
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalytic esterification of corn starches towards enhanced thermal stability and moisture resistance†
Balázs
Imre
 and
Francisco
Vilaplana
*
and
Francisco
Vilaplana
*
Division of Glycoscience, Department of Chemistry, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, Stockholm, Sweden. E-mail: franvila@kth.se
First published on 1st July 2020
Abstract
Most commercial starch esters are currently produced under harsh alkaline conditions. Triggered by environmental concerns, organocatalysis has recently emerged as a sustainable alternative offering mild reaction conditions, reduced waste formation and high selectivity. In this paper, the catalytic effect of several organic acids for the heterogeneous esterification of corn starch is compared, followed by a systematic analysis of tartaric acid-catalysed reactions on substrates with varying amylose/amylopectin ratios using a range of carboxylic acid and anhydride reagents of different chain lengths. Reaction conditions (temperature, time and catalyst concentration) were optimized and products were thoroughly characterized in terms of degree of substitution, chemical structure, morphology, moisture uptake and thermal stability. Organocatalysis proved to be competitive with conventional, alkaline methods. Our study also demonstrates that sustainability can further be improved by the direct recycling of the liquid phase. Notably, however, the tartaric acid catalyst participates in substitution reactions with starch. Despite recent claims to the opposite, reactivity of the carboxylic acid derivatives is primarily determined by carbon chain-length, while reagent type and therefore water content in the reaction mixture has a crucial effect on the final properties of the product. Our results highlight the importance of selecting appropriate synthesis methods and conditions as well as characterization techniques in order to create materials with the desired property profile according to the targeted application.
Introduction
Along with other plant polysaccharides such as cellulose and hemicelluloses, starch is among the most abundant macromolecules present in biomass,1 thus serving as a fundamental renewable resource for the replacement of fossil-based polymeric materials in commodity and engineering applications. Native starch, however, has several disadvantages compared with its synthetic counterparts that hinder its direct application. It is not intrinsically thermoplastic, i.e. cannot be processed in the melt state without chemical modification and/or plasticization. Plasticizers, however, have a tendency of migrating to the surface, resulting in unstable mechanical characteristics over time. Thermoplastic starch is also prone to slow recrystallisation after processing that leads to the progressive embrittlement of the material.2 Another consequence of its rather polar, hydrophilic nature is moisture sensitivity; starch generally contains a significant amount of water, depending on environmental conditions such as temperature and humidity. This can lead to considerable hydrolysis and molar mass decrease during processing and to unstable properties during application.The recalcitrant nature of plant polysaccharides is a consequence of strong intra- and intermolecular interactions often accompanied by a high degree of crystallinity. Starch is a semicrystalline polysaccharide that consists of two types of macromolecules, both having mainly α-D-(1 → 4) anhydroglucose repeating units (Fig. 1a). Amylose, an almost-linear polymer with a molar mass in the range of 105–106 g mol−1, is typically the minor component. Amylopectin, on the other hand, has a hyperbranched structure with α-D-(1 → 6) glycosidic linkages at the branching points and can reach very large molar masses over 108 g mol−1. The ratio of amylose in natural starches is typically ca. 20–25%, but other varieties with higher amylose contents (high-amylose starches) or with virtually no amylose (waxy starches) are also common.
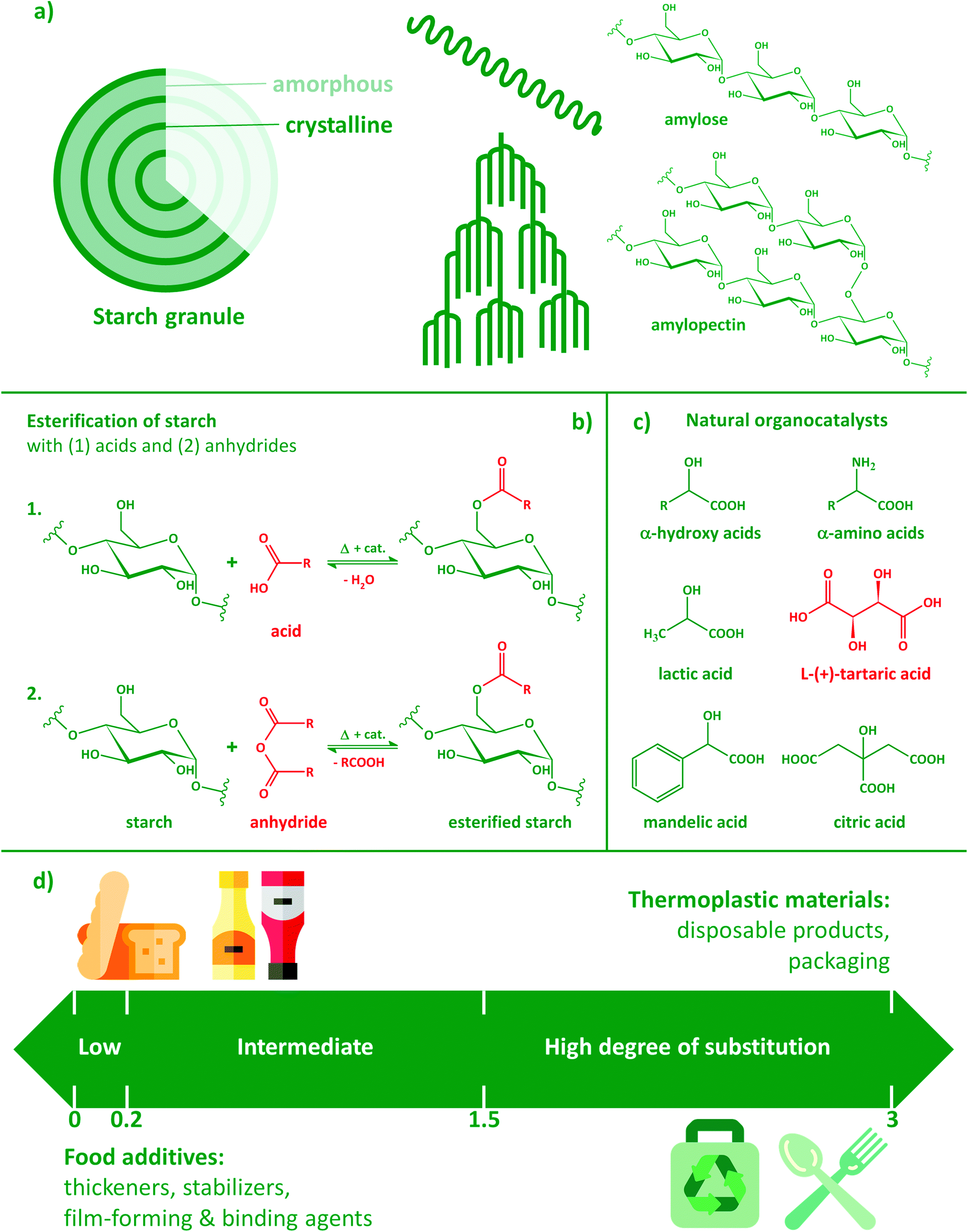 | ||
| Fig. 1 Native starch and its esterification: (a) the granular structure of native starch; (b) esterification with acids and anhydrides; (c) natural organocatalysts; (d) application areas of modified starches with different degrees of substitution. | ||
The crystalline polymorphs of starch can be classified into A, B and C forms, the latter being a mixture of the first two.3,4 The main difference between A and B type crystallites is that the unit cell of the A-type polymorph is monoclinic, whereas that of the B type is hexagonal.5,6 Therefore, the former adopts a close-packed arrangement with water molecules between each double helical structure, while the B-type is more open and contains more water, most of which is located in a central cavity surrounded by six double helices. The formation of different polymorphs is attributed to the amylopectin branch chain-lengths of starches from different sources.7 Another important factor is the ratio of amylose and amylopectin: high-amylopectin (waxy) starches mainly consist of A-type crystallites, while the ratio of B-type crystallites increases as a function of amylose-content.
Polysaccharides are often applied in a derivatized form and/or in combination with other biobased polymers. The substitution of hydroxyl groups of plant polysaccharides has a long history: cellulose acetate (CA) was first described as early as 1865,8,9 while extensive studies on the chemical modification of starch were performed in the early 1940s.10 The origin of the polysaccharide substrate, methods, conditions and types of reagents all have a significant effect on the outcome of the modification. Products are often characterized by their degree of substitution (DS), i.e. the average number of hydroxyls replaced by other moieties per repeating unit. In the case of starch, this value ranges from 0 to 3, a DS of 3 meaning that on average all three hydroxyl groups of the anhydroglucose repeating units are substituted. Starches having a low DS of about 0.01–0.2 are widely used as adhesive, thickening, texturizing, film-forming, stabilizing, and binding agents and find numerous applications in the food industry (Fig. 1d). Starches esterified with short chain fatty acids (SCFA) such as acetate, propionate, and butyrate have the potential to support the maintenance of a healthy gut and to reduce risk of gut inflammation as well as colorectal cancer.11,12 Acetylated starches with intermediate (0.2–1.5) and high DS (1.5–3.0) have been reported to be readily soluble in acetone and chloroform, and can be used as thermoplastic materials.13
Esterification is typically performed using acids, acid chlorides or anhydrides (Fig. 1b). With the increasing proportion of acetyl moieties or longer nonpolar side-groups, besides becoming more hydrophobic, starch may also lose its original, highly crystalline granular structure. Due to the disruption of inter- and intra-molecular hydrogen bonds induced by the progressive replacement of –OH groups, the surface of the particles first starts to show increased roughness. Finally, the complete disruption of the granular structure and the fusion of the particles might also occur.
Currently, most commercial starch esters are produced by the reaction of starch with anhydrides and sodium hydroxide as catalyst in aqueous medium at pH 7–9.14 Methodologies based on esterification catalysed by pyridine are also well-known, although the high cost and toxicity of the catalyst limits the commercial development of this technology.15 Triggered by environmental concerns, many current research efforts in organic synthesis are focused on the development of more sustainable chemical processes.16 Organocatalysis has emerged during the last decade as a concept with a significant number of synthetic applications. Important driving forces for this growth are rather mild reaction conditions, reduced waste formation and potentially high selectivity.17 Therefore, organocatalysts have been described as “minimal hydrolases”, displaying catalytic performances analogous to lipases or esterases for synthetic purposes such as transesterification or esterification.
Organocatalysts may have various chemical structures, including molecules such as 4-dimethylaminopyridine (DMAP), N-heterocyclic carbenes, guanidines, and bifunctional thiourea-amines. Compounds directly derived from natural resources, such as α-hydroxycarboxylic acids and amino acids (Fig. 1c), show particular promise as catalysts due to several inherent advantages. Many of these molecules (e.g., lactic, citric and tartaric acids [TA], or amino acids) can be directly derived from natural resources or produced at large scale by biotechnological routes in a straightforward and cost-effective manner.17 Moreover, they are fully biodegradable and neither toxic nor harmful to the environment.
The organocatalytic esterification of starch has been recently explored by several studies, focusing exclusively on tartaric acid12,18,19 as a sustainable catalyst of natural origin, based on its favourable effect on the ring-opening polymerization (ROP) of lactones.20 For the first time, we report here a comparison of the catalytic effect of TA and a range of other natural organic acids specifically for the heterogeneous esterification of corn starch. We systematically explore the role of starch substrate structure and reagent type in TA-catalysed esterification processes, providing an in-depth analysis of chemical structure, crystallinity, micro-scale morphology, and thermal stability of the modified starches, using corn starch grades with different crystalline structures and amylose/amylopectin ratios as well as several acid and anhydride reagents of different aliphatic chain lengths. By tightly controlling the synthesis process, the macroscopic properties of esterified starch can be tailored for various applications including food additives, surfactants as well as structural materials in combination with thermoplastic polymers.
Results and discussion
Comparison of potential natural organocatalysts
Natural organocatalysts are commonly considered for the ROP of lactones; their potential for the direct esterification of polymers such as polysaccharides, however, is seldom investigated. In this study, eight carboxylic acids of natural origin (fumaric,21 glycolic, L-lactic,20L-malic,22L-tartaric,18,20,23 citric,20L-aspartic24 acids and L-proline20,25) were selected to evaluate their effect on the esterification of corn starch with acetic anhydride based on earlier publications suggesting them as potential organocatalysts, while two more were added to the list of candidates due to their similar structures (maleic and L-glutamic acids). Among them, one can distinguish dicarboxylic acids, hydroxy mono-, di and tricarboxylic acids as well as amino acids. Their chemical structures as well as the degrees of acetylation of esterified corn starches prepared using these compounds as catalysts are presented in Fig. 2a.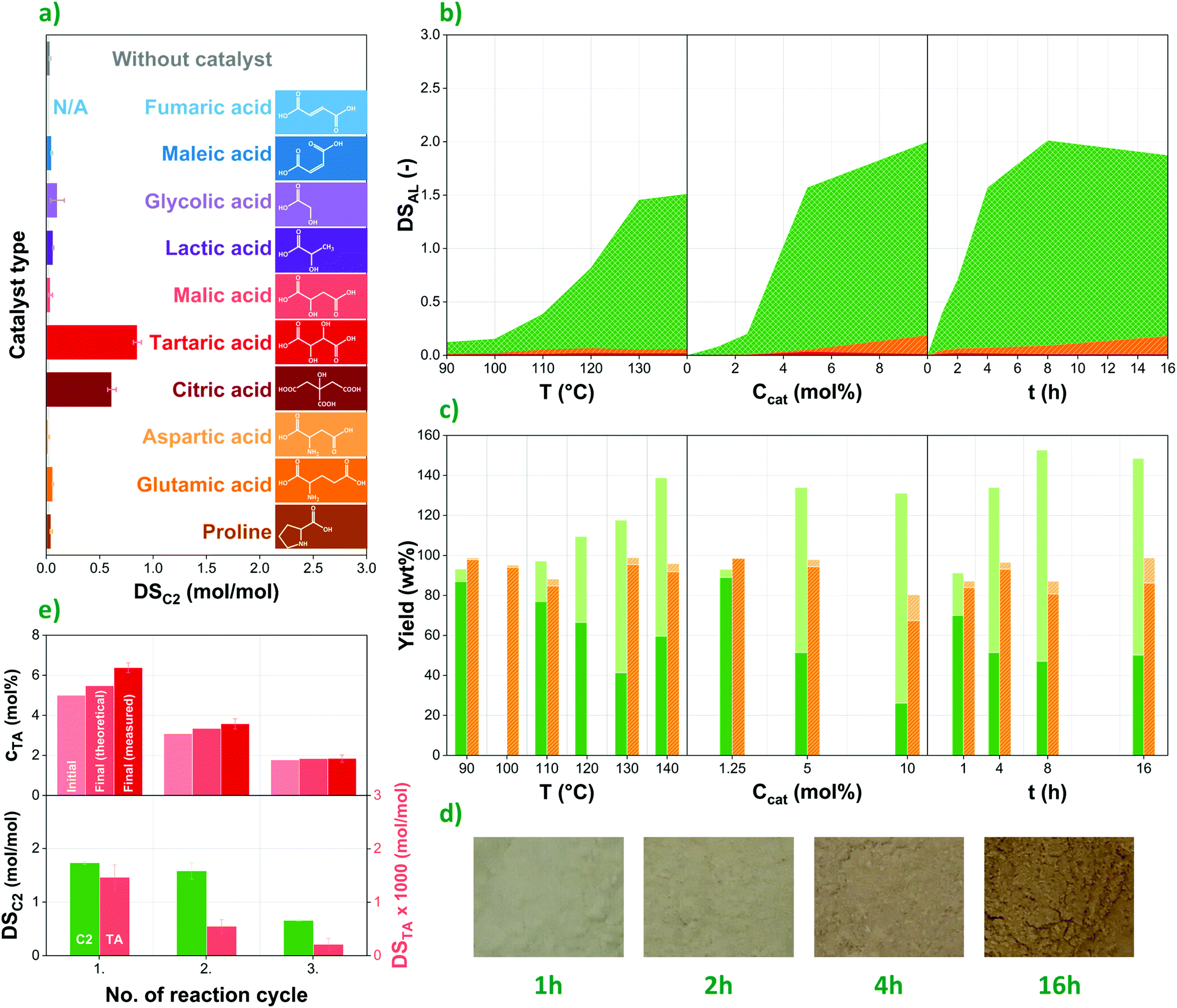 | ||
| Fig. 2 Organocatalytic esterification of starch: (a) the catalytic effect of organic acids as potential organocatalysts (acetic anhydride reagent, 120 °C, 5 mol% catalyst content and 4 h reaction time); (b) DSAL as a function of temperature (5 mol% catalyst, 4 h), catalyst concentration (130 °C, 4 h), and reaction time (5 mol% catalyst, 130 °C) during organocatalytic esterification performed with acetic (green, diagonal grid), propionic (orange, diagonally striped), and butyric anhydride (red, solid); (c) Net (dark) and total yield (light) during esterification with acetic (green, solid) and propionic anhydride (orange, diagonally striped); (d) appearance of the samples after 1, 2, 4 and 16 h acylation in acetic anhydride (at 5 mol% catalyst concentration and 130 °C); (e) recyclability of the liquid phase in terms of catalyst concentration and degrees of substitution in the respective reaction cycles. | ||
Degree of acetylation (DSC2), i.e. the number of acetyl groups per anhydroglucose units can range between 0 and 3 in modified corn starch. Among the potential organocatalysts tested, only tartaric and citric acids showed considerable catalytic effect, reaching DSC2 values of 0.85 and 0.62, respectively. In all other cases, the degree of acetylation does not significantly exceed the one achieved without using any catalyst (0.04), with the possible exception of glycolic (0.10), lactic (0.07) and glutamic acids (0.06). This might suggest that the presence of multiple carboxyl groups and hydroxyl moieties in α-position facilitate the esterification process. Still, very low DS values were achieved with malic acid, for instance, despite its similar structure, highlighting the challenges with establishing a clear connection between chemical structure and catalytic effect. Therefore, at this point we refrain from drawing more general conclusions. Nevertheless, the results presented in Fig. 2a highlight the necessity of more detailed investigations into the catalytic effect of natural organic acids specifically for esterification purposes, as several of these compounds perform rather differently in the ROP of lactones.20
Optimization of reaction conditions
Tartaric acid was selected to study the effect of various parameters on the organocatalytic esterification of corn starch, due to its high catalytic effect as well as itself not being reactive with starch to a large extent, unlike citric acid that is commonly used as a cross-linking agent in polysaccharides.26,27 In order to investigate the effect of reaction conditions on the final degree of esterification, reaction temperature, catalyst concentration in the liquid phase as well as reaction time were changed systematically. The results collected in Fig. 2b suggest that DSAL (DS of the respective alkanoyl groups, depending on the reagent used) increases as a function of all these factors, in accordance with earlier findings.19 Reactivity rises greatly between 110 and 130 °C, while minimal improvement can be achieved by further elevating temperature. Naturally, this parameter is also limited by the potential degradation and thus molar mass decrease of the polymer. This latter is to some degree reflected in the yield data presented in Fig. 2c. A similar correlation can be observed as a function of catalyst content. Notably, no esterification at all was observed in the absence of tartaric acid, confirming the catalytic effect of this natural organic compound. Modified starches tend to have a beige to light dark colour that turns gradually darker as the reaction proceeds (Fig. 2d).The degree of esterification continuously increases over time up to 8 h, reaching a maximum DS of ca. 2 (67%) when acetic anhydride is used as reagent, far exceeding 0.15 (5%) and 0.05 (1.6%) measured for propionic and butyric anhydrides, respectively. Such a trend was anticipated based on earlier studies that show decreasing reactivity of carboxylic acids in esterification reactions as a function of carbon chain length.28 Nevertheless, the drastic reduction of final DS values for propionic and butyric anhydrides suggest that tartaric acid-catalysed esterification is only able to provide medium- to high-DS products when acetic anhydride or similar, highly reactive reagent is used.
The comparison of product yields achieved with acetic (Fig. 2c, green, solid) and propionic anhydride (Fig. 2c, orange, diagonally striped) provides further insight into the performance of the reagents. Total yields (light columns in Fig. 2c) are based on the weight of the esterified starch product compared with that of unmodified starch at the beginning of the reaction. Therefore, this value includes the molar mass increase of the polymer due to substituting its hydroxyl groups with acetyl and propionyl moieties, respectively. Due to the higher degrees of substitution achieved, total yields are around or above 100% in the case of acetic anhydride and tend to increase as a function of all three reaction parameters. With propionic anhydride that has much lower reactivity, on the other hand, total yields do not show the same increasing trend and fluctuate between 80 and 100%, showing rather similar values to those of net yield (dark columns in Fig. 2c). This latter parameter does not include the molar mass increase of starch, and thus better reflects the amount of polysaccharide lost due to degradation and during the purification process. The results suggest that the use of acetic anhydride as reagent, besides higher DS values, causes more degradation as well, which is in accordance with the molar mass data presented in Fig. 4e. Net yields for acetylated starches range between 20 and 80%, reaching their lowest values under harsh reaction conditions. One must also keep in mind, however, that in part this might be caused by morphological changes, i.e. the breaking down of starch granules during the reaction, and thus losing a higher proportion of smaller particles during membrane filtration. While inefficient transportation and separation methods under laboratory conditions surely add to the amount of starch lost in the modification process, low yields remain a reason for concern, especially when high degrees of esterification are desired, necessitating harsher conditions and longer reaction times.
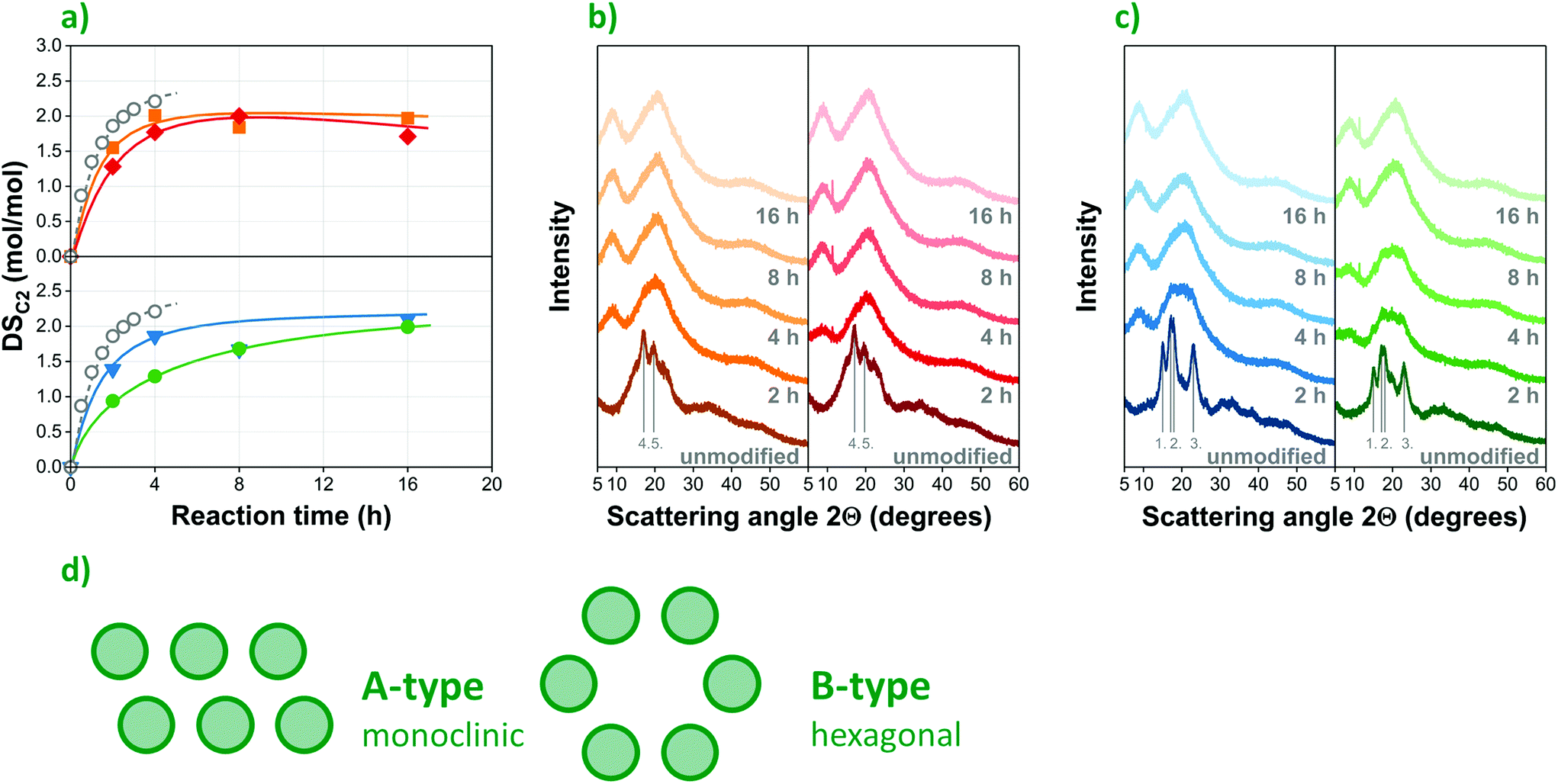 | ||
| Fig. 3 Organocatalytic acetylation of corn starch substrates with different amylose/amylopectin ratios: (a) progression of DSC2 over time for waxy (▼) native (●) and high-amylose (■, ◆) starches compared to conventional acetylation under alkaline aqueous conditions14 (○); X-ray diffraction (XRD) spectra of (b) high-amylopectin (left: waxy, right: native) and (c) high-amylose (left: A1, right: A2) starches; (d) the schematic structure of A and B-type crystalline polymorphs. | ||
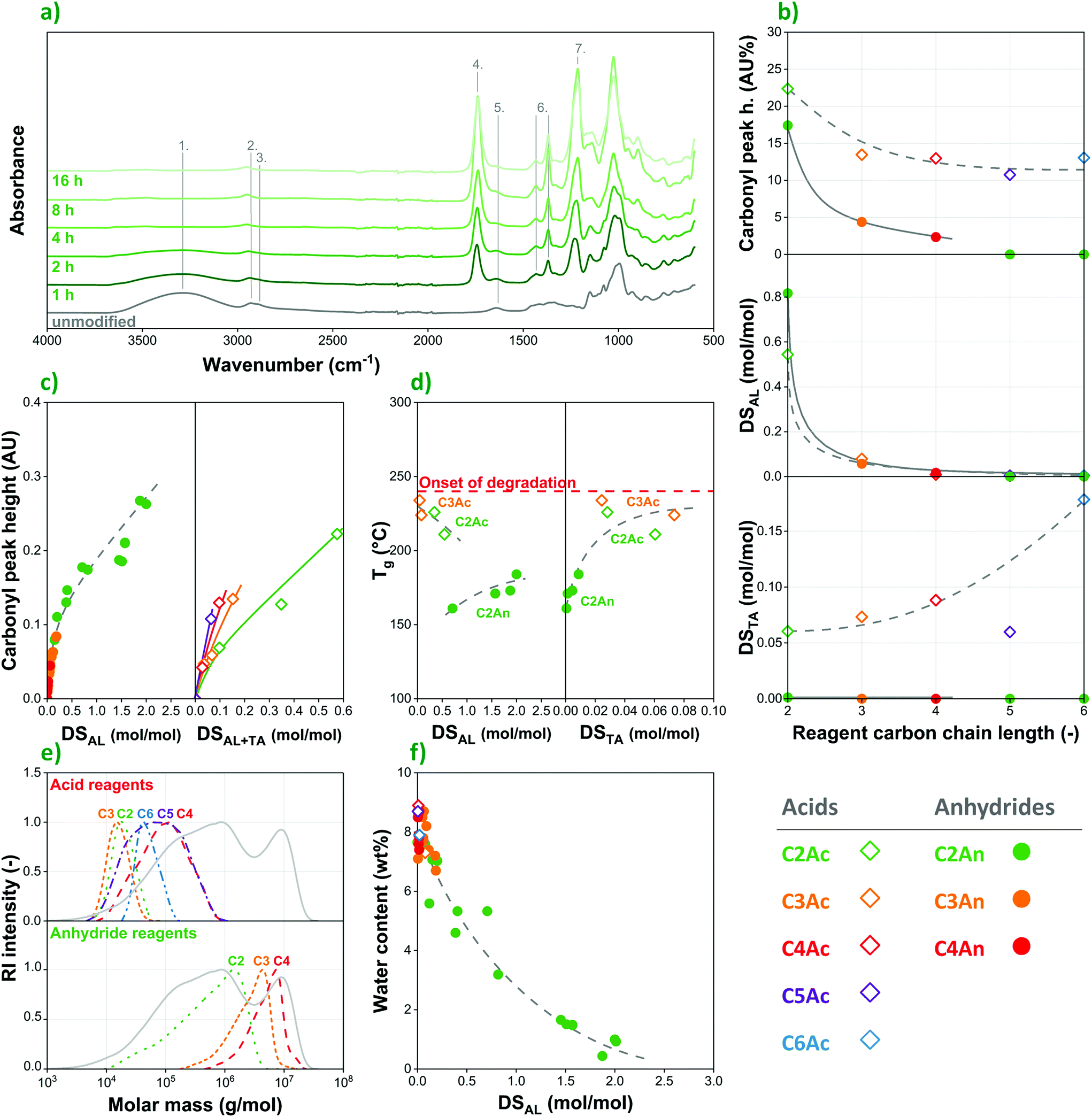 | ||
Fig. 4 Characteristics of acid and anhydride-modified native corn starches: (a) Fourier-transform infrared (FTIR) spectra of acetylated starches (acetic anhydride reagent, 130 °C, 5 mol% catalyst), labelled absorption bands: O–H stretch (1), CH2 asymmetric stretch (2), CH2 symmetric stretch (3), H-bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (4), amide I (5), alkyl CH3 bend (6), ester C–O stretch (7); (b) carbonyl peak height compared with the DS of alkanoyl (DSAL) and tartaryl (DSTA) side groups at different carbon chain lengths; (c) carbonyl peak height as a function of DS determined by HPLC; (d) glass transition temperatures; (e) molar mass distributions; (f) equilibrium water content at 42% RH. O (4), amide I (5), alkyl CH3 bend (6), ester C–O stretch (7); (b) carbonyl peak height compared with the DS of alkanoyl (DSAL) and tartaryl (DSTA) side groups at different carbon chain lengths; (c) carbonyl peak height as a function of DS determined by HPLC; (d) glass transition temperatures; (e) molar mass distributions; (f) equilibrium water content at 42% RH. | ||
It is also worthy to note that the degree of esterification does not exceed 2 (67%) in any of the reactions performed, regardless of reaction conditions and type of reagent. These results are in agreement with the values obtained by Tupa et al.19 for dry starch, suggesting that one hydroxyl group has lower reactivity and is therefore not esterified in the process. On the other hand, as we discuss below in more detail, the back-titration method applied by Tupa et al. is prone to overestimate degrees of esterification at higher moisture contents due to substitution reactions between starch and the tartaric acid catalyst.
Recyclability
Experiments were performed also to investigate the direct recyclability of the liquid phase, i.e. both the catalyst and the reagent, thereby improving the sustainability of the process. Three subsequent cycles of reactions were performed, in each new cycle reusing the liquid phase (tartaric acid/acetic anhydride solution) recovered from the previous one. The liquid lost during the reaction and filtration was compensated by adding an appropriate amount of fresh acetic anhydride, keeping the amount of liquid phase constant in each cycle, while also resulting in gradually decreasing tartaric acid concentrations. This latter parameter was measured as ca. 5, 3, and 2 mol% in the first, second, and third reactions, respectively (see Fig. 2e, above). Within one cycle, on the other hand, catalyst concentration increases in the liquid phase, as mainly acetic anhydride is consumed in the reaction. Final TA concentrations determined by HPLC slightly exceed theoretical values calculated from anhydride consumption, suggesting that the reagent is lost in higher amounts due to evaporation than the catalyst.Most importantly, however, results suggest that the catalyst as well as the reagent can be recovered and remain active in the recycled liquid phase to be used for esterification in subsequent reaction cycles. The decreasing degrees of substitution with respect to acetyl (DSC2) as well as tartaryl groups (DSTA) in Fig. 2e (below) seem to be primarily determined by catalyst concentration and follow a similar trend to the one observed in the case of using fresh raw materials (Fig. 2b). The somewhat higher degrees of acetylation achieved with the recycled liquid phase are likely due to the increasing concentration of water facilitating the esterification process. Nevertheless, although directly recycling the liquid phase might be a convenient and cost-effective way of improving sustainability, better control of the process can probably be achieved by including separation steps in order to recover components in their pure forms. Further information on the recycling experiments is available in Fig. S1 in the ESI.†
Esterification of corn starch substrates with different amylose/amylopectin ratios
Differences in the chemical composition as well as the crystalline structure of the starch substrate may also affect the outcome of chemical modifications29,30 such as organocatalytic esterification. The DSC2 values of acetylated corn starches determined by saponification followed by HPLC measurements at different reaction times largely confirm this. The results collected in Fig. 4a provide a comparison of the acetylation of four substrates, i.e. waxy (W, ▼), native (N, ●) and two high-amylose (A1, ■ and A2, ◆) corn starches with amylose contents ranging from 0 to 70%, as listed in Table S1.† The respective average molar masses and molar mass distributions of the commercial starches investigated are presented in Fig. S2 in the ESI.† Based on the optimized reaction conditions previously presented, a constant reaction temperature of 130 °C and a catalyst concentration of 5 mol% were selected for investigating the progression of esterification over time. Samples were prepared with reaction times between 1 and 16 hours, using acetic anhydride as reagent. For the sake of comparison, a fifth dataset (○) is also included, showing the results of a conventional acetylation process performed under alkaline conditions, as published by Xu, Miladinov & Hanna.14Remarkably, the DSC2 values obtained in the organocatalytic process (▼, ●, ■, ◆) are rather close to the outcome of conventional esterification (○) within the same timeframe, signalling the efficiency of tartaric acid as an environmentally friendly organocatalyst. This seems even more to be the case when high-amylose corn starches (Fig. 3a, above) are used as substrates. While further optimization might lead to improved reaction rates, any difference observed might also partially result from the different measurement techniques used to determine the degree of esterification, as the data published by Xu et al.14 was obtained by back titration with hydrochloric acid, while HPLC was used in this study.
Although the maximum degree of substitution is similar for the 4 starches, significant differences can be observed in the progression of acetylation over time, i.e. in reaction kinetics. The esterification of the high-amylose (■, ◆) samples proceeds at a higher rate, and reaches its maximum (ca. 2 or 67%) at low reaction times (ca. 4 h). Waxy corn starch (▼) is esterified at a slightly slower rate, while the native sample (●) proved to be the most recalcitrant, reaching the same maximum DSC2 only after 16 h reaction time.
By all likelihood, the observed differences in reaction kinetics are caused by the different amylose/amylopectin ratios and the resulting structural diversity of the substrates. While the rapid progress of acetylation in high-amylose starches with a looser hexagonal (B-type, see Fig. 3d) crystalline structure was anticipated, the similar kinetics observed in the case of waxy starch with 100 wt% amylopectin content and a more compact monoclinic (A-type in Fig. 3d) arrangement seems counterintuitive at first. One would expect the latter substrate to be even more recalcitrant than its native counterpart that has a similar crystalline structure but contains less amylopectin (75 wt%). This trend can be explained by further structural differences. In the starch granule, amylopectin and amylose molecules are radially oriented in alternating crystalline and amorphous regions from the hilum to the periphery, contributing to the semi-crystalline structure of the particles. The branch-chains of amylopectin are assumed to be organized in parallel (as depicted in Fig. 1a) to form crystalline regions31,32 stabilized by hydrogen bonds and hydrophobic interactions between double helices of branch-chains.33 Amylose, on the other hand, is interdispersed among amylopectin molecules, contributing to the integrity of the granular structure by intertwining with amylopectin.34 The important role that amylose plays in stabilizing the semicrystalline structure of starch provides a plausible explanation for the apparent recalcitrance of native starch compared with its waxy counterpart consisting of 100% amylopectin.
This is further corroborated by the gradual change of crystallinity followed by X-ray diffraction (XRD), as presented in Fig. 3b and c for high-amylose and high-amylopectin starches, respectively. Its high degree of crystallinity initially makes starch resilient towards chemical modification. The elevated temperature and acidic environment applied during acylation, however, gradually break down the crystallites, making the hydroxyl groups more accessible to the acylating agent. In the XRD spectra, both native and waxy starch (Fig. 3c) show a typical A-type crystalline structure, distinguished by sharp and intense diffraction peaks at 15° and 23° 2θ angles as well as a characteristic double peak at 17°. High-amylose starches (Fig. 3b), on the other hand, have a single peak at 17° in their spectra accompanied by another intense peak close to 20°, indicating the presence of more loosely arranged B-type crystallites. Gradually reducing peak intensities suggest the breaking down of the intrinsic semicrystalline structure of starch due to the acylation process. This, however, takes place at different rates, in accordance with the reaction kinetics shown in Fig. 3a: modification results in a complete loss of crystallinity in the high-amylose samples (Fig. 3b) already after two hours of reaction, while high-amylopectin samples (Fig. 3c), and native starch in particular, proved to be more resilient. Although unmodified waxy starch shows more intense peaks suggesting a higher degree of crystallinity initially, its crystalline structure breaks down completely after 4 hours of esterification, while native corn starch requires ca. 8 hours to become fully amorphous. These results clearly confirm the importance of structural phenomena in determining the kinetics of the reaction and highlight the significant role amylose plays in stabilizing the semicrystalline structure of granular starch.
Comparison of anhydride- and acid-type reagents in the organocatalytic acylation of starch
In order to investigate the relative reactivity of different reagent types in tartaric acid-catalyzed esterification, trials with a series of 1-substituted monocarboxylic acid (carbon chain length: 2–6) and anhydride (carbon chain-length 2–4) derivatives of n-alkanes have been performed, as listed in Table 1. The main difference between the two series of reagents is that while acylation with an anhydride results in the formation of the respective acid as side-product, acylation with an acid is accompanied by the elimination of water (see Fig. 1b). A desirable effect of water can be that it is able to plasticize starch, effectively breaking down its highly crystalline, granular structure, thereby enhancing the accessibility of its hydroxyl groups to the reagent and accelerating the acylation reaction. Nevertheless, acidic environment, high temperature and moisture content can also lead to extensive hydrolytic degradation of the polysaccharide substrate. Therefore, besides examining morphological changes and determining the degree of substitution, molar mass of the product also needs to be considered in every case. In the below sections, all these aspects of the tartaric acid-catalysed esterification of native corn starch are discussed in detail.| Reagent | Abbreviation | Carbon-chain length | pKa |
|---|---|---|---|
| Acetic acid | C2Ac | 2 | 4.76 |
| Propionic acid | C3Ac | 3 | 4.88 |
| Butyric acid | C4Ac | 4 | 4.82 |
| Valeric acid | C5Ac | 5 | 4.82 |
| Hexanoic acid | C6Ac | 6 | 4.88 |
| Acetic anhydride | C2An | 2 | — |
| Propionic anhydride | C3An | 3 | — |
| Butyric anhydride | C4An | 4 | — |
O) appears at 1740 cm−1. The substitution reaction is further confirmed by the appearance of alkyl –CH3 bands (6) at 1430 and 1370 cm−1 as well as an ester C–O stretch peak (7) at 1210 cm−1. The latter absorption bands are absent in the spectrum recorded on the unmodified polysaccharide, while their intensity increases significantly as a function of reaction time.
In order to accurately interpret the outcome of esterification reactions, one must consider that, despite their convenience, infrared spectroscopy and back titration also have some important shortcomings. Most notably, these methods are not able to easily differentiate between carboxylic acid species and the related side-groups on the polymer backbone. Since not only the carboxylic acid and anhydride reagents, but also the tartaric acid catalyst can participate in substitution reactions, HPLC measurements were performed on saponified samples in order to determine the exact nature and amount of the side-groups attached to the starch substrate. For the easier comparison with FTIR data, these results are also included in Fig. 4b. According to HPLC measurements, DSAL values for the respective alkanoyl groups of the reagent are largely the same in starches modified with acids or anhydrides, while reactivity is in both cases primarily determined by carbon chain-length.
Furthermore, the data clearly confirm that the tartaric acid catalyst participates in substitution reactions with starch, competing with the acid reagents, and to a much lesser extent also with anhydrides. The amount of tartaryl side-groups (DSTA in Fig. 4b) increases as reactivity of the reagent decreases, i.e. as a function of carbon chain-length, although DSTA remains rather low, under 0.2 even in the case of hexanoic acid. Nevertheless, due to its chemical structure (TA being a dicarboxylic acid), both titration methods and FTIR are prone to overestimate the DS of modified starches prepared according to the organocatalytic approach. Recently, Nielsen et al.12 came to a similar conclusion based on nuclear magnetic resonance (NMR) measurements. This phenomenon leads to artificially high DS values produced by these techniques especially when acid reagents or higher concentrations of water (resulting in hydrolysis of the anhydride reagent and thus acid formation) are applied that facilitate the formation of tartaryl side-groups on starch.
The information provided by FTIR using an attenuated total reflection (ATR) setup and HPLC differs also in that the former provides information on the surface of starch granules, while the latter determines DS in the bulk. If one plots carbonyl peak height in the infrared spectra as a function of DS determined by HPLC, the two values correlate rather well for the anhydride reagents (Fig. 4c, left), in which the concentration of tartaryl side-groups is largely negligible. It is worthy to note, however, that this trend is not linear: the infrared absorption of the carbonyl peaks increases rapidly at lower DS values, while the rise of the curve decreases at higher degrees of substitution. In other words, the results indicate more rapid esterification on the surface initially, while hydroxyl groups in the inside of the granules are exchanged at a lower rate. On the other hand, carbonyl peak height – DS correlations do not follow a single trendline in the case of the acid reagents (Fig. 4c, right). Infrared absorption of the carbonyl moieties increases at a higher rate for reagents with longer carbon chains, i.e. in the following order: acetic < propionic < butyric < valeric acid. This is due to the lower reactivity of the larger molecules, and thus the increasing concentration of tartaryl side-groups.
 | ||
| Fig. 5 Morphology of acid and anhydride-modified starches: scanning electron micrographs (2500× magnification) of (a) unmodified, (c) acetic (d) propionic, and (e) butyric anhydride as well as (f) acetic (g) propionic, and (h) butyric acid-treated native corn starch (reactions performed at 120 °C, 5 mol% catalyst content, 4 h reaction time); the respective XRD spectra of (b) unmodified as well as (i) acetic, (j) propionic, and (k) butyric acid-esterified starch. | ||
Unlike anhydrides, acid reagents drastically reduce the molar mass of corn starch. By all likelihood this can be explained by the presence and increasing amount of water forming as a side-product in substitution reactions with acids, resulting in the hydrolytic degradation of the polysaccharide. The extent of molar mass decrease depends on the carbon chain length of the carboxylic acid, although the correlation is not as clear as in the case of anhydrides. Weight average molar mass values under 105 g mol−1 were achieved in each case, which is anticipated to hinder the application of acid-modified starches as structural materials.
On the contrary, treatment with acid reagents results in highly amorphous, elongated particles that gradually coalesce and fuse with each other, even forming a fibrous structure in the case of acetic acid (Fig. 5f), similar to the one observed by Xu et al.14 Carbon chain-length plays an important role: reagents with shorter aliphatic chains have a much more pronounced effect on the morphology of the polysaccharide. This correlation can be attributed to the order of reactivity of the reagents; nevertheless, the considerably different morphology of acid- and anhydride-treated samples suggests that the loss of granular structure is more related to plasticization phenomena caused by water (and to a lesser extent by the respective carboxylic acid) during modification rather than the esterification process itself.
Although Singh et al.41 claims that the fusion of starch granules due to esterification could be attributed to the introduction of acetyl groups to starch resulting in an increase in hydrogen bonding, this explanation is rather counterintuitive. Acetylation creates increasingly nonpolar surfaces, thus reduced hydrogen bonding can be anticipated. The same authors, however, also mention the possibility of gelatinization due to the modification being conducted in aqueous medium in the presence of sodium hydroxide, which seems more plausible. Similar morphological features have been observed in studies that involve esterification of starch in aqueous media14,40 or using acid reagents,18 while no loss of granular structure took place when the modification was conducted in the absence of water18 such as in our study. This fact further corroborates that it is indeed plasticization and potential molar mass decrease that play a major role in determining the morphology of esterified starches, as opposed to esterification alone. Therefore, besides degree of substitution, other characteristics of the product as well as reaction conditions also need to be taken into consideration to a greater degree when tailoring the properties of modified starches.
The length of the aliphatic chain of the reagent plays a role in this case as well: while using acetic acid as reagent leads to the complete destruction of crystalline structure, acids with longer aliphatic chains do not affect crystallinity to the same extent. Notably, despite their significantly altered morphology indicated by the SEM micrographs presented Fig. 5g and h, propionic and butyric acid-modified starches both partially retain their crystalline structure. Besides the increasingly nonpolar nature of these compounds, this difference might also be explained by the stronger acidity of acetic acid (pKa: 4.76) compared with the other acid reagents tested (pKa: 4.88, 4.82, 4.82, and 4.88 for propionic, butyric, valeric, and caproic acid, respectively).
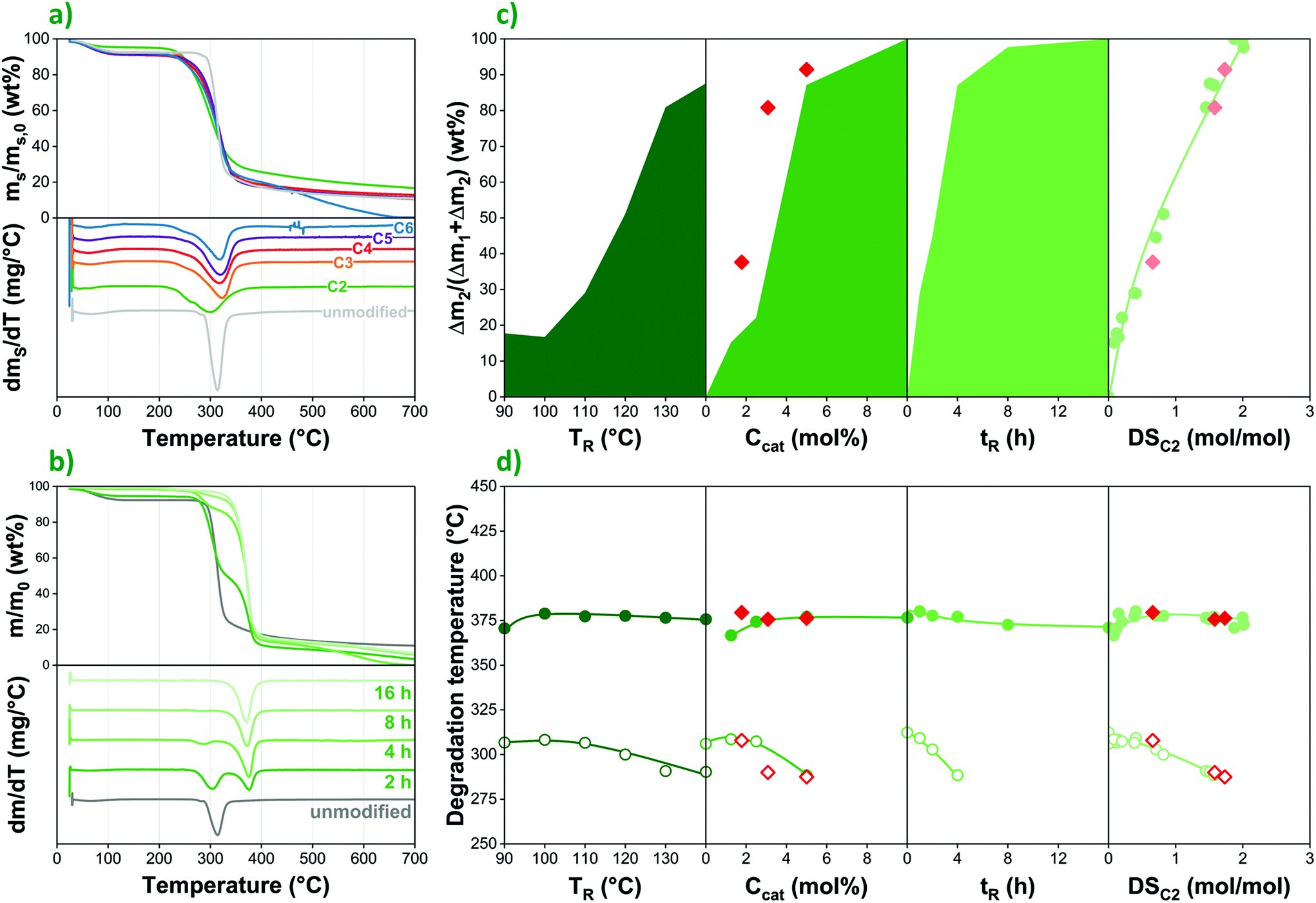 | ||
| Fig. 6 The thermal stability of esterified starches investigated by thermo-gravimetric analysis (TGA): mass loss and derivative mass loss of (a) acid-modified starches and (b) acetic anhydride-modified starches; (c) relative mass loss in the second degradation step; (d) degradation temperatures as a function of reaction temperature, catalyst concentration, time, and DS in acetic anhydride-modified native starch; ● and ○ refer to modified starches prepared with fresh raw materials, while ◆ and ◇ indicate using recycled liquid phase. | ||
In native corn starch esterified with an anhydride reagent (acetic anhydride in this case), one can observe a rather different thermal behaviour (Fig. 6b). In the modified samples, a second weight loss step appears at a higher temperature (ca. 370 vs. 320 °C peak temperature), which is assigned to the degradation of substituted anhydroglucose units.42 Accordingly, as reaction time and thus the degree of esterification increases, the lower temperature degradation step gradually disappears, and the weight loss process at ca. 370 °C becomes dominant. This can be seen in more detail as a function of reaction conditions as well as DS in Fig. 6c. The correlations clearly confirm that thermal degradation behaviour in the anhydride-treated samples is indeed determined primarily by the degree of esterification, regardless of the effect of other factors such as temperature or reaction time. The same trends can be observed in the case of acetylated starch prepared using the recycled liquid phase (◆ in Fig. 6c), providing further confirmation that both the catalyst and the reagent are directly recyclable; subsequent reaction cycles yield products with similar characteristics to the ones prepared using fresh raw materials.
Reaction parameters have a more pronounced effect on the temperature at which the degradation of unmodified repeating units occurs (labelled as TDU, while TDE stands for esterified repeating units). In Fig. 6d, degradation temperatures are plotted as a function of reaction conditions. While TDE is largely constant or only mildly affected by DS as well as other factors, the lower temperature process shifts to even lower temperatures, indicating that the molar mass decrease and the loss of crystalline structure taking place during modification makes unsubstituted chain sections and repeating units more prone to thermal degradation. Therefore, unless a high DS is achieved and thus the lower temperature degradation step is eliminated completely, considerable molar mass decrease can be expected during the processing of modified starches at elevated temperatures, despite the presence of an increasing amount of more thermally stable esterified repeating units.
Conclusions
In our present study, the organocatalytic esterification of corn starch grades with different amylose/amylopectin ratios was performed using a series of organic acid catalysts as well as monocarboxylic acid and anhydride reagents. Among the compounds investigated, tartaric acid showed the highest catalytic effect, providing similar degrees of substitution as conventional methods performed under alkaline, aqueous conditions. Reaction kinetics were found to be similar for the different starch substrates, although crystalline structure clearly affects reaction rates. The apparent recalcitrance of native starch highlights the important role of amylose in stabilizing the semicrystalline structure of starch. Despite claims to the opposite, carboxylic acids were not found to be more efficient as reagents than anhydrides, while reactivity is primarily determined by carbon chain-length. On the other hand, the tartaric acid catalyst appears to participate in substitution reactions with starch, competing with acid reagents, and to a much lesser extent also with anhydrides. Therefore, measurement techniques such as FTIR and back titration were found not to be appropriate for determining the DS of organocatalytically modified starches, and the use of HPLC is recommended instead.Unlike anhydrides, acid reagents drastically reduce the molar mass of corn starch due to the increasing amount of water forming as a side-product in substitution reactions with acids. The presence or absence of water in the reaction mixture also has a drastic effect on morphology and crystalline structure. While surface roughness increases upon acylation, even relatively high-DS anhydride-treated samples tend to retain their distinct granular structure in the absence of a significant amount of water. The thermal stability of acid-treated corn starch is largely determined by its reduced molar mass and lower degree of crystallinity, while a higher temperature degradation process assigned to the degradation of substituted anhydroglucose units appears in anhydride-modified ones. In these latter, thermal stability is improved by acylation, and correlates well with the degree of esterification.
The improved thermal stability and reduced moisture sensitivity of esterified starches might facilitate their application as biobased fillers in sustainable polyamide and polyester matrices for demanding structural applications. The use of benign, easily recoverable reagents and catalysts as opposed to conventional methods, on the other hand, highlights the possibility of implementing green chemistry principles into the production of starch esters.
Experimental
Materials
Four commercial corn starches with different amylose/amylopectin ratios were provided by Cargill Deutschland GmbH (Malchin, Germany). The waxy (CG 04201), native (CG 03401) and high-amylose (AG 03001 and AG 03003) products are labeled throughout the paper as W, N, A1 and A2, respectively. The four grades have amylose contents of 0, 25, 55 and 70 wt%, respectively, as collected in Table S1 in the ESI.† The tartaric acid (99.5%) used to catalyze the esterification reactions was supplied by VWR International, LLC. Acetic acid (99.8%) was purchased from Acros Organics, while propionic (99%), butyric (99%), valeric (99%), and hexanoic (98%) acids as well as acetic (99%), propionic (97%), and butyric (98%) anhydrides were supplied by Alfa Aesar. The abbreviations and carbon chain lengths of all reagents used in the study are collected in Table 1. Risks and hazards associated with the use of all the acid and anhydride reagents are also available in the ESI.†Organocatalytic acylation of the starch substrates
The desired amount of the catalyst (0–20 mmol) was first mixed with the anhydride (100 mmol) or acid reagent (200 mmol) in an oven-dried 25 ml glass round-bottom flask equipped with a magnetic stir bar and an Allihn-type reflux condenser. The flask was then submersed in a thermostatized oil bath (120 °C) for ca. 5 min in order to dissolve the catalyst. Subsequently, temperature was set to the desired value (90–150 °C), and freeze-dried corn starch (5 mmol) was added to the mixture. The addition of starch was considered as the start of the reaction, which was then conducted for 1–16 hours.The solid product was separated by vacuum filtration using a 0.2 μm PES membrane filter. Several washings of the recovered solid with ethanol were performed followed by dialysis in distilled water (48 h, 3.5 kDa membrane) in order to ensure the complete removal of the catalyst as well as any unreacted reagent. The dry solid product was obtained by freeze-drying.
Characterization of the modified starches
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was performed under the framework of the BARBARA project that has received funding from the Bio-Based Industries Joint Undertaking under the European Union's Horizon 2020 research and innovation programme under grant agreement no. 745578.References
- C. B. Field, M. J. Behrenfeld, J. T. Randerson and P. Falkowski, Science, 1998, 281, 237–240 CrossRef CAS PubMed.
- M. A. Huneault and H. Li, Polymer, 2007, 48, 270–280 CrossRef CAS.
- A. Imberty, A. Buléon, V. Tran and S. Péerez, Starch – Stärke, 1991, 43, 375–384 CrossRef CAS.
- A. Buleon, D. J. Gallant, B. Bouchet, G. Mouille, C. D'Hulst, J. Kossmann and S. Ball, Plant Physiol., 1997, 115, 949–957 CrossRef CAS PubMed.
- A. Buléon, P. Colonna, V. Planchot and S. Ball, Int. J. Biol. Macromol., 1998, 23, 85–112 CrossRef.
- H. W. Hsien-Chih and A. Sarko, Carbohydr. Res., 1978, 61, 27–40 CrossRef.
- J. Jane, in Chemical and Functional Properties of Food Saccharides, ed. P. Tomasik, CRC Press, New York, 2004, pp. 81–101 Search PubMed.
- P. Schützenberger, C. R. Acad. Hebd. Seances Acad. Sci., 1865, 61, 484–487 Search PubMed.
- J. Wisniak, Educ. Quim., 2015, 26, 57–65 CAS.
- R. L. Whistler, in Advances in Carbohydrate Chemistry, 1945, pp. 279–307 Search PubMed.
- F. Brouns, B. Kettlitz and E. Arrigoni, Trends Food Sci. Technol., 2002, 13, 251–261 CrossRef CAS.
- T. Nielsen, N. Canibe and F. Larsen, Foods, 2018, 7, 79 CrossRef PubMed.
- Zia-ud-Din, H. Xiong and P. Fei, Crit. Rev. Food Sci. Nutr., 2017, 57, 2691–2705 CrossRef CAS PubMed.
- Y. X. Xu, V. Miladinov and M. A. Hanna, Cereal Chem., 2004, 81, 735–740 CrossRef CAS.
- Đ. Ačkar, J. Babić, A. Jozinović, B. Miličević, S. Jokić, R. Miličević, M. Rajič and D. Šubarić, Molecules, 2015, 20, 19554–19570 CrossRef PubMed.
- Q. Chen, H. Yu, L. Wang, Z. ul Abdin, Y. Chen, J. Wang, W. Zhou, X. Yang, R. U. Khan, H. Zhang and X. Chen, RSC Adv., 2015, 5, 67459–67474 RSC.
- P. Domínguez de María, ChemCatChem, 2010, 2, 487–492 CrossRef.
- M. Tupa, L. Maldonado, A. Vázquez and M. L. Foresti, Carbohydr. Polym., 2013, 98, 349–357 CrossRef CAS PubMed.
- M. V. Tupa, J. A. Ávila Ramírez, A. Vázquez and M. L. Foresti, Food Chem., 2015, 170, 295–302 CrossRef CAS PubMed.
- J. Casas, P. V. Persson, T. Iversen and A. Córdova, Adv. Synth. Catal., 2004, 346, 1087–1089 CrossRef CAS.
- F. Zeng, H. Lee, M. Chidiac and C. Allen, Biomacromolecules, 2005, 6, 2140–2149 CrossRef CAS PubMed.
- S. B. Kasar and S. R. Thopate, Curr. Green Chem., 2018, 5, 177–184 CrossRef CAS.
- J. Hafrén and A. Córdova, Macromol. Rapid Commun., 2005, 26, 82–86 CrossRef.
- A. Ghorbani-Choghamarani, M. Hajjami, F. Gholamian and G. Azadi, Russ. J. Org. Chem., 2015, 51, 352–356 CrossRef CAS.
- J. Liu and L. Liu, Macromolecules, 2004, 37, 2674–2676 CrossRef CAS.
- N. Reddy and Y. Yang, Food Chem., 2010, 118, 702–711 CrossRef CAS.
- E. Olsson, M. S. Hedenqvist, C. Johansson and L. Järnström, Carbohydr. Polym., 2013, 94, 765–772 CrossRef CAS PubMed.
- Y. Liu, E. Lotero and J. G. Goodwin, J. Catal., 2006, 243, 221–228 CrossRef CAS.
- Z.-G. Luo and Y.-C. Shi, J. Agric. Food Chem., 2012, 60, 9468–9475 CrossRef CAS PubMed.
- R. Colussi, V. Z. Pinto, S. L. M. El Halal, N. L. Vanier, F. A. Villanova, R. Marques e Silva, E. da Rosa Zavareze and A. R. G. Dias, Carbohydr. Polym., 2014, 103, 405–413 CrossRef CAS PubMed.
- D. J. Gallant, B. Bouchet and P. M. Baldwin, Carbohydr. Polym., 1997, 32, 177–191 CrossRef CAS.
- H. Yangcheng, Characterization of normal and waxy corn starch for bioethanol production, Iowa State University, 2012 Search PubMed.
- A. Imberty, H. Chanzy, S. Pérez, A. Bulèon and V. Tran, J. Mol. Biol., 1988, 201, 365–378 CrossRef CAS PubMed.
- J. Jane, S. A. S. Craig, P. A. Seib and R. C. Hoseney, Starch – Stärke, 1986, 38, 258–263 CrossRef CAS.
- K. Ogawa, I. Hirai, C. Shimasaki, T. Yoshimura, S. Ono, S. Rengakuji, Y. Nakamura and I. Yamazaki, Bull. Chem. Soc. Jpn., 1999, 72, 2785–2790 CrossRef CAS.
- A. Biswas, R. L. L. Shogren, G. Selling, J. Salch, J. L. L. Willett and C. M. Buchanan, Carbohydr. Polym., 2008, 74, 137–141 CrossRef CAS.
- A. Mizuno, M. Mitsuiki and M. Motoki, J. Agric. Food Chem., 1998, 46, 98–103 CrossRef CAS PubMed.
- D. Heins, W. M. Kulicke, P. Kauper and H. Thielking, Starch-Starke, 1998, 50, 431–437 CrossRef CAS.
- N. Singh, D. Chawla and J. Singh, Food Chem., 2004, 86, 601–608 CrossRef CAS.
- S. Garg and A. K. Jana, Carbohydr. Polym., 2011, 83, 1623–1630 CrossRef CAS.
- J. Singh, L. Kaur and N. Singh, Starch – Stärke, 2004, 56, 586–601 CrossRef CAS.
- S. Thiebaud, J. Aburto, I. Alric, E. Borredon, D. Bikiaris, J. Prinos and C. Panayiotou, J. Appl. Polym. Sci., 1997, 65, 705–721 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc00681e |
| This journal is © The Royal Society of Chemistry 2020 |