
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning regioselective oxidation toward phenol via atomically dispersed iron sites on carbon†
Yuxiao
Ding
ab,
Pengfei
Zhang
 b,
Hailong
Xiong
c,
Xiaoyan
Sun
ad,
Alexander
Klyushin
e,
Bingsen
Zhang
f,
Zigeng
Liu
g,
Jinshui
Zhang
b,
Huiyuan
Zhu
b,
Zhen-An
Qiao
*c,
Saskia
Heumann
*a and
Sheng
Dai
*b
b,
Hailong
Xiong
c,
Xiaoyan
Sun
ad,
Alexander
Klyushin
e,
Bingsen
Zhang
f,
Zigeng
Liu
g,
Jinshui
Zhang
b,
Huiyuan
Zhu
b,
Zhen-An
Qiao
*c,
Saskia
Heumann
*a and
Sheng
Dai
*b
aMax Planck Institute for Chemical Energy Conversion, Stiftstrasse 43-36, Mülheim an der Ruhr 45470, Germany. E-mail: saskia.heumann@cec.mpg.de
bChemical Science Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: dais@ornl.gov
cState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, China. E-mail: qiaozhenan@jlu.edu.cn
dInstitut für Chemie, Technische Universität Berlin, Straße des 17. Juni 135, Berlin 10623, Germany
eFritz-Haber-Institut der Max-Planck Gesellschaft, Faradayweg 4-6, Berlin 14195, Germany
fInstitute of Metal Research, Chinese Academy of Sciences, Shenyang 110016, China
gInstitute of Energy and Climate Research, Forschungzentrum Jülich GmbH, Ostring O10, Jülich 52425, Germany
First published on 13th July 2020
Abstract
The development of environmentally benign catalysts for highly regioselective hydroxylation of phenol remains an unsolved challenge in both industry and academia because the electrophilic substitution of phenol simultaneously occurs on both ortho- and para-positions. Herein, we report a designed atomically dispersed iron-based heterogeneous catalyst, in which the iron species is coordinated by a functionalized ionic liquid monolayer on carbon nanotubes. The catalyst exhibits an unprecedented level of regioselectivity (>99%) towards the hydroxylation of phenol and displays a much better activity (TOF towards catechol productivity, 1.79 s−1) compared to the homogeneous free ion system (TOF towards catechol productivity, 0.44 s−1). Both experimental and theoretical investigations confirm that the catalytic oxidation with hydroperoxide undergoes a non-radical addition process and substitutes only the ortho-positions of phenol. This finding provides not only a quite active and selective catalyst for industrially very important reactions, but also a promising methodology of designing biomimetic iron-based heterogeneous catalysts at the atomic level.
Introduction
In industry, dihydroxybenzenes are desirably being produced by the direct hydroxylation of phenol with the environmentally benign oxidant hydrogen peroxide.1,2 The products show both high scientific importance and commercial value due to their applications in pharmaceuticals, polymerization inhibitors, and coatings.3,4 The generally accepted Fenton-like process for the hydroxylation reaction with hydrogen peroxide is initiated by the formation of hydroxyl radicals,5,6 which have no selectivity to attack the aromatic carbon on the phenolic ring. The electrophilic substitution of phenol always occurs in both the ortho- and para-positions of the electron donating hydroxyl group, since they show quite similar electronic density. Subsequently, the products of phenol oxidation are always a mixture of hydroquinone (HQ) and catechol (CAT),7–9 which have to go through an extra much more energy-consuming separation step. In addition, deep oxidation of the products also produces some other byproducts such as benzoquinones.10,11 Exposure to quinones is a toxicological concern because their electrophilic character leads to cellular damage.12 Therefore, designing highly selective catalysts for the hydroxylation of phenol affording only the desirable products is significant but quite challenging.The scarcity of precious metals has led to a quick development of sustainable strategies for using iron-based heterogeneous catalysts owing to their low cost, abundance, ready availability, and very low toxicity.13–15 However, the intense reactions between iron and oxidants often lead to strong self-decomposition of the oxidants and cause a relatively low reaction rate of the desired oxidation process. Additionally, their selectivity during catalytic reactions is generally uncontrollable due to the complexity of the active iron species and the radical mediated reaction pathways.16,17 Despite all these difficulties, ideal iron-based catalysts are still well established in nature. Iron enzymes exhibit very high levels of chemo-, regio-, and stereo-selectivity in hydrocarbon oxidation reactions, inspiring chemists to develop biomimetic iron-based catalysts to achieve remarkable activities toward C–H bond oxidation for decades.18,19 Among different candidates for modifying catalytic sites, ionic liquids (ILs) have drawn significant attention as their properties can be easily tuned by the variation of cations and anions.20–22 The successful cases of coating ionic liquid on carbon materials23,24 also show a promising direction to combine the advantages of both homogeneous and heterogeneous catalysis processes.25–27
Inspired by these ideas, herein, we design atomically dispersed iron ionic sites on carbon nanotubes as heterogeneous catalysts for phenol oxidation to achieve high activity and selectivity. ILs are used to modify the surface properties of the carbon for a better dispersion in aqueous solvent and also provide coordinated sites for the iron ions. The obtained catalyst exhibits better activity than the homogeneous free iron ion system, with turnover frequencies (TOF towards CAT productivity) of 1.79 s−1 and 0.44 s−1, respectively. More surprisingly, the catalyst achieves >99% selectivity to CAT, which is reported for the first time to our knowledge. According to the thorough experimental and theoretical investigations, the catalytic mechanism is confirmed as a non-radical oxidative addition process. In contrast, conventional free iron ion systems show relatively low CAT selectivity, which is caused by the involvement of the uncontrollable radical pathways. The designed catalyst brings some new insights into the understanding of the iron involving oxidation process in the area and provides a promising direction for designing more effective catalysts in industry.
Results and discussion
Design and fabrication of the atomically dispersed iron catalyst
The fabrication process of the catalyst, Fe-based polymerized ionic liquid on carbon nanotubes (Fe-PIL/CNTs), is illustrated in Fig. 1. Specifically, a functionalized IL with an imidazolium cation bearing both acetylacetone and vinyl groups is loaded onto the surface of CNTs via the strong interaction between the π network of the graphitic structure and the cationic ions of the ILs (mainly a static-assisted CH–π hydrogen bond).28 Then the mixture undergoes radical polymerization at 100 °C with 2,2-azobisisobutyronitrile (AIBN) as an initiator29 to form PIL/CNTs. The detailed synthesis protocol is shown in ESI Fig. 1.† The surface PIL provides acetylacetone groups on the CNT surface, which reversibly form negative ion sites in the water phase. With this property, different transition metal ions can be coordinated on the carbon surface by the groups. In the case of iron ions, 80 °C is needed to obtain a stabilized Fe-PIL/CNT, in which the iron ions are singly dispersed on the CNT surface as shown in Fig. 1. According to the design, the iron sites are fully exposed to the reactants when being used as catalysts for catalytic reactions.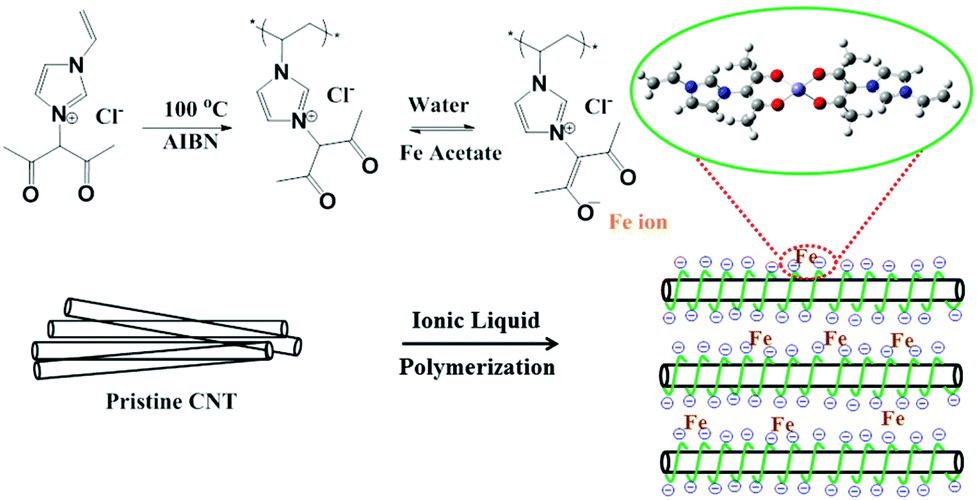 | ||
| Fig. 1 Schematic illustration of the designed catalyst. Schematic illustration of the fabrication process of the Fe-PIL/CNTs. The model structure in the green cycle shows the coordinated sphere of the isolated iron species in the final product. | ||
The morphology of the PIL/CNTs is examined by high-resolution transmission electron microscopy (HRTEM), which presents the monolayer of the tangled PIL on the CNT surface (Fig. 2a). In Fig. 2b, the high-angle annular dark field (HAADF) scanning TEM-energy-dispersive X-ray spectroscopy (STEM-EDS) mapping of the PIL/CNTs shows good accordance with the nitrogen and oxygen distribution in the PIL throughout the carbon framework. Scanning electron microscopy (SEM, Fig. 2c and d) images show the morphologies of pristine CNTs and PIL/CNTs, in which the CNTs are severely tangled together while the PIL/CNTs show a good dispersion. It is worth mentioning that with the involvement of the π–π interaction, CNTs are always tangled together as bundles.30 The good dispersion of the PIL/CNTs suggests that the dielectric constants of the ILs can prevent the detached CNTs from rebundling, which is favourable for their contact with the reactants during reactions, and it also demonstrates that the PILs are well covered on the CNT surface. Furthermore, an obvious Tyndall effect of the PIL/CNTs in water also proves that the surface of the CNTs is finely covered by the PIL (Fig. 2e), while in the case of the pure CNT system (ESI Fig. 4†), the dispersion of the CNTs barely occurs in water. The hydrophilic functional groups of the PIL help to suspend the CNTs in water, which is favourable for applications in the aqueous phase. The IL thickness on the CNT surface can be controlled by the IL content.31
 | ||
| Fig. 2 (a) HRTEM image of the PIL/CNTs (red arrows point to the PIL layer). (b) HAADF STEM-EDS mapping of C, N, and O, as labelled, measured in the white box of the PIL/CNTs. SEM images of the (c) pristine CNT and (d) PIL/CNTs. (e) Photograph images of PIL/CNT dispersions in water (the weight of the sample is 1 mg). (f) The STEM image of the sample, which shows the distribution of the iron sites on the CNT surface. | ||
The STEM image in Fig. 2f exhibits an atomical distribution of the iron species (white spots) on the CNT surface. A small amount of iron species aggregates to clusters, but still exposes all the sites to the surface. No crystalline iron species are detected from both TEM and X-ray powder diffraction (ESI Fig. 5†), which is in agreement with the schematic illustration of the designed catalyst shown in Fig. 1. Based on the above characterization, it can be concluded that the designed catalyst is successfully synthesized. In a catalytic process, the reactions occur in the PIL phase, through which the catalysis micro-environment can be well adjusted by changing the cations and anions. Considering the diversity of ILs and carbon materials,32 the method can therefore be used for designing totally different materials for different functions.
Structural and surface properties of the catalyst
Since the CNTs have closed ends, the pore size of the CNTs is maintained (ESI Fig. 2†) and the IL can only wrap the outer surface of the CNTs. The surface area determined by nitrogen adsorption and BET analysis of the CNTs is 265 m2 g−1 (ESI Fig. 3 and Table S1†) and the theoretical surface area of the PIL/CNTs (with one layer of PIL) is 195 m2 g−1 (ESI†). The measured surface area of the PIL/CNTs is 219.8 m2 g−1, indicating a loss of 23% of the IL during the synthesis. It also means that the CNT surface is fully covered and some graphitic structure is still exposed. X-ray photoelectron spectroscopy (XPS) and elemental analysis provided surface and bulk elemental information of the PIL/CNTs (ESI Tables S2 and S3†). Thermal analysis reveals thermal stabilities of the different samples (Fig. 3a). The PIL on the CNT decomposes (when the TG values start to show a decrease of 1%) at 200 °C, while the pure CNT decomposes at 536 °C. The nice combination between the CNT and the IL leads to a lower CNT combustion temperature. For the Fe-PIL/CNT sample, iron catalyzes the thermal decomposition, leading to a much lower combustion temperature. The residue of 6.1 wt% could be a mixture of ferroferric oxide and the ash content of the CNT (1.9%), from which an iron content of around 3% in the Fe-PIL/CNTs can be obtained (ESI†). This fits well with the elemental analysis data of 3.8% Fe content. In addition, the samples containing PIL show the same decomposition temperature in inert gas (ESI Fig. 6†) as in air, which means that the PIL is stable under oxidative conditions at temperatures lower than 200 °C. | ||
| Fig. 3 Properties of the samples. (a) Thermogravimetric curves (gas: air, 20 mL min−1; argon, 10 mL min−1) of the CNTs, PIL/CNTs and Fe-PIL/CNTs. (b) ATR spectra of different samples. The difference spectra were obtained by subtraction of the CNT background spectrum. (c) EELS results of the N and O K edge of the Fe-PIL/CNTs. (d) O 1s XPS spectra and distribution diagrams of different species from the PIL/CNTs and Fe-PIL/CNTs. Images of water contact angle measurements on (e) CNTs and (f) Fe-PIL/CNTs. | ||
Raman spectra (ESI Fig. 7†) show no obvious change in the peak shape and position in the PIL/CNT spectrum compared with the CNTs, revealing that the polymerization process might not form chemical bonds with the CNTs.28 Since attenuated total reflectance (ATR) spectra (Fig. 3b) can directly monitor the bond vibrations, the differential ATR spectra of the prepared samples obtained by subtracting the CNT background are used to reflect the evolution of the bonds. The bonds of the PIL/CNTs in the 1600–1780 cm−1 region correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the acetylacetone group. Accordingly, the introduction of the Fe ions causes a dramatic decrease in the CO bond while a relative C–O– bond at 1014 cm−1 increases.33,34 The electron energy loss spectra (EELS) of the PIL/CNTs (Fig. 3c) confirm the incorporation of nitrogen and oxygen atoms in the carbon-based materials as the original forms. The acetylacetone groups on the PIL/CNTs provide versatile coordinating sites for iron ions to form the complex of Fe-PIL/CNTs. The deconvolution results of O 1s reveal the alteration of the oxygen in the complexing process (Fig. 3d). The main tendency is that a CO (36.3% and 36.9%) bond transforms into a C–O bond (41.2% and 33.5%),35 which is consistent with the ATR results. Moreover, the contact angle measurements (Fig. 3e–f) with water show that the contact angle reduces from 152.7° for the CNTs to 24.1° for the Fe-PIL/CNTs, indicating that the introduction of the acetylacetone groups makes the super hydrophobic carbon surface highly hydrophilic, which is favorable for application with solid–water interfaces. This explains the good dispersion of the PIL/CNT sample in water and demonstrates the introduction of the iron sites having no obvious influence on the coverage of the PIL on the CNT surface. NMR spectra of different samples also demonstrate the coating (ESI Fig. 8†). Due to the influence of the CNT signal, only broad peaks from the carbon support can be observed with 13C spectra. However, the 1H spectra can confirm the coating, from which the coordination of the iron with the OH group can also be confirmed.
O stretching vibration of the acetylacetone group. Accordingly, the introduction of the Fe ions causes a dramatic decrease in the CO bond while a relative C–O– bond at 1014 cm−1 increases.33,34 The electron energy loss spectra (EELS) of the PIL/CNTs (Fig. 3c) confirm the incorporation of nitrogen and oxygen atoms in the carbon-based materials as the original forms. The acetylacetone groups on the PIL/CNTs provide versatile coordinating sites for iron ions to form the complex of Fe-PIL/CNTs. The deconvolution results of O 1s reveal the alteration of the oxygen in the complexing process (Fig. 3d). The main tendency is that a CO (36.3% and 36.9%) bond transforms into a C–O bond (41.2% and 33.5%),35 which is consistent with the ATR results. Moreover, the contact angle measurements (Fig. 3e–f) with water show that the contact angle reduces from 152.7° for the CNTs to 24.1° for the Fe-PIL/CNTs, indicating that the introduction of the acetylacetone groups makes the super hydrophobic carbon surface highly hydrophilic, which is favorable for application with solid–water interfaces. This explains the good dispersion of the PIL/CNT sample in water and demonstrates the introduction of the iron sites having no obvious influence on the coverage of the PIL on the CNT surface. NMR spectra of different samples also demonstrate the coating (ESI Fig. 8†). Due to the influence of the CNT signal, only broad peaks from the carbon support can be observed with 13C spectra. However, the 1H spectra can confirm the coating, from which the coordination of the iron with the OH group can also be confirmed.
Catalytic performance of the catalyst
The selective oxidation of organic compounds is a primary and essential transformation in organic synthesis and industrial chemistry. The Fe-PIL/CNTs with multiunique characteristics in principle could afford some exceptional performance, and therefore oxidation of phenol by hydrogen peroxide in the aqueous phase is studied over the catalyst. Table 1 shows the activities of different samples for phenol oxidation. Without a catalyst, no oxidation of phenol occurs. When CNTs and PIL/CNTs are used as catalysts, the conversion of phenol is still negligible. For the Fe-PIL/CNTs, the selectivity of the CAT could reach >99% and the TOF is determined as 1.79 s−1. However, generally in this reaction, the hydroquinone is always unavoidable and this has been widely accepted since a long time ago.1 The unprecedentedly high selectivity is obviously from some pathways different from that on previous catalysts. The ratio of the obtained catechol and the used H2O2 is around 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. As not all H2O2 is consumed, the efficiency of the hydrogen peroxide is therefore higher than 33.3%. For further comparison, commercial Fe(CH3COO)2 was chosen as the catalyst to convert phenol under identical conditions. Although displayed as a homogeneous catalyst, the catalytic activity of the free ion system (TOF towards CAT, 0.44 s−1) is even lower than that of the Fe-PIL/CNTs and shows lower selectivity towards CAT (55.6%). The reason for the excellent catalytic performance will be discussed in the following part. As the catalyst and the reactant are not in the same phases, the catalyst can be separated easily with a centrifugation treatment. This is one of the most important advantages of the heterogeneous catalysis system. In a reuse process, the catalyst was centrifuged out firstly. Then the obtained catalyst was washed with ethanol twice to remove the phenol residue from the catalyst and dried for reuse. Due to the loss from the transfer process, the catalyst shows a slight decrease in the conversion of the phenol to 24.4% (the second cycle) and 22.5% (the third cycle). No iron leaching was detected during the recycling procedures. The selectivity to the final product is still >99%. This proves the good cycling ability of the heterogeneous catalyst.
:3. As not all H2O2 is consumed, the efficiency of the hydrogen peroxide is therefore higher than 33.3%. For further comparison, commercial Fe(CH3COO)2 was chosen as the catalyst to convert phenol under identical conditions. Although displayed as a homogeneous catalyst, the catalytic activity of the free ion system (TOF towards CAT, 0.44 s−1) is even lower than that of the Fe-PIL/CNTs and shows lower selectivity towards CAT (55.6%). The reason for the excellent catalytic performance will be discussed in the following part. As the catalyst and the reactant are not in the same phases, the catalyst can be separated easily with a centrifugation treatment. This is one of the most important advantages of the heterogeneous catalysis system. In a reuse process, the catalyst was centrifuged out firstly. Then the obtained catalyst was washed with ethanol twice to remove the phenol residue from the catalyst and dried for reuse. Due to the loss from the transfer process, the catalyst shows a slight decrease in the conversion of the phenol to 24.4% (the second cycle) and 22.5% (the third cycle). No iron leaching was detected during the recycling procedures. The selectivity to the final product is still >99%. This proves the good cycling ability of the heterogeneous catalyst.
| Catalyst | Con. (%) | Sel. (%) | TOF (s−1) |
|---|---|---|---|
| Experimental conditions: T = 60 °C, m (phenol) = 9.4 g, V (H2O) = 10 mL, V (H2O2) = 10 mL, t = 60 min, m (catalyst) = 10 mg (details in the ESI†). The selectivity indicates the selectivity of catechol; the other product is hydroquinone. * and ** represent the second- and third-time recycling data of the sample. | |||
| None | 0 | — | — |
| CNT | 0.8 | 29.4 | — |
| PIL/CNT | 0.6 | 25.8 | — |
| Fe-PIL/CNT | 25.7 | >99 | 1.79 |
| Fe-PIL/CNT* | 24.4 | >99 | 1.70 |
| Fe-PIL/CNT** | 22.5 | >99 | 1.56 |
| Fe(CH3COO)2 | 34.5 | 55.6 | 0.44 |
The investigation of the mechanism
Ferric-hydroperoxo complexes have been identified as intermediates in the catalytic cycle of biological oxidants with a following radical formation process.36,37 To shed more light on the catalytic process with such a high selectivity towards CAT, electron paramagnetic resonance (EPR) spectroscopy is used to study the intermediates. Interestingly, Fe(CH3COO)2 and Fe-PIL/CNTs show almost identical EPR peaks before the reaction (Fig. 4a), which is in agreement with the Mössbauer spectra (ESI Fig. 9†). After adding hydroperoxide, a radical peak arises in the Fe(CH3COO)2 spectrum, while the Fe-PIL/CNTs show no radical peak at all (Fig. 4b). This indicates that the iron species in the Fe-PIL/CNT sample exhibit a non-radical process upon reaction with H2O2, which seldom occurs in an iron–H2O2 system. This suggests that the coordination environment of the Fe-PIL/CNTs might stabilize the formed ferric-hydroperoxo from deep oxidation and might provide a chance for regioselective oxidation towards phenol. H2O2 generally undergoes a fast self-decomposition process to form oxygen when being exposed to heat or a metal-based catalyst. In an oxidation reaction, the self-decomposition of the oxidant ruins the oxidation process but occurs at the same time as a competition reaction. In Fig. 4c, we compared the decomposition rate of the oxidant when being exposed to free iron ions (iron acetate) and the Fe-PIL/CNTs. After a short overlap of the curves, the decomposition rate in the Fe-PIL/CNT system decreased after 6 min, which indicates that the coordinated iron with ionic liquid is able to bond the hydrogen peroxide and protect it from high decomposition after an initial step; while in the free ion system, the decomposition rate shows a sudden increase after the 12 minutes initial step, which should be followed by a Fenton-like process to continuously form oxygen. The results confirm that the coordinated iron compared to the free iron ion has better ability to preserve the formed iron peroxides during the oxidation process, which is in good accordance with the non-radical reaction pathway.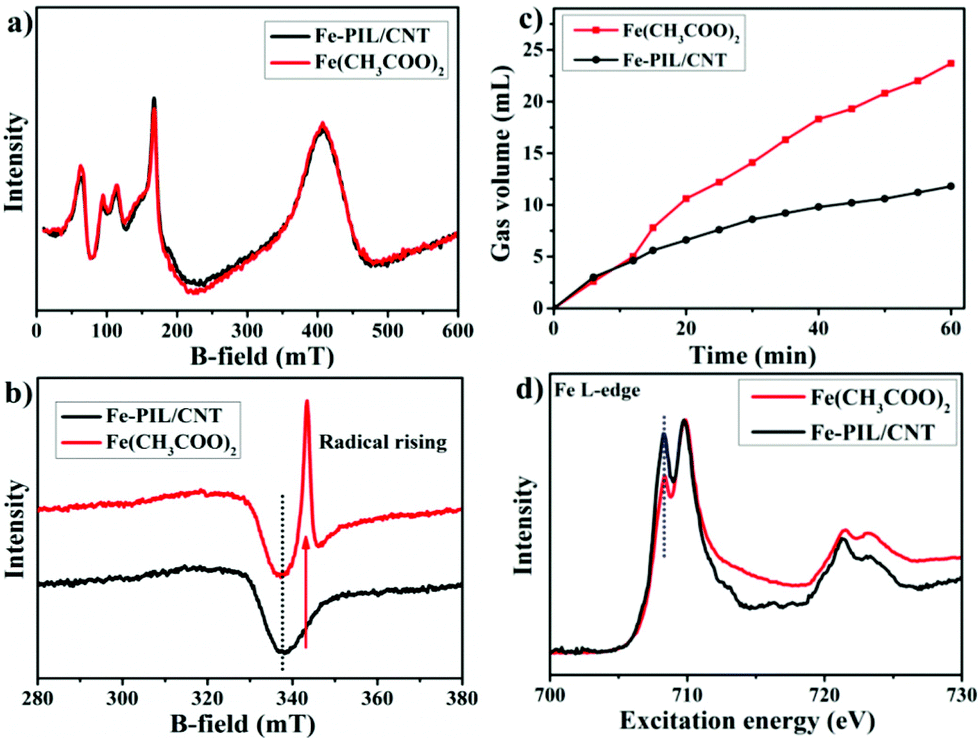 | ||
| Fig. 4 Catalytic behaviour comparison between the free ion system and the Fe-PIL/CNT system. (a) EPR curves of Fe(CH3COO)2 and Fe-PIL/CNTs and (b) with the addition of H2O2. (c) Gas accumulation via the oxidant decomposition with the addition of the Fe-PIL/CNTs and Fe(CH3COO)2. Conditions: water (3 mL), 30% hydrogen peroxide solution (2 mL) and 18 mg solid sample. (d) Fe L-edge NEXAFS spectra of the Fe-PIL/CNTs and Fe(CH3COO)2. | ||
The Fe L-edge NEXAFS results of the Fe-PIL/CNTs and Fe(CH3COO)2 (Fig. 4d) shows that the Fe configuration changed from tetrahedral to octahedral (the dashed line position)38 after the synthesis process. The oxidation state of the iron species remains the same from NEXAFS results, which can also be confirmed by the Mössbauer spectra of the two samples. As the Fe2+ ions are inevitably being oxidized, the mixture of Fe2+/Fe3+ exists in both the precursor and the Fe-PIL/CNTs. Due to the protection of the ligand, the octahedral configuration exposes less electron density to the oxidant than the tetrahedral configuration, which might be the reason why the Fe-PIL/CNTs are able to protect the oxidant from self-decomposition. In contrast, from the comparison of the Fe 2p spectra of the two samples (ESI Fig. 10†), the PIL lowers the binding energy of the Fe 2p electron, making it easier to react with the oxidizing agent and to form active intermediates for the reaction.
Density functional theory (DFT) calculations are performed to reveal the possible reaction mechanism. The electronic structure of the ionic coordinated iron species was confirmed by theoretical calculation, indicating the presence of a highly electrophilic Fe center site (ESI Fig. 11†). Therefore, the formation of the ferric-hydroperoxo is quite thermodynamically favourable by 51 kcal mol−1 and can occur directly when the iron species come into contact with H2O2 (Fig. 5a). As shown in Fig. 5b, the approach of phenol at the hydroperoxo group of the [(L2)FeIII(OOH)]2+ complex can lead to the direct oxidative addition of an OH group on the ortho-carbon atom via a transition state, TS1. From intermediate I1, the hydrogen atom transfer can occur to give rise to intermediate I3, in which two OH groups are coordinated to the Fe center. Subsequently, by overcoming a small transition state barrier TS4, the product complex PC with a CAT unit is finally generated. In addition, several spin-crossings between the ground sextet state and excited quartet state could occur along the reaction pathway as they are similar in energy. It is important to note that this direct oxidative addition mechanism is inaccessible for forming a para-isomer, which, therefore, explains the observed selective hydroxylation of phenol towards CAT. Furthermore, the CNTs as support materials may also play a promotion role in the reaction, which is not considered in the calculation.39 For a better understanding of the mechanism, a simple schematic catalytic cycle was also presented in ESI Fig. 12.†
 | ||
| Fig. 5 Reaction pathways by theoretical calculations. (a) The thermodynamics of the reaction between ionic liquid-coordinated iron species and the deprotonated peroxide. (b) DFT calculated potential energy surface and selected structural information for the reaction between the iron(III)–hydroperoxo complex and hydroxybenzene. For the sake of clarity, the complete structure of ligands is not shown. Details for all transition states are available in ESI Fig. 13.† Bond lengths are given in Å. The zero-point corrected energies are given in kcal mol−1. | ||
Based on the above results, the high activity of the Fe-PIL/CNTs is due to the following reasons: (i) metal ions are highly dispersed in the reaction system when the reaction occurred, which allows a homogeneous-like catalytic reaction to occur on the heterogeneous sites; (ii) the transformation of the configuration from tetrahedral to octahedral (Fig. 4d) contributes to the protection of the oxidant decomposition (Fig. 4c), thus improving the oxidation efficiency; the PILs lower the binding energy of the Fe 2p electron (ESI Fig. 10†), making it easier to react with the oxidizing agent to form active intermediates; and (iii) the PIL/CNT possesses great capability to adsorb phenol; the CNT, as a support for the heterogeneous catalytic system, not only provides a large interface for the reaction, but also provides a π network for adsorbing the reactant, which enriches the concentration for facilitating the reaction. The high selectivity is caused by the non-radical reaction pathway (Fig. 4b), which can be confirmed by the DFT results (Fig. 5). The high reaction rate is also favorable for the high selectivity as the competing reactions are relatively restricted. Compared to the homogeneously distributed free ion system, the Fe-PIL/CNT system shows superiority during the recycling process.
Conclusions
In summary, isolated dispersed iron species are bridged on the CNT by an acetylacetone functionalized IL polymer and exhibit surprisingly high activity and regioselectivity to phenol oxidation. The tangled PIL on the CNTs stabilizes the iron species on the carbon surface, shields the detached CNTs from rebundling in the aqueous phase and influences the reaction by providing a proper coordinated environment for the iron sites. These unique characteristics lead to higher activity of the Fe-PIL/CNTs than the homogeneous system. Moreover, the ferric–hydroperoxo complex formed during the reaction is confirmed to undergo a non-radical process with >99% selectivity of CAT when oxidizing phenol. The current work has brought some new perspectives in understanding the role of iron species in selective oxidation, and our insights into the correlation of the catalytic behaviors of iron species with the coordinated environment will advance the future design of highly selective and efficient iron-based heterogeneous catalysts. In addition, ILs have been identified as “designable solvents”, which is promising to tune carbons with different properties for different catalytic processes.Methods
Characterization
The STEM image in Fig. 1 was taken on a Hitachi FE-S-TEM system at 200 kV. The other TEM images and EELS were observed on a Tecnai G2 F20 S-TWIN operated at 200 kV and a Philips CM200 FEG operated at 200 kV. Thermogravimetry was performed on a NETZSCH STA 449 F3. The NEXAFS experiments were performed at BESSY II of the Helmholtz Zentrum Berlin. The NEXAFS spectra were obtained by recording the total electron yield (TEY). The energy resolution of the monochromator in the range of the Fe L-edge was 0.3 eV. The energies were calibrated using the C 1s first and second order peaks. The accuracy of energy calibration was estimated to be around 0.1 eV. XPS characterization was performed on an ESCALAB 250 instrument with Al Kα X-rays (1489.6 eV). ATR-infrared spectroscopy was performed on a Thermo Scientific Nicolet IS10 Fourier transform infrared spectrometer. N2 physisorption was measured at −196 °C using a Micrometrics ASAP 2020 instrument. Raman spectroscopy was conducted with a LabRam high-resolution 800 spectrometer using 633 nm excitation at 25 mW laser power with a 2.5 cm−1 resolution. X ray diffraction measurements were performed on a D/max 2400 diffractometer (JEOL Ltd, Japan) at a scanning rate of 1° min−1, with graphite monochromatized Cu Kα radiation (l = 0.1506 nm).Catalyst preparation and evaluation
The stability of the PIL/CNTs is of great importance for their application in catalysis. The results from gravimetric determination of the PIL content after 5, 10 and 15 cycles of washing with ethanol were 11.9%, 12.4% and 12.3%, respectively, which means the PIL remained steadily on the CNT surfaces even in the liquid phase.
DFT calculations
All theoretical work was performed using the Gaussian 09 program.40 The BP86 functional,41,42 including DFT-D3 dispersion correction,43,44 was used for the structural optimization and frequency analysis. The reliability of this functional in calculating iron-containing systems has been demonstrated previously.19,45,46 The double-ζ type LANL2DZ basis set47 was employed for iron, and 6-31G(d)48 was used for the remaining atoms, basis set BSI. Our model uses an iron(III)–hydroperoxo complex with chelating ligands L2 (Fig. 4). The geometry of [(L2)FeIII(OOH)]2+ was initially optimized without constraints. The doublet potential energy surfaces have also been calculated whereas they are relatively higher in energy than the sextet and quartet states, which are not considered in Fig. 4.The energy is further refined with a second set of calculations using a triple-ξ LANL2TZ(f) basis set49 on iron and 6-311G(d)50 on the remaining atoms, basis set BSII. Solvent corrections with water were obtained through a single point calculation using the solvation model based on density (SMD)51 and with dispersion corrected BP86. Intrinsic reaction coordinate (IRC)52–54 calculations were performed to link transition structures with the respective intermediates. Unscaled vibrational frequencies were used to correct the relative energies for zero-point vibrational energy (ZPVE) contributions. Natural bond orbital (NBO)55–60 calculations were performed to obtain further information on selected stationary points along the reaction paths.
Author contributions
Y. D. conceived the research, and carried out material treatment and basic characterization. P. Z. carried out catalytic tests. X. S. revealed the mechanism and the related calculation part. A. K. performed and analyzed NEAFS and XPS. B. Z. contributed to the TEM related characterization. Z. L. contributed to EPR analysis. H. X., H. Z. and Z. Q. contributed to the other characterization. S. D. supervised the project. Y. D. wrote the manuscript with input from J. Z., Z. Q. and S. H. All authors contributed to data analysis and manuscript revision.Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
The authors acknowledge the funding support from the Max Planck Society. Zhen-An Qiao was supported by the National Natural Science Foundation of China (21671073 and 21621001), the “111” Project of the Ministry of Education of China (B17020), and the Program for JLU Science and Technology Innovative Research Team. Prof. Dr Shengfa Ye is thanked for the fruitful discussion. The authors thank Prof. Dr Robert Schlögl for the useful suggestions. Ding thanks Prof. Dr Dang Sheng Su for his support in the manuscript preparation and guidance in his scientific career. Open Access funding was provided by the Max Planck Society.References
- H. Fiege, H. Voges, T. Hamamoto, S. Umemura, T. Iwata, H. Miki, Y. Fujita, H. Buysch, D. Garbe and W. Paulus, in Ullmann's Encyclopedia of Industrial Chemistry, 2000, DOI:10.1002/14356007.a19_313.
- C. Perego, A. Carati, P. Ingallina, M. A. Mantegazza and G. Bellussi, Appl. Catal., A, 2001, 221, 63–72 CrossRef CAS.
- Kirk-Othmer, in Encyclopedia of Chemical Technology, ed. Kirk-Othmer, 2005, vol. 13 Search PubMed.
- J. Saiz-Poseu, J. Mancebo-Aracil, F. Nador, F. Busqué and D. Ruiz-Molina, Angew. Chem., Int. Ed., 2019, 58, 696–714 CrossRef CAS PubMed.
- E. Brillas, I. Sirés and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed.
- Z. Tang, Y. Liu, M. He and W. Bu, Angew. Chem., Int. Ed., 2019, 58, 946–956 CrossRef CAS PubMed.
- F. Shi, L. Mu, P. Yu, J. Hu and L. Zhang, J. Mol. Catal. A: Chem., 2014, 391, 66–73 CrossRef CAS.
- S. Song, W. Zhao, L. Wang, J. Chu, J. Qu, S. Li, L. Wang and T. Qi, J. Colloid Interface Sci., 2011, 354, 686–690 CrossRef CAS PubMed.
- M. G. Clerici, in Metal Oxide Catalysis, 2009, pp. 705–754, DOI:10.1002/9783527626113.ch18.
- C. Prasse, B. Ford, D. K. Nomura and D. L. Sedlak, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2311–2316 CrossRef CAS PubMed.
- D. Schilter, Nat. Rev. Chem., 2018, 2, 0129 CrossRef CAS.
- J. L. Bolton, M. A. Trush, T. M. Penning, G. Dryhurst and T. J. Monks, Chem. Res. Toxicol., 2000, 13, 135–160 Search PubMed.
- G. Jaiswal, V. G. Landge, D. Jagadeesan and E. Balaraman, Nat. Commun., 2017, 8, 2147 Search PubMed.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS PubMed.
- D. Wei and C. Darcel, Chem. Rev., 2019, 119, 2550–2610 CrossRef CAS PubMed.
- W. P. Liu, W. Li, A. Spannenberg, K. Junge and M. Beller, Nat. Catal., 2019, 2, 523–528 CrossRef CAS.
- R. A. Watile, A. Bunrit, J. Margalef, S. Akkarasamiyo, R. Ayub, E. Lagerspets, S. Biswas, T. Repo and J. S. M. Samec, Nat. Commun., 2019, 10, 3826 CrossRef PubMed.
- M. Borrell, E. Andris, R. Navratil, J. Roithova and M. Costas, Nat. Commun., 2019, 10, 901 CrossRef PubMed.
- B. Mondal, F. Neese, E. Bill and S. F. Ye, J. Am. Chem. Soc., 2018, 140, 9531–9544 CrossRef CAS PubMed.
- J. D. Scholten, B. C. Leal and J. Dupont, ACS Catal., 2012, 2, 184–200 CrossRef CAS.
- H. P. Steinruck and P. Wasserscheid, Catal. Lett., 2015, 145, 380–397 CrossRef.
- Q. H. Zhang, S. G. Zhang and Y. Q. Deng, Green Chem., 2011, 13, 2619–2637 RSC.
- M. Tunckol, J. Durand and P. Serp, Carbon, 2012, 50, 4303–4334 CrossRef CAS.
- Y. X. Ding, A. Klyushin, X. Huang, T. Jones, D. Teschner, F. Girgsdies, T. Rodenas, R. Schlogl and S. Heumann, Angew. Chem., Int. Ed., 2018, 57, 3514–3518 CrossRef CAS PubMed.
- Y. X. Ding, B. S. Zhang, N. Gupta and D. S. Su, Green Chem., 2015, 17, 1107–1112 RSC.
- B. H. Wu, D. Hu, Y. J. Kuang, B. Liu, X. H. Zhang and J. H. Chen, Angew. Chem., Int. Ed., 2009, 48, 4751–4754 CrossRef CAS PubMed.
- M. Qiao, C. Tang, L. C. Tanase, C. M. Teodorescu, C. M. Chen, Q. Zhang and M. M. Titirici, Mater. Horiz., 2017, 4, 895–899 RSC.
- Y. X. Ding and D. S. Su, ChemSusChem, 2014, 7, 1542–1546 CrossRef CAS PubMed.
- M. Choi and R. Ryoo, Nat. Mater., 2003, 2, 473–476 CrossRef CAS PubMed.
- T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N. Ishii and T. Aida, Science, 2003, 300, 2072–2074 CrossRef CAS PubMed.
- F. Heym, C. Kern, J. Thiessen and A. Jess, in Supported Ionic Liquids, ed. R. Fehrmann, A. Riisager and M. Haumann, 2014, pp. 95–104 Search PubMed.
- J. Liu, T. Yang, D.-W. Wang, G. Q. Lu, D. Zhao and S. Z. Qiao, Nat. Commun., 2013, 4, 2798 CrossRef.
- E. Fuente, J. A. Menéndez, M. A. Díez, D. Suárez and M. A. Montes-Morán, J. Phys. Chem. B, 2003, 107, 6350–6359 CrossRef CAS.
- M. Smith, L. Scudiero, J. Espinal, J.-S. McEwen and M. Garcia-Perez, Carbon, 2016, 110, 155–171 CrossRef CAS.
- G. Levi, O. Senneca, M. Causà, P. Salatino, P. Lacovig and S. Lizzit, Carbon, 2015, 90, 181–196 CrossRef CAS.
- A. Thibon, V. Jollet, C. Ribal, K. Sénéchal-David, L. Billon, A. B. Sorokin and F. Banse, Chem. – Eur. J., 2012, 18, 2715–2724 CrossRef CAS PubMed.
- A. S. Faponle, M. G. Quesne, C. V. Sastri, F. Banse and S. P. de Visser, Chem. – Eur. J., 2015, 21, 1221–1236 CrossRef CAS PubMed.
- O. Lobacheva, M. Chavarha, Y.-M. Yiu, T. K. Sham and L. Goncharova, J. Appl. Phys., 2014, 116, 013901–013901 CrossRef.
- Q. L. Zhu and Q. Xu, Chem, 2016, 1, 220–245 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- C. Loschen and G. Frenking, Inorg. Chem., 2004, 43, 778–784 CrossRef CAS PubMed.
- S. F. Ye, T. Tuttle, E. Bill, L. Simkhovich, Z. Gross, W. Thiel and F. Neese, Chem. – Eur. J., 2008, 14, 10839–10851 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS.
- L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS PubMed.
- M. P. Andersson and P. Uvdal, J. Phys. Chem. A, 2005, 109, 2937–2941 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61–69 CrossRef CAS PubMed.
- D. G. Truhlar, N. J. Kilpatrick and B. C. Garrett, J. Chem. Phys., 1983, 78, 2438–2442 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- J. E. Carpenter and F. Weinhold, J. Mol. Struct.: THEOCHEM, 1988, 46, 41–62 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1985, 83, 1736–1740 CrossRef CAS.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef CAS.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc01717e |
| This journal is © The Royal Society of Chemistry 2020 |