ > 1232↑ > 1283↑ > 1574↑ > 1149 cm−1↑. As evident from the initial stages of degradation, carbonyls are separated into 1717, 1731, and 1654 cm−1, which correspond to CO adjacent to the furan ring, CO from the aliphatic segment, and carbonyl from carboxylic acid, respectively. The carboxylic acid is formed because of the ester hydrolysis of FDCA. The interesting aspect of this spectral sequence is that the change in carbonyl bonds near the furan ring triggered the entire degradation of PBAF50. In other words, the participation of the ester groups on the ring triggers the entire hydrolysis, giving rise to enzymatic specificity. Conventionally, the enzymatic degradation of some copolyesters has been exclusively explained by the hydrolysis of the aliphatic region, while ignoring the role of the aromatic phase. However, this study confirms that if the aromatic ring segment can actively participate in degradation owing to a lower Ea value for the conformational change, as in PBAF50, the hydrolysis of ester groups neighboring the ring structure is the first step in the enzymatic degradation.
Conclusions
In summary, it is shown that PBAF has significant merits in terms of elastic performance as well as biodegradability, compared to its terephthalic counterpart, PBAT. Upon uniaxial stretching, PBAT lost its mechanical properties due to permanent deformation, while PBAF exhibited remarkable recovery upon release, ensuring its long-term durability. The advantages of PBAF was further highlighted in its enzymatic degradation with a ring fraction of 50 mol%. In the TLL-induced degradation, PBAF solely underwent hydrolysis, highlighting the enzymatic specificity. Such remarkable elasticity and enzymatic degradability of PBAF can be attributed to the structural characteristics of the furan ring. The rheological analyses confirmed that the asymmetric and semi-rigid FDCA-based PBAF underwent a larger number of chain entanglement than the rigid TPA-based PBAT, ultimately developing stronger elasticity. During the degradation, the ease of the conformational change of the PBAF chains, owing to its low Ea, enabled the hydrolysis of the ester groups neighboring the furan ring, thereby triggering the successive enzymatic degradation. At the current state, PBAF production is incomparable to PBAT since FDCA is in economic disadvantage to TPA in terms of cost, production and technology. The importance of successful petroleum counterparts, however, emphasizes the potential of FDCA to continue its research and become a potential core component in biodegradable plastics.Experimental
Materials
Dimethyl-2,5-furandicarboxylate (DMFD, 99.7%) was purchased from Apollo Scientific (UK). Adipic acid (AA, 99%), dimethyl terephthalate (DMT, 99%), 1,4-butanediol (BD, 99.5%), titanium(IV) butoxide (TBT, 97%), deuterated chloroform (CDCl3, 99.5 atom%), trifluoroacetic acid-d (TFA, 99.5 atom%), high purity chloroform (HPLC grade, CHCl3), and Thermomyces lanuginosus lipase were purchased from Sigma-Aldrich (USA). Sodium phosphate monobasic and dibasic (98.0%–102.0%) were purchased from GENERAY Biotech (China). All the chemicals were used as received without further purification.Synthesis of PBAT and PBAF copolyesters
PBAF and PBAT copolyesters were synthesized by a conventional two-step melt polymerization method. For the synthesis of PBAF50, DMFD (0.183 mol, 33.64 g), AA (0.183 mol, 26.70 g), and BD (0.585 mol, 52.69 g) were injected into a 1 L reactor placed on a heating mantle equipped with a condenser, N2 line, and mechanical stirrer. The reactor was purged with N2 gas to remove H2O and O2, in order to prevent oxidation and depolymerization. The esterification was initiated by heating the mixture to 140 °C, along with mechanical stirring at 150 rpm. When the monomers were fully molten, TBT (0.05 mol% relative to the total molarity of diacids) was added. The temperature was gradually increased to 180 °C at 2 °C min−1 and held for 2 h. Following this, the temperature was increased to 210 °C and held at this temperature for another 2 h. The esterification product was directly transferred to a customized glass reactor equipped with a mechanical stirrer and a vacuum line. The reactor was placed in an oil bath to initiate the polycondensation. Starting at 160 °C with a mechanical stirring of 50 rpm, the temperature was gradually increased to 240 °C at a rate of 2 °C min−1 while the pressure was gradually decreased. The reaction was held for 2 h while maintaining the vacuum at 100 mTorr. The resulting product was quenched in cold water and dried in a vacuum oven at 60 °C for 24 h. All the samples were analyzed without any further purification, and no anti-oxidants and anti-discolorants were used.Characterization
The number-average molecular weight (Mn), weight average molecular weight (Mw), and polydispersity index (PDI = Mn/Mw) were determined using size exclusion chromatography (SEC, ACQUITY APC XT, Waters Corp., USA); the instrument was equipped with a ACQUITY refractive index detector. Chloroform was used as an eluent at a flow rate was 0.6 mL min−1, and the column temperature was fixed at 40 °C. Mn, Mw, and PDI were calculated after calibrating with polystyrene (PS) standards. 1H NMR spectra were obtained on an AVANCE NEO 500 (Bruker, USA) spectrometer at 500 MHz. CDCl3 and tetramethylsilane (TMS) were used as the solvent and internal standard, respectively. Due to the poor solubility of PBAT80 in CDCl3, TFA was used as the solvent. Sixteen scans with 65536 data points were acquired for each 1H NMR spectrum.
The mechanical properties of the synthesized polyesters were measured by a universal testing machine (UTM, Instron 5943, USA). The synthesized copolyesters were hot-pressed at 150–210 °C at 100 bar for 5 min and quenched in cold water. After drying, films (0.5 mm thickness) were cut in the shape of a dumbbell using a cutting die (ASTM D412). All the specimens were conditioned at 25 °C and 20% relative humidity for 48 h; measurements were carried out under the same conditions. The crosshead speed was 100 mm min−1, and each specimen was tested more than 10 times. In order to evaluate the elastic recovery behavior, each dumbbell-shaped specimen was stretched up to 400% using a customized stretching machine and then conditioned at 30 °C for 24 h while maintaining the stretched state. After annealing, the stretched sample was released in an ice water bath for quenching, to minimize the instantaneous recovery. Finally, the extents of elastic recovery of the quenched samples were measured after stress relaxation at 30 or 70 °C for 3 h. For comparing the instant elastic recovery and hysteresis of PBAF50 and PBAT50, the dumbbell-shaped specimens were stretched up to 800% using UTM at a crosshead speed of 100 mm min−1 and then released immediately. Particularly for the highly elastic PBAF50, the cyclic hysteresis test was conducted with 10 cycles of 200%, 400%, and 600% strains. The crosshead speed of each hysteresis test was fixed at 100 mm min−1.
Thermal behaviors of the synthesized copolyesters were investigated by differential scanning calorimetry (TA instrument, Q200, USA) under a N2 atmosphere. The temperature was varied from −50 to 250 °C at a scan rate of 10 °C min−1. The measurements were carried out with 5 mg of each sample. The dynamic mechanical properties of the samples were analyzed using a PerkinElmer DMA 8000 (USA) in the temperature range of −50–150 °C at a heating rate of 3 °C min−1. All the measurements were carried out in tension mode at fixed frequencies of 0.1, 1, 5, and 20 Hz, and at 0.02% strain. The specimens were prepared in the shape of a rectangular bar of dimensions 5 × 0.5 × 10 mm by a hot-pressed method, mentioned in the UTM analysis. The chemical structures of the synthesized and degraded copolyesters were analyzed by FTIR (iS50-FTIR, USA). For each spectrum, 258 scans were collected with a spectral resolution of 4 cm−1. The background of air was subtracted before each measurement. Before analysis, each spectra were smoothed to 5 points and baseline correction was done with 11 points. Then the data were normalized in accordance with PBAT50 and PBAF50 for each series, respectively. For the attenuated total reflection (ATR) measurements, the film or dumbbell-type specimen was placed on the surface of a diamond crystal plate. 2DCOS analysis was conducted using 2DShige program (freely downloadable software developed by Prof. Shigeaki Morita, Osaka Electro-Communication University, Japan). Field-emission SEM was performed on a Tescan MIRA3 (Czech Republic) microscope to view the degradation morphologies of the synthesized copolyesters. Before the analysis, the samples were coated with platinum for fine observation. The rheological properties of PBAT50 and PBAF50 were measured by the dynamic frequency sweep test using a rotational rheometer MCR 302 (Anton Paar, Austria) with a 25 mm parallel plate. The gap distance between the plates and the strain were 1 mm and 10%, respectively. The oscillation frequency sweep was carried out at 220 °C in the angular frequency range of 0.05–500 rad s−1. The disc type specimens were prepared by the hot-press method as mentioned above.
Chemical and enzymatic degradation analysis
All the samples were prepared as 0.2 mm thick films using the hot-press method mentioned in UTM analysis. The specimens were cut in the shape of a dumb-bell using a cutting die (ISO 37) to maintain the same surface area. The weight was fixed at 0.25 ± 0.05 g. Chemical degradation was conducted in aqueous 1 M NaOH solution at pH 13.4. Enzymatic degradation was performed in 0.1 M phosphate buffer of pH 6.5 using lipase from Thermomyces lanuginosus.For each medium, the specimen was placed in a vial (5 ml) and shaken at 50 °C at 80 rpm for 6 days. For the enzymatic degradation, the medium was replenished every day to ensure the maximum enzyme activity. The degradation rate was determined by weight and thickness losses, and the structural transition during the degradation was observed through FT-IR spectroscopy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Korea Research Institute of Chemical Technology (KRICT) Core Project (SS2042-10), Bio-Industrial Technology Development Program (20008628) funded by the Ministry of Trade, Industry & Energy (MI, Korea), the National Research Foundation of Korea (NRF-2020R1C1C1009340, 2020R1C1C1003665) and the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (No. 20194010201840).Notes and references
- R. Siddique, J. Khatib and I. Kaur, Waste Manage., 2008, 28, 1835 CrossRef CAS PubMed.
- S. Wong, N. Ngadi, T. Abdullah and I. Inuwa, Renewable Sustainable Energy Rev., 2015, 50, 1167 CrossRef CAS.
- Y. Zheng, E. K. Yanful and A. S. Bassi, Crit. Rev. Biotechnol., 2005, 25, 243 CrossRef CAS PubMed.
- M. Eriksen, L. C. Lebreton, H. S. Carson, M. Thiel, C. J. Moore, J. C. Borerro, F. Galgani, P. G. Ryan and J. Reisser, PLoS One, 2014, 9, e111913 CrossRef PubMed.
- Y. Chae and Y.-J. An, Environ. Pollut., 2018, 240, 387 CrossRef CAS PubMed.
- S. Curia, A. Biundo, I. Fischer, V. Braunschmid, G. M. Gübitz and J. F. Stanzione III, ChemSusChem, 2018, 11, 2529 CrossRef CAS PubMed.
- S. Lambert and M. Wagner, Chem. Soc. Rev., 2017, 46, 6855 RSC.
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50 CrossRef CAS PubMed.
- Y. Tokiwa and T. Suzuki, Nature, 1977, 270, 76 CrossRef CAS PubMed.
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737 RSC.
- T. Kim, J. M. Koo, M. H. Ryu, H. Jeon, S.-M. Kim, S.-A. Park, D. X. Oh, J. Park and S. Y. Hwang, Polymer, 2017, 132, 122 CrossRef CAS.
- S.-A. Park, H. Jeon, H. Kim, S.-H. Shin, S. Choy, D. S. Hwang, J. M. Koo, J. Jegal, S. Y. Hwang, J. Park and D. X. Oh, Nat. Commun., 2019, 10, 1 CrossRef PubMed.
- J. M. Koo, J. Kang, S.-H. Shin, J. Jegal, H. G. Cha, S. Choy, M. Hakkarainen, J. Park, D. X. Oh and S. Y. Hwang, Compos. Sci. Technol., 2020, 185, 107885 CrossRef CAS.
- S. Rameshkumar, P. Shaiju, K. E. O'Connor and R. Babu, Curr. Opin. Green Sustainable. Chem., 2020, 21, 75 CrossRef.
- R. Herrera, L. Franco, A. Rodríguez-Galán and J. Puiggalí, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4141 CrossRef CAS.
- L. Zhao and Z. Gan, Polym. Degrad. Stab., 2006, 91, 2429 CrossRef CAS.
- F. V. Ferreira, L. S. Cividanes, R. F. Gouveia and L. M. Lona, Polym. Eng. Sci., 2019, 59, E7 CrossRef CAS.
- J. Jian, Z. Xiangbin and H. Xianbo, Adv. Ind. Eng. Polym. Res., 2020, 3, 19 Search PubMed.
- H. Moustafa, C. Guizani, C. Dupont, V. Martin, M. Jeguirim and A. Dufresne, ACS Sustainable Chem. Eng., 2017, 5, 1906 CrossRef CAS.
- A. M. Díez-Pascual and A. L. Díez-Vicente, RSC Adv., 2015, 5, 93095 RSC.
- O. Weizman, A. Dotan, Y. Nir and A. Ophir, Polym. Adv. Technol., 2017, 28, 261 CrossRef CAS.
- T. Kijchavengkul, R. Auras, M. Rubino, M. Ngouajio and R. T. Fernandez, Chemosphere, 2008, 71, 942 CrossRef CAS PubMed.
- F. Touchaleaume, L. Martin-Closas, H. Angellier-Coussy, A. Chevillard, G. Cesar, N. Gontard and E. Gastaldi, Chemosphere, 2016, 144, 433 CrossRef CAS PubMed.
- A. P. Bilck, M. V. Grossmann and F. Yamashita, Polym. Test., 2010, 29, 471 CrossRef CAS.
- K. Fukushima, M.-H. Wu, S. Bocchini, A. Rasyida and M.-C. Yang, Mater. Sci. Eng., C, 2012, 32, 1331 CrossRef CAS PubMed.
- G. F. Santana-Melo, B. V. Rodrigues, E. da Silva, R. Ricci, F. R. Marciano, T. J. Webster, L. M. Vasconcellos and A. O. Lobo, Colloids Surf., B, 2017, 155, 544 CrossRef CAS PubMed.
- A. Arslan, S. Çakmak, A. Cengiz and M. Gümüşderelioğlu, J. Biomater. Sci., Polym. Ed., 2016, 27, 1841 CrossRef CAS PubMed.
- D. Savostianoff and S. Didier, Inf. Chim., 1993, 352, 119 Search PubMed.
- R. Wolkowski-Tyl, T. Chin and H. D. A. Heck, Drug Metab. Dispos., 1982, 10, 486 CAS.
- P. Engel, K. K. Brandt, L. H. Rasmussen, R. G. Ovesen and J. Sørensen, Chemosphere, 2007, 67, 202 CrossRef CAS PubMed.
- S. Qi, X. Wang, X. Xu and Q. Xiang, J. Hyg. Res., 2002, 31, 10 CAS.
- N. Scholz, Chemosphere, 2003, 53, 921 CrossRef CAS.
- B.-J. Chen, Y.-X. Yuan and H.-P. Wang, J. Fish. Sci. China, 2001, 8, 73 CAS.
- A. Eerhart, A. Faaij and M. K. Patel, Energy Environ. Sci., 2012, 5, 6407 RSC.
- G. Z. Papageorgiou, D. G. Papageorgiou, Z. Terzopoulou and D. N. Bikiaris, Eur. Polym. J., 2016, 83, 202 CrossRef CAS.
- A. Sousa, N. Guigo, M. Pożycka, M. Delgado, J. Soares, P. Mendonca, J. Coelho, N. Sbirrazzuoli and A. Silvestre, Polym. Chem., 2018, 9, 722 RSC.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. Freire, G.-J. M. Gruter, J. F. Coelho and A. J. Silvestre, Polym. Chem., 2015, 6, 5961 RSC.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497 RSC.
- R. De Clercq, M. Dusselier and B. Sels, Green Chem., 2017, 19, 5012 RSC.
- S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Macromolecules, 2014, 47, 1383 CrossRef CAS.
- F. A. Kucherov, E. G. Gordeev, A. S. Kashin and V. P. Ananikov, Angew. Chem., Int. Ed., 2017, 56, 15931 CrossRef CAS.
- S. Peng, L. Wu, B.-G. Li and P. Dubois, Polym. Degrad. Stab., 2017, 146, 223 CrossRef CAS.
- S. Weinberger, K. Haernvall, D. Scaini, G. Ghazaryan, M. T. Zumstein, M. Sander, A. Pellis and G. M. Guebitz, Green Chem., 2017, 19, 5381 RSC.
- L. Papadopoulos, A. Magaziotis, M. Nerantzaki, Z. Terzopoulou, G. Z. Papageorgiou and D. N. Bikiaris, Polym. Degrad. Stab., 2018, 156, 32 CrossRef CAS.
- H. Hu, R. Zhang, J. Wang, W. B. Ying and J. Zhu, Eur. Polym. J., 2018, 102, 101 CrossRef CAS.
- K. Haernvall, S. Zitzenbacher, H. Amer, M. T. Zumstein, M. Sander, K. McNeill, M. Yamamoto, M. B. Schick, D. Ribitsch and G. M. Guebitz, Biotechnol. J., 2017, 12, 1600741 CrossRef.
- I. Noda, J. Mol. Struct., 2016, 1124, 29 CrossRef CAS.
- Y. Park, S. Jin, I. Noda and Y. M. Jung, J. Mol. Struct., 2018, 1168, 1 CrossRef CAS.
- H. Moon, S. Choy, Y. Park, Y. M. Jung, J. M. Koo and D. S. Hwang, Mar. Drugs, 2019, 17, 318 CrossRef CAS.
- R. Herrera, L. Franco and A. Rodríguez-Galán, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4141 CrossRef CAS.
- J. C. Randell, Polymer Sequence Determination, Academic Press, New York, 1977 Search PubMed.
- Y. Eom and B. C. Kim, Polymer, 2014, 55, 2570 CrossRef CAS.
- J. H. Yoon, S.-M. Kim, Y. Eom, J. M. Koo, H.-W. Cho, T. H. Lee, K. G. Lee, H. J. Park, Y. K. Kim, H.-J. Yoo, S. Y. Hwang, J. Park and B. G. Choi, ACS Appl. Mater. Interfaces, 2019, 11, 46165 CrossRef CAS.
- F. Kim, B. Kwon, Y. Eom, J. E. Lee, S. Park, S. Jo, S. H. Park, B.-S. Kim, H. J. Im, M. H. Lee, T. S. Min, K. T. Kim, H. G. Chae, W. P. King and J. S. Son, Nat. Energy, 2018, 3, 301 CrossRef CAS.
- R. Fernandez-Lafuente, J. Mol. Catal. B: Enzym., 2010, 62, 197 CrossRef CAS.
- Y. Marcus and R. Fernandez-Lafuente, J. Chem. Phys., 2012, 137, 154501 CrossRef PubMed.
- J.-E. Lee, Y. Eom, Y.-E. Shin, S.-H. Hwang, H.-H. Ko and H. G. Chae, ACS Appl. Mater. Interfaces, 2019, 11, 13665 CrossRef CAS PubMed.
- A. F. Lewis, J. Polym. Sci., Part B: Polym. Lett., 1963, 1, 649 CrossRef.
- S.-A. Park, Y. Eom, H. Jeon, J. M. Koo, E. S. Lee, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Green Chem., 2019, 21, 5212 RSC.
- J.-E. Lee, Y. Eom, Y.-E. Shin, S.-H. Hwang, H.-H. Ko and H. G. Chae, ACS Appl. Mater. Interfaces, 2019, 11, 13665 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and complete characterization data. See DOI: 10.1039/d0gc01688h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |