
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoreforming of food waste into value-added products over visible-light-absorbing catalysts†
Taylor
Uekert
 ,
Florian
Dorchies
,
Christian M.
Pichler
and
Erwin
Reisner
*
,
Florian
Dorchies
,
Christian M.
Pichler
and
Erwin
Reisner
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk
First published on 28th April 2020
Abstract
Approximately 1.3 billion tons of food waste are generated each year, resulting in societal, economic and environmental repercussions across the globe. While efforts to minimise losses and redistribute resources are underway, vast quantities of food waste must still be managed. Photoreforming offers a simple, sunlight-driven method for transforming food waste into valuable chemicals and clean H2 fuel, but the minimal previous research on this topic relied on expensive and UV-absorbing catalysts. Here, we utilise two precious-metal-free and visible-light-driven photocatalytic systems (CdS quantum dots in alkaline solution and carbon nitride with co-catalyst Ni2P under pH neutral conditions) to photoreform a variety of carbohydrates, fats, proteins and real-world mixed wastes into H2 and organic products such as formate. CdS offers higher efficiencies in alkaline media than a benchmark TiO2|RuO2-Pt catalyst, but carbon footprint calculations suggest that photoreforming with carbon nitride|Ni2P in pH neutral H2O offers a more sustainable route towards real-world application.
1. Introduction
One-third of all food produced for human consumption is lost or wasted each year.1–3 This results in food insecurity, economic losses (∼$940 billion USD per year)4 and environmental impacts; the carbon footprint of food wastage is approximately 4.4 Gt CO2 equivalents per year, or 8% of total global greenhouse gas emissions.1 Food waste arises at all stages of the supply chain, from agricultural production to household consumption, and although the exact loss mechanisms differ between regions, the issue remains a global challenge.2,3Much food waste is avoidable and can be addressed by improved infrastructure, knowledge transfer and marketing techniques.5 However, even if the goal outlined by the United Nations to halve food losses by 2030 is achieved,6 billions of tons of material will still go to waste. The majority of this waste is currently sent to landfill or incinerated, resulting in greenhouse gas emissions and a loss of energy and nutrients.7,8 One promising alternative is anaerobic digestion, in which microorganisms produce biogas (a mixture of CH4, CO2 and H2) from food waste. While this process allows for energy recovery, it has high initial capital costs, cannot use mixed waste and often produces impure biogas.7 New technologies are therefore required to reclaim the economic and material value of food waste.
Photoreforming (PR) is one such option. In PR, electrons in a semiconductor are excited to the conduction band (CB) by sunlight and reduce water to H2, while the photogenerated holes in the valence band (VB) drive the oxidation of an organic substrate (Fig. 1). It can therefore be considered a hybrid process between photocatalytic water splitting and organic photo-redox catalysis. PR has several unique benefits, including no external energy input beyond sunlight, applicability to small off-grid systems, compatibility with mixed and wet waste, and ability to produce pure, fuel-cell-grade H2. This technology is thus a suitable candidate for simultaneous food waste management and fuel generation.
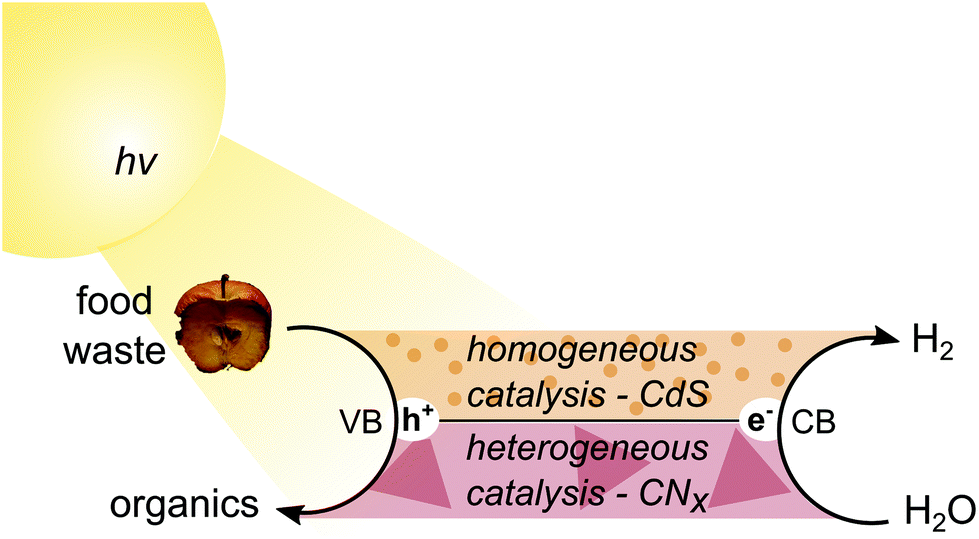 | ||
| Fig. 1 Schematic diagram of food waste photoreforming over CdS/CdOx quantum dots or H2NCNx|Ni2P. | ||
While PR of simple organic molecules,9 sugars,10,11 biomass12,13 and even plastic14–16 has been reported, there has been limited research on food or mixed waste. The few publications on PR of food waste all utilised a TiO2 photocatalyst coupled with noble metal co-catalysts such as Pt.16,17 The efficiency and real-world applicability of these systems were therefore limited by their ultraviolet-only absorption and expense.
Here, we report visible-light-driven and noble-metal-free PR of food and mixed wastes. We select two different types of catalysts – water-soluble CdS/CdOx quantum dots (QDs) and heterogeneous carbon nitride with a nickel phosphide co-catalyst (H2NCNx|Ni2P) – to reform a range of substrates including carbohydrates, proteins and fats into H2 and organic products under both neutral and alkaline aqueous conditions (Fig. 1). CdS/CdOx is shown to exhibit higher activity for both H2 evolution and formate production in alkaline conditions, but H2NCNx|Ni2P offers greater versatility over a wide pH range. Finally, we apply PR to real-world mixed wastes and provide preliminary carbon footprint calculations for the different photocatalytic systems, thereby highlighting the potential of PR to sustainably transform food and mixed waste into renewable fuel and chemicals.
2. Photocatalysis for H2 generation
CdS/CdOx QDs and H2NCNx|Ni2P were selected for their visible light absorption, lack of noble metals and known applicability to relevant photocatalytic processes such as H2 evolution, pollutant degradation and organic transformations.18,19 CdS/CdOx QDs feature a band gap of 2.4 eV (λ < 515 nm), favourable band positions (CB −0.5 V vs. NHE, VB +1.9 V vs. NHE at pH 7),20 and have been utilised previously to photoreform both biomass21 and plastics14 under alkaline conditions. Carbon nitride (H2NCNx) has a band gap of 2.7 eV (λ < 460 nm) and suitable band positions for PR (CB −0.85 V vs. NHE, VB +1.85 V vs. NHE at pH 7).22,23 When coupled with an appropriate H2 evolution co-catalyst, carbon nitride has reformed biomass (with a molecular Ni catalyst)24 and plastics (with Ni2P).15 The different form factors of CdS and H2NCNx also enable analysis of the impact of homogeneity versus heterogeneity on PR efficacy.CdS QDs were prepared by hot-injection synthesis as described previously25 (diameter ∼ 4.7 nm, λmax ∼ 460 nm, Fig. S1†). Ligand-free QDs were used for PR as the exposed surfaces offer higher catalytic performance than ligand-capped QDs.25 When dispersed in alkaline aqueous solution, the QDs form a thin Cd oxide/hydroxide shell (CdOx) that prevents photocorrosion.21,26H2NCNx was prepared from melamine at 550 °C,27 and then loaded with a Ni2P co-catalyst (2 wt%) as reported previously.15 Inductively-coupled plasma optical emission spectrometry (ICP-OES) confirms that Ni2P is present at the expected weight percentage (Table S1†). Upon addition of the co-catalyst, the visible light absorption of H2NCNx is retained (Fig. S2a†) and the fluorescence emission is quenched slightly, indicating reduced radiative charge recombination (Fig S2b†). The bulk chemical properties of H2NCNx are unaffected by co-catalyst addition (Fourier-transform infrared spectroscopy – Fig. S2c,† X-ray powder diffraction – Fig. S2d,† and X-ray photoelectron spectroscopy – Fig. S3†) and Ni2P is present as agglomerates of nanoparticles on the photocatalyst surface (scanning electron microscopy and energy dispersive X-ray spectroscopy – Fig. S4†). Cyanamide-functionalised carbon nitride (NCNCNx), which was previously shown to enhance charge separation and photocatalytic efficiency,24,28 was also investigated for PR with a Ni2P co-catalyst, but offered no substantial improvement (Table S2†).
We then applied the photocatalysts to PR of food-derived substrates. All conditions, including photocatalyst15,21 and substrate concentrations (Table S3†), were optimised for maximal H2 evolution. Experiments with CdS/CdOx QDs were conducted in 10 M aq. KOH, as CdS photo-corrodes at neutral or acidic pH (Table S4†).21,26 On the other hand, H2NCNx|Ni2P functions at highly alkaline (10 M KOH), neutral (H2O) and acidic (1 M H2SO4) pH, with PR in KOH outperforming H2O by at least three times (Table S2†). It was therefore decided to study H2NCNx|Ni2P at both alkaline (for highest performance and direct comparison to CdS/CdOx) and pH neutral (for enhanced sustainability) conditions.
In a typical optimised experiment, the substrate was pre-treated (24 h with stirring in the dark at 40 °C in KOH or 80 °C in H2O) to initiate substrate breakdown and enhance substrate–catalyst interaction for improved PR performance.14,15 While pre-treatment has little effect on soluble substrates such as fructose, it increases activity for recalcitrant samples like starch (Table S5 and Fig. S5†). Slightly higher pre-treatment temperatures were required in H2O to promote solubilisation, as evidenced by a sharpening in the characteristic 1H-Nuclear Magnetic Resonance (1H-NMR) spectroscopy peaks of starch at 80 °C (Fig. S5†). The pre-treated mixture was combined with either CdS/CdOx QDs or an ultrasonicated suspension of H2NCNx|Ni2P (ultrasonication has been shown to increase carbon nitride surface area and improve PR efficiency).24 The samples were then exposed to simulated solar light (AM 1.5G, 100 mW cm−2) at 25 °C under N2 (atmospheric pressure). All H2 measurements are background-corrected by yield without substrates (<10% of H2 produced during PR, Table S6†), and no H2 is detected without the photocatalyst or light (Table S7†).
A variety of carbohydrates (glucose, fructose, galactose, sucrose and starch), the amino acid glutamic acid, proteins (casein, bovine serum albumin (BSA) and beef extract) and fats (glycerol, castor oil and soybean oil) were shown to be active for PR over CdS/CdOx in 10 M KOH and H2NCNx|Ni2P in H2O (Fig. 2 and Table S8†). Simple soluble molecules such as sugars, glutamic acid and glycerol offer the highest H2 yields with both catalytic systems. As the complexity of the substrate increases, activities tend to decrease. This is due to low solubilities and limited percentages of oxygenated regions that can be photoreformed. For instance, beef extract is a mixture of peptides, nucleotides and vitamins and offers yields of only 10.2 ± 2.0 μmolH2 gsub−1 over CdS/CdOx and 0.51 ± 0.02 μmolH2 gsub−1 over H2NCNx|Ni2P.
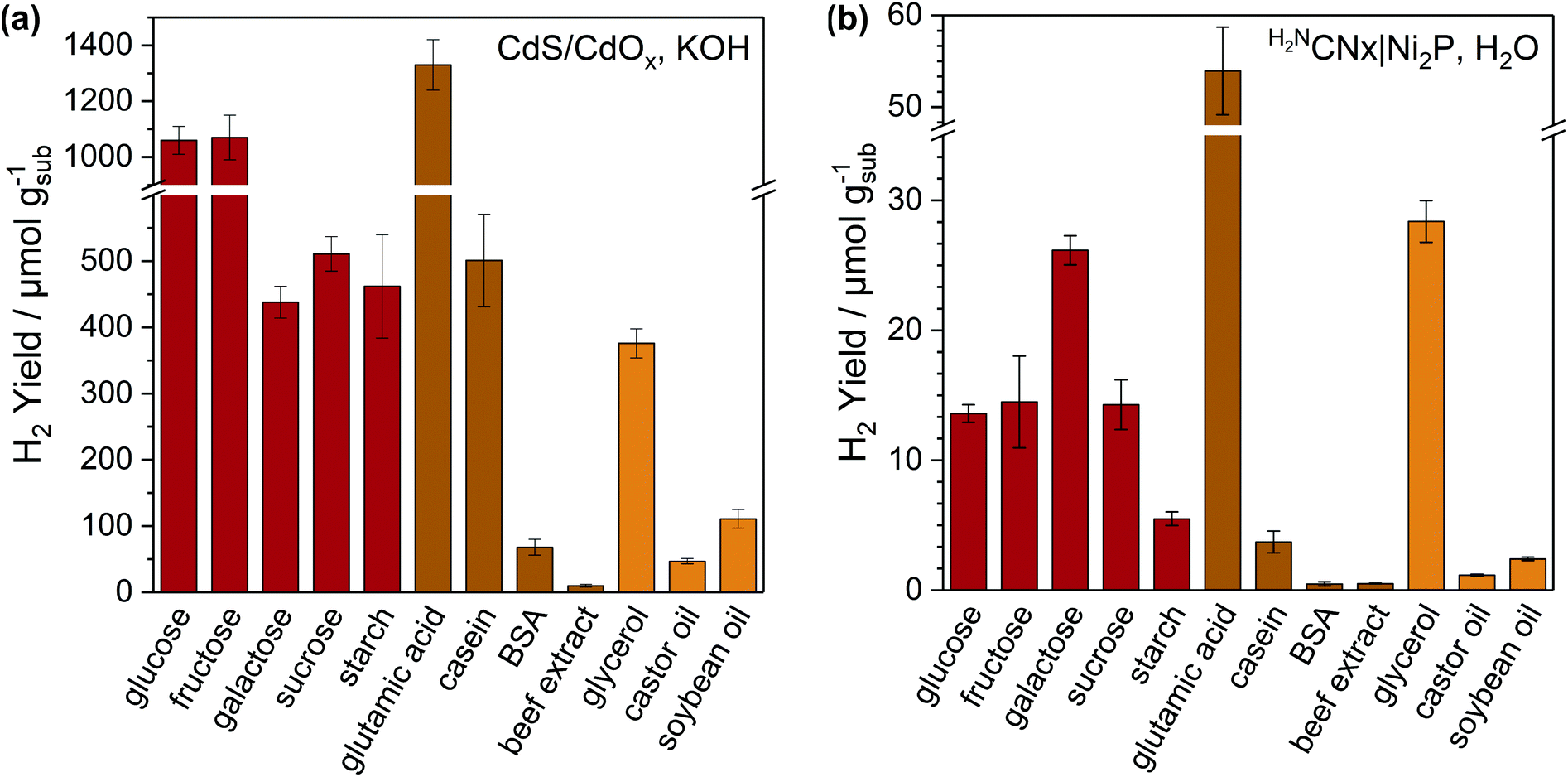 | ||
| Fig. 2 Photoreforming of food-derived molecules with (a) CdS/CdOx QDs in alkaline solution and (b) H2NCNx|Ni2P in H2O. Conditions: CdS/CdOx QDs (1 nmol) in 10 M aq. KOH (2 mL) or H2NCNx|Ni2P (1.5 mg mL−1) in H2O (2 mL); substrate (25 mg mL−1) pre-treated (24 h with stirring) in 10 M KOH at 40 °C for (a) or H2O at 80 °C for (b); simulated solar light (20 h, AM 1.5G, 100 mW cm−2, 25 °C). | ||
Under identical alkaline conditions, CdS/CdOx outperforms H2NCNx|Ni2P by 10–20 times, especially with complex substrates such as casein and starch. The homogeneous nature of CdS/CdOx QDs likely promotes access to insoluble substrates, whereas charge transfer between insoluble samples and heterogeneous H2NCNx|Ni2P is less favourable. CdS/CdOx QDs also benefit from wider light absorption and high charge extraction efficiency.20 H2 yields with H2NCNx|Ni2P are lower in H2O than KOH (∼10–100 and ∼2–4 times less than CdS/CdOx and H2NCNx|Ni2P in KOH, respectively). This is due to differences in substrate solubility and breakdown, as well as the lower efficiency of Ni2P in neutral versus alkaline solution.29 However, H2NCNx|Ni2P remains active and reforms all tested substrates in H2O with yields 4–15 times greater than CdS under the same conditions (Table S4†).
Casein, fructose and starch were selected for further study due to their presence in commonly discarded food items (cheese, apples and bread),30 defined molecular formulas and range of solubilities. After 5 days of irradiation, H2 conversions (measured versus theoretical H2 yield) of 16–27% were achieved with CdS/CdOx in KOH, 3–7% with H2NCNx|Ni2P in KOH, and 1–4% with H2NCNx|Ni2P in H2O (Table S9†). These values are competitive with previous reports of PR with cellulose (9.7%)21 and polyethylene terephthalate (PET, 16.6%)14 over CdS/CdOx in 10 M KOH, as well as PET PR (4.4%)15 with NCNCNx|Ni2P in 1 M KOH. The external quantum yields with fructose – 2.73% for CdS/CdOx and 0.026% and 0.005% for H2NCNx|Ni2P in KOH and H2O, respectively (Table S10†) – are also comparable to those reported for CdS/CdOx with cellulose (1.2%)21 and NCNCNx|Ni2P with PET (0.035%).15 All systems remained active after 5 days, suggesting that higher total conversions could be achieved at longer timescales.
Although CdS/CdOx QDs agglomerate during long-term PR (transmission electron microscopy, Fig. S6†), they appear to remain chemically robust as only 3.5% of the Cd content leaches into solution after 5 days (ICP-OES, Table S1†). The stability of H2NCNx|Ni2P, on the other hand, differs greatly depending on the aqueous conditions utilised. In KOH, only 4% of Ni dissolves into solution after 5 days of PR (Table S1†). The Ni content is likely stabilised in alkaline conditions by the formation of Ni(OH)2 on the Ni2P surface.29,31 In contrast, 60% of Ni leaches into solution during PR in H2O (Table S1 and Fig. S7†). Yet this does not appear to affect efficiency, as evidenced by the nearly constant activity of H2NCNx|Ni2P during long-term fructose PR (Table S11†). Improved interaction between H2NCNx and Ni2P would help prevent leaching and promote catalyst recyclability. In the future, heterogeneous H2NCNx|Ni2P could be easily separated from the PR solution by centrifugation and re-used,15 whereas immobilisation of the water-soluble CdS/CdOx QDs on a substrate could promote facile recycling.
For comparison, H2NCNx|Pt, TiO2|Ni2P and TiO2|RuO2-Pt (which was used in the first report of carbohydrate PR)10 were prepared and studied under identical conditions (Tables 1, S12 and S13†). CdS/CdOx remains the best-performing photocatalyst under alkaline conditions, whereas TiO2|RuO2-Pt offers the highest activity in H2O. Of the noble-metal-free options in pH neutral solution, however, H2NCNx|Ni2P yields the most H2. Furthermore, none of the TiO2-based photocatalysts perform under visible-light-only irradiation (λ > 410 nm), whereas CdS/CdOx and H2NCNx|Ni2P maintain 60% and 16% of their activity, respectively (Tables 1, S7 and S12†). While further activity enhancements are necessary in the future, the application of CdS/CdOx QDs and H2NCNx|Ni2P to a wide range of food-derived substrates is an encouraging proof-of-concept for efficient visible-light-driven and noble-metal-free PR of food waste.
| H2 yield (μmol gsub−1) | |||||||
|---|---|---|---|---|---|---|---|
| Light | Substrate | Aqueous condition | CdS/CdOx | H2NCNx|Ni2P | H2NCNx|Pt | TiO2|RuO2-Pt | TiO2|Ni2P |
| Full spectrum | Casein | KOH | 501 ± 70 | 19.6 ± 3.3 | 65.4 ± 3.3 | 387 ± 19 | 21.8 ± 1.1 |
| H2O | 0.80 ± 0.06 | 3.72 ± 0.83 | 0.84 ± 0.04 | 12.4 ± 0.6 | 0.30 ± 0.02 | ||
| Fructose | KOH | 1070 ± 80 | 57.3 ± 5.8 | 84.7 ± 4.2 | 380 ± 19 | 53.2 ± 2.7 | |
| H2O | 1.00 ± 0.05 | 14.5 ± 3.5 | 271 ± 13 | 449 ± 22 | 11.2 ± 0.6 | ||
| Starch | KOH | 462 ± 78 | 37.4 ± 1.6 | 23.2 ± 1.2 | 219 ± 11 | 23.8 ± 1.2 | |
| H2O | 1.30 ± 0.08 | 5.50 ± 0.53 | 69.3 ± 3.5 | 159 ± 8 | 0.82 ± 0.05 | ||
| λ > 410 nm | Fructose | KOH | 644 ± 36 | 8.97 ± 0.45 | n.m. | n.d. | n.d. |
| H2O | 0.58 ± 0.03 | 2.34 ± 0.20 | n.m. | n.d. | n.d. | ||
3. Substrate oxidation
Complete substrate conversion yields H2 and CO2, but the economic feasibility of PR would be enhanced if substrate oxidation generated value-added chemicals rather than CO2. 1H-NMR spectroscopy was used to identify the liquid oxidation products of casein, fructose and starch (Fig. 3 and S8†). All peak assignments were determined by comparison to authentic samples (Fig. S9†). Unidentified oxidation products are labelled (x) and unlabelled peaks are from the substrate itself. Where possible, 13C-NMR spectroscopy (Fig. S10†) and high-performance liquid chromatography (HPLC, Fig. S11†) were utilised to verify the liquid oxidation products, and mass spectrometry was used to analyse any additional gaseous products (Fig. S12†).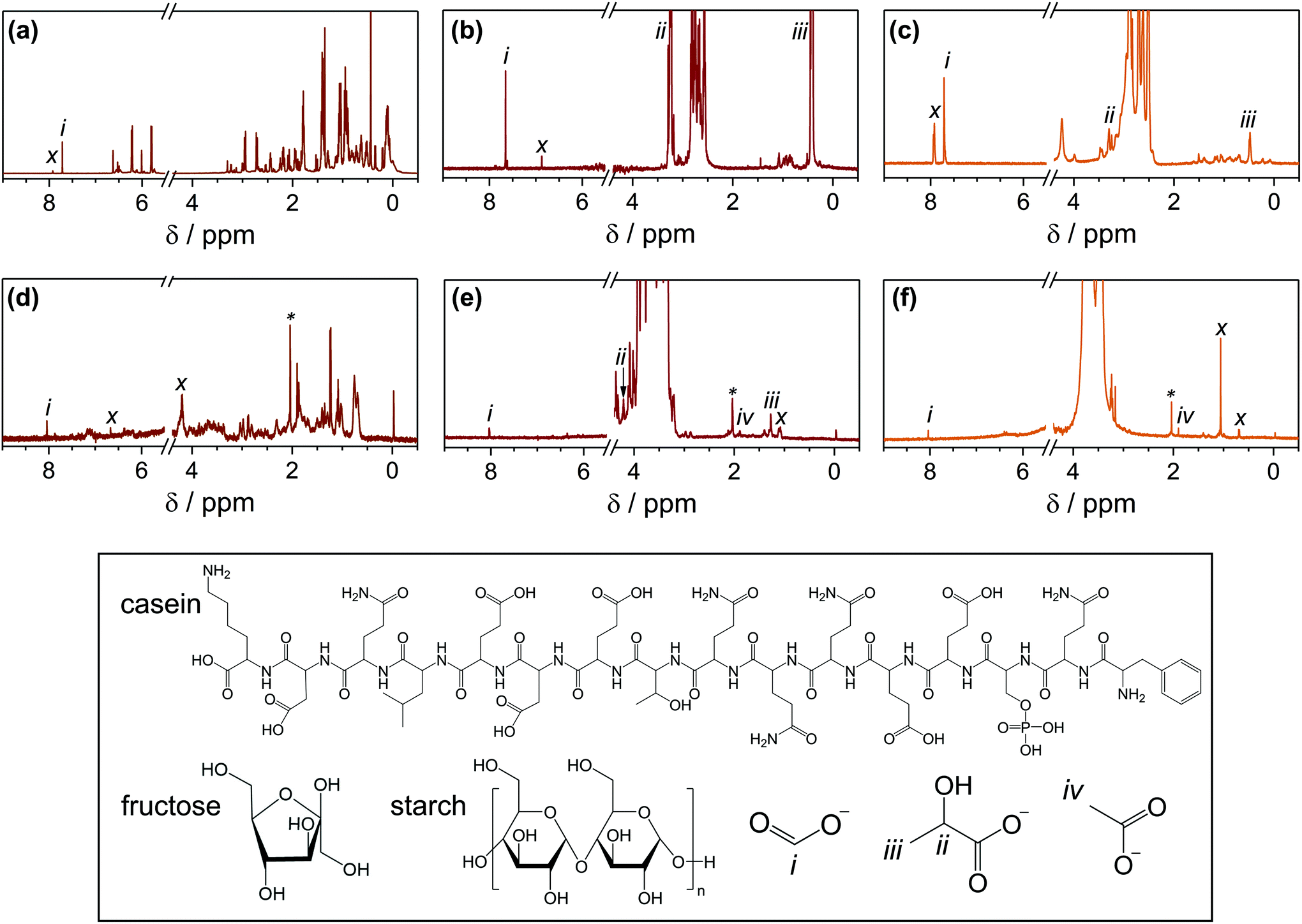 | ||
| Fig. 3 1H-NMR spectroscopy of (a) casein, (b) fructose and (c) starch after photoreforming with CdS/CdOx QDs in 10 M NaOD; and (d) casein, (e) fructose and (f) starch after photoreforming with H2NCNx|Ni2P in D2O. Chemical structures of casein, fructose, starch, formate, lactate and acetate, and their peak assignments are shown at the bottom. (x) are unassigned oxidation products, (*) are trace amounts of acetone contamination, and unlabelled peaks are from the substrate structure. Photoreforming conditions: CdS/CdOx QDs (0.5 nmol) in NaOD (10 M) in D2O (1 mL) or H2NCNx|Ni2P (1.5 mg mL−1) in D2O (1 mL), pre-treated substrate (25 mg mL−1), simulated solar light (4 days, AM 1.5G, 100 mW cm−2, 25 °C). | ||
After 4 days of PR with CdS/CdOx QDs in alkaline conditions, casein oxidises to formate and other unidentified products (Fig. 3a and Table 2); these molecules likely originate from oxidisable amino acids such as glutamic acid within the casein structure.32 Fructose analysis is more complex because alkaline pre-treatment yields a range of substrates,33,34 with glucose, mannose, arabinose, erythrose, lactate (ii, iii) and formate (i) identifiable by NMR spectroscopy (Fig. S5 and Table S14†) and/or HPLC (Fig. S11b, also see ESI† for mechanistic details). It is thus challenging to differentiate between hydrolysis and oxidation products. However, quantitative 1H-NMR spectroscopy shows that formate concentrations increase after fructose PR with CdS/CdOx QDs, indicating that formate is also an oxidation product (Fig. 3b and Table 2). Finally, starch hydrolyses to its monomer glucose, oligomers such as maltose and maltotriose, lactate and gluconate (Fig. S11d†), with formate detected as an oxidation product (Fig. 3c and Table 2).
| Catalyst | Aqueous conditions | Substrate | Formate (μM) | Formate rate (μmol gsub−1 h−1) |
|---|---|---|---|---|
| CdS/CdOx | NaOD | Casein | 2960 | 1.23 |
| Fructose | 6280 | 2.62 | ||
| Starch | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 800 |
4.92 | ||
| H2NCNx|Ni2P | NaOD | Casein | 328 | 0.137 |
| Fructose | 2800 | 1.17 | ||
| Starch | 640 | 0.267 | ||
| H2NCNx|Ni2P | D2O | Casein | 48 | 0.020 |
| Fructose | 100 | 0.042 | ||
| Starch | 56 | 0.023 |
Formate tends to photoreform over CdS/CdOx more slowly than the initial substrates (147 μmolH2 gsub−1 after 20 h versus 501, 1070 or 462 μmolH2 gsub−1 with casein, fructose or starch, respectively; Table S8†), which accounts for its accumulation in solution. This behaviour can perhaps be attributed to repulsion from the negatively-charged catalyst surface (Fig. S13†).21 The presence of large quantities of formate indicates that partial substrate conversion is a common pathway. Nevertheless, some complete conversion is also achieved, as CO32− is evident in 13C-NMR spectroscopy (Fig. S10a†). The same array of products is observed after PR with H2NCNx|Ni2P in 10 M NaOD, although formate concentrations tend to be less than with CdS/CdOx due to the lower efficiency of the carbonaceous catalyst (Fig. S8, S10b† and Table 2).
Under neutral PR conditions with H2NCNx|Ni2P, the oxidation products CO2 (Fig. S12†) and formate (Fig. 3 and Table 2) are observed from casein, fructose and starch. However, the mechanism varies in H2O versus KOH. In contrast to the alkaline case, pre-treatment of fructose in H2O does not alter the original sugar (Fig. S11a†). PR of the pure fructose in H2O may proceed by ring-opening followed by C–C cleavage to shorter aldoses, a process that releases large quantities of formate (see ESI† for mechanistic details).35 It is thus expected that the oxidation products of fructose observed by 1H-NMR spectroscopy (Fig. 3e and Table 2) and HPLC analysis (Fig. S11a†) include formate (i), as well as lactate (ii, iii), acetate (iv) and gluconate.36 40% of the produced formate can be extracted with heptanol by a facile procedure37 (Table S14†). Starch remains an oligomer after pre-treatment in H2O, as only species with higher molecular weights than the glucose trimer maltotriose are observed by HPLC (Fig. S11c†). PR then proceeds by the same mechanism as for fructose,35 with formate (i) and acetate (iv) again apparent as oxidation products (Fig. 3f, S11c† and Table 2).
The presence of different species after pre-treatment (pure sugar in H2O versus a mixture of hydrolysis products in KOH) as well as different consumption rates of the oxidation intermediates (e.g. formate reforms much faster in H2O than KOH) likely account for some of the observed variations in PR efficacy between neutral and alkaline conditions (Table S8, also see cell potentials on page S2 of the ESI†). Future work will utilise this initial understanding of the oxidation half-reaction to alter and improve its selectivity towards value-added chemicals.
4. PR of real-world waste
Having demonstrated PR of food-derived substrates, we next studied the applicability of the photocatalytic systems to real-world food and mixed wastes (Fig. 4 and Table S15†). Pre-treated apples, bread and cheese – three of the mostly commonly discarded food items in the UK30 – were tested for PR (Fig. 4a and Table S15†). As expected, CdS/CdOx has 8–15 times higher activity than H2NCNx|Ni2P in alkaline conditions, but H2NCNx|Ni2P also photoreforms waste in H2O at moderate yields.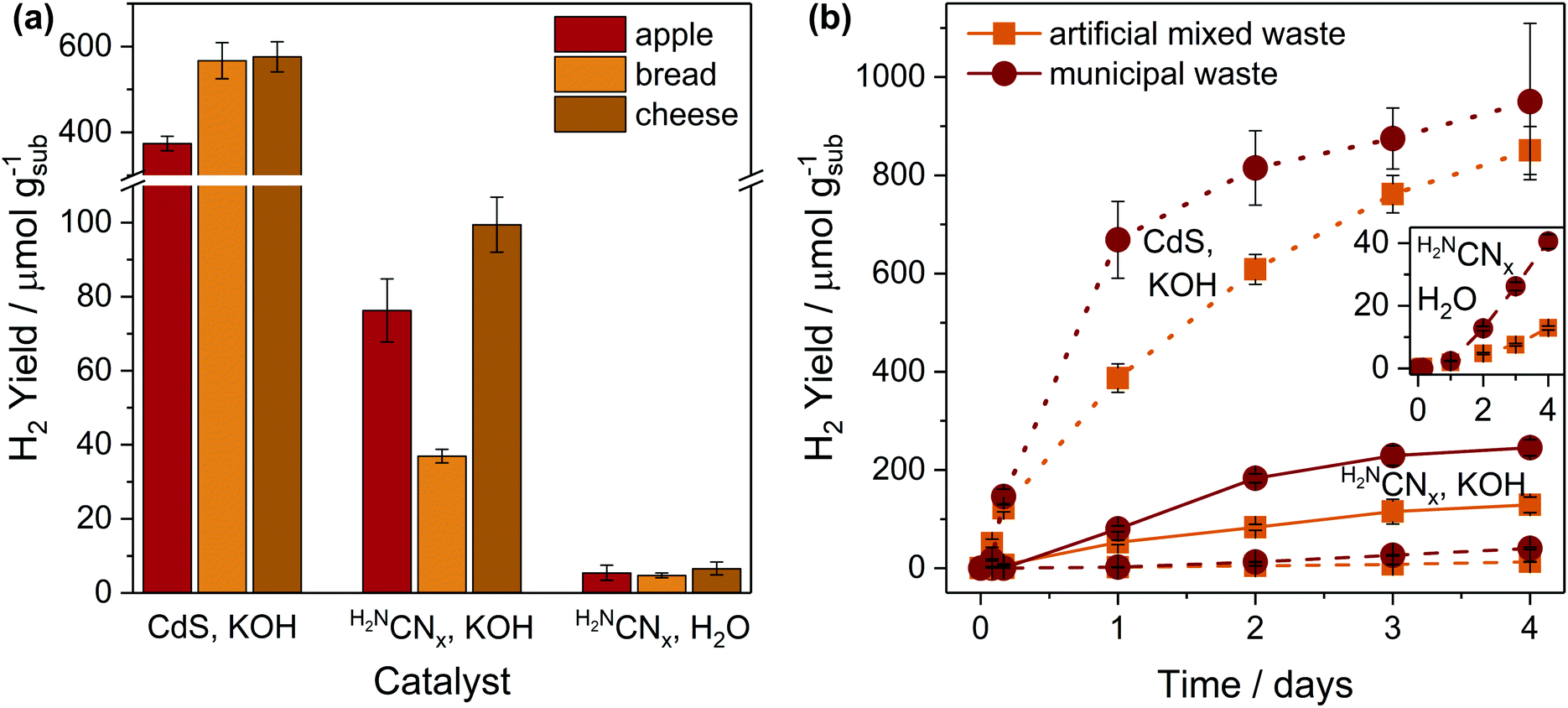 | ||
| Fig. 4 Photoreforming of real-world waste, including (a) apple, bread and cheese, and (b) artificial mixed and municipal wastes; inset shows the bottom two traces (H2NCNx|Ni2P in H2O) in more detail. Artificial mixed waste consists of 5 mg mL−1 each of apple, bread, cheese, cardboard and polyethylene terephthalate bottle. Conditions: CdS/CdOx QDs (1 nmol) in 10 M aq. KOH (2 mL) or H2NCNx|Ni2P (1.5 mg mL−1) in either 10 M aq. KOH (2 mL) or H2O (2 mL); pre-treated substrate (25 mg mL−1 apple, bread, cheese or artificial mixed waste or 12.5 mg mL−1 municipal waste); simulated solar light (20 h for a, 4 days for b, AM 1.5G, 100 mW cm−2, 25 °C). | ||
In all cases, fructose performs better (∼3×) than apples, which is expected due to the low fructose concentration in apples (<20 wt%).38 H2 yields with starch and bread are nearly identical (462 ± 78 starch and 567 ± 42 μmolH2 gsub−1 bread with CdS/CdOx in KOH; 5.50 ± 0.53 starch and 4.76 ± 0.64 μmolH2 gsub−1 bread with H2NCNx|Ni2P in H2O), as the flour in bread contains ∼74–86% starch.39 Finally, cheese performs slightly better than casein, likely because the additional carbohydrates (e.g. lactose)40 in cheese reform more rapidly than casein (Table S8†). The close match between the activities of these samples and relevant pure substrates indicates that “model” molecules can predict PR performance with real-world waste.
A drawback of existing food waste management technologies is their incompatibility with mixed waste. We therefore conducted long-term PR with artificial mixed waste (equal parts apple, bread, cheese, cardboard and PET bottle) and real-world municipal waste (received from University of Leoben, Austria). Both samples were pre-treated under the conditions described previously, but any insoluble portions were removed by centrifugation to reduce light absorption and scattering by the solid residues (see Fig. S14† for 1H-NMR spectra of the pre-treated samples). A lower concentration of municipal waste (12.5 mg mL−1) was used since the sample was otherwise highly scattering and gelatinous. All other experimental conditions were identical to those described above.
Both artificial mixed waste and municipal waste can be photoreformed under all conditions (Fig. 4b and Table S15†). H2 evolution initially proceeds much faster with CdS/CdOx than with H2NCNx|Ni2P under all aqueous conditions, suggesting that the homogeneous nature of CdS/CdOx might promote interaction with the substrates. Although the conversion rates remain low (∼50–225 μmolH2 gsub−1 day−1 in 10 M KOH), they already approach rates reported for bio-hydrogen production from dark fermentation of food waste (∼400–4000 μmolH2 gsub−1 day−1).41 In H2O, the overall yields of real waste PR with H2NCNx|Ni2P are up to 10 times lower than in KOH, but neutral pH offers an interesting possibility for waste separation. Plastic is not reformed in H2O, meaning that neutral PR could potentially generate H2 from food or cellulosic waste while simultaneously cleaning plastic. These experiments showcase the unique applicability of PR to mixed waste that is otherwise non-recyclable.
Finally, the carbon footprints (g CO2 per kW h H2) of PR under various conditions were estimated (see ESI† for assumptions and details). At the current status quo, PR with CdS/CdOx in 10 M KOH (22% conversion after 3 days, no formate extracted, CO32− captured in solution) has a carbon footprint of 44600 g CO2 per kW h H2, with KOH accounting for 96% of that value. Unless efficient chemical recovery is implemented, the footprint of KOH is prohibitively high. PR with H2NCNx|Ni2P in H2O (1.9% conversion after 3 days, formate extracted, no CO2 captured) has a carbon footprint of 68800 g CO2 per kW h H2, with the energy required for stirring and pre-treatment as the largest CO2 contributors. Using renewable sources to provide energy for stirring and pre-treatment (e.g. solar water heating) results in a drop to −450 g CO2 per kW h H2. Furthermore, an ideal PR scenario in H2O (100% conversion to H2 and formate, renewable energy for stirring and pre-treatment) has a negative carbon footprint of −3200 g CO2 per kW h H2. These values compare favourably to existing H2 evolution technologies, including steam methane reforming (23–150 g CO2 per kW h H2 with carbon capture), electrolysis (24–178 g CO2 per kW h H2 with low-carbon energy) and biomass gasification (504 g CO2 per kW h H2).42 While economics will ultimately depend on catalyst safety, efficiency, selectivity and reusability, these preliminary carbon footprint calculations highlight the potential of PR as an environmentally-friendly method for obtaining value-added products from waste.
5. Conclusion
We have demonstrated the visible-light-driven photoreforming of food waste over two precious-metal-free photocatalytic systems. Both CdS/CdOx QDs and H2NCNx|Ni2P reformed a variety of carbohydrates, proteins and fats into H2, formate and CO2 or carbonate. CdS/CdOx offered significantly higher efficiencies, especially with insoluble substrates such as casein, whereas H2NCNx|Ni2P benefited from non-toxicity, applicability to benign (neutral pH) aqueous conditions, and a smaller carbon footprint. Mixed and municipal wastes – comprising a range of food, biomass and plastic materials – were also reformed, highlighting a key advantage of photoreforming in comparison to other food waste management technologies. With enormous quantities of food waste generated every year, photoreforming offers a unique sunlight-driven platform for transforming this resource – even when combined with other types of waste – into both valuable H2 and organic chemicals.6. Experimental section
Reagents
Bovine serum albumin (BSA), castor oil, D-(−)-fructose, D-(+)-glucose, maleic acid, NaOD (40 wt% in D2O), NiCl2·6H2O, NaH2PO2·H2O and potassium thiocyanate were purchased from Fischer Scientific. Casein, D-(+)-galactose, L-(+)-glutamic acid, and starch were obtained from Acros Organics. Chloroplatinic acid (8 wt%), KOH (semiconductor grade), L-(+)-lactic acid, melamine, RuO2 and sucrose were purchased from Sigma-Aldrich. Beef extract powder and glycerol were obtained from VWR Chemicals. D2O (99.96 atom% D) and soybean oil were obtained from Euriso-Top and Alfa Aesar, respectively. Apples (organic royal gala apples) and bread (soft multiseed wholemeal) were purchased from Sainsbury's, cheese (mature cheddar) was purchased from Tesco Superstore, and the plastic water bottle (still Scottish mountain water) was purchased from Marks and Spencer Simply Food. Municipal waste was received from the University of Leoben, Austria.Catalyst synthesis
Ligand-free CdS QDs were synthesised using a literature procedure reported previously.21,25 The particle size and concentration of the CdS QDs were determined by a UV-Vis procedure based on the position and intensity of the absorption maximum.43Unfunctionalised carbon nitride (H2NCNx) was prepared by heating melamine to 550 °C for 3 h under air according to a modified literature procedure.27 The obtained powder was ground with a pestle and mortar. Cyanamide-functionalized carbon nitride (NCNCNx) was prepared according to a literature procedure.28H2NCNx|Ni2P, NCNCNx|Ni2P and TiO2|Ni2P were prepared as reported previously.15
Physical characterisation
Emission spectra (λex = 360 nm, λem = 450 nm) were recorded on an Edinburgh Instruments FS5 spectrofluorometer equipped with a Xe lamp and integrating sphere. All samples were prepared at a concentration of 1.5 mg mL−1 in H2O in a quartz glass cuvette (1 cm path length). UV-vis spectra were recorded on a Varian Cary 50 UV-vis spectrophotometer (with a diffuse reflectance accessory for CNx samples). Fourier transform infrared spectroscopy (FTIR) spectra were collected on a Thermo Scientific Nicolet iS50 FTIR spectrometer (ATR mode). Powder X-ray diffraction (XRD) was conducted on a PANalytical Empyrean Series 2 instrument using Cu Kα irradiation. Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDX) were conducted on a TESCAN MIRA3 FEG-SEM. Samples were sputter-coated with a 10 nm layer of Cr prior to microscopy. Transmission electron microscopy (TEM) was conducted on a Thermo Scientific (FEI) Talos F200X G2 TEM. All samples were drop-cast on carbon-coated Cu grids.Samples for X-ray photoelectron spectroscopy (XPS) were dispersed in ethanol (concentration of 5 mg mL−1) and drop-cast (50 μL, 10×) onto clean FTO glass slides and dried. XPS was performed on a Thermo Fisher Scientific K-alpha+ spectrometer. Samples were analysed using a microfocused monochromatic Al X-ray source (72 W) over an area of ∼400 μm. Data was recorded at pass energies of 150 eV for survey scans and 40 eV for high resolution scans with 1 eV and 0.1 eV step sizes respectively. Charge neutralisation of the sample was achieved using a combination of both low energy electrons and argon ions. Two well-separated areas were selected on each sample for analysis to examine any surface heterogeneity. Data analysis was performed in CasaXPS using a Shirley type background and Scofield cross sections, with an energy dependence of −0.6.
Inductively coupled plasma optical emission spectrometry (ICP-OES) measurements were completed by the Microanalysis Service at the University of Cambridge (Department of Chemistry) on a Thermo Scientific iCAP 700 spectrometer.
Nuclear magnetic resonance (NMR) spectroscopy
1H- and 13C-NMR spectra were collected on either a 400 or 500 MHz Bruker Avance spectrometer equipped with a smart probe. For peak determination, samples were compared to and/or spiked with pure authentic molecules. For quantitative 1H-NMR spectroscopy, samples were spiked with a known quantity of a standard solution (50 mg mL−1 maleic acid in D2O for samples at neutral pH or 50 mg mL−1 of potassium hydrogen phthalate in D2O for samples at alkaline pH) after photoreforming. The quantity of analyte (manalyte) was determined with eqn (1):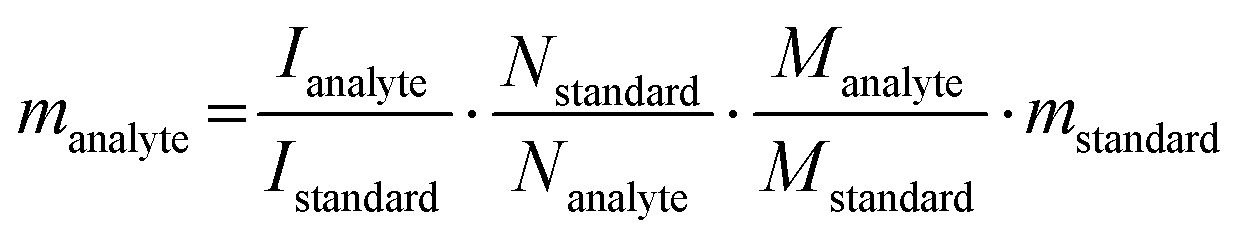 | (1) |
Substrate pre-treatment
Following a reported procedure,14,15 substrates (typically 50 mg mL−1) were soaked in aq. KOH at 40 °C or H2O at 80 °C for 24 h with stirring at 500 rpm in air. The solution – including any undissolved pieces – was then used for catalysis as below. Real waste samples were centrifuged to remove insoluble components, as otherwise the opacity of the solution prevented light absorption by the photocatalyst.Photocatalytic generation of H2
For CdS samples, 1 nmol of QDs were transferred to a Pyrex glass photoreactor vial (internal volume of 7.91 mL) and the solvent was removed under vacuum with stirring. For experiments with untreated substrate, the substrate (25 mg mL−1) and 2 mL of 10 M aq. KOH were added. For experiments with pre-treated substrate, 1 mL of the pre-treated mixture and 1 mL 10 M KOH were added.For CNx samples, a dispersion of the catalyst (H2NCNx|Ni2P or NCNCNx|Ni2P) in H2O (5 mg mL−1) was ultrasonicated as described previously (10 min, pulses of 30 s at 100% amplitude followed by 5 s pauses).24 For experiments at neutral pH, the ultrasonicated mixture (0.6 mL) was combined with H2O (1.4 mL) and substrate (25 mg mL−1) for untreated samples, or combined with the pre-treated substrate mixture (1 mL) and H2O (0.4 mL) for pre-treated samples. For experiments at alkaline pH, the ultrasonicated mixture (0.6 mL) was combined with 15 M KOH (1.33 mL), H2O (0.07 mL) and substrate (25 mg mL−1) for untreated samples, or combined with the pre-treated substrate mixture (1 mL), 15 M KOH (0.33 mL) and H2O (0.07 mL) for pre-treated samples. Final conditions were 2 mL of either H2O or 10 M aq. KOH, 1.5 mg mL−1 catalyst, and 25 mg mL−1 substrate. H2NCNx|Pt was prepared by ultrasonicating H2NCNx according to the above procedure and then adding H2PtCl6 as a precursor, which forms Pt by in situ photodeposition. For TiO2|Ni2P samples, the photocatalyst was added directly (no ultrasonication) to the pre-treated or untreated solution. For TiO2|RuO2-Pt samples, TiO2 and RuO2 were ground at a ratio of 10:1 with a mortar and pestle. 15 mg of the mixed catalyst were combined with 16.4 μL of H2PtCl6 (Pt precursor) for a final weight ratio of 100:10:5 TiO2:RuO2:Pt.10
All prepared samples were added to Pyrex glass photoreactor vials, capped with rubber septa, and purged at ambient pressure for 10 min with N2 containing 2% CH4 for gas chromatographic (GC) analysis. The samples were then irradiated by a solar light simulator (Newport Oriel, 100 mW cm−2) equipped with an air mass 1.5 global (AM 1.5G) filter and a water filter to remove infrared radiation. Visible-light-only experiments were conducted by adding a λ > 410 nm cut-off filter. All samples were stirred at 600 rpm and kept at a constant temperature of 25 °C during irradiation. H2 generation was monitored periodically by analysing samples of the reactor headspace gas (50 μL) by GC (see below). Overpressure within the vial is minimal.
Gas analysis
The accumulation of H2 was measured by GC with an Agilent 7890A gas chromatograph equipped with a thermal conductivity detector and HP-5 molecular sieve column using N2 as the carrier gas. Methane (2% CH4 in N2) was used as an internal standard after calibration with different mixtures of known amounts of H2/N2/CH4. No CH4 was detected after photocatalysis without the internal standard. CO2 detection was carried out by mass spectrometry on a Hiden Analytical HPR-20 benchtop gas analysis system fitted with a custom-designed 8-way microflow capillary inlet to a HAL 101 RC electron impact quadrupolar mass spectrometer with a Faraday detector.High performance liquid chromatography (HPLC)
Chromatographic separations were conducted with either a Pheomenex Rezex RCM Monosaccharide 8% Ca2+ column or a Phenomenex Rezex ROA-Organic Acid H+ column. Samples were analysed in the isocratic flow mode (flow rate 0.5 mL min−1) using a Shimadzu LC 20 equipped with refractive index (RID-10A) and diode array UV-Vis (λ = 190 nm) detectors. To identify particular substances in the pre-treated or photoreformed samples, retention times were compared to those of authentic samples.Treatment of data
All analytical measurements were performed in triplicate, unless otherwise stated, and are given as the unweighted mean ± standard deviation (σ). All measurements are listed as H2 yield per weight of substrate (μmolH2 gsub−1) and activity per weight of catalyst (μmolH2 gcat−1 h−1). σ was calculated using eqn (2).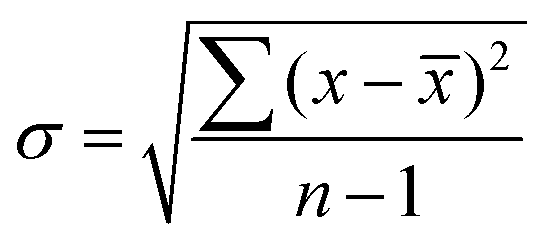 | (2) |
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) – unweighted mean of the measurements.
– unweighted mean of the measurements.
σ was increased to 5% of in the event that the calculated σ was below this threshold.
Stoichiometric H2 conversion calculations
Samples with 1 mg substrate in either 10 M aq. KOH or H2O (2 mL) were prepared for photocatalysis and irradiated as described above. H2 Conversion (%) was calculated as described in eqn (3):15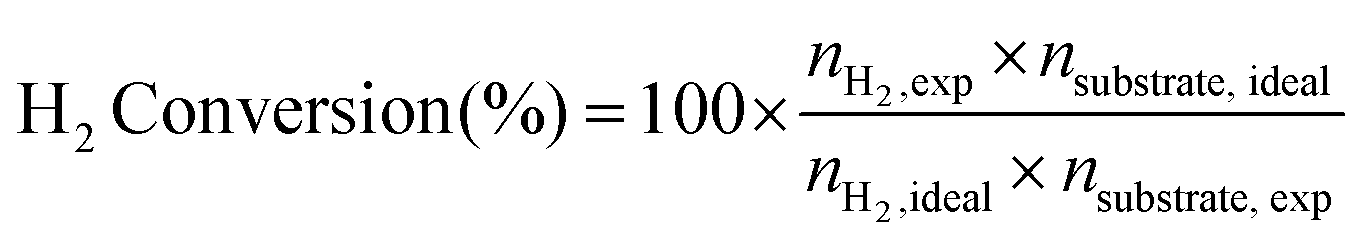 | (3) |
External quantum yield (EQY) determination
Samples were prepared for photocatalysis as described above and added to a quartz cuvette (1 cm path length) that was then sealed with a rubber septum. The cuvette was purged with N2 containing 2% CH4 for 10 min and irradiated by a Xe lamp (LOT LSH302) fitted with a monochromator (LOT MSH300) focused at a single wavelength of λ = 430 nm (accurate to a full width at half-maximum of 5 nm). The light intensity was adjusted to ∼1000 μW cm−2, as measured with a power meter (ILT 1400, International Light Technologies). The cuvette was irradiated across an area of 0.28 cm2. The evolved headspace gas was analysed by gas chromatography and the EQY (%) calculated with eqn (4).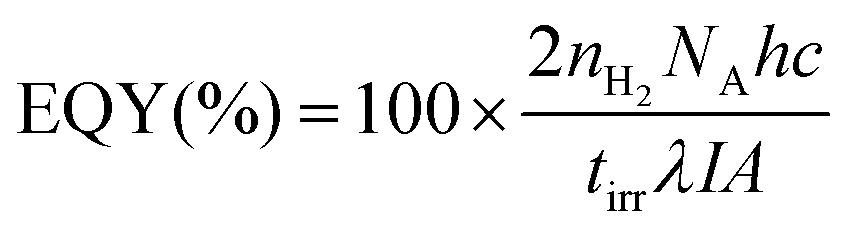 | (4) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Austrian Science Fund (Schrödinger Scholarship J-4381), the UKRI Cambridge Creative Circular Plastics Centre (CirPlas, EP/S025308/1), École Normale Supérieure Paris-Saclay and Bourse Mobilité Île-de-France (AMIS scholarship), EPSRC (NanoDTC, EP/L015978/1 and EP/S022953), and the OMV Group. XPS data collection was performed at the EPSRC National Facility for XPS (HarwellXPS), operated by Cardiff University and UCL under Contract PR 16195. We thank Dr Heather Greer for assistance with electron microscopy, Dr Annika Eisenschmidt for assistance with NMR spectroscopy, Dr Ana Belenguer for assistance with HPLC, and Ava Lage, Constantin Sahm, Dr Moritz Kuehnel and Dr Teresa Schubert for useful discussions.References
- Food and Agriculture Organization of the United Nations, Food wastage footprint and climate change, Rome, 2015 Search PubMed.
- J. Gustavsson, C. Cederberg, U. Sonesson, R. Van Otterdijk and A. Meybeck, Global Food Losses and Food Waste, Food and Agriculture Organization of the United Nations, Rome, 2011 Search PubMed.
- M. van den B. Verma, L. de Vreede, T. Achterbosch and M. M. Rutten, PLoS One, 2020, 15, e0228369 CrossRef CAS PubMed.
- W. Britz, H. Dudu, I. Fusacchia, Y. Jafari, R. Roson, L. Salvatici and M. Sartori, Economy-wide analysis of food waste reductions and related costs: A global CGE analysis for the EU at NUTS-II level, Luxembourg, 2019 Search PubMed.
- J. Aschemann-Witzel, Science, 2016, 352, 408–409 CrossRef CAS PubMed.
- K. Flanagan, K. Robertson and C. Hanson, Reducing food loss and waste: Setting a global action agenda, World Resources Institute, Washington, D.C., 2019 Search PubMed.
- S. Jain, D. Newman, R. Cepeda-Marquez and K. Zeller, Global food waste management: An implementation guide for cities, World Biogas Association, London, 2018 Search PubMed.
- A. Cerda, A. Artola, X. Font, R. Barrena, T. Gea and A. Sánchez, Bioresour. Technol., 2018, 248, 57–67 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- T. Kawai and T. Sakata, Nature, 1980, 286, 474–476 CrossRef CAS.
- D. I. Kondarides, V. M. Daskalaki, A. Patsoura and X. E. Verykios, Catal. Lett., 2008, 122, 26–32 CrossRef CAS.
- M. F. Kuehnel and E. Reisner, Angew. Chem., Int. Ed., 2018, 57, 3290–3296 CrossRef CAS PubMed.
- A. V. Puga, Coord. Chem. Rev., 2016, 315, 1–66 CrossRef CAS.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- T. Kawai and T. Sakata, Chem. Lett., 1981, 81–84 CrossRef CAS.
- T. Sakata and T. Kawai, J. Synth. Org. Chem., Jpn., 1981, 39, 589–602 CrossRef CAS.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS.
- L. Cheng, Q. Xiang, Y. Liao and H. Zhang, Energy Environ. Sci., 2018, 11, 1362–1391 RSC.
- Y. Xu, Y. Huang and B. Zhang, Inorg. Chem. Front., 2016, 3, 591–615 RSC.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- J. Wirth, R. Neumann, M. Antonietti and P. Saalfrank, Phys. Chem. Chem. Phys., 2014, 16, 15917–15926 RSC.
- H. Kasap, D. S. Achilleos, A. Huang and E. Reisner, J. Am. Chem. Soc., 2018, 140, 11604–11607 CrossRef CAS PubMed.
- C. M. Chang, K. L. Orchard, B. C. M. Martindale and E. Reisner, J. Mater. Chem. A, 2016, 4, 2856–2862 RSC.
- T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrović, D. Volbers, R. Wyrwich, M. Döblinger, A. S. Susha, A. L. Rogach, F. Jäckel, J. K. Stolarczyk and J. Feldmann, Nat. Mater., 2014, 13, 1013–1018 CrossRef CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- V. W. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- Z. Zhou, L. Wei, Y. Wang, H. E. Karahan, Z. Chen, Y. Lei, X. Chen, S. Zhai, X. Liao and Y. Chen, J. Mater. Chem. A, 2017, 5, 20390–20397 RSC.
- Waste & Resources Action Programme, Estimates of food surplus and waste arisings in the UK, Banbury, 2017 Search PubMed.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K.-C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2012, 51, 12495–12498 CrossRef CAS PubMed.
- B. H. Lauer, B. E. Baker and B. E. Baker, Can. J. Zool., 1977, 231–236 CrossRef CAS PubMed.
- T. Vuorinen, Carbohydr. Res., 1985, 141, 319–322 CrossRef CAS.
- H. G. J. De Wilt and I. Lindhout, Carbohydr. Res., 1971, 23, 333–341 CrossRef.
- K. E. Sanwald, T. F. Berto, W. Eisenreich, A. Jentys, O. Y. Gutiérrez and J. A. Lercher, ACS Catal., 2017, 7, 3236–3244 CrossRef CAS.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- J. Reichert, B. Brunner, A. Jess, P. Wasserscheid and J. Albert, Energy Environ. Sci., 2015, 8, 2985–2990 RSC.
- S. Musacchi and S. Serra, Sci. Hortic., 2018, 234, 409–430 CrossRef.
- R. Andersson, E. Westerlund, A. C. Tilly and P. Aman, J. Cereal Sci., 1992, 17, 183–189 CrossRef.
- S. Milewski, K. Zbek, Z. Antoszkiewicz, Z. Tański and A. Sobczak, Emirates J. Food Agric., 2018, 30, 107–114 Search PubMed.
- Y. M. Yun, M. K. Lee, S. W. Im, A. Marone, E. Trably, S. R. Shin, M. G. Kim, S. K. Cho and D. H. Kim, Bioresour. Technol., 2018, 248, 79–87 CrossRef CAS PubMed.
- The Royal Society, Options for producing low-carbon hydrogen at scale - Policy briefing, London, 2018 Search PubMed.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc01240h. Raw data related to this publication are available at the University of Cambridge data repository: https://doi.org/10.17863/CAM.51830 |
| This journal is © The Royal Society of Chemistry 2020 |