
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards an environmentally and economically sustainable biorefinery: heavy metal contaminated waste wood as a low-cost feedstock in a low-cost ionic liquid process†
Florence J. V.
Gschwend
,
Louis M.
Hennequin
,
Agnieszka
Brandt-Talbot
 ,
Franky
Bedoya-Lora‡
,
Geoffrey H.
Kelsall
,
Karen
Polizzi
,
Paul S.
Fennell
* and
Jason P.
Hallett
*
,
Franky
Bedoya-Lora‡
,
Geoffrey H.
Kelsall
,
Karen
Polizzi
,
Paul S.
Fennell
* and
Jason P.
Hallett
*
Department of Chemical Engineering, Imperial College London, Exhibition Road, London, SW7 2AZ, UK. E-mail: P.fennell@imperial.ac.uk; j.hallett@imperial.ac.uk
First published on 20th May 2020
Abstract
In the present study, we used a low-cost protic ionic liquid, 1-methylimidazolium chloride, to simultaneously fractionate heavy metal contaminated wood and extract the metals from the wood at elevated temperature and short reaction time. This treatment selectively dissolves the lignin and hemicellulose in the biomass, leaving a solid cellulose-rich pulp, while coordinating and extracting 80–100% of the metal species present in the wood in a one-pot process. The lignin stream was recovered from the liquor and the cellulose was hydrolysed and then fermented into ethanol. The ionic liquid was recycled 6 times and the metals were recovered from the liquor via electrodeposition. This is the first time that highly contaminated waste wood has been integrated into a process which does not produce a contaminated waste stream, but instead valorises the wood as a feedstock for renewable chemicals, materials and fuels, while efficiently recovering the metals, converting a toxic environmental hazard into a rich source of biorenewables. We have therefore used an otherwise problematic waste as a low-cost lignocellulsoic feedstock for a circular bioeconomy concept.
Introduction
The production of renewable fuels and chemicals from lignocellulosic biomass in a biorefinery presents an attractive alternative to the fossil-based fuels, chemicals and materials upon which we currently rely. Despite their conceptual advantages, biorefineries unfortunately suffer economically when compared to petrochemical refining, due in part to very high feedstock costs and difficulties with feedstock acquisition at large scale. Furthermore, various factors, including direct and indirect land-use change and fertilizer use have to be considered if an environmental benefit compared to petrochemicals and fuels is to be achieved.1,2 Indeed, a 2008 study published in Science found that land-use change was the single largest contributing factor to GHG emissions from corn and cellulosic ethanol, causing GHG emissions to increase by 93 and 50% versus petrol for corn and cellulosic ethanol, respectively.1 Without land-use change, however, GHG emissions from cellulosic ethanol are lowered by 70% versus regular petrol, according to the study, indicating that switching to a post-consumer waste biomass feedstock for biofuel or bioechemical production could afford large CO2 savings versus regular petrol and other bio-derived alternatives. In a similar way, the exergy efficiency balance, as carried out by Kang and Tan3 for the case of bio-ethanol produced from corn, could be drastically improved if the feedstock were switched to a post-consumer waste. The calculation included the total non-renewable energy input needed for the biofuel production, covering seeds, fertilizers and herbicides for cultivation and fuel for transport of the corn, as well as ethanol production and recovery. In the case of a diversion of a waste from landfill, the fossil fuel inputs for cultivation can be argued to be equal to 0 and hence the exergy efficiency would be significantly improved. Feedstock cultivation, also in the case of bioenergy crops such as switchgrass and Miscanthus, includes sowing, tilling, spreading of fertilizers, harvesting and baling, causing non-negligible cost and associated GHG emissions.4A study comparing six different pretreatment technologies for the production of bio-ethanol from switchgrass and the consequent minimum ethanol selling price (MESP) found that feedstock cost is 45–53% of the MESP across the six investigated pretreatment technologies, with switchgrass costing ca. $79 per dry tonne.5 At the same time gate fees for disposal, treatment and recycling reached up to £82 per tonne in the UK in 2015/16 for treated but not hazardous wood waste.6 Landfilling attracts, in addition to the gate fee, a landfill tax which increased the median disposal cost in the UK in 2015/16 to over £100 per tonne. Chromated copper arsenate (CCA) treated wood has been used extensively in Europe,7 the US,8 and Japan.9 It contains copper, chromium and arsenic in the range of 1000–5000 ppm and presents a pernicious environmental issue at the end of its life. This hazardous waste material is either disposed of within landfills or combusted for energy recovery,10 raising not only economic, but also ecological concerns about the release of toxic compounds into the environment.11,12 The use of post-consumer and hazardous waste wood as a negative-cost biorefinery feedstock therefore represents an economically attractive opportunity, as it is already preserved, collected, transported and stored prior to disposal, were it not for its common heavy metal and organic preservatives preventing its bioconversion to e.g. ethanol or lactic acid.
Ionic liquids (ILs) have been studied extensively for lignocellulose pretreatment in the context of biorefining and biofuel production.13–15 Recently, we have demonstrated that low-cost protic ionic liquids can be used to effectively fractionate even highly recalcitrant softwoods, resulting in near-quantitative glucose release during enzymatic saccharification.16 The process, called ionoSolv, selectively dissolves lignin and hemicelluloses, leaving behind an easily digestible cellulose pulp. The lignin can be precipitated using water as an anti-solvent. Ionic liquids are well documented also in electrochemistry due to their wide electrochemical window and their ability to dissolve various metals. Electrodeposition from ionic liquids has been shown to be possible for many different metals and some metal alloys.17–21 The use of ILs offers certain advantages over electrodeposition from aqueous or organic solutions, such as the possibility to use them in an open atmosphere and at elevated temperatures due to their low vapour pressure.19 While most electrochemical applications make use of aprotic ionic liquids, protic ionic liquids have been studied to a minor extent. Their ionic conductivity is in many cases low due to their high viscosity22 and their potential window is smaller than for aprotic ILs although still larger than for molecular protic sovents.22 However, protic ILs often show higher solubility of metal salts than their aprotic analogues and have successfully been used for electrodeposition of different metals.23,24 In certain cases, the use of ILs over aqueous systems has further advantages. For example chromium deposition from aqueous solutions is typically achieved from hexavalent chromium, which is very toxic.25 Trivalent chromium, although less toxic, is not typically used for chromium plating. This is due to the high stability of the hexaaquachromium(III) complex formed in aqueous solutions which makes plating impossible. However, chromium deposition from imidazolium based ILs ([C2C1im]Cl, [C4C1im]Cl and [C6C1im]Cl) from chromium(III), even in the presence of up to 18 equivalents of water, has been shown to be possible.26
In order to achieve an economically viable process, especially when working with relatively costly chemicals and solvents, recovery and reuse of the solvent and/or chemicals are crucial.27 Owing to their low vapour pressure, quantitative recovery of ILs is in theory possible without the need for enclosed systems and/or condensers. This is, however, only the case if the IL in question is thermally stable under the processing conditions. The IL often used for biomass applications, [C2C1im][OAc], is not thermally stable in the long term.28 A low IL recovery (maximum 96%) was reported for 1-allyl-3-methylimidazolium chloride [AMim]Cl after microwave assisted pretreatment of Pinus radiata.29 Even though the IL structure was found to be unaltered, a gradual decrease in the IL's ability to dissolve the pine was observed, resulting in reduction of the fractionation yields as well as a variation in the composition and crystallinity of the produced fractions. The study, however, did not investigate the effects on enzymatic hydrolysis and it is therefore impossible to make a statement on the performance of the IL, especially since full dissolution of biomass is not always required in order to achieve good hydrolysis yields.30 Nevertheless, the authors proposed29 that a cleaning step might be necessary after a certain number of cycles where an antisolvent would regenerate the IL by removing biomass-derived impurities.29 A study looking at the reuse of [C2C1im][OAc] over 8 cycles reported fluctuating glucose yields from both oak and spruce, ranging from 65.4% in cycle 7 and 44.7% in cycle 6 for spruce and 63.7% in cycle 6 and 51.1% in cycle 2 for oak.31 The lignin was not precipitated after the pretreatment but left in the IL to build up. Meanwhile, we have previously demonstrated recycling of the low-cost protic ionic liquid triethylammonium hydrogen sulfate [TEA][HSO4] over 4 cycles, recovering the ionic liquid quantitatively while maintaining enzymatic saccharification yields.32
The present study looks at the opportunity of using a negative-cost feedstock combined with a low-cost recyclable solvent as a potentially economically and environmentally sustainable way of producing bio-renewable chemicals and fuels. Its previously shown high effectiveness makes the ionoSolv process a promising candidate for tackling this waste disposal problem, as it has been reported to be suitable for various feedstocks including softwoods, and we hypothesised that this could be combined with numerous accounts of ILs’ ability to leach33,34 and extract metals35–37 and the possibility to recover the dissolved metals via electrodeposition.25,38
Experimental
Ionic liquid synthesis
Starting materials for ionic liquid synthesis were purchased from Sigma Aldrich and, unless stated otherwise, used as received. The minimum purity of the starting material for 1-methylimidazole was 99%. HCl was obtained as a concentrate containing 1 mole FIXANAL (Fluka Analytical).The protic IL was synthesised according to the standard operating procedure of our laboratory.39
1H, 13C and HMQC NMR spectra were recorded on a Bruker 400 MHz spectrometer. Chemical shifts (δ) are reported in ppm, referenced to the DMSO signal at 2.500 (1H dimension) and 39.520 (13C dimension). Mass spectrometry was performed by Dr Lisa Haigh (Imperial College London, Department of Chemistry) on a Micromass Premier spectrometer.
Synthesis of 1-methylimidazolium chloride [HC1im]Cl
1-Methylimidazole (82.1 g, 1.0 mol) was cooled with an ice bath in a 500 mL round-bottom flask. Under stirring, 1 mole (1 equivalent) of aqueous HCl was added dropwise. Excess water was removed using a rotary evaporator until a water content of below 20% by mass was obtained. The ionic liquid water mixture was recovered as a yellowish liquid. 1H NMR: δH (400 MHz, DMSO-d6)/ppm: 9.16 (s, 1H, H-2), 7.71 (s, 1H, H-4/5), 7.64 (s, 1H, H4/5), 4.5–3.5 (br, H2O, N-H+), 3.87 (s, 3H, N-CH3). 13C NMR: δC (101 MHz, DMSO-d6)/ppm: 135.63 (C-2), 123.21 (C-4/5), 119.55 (C-4/5), 35.48 (N-CH3).Feedstock
Pinus sylvestris was obtained from Metla (Finish Forest Research Institute). CCA treated wood was received from (Tianjin Dongli District Yilin wood preservative processing plant, CN). All biomass was air-dried, chopped (Retch SM 2000) and sieved (Retsch AS 200) (180–850 μm, 20 + 80 US mesh scale) prior to use and stored in plastic bags at room temperature in the dark.
(Tianjin Dongli District Yilin wood preservative processing plant, CN). All biomass was air-dried, chopped (Retch SM 2000) and sieved (Retsch AS 200) (180–850 μm, 20 + 80 US mesh scale) prior to use and stored in plastic bags at room temperature in the dark.
Fractionation of biomass
Pretreatments and determination of oven dried weight and ionic liquid water content measurements were conducted according to the standard operating procedure from our laboratory40 in triplicate. A biomass to solvent ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 to 1:2 g g−1 was used on an oven dried basis. Pretreatments were conducted with a final water content of 20% by mass according to eqn (1):
:20 to 1:2 g g−1 was used on an oven dried basis. Pretreatments were conducted with a final water content of 20% by mass according to eqn (1):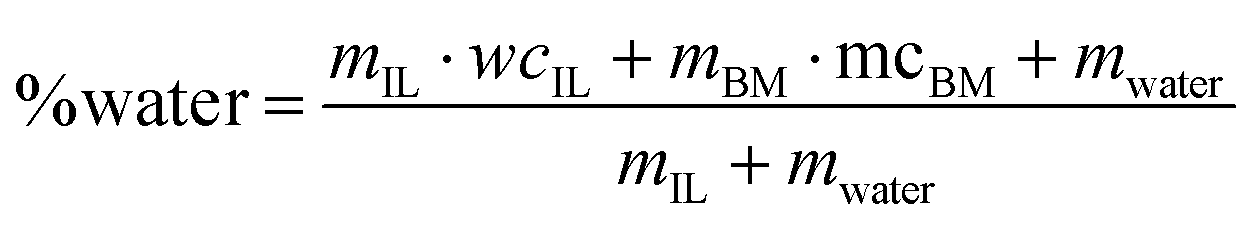 | (1) |
The pulp containing thimbles were removed from the Soxhlet apparatus after completion of the extraction and put into 50 mL falcon tubes which were immediately filled with DI water. The pulp was left in the thimble inside the falcon tube for at least an hour. The thimble was then taken out of the falcon tube and the pulp transferred back to the falcon tube using a spatula. The tubes were centrifuged for 30 min at 3000 rpm or 2000g and the supernatant decanted. The pulp was weighed, its moisture content determined immediately and saccharifications started the following day.
Compositional analysis
Compositional analysis was carried out according to a published procedure by the National Renewable Energy Laboratory (NREL).41 300 mg (calculated on ODW basis) of air-dry biomass or pulp was weighed out into a pressure tube and the weight recorded. 3 mL of 72% sulfuric acid (Fluka) were added, the samples stirred with a Teflon stir rod and the pressure tubes placed into a preheated water bath at 30 °C. The samples were stirred again every 15 min for one hour. They were then diluted with 84 mL distilled water and closed. The samples were autoclaved (Sanyo Labo Autoclave ML5 3020 U) for 1 hour at 121 °C and left to cool to close to ambient temperature. The samples were then filtered through filtering ceramic crucibles of a known weight. The filtrate was stored in two Falcon tubes and the remaining black solid washed with distilled water. The crucibles were placed into a convection oven (VWR Venti-Line 115) at 105 °C for 24 ± 2 h. They were then taken out and placed in a desiccator for 15 min before weighing, and the weight recorded. They were then placed into a muffle oven (Nabertherm + controller P 330) and ashed to constant weight at 575 °C. The weight after ashing was recorded. The content of acid insoluble lignin (AIL) was determined according to eqn (2). The content of one of the Falcon tubes was used for the determination of acid soluble lignin content (ASL) by UV analysis at 240 nm (eqn (3)) (Perkin Elmer Lambda 650 UV/Vis spectrometer).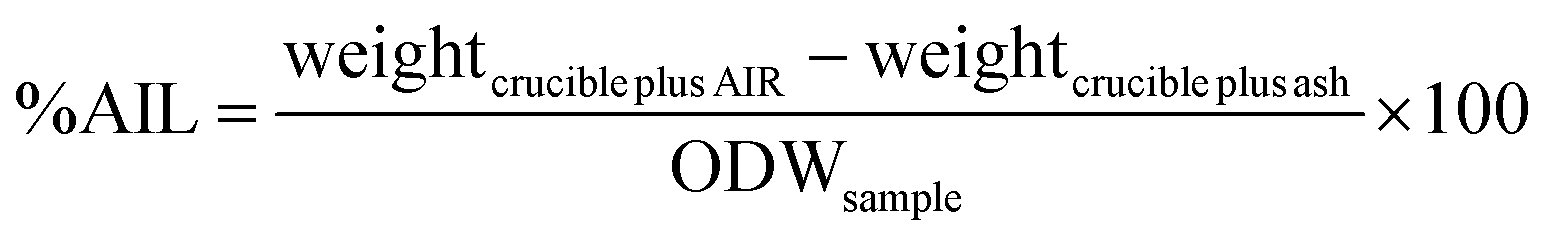 | (2) |
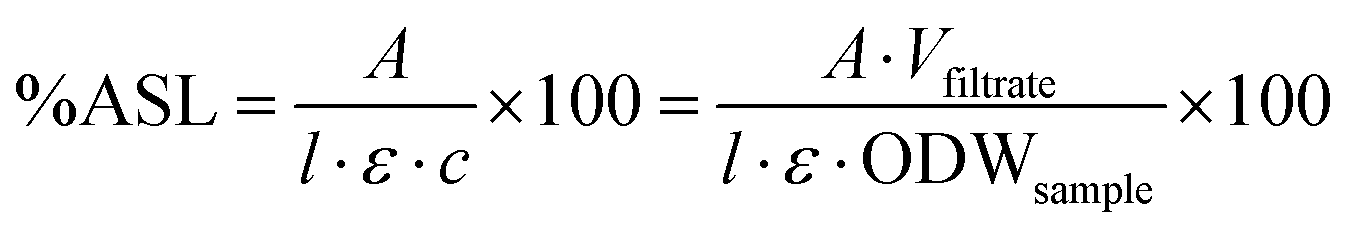 | (3) |
Calcium carbonate was added to the content of the second Falcon tube until the pH reached 5. The liquid was passed through a 0.2 μm PTFE syringe filter and subsequently submitted to HPLC analysis (Shimadzu, Aminex HPX-87P from Bio-Rad, 300 × 7.8 mm, purified water as mobile phase at 0.6 ml min−1, column temperature 85 °C) for the determination of total sugar content. Calibration standards with concentrations of 0.1, 1, 2 and 4 mg mL−1 of glucose, xylose, mannose, arabinose and galactose were used. Sugar recovery standards were prepared using the respective sugars obtained from Sigma Aldrich and dissolved in purified water to produce 10 mL aqueous solutions close to the expected sugar concentration of the samples and transferred to pressure tubes. 278 μL 72% sulfuric acid was added, the pressure tube closed and autoclaved and the sugar content determined as described above. The sugar recovery coefficient (SRC) was determined according to eqn (4) and the sugar content of the analysed sample using eqn (5):
 | (4) |
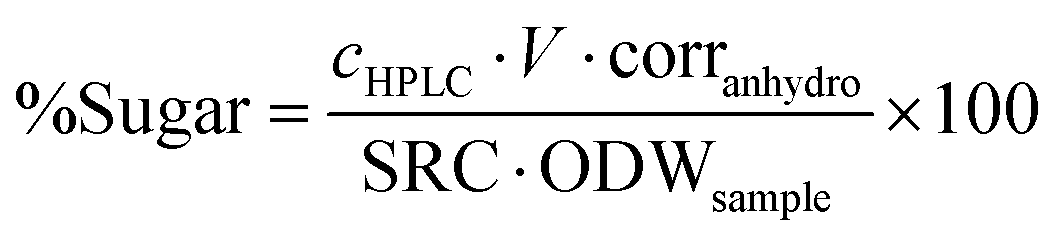 | (5) |
Correlations between delignification and saccharification yields were calculated using least squares linear regression in Excel.
Saccharification assay
Saccharification assays were carried out according to an adapted procedure by the NREL42 in triplicate with blanks (also as triplicates). All reagents were purchased from Sigma Aldrich.For wet samples, moisture contents were determined again directly prior to saccharification. 100 ± 10 mg (on and ODW basis) of air-dried or wet biomass were placed into a Sterilin tube and the weight recorded. Three blanks were run with 100 μL of purified water instead of biomass in order to correct for sugar residues present in the enzyme solutions. The water contained in the biomass sample was calculated using its moisture content and the total sample mass and subtracted from 1.5 mL. The difference was added as water using a pipette. 8.4 mL solution consisting of 5 mL 1 M sodium citrate buffer at pH 4.8, 40 μL tetracyline antibiotic solution (10 mg mL−1 in 70 v/v% ethanol), 30 μL cycloheximide antibiotic solution (10 mg mL−1 in purified water), 3.38–3.41 mL purified water and 20–50 μL of Novozymes experimental enzyme mixture NS-22201 (kindly provided directly by Novozymes) were added, the tubes closed and placed into an Stuart Orbital Incubator (S1500) for 7 days at 50 °C and 250 rpm.
Saccharification yields were obtained after filtering 1 mL of the hydrolysate through a PTFE syringe filter. Samples were run on Shimadzu HPLC with an Aminex HPX-87P column (Bio-Rad, 300 × 7.8 mm) with purified water as mobile phase (0.6 mL min−1). The column temperature was 85 °C and acquisition was run for 20 min. A representative chromatogram can be found in the ESI.† Calibration standards with concentrations of 0.1, 1, 2 and 4 mg mL−1 of glucose, xylose, mannose, arabinose and galactose and 8 mg mL−1 of glucose were used.
Trace element analysis
ICP-OES measurements were performed on a Perkin Elmer Optima 2000. Standards were prepared using Multielement standard solution 4 for ICP in 10% nitric acid (TraceCERT). The standard solution was diluted with 5% nitric acid as required to obtain at least 3 different concentrations for calibration with no metal at a concentration higher than 25 ppm. Ionic liquid liquors were diluted to approximately 1% ionic liquid (by mass) concentration with 5% nitric acid. In the case of IL liquors with high concentrations of metals, this solution was further diluted to obtain a maximum concentration of each metal of no more than 25 ppm. All solutions were filtered through a 0.4 μm syringe filter prior to analysis.For the analysis of the recycled IL, a blank IL without any biomass was subjected to the same experimental procedures as the working IL to account for eventual accumulation of metals from external sources, such as metallic dust found on stirrer bars and chromium leaching from stainless steel spatulas. It was used as a blank in the ICP-measurements and its metal concentrations (below detection limit for As and Cu, <1 ppm for Cr) subtracted from the ones measured for the working ILs.
For analysis of solid samples (biomass, pulp and lignin), approximately 100 mg of sample was weighed out and the exact weight recorded (±0.1 mg, Mettler Toledo NewClassic MS). The samples were digested in 1 mL 69% nitric acid in a closed PTFE vessel (MARSXpress) vessels and microwave with power/time control by CEM with the following sequence: 300 W at 83% power for 5 min, 600 W at 66% power for 5 min and 1200 W at 58% power for 6 min. The obtained solution was cooled in a freezer for one hour before diluting to 10 mL with 5% nitric acid and filtration through a 0.4 μm PTFE syringe filter.
Percentage recovery of metal M in the pulp was calculated according to eqn (6), in IL liquor according to eqn (7), and in lignin according to eqn (8):
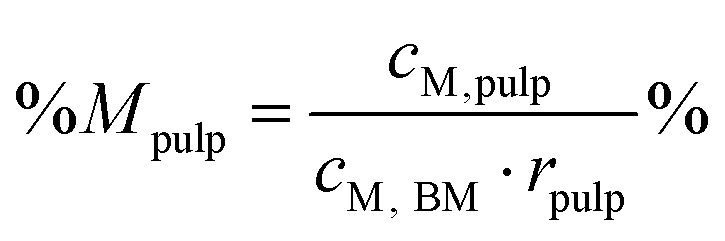 | (6) |
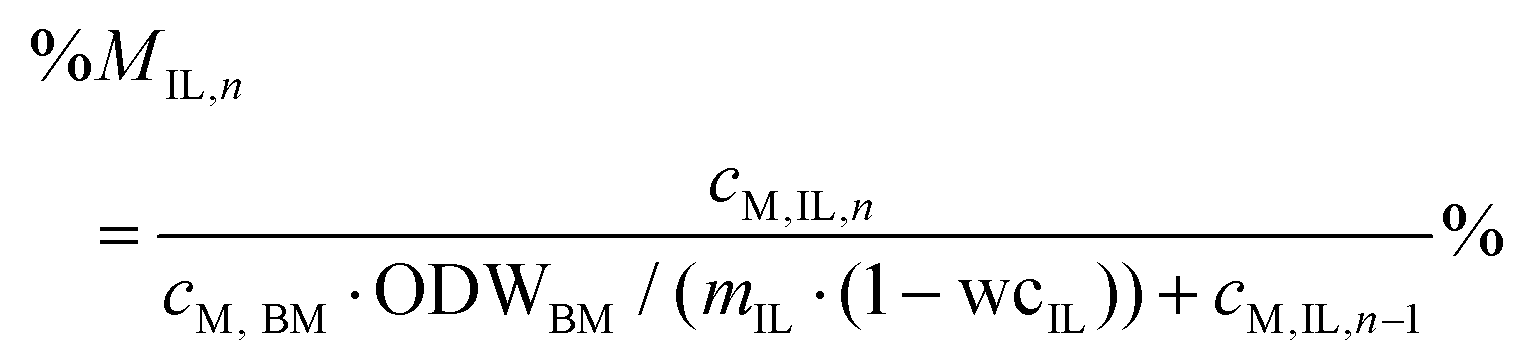 | (7) |
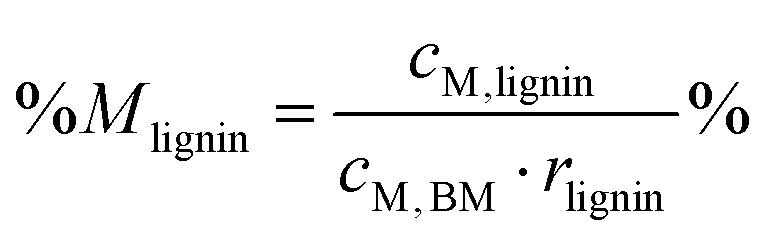 | (8) |
Fermentation
Saccharomyces cerevisiae BY4741 [MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0] was used for fermentation studies. Single colonies were used to inoculate 10 mL of YPD medium in 250 mL baffled flasks in triplicate and grown for 24 hours at 30 °C with shaking at 250 rpm. The optical density (absorbance) of these cultures was measured and used to inoculate 3 mL of YPD containing varying concentrations of metal salts (CrIII, CuII and AsV ranging from 128 mM to 100 nM) at an initial optical density of 0.1. 3 mL medium containing cells was then transferred to individual falcon tubes. The cells were cultured anaerobically at 30 °C, 250 rpm for 48 hours. The cultures were pelleted by centrifugation and the supernatant was retained for HPLC analysis.Fermentation of hydrolysed wood samples was carried out in 10 mL centrifuge tubes (Falcon). Saccharification was carried out to obtain final sugar concentrations of around 20 mg mL−1: around 330 mg of pretreated samples and 1.6 g of non-pretreated samples were incubated in a medium consisting of 4 mL of 5 M sodium citrate buffer, 4 mL of DI water and 400 μL of CTec2 enzyme mix. It should be noted that the enzymatic hydrolysis was run using an excess of enzymes in order to achieve high enough glucose concentrations from the non-pretreated biomass (VP-UT). Saccharified wood samples were centrifuged to remove the solids and the resulting glucose containing solution was used to make a medium by mixing with 1X yeast nitrogen base with amino acids (Sigma Aldrich). Samples were standardized to the amount of glucan added to the saccharification reaction. Controls containing metals (Cr, Cu, As from chromium sulfate, copper sulfate and arsenic(V) in water respectively – all purchased from Sigma) were prepared as follows: for samples of CCA-treated wood that had undergone extraction, the metal content was readjusted to the amount of Cr, Cu and As in the pulp after pretreatment if no extraction had occurred (as a non-extractive control); in regards to the pulp yield after pretreatment. In this instance the metal content of the CCA treated wood was approximated as 4000 ppm (mg kg−1) of CrIII/AsV and 2000 ppm (mg kg−1) of CuII. All liquid samples were analysed in triplicate through the ICP-OES to check the concentration of metals.
For the assay, single colonies were used to inoculate YPD medium as described above. After 24 hours of growth, cultures were diluted to an OD600 of 0.1 in PBS. 3 mL was transferred to each centrifuge tube, cells were pelleted by centrifugation, the supernatant was removed, and the cell pellet was resuspended in 3 mL of the appropriate wood medium. Cells were grown at 30 °C with shaking at 250 rpm in a Vortemp™ 56 shaking incubator (Labnet International) for 48 hours. The OD600 of cultures was measured against a blank of the same type of medium and then the cells were pelleted by centrifugation and the supernatant retained for HPLC analysis.
The supernatant was pipetted into a HPLC vial and submitted for analysis on a Shimadzu HPLC system with RI and UV/Vis detector and an Aminex HPX-87H column (BioRad, 300 × 7.8 mm) with 0.005 M H2SO4 as mobile phase (0.6 mL min−1). The column temperature was 55 °C and acquisition was run for 35 min. Calibration was carried out using standards with concentrations of 0.1, 0.4, 1, 2, 4, 10, 20 and 30 mg mL−1 of HPLC grade ethanol. Ethanol concentrations in the HPLC sample were calculated using the resulting calibration curves.
Cyclic voltammetry
Cyclic voltammograms of the fresh IL and recycled IL liquors were performed using printed electrodes obtained from DropSens (DRP-110, Spain) with carbon as working (12.56 mm2) and counter electrode and silver as a quasi-reference electrode. The cell was connected to the potentiostat (BioLogic SP300, using EC-Lab Software) with a cable connector for screen-printed electrodes (DRP-CAC, DropSens, Spain). A drop of 50 μL was placed on the chip, making sure it covered all three electrodes. Three consecutive voltammograms with a scan rate of 10 mV s−1 were performed over a potential window of −1 to +1 V.Potentiostatic electrodeposition of metals
Metal depletion was performed potentiostatically using the DropSens printed electrodes. A potential of −0.7 V was applied for a defined time. Most of the drop was collected using an Eppendorf pipette and its weight recorded. It was diluted to 10 mL using 5% HNO3 and analysed by ICP-OES.Results and discussion
Fractionation and saccharification
The chromated copper arsenate (CCA) treated wood used in this study contained 43.8% glucose and 31.1% lignin (by mass). Its composition is displayed in Fig. S1 of the ESI.† The CCA wood was successfully pretreated with an ionic liquid-based fractionation approach, based on previously successful conditions for softwood.16,43 The protic ionic liquid (IL), 1-methylimidazolium chloride [HC1im]Cl, is a relatively low-cost IL because of its simple one step synthesis from an acid and a (stoichiometric) base.44 It was used for 6 subsequent treatments and the IL recovery was found to be 99.06 ± 0.72% overall, showing it can be recycled with minimal losses. After pretreatment, the isolated pulps (solid fraction) were subjected to enzymatic hydrolysis and the lignin was recovered by the addition of an anti-solvent into the ionic liquid liquor (Fig. 1). Pulp yields remained stable in the region of 58% (42% of the mass dissolved into the IL). Lignin yields, calculated using the initial amount of lignin in the biomass, increased monotonically from 39% in cycle 1 to 69% in cycle 6. This can be explained by a significant amount of lignin being in the form of small water-soluble fragments which re-condense during following cycles until they eventually precipitate out as discussed in more detail below. Saccharification (sugar) yields, starting at 67% after the first cycle, gradually decreased over the subsequent 3 cycles to 55% in cycle 4 before increasing to 75% in the 6th cycle – a 15-fold increase compared to raw non-pretreated waste wood. This shows the importance of such pretreatment for any subsequent chemical valorisation of wood biomass. The ionic liquid pretreatment is both efficient and robust, with glucose yields increasing slightly across multiple re-uses, showing that the solvent can be recycled efficiently for at least 6 cycles.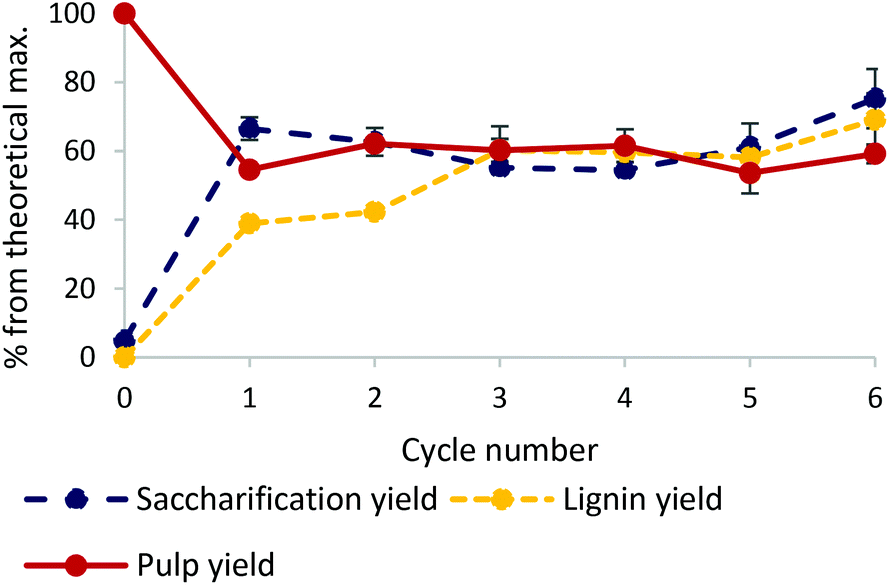 | ||
| Fig. 1 Lignin, pulp and 7-day saccharification yield for untreated and pretreated CCA treated wood over 6 cycles. CCA treated wood was pretreated with [HC1im]Cl for 30 min at 170 °C at a biomass to solvent ratio of 1:5 g g−1 and a final water content of 20% by mass. Standard errors were calculated for triplicate measurements. | ||
Changes in lignin structure
Changes occurring in the lignin structure during ionoSolv pretreatment have been reported for Miscanthus45 and pine and for up to 4 cycles of IL use.32,46 Here we analysed the lignin obtained after the 1st, 2nd and 6th cycle. The results are displayed in Fig. S2 of the ESI.† The data indicate that recycling of the IL has a similar effect on lignin properties as a prolonged pretreatment:12–14,25 an increase in condensation was evidenced by a shift of the G2 signal (see G2vs. G2 condensed) and a decrease in G6 signal intensity, accompanied by a weaker signal for ether bonds in the isolated lignin. Indeed, part of the lignin precipitating from the liquor at the second cycle will have been in the IL for two cycles, but was in the form of water soluble fragments after the first cycle. Precipitating after the 2nd cycle, those fragments are likely to have had their ether bonds broken and a significant amount of condensation between aromatic units would have occured, resulting in a decreased signal intensity in the ether and G2 areas, and an increased signal in G2,cond. The same, but in a more pronounced way, can be observed for lignin precipitated after cycle 6. The signal intensities for the β-O-4′ linkage and G6 changed significantly between cycles 2 and 6, potentially indicating that lignin fragments can spend several cycles in the IL before precipitating. The G5 signal remained relatively stable, indicating that the incorporation of carbohydrate degradation products increases signal intensity in this area while the condensation occurring in G5 decreases it in a complementary fashion.Metal extraction
For the produced sugars to be fermentable, CuII (and CrIII and AsV), a potent biocide,47 must be removed from the pulp. The isolated pulp, recycled IL and lignin precipitate were analysed for Cu, Cr and As content (Fig. 2 and Table S1 of the ESI†). CuII extraction from the pulp was very high throughout the 6 cycles, reaching at least 98% for each cycle. CrIII and AsV extractions fluctuated slightly between each cycle.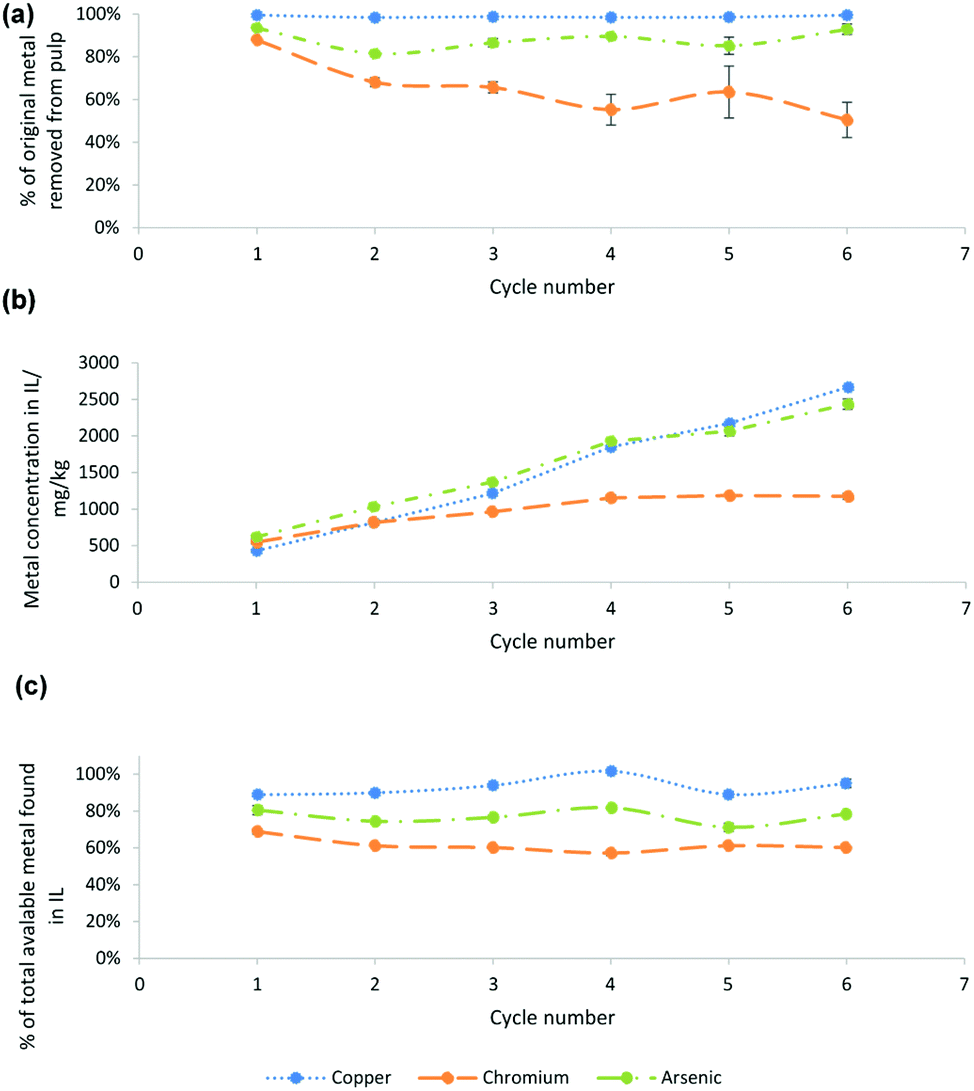 | ||
| Fig. 2 (a) Metal extractions from the pulp as calculated from the metal content in the pulp, measured by ICP-OES, and the pulp yield obtained after pretreatment. (b) Metal ppm found in the IL brown liquor after pretreatment for n cycles as analysed by ICP-OES. (c) Percentage of CuII, CrIII and AsV found in the IL brown liquor after n cycles based on the metal concentration prior to the cycle and the amount of new metal entering the system. CCA treated wood was pretreated with [HC1im]Cl for 30 min at 170 °C at a biomass to solvent ratio of 1:5 g g−1 and a final water content of 20% by mass. Standard errors were calculated for triplicate measurements. | ||
As can be seen from Fig. 2(b), CuII and AsV concentrations steadily increased in the IL over the 6 cycles at a relatively stable rate. Despite there being almost twice as much AsV in the wood as CuII, concentrations in the IL were found to be similar. CrIII concentration however only increased slightly over the first 4 cycles and then remained stable at around 1200 ppm. These findings suggest that a large proportion of CrIII and smaller amounts of AsV and CuII precipitate out during the lignin recovery, as lignin is known to be a promising sorbent for transition metals. Maintaining high metal concentrations has the advantage of a relatively easier electrodeposition versus a case where the deposition needs to take place at very low concentrations of metals if the IL cannot solubilise the metals throughout processing.
Fig. 2(c) shows the percentage of the different metals found in the IL after each stage, taking into account the metal in the IL from the previous cycle. CuII recovery into the IL was initially ∼90%, rising to ∼95%. AsV extraction started off at 81% and subsequently fluctuated at around 75%. Competition for ligands is likely an underlying cause, e.g. wash solvent addition might cause some lignin precipitation, leading to adsorption of metals to the pulp. An altered washing protocol where less wash solvent is used, or where recycled IL that contains less lignin is used, may yield better metal extractions over many cycles.
Between 90 and 99% of the metals can be recovered from CCA treated wood using traditional leaching processes, though at a cost of ca. $120–$180 per ton of wood.7 Here we provide a cost-effective solution that does not produce a waste stream and contributes to the production of clean chemicals (cellulose, lignin, glucose, ethanol). The mass balance of metal extraction into the different fractions is discussed in the ESI.†
Metal recovery
Cyclic voltammetry of a carbon electrode in IL alone and in IL that had been recycled 6 times (Fig. 1), was used to define electrode potentials for metal deposition, within the potential range −1 V to +1 V (vs. AgCl|Ag), so avoiding excessive rates of hydrogen and chlorine evolution at lower and higher electrode potentials, respectively.48 In the chloride-containing IL, the Ag reference electrode was assumed to have formed AgCl on its surface. AgCl|Ag|aqueous solution with unit Cl− activity has an electrode potential of ca. 0.22 V vs. standard hydrogen electrode (SHE). Assuming that the thermodynamic properties of IL-aqueous solutions and the speciation therein approximate to those in aqueous solutions, then equilibrium potentials calculated from critically reviewed thermodynamic data49 can be used to aid assigning reactions to current responses in voltammograms. In IL alone, significant current densities were measured only at electrode potentials <−0.5 V vs. AgCl|Ag, as expected for the hydrogen evolution reaction.From the start potential of 0.928 V (vs. AgCl|Ag), the positive-going potential scan to 1.0 V at 10 mV s−1 resulted in significantly increased current densities above those in the IL alone, probably due to oxidation of sugars (which were present in large (ca. 100 times) excess of the metal concentrations); onset of oxidation of the carbon electrode and of chlorine evolution were not evident in voltammograms of the IL alone. As sugars were present in excess of dissolved metal concentrations in the IL solutions, probably CuI and AsIII would have predominated over CuII and AsV species. On the subsequent negative-going potential scan, at potentials <0.72 V (vs. AgCl|Ag) cathodic current densities were measured, leading to a peak at 0.4 V (vs. AgCl|Ag), probably due to the reaction:
| Cu2+ + 2Cl− + e− → CuCl2− | (9) |
| H2AsO4− + 3H+ + 2e− ↔ H3AsO3 + H2O | (10) |
| CuCl2− + e− → Cu + 2Cl− | (11) |
Coupled with AsIII reduction51,52 by:
| H3AsO3 + 3H+ + 3e− ↔ As + 3H2O | (12) |
Hence, Cu–As alloys may have been electrodeposited, with the Cu electrodeposition at least inhibiting possible AsH3 (g) formation53 at potentials <ca. −0.5 V (AgCl|Ag). At electrode potentials <−0.65 V (AgCl|Ag), reduction of CrIII species by the reaction:
| Cr3+ + e− → Cr2+ | (13) |
On the subsequent positive-going potential scan from the negative potential limit of −1.0 V vs. AgCl|Ag, an oxidation peak at ca. −0.2 V occurred, due to the reverse of reaction (13) and (12), with an additional current peak at ca. 0.0 V, due to the reverse of reaction (11) causing dissolution of CuI species. Their further oxidation led to a current peak at ca. 0.5 V due to the reverse of reaction (9) producing CuII species, convoluted with the reverse of reaction (10) producing AsV ions.
Cycling between the potential limits resulted in a slight growth after 2 cycles, of the oxidation current density peak at ca. 0 V vs. AgCl|Ag, shown in Fig. 3, probably due to more facile growth and subsequent dissolution of Cu on the carbon electrode.
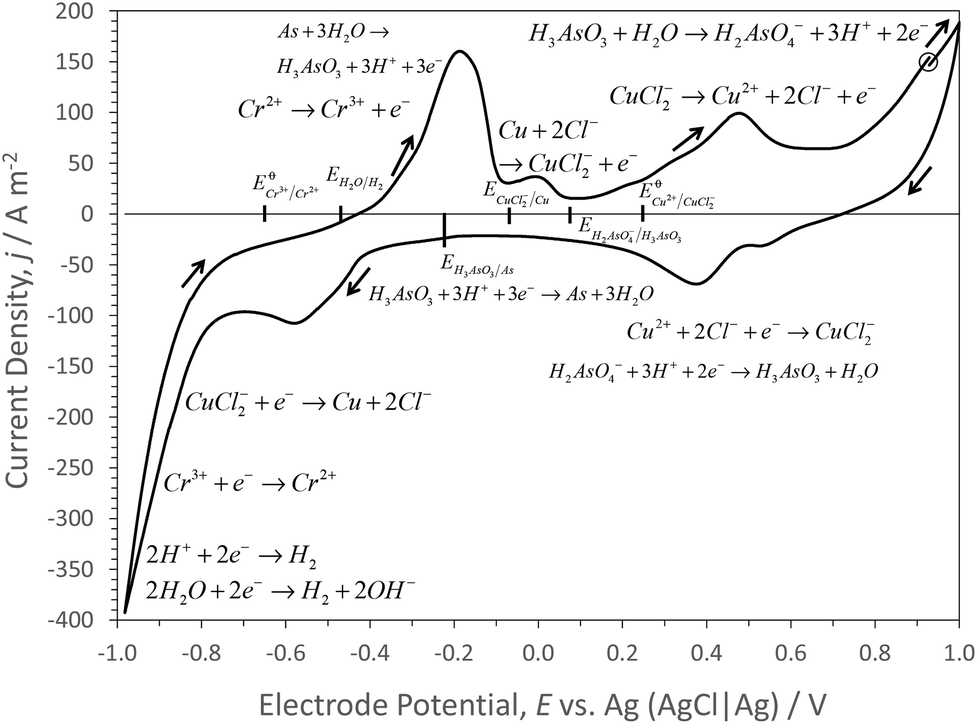 | ||
| Fig. 3 Cyclic voltammograms of printed carbon electrode with a silver quasi reference electrode in recycled [HC1im]Cl containing 17.7% water by mass obtained from 6 cycles of pre-treatment of CCA treated timber for 30 min at 170 °C at a biomass to solvent ratio of 1:5 g g−1 and a final water content of 20% by mass (48.3 mol m−3 copper, 44.2 mol m−3 arsenic, 96.2 mol m−3 chromium). | ||
From the voltammetric behaviour in Fig. 3, a constant potential of −0.7 V vs. AgCl|Ag was chosen to deplete dissolved metals by electrodeposition from the recycled IL. Fig. 4 shows concentrations, determined by ICP-OES and normalised by initial concentrations, after 2, 5 and 10 minutes of deposition. The numerical values of the relative amounts remaining in solution after deposition are in Table S2 of the ESI.† The content of all three analysed metals appeared to decrease slightly over the first 2 min of deposition. Thereafter, changes were minimal; it should be noted that the error bars at this point are almost equal to the changes determined. Dissolved metal concentrations after 10 minutes of deposition corresponded to 85–92% of their initial concentrations. Unfortunately, the extent of their depletion was limited by the design of the printed electrochemical cell, with its small droplet of solution, cathode undivided from the counter electrode by an ion-permeable membrane, and restricted mass transport rate coefficient. As chromium electrodeposition in ionic liquids has been shown to require more negative electrode potentials, it is unclear if its depletion, as analysed by ICP, is attributable to electrodeposition. Hence, these experiments will be repeated with more appropriate equipment in the future. For reduction e.g. of CuI species under transport control at the carbon cathode of area A in a batch reactor with solution volume V and with a mass transport rate coefficient of km (ca. 10−6 m s−1), concentrations would be expected to decrease exponentially with time t from their initial value at t = 0:
| [CuI]t = [CuI]t=0exp{−kmAt/V} | (14) |
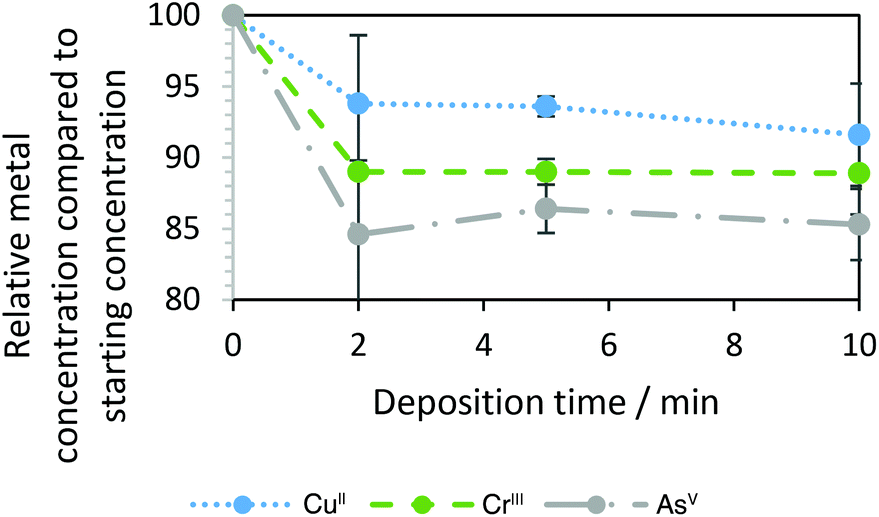 | ||
| Fig. 4 Dissolved metal concentrations determined in the brown liquor (50 μL) before and after applying an electrode potential of −0.7 V vs. AgCl|Ag on a screen-printed carbon electrode. Recycled [HC1im]Cl was obtained from 6 cycles of pre-treatment of CCA treated timber for 30 min at 170 °C at a biomass to solvent ratio of 1:5 g g−1 and a final water content of 20% by mass (48.3 mol m−3 copper, 44.2 mol m−3 arsenic, 96.2 mol m−3 chromium). Standard errors were calculated for triplicate measurements. | ||
Fermentation of glucose
To assess the impact of the metal removal (vs. the general pretreatment effect) from CCA treated wood pulp on saccharification, Cu, Cr and As were added to CCA treated wood pulp to mimic the original metal content in the wood. The obtained hydrolysates were used to grow yeast cells (Saccharomyces cerevisiae BY4741) for ethanol production, together with controls consisting of enzymatic hydrolysates of non-pretreated virgin pine (VP-UT), pretreated virgin pine (VP-PT), pretreated CCA treated wood (CCA) and glucose solutions of equivalent concentrations. Ethanol production was observed for all feedstocks (Fig. 5), but the findings are most significant for the potential valorisation of waste wood. Heavy metals are clearly a detriment to fermentation using Saccharomyces cerevisiae: 10.1% ethanol yield was obtained for CCA wood pulp, compared to 1.3% from CCA treated wood pulp in the presence of metals. This shows that CCA treated wood would not be suitable for the production of bioethanol or other fermentation products without an additional decontamination or detoxification process. The decrease of only 1.2% in ethanol yield for CCA treated wood pulp compared to pretreated virgin pine (VP-PT) demonstrates that the difference in the quality of the pulp for fermentation is minimal. The ethanol yields obtained from lignocellulosic hydrolysates are higher than the ones from untreated pine (VP-UT) and glucose medium. This might be due to the presence of other sugars (e.g. mannose) and other carbon and nitrogen compounds from the hydrolysate of the pretreated pulp, that enhance the growth and fermentation of the yeasts.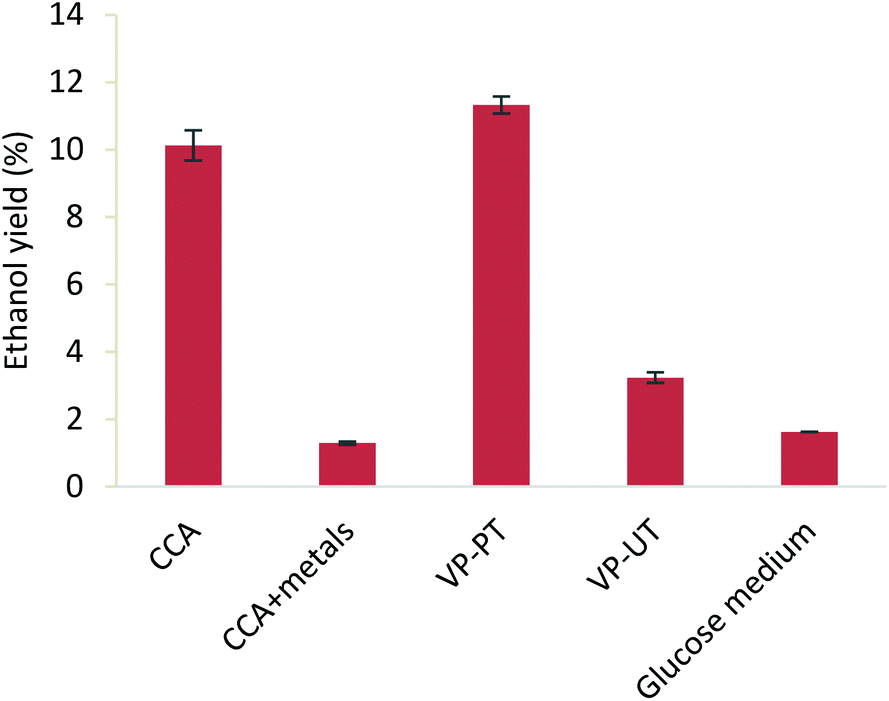 | ||
| Fig. 5 Ethanol yields (below) from CCA treated wood pulp pretreated with [HC1im]Cl, CCA treated wood pulp with added metals back in, virgin pine pulp pretreated with [HC1im]Cl, untreated virgin pine and glucose medium after 48 hours of fermentation. Ethanol yields were standardised to the amount of glucan added to the saccharification reaction. | ||
Finally, the untreated virgin pine showed a low yield of 3.2% due to the high amount of biomass that was necessary to achieve a sufficient sugar concentration for the fermentation (Fig. 5).
Discussion
The largest obstacle to commercialisation of lignocellulosic biorefineries is feedstock cost. Virgin (grown) biomass or agricultural residues must be harvested or collected, transported and stored (with silage to prevent rotting) to overcome the limited supply radius and seasonal production. Approximately 4.5 million tonnes of waste wood are produced in the UK annually (and collected, transported and stored, without any need for silage) and would be ready to be recycled.54 This ionic liquid pretreatment of waste wood is capable of processing these heavy metal contaminated feedstocks to produce second-generation lignocellulosic biofuels while simultaneously recovering the heavy metals. This could drastically reduce the cost of glucose from waste wood at zero net cost, compared to a previously reported gate fee of $90 per tonne or more for energy crops.32 This may lead to second-generation bioethanol that is both carbon neutral and lower in price than first-generation biofuels produced from sugarcane or corn starch. Bioethanol conversion was chosen as an example glucose derived product, but any alternative bio-based products such as bioplastics or cellulosic materials can also be investigated. This study can hence provide a roadmap for possible inroads to the circular economy by rehabilitating a toxic waste material.Conclusions
We have demonstrated that the ionoSolv process is capable of efficiently purifying the cellulose contained in heavy metal-contaminated waste wood by simultaneously complexing and removing the heavy metal contaminants. Enzymatic glucose yields of up to 75% were possible after pretreatment, a 15-fold increase over the raw biomass. The resulting sugar hydrolysate was readily fermentable to bioethanol at levels commensurate or even higher than with sugars derived from traditional biomass sources. The concentrations of the heavy metal poisons copper, chromium and arsenic in the wood were all reduced by 80–100% and the metals could be recovered from the ionic liquid liquor via electrodeposition. 99% of the ionic liquid solvent was recovered and recycled 6 times with retention of efficacy. These findings open new strategies for the utilisation of toxic waste as a negative-cost feedstock for the production of economically attractive, sustainable fuels, chemicals and materials, thereby using one environmental problem as the solution for a greater one.Conflicts of interest
The authors wish to declare that FG, ABT, GK, PF and JH are co-founders and shareholders of Lixea Limited, a company dedicated to the industrialisation and commercialisation of protic IL lignocellulose fractionation.Acknowledgements
We thank the Grantham Institute for Climate Change for studentships for F. J. V. G. and L. M. H. Additional funding came from Climate-KIC and the EPSRC (EP/S000771/1 and EP/K038648/1).Notes and references
- T. Searchinger, R. Heimlich, R. A. Houghton, F. Dong, A. Elobeid, J. Fabiosa, S. Tokgoz, D. Hayes and T.-H. Yu, Science, 2008, 319, 1238–1240 CrossRef CAS PubMed.
- R. Melamu and H. Von Blottnitz, J. Cleaner Prod., 2011, 19, 138–144 CrossRef.
- Q. Kang and T. Tan, Sustainability, 2016, 8, 76 CrossRef.
- F. Cherubini and G. Jungmeier, Int. J. Life Cycle Assess., 2010, 15, 53–66 CrossRef CAS.
- L. Tao, A. Aden, R. T. Elander, V. R. Pallapolu, Y. Y. Lee, R. J. Garlock, V. Balan, B. E. Dale, Y. Kim, N. S. Mosier, M. R. Ladisch, M. Falls, M. T. Holtzapple, R. Sierra, J. Shi, M. a. Ebrik, T. Redmond, B. Yang, C. E. Wyman, B. Hames, S. Thomas and R. E. Warner, Bioresour. Technol., 2011, 102, 11105–11114 CrossRef CAS PubMed.
- H. Dick, A. Hennig and P. Scholes, Gate fees Report 2015/16: Comparing the cost of alternative waste treatment options, 2016 Search PubMed.
- T. G. Mercer and L. E. Frostick, Sci. Total Environ., 2012, 427–428, 165–174 CrossRef CAS PubMed.
- J. Jambeck, K. Weitz, H. Solo-Gabriele, T. Townsend and S. Thorneloe, Waste Manage., 2007, 27, 21–28 CrossRef PubMed.
- N. Ohgami, O. Yamanoshita, N. D. Thang, I. Yajima, C. Nakano, W. Wenting, S. Ohnuma and M. Kato, Environ. Pollut., 2015, 206, 456–460 CrossRef CAS PubMed.
- F.-C. C. Chang, Y.-N. N. Wang, P.-J. J. Chen and C.-H. H. Ko, J. Environ. Manage., 2013, 122, 42–46 CrossRef CAS PubMed.
- A. Nzihou and B. Stanmore, J. Hazard. Mater., 2013, 256–257, 56–66 CrossRef CAS PubMed.
- A. Janin, J.-F. Blais, G. Mercier and P. Drogui, J. Hazard. Mater., 2009, 169, 136–145 CrossRef CAS PubMed.
- E. Sundstrom, J. Yaegashi, J. Yan, F. Masson, G. Papa, A. Rodriguez, M. Mirsiaghi, L. Liang, Q. He, D. Tanjore, T. R. Pray, S. Singh, B. Simmons, N. Sun, J. Magnuson and J. Gladden, Green Chem., 2018, 20, 2870–2879 RSC.
- C. L. Chambon, T. Y. Mkhize, P. Reddy, A. Brandt-Talbot, N. Deenadayalu, P. S. Fennell and J. P. Hallett, Biotechnol. Biofuels, 2018, 11, 247 CrossRef CAS PubMed.
- F. J. V. Gschwend, F. Malaret, S. Shinde, A. Brandt-Talbot and J. P. Hallett, Green Chem., 2018, 20, 3486–3498 RSC.
- F. J. V. Gschwend, C. L. Chambon, M. Biedka, A. Brandt-Talbot, P. S. Fennell and J. P. Hallett, Green Chem., 2019, 21, 692–703 RSC.
- R. Bomparola, S. Caporali, A. Lavacchi and U. Bardi, Surf. Coatings Technol., 2007, 201, 9485–9490 CrossRef CAS.
- J. Vaughan, Miner. Process. Extr. Metall., 2008, 117, 113–117 CrossRef CAS.
- F. Endres, D. MacFarlane and A. Abbott, Electrodeposition from Ionic Liquids, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008 Search PubMed.
- F. Endres, ChemPhysChem, 2002, 3, 145–154 CrossRef PubMed.
- Q. Zhang, Q. Wang, S. Zhang, X. Lu and X. Zhang, ChemPhysChem, 2016, 17, 335–351 CrossRef CAS PubMed.
- C. Zhao, G. Burrell, A. A. J. Torriero, F. Separovic, N. F. Dunlop, D. R. MacFarlane and A. M. Bond, J. Phys. Chem. B, 2008, 112, 6923–6936 CrossRef CAS PubMed.
- B. H. R. Suryanto, C. a. Gunawan, X. Lu and C. Zhao, Electrochim. Acta, 2012, 81, 98–105 CrossRef CAS.
- C. a. Gunawan, B. H. R. Suryanto and C. Zhao, J. Electrochem. Soc., 2012, 159, D611–D615 CrossRef CAS.
- X. He, Q. Zhu, B. Hou, C. Li, Y. Jiang, C. Zhang and L. Wu, Surf. Coatings Technol., 2015, 262, 148–153 CrossRef CAS.
- L. Sun and J. F. Brennecke, Ind. Eng. Chem. Res., 2015, 54, 4879–4890 CrossRef CAS.
- N. R. Baral and A. Shah, Biofuels, Bioprod. Biorefin., 2016, 10, 70–88 CrossRef CAS.
- M. T. Clough, K. Geyer, P. a. Hunt, J. Mertes and T. Welton, Phys. Chem. Chem. Phys., 2013, 15, 20480–20495 RSC.
- V. Rigual, T. M. Santos, J. C. Domínguez, M. V. Alonso, M. Oliet and F. Rodriguez, ACS Sustainable Chem. Eng., 2017, 5, 2384–2392 CrossRef CAS.
- A. Brandt, M. J. Ray, T. Q. To, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2011, 13, 2489 RSC.
- T. Auxenfans, S. Buchoux, D. Larcher, G. Husson, E. Husson and C. Sarazin, Energy Convers. Manage., 2014, 88, 1094–1103 CrossRef CAS.
- A. Brandt-Talbot, F. J. V. Gschwend, P. S. Fennell, T. M. Lammens, B. Tan, J. Weale and J. P. Hallett, Green Chem., 2017, 19, 3078–3102 RSC.
- J. a. Whitehead, J. Zhang, N. Pereira, a. McCluskey and G. a. Lawrance, Hydrometallurgy, 2007, 88, 109–120 CrossRef CAS.
- J. a. Whitehead, G. a. Lawrance and A. McCluskey, Green Chem., 2004, 6, 313–315 RSC.
- C. H. C. Janssen, N. A. Macías-Ruvalcaba, M. Aguilar-Martínez and M. N. Kobrak, Sep. Purif. Technol., 2016, 168, 275–283 CrossRef CAS.
- M. Abai, M. P. Atkins, A. Hassan, J. D. Holbrey, Y. Kuah, P. Nockemann, A. a. Oliferenko, N. V. Plechkova, S. Rafeen, A. a. Rahman, R. Ramli, S. M. Shariff, K. R. Seddon, G. Srinivasan and Y. Zou, Dalton Trans., 2015, 44, 8617–8624 RSC.
- S. Rabieh, M. Bagheri and B. Planer-Friedrich, Microchim. Acta, 2013, 180, 415–421 CrossRef CAS.
- A. P. Abbott and K. J. McKenzie, Phys. Chem. Chem. Phys., 2006, 8, 4265–4279 RSC.
- P. Verdía, A. Brandt, J. P. Hallett, M. J. Ray and T. Welton, Green Chem., 2014, 16, 1617 RSC.
- F. J. V. Gschwend, A. Brandt, C. L. Chambon, W.-C. Tu, L. Weigand and J. P. Hallett, J. Visualized Exp., 2016, e54246–e54246 Search PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of Structural Carbohydrates and Lignin in Biomass, 2012 Search PubMed.
- M. G. Resch, J. O. Baker and S. R. Decker, Low Solids Enzymatic Saccharification of Lignocellulosic Biomass Laboratory Analytical Procedure (LAP), 2015 Search PubMed.
- B. J. Cox and J. G. Ekerdt, Bioresour. Technol., 2013, 134, 59–65 CrossRef CAS PubMed.
- L. Chen, M. Sharifzadeh, N. Mac Dowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098 RSC.
- A. Brandt, L. Chen, B. E. van Dongen, T. Welton and J. P. Hallett, Green Chem., 2015, 17, 5019–5034 RSC.
- F. J. V. Gschwend, C. L. Chambon, M. Biedka, A. Brandt-Talbot, P. S. Fennell and J. P. Hallett, Green Chem., 2019, 21, 692–703 RSC.
- S. Labbe, Z. Zhu and D. J. Thiele, J. Biol. Chem., 1997, 272, 15951–15958 CrossRef CAS PubMed.
- L. Yu, H. Sun, J. He, D. Wang, X. Jin, X. Hu and G. Z. Chen, Electrochem. Commun., 2007, 9, 1374–1381 CrossRef CAS.
- Standard Potentials in Aqueous Solution, ed. A. J. Bard, R. Parsons and J. Jordan, Marcel Dekker, New York, 1985 Search PubMed.
- S.-Q. Huang, Z. Huang, T.-A. Gu, Q.-J. Xie and S.-Z. Yao, Chin. J. Anal. Chem., 2011, 39, 978–984 CAS.
- I. A. Menzies and L. W. Owen, Electrochim. Acta, 1966, 11, 251–265 CrossRef.
- F. Brusciotti and P. Duby, Electrochim. Acta, 2007, 52, 6644–6649 CrossRef CAS.
- H. Cao, H. Shan, H. Ruan and G. Zheng, Electrochem. Commun., 2012, 23, 44–47 CrossRef CAS.
- Wood waste: A short review of recent research, Department of the Environment, Food and Rural Affairs (DEFRA), 2012, pp. 1–29 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Compositional analysis, lignin analysis and metal recovery on recycling. See DOI: 10.1039/d0gc01241f |
| ‡ Current address: Universidad de Antioquia, SIU, Calle 62 No 52-59, Torre 2, Lab. 330. Medellin, Colombia. |
| This journal is © The Royal Society of Chemistry 2020 |