
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards closed-loop recycling of multilayer and coloured PET plastic waste by alkaline hydrolysis†
Sibel
Ügdüler
 a,
Kevin M.
Van Geem
b,
Ruben
Denolf
a,
Martijn
Roosen
a,
Nicolas
Mys
ac,
Kim
Ragaert
c and
Steven
De Meester
*a
a,
Kevin M.
Van Geem
b,
Ruben
Denolf
a,
Martijn
Roosen
a,
Nicolas
Mys
ac,
Kim
Ragaert
c and
Steven
De Meester
*a
aLaboratory for Circular Process Engineering, Department of Green Chemistry and Technology, Ghent University, Graaf Karel De Goedelaan 5, 8500 Kortrijk, Belgium. E-mail: Steven.DeMeester@UGent.be
bLaboratory for Chemical Technology, Department of Materials, Textiles and Chemical Engineering, Faculty of Engineering & Architecture, Ghent University, Technologiepark 121, B-9052 Zwijnaarde, Belgium
cCenter for Polymer and Material Technologies (CPMT), Department of Materials, Textiles and Chemical Engineering, Faculty of Engineering and Architecture, Ghent University, Technologiepark 130, B-9052 Zwijnaarde, Belgium
First published on 27th July 2020
Abstract
The vast increase in the generation of post-consumer PET plastic waste, as well as fast increasing pledges of brand owners around the world to include recycled content have resulted in a pressing need for efficient recycling processes, such as chemical depolymerization. Although recycling rates of PET bottles are high, those of PET trays and films are still significantly lower due to the broad range of colours and multilayer structures, as well as due to a much poorer collection. In this study, a two-step aqueous alkaline hydrolysis was carried out on different types of real PET plastic waste under mild conditions (≤80 °C under atmospheric pressure). Reaction conditions such as temperature (50–80 °C), ethanol to water ratio (20–100 vol%), NaOH amount (5–15 wt%) and stirring rate (250–500 rpm) have been optimized by using pure PET pellets in order to maximize the product yield. At optimal conditions (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 vol% EtOH:H2O, 5 wt% NaOH and at 80 °C) product yields on a mass basis of approximately 95% have been achieved in less than 20 minutes. The purity of the obtained monomers, ethylene glycol (EG) and terephthalic acid (TPA), was characterized by NMR, UV-VIS and FTIR measurements. The experimental kinetic data are represented adequately using the diffusion model. Experiments performed at optimal conditions with different types of post-consumer plastic waste, revealed that the degradation rate increases inversely proportional to the particle size. Furthermore, the increased thickness of the samples and the presence of multilayers reduce the decomposition yield with a factor two as observed for monolayer (80%) versus multilayer PET trays (45%). In addition to transparent multilayer PET samples, by using the optimized alkaline hydrolysis with further cleaning processes different types of colours, including carbon black are removed from the hydrolysate successfully. A life cycle assessment (LCA) shows that the key to lower the carbon emissions is keeping the energy consumption low by increasing the solid/liquid (S/L) ratio and avoiding excess water addition during monomer purification.
:40 vol% EtOH:H2O, 5 wt% NaOH and at 80 °C) product yields on a mass basis of approximately 95% have been achieved in less than 20 minutes. The purity of the obtained monomers, ethylene glycol (EG) and terephthalic acid (TPA), was characterized by NMR, UV-VIS and FTIR measurements. The experimental kinetic data are represented adequately using the diffusion model. Experiments performed at optimal conditions with different types of post-consumer plastic waste, revealed that the degradation rate increases inversely proportional to the particle size. Furthermore, the increased thickness of the samples and the presence of multilayers reduce the decomposition yield with a factor two as observed for monolayer (80%) versus multilayer PET trays (45%). In addition to transparent multilayer PET samples, by using the optimized alkaline hydrolysis with further cleaning processes different types of colours, including carbon black are removed from the hydrolysate successfully. A life cycle assessment (LCA) shows that the key to lower the carbon emissions is keeping the energy consumption low by increasing the solid/liquid (S/L) ratio and avoiding excess water addition during monomer purification.
1. Introduction
Polyethylene terephthalate (PET) exists both as a semi-crystalline and an amorphous thermoplastic polyester mainly used in textiles and packaging due to its good physicochemical properties such as lightweight, good heat resistance and dimensional stability, resistance towards chemicals, among others.1 PET is best known as a monolayer “clear plastic” widely used for (carbonated) beverage containers. Beside monolayer bottles, PET is extensively laminated with other types of polymers such as polyolefins and ethylene vinyl alcohol (EVOH) especially in food packaging applications, as it has a limited properties related to permeation of oxygen and sealing.2Generally, post-consumer plastics have very low recycling rates. However, PET is one of the most recycled materials; in 2017 more than 57% of PET bottles were recycled in Europe.3 Especially transparent PET bottles have high collecting and recycling rates over Europe, but recycling rates for opaque PET bottles, PET trays and films are significantly lower due to the broad range of colours, additives, multilayer structure, labels and other complexities.4 Therefore, they are mainly disposed of in landfill or incineration because it is not possible to obtain a secondary raw material with a high value by using the typical mechanical recycling processes.5,6
PET can be recycled or recovered via four pathways, namely primary recycling, secondary recycling, chemical recycling and incineration.7 Primary recycling focuses on mainly uncontaminated industrial scrap that can be recycled purely or mixed with a virgin material to increase the product quality. Secondary recycling is a mechanical recycling process which passes through a series of contaminant removal, drying and reprocessing steps. This process is common practice for closed-loop recycling of PET bottles, or recycling of bottles to fibers. Although this process is simple and requires relatively low investments, the generation of cyclic and linear oligomers during the melting processing causes around 30% reduction in PET melt viscosity.8 In addition, these mechanical pretreatment steps are often imperfect, which can lead to quality deteriorations. Especially for the more complex PET waste streams such as PET trays, mechanical recycling is even hardly possible, or only towards open-loop recycling (often called downcycling) applications. Incineration is an option for energy recovery, but whereas this method generates a certain amount of energy, it does not fit in the circular economy strategy.9 It is thus clear that there is a need for an efficient recycling of complex PET plastics waste. Chemical recycling is therefore a promising option in which polymers are broken down into its monomers, as such allowing the production of virgin PET which can be used in closed-loop applications after depolymerization. Within chemical recycling processes of PET, several options are possible such as hydrolysis, alcoholysis, aminolysis depending on the type of reagent used during chemical degradation.10 There are also other techniques where e.g. pyrolysis, supercritical fluids, enzymes are used, but they are currently at an early research stage.11 Among the chemical methods, glycolysis which is a method of alcoholysis using ethylene glycol, is the oldest and most common method in industry.11,12 For example, Ioniqa (The Netherlands) is developing a glycolysis technology for PET bottles and polyester fibers by using magnetic ionic fluids and a catalyst.13 Although Ioniqa process offers an alternative to incineration, the results of a first screening life cycle assessment (LCA) show that it is not easy to decrease the environmental impact of the process compared to mechanical recycling.14 Similarly Garbo (Italy), developed in 2017 a glycolysis technology with a specific purification system, called ChemPET, to depolymerize PET waste including fabrics.5 IFPEN Axens (France), JEPLAN (Japan) and PerPETual Global Technologies (UK) also recycle PET waste through glycolysis.15–17 Methanolysis is another commonly used alcoholysis method which is based on degradation of PET by using methanol at high temperatures under high pressures.10 For instance, Loop industries (Canada) use methanolysis to depolymerize PET waste to PET resins and fibers.18 Eastman (USA) is also currently performing feasibility tests to commercialize a methanolysis facility to recycle PET waste.19 In addition to glycolysis and methanolysis, DEMETO technology used by GR3N (Switzerland) depolymerizes a broad range of different types of PET through hydrolysis by using microwaves.20 In contrast to using simple alcohols and/or water as a reagent for PET degradation, Rampf Eco Solutions (Germany) uses polyols to degrade PET to high quality and multi-functional PET-based polyols with potential environmental and economic benefits.21
Although chemical recycling of PET is increasing to enable for the production of PET designated for high-end applications, even at industrial scale, little fundamental information is available in the scientific literature related to these type of reactions. Especially the kinetics of chemical recycling on real plastic waste streams is not commonly understood. Among PET degradation methods, kinetics of the PET glycolysis have been studied vastly.22–26 According to these studies, after glycolysis, monomer recoveries are low (∼25%) even in the presence of catalysts.11 Moreover, the purification of monomers is problematic due to the occurrence of oligomers during degradation.27 On the other hand, high recoveries (∼90%) can be obtained through methanolysis, but generally very harsh degradation conditions are used and purification step is not always straightforward due to complex mixture of glycols, phthalate derivatives and alcohols in the reaction medium.28,29 In addition, the presence of water in the methanolysis process causes deterioration of the catalyst and formation of various azeotropes resulting in a decrease in the monomer purity.30,31 Hydrolysis is an alternative method, which is sometimes discarded due to inevitable formation of salt, but on the other hand it can be performed in mild conditions and it can tolerate highly contaminated post-consumer waste. Furthermore, the purity and yield of obtained monomers is often higher.11 An overview of yields and conditions of different chemical depolymerization methods of PET is shown in Table 1.
| Method | Catalyst | Temperature (°C) | Pressure (bar) | Time (h) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| Hydrolysis | None | 200 | 1 | 1 | 97.9 | 27 |
| Cyclo hexylamine | 90 | 1 | 2 | 85.1 | 32 | |
| None | 110 | 1 | 0.5 | 89 | 33 | |
| None | 99 | 1 | 2.5 | 85 | 34 | |
| Tetrabutyl ammonium bromide | 90–98 | 1 | <1 | 99 | 35 | |
| Glycolysis | Zinc acetate | 196 | 1 | 2 | 66 | 36 |
| Tetragonal | 260 | 5 | 1.5 | 92.2 | 37 | |
| None | 300 | 11 | 0.4–0.8 | 0.3 | 38 | |
| Zinc sulfate | 80–200 | 1 | 15 | 25 | 39 | |
| Didymium chloride | 196 | 1 | 9 | 72 | 40 | |
| Methanolysis | None | 270 | 1–150 | 1.5 | 60 | 41 |
| None | 300–350 | 200 | 2 | 80 | 42 | |
| Zinc acetate | 250–270 | 85–140 | 1 | 60–95 | 43 | |
| None | 300 | 9.8 | 1.5 | 80 | 44 | |
| Aluminium triisopropoxide | 200 | Not given | 2 | 88 | 29 | |
The hydrolysis of PET can be categorized as neutral, acidic and alkaline hydrolysis.10 Acidic hydrolysis generally gives high yields of terephthalic acid (TPA) monomer, but the use of high amounts of acids makes the process very costly and also affects the purity of ethylene glycol (EG) adversely.12 Similarly, in the eco-friendly neutral hydrolysis all mechanical impurities present in the polymer stay in the TPA, as such affecting the purity of TPA.10,11 Therefore, as an alternative, alkaline hydrolysis of PET has been studied. In many of these studies either extreme degradation conditions e.g. high temperatures and pressure are applied or catalysts are used.27,32,34,45–49 To the best of our knowledge there is no detailed kinetic study available on the alkaline hydrolysis of PET in mild degradation conditions. Furthermore, in many scientific studies pure PET pellets are used related to chemical degradation of PET, which is typically different compared to more complex real plastic waste streams, that differ in composition and shape, amongst others.
The purposes of this study:
• To investigate the parameters affecting the PET degradation rate such as temperature, ethanol to water ratio, weight percentage of NaOH and stirring rate, among others via GC-FID measurements by following the concentration increase of EG in the solution during alkaline hydrolysis. After hydrolysis, the purity of the monomers is also characterized via NMR, UV-VIS and FTIR measurements.
• To determine the best-fit kinetic model for the experimental kinetic data of PET hydrolysis. By using this kinetic model, the effect of particle size, thickness of the PET samples and the presence of multilayers on the kinetic rate constant is shown.
• To present a holistic study on mild hydrolysis of PET, studying reaction kinetics of different types of real PET waste streams including monolayer and multilayer PET plastic waste. The advantage of using the proposed mild conditions is that ethylene glycol (EG) and terephthalic acid (TPA), are obtained, whilst maintaining other polymers e.g. polyolefins that are often present in multilayer structures with PET and that would cause problems due to e.g. melting in high temperature processes such as glycolysis. Our proposed process is thus able to work on PET waste streams such as bottles, as well as on multilayer structures such as films and trays, often containing PE.
• To test the alkaline hydrolysis at optimal experimental conditions on the highly coloured PET plastic waste including carbon black to show its potential towards closed-loop recycling of complex PET waste. In order to evaluate the carbon footprint of this proposed PET hydrolysis process, a life cycle assessment (LCA) has also been performed on two hydrolysis scenarios at different solid/liquid (S/L) ratios based on performed experiments and ASPEN simulations.
2. Materials and methods
2.1. PET samples, chemicals and reagents
For the baseline experiments, PET pellets (Lighter™ C93) were obtained from Equipolymers. For further experiments, flakes were prepared from transparent, coloured, mono and multilayer post-consumer PET trays and films and also from clear PET water bottles whose caps, labels and glue had been removed. Samples were first washed with a detergent to remove any surface impurities and oil components and subsequently washed with water and dried in an oven overnight at 50 °C. All the samples were cut with a cryogenic rotary cutter (MDS 340/150 Hellweg Maschinenbau) to reduce the particle size to 5 mm. These samples were further separated to a range of particle sizes as lower than 0.5 millimeter (mm), 0.5–1 mm, 1–1.6 mm, 2–2.5 mm, 2.5–3.15 mm and higher than 3.15 mm by using sieves with different mesh sizes. Post-consumer PET samples with 1 cm and 4 cm were cut manually. The intrinsic viscosity of PET pellets was measured via Ubbelohde viscometer at 25 °C in a solution consisting of 60/40 (w/w) phenol/1,1,2,2-tetrachloroethane mixture. From the intrinsic viscosity (IV) (dL g−1), the number average molecular weight (M) was calculated from the following equation:50| M = 3.61 × 102 IV1.46 | (1) |
The anhydrous EG, 1,3-propanediol, dimethyl sulfoxide (DMSO), sulphuric acid, ethanol, sodium hydroxide (NaOH), potassium bromide (KBr) were supplied by Sigma Aldrich (Merck). These chemical compounds were used without any purification.
2.2. Alkaline hydrolysis experiments
PET flakes were hydrolyzed in an aqueous alkaline medium without a catalyst at atmospheric pressure to yield disodium terephthalate (Na2TP) salt and EG. Afterwards, the solution was acidified to transform the Na2TP salt to TPA monomer, based on the chemical reaction as shown in Scheme 1.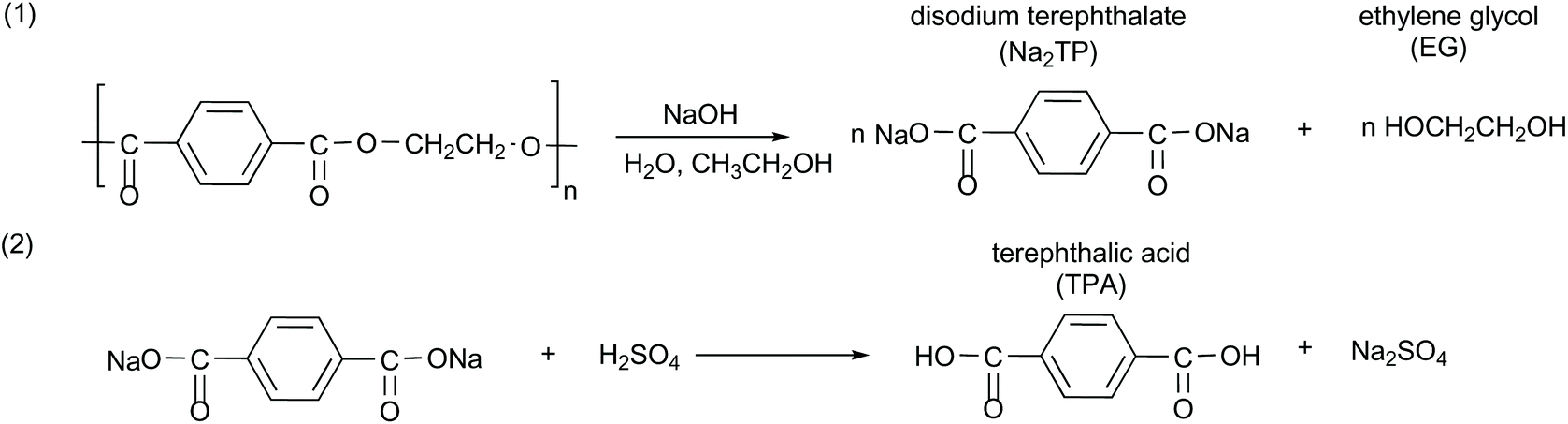 | ||
| Scheme 1 Alkaline hydrolysis of PET in the presence of NaOH, EtOH and water. | ||
PET alkaline hydrolysis experiments were carried out in a three-necked round bottom flask equipped with a condenser and an agitator for stirring. The 100 mL flask containing sodium hydroxide (NaOH) and water:ethanol mixture was placed into an oil bath at room temperature and preheated to the selected temperature prior to the addition of PET flakes in order to minimize the delays to reach the specified temperature at atmospheric pressure. After a specified time interval, the flask was removed from the oil bath and quenched in an ice bath to stop the progress of the PET hydrolysis. Afterwards, the residual PET flakes were separated by filtration, washed, dried overnight at 60 °C and weighed. Sulfuric acid was added to the filtrate in order to convert disodium terephthalate to solid terephthalic acid (TPA) monomer and then separated via filtration. The solid TPA is further washed with deionized water and dried at 60 °C. This experimental set-up is shown in Scheme 2.
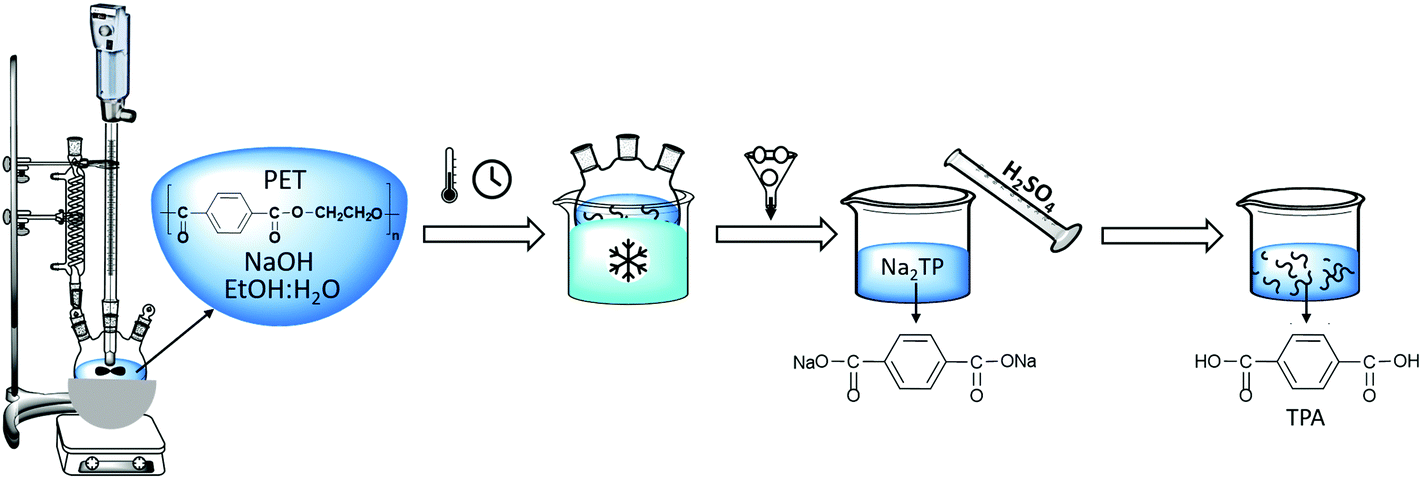 | ||
| Scheme 2 Experimental set-up of PET alkaline hydrolysis. | ||
In order to investigate the effect of degradation conditions, kinetic studies were performed by conducting alkaline hydrolysis at different experimental conditions as shown in Table 2. In these screening experiments, low temperatures (50 and 80 °C) were used in order to be able to separate polyolefins without causing any degradation or melting in case of multilayer PET samples. In order to achieve high conversion yields, NaOH amount was also optimized by testing three different NaOH concentrations: 5, 10 and 15 wt%. In addition, ethanol is mixed with water at different volume ratios, 20, 60 and 100 vol%, in order to assess its efficiency as a co-solvent. The effect of stirring rate was also tested under these experimental conditions by stirring with a magnetic stirrer at 250 rpm and with an agitator at 500 rpm.
| # of experiment | T (°C) | NaOH wt% | EtOH vol% |
|---|---|---|---|
| 1 | 80 °C | 10 | 60 |
| 2 | 80 °C | 5 | 60 |
| 3 | 80 °C | 5 | 100 |
| 4 | 80 °C | 15 | 60 |
| 5 | 80 °C | 10 | 20 |
| 6 | 80 °C | 10 | 100 |
| 7 | 80 °C | 15 | 20 |
| 8 | 80 °C | 5 | 20 |
| 9 | 50 °C | 15 | 60 |
| 10 | 50 °C | 10 | 20 |
| 11 | 50 °C | 10 | 100 |
| 12 | 50 °C | 5 | 60 |
During the kinetic study, at every specific time interval, an aliquot of liquid sample was collected from the hydrolysis solution to measure the ethylene glycol concentration in order to follow the hydrolysis rate. The sample was then transferred into a vial and immersed in an ice bath to interrupt the hydrolysis process. Afterwards, 1,3-propanediol as an internal standard was added to each vial and injected to the GC-FID for analysis. Instrumental conditions are indicated in section 2.3. The resulted chromatogram, shown in Appendix, Fig. A2† was elaborated with known EG concentrations to quantify the amount of EG formed by time during kinetic studies. The sequence of kinetic studies is shown in Scheme 3. Based on obtained kinetic data, the yield (Y) was calculated according to the following equation:
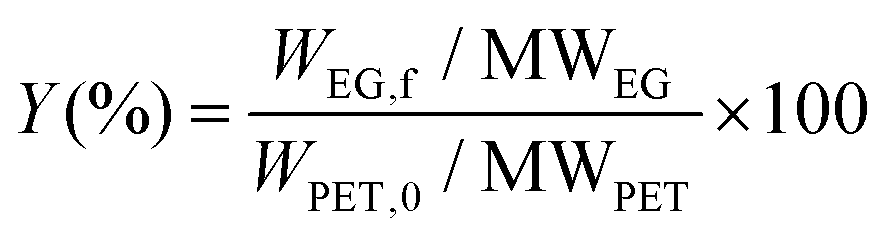 | (2) |
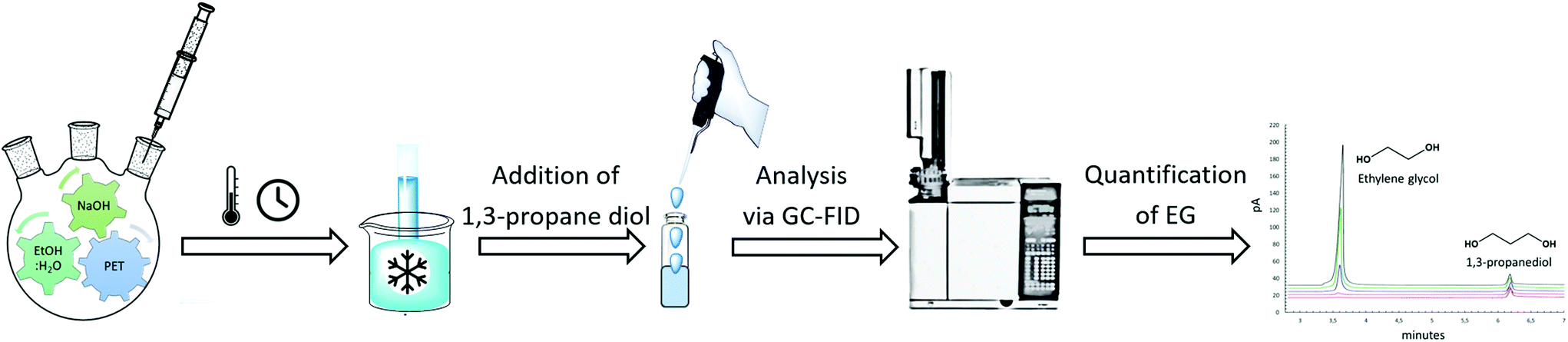 | ||
| Scheme 3 Sequence of kinetic studies performed during alkaline hydrolysis to quantify ethylene glycol. | ||
In most studies the PET degradation yield is determined gravimetrically through weights of residual PET or TPA obtained after purification by using the following formula:
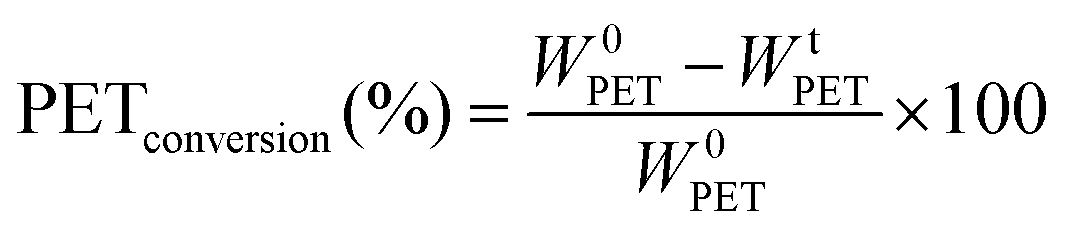 | (3) |
Compared to a chromatographic analysis a gravimetric method has difficulties to quantify the amount of solid product obtained at a specific time and also generally lower yields are obtained due to inevitable weight losses during filtration. To confirm our chromatographic analysis the yields of some experiments were measured by using both methods and it is confirmed that yields obtained through gravimetric method is lower, but still our results are in the same order of magnitude, as shown in Table 3. Therefore, in this study the yield is calculated through GC-FID measurements by following the increase in EG concentration during degradation. In this way faster and more accurate data is obtained at different time intervals.
| Reaction time (min) | PET conversion (%) (gravimetric determination) | PET conversion (%) (chromatographic determination) |
|---|---|---|
| # 1 (10 wt% NaOH, 60 vol% EtOH at 80 °C) | ||
| 10 | 57 | 60 |
| 15 | 60 | 63 |
| 20 | 64 | 68 |
| # 2 (5 wt% NaOH, 60 vol% EtOH at 80 °C) | ||
| 10 | 85 | 88 |
| 15 | 89 | 91 |
| 20 | 93 | 95 |
| # 12 (5 wt% NaOH, 60 vol% EtOH at 50 °C) | ||
| 20 | 63 | 65 |
Once the degradation conditions were optimized based on the results of the conditions indicated in Table 2, kinetic studies were performed on real post-consumer samples from bottles, transparent monolayer and multilayer PET with different particle sizes. In order to investigate the effect of particle size on the degradation rate, shredded samples were grouped into seven different particle size ranges: lower than 0.05 cm, 0.05–0.1 cm, 0.1–0.16 cm, 0.2–0.25 cm, 0.25–0.315 cm, 1 cm and 4 cm. A broad range of PET particle sizes was chosen to test the efficiency of current PET bottle recycling plants which typically use 1.2–1.8 cm of PET flakes.51 For each sample, two-step alkaline hydrolysis were conducted at optimal degradation conditions. During hydrolysis, liquid samples were collected at every specific time interval to be injected into GC-FID. Based on the measurements, PET conversion was calculated for each sample by using eqn (2). Regarding multilayer PET samples, since the measured weight includes PET and also other polymer layers (LDPE and EVOH), correction on the weight of PET sample was made by considering the thickness of the PET layer obtained through POM images (section 2.3). In addition, during manufacturing of PET samples, some additives might be added to improve their physico-chemical properties. Therefore, additive adjustment was also applied by subtracting 5 wt% from the total weight of PET samples based on the literature.52,53 The total weight correction applied on the multilayer PET samples to calculate the theoretical PET weight undergoing hydrolysis is calculated via following formula:
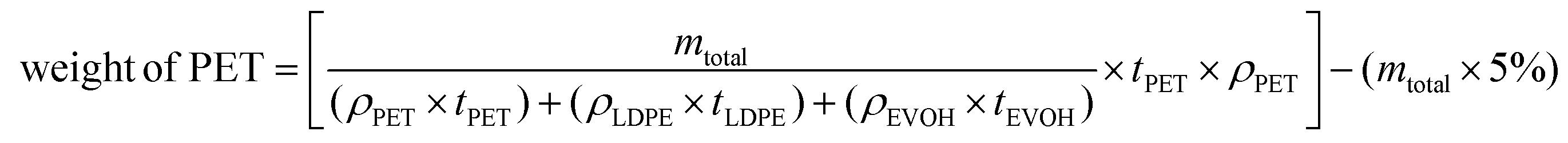 | (4) |
After alkaline hydrolysis of PET waste, unreacted LDPE polymer films were separated from the solution via vacuum filtration and the filtrate was acidified with concentrated sulphuric acid. In the case of black PET samples, the black pigments were removed due to their insolubility in an aqueous medium. A sufficient amount of water was added to the solution to solubilize the precipitated Na2TP and then the solution was centrifuged at 2000 rpm for 20 min or filtered by using a cellulose membrane with 0.1 μm pore size. This allows to separate black pigments having particle size typically between 8 and 100 μm, from the solution which was thereafter acidified to obtain pure white TPA monomer.54
2.3. Characterization of depolymerized monomers and PET waste
The concentration and the quality of EG were investigated by using an Agilent Technologies 7820A Gas Chromatography-Flame Ionization Detector (GC-FID). EZ Chrom Elite software was used to automatically control the instrument, integrate the peak area and calculate concentrations. A J&W 121-7022 DB-Wax column, 20 m × 0.18 mm ID × 0.18 μm DF from Agilent and Sigma-Aldrich 2048605 4 mm ID split/splitless wool packed inlet liner were used. The oven temperature program was adjusted by a hold at 130 °C for 6 min, then the temperature ramped from 130 °C to 250 °C at 10 °C min−1, followed by a hold at 250 °C for 2 min. Inlet and detector temperatures were 260 °C; the helium flow through the column was 25 ml min−1; the flow rates in the detector were 300 mL min−1 for air and 30 mL min−1 for hydrogen. The flow rate for helium makeup gas was 5 mL min−1 and the sample injection volume was 1 μL. Under these parameters, peak times for EG and the 1,3-propanediol internal standard were 3.6 min, 6.2 min, respectively. Total time required for the analysis was 20 minutes. Relative standard deviation has been calculated between duplicate measurements and they are shown in the PET conversion figures.The chemical structure of the obtained TPA was verified via proton-Nuclear Magnetic Resonance (1H-NMR) spectra recorded on Bruker Avance 300 Ultrashield at room temperature by using DMSO-d6 as a solvent. In addition, Shimadzu UV-1280 UV-VIS spectrophotometer was used to qualify the obtained TPA monomer and also to investigate the solubility of Na2TP in function of temperature under the optimal degradation conditions. Furthermore, the purity of TPA was investigated through determination of its acid value by using the following formula:34
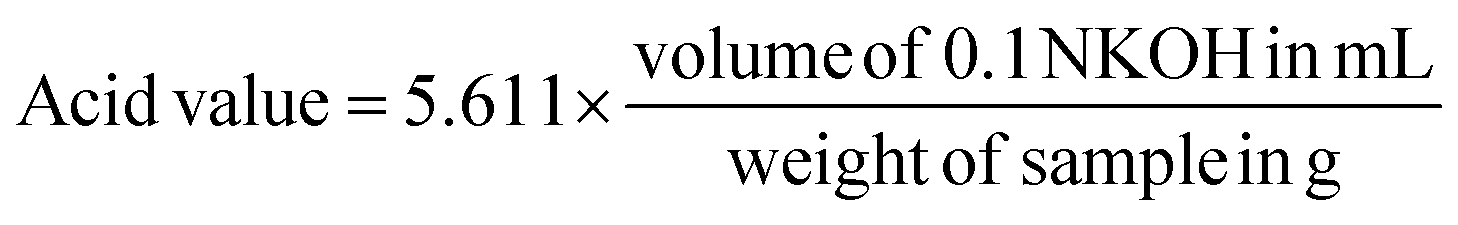 | (5) |
To do this, around 1 g of TPA is weighed to into a 250 ml conical flask and dissolved with 25 mL of pyridine under controlled heating. In addition to pyridine, alternative green solvents can be used, such as dimethyl sulfoxide (DMSO) or ionic liquids e.g. 1-butyl-3-methylimidazolium acetate and 1-ethyl-3-methylimidazolium diethylphosphate.55,56 Afterwards the content of the flask is titrated with 0.5 N KOH solution to a phenolphthalein endpoint. 1H-NMR and UV-VIS spectra of obtained TPA after degradation are shown in Appendix, Fig. A1.†
Regarding multilayer PET samples, the composition of the samples and the thickness of each polymer layer was determined by making microtome cuts of 15 μm using a Leica RM 2245 microtome and then by placing the samples in Canada balsam and conditioning them for 24 h under a bench press. The samples were thereafter analysed using Polarized Optical Microscopy (POM) on a Keyence VHX-500F microscope. In addition, crystallinity of each sample was calculated through Differential Scanning Calorimetry (DSC) measurements by using a NETZSCH Polyma DSC 214 under N2 atmosphere with a flow of 20 mL min−1. Each sample was heated starting from 20 °C till 300 °C then cooled to 50 °C and again heated to 300 °C at a heating/cooling rate of 10 °C min−1. The crystallinity of PET samples was calculated via following formula:57
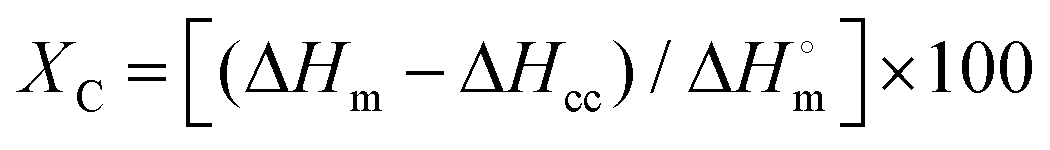 | (6) |
Based on these measurements, the total thickness of each PET sample and their specific surface area are shown in Table 4 together with their crystallinity.
| Type of PET | Thickness (mm) | Specific surface area (m2 g−1) | Crystallinity (%) |
|---|---|---|---|
| Multilayer tray | 0.35 | 0.0025 | 7.26 |
| Multilayer film | 0.045 | 0.0192 | 12.49 |
| Bottle | 0.27 | 0.0062 | 33.15 |
| Monolayer tray | 0.16 | 0.0052 | 11.73 |
| Monolayer film | 0.035 | 0.0213 | 30.77 |
| Pure PET pellets | 2.5 | 0.0003 | 41.28 |
The outer layers of the PET samples were also confirmed via Fourier-Transform Infrared spectroscopy (FTIR) on a Bruker Tensor 27 FTIR spectrometer. The results of these measurements are indicated in Fig. 1.
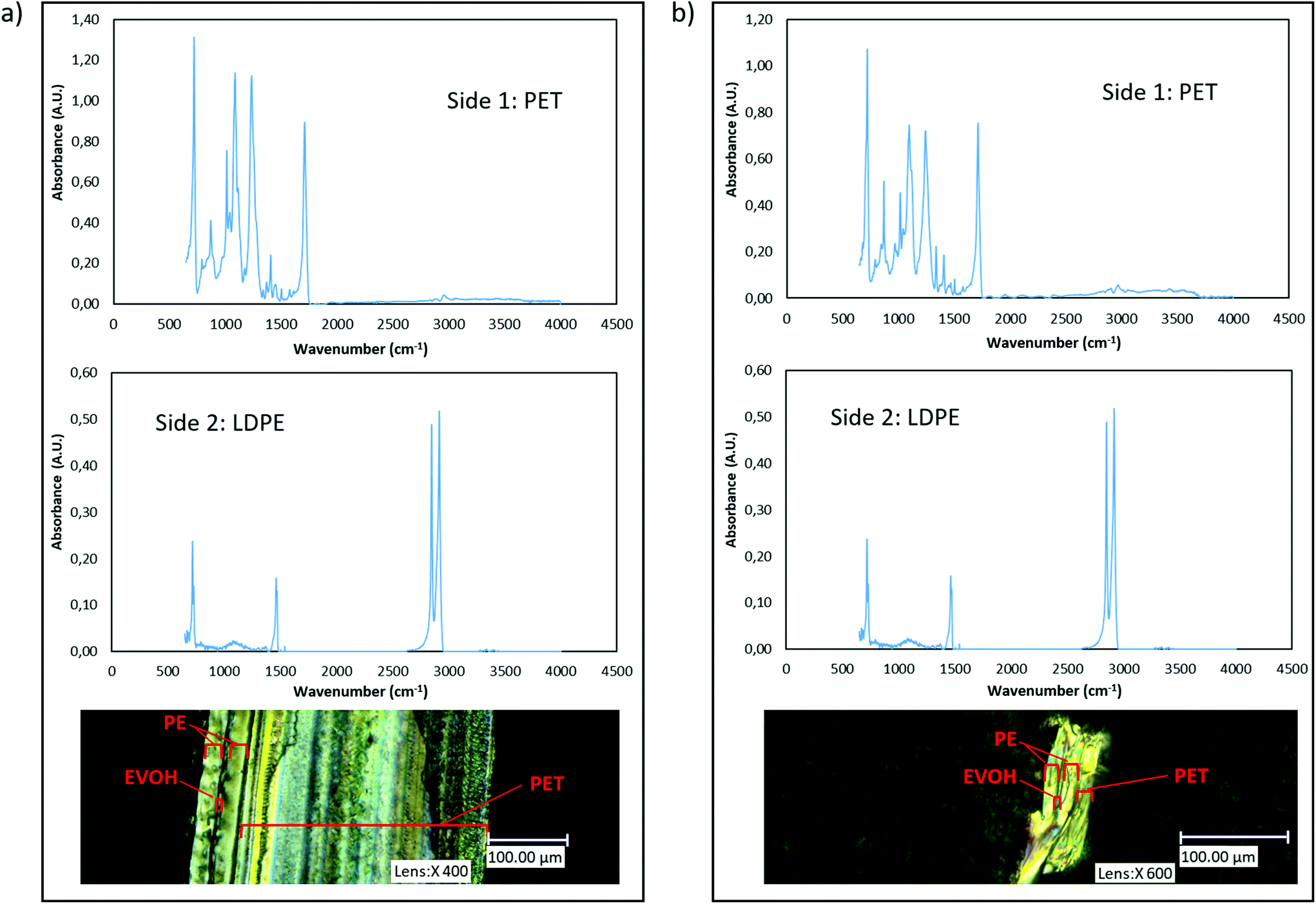 | ||
| Fig. 1 (a) FTIR spectra of both sides of multilayer PET tray and its POM image (b) FTIR spectra of both sides of multilayer PET film and its POM image. | ||
The transformation of ester bonds in Na2TP to the TPA after acidification was confirmed via FTIR spectrometer using KBr pellets. Pellets were prepared by using 0.250 ± 0.010 g fine KBr powder which was put into a pellet-forming die and subsequently subjected to a force of approximately 10 tons. By release of force, transparent 13 mm-diameter pellets were obtained. Afterwards, 35 μl aliquot of liquid samples, collected from the reaction medium at every specific time interval, were placed on each prepared KBr pellet and dried for 15 minutes using an infrared lamp. The TPA sample was prepared by mixing with pure KBr powder in 1:100 weight ratio. The FTIR measurements were recorded using the Omnic software in the range of 4000–400 cm−1, at resolution of 4 cm−1 and with 32 scans. For each FTIR analysis, automatic smooth and baseline correction was applied. The obtained spectra are shown in Fig. A6.†
3. Results and discussion
3.1. Effect of the reaction parameters on the PET degradation rate
In order to investigate the optimal degradation conditions, 2 g of PET flakes with 500 μm particle size were depolymerized in 100 mL of a sodium hydroxide solution under continuous stirring with a magnetic stirrer at 250 rpm based on the experimental conditions as shown in Table 2. For each experiment, the obtained PET conversions were plotted as a function of reaction time and shown in Fig. 2.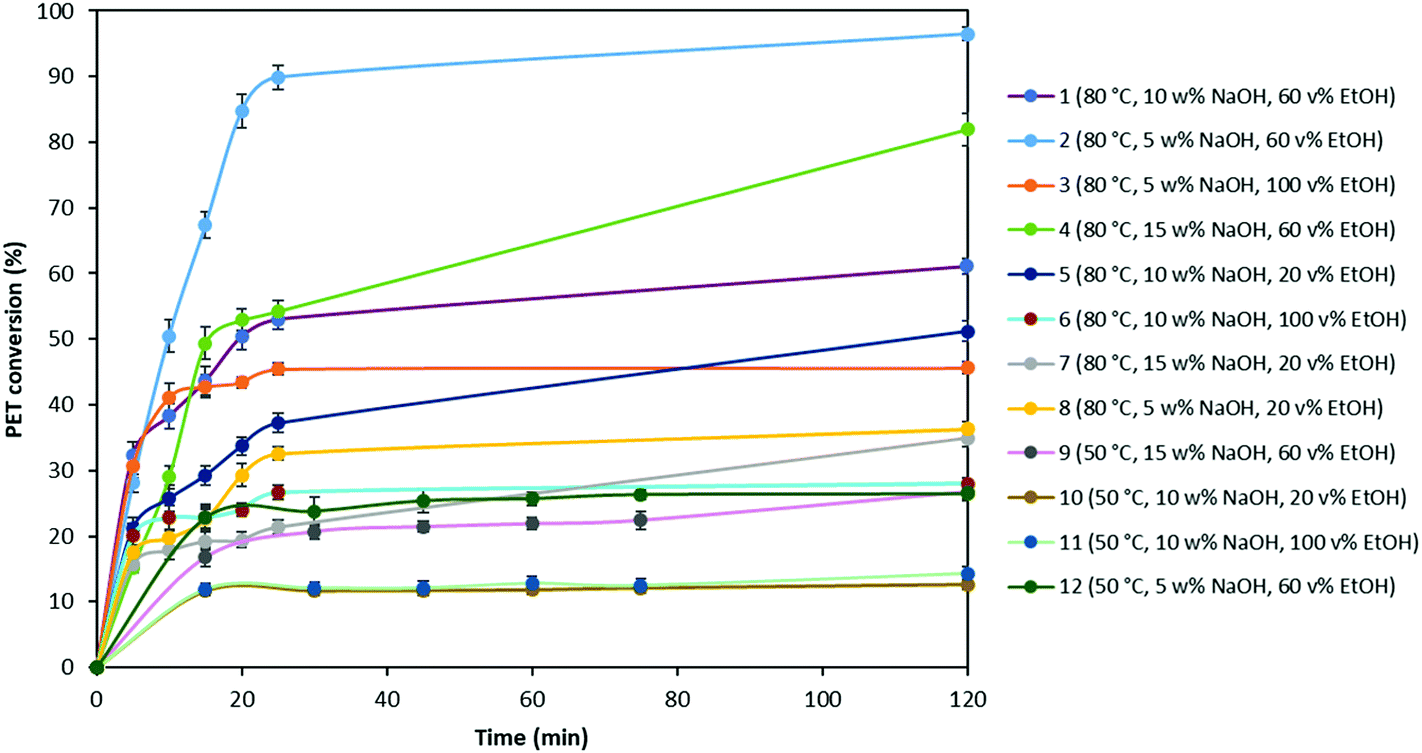 | ||
| Fig. 2 PET conversion at different experimental conditions (0.02 g mL−1 PET flake with 500 μm particle size, stirring at 250 rpm with a magnetic stirrer). | ||
Fig. 2 shows that the amount of PET converted to its monomers increases as the temperature rises from 50 °C to 80 °C. For example, PET conversion yield at 50 °C with 10 wt% of NaOH and 20 vol% of EtOH (#10) is 10% after 2 h, while this amount increases to 48% at 80 °C (#5). The NaOH concentration was also found to be a critical parameter in the hydrolysis. When experiments # 5, 7 and 8 conducted with 10, 15 and 5 wt% of NaOH respectively (at 80 °C and 20 vol% EtOH), are compared, it is observed that PET degradation yield increases from 35% to 58% with the increase in NaOH concentration from 5 to 10 wt%. However, further increase in the NaOH wt% results in decrease of the PET conversion yield. This might be due to possible deposition of excess NaOH on the PET sample surface, which acts as a blocking organic film during the hydrolysis reaction, making it difficult for hydroxide ions to access new carbonyl carbons in unreacted PET and, consequently, reducing the efficiency of hydrolysis reaction. Furthermore, Fig. 2 shows that the EtOH to water volume ratio also has an important effect on the PET conversion. With a change in the volume percentage of EtOH from 20 vol% to 60 vol%, the yield increases significantly as from 30% to 95% (#2 and 8, respectively), most likely due to higher solubility of EG in EtOH compared to water. However, it is noticed that using 100 vol% EtOH during PET degradation causes lower yields compared to using 60 vol% of EtOH. This might be due to higher solubility of Na2TP in water, which is 13.26 wt% at 40 °C.59 A mix of both solvents thus increase the degradation rate.
During PET degradation, mass transfer between the solid and liquid phase might also be a limiting factor on the hydrolysis rate. To support this hypothesis, the same experiments as mentioned in Table 2 were also performed by stirring with an agitator at 500 rpm and the obtained results were compared with those obtained by stirring with a magnetic stirrer at 250 rpm (Table 5).
| # of experiment | Experimental conditions | PET conversion (%) with 250 rpm | PET conversion (%) with 500 rpm |
|---|---|---|---|
| 1 | 80 °C, 10 wt% NaOH, 60 vol% EtOH | 50.44 | 77.49 |
| 2 | 80 °C, 5 wt% NaOH, 60 vol% EtOH | 84.69 | 95.23 |
| 3 | 80 °C, 5 wt% NaOH, 100 vol% EtOH | 43.40 | 72.11 |
| 4 | 80 °C, 15 wt% NaOH, 60 vol% EtOH | 52.93 | 81.41 |
| 5 | 80 °C, 10 wt% NaOH, 20 vol% EtOH | 33.75 | 70.47 |
| 6 | 80 °C, 10 wt% NaOH, 100 vol% EtOH | 24.01 | 46.90 |
| 7 | 80 °C, 15 wt% NaOH, 20 vol% EtOH | 19.40 | 48.44 |
| 8 | 80 °C, 5 wt% NaOH, 20 vol% EtOH | 29.30 | 68.32 |
| 9 | 50 °C, 15 wt% NaOH, 60 vol% EtOH | 16.80 | 32.45 |
| 10 | 50 °C, 10 wt% NaOH, 20 vol% EtOH | 11.64 | 20.20 |
| 11 | 50 °C, 10 wt% NaOH, 100 vol% EtOH | 11.90 | 28.10 |
| 12 | 50 °C, 5 wt% NaOH, 60 vol% EtOH | 22.78 | 34.45 |
As seen in Table 5, due to facilitating mass transfer with rigorous stirring, the rate of PET conversion increased substantially compared to that obtained with a magnetic stirrer at 250 rpm. It is also noticed that PET conversion at 50 °C did not show significant changes at higher stirring rate due to insufficient thermal energy to activate PET hydrolysis. In addition, since glass transition temperature (Tg) of PET is around 80 °C, at 50 °C amorphous region of the PET sample will not be able to undergo transition from glassy to rubbery state, which might also have an effect on the degradation rate. Based on these performed screening experiments, it can be concluded that all studied parameters, temperature, NaOH and EtOH concentration and turbulence influence the reaction rate. Among these parameters, ethanol to water ratio and stirring rate has more dominant effect on the hydrolysis rate. Furthermore, since Na2TP salt might precipitate during degradation depending on the amount of PET used, we have validated that precipitation of the salt is not a limiting factor on diffusion and PET degradation rate depends only on the reactants (Appendix A.1). Based on these results, the highest yield is obtained with 500 rpm stirring rate via an agitator, at 80 °C and in a solution containing 5 wt% NaOH and 60:40 EtOH:H2O mixture. These conditions will therefore be used in the alkaline hydrolysis of post-consumer PET plastic waste.
3.2. Best-fit kinetic model
Polymer degradation is a complex process in which chemical reactions or physical changes are occurring simultaneously. Most of the kinetic studies on PET depolymerization are based on an reversible polycondensation reaction. Therefore, often conventional nth-order kinetics describing the polymerization to form PET ester linkages, are used for hydrolysis. Although adopting the kinetic model of PET polycondensation for the depolymerization simplifies the kinetic rate calculations, these homogeneous nth-order kinetic models discard possible consequences of a polymeric and heterogeneous system.60 Therefore, in this study we use solid-state kinetic models for PET depolymerization instead of other heterogeneous kinetic modeling approaches. In solid-state kinetics, mechanistic interpretations are made by identifying a reasonable reaction model.61 These models are generally categorized based on the graphical shape of mechanistic assumptions. Due to these graphical presentations so called “master plots”, the most appropriate model can easily be determined based on the best superimposition of a particular data set on a kinetic model. The model is derived based on different reaction mechanisms which include reaction-order, diffusion, nucleation and geometrical contraction.60 Šesták and Berggren62 have suggested the following general expression:| g(α) = αm(1 − α)n(−ln(1 − α))p | (7) |
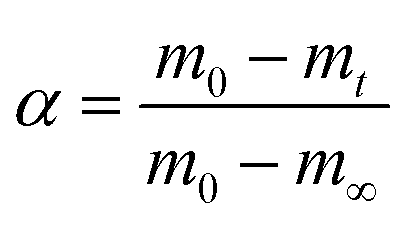 | (8) |
Since kinetic equations depend on the extent of conversion α, kinetic data obtained under different experimental conditions can be compared with those of known kinetic models by plotting the kinetic data and reduced generalized rates against the conversion kinetic models.63,64 Based on the type of experimental conditions and also the kinetic data, these theoretical plots can be expressed in different forms such as differential, integral, and differentio-integral.65 In this study, hydrolysis kinetics of PET were performed in terms of batch-mode experiments, thus the integral form of the theoretical curves, g(α), as indicated in Table 6 becomes the most suitable choice. These integral forms of the generalized kinetic plots are superimposed in Fig. 3 together with some representative experimental kinetic data.
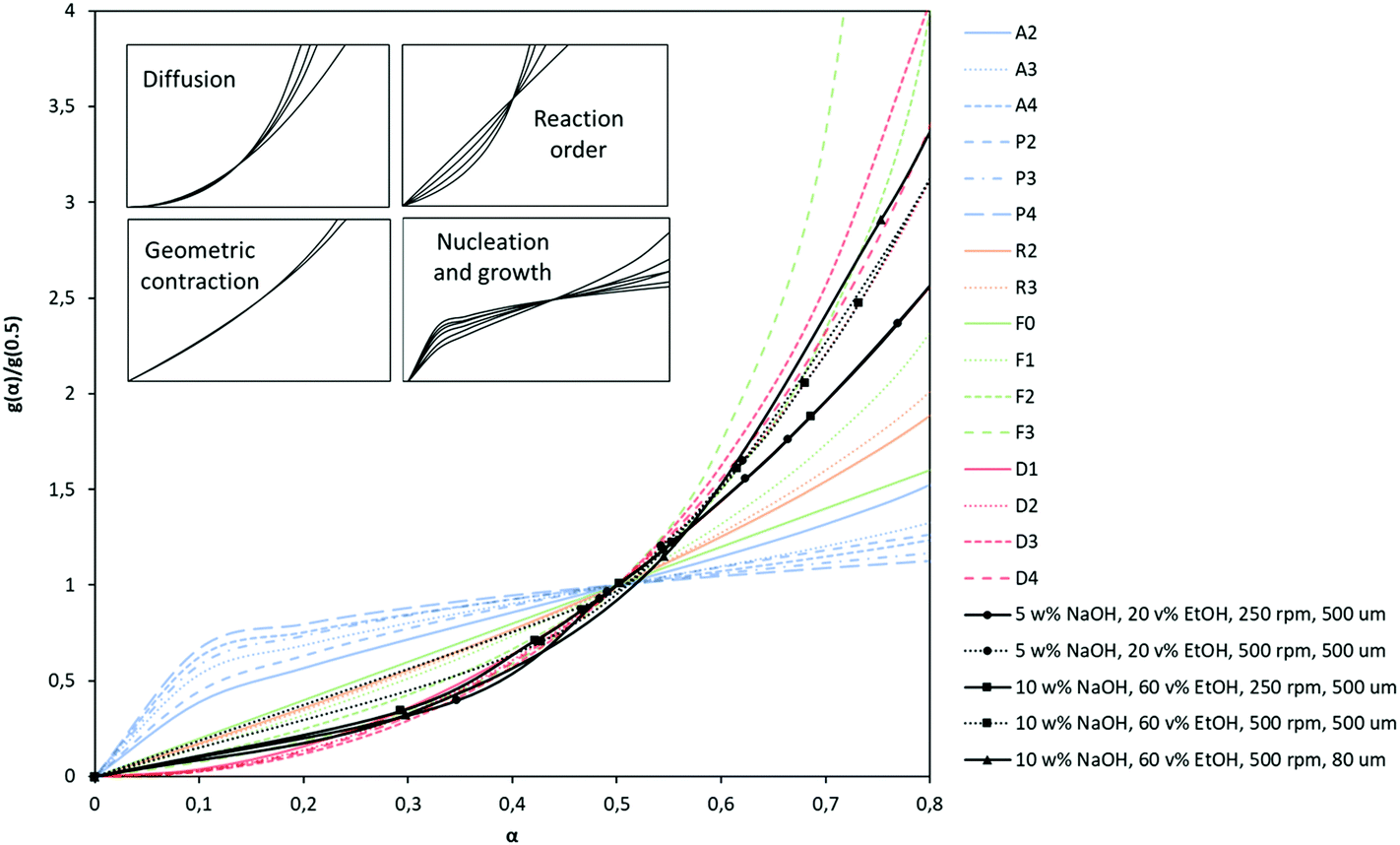 | ||
| Fig. 3 Theoretical kinetic master curves plotted as integral form of reaction models at isothermal condition (g(α)/g(0.5)) versus conversion fraction (α) and superimposition of experimental kinetic data on these kinetic models (R2 values and best-fit diffusion kinetic models can be found in Appendix, Table A-1 and Fig. A-3). | ||
| Symbol | Reaction model | Integral form g(α) = kt |
|---|---|---|
| a Jander equation. b Ginstling–Brounshtein equation. | ||
| Diffusion models | ||
| D1 | 1-D diffusion | α 2 |
| D2 | 2-D diffusion | [(1 − α)ln(1 − α)] + α |
| D3 | 3-D diffusiona | [1 − (1 − α)1/3]2 |
| D4 | 3-D diffusionb | 1 − 2α/3 − (1 − α)2/3 |
| Geometric contraction models | ||
| R2 | Contracting area | 1 − (1 − α)1/2 |
| R3 | Contracting volume | 1 − (1 − α)1/3 |
| Reaction order models | ||
| F0 | Zero-order | α |
| F1 | First-order | −ln(1 − α) |
| F2 | Second-order | (1 − α)−1 − 1 |
| F3 | Third-order | 1/2[(1 − α)−2 − 1] |
| Nucleation and growth models | ||
| P2 | Power-law | α 1/2 |
| P3 | Power-law | α 1/3 |
| P4 | Power-law | α 1/4 |
| A2 | Avrami-Erofe'ev | [−ln(1 − α)]1/2 |
| A3 | Avrami-Erofe'ev | [−ln(1 − α)]1/3 |
| A4 | Avrami-Erofe'ev | [−ln(1 − α)]1/4 |
As seen in Fig. 3, experimental kinetic data obtained at different experimental conditions at 80 °C matches closely with the diffusion kinetic models (D). Although PET hydrolysis is commonly explained via reaction-order kinetic models,27,32,36 the experimental data do not show significant scatter especially at higher conversion rates. Generally in diffusion-controlled reactions, the rate of product formation increases proportionally with agitation. This is also confirmed by our kinetic experiments of alkaline hydrolysis at two different agitation speeds. When agitation speed is increased from 250 rpm to 500 rpm, the kinetic data still exhibit a diffusion kinetic behavior. It is interesting to note that at higher agitation, the data is better represented by two-dimensional diffusion kinetics (D2) instead of one-dimensional diffusion kinetics (D1). In the D2 model, solid particles are considered as cylindrical and diffusion occurs through a cylindrical shell with an increasing reaction zone. Therefore, it is obvious to observe D2 model at higher agitation rate since the reaction rate increases due to higher mass transfer. Using different agitation rates thus allowed us to calculate rate constant with minimum standard deviation although obtaining higher yield with higher agitation is obvious. Moreover, the kinetic data obtained at lower particle size (80 μm) fit better on three-dimensional diffusion model (D4). This kinetic model is based on assumption of spherical solid particles and thickness of these particles has a significant effect on the rate of reaction. Therefore, D4 kinetic model represents the data better at lower particle size. D3 is also a three-dimensional diffusion model, but Ginstling–Brounshtein have shown that the Jander model is oversimplified and holds only at low conversion values.60 Based on these comparisons, the diffusion models supports the obtained kinetic data and thus it is selected as a best-fit kinetic model for the hydrolysis of PET and it is also used to explain the kinetic data of post-consumer PET waste.
3.3. Degradation kinetics of post-consumer PET waste at optimal degradation conditions
Post-consumer PET waste generally consists of multilayer polymer layers such as polyolefins and EVOH to improve the sealing and also gas barrier properties of packaging. Although these multilayers allow to increase physico-chemical properties of plastics, they impede recycling. Whereas the previous part lays the baseline of this work with virgin grade PET, in this part degradation kinetics of transparent PET bottles and also monolayer and multilayer PET trays and films were investigated through alkaline hydrolysis at optimal experimental conditions. Kinetic studies were performed on these different post-consumer PET waste having different thicknesses, specific surface area and crystallinity (Table 4) and also particle sizes ranging from 0.05 cm to 4 cm. Based on obtained kinetic data, PET conversions were calculated for each sample and plotted as a function of reaction time as shown in Fig. 4. Similar to this study, in the patent of Loop Industries PET bottles were hydrolyzed in an aqueous alkaline medium including potassium hydroxide (KOH), methanol and a non-polar solvent for swelling of the polymer, at ambient temperature and atmospheric pressure. In this work, we have optimized the aqueous alkaline reaction conditions and we showed that an increase in temperature affects the degradation rate positively. In addition, we have extended the degradation study to post-consumer PET waste including multilayer and coloured samples. This study will help to further improve alkaline hydrolysis processes such as the one developed by Loop Industries.66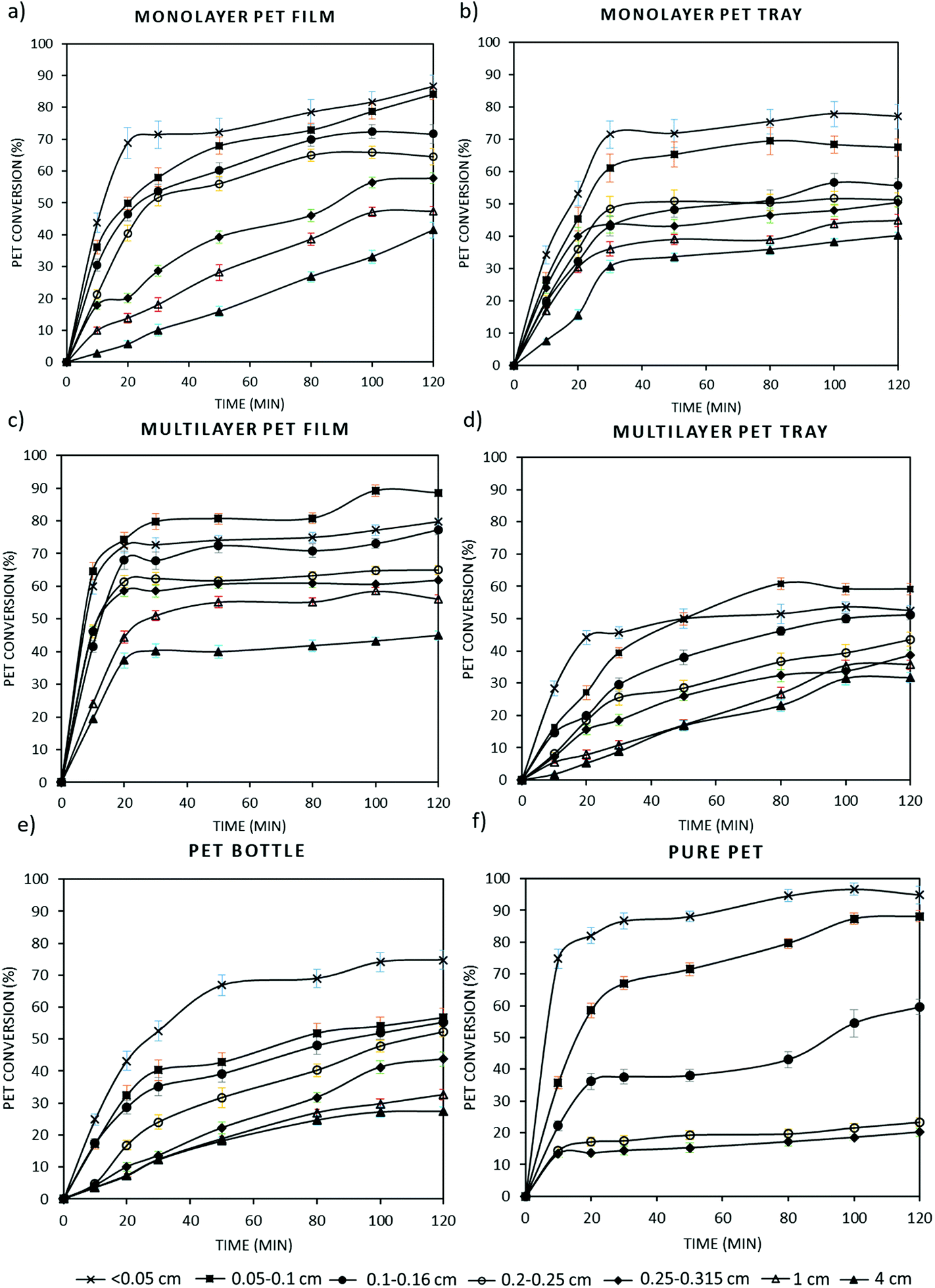 | ||
| Fig. 4 PET conversion (%) versus time graph of (a) monolayer PET film; (b) monolayer PET tray; (c) multilayer PET film; (d) multilayer PET tray; (e) PET bottle; (f) pure PET grade, at different particle sizes (0.02 g mL−1 PET flake at 500 rpm stirring rate via an agitator). | ||
As seen in Fig. 4, the highest PET conversion (∼95%) is obtained with pure PET at the smallest particle size (<0.05 cm). This is followed by PET films, bottles and trays, respectively. Among all PET samples, conversion is the slowest with multilayer trays due to their higher thickness and lower specific surface area compared to the other samples and the fact that only one side of the particle is accessible for hydrolysis, obviously limiting degradation rate. Regarding all type of samples, PET conversion percentage decreases with an increase in the particle size. For example, while PET bottle conversion with the smallest particle size (<0.05 cm) is around 70%, this decreases up to 20% at the highest particle size (4 cm). In plastic recycling industry, generally 1.2–1.8 cm of PET bottle flakes are used.51 Based on the experimental results, we can conclude that the efficiency of industrial PET recycling can be improved substantially if particle size is decreased further, even lower than 0.05 cm. Bigger particle sizes have a lower specific surface area (SSA), which is thus a limiting factor for the PET conversion. For instance, although higher conversions are expected with degradation of pure PET pellets compared to the other multilayer samples, at high particle size conversion of PET pellets was very low (∼15%) due to small SSA as indicated in Table 4. It is thus clear that smaller particle sizes might be interesting; however, powders might be more difficult to handle and micronizing polymers at large scale is not so common practice in the current recycling industry. It is also interesting to note that in multilayer PET samples, the conversion obtained with the particle size smaller than 0.05 cm is lower than that obtained with the particle size between 0.05 and 1 cm. The probable explanation is that hydrolysis resistant polymer layers e.g. LDPE become more important in the multilayer structure when PET degrades, which increases the floating probability of PET flakes due to decrease in the total weight.
By using these PET conversions, a master plot is elaborated for each PET sample in order to investigate the best-fit kinetic model as it is performed with pure PET resins in section 3.2. It is observed that experimental kinetic data of all PET samples match closely with the diffusion kinetic model. Similar to the kinetics of pure PET grades, data obtained with higher degradation rates, typically with lower particle sizes, fit in the D4 diffusion model, whereas lower degradation rates fit better in the D2 diffusion model with high coefficient of determination values (R2) as shown in Appendix, Table A1.† By using these diffusion kinetic models, rate constants (k) were obtained for each sample and the trend of the k value based on the particle size and type of sample is shown in Fig. 5.
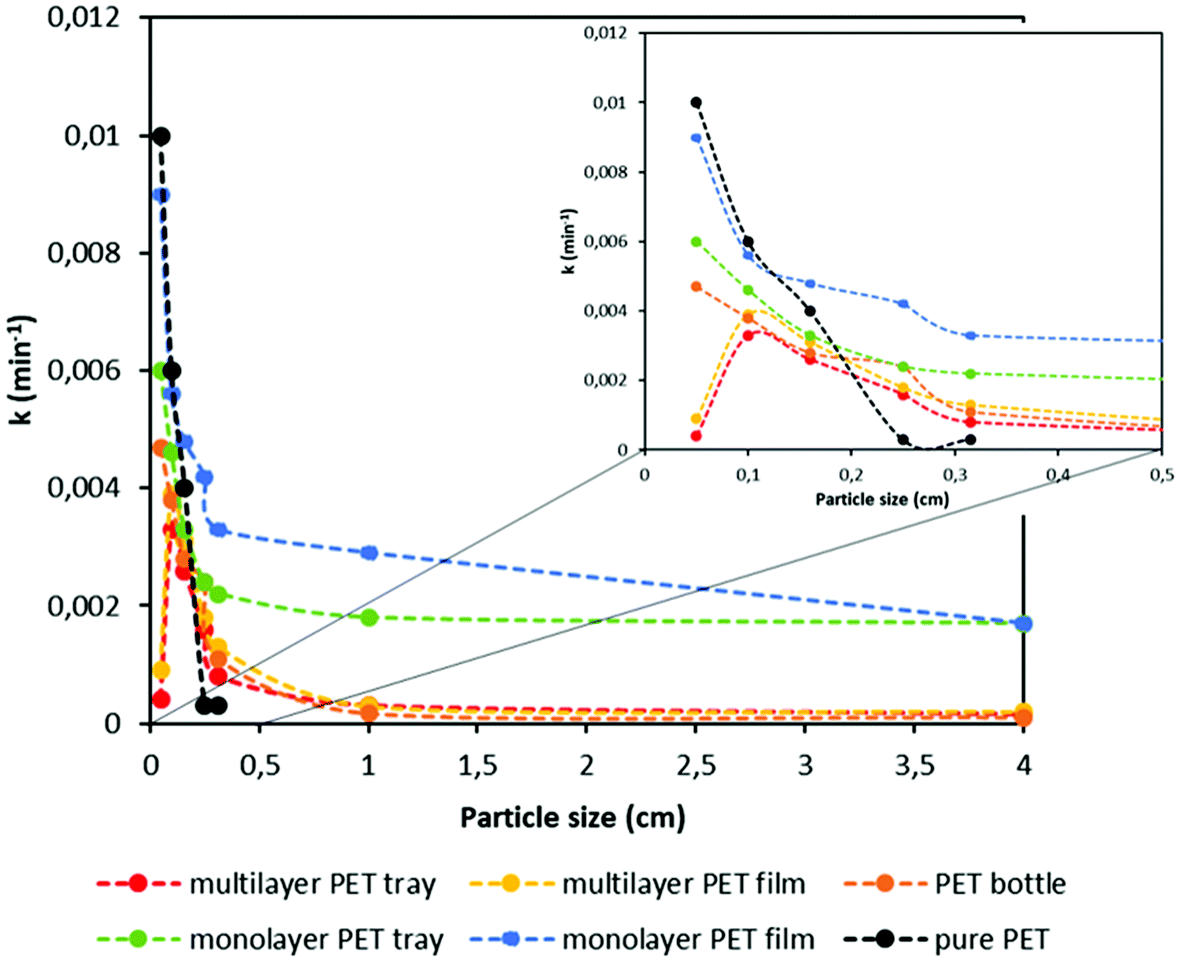 | ||
| Fig. 5 Rate constant (k) value versus particle size graph for different PET samples at different particle sizes. | ||
Similar to the PET conversion, rate constant shows a similar trend as it decreases with increasing particle size. In addition, it is notable that existence of additional polymer layers has a significant effect on the rate constant. While the k values of monolayer PET samples drops substantially with an increase in the particle size, those of multilayer samples do not change considerably. Since hydrolysis is diffusion controlled, existence of the other polymer layers limits reagent diffusion for degradation of PET. For example, the rate constant (k) of monolayer PET film with a particle size lower than 0.05 cm decreases around 40% when its size is doubled. As mentioned earlier, rate constants of multilayer PET tray and film at particle size lower than 0.05 cm are smaller than those at particle size between 0.05 and 0.1 cm, potentially due to higher floating tendency of smaller particles, causing diffusion limitations. This can be solved industrially by better reactor design though. Another important factor affecting the rate constant is the thickness of the PET samples. It is well known that the diffusion rate is inversely proportional to the thickness of the material. Since the thicknesses of the PET trays is more than 5-fold higher compared to that of PET films as shown in Table 4, 35% lower k values are obtained with PET trays compared to PET films at particle size lower than 0.05 cm. Likewise, the thickness of a PET bottle is higher than multilayer PET film, thus at higher particle size the k value of PET bottle is lower, although the bottle consists of a single polymer layer. Above 0.2 cm particle size, the thickness of pure PET pellets is larger, which causes to obtain the lowest k values with pure PET compared to other multilayer and monolayer PET samples. This can be explained by the fact that pure PET pellets are more spherical, and thus have a lower specific surface area at large particle size. In addition to thickness, crystallinity of the sample plays an important role on hydrolysis by affecting the solvent diffusion. As the crystallinity of the polymer increases, solvent penetration through tightly packed chains in the crystalline domain becomes slower, as such the degradation rate decreases.67,68 As shown in Table 4, bottles and pure PET pellets have higher crystallinity, thus at high particle size lower degradation rates were obtained with those samples compared to the multilayer PET samples. However, based on the experimental results it is seen that particle size and thickness have more dominant effect on the hydrolysis rate. Furthermore, it is observed that for all the PET samples, k value does not change considerably beyond 1 cm particle size. Decreasing particle size thus only makes sense if it is decreased below 0.5 cm. As seen, particle size, thickness and specific surface area affect the PET conversion based on the type of PET sample. By adjusting these parameters well and solving the diffusion limitations, the highest PET degradation yields can be obtained even with multilayer PET plastic waste.
3.4. Scale-up assessment
In addition to multilayer structures, also different additives such as colours impede closed-loop recycling of PET plastic waste. Especially carbon black is one of the biggest bottlenecks during recycling due to its insolubility in an aqueous medium. In the meantime, excess Na2TP salt might also start to precipitate and mixes with carbon black pigments causing a decrease in purity of the monomers. To avoid these problems, a potential process to obtain pure monomers from alkaline hydrolysis of multilayer and coloured PET trays including black PET samples with 0.25 cm particle size was shown in Fig. 6 and then a life cycle assessment (LCA) has been performed on this process in order to investigate its carbon footprint.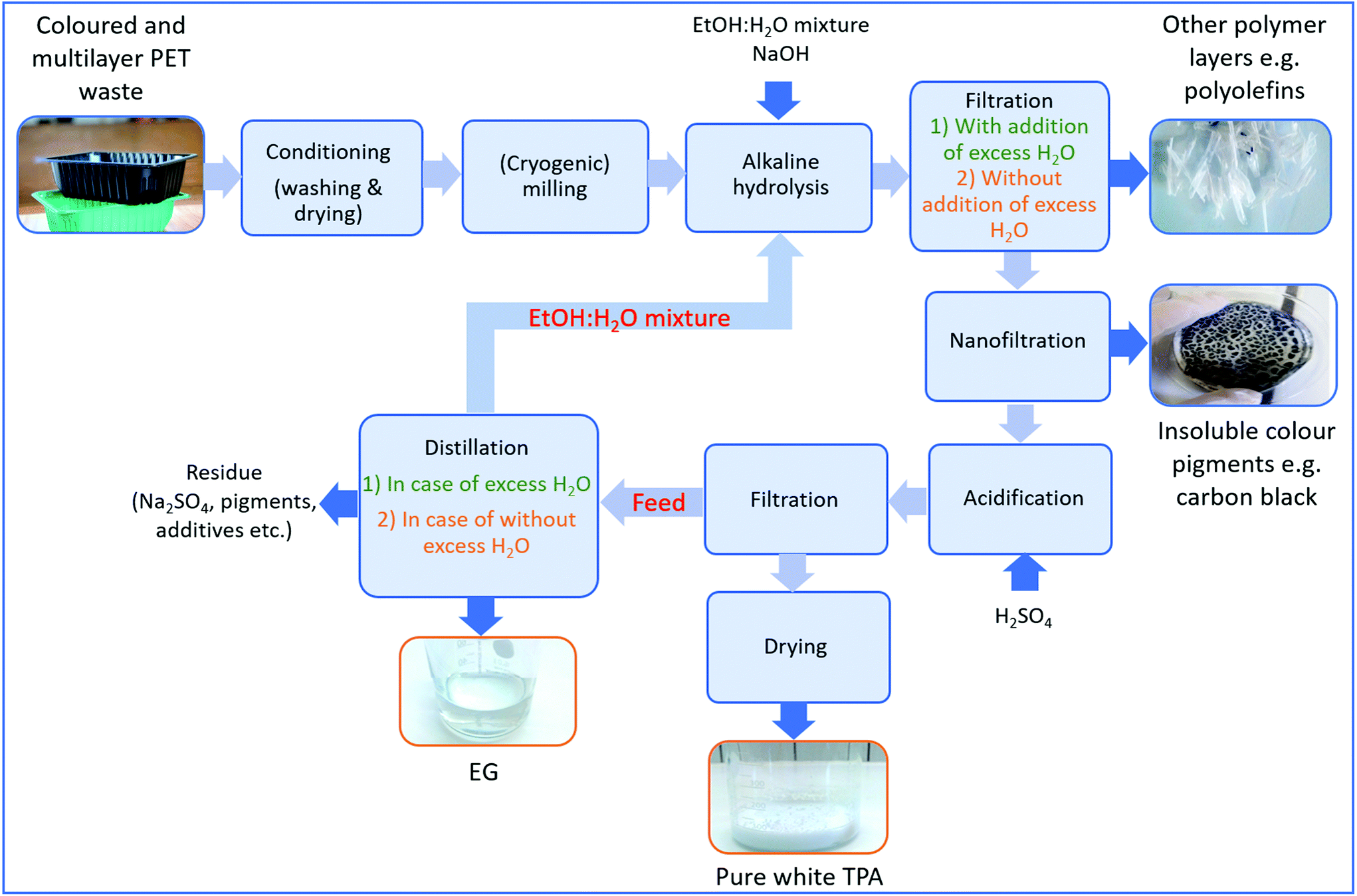 | ||
| Fig. 6 Possible flowsheet for separation of colours and polyolefins while obtaining pure PET monomers. | ||
First of all, post-consumer plastics are conditioned to remove traces from residual products such as food. Afterwards, their particle size is reduced to the desired size (e.g. 0.25 cm) and then subjected to alkaline hydrolysis at the previously described optimal experimental conditions (60:40 vol% EtOH:H2O, 5 wt% NaOH and at 80 °C). Since a low temperature is applied during PET hydrolysis, other polymer layers such as polyolefins are not affected by the process, thus they can be recovered from the solution via filtration after degradation is completed. Afterwards, more water is added to the medium to solubilize the precipitated Na2TP salt, as such separating insoluble black pigments having particle size typically between 8 and 100 μm (ref. 54) via centrifugation or by using a membrane. The advantage of using hydrolysis in this case is also the relatively low viscosity of the water/ethanol mixture which is beneficial for many separation steps such as filtration and centrifugation. Since the use of excess water makes the process costly and increases the environmental impact, any insoluble colour pigments can be separated from the precipitated Na2TP salt by utilizing their differences in density and particle size. For example, since the particle size range of Na2TP at its saturation point (a length of about ∼8 μm, a diameter of ∼2 μm)69 is smaller than that of carbon black, they might also be separated via selective membrane filtration without solubilizing the precipitated salt.
Since TPA monomers are insoluble in an aqueous media, they will precipitate upon acidification while the soluble pigments stay in the solution. After unreacted polymer layers such as polyolefins and insoluble pigments are removed, the filtrate is acidified and the precipitated monomer is subjected to the second filtration. The new filtrate is then dried to obtain pure white TPA monomer. In case the PET stream would contain only water soluble colour pigments e.g. green or blue PET food trays, the aqueous solution can be acidified directly without separating the non-water soluble or inorganic pigments beforehand. In addition to the TPA monomer, EG can also be recovered from the solution through distillation. This could be achieved in two steps. First, the water/ethanol mixture is flashed, which can then be reused in the alkaline hydrolysis or during filtration, reducing the need of fresh solvent. Second, EG can be separated, leaving a residue containing a whole mix of components, amongst others additives. In order to assess the feasibility of reaction medium purification, simulations have been performed in Aspen Plus 10 based on two hydrolysis scenarios, one considering excess water addition to dissolve precipitated Na2TP salt and the other one considering filtration of precipitated Na2TP salt without use of excess water. The latter simulation is shown in Scheme 4 and the former one is given in Appendix, Scheme A1.†
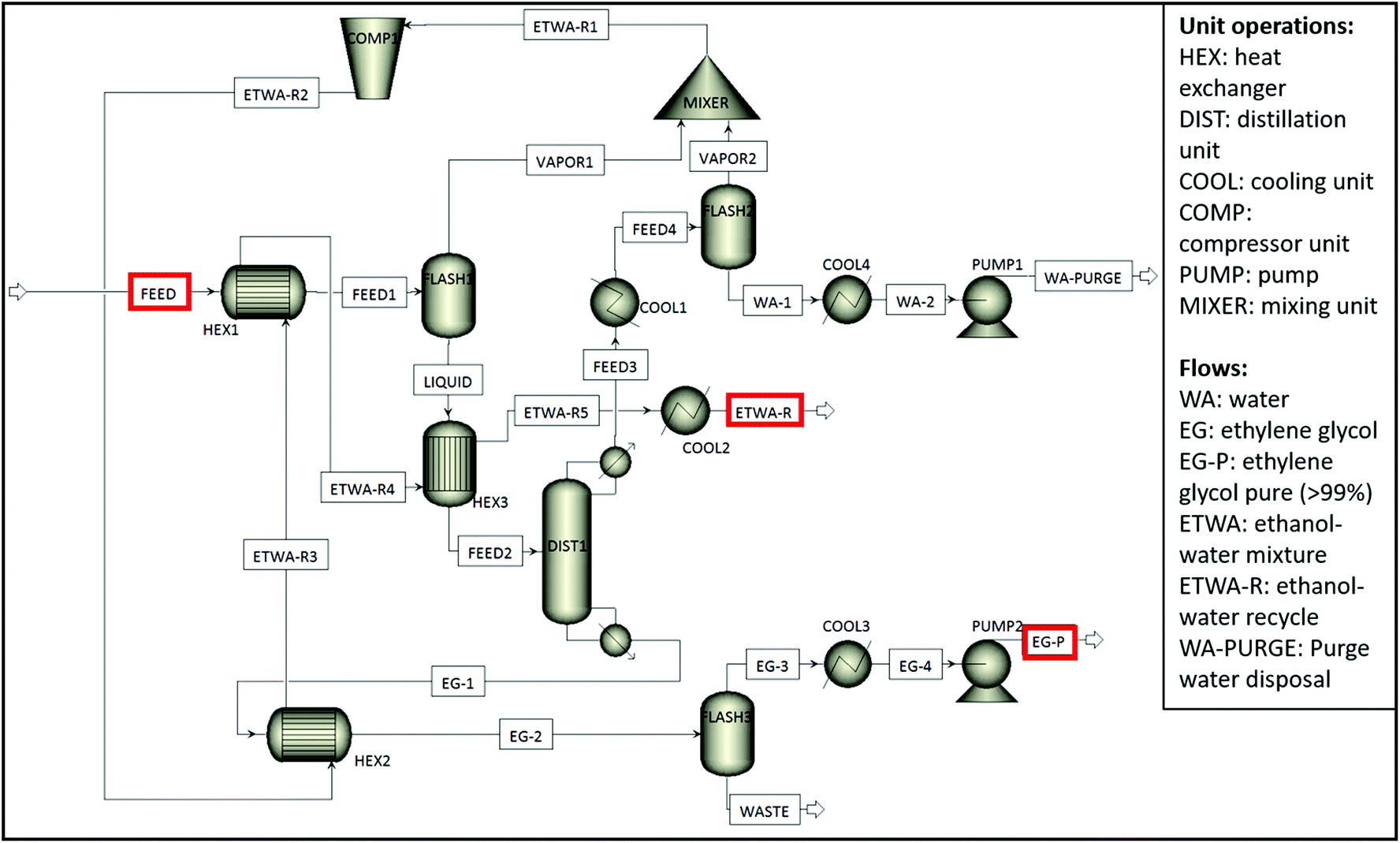 | ||
| Scheme 4 Aspen Plus process flow diagram of the recovery section for the ethylene glycol and water:ethanol separation plant (hydrolysis scenario 1: no excess addition of water). | ||
:water mixture at 60:40 vol% to be reused in hydrolysis. Both simulations have been performed with different solid/liquid (S/L) ratios: 0.02 g mL−1 (used in this study), 0.03 g mL−1, 0.04 g mL−1 and 0.05 g mL−1 which corresponds to 1000 kg, 1500 kg, 2000 kg and 2500 kg of post-consumer PET waste, respectively, in 50000 liters of water:ethanol solution. The S/L ratio can be increased further by adjusting the amount of NaOH. Since in the first step of PET alkaline hydrolysis sodium terephthalate salt is formed, sufficient NaOH should be present in the medium to react with all PET flakes. Therefore, the concentration of PET is mainly determined by NaOH concentrations which can be increased until its saturation point. In order to confirm that depolymerization still occurs at high PET concentrations considered in Aspen simulations, PET hydrolysis was also performed at 0.05 g mL−1 of PET concentration under optimal degradation conditions (Fig. A4†). It is observed that although the hydrolysis rate is affected by the PET concentration, higher PET concentrations hardly limit the depolymerization yield. In each scenario degradation conditions were kept at their optimal values obtained through kinetic studies of pure PET pellets (at 80 °C with 5 wt% NaOH and 60:40 vol% EtOH:water). The amounts of inputs/outputs such as H2SO4, excess water used and formed monomers were calculated for each scenario (Appendix, Table A-2). By using these calculated values, energy recovery and consumption values of each process step were obtained through Aspen Plus simulations as given in Appendix (Tables A-2 to A-5) and then these values were used to perform a life cycle assessment (LCA) for the proposed PET alkaline hydrolysis process.
The LCA was performed using the OpenLCA software for both Aspen simulations, based on PET alkaline hydrolysis scenarios with and without excess addition of water during purification of monomers, at different S/L ratios. The result has been expressed in terms of carbon dioxide equivalent (CO2-eq) per kg of PET waste via the ReCiPE Midpoints (H) impact assessment method. The individual carbon footprint for all inputs/outputs of the analysed process were extracted from the Ecoinvent Database v3.1. For both hydrolysis scenarios, end-of-life processing of Na2SO4 salt formed after acidification of the reaction medium has not been taken into account since it can, for instance, be used to recover H2SO4 and NaOH through bipolar membranes instead of being incinerated.70 In addition, additives that stay in the EG fraction also have not been considered due to their small amounts. Moreover, two flash distillations (Flash 1 and 3) and one fractional distillation (Dist1) were considered as shown in Scheme 4 in order to recover ethanol:water mixture and EG with a purity above 99%. For the filtration step, two microfiltrations and one nanofiltration have been considered for each scenario. First microfiltration is considered to separate unreacted polyolefins and then via nanofiltration Na2TP is separated from inorganic colorants e.g. carbon black, followed by second microfiltration to obtain TPA monomer with a mean particle diameter is between 50–150 μm.71 In case of a hydrolysis with excess water addition, the total volume has been considered to calculate the impact of the filtration steps on the LCA results. Depending on the type of filtration, different values for the processes’ electricity demand were used.72 Based on greenhouse gas (GHG) emission of each step (Appendix, Table A-7), the total carbon footprint of obtaining pure monomers through PET alkaline hydrolysis is calculated by using the following equation:
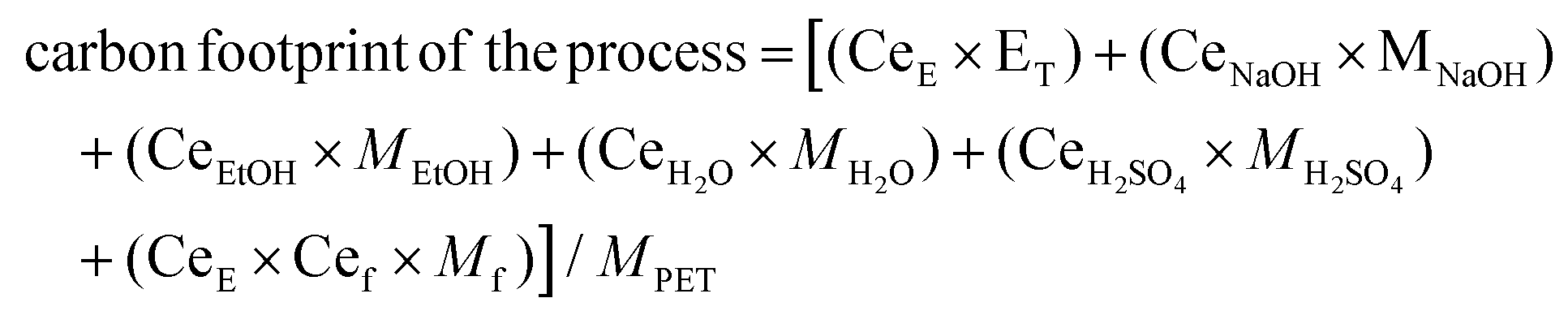 | (9) |
Based on eqn (9), the GHG emission per kg of PET is calculated for both hydrolysis scenarios, with and without excess water addition during purification, at different S/L ratios. The GHG emission of incineration of PET with energy recovery73 together with production of equivalent amount of PET monomers is taken as a reference value and the results are shown in Fig. 7.
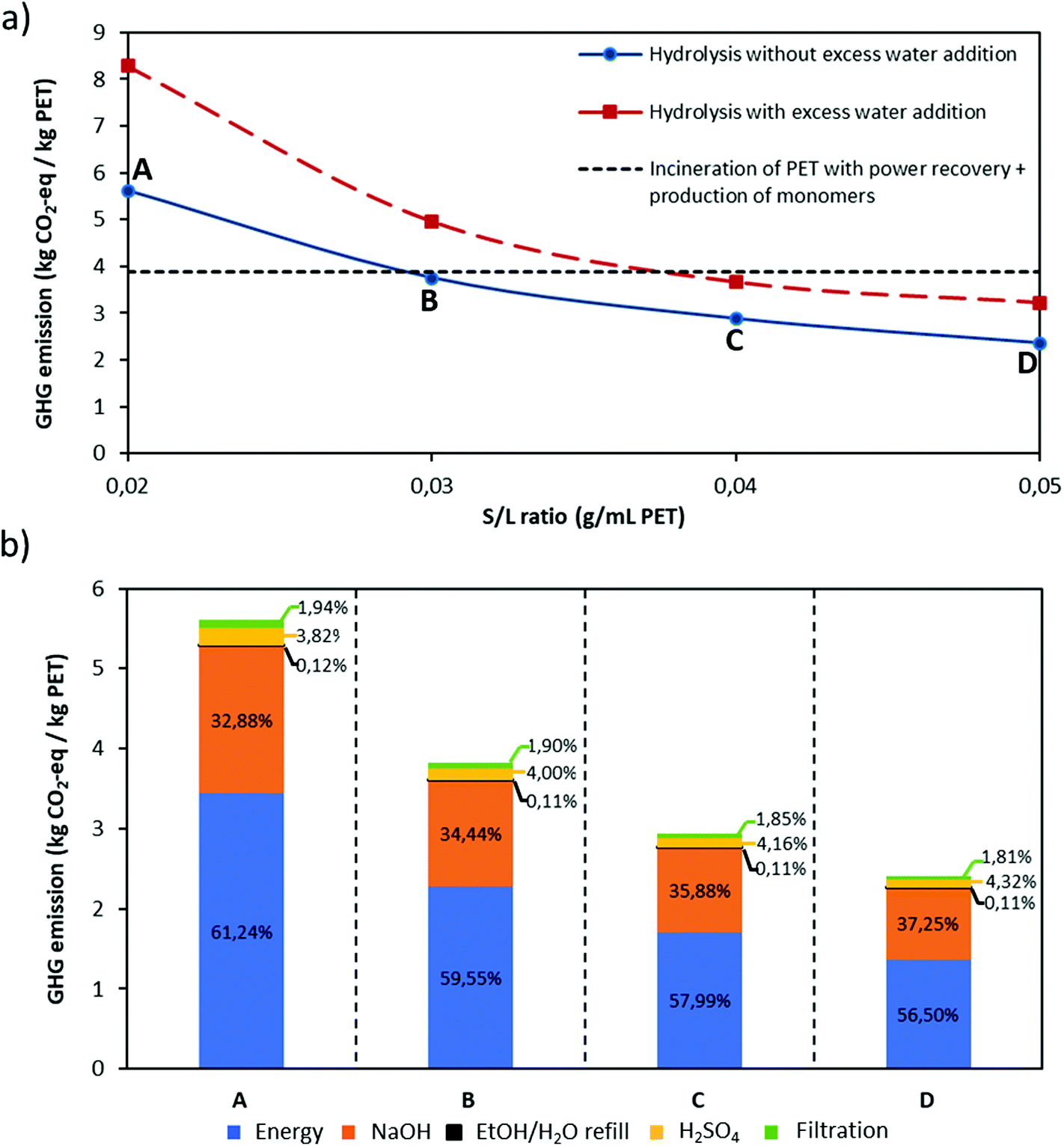 | ||
| Fig. 7 (a) GHG emission of PET alkaline hydrolysis (kg CO2-eq per kg PET) with and without excess water addition at different S/L ratios (g mL−1 PET) and comparison with GHG emission of incineration of PET with energy recovery together with production of equivalent amount of PET monomers; (b) contribution of different steps/inputs to the total GHG emission of the PET hydrolysis process without excess water addition (%) at each S/L ratio. | ||
4. Conclusion
The two-step aqueous alkaline hydrolysis of pure PET monomers, resulting in ethylene glycol (EG) and terephthalic acid (TPA), was optimized. As a starting point we investigated the optimal degradation conditions. Based on obtained kinetic data through GC-FID measurements, PET depolymerization was carried out with the highest yield (∼95%) at 80 °C with particle size lower than 500 μm in a solution containing 60:40 vol% EtOH:H2O and 5 wt% NaOH in 20 minutes. As expected, the increase of temperature and the decrease of particle size leads to higher PET depolymerization. Afterwards, the best-fit kinetic model has been determined. Based on experimental data, we have shown that the diffusion model describes the kinetic behavior of PET alkaline hydrolysis adequately. Within this diffusion model, our experimental kinetic data showed better fit with the D2 and D4 models based on the conversion rates. Next, we explored the validity of the degradation conditions and the kinetic model on real PET waste streams by using post-consumer PET samples including bottles and also multilayer and monolayer PET trays and films at different particle sizes. It is shown that PET conversion rate decreases with an increase in the particle size. Among these samples, the highest PET conversion (∼90%) was obtained with monolayer PET films at the smallest particle size (<500 μm), followed by monolayer PET trays, bottles and multilayer PET samples, respectively. Similar to pure PET pellets, kinetic data obtained with these post-consumer PET samples showed good agreement with the diffusion kinetic model. By using this model, the effect of particle size and the type of sample on the rate constant (k) was investigated. It is observed that the increased particle size, thickness and crystallinity of the samples and also the existence of other polymer layers results in lower degradation yields, thus lower rates. Especially with PET bottles and multilayer trays, degradation yield did not exceed 70%. To our knowledge, it is one of the first times that it is quantitatively proven that hydrolysis of real plastic waste, including multilayers, occurs slower compared to virgin pellets. Since solvent diffusion is limited to one side of plastic waste, especially for multilayer samples, further research could focus on enhancing diffusion to improve yields and rates for real plastics, for example by creating more shear during the hydrolysis. In addition, the effect of additives, ink layers, adhesives, amongst others is expected to play a role as well and could thus be investigated to bridge the gap between yields and rates.
In this study a potential process scheme to scale up this process is proposed, including removal of colours from post-consumer plastic waste. It is shown that via this proposed process it is possible to obtain pure monomers even from black coloured PET samples and due to mild degradation conditions, constituent polymer layers of multilayer PET samples such as polyolefins can be recovered without any degradation. In addition to technical feasibility, environmental impact of the process has been assessed by an LCA at different S/L ratios for the hydrolysis scenarios with and without excess water addition during purification of the monomers. Based on this assessment, increase in the total volume of the solution due to excess water addition causes higher energy consumption during EG recovery, as such increasing the carbon footprint of the process. Therefore, using selective filtration without excess water would make the process more eco-friendly. Likewise, at low S/L ratio, higher amount of energy is consumed per kg of post-consumer PET treated despite of lower amount of inputs e.g. NaOH, H2SO4 are used, causing higher carbon footprints. All in all, we can state that our proposed alkaline hydrolysis process is promising towards chemical recycling of complex PET plastic waste if the parameters e.g. S/L ratio, amount of solvent etc. have been adjusted for the environmental sustainability.
Abbreviations
| CO2-eq | Carbon dioxide equivalent |
| cm | Centimeter |
| DSC | Differential scanning calorimetry |
| DMSO | Dimethyl sulfoxide |
| Na2TP | Disodium terephthalate |
| EtOH | Ethanol |
| EG | Ethylene glycol |
| EVOH | Ethylene vinyl alcohol |
| FTIR | Fourier-transform infrared spectroscopy |
| GC-FID | Gas chromatography-flame ionization detector |
| T g | Glass transition temperature |
| g | Gram |
| GHG | Greenhouse gas |
| IV | Intrinsic viscosity |
| kW h | Kilowatt-hour |
| LCA | Life cycle assessment |
| LDPE | Low density olyethylene |
| μm | Micrometer |
| mm | Millimeter |
| POM | Polarized optical microscopy |
| PE | Polyethylene |
| PET | Polyethylene terephthalate |
| KOH | Potassium hydroxide |
| 1H-NMR | Proton-nuclear magnetic resonance |
| k | Rate constant |
| SSA | Specific surface area |
| NaOH | Sodium hydroxide |
| S/L | Solid/liquid |
| H2SO4 | Sulphuric acid |
| TPA | Terephatalic acid |
| UV-VIS | Ultraviolet–visible spectroscopy |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Joël Hogie for his valuable feedback on Aspen simulations. This work has received support from the European Regional Development Fund (ERDF) via the PSYCHE project (Interreg France-Wallonie-Vlaanderen) with co-financing from the provinces of East-Flanders and West-Flanders.References
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, L. Flores-Giraldo and J. I. Gutiérrez-Ortiz, Kinetics of catalytic glycolysis of PET wastes with sodium carbonate, Chem. Eng. J., 2011, 168, 312–320 CrossRef.
- K. Kaiser, M. Schmid and M. Schlummer, Recycling of polymer-based multilayer packaging: A review, Recycling, 2018, 3, 1 CrossRef.
- Plastic Recyclers Europe, 2018. Available at: https://www.plasticsrecyclers.eu/pet-recycling-industry-installed-capacity-reviewed, (Accessed: 4th December 2019).
- M. Crippa, et al., A circular economy for plastics – Insights from research and innovation to inform policy and funding decisions, 2019 Search PubMed.
- Garbo. ChemPET Project, 2019. Available at: http://www.garbosrl.net/chempet-project/?lang=en. (Accessed: 7th December 2019).
- Petcore Europe. Recycling of PET thermoforms, 2016. Available at: https:// http://www.foodpackagingforum.org/news/recycling-of-pet-thermoforms. (Accessed: 14th February 2020).
- C. Papaspyrides and J. Poulakis, Encyclopedia of Polymeric Materials, CRC Press, Boca Raton, FL, 1996 Search PubMed.
- K. Ragaert, L. Delva and K. Van Geem, Mechanical and chemical recycling of solid plastic waste, Waste Manage., 2017, 69, 24–58 CrossRef CAS PubMed.
- S. Muznik, 9 reasons why we better move away from waste-to-energy, and embrace zero waste instead, 2018. Available at: https://zerowasteeurope.eu/2018/02/9-reasons-why-we-better-move-away-from-waste-to-energy-and-embrace-zero-waste-instead/. (Accessed: 14th February 2020) Search PubMed.
- T. Spychaj, Chemical Recycling of PET: Methods and Products, in Handbook of Thermoplastic Polyesters: Homopolymers, Copolymers, Blends, and Composites, ed. D. S. Stoyko Fakirov, 2002. DOI:10.1002/3527601961.ch27.
- B. Geyer, G. Lorenz and A. Kandelbauer, Recycling of poly(ethylene terephthalate) – A review focusing on chemical methods, eXPRESS Polym. Lett., 2016, 10, 559–586 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, G. Eshaq, A. M. Rabie and A. E. ElMetwally, Greener routes for recycling of polyethylene terephthalate, Egypt. J. Pet., 2016, 25, 53–64 CrossRef.
- Ioniqa. Ioniqa's Circular Solution, 2019. Available at: https://ioniqa.com/applications/, (Accessed: 7th December 2019).
- Zero Waste Europe, El Dorado of Chemical Recycling, State of play and policy challenges, 2019 Search PubMed.
- Perpetual. Technology, 2008. Available at: https://www.perpetual-global.com/manufacturing/#polygenta, (Accessed: 7th December 2019).
- IFPEN, Plastics Recycling, 2015. Available at: https://www.ifpenergiesnouvelles.com/innovation-and-industry/our-expertise/climate-and-environment/plastics-recycling/our-solutions. (Accessed: 7th December 2019) Search PubMed.
- JEPLAN, Polyester Recycle, 2019. Available at: http://www.jeplan.co.jp/en/technology/polyester_recycle/, (Accessed: 7th December 2019) Search PubMed.
- Loop Industries, Revolutionary Technology, 2014. Available at: http://www.loopindustries.com/en/tech, (Accessed: 7th December 2019) Search PubMed.
- Eastman, Eastman offers innovative recycling technology for polyesters, 2019. Available at: https://www.eastman.com/Company/News_Center/2019/Pages/Eastman-offers-innovative-recycling-technology-for-polyesters.aspx. (Accessed: 7th December 2019) Search PubMed.
- Gr3n. Gr3n Project, 2014. Available at: http://gr3n-recycling.com/. (Accessed: 7th December 2019).
- Rampf Eco Solutions, Multi-functional plants for polyol production, 2020. Available at: https:// http://www.rampf-group.com/en/products-solutions/engineering/plant-engineering/, (Accessed: 9th April 2020) Search PubMed.
- S. Baliga and W. T. Wong, Depolymerization of poly(ethylene terephthalate) recycled from post-consumer soft-drink bottles, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2071–2082 CrossRef CAS.
- J. Y. Chen, C. F. Ou, Y. C. Hu and C. C. Lin, Depolymerization of poly(ethylene terephthalate) resin under pressure, J. Appl. Polym. Sci., 1991, 42, 1501–1507 CrossRef CAS.
- P. I. Johnson and D. Teeters, Kinetic study of the depolymerization of poly(ethylene terephthalate) recycled from soft-drink bottles, in American Chemical Society, Polymer Preprints, Division of Polymer Chemistry, 1991 Search PubMed.
- J. R. Campanelli, M. R. Kamal and D. G. Cooper, Kinetics of glycolysis of poly(ethylene terephthalate) melts, J. Appl. Polym. Sci., 1994, 54, 1731–1740 CrossRef CAS.
- S.-C. Lee, Y.-W. Sze and C.-C. Lin, Polyurethanes synthesized from polyester polyols derived from PET waste. II. Thermal properties, J. Appl. Polym. Sci., 1994, 52, 869–873 CrossRef CAS.
- G. P. Karayannidis, A. P. Chatziavgoustis and D. S. Achilias, Poly(ethylene terephthalate) recycling and recovery of pure terephthalic acid by alkaline hydrolysis, Adv. Polym. Technol., 2002, 4, 250–259 CrossRef.
- B. L. Smith and G. E. Wilkins, Process for recovering dimethyl terephthalate, 1996 Search PubMed.
- H. Kurokawa, M. A. Ohshima, K. Sugiyama and H. Miura, Methanolysis of polyethylene terephthalate (PET) in the presence of aluminium tiisopropoxide catalyst to form dimethyl terephthalate and ethylene glycol, Polym. Degrad. Stab., 2003, 79, 529–533 CrossRef CAS.
- D. Paszun and T. Spychaj, Chemical Recycling of Poly(ethylene terephthalate), Ind. Eng. Chem. Res., 1997, 36, 1373–1383 CrossRef CAS.
- J. Scheirs, Polymer Recycling, Wiley, Sussex, 1998, pp. 119–182 Search PubMed.
- S. Mishra and A. S. Goje, Chemical recycling, kinetics, and thermodynamics of alkaline depolymerization of waste poly(ethylene terephthalate) (PET), Polym. React. Eng., 2003, 11, 963–987 CrossRef CAS.
- M. Yamashita and H. Mukai, Alkaline Hydrolysis of Polyethylene Terephthalate at Lower Reaction Temperature, Sci. Eng. Rev. Doshisha Univ., 2011, 52, 143–148 CAS.
- S. Mishra, V. S. Zope and A. S. Goje, Kinetic and thermodynamic studies of depolymerisation of poly(ethylene terephthalate) by saponification reaction, Polym. Int., 2002, 51, 1310–1315 CrossRef CAS.
- H. I. Khalaf and O. A. Hasan, Effect of quaternary ammonium salt as a phase transfer catalyst for the microwave depolymerization of polyethylene terephthalate waste bottles, Chem. Eng. J., 2012, 192, 45–48 CrossRef CAS.
- R. López-Fonseca, M. P. González-Marcos, J. R. González-Velasco and J. I. Gutiérrez-Ortiz, A kinetic study of the depolymerisation of poly(ethylene terephthalate): By phase transfer catalysed alkaline hydrolysis, J. Chem. Technol. Biotechnol., 2009, 84, 92–99 CrossRef.
- M. Imran, D. H. Kim, W. A. Al-Masry, A. Mahmood, A. Hassan, S. Haider and R. Shadid Mahmood, Manganese-, cobalt-, and zinc-based mixed-oxide spinels as novel catalysts for the chemical recycling of poly(ethylene terephthalate) via glycolysis, Polym. Degrad. Stab., 2013, 98, 904–915 CrossRef CAS.
- M. Imran, B. K. Kim, M. Han, B. G. Cho and D. H. Kim, Sub-and supercritical glycolysis of polyethylene terephthalate (PET) into the monomer bis(2-hydroxyethyl) terephthalate (BHET), Polym. Degrad. Stab., 2010, 95, 1686–1693 CrossRef CAS.
- M. Ghaemy and K. Mossaddegh, Depolymerisation of poly(ethylene terephthalate) fibre wastes using ethylene glycol, Polym. Degrad. Stab., 2005, 90, 570–576 CrossRef CAS.
- H. A. Essawy, M. E. Tawfik and N. H. Elsayed, Effect of addition of glycolysis products of poly(ethyleneterephthalate) wastes to urea-formaldehyde resin on its adhesion performance to wood substrates and formaldehyde emission, J. Appl. Polym. Sci., 2012, 123, 2377–2383 CrossRef CAS.
- M. Genta, T. Iwaya, M. Sasaki, M. Goto and T. Hirose, Depolymerization mechanism of poly(ethylene terephthalate) in supercritical methanol, Ind. Eng. Chem. Res., 2005, 44, 3894–3900 CrossRef CAS.
- M. Goto, H. Koyamoto, A. Kodama, T. Hirose and S. Nagaoka, Depolymerization of polyethylene terephthalate in supercritical methanol, J. Phys.: Condens. Matter, 2002, 14, 11427–11430 CrossRef CAS.
- Y. Yang, Y. Lu, H. Xiang, Y. Xu and Y. Li, Study on methanolytic depolymerization of PET with supercritical methanol for chemical recycling, Polym. Degrad. Stab., 2002, 75, 185–191 CrossRef CAS.
- M. Genta, T. Iwaya, M. Sasaki and M. Goto, Supercritical methanol for polyethylene terephthalate depolymerization: Observation using simulator, Waste Manage., 2007, 27, 1167–1177 CrossRef CAS PubMed.
- A. R. Caparanga, B. A. Basilia, K. B. Dagbay and J. W. L. Salvacion, Factors affecting degradation of polyethylene terephthalate (PET) during pre-flotation conditioning, Waste Manage., 2009, 29, 2425–2428 CrossRef CAS PubMed.
- G. P. Karayannidis and D. S. Achilias, Chemical recycling of poly(ethylene terephthalate), Macromol. Mater. Eng., 2007, 292, 128–146 CrossRef CAS.
- V. A. Kosmidis, D. S. Achilias and G. P. Karayannidis, Poly(ethylene terephthalate) recycling and recovery of pure terephthalic acid. Kinetics of a phase transfer catalyzed alkaline hydrolysis, Macromol. Mater. Eng., 2001, 286, 640–647 CrossRef CAS.
- C. Y. Kao, W. H. Cheng and B. Z. Wan, Investigation of alkaline hydrolysis of polyethylene terephthalate by differential scanning calorimetry and thermogravimetric analysis, J. Appl. Polym. Sci., 1998, 70, 1939–1945 CrossRef CAS.
- B. Z. Wan, C. Y. Kao and W. H. Cheng, Kinetics of depolymerization of poly(ethylene terephthalate) in a potassium hydroxide solution, Ind. Eng. Chem. Res., 2001, 40, 509–514 CrossRef CAS.
- W. L. Hergenrother and C. J. Nelson, Viscosity-Molecular Weight Relationship for Fractionated Poly(Ethylene Terephthalate), J. Polym. Sci., Part A-1: Polym. Chem., 1974, 12, 2905–2915 CAS.
- Plastic Recycling Machine, PET bottle recycling, 2013. Available at: https://www.petbottlewashingline.com/pet-bottle-recycling/, (Accessed: 18th February 2020) Search PubMed.
- P. Salminen, Using recycled polyethylene terephthalate (PET) in the production of bottle trays, Arcada, 2013 Search PubMed.
- S. Ügdüler, K. M. Van Geem, M. Roosen, E. I. P. Delbeke and S. De Meester, Challenges and opportunities of solvent-based additive extraction methods for plastic recycling, Waste Manage., 2020, 104, 148–182 CrossRef PubMed.
- Birla Carbon, Carbon black 101, 2020. Available at: https://birlacarbon.com/whats-trending/carbon-black/. (Accessed: 18th February 2020) Search PubMed.
- T. P. Thuy Pham, C. W. Cho and Y. S. Yun, Environmental fate and toxicity of ionic liquids: A review, Water Res., 2010, 44, 352–372 CrossRef PubMed.
- K. Matuszek, E. Pankalla, A. Grymel, P. Latos and A. Chrobok, Studies on the solubility of terephthalic acid in ionic liquids, Molecules, 2020, 25, 1–10 Search PubMed.
- G. W. H. Hohne, From DSC curve to thermodynamic potential function, Thermochim. Acta, 1991, 187, 283–292 CrossRef.
- J. Fabia, A. Gawłowski, T. Graczyk and C. Ślusarczyk, Changes of crystalline structure of poly(ethylene terephthalate) fibers in flame retardant finishing process, Polimery, 2014, 7, 557 CrossRef.
- J. L. Ellingboe and J. H. Runnels, Solubilities of Disodium Terephthalate in Aqueous Solutions of Sodium Carbonate and Sodium Bicarbonate, J. Chem. Eng. Data, 1966, 11, 185–187 CrossRef CAS.
- A. Khawam and D. R. Flanagan, Solid-state kinetic models: Basics and mathematical fundamentals, J. Phys. Chem. B, 2006, 110, 17315–17328 CrossRef CAS PubMed.
- M. E. Brown and A. K. Galwey, The distinguishability of selected kinetic models for isothermal solid-state reactions, Thermochim. Acta, 1979, 29, 129–146 CrossRef CAS.
- J. Šesták and G. Berggren, Study of the kinetics of the mechanism of solid-state reactions at increasing temperatures, Thermochim. Acta, 1971, 3, 1–12 CrossRef.
- F. J. Gotor, C. M. José, J. Malek and N. Koga, Kinetic analysis of solid-state reactions: The universality of master plots for analyzing isothermal and nonisothermal experiments, J. Phys. Chem. A, 2000, 104, 10777–10782 CrossRef CAS.
- J. H. Sharp, G. W. Brindley and B. N. N. Achar, Numerical Data for Some Commonly Used Solid State Reaction Equations, J. Am. Ceram. Soc., 1966, 49, 379–382 CrossRef CAS.
- A. Sangalang, L. Bartolome and D. H. Kim, Generalized kinetic analysis of heterogeneous PET glycolysis: Nucleation-controlled depolymerization, Polym. Degrad. Stab., 2015, 115, 45–53 CrossRef CAS.
- H. Essaddam, Polyethylene terephthalate depolymerization, 2015 Search PubMed.
- D. R. Sturm, K. J. Caputo, S. Liu and R. P. Danner, Diffusivity of solvents in semi-crystalline polyethylene using the Vrentas-Duda free-volume theory, J. Polym. Eng., 2018, 38, 925–931 CAS.
- E. Pirzadeh, A. Zadhoush and M. Haghighat, Hydrolytic and thermal degradation of PET fibers and PET granule: The effects of crystallization, temperature, and humidity, J. Appl. Polym. Sci., 2007, 106, 1544–1549 CrossRef CAS.
- Y. Li, X. Hu and H. Tang, Size effect on electrochemical performance of sodium terephthalate as anode material for sodium-ion batteries, Int. J. Electrochem. Sci., 2018, 13, 7175–7182 CrossRef CAS.
- J. Kroupa, J. Kinčl and J. Cakl, Recovery of H2SO4 and NaOH from Na2SO4 by electrodialysis with heterogeneous bipolar membrane, Desalin. Water Treat., 2015, 56, 1–9 CrossRef.
- Y. Toyosawa, T. Izumisawa and A. Kawahara, Process for producing terephthalic acid, 1995 Search PubMed.
- M. K. Patel, et al., The BREW Project - Medium and Long-term Opportunities and Risks of the Biotechnological Production of Bulk Chemicals from Renewable Resources - The Potential of White Biotechnology, Utrecht University - Department of Science, Technology and Society (STS)/Copernicus Institute, Utrecht, the Netherlands, 2006. DOI:10.1111/j.1442-8903.2004.00172.x.
- T. Chilton, S. Burnley and S. Nesaratnam, A life cycle assessment of the closed-loop recycling and thermal recovery of post-consumer PET, Resour., Conserv. Recycl., 2010, 54, 1241–1249 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc00894j |
| This journal is © The Royal Society of Chemistry 2020 |