
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocatalytic reduction of α,β-unsaturated carboxylic acids to allylic alcohols†
Godwin A.
Aleku
 ,
George W.
Roberts
and
David
Leys
*
,
George W.
Roberts
and
David
Leys
*
Manchester Institute of Biotechnology, School of Chemistry, University of Manchester, Manchester, UK. E-mail: David.Leys@manchester.ac.uk
First published on 1st June 2020
Abstract
We have developed robust in vivo and in vitro biocatalytic systems that enable reduction of α,β-unsaturated carboxylic acids to allylic alcohols and their saturated analogues. These compounds are prevalent scaffolds in many industrial chemicals and pharmaceuticals. A substrate profiling study of a carboxylic acid reductase (CAR) investigating unexplored substrate space, such as benzo-fused (hetero)aromatic carboxylic acids and α,β-unsaturated carboxylic acids, revealed broad substrate tolerance and provided information on the reactivity patterns of these substrates. E. coli cells expressing a heterologous CAR were employed as a multi-step hydrogenation catalyst to convert a variety of α,β-unsaturated carboxylic acids to the corresponding saturated primary alcohols, affording up to >99% conversion. This was supported by the broad substrate scope of E. coli endogenous alcohol dehydrogenase (ADH), as well as the unexpected C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond reducing activity of E. coli cells. In addition, a broad range of benzofused (hetero)aromatic carboxylic acids were converted to the corresponding primary alcohols by the recombinant E. coli cells. An alternative one-pot in vitro two-enzyme system, consisting of CAR and glucose dehydrogenase (GDH), demonstrates promiscuous carbonyl reductase activity of GDH towards a wide range of unsaturated aldehydes. Hence, coupling CAR with a GDH-driven NADP(H) recycling system provides access to a variety of (hetero)aromatic primary alcohols and allylic alcohols from the parent carboxylates, in up to >99% conversion. To demonstrate the applicability of these systems in preparative synthesis, we performed 100 mg scale biotransformations for the preparation of indole-3-aldehyde and 3-(naphthalen-1-yl)propan-1-ol using the whole-cell system, and cinnamyl alcohol using the in vitro system, affording up to 85% isolated yield.
C bond reducing activity of E. coli cells. In addition, a broad range of benzofused (hetero)aromatic carboxylic acids were converted to the corresponding primary alcohols by the recombinant E. coli cells. An alternative one-pot in vitro two-enzyme system, consisting of CAR and glucose dehydrogenase (GDH), demonstrates promiscuous carbonyl reductase activity of GDH towards a wide range of unsaturated aldehydes. Hence, coupling CAR with a GDH-driven NADP(H) recycling system provides access to a variety of (hetero)aromatic primary alcohols and allylic alcohols from the parent carboxylates, in up to >99% conversion. To demonstrate the applicability of these systems in preparative synthesis, we performed 100 mg scale biotransformations for the preparation of indole-3-aldehyde and 3-(naphthalen-1-yl)propan-1-ol using the whole-cell system, and cinnamyl alcohol using the in vitro system, affording up to 85% isolated yield.
Introduction
Devising sustainable, cost-effective and environmentally friendly catalytic methods is pivotal to achieving green manufacturing goals. An important strategy is to harness abundant and replenishable resources such as non-food lignocellulosic biomass to derive raw materials for industrial chemical production. Carboxylic acids readily occur in biomass and recent efforts towards developing biomass utilisation technologies can further enhance their availability.1–3 Given the versatility of carboxylic acids as substrates in organic synthesis,4 there is an attractive opportunity to harness biomass-derived carboxylates as renewable raw materials for sustainable chemical production. Hence, synthetic methods for the derivatization of these carboxylates to provide access to a variety of chemical entities such as aldehydes, alcohols, esters, acyl chlorides and amides under mild reaction conditions are highly desired.The role of biocatalysis is crucial to the development of clean manufacturing technologies for today and tomorrow. A number of biocatalytic synthetic routes are emerging as methods of choice in many industrial processes.5,6 This is largely a result of significant progress achieved in the identification and development of biocatalysts for various biocatalytic functional group interconversions,7–12 including those acting on carboxylic acids and their derivatives.13–18 One such class of enzymes are the carboxylic acid reductases (CARs), which catalyse the selective one-step (two-electron) reduction of carboxylic acids to the corresponding aldehydes at the expense of ATP and NADPH cofactors.13,16 These multi-domain enzymes (comprising the adenylation domain, PCP phosphopantetheine linker and the terminal reductase domain) mediate carboxylate reduction via a multi-step process. At the adenylation domain, an ATP-dependent activation of the carboxylate occurs, generating the corresponding acyl adenylate, followed by the transfer of the acyl group onto the PCP phosphopantetheine linker. The reduction of the acyl-thioester occurs at the terminal reductase domain to yield the final aldehyde product. Recent structural insights reveal domain dynamics underpins strict selective two electron reduction.13,19
The use of CAR enzymes in synthetic processes is impeded by requirement to supply stoichiometric quantities of expensive cofactors NADPH and ATP.20 To circumvent this, application in whole-cell preparations has been considered, as they afford in situ cofactor regeneration system in place. However, in vitro CAR biotransformation is preferred where conversion exclusively to the aldehyde is desired. In this regard, co-factor recycling systems have been coupled to the CAR reaction.21–25
There are limited examples demonstrating biocatalytic carboxylate reduction to the corresponding alcohols.26–28 Akhtar et al. employed E. coli cells expressing Mycobacterium marinum CAR and a native aldehyde reductase (AHR) to convert fatty acids to the corresponding alcohols, Fig. 1a.27 Similarly, Kramer et al. have recently developed a whole-cell CAR-based pathway for the conversion of a panel of short-chain dicarboxylic acids and hydroxy acids to the corresponding diols, employing yahK-encoded aldehyde reductase for carbonyl reduction,28Fig. 1b. In this work, we aimed to develop CAR-based in vivo and in vitro systems for the conversion of as yet unexplored substrate space: benzo-fused (hetero)aromatic carboxylic acids and acrylic acid derivatives to provide access to (hetero)aromatic alcohols and allylic alcohols. These compounds are prevalent scaffolds in industrial chemicals and pharmaceuticals, Fig. 1c–f. We envisage that development of such methods can provide an alternative green synthetic route to the traditional chemical methods employing metal hydrides (e.g. LiAlH4 or Zn(BH4)2), borane-reducing agents (e.g. BH3·SMe2),29,30 and transition metal homogeneous and heterogeneous catalysts.31,32 More so, as metal hydrides are required in stoichiometric amounts, prone to inactivation by air and moisture, generate inorganic waste and are poorly selective. In addition, homogeneous and heterogeneous metal catalysts are expensive and require harsh reaction conditions including elevated temperatures and pressures.
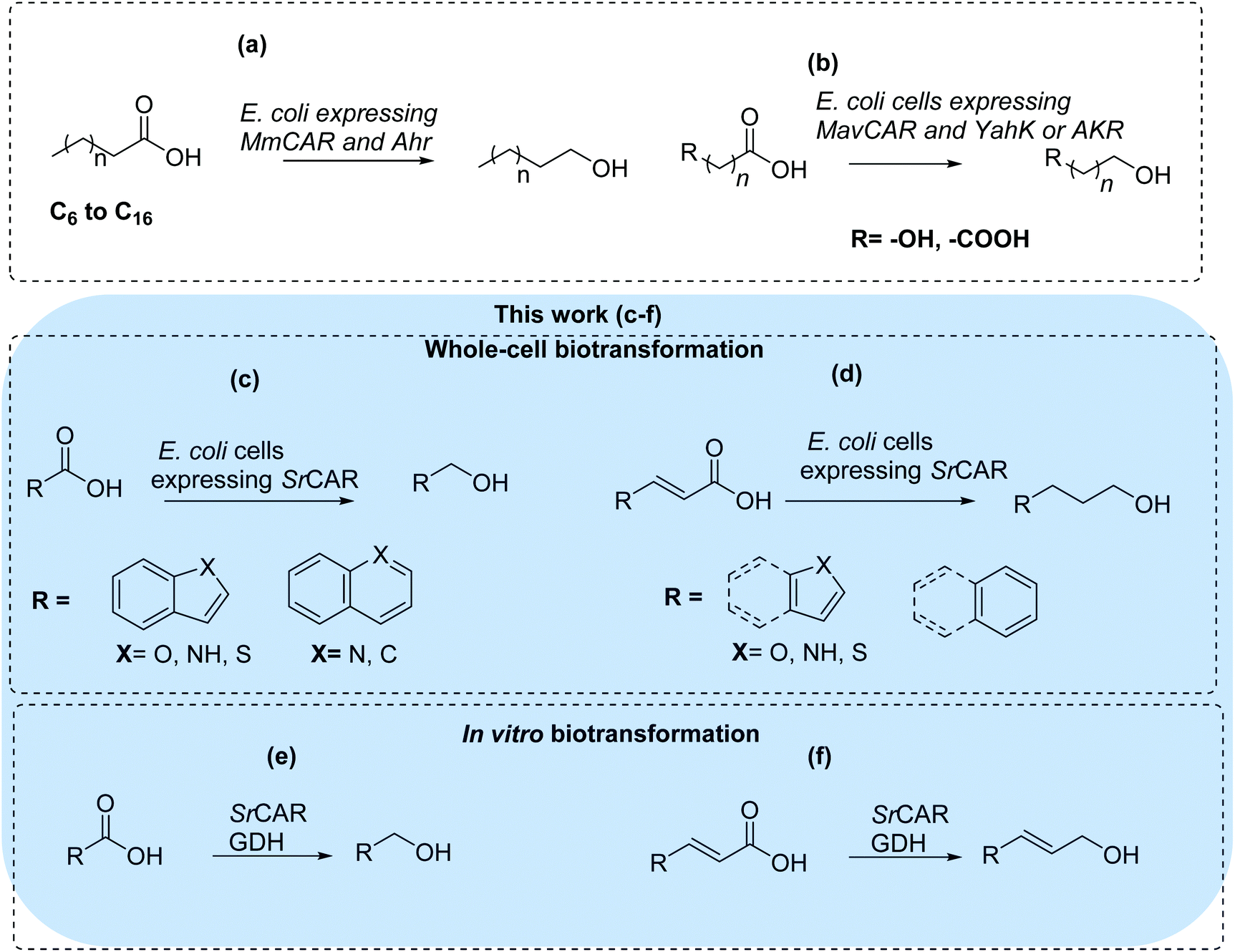 | ||
| Fig. 1 Biocatalytic conversion of carboxylic acids to alcohols applying carboxylic acid reductases (CARs) for carboxylate reduction. (a) Route for conversion of fatty acids to fatty alcohol employing recombinant aldehyde reductase (AHR) for the carbonyl reduction step.27 (b) Route for conversion of hydroxyl or dicarboxylic acids to diols employing lactaldehyde reductase (YahK) or aldo-keto reductases (AKR) for the carbonyl reduction step.28 | ||
Results and discussion
Profiling unexplored substrate space: CAR activity on benzofused (hetero)aromatic and α,β-unsaturated acids
To assess the activity of CARs towards benzo-fused (hetero)aromatic carboxylic acids and acrylic acid derivatives, we selected two CAR enzymes: Tsukamurella paurometabola carboxylic acid reductase (TpCAR) which was identified as a substrate promiscuous CAR,33 and the structurally characterised Segniliparus rugosus CAR (SrCAR).34 The CAR enzymes were each co-expressed with Bacillus subtilis phosphopantetheinyl transferase (sfp) in E. coli to ensure posttranslational modification and activation. We then set out to investigate CAR activity towards a variety of bulky benzo-fused (hetero)aromatic carboxylic acids and acrylic acids, initially using whole-cell systems. Preliminary profiling for TpCAR and SrCAR revealed similar substrate spectrum and comparable conversion values for the two enzymes, ESI Fig. S1.† Therefore, we selected SrCAR for further study since significant structural and in silico information are available for this enzyme34,35 which may guide future engineering efforts.To assess the substrate scope and the relative reactivity of the substrates, we determined specific activity of isolated SrCAR from initial rate of carboxylate reduction. Two distinct compound libraries were constructed containing structurally diverse benzo-fused (hetero)aromatic carboxylic acids, and α,β-unsaturated carboxylic acids bearing a wide range of substituents at the β- and α-carbons.
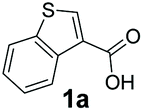
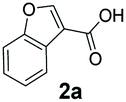
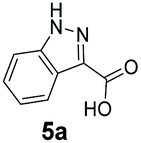
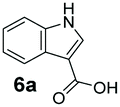
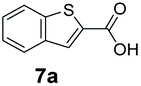
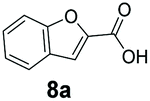
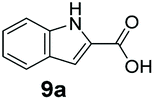
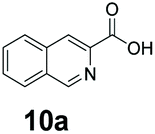
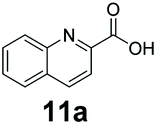
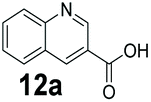
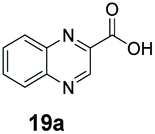
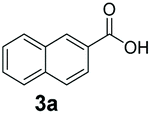
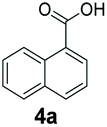
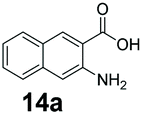
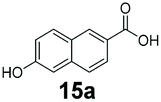
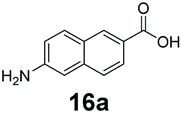
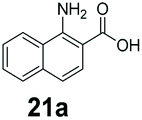
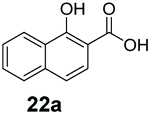
An assessment of the reactivity pattern for benzo-fused (hetero)aromatic acids reveals that benzo-fused S-, O-, and N-heteroaromatic carboxylates (1a, 2a, 5a & 6a) and naphthoic acids 3a & 4a (group I) displayed superior specific activity (0.9–1.4 U mg−1) compared to the corresponding 2-carboxylates, 7a, 8a & 9a and isoquinoline derivate 10a, (group II, specific activity = 0.4–0.8 U mg−1). Further down the specific activity scale are quinolone-2-carboxylic acid 11a and 2-naphthoic acid derivatives bearing a hydroxyl or an amino group at 3- or 6-position of the aromatic system (13a–16a), displaying specific activity between 0.20 and 0.40 U mg−1 with a significantly slower rate observed for a 1-hydroxy substituent (22a). The presence of an additional heteroatom significantly decreases activity; for example, the least reactive group IV (specific activity <0.2 U mg−1) features benzo-fused heteroaromatic carboxylates containing two heteroatoms (17a–19a). Similarly, 1-naphthoic acid derivatives bearing a hydroxyl or an amino group at 2- or 6-position of the aromatic system (20a–22a) exhibited weak reactivity. Marked decrease in reactivity was also observed with diheteroatom-containing carboxylates (vs. the mono-heteroatom substrates).
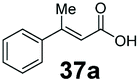
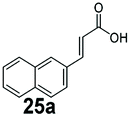
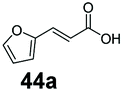
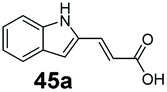
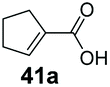
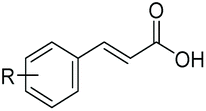
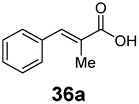
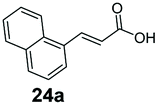
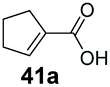
SrCAR also displayed broad biocatalytic scope for α,β-unsaturated carboxylic acids, exhibiting high specific activity for acrylic acids bearing a naphthyl group (24a and 25a), as well as cinnamic acid 26a and derivatives bearing weakly e−-donating group at the α or β-carbon to the carboxylate (e.g. α-Me 36a, β-Me 37a), Fig. 2b, (group I, specific activity 1.50–2.30 U mg−1). In comparison, cinnamic acid derivatives bearing small weak e−-withdrawing group as α-F 38a (group III), or bulky substituents at the α-carbon such as α-Ph, α-NHCOMe 39a, 40a (group IV) displayed an order of a magnitude lower reactivity. This trend highlights the importance of both steric and electronic effects of substituents attached to the α-carbon. The effect of substituents on the phenyl ring of cinnamic acid was also investigated, revealing that the steric effect of para-substituted bulky substituents was less profound. For example, SrCAR displayed a ∼2-fold lower activity towards cinnamic acids bearing bulkier p-substituents such as Me, Br, Ph, carboxy (28a–31a, group II, 0.6–1.0 U mg−1) when compared to a small para substituent (p-fluorocinnamic acid 27a, specific activity 1.7 U mg−1). Acrylic acids bearing heteroaromatic systems at the β-carbon (43a–45a) were reactive, albeit with lower rates (group III), whereas α,β-unsaturated monocyclic carboxylic acids 41a, 42a (group II) showed good reactivity (specific activity, 0.8–1.1 U mg−1).
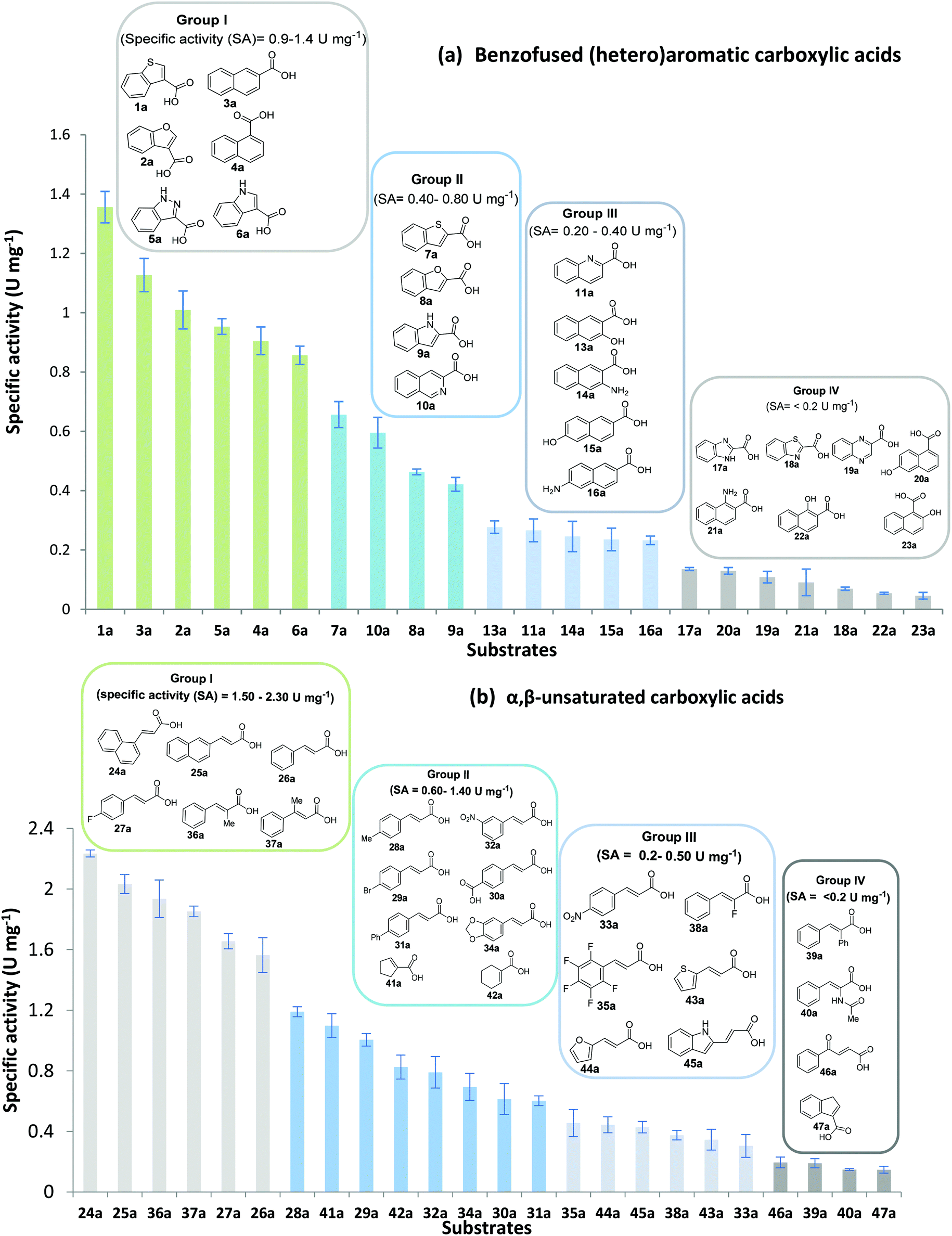 | ||
| Fig. 2 Substrate profiling studies for Segniliparus rugosus CAR (SrCAR) against unexplored substrate groups: (a) benzo-fused (hetero)aromatic carboxylic acids; (b) α,β-unsaturated carboxylic acids. SA = specific activity determined from initial rates. Assays were performed in triplicate and the error bars represent standard deviation from the mean specific activity. A unit of activity = the amount of pure enzyme required to consume 1 μmol NADPH per min. | ||
While previous substrate profiling studies of CARs have examined scope for monocyclic aromatic carboxylic acids, fatty acids and linear aliphatic carboxylates,26–28,33 this work represents the first systematic investigation into the scope of CAR enzymes towards bulky benzo-fused (hetero)aromatic carboxylates as well α,β-unsaturated carboxylic acids. In general, we find SrCAR an efficient catalyst for the reduction of a broad range of benzo-fused (hetero)aromatic carboxylic acids. Similarly, a large collection of previously un-investigated acrylic acids bearing structurally diverse groups at α- or β-carbon to the carboxylate were efficiently reduced by SrCAR. In view of the relatively small acid substrate binding pocket of the SrCAR A-domain,34 the broad substrate tolerance towards these relatively bulky benzo-fused aromatic compounds suggests that significant intradomain and interdomain motions occur during catalysis to accommodate and orientate substrates for catalysis. In addition, the efficiency of the CAR enzymes towards these benzofused (hetero)aromatic carboxylates suggests a role for the benzo component in facilitating productive interaction between the substrate and substrate binding site residues of SrCAR A-domain (and by extension other CARs) which are mostly hydrophobic.22,34 Taken together, the results from Fig. 2 reveal the following substrate structure–activity determinants: (i) for benzo-fused heteroaromatic carboxylic acids, the reactivity differs with the position of the carboxylate on the benzofused heteroaromatic ring system; substrates bearing 2-carboxylate displayed lower reactivity when compared with the 3-carboxylate regio-isomer. (ii) The type of heteroatom (O, N, S) in the benzo-fused 5-memebered heteroaromatic system influenced reactivity in line with the degree of aromatic stability of the systems. For example, SrCAR displayed highest activity towards benzothiophene derivatives whereas indole derivatives where least reactive. (iii) Steric hindrance resulting from bulky substituents was the most important factor contributing to weak reactivity, especially when such substituents are adjacent to the carboxylate group. This is consistent with previously established trend with benzoic acid derivatives bearing sterically demanding ortho-substituents.33,36
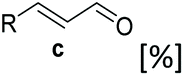
Whole cell biotransformation for the conversion of unexplored carboxylic acid substrates to alcohols
To assess the biocatalytic scope of recombinant E. coli whole cells containing SrCAR for the conversion of sterically demanding benzo-fused (hetero)aromatic carboxylic acids and acrylic acid derivatives to the corresponding alcohols, we set out to investigate CAR in vivo activity towards a variety of these acids. Simultaneously, carbonyl reductase activity of E. coli endogenous alcohol dehydrogenase (EcADH) on corresponding aldehydes to generate the corresponding primary alcohols was monitored. First, we performed biotransformation for the reduction of benzothiophene-2-carboxylic acid 1a using E. coli whole-cells containing SrCAR, with glucose added to enable in situ cofactor recycling. Conversion of >99% to the corresponding alcohol was obtained, indicating that the carboxylate and the corresponding aldehyde are good substrates for the SrCAR and EcADH respectively. Encouraged by this initial result, we investigated the two-step reduction of a wide range of carboxylic acids to the corresponding alcohols using E. coli whole-cells expressing SrCAR.Scope for conversion of benzofused (hetero)aromatic carboxylic acids to corresponding alcohols
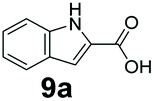
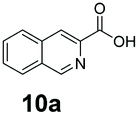
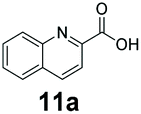
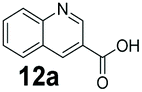
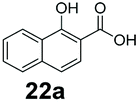
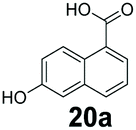
We found a structurally diverse set of (hetero)aromatic carboxylic acids were converted to the corresponding alcohols, affording moderate to excellent conversion in most cases, with no requirement for recombinant overexpression of a carbonyl reductase. Benzo-fused furan and thiophene 3-carboxylates 1a, 2a or 2-carboxylates 7a, 8a were converted to the corresponding alcohols with excellent conversion values (96–>99%) and no accumulated aldehyde intermediate; Table 1, entries 1–7. However, lower conversion values were achieved with indole-2-carboxylate 9a, yielding the corresponding alcohol at 48% conversion. Interestingly, the regio-isomer indole-3-carboxylate 6a was an excellent CAR substrate, but yielded indole-3-carboxyaldehyde (96% conversion) as the latter was not converted by EcADH. Indazole-3-carboxylic acid 5a yielded the corresponding alcohol in up to 96% conversion. The system also displayed tolerance towards six-membered benzo-fused N-heteroaromatic carboxylates (such as quinoxaline, quinolone and isoquinoline derivatives 10a–12a), yielding the corresponding alcohols in a range between 15 to >99%. In particular, isoquinoline-3-carboxylate 10a was an excellent substrate. We then examined a range of 1-naphthoic and 2-napthoic acid derivatives Table 1, entries 12–19. Unsubstituted naphthoic acids 3a & 4a were good substrates, generating the corresponding alcohol in high conversion of up >99%. Substitution at different positions of the aromatic ring system with strong e−-donating such as –OH, –NH2 were tolerated by both CAR and EcADH. However, 1-hydroxy-2-naphthoic acid 22a yielded the aldehyde (83%), indicating that the corresponding aldehyde was unreactive with EcADH.|
|
|||
|---|---|---|---|
| Entry | Substrates | Conversion [%] | |
|
|
|
||
| Reaction contained 5 mM substrate, 20 mM D-glucose, 10 mM MgCl2, 2% v/v DMSO and fresh resting E. coli cells containing overexpressed SrCAR at OD600 = 30, all in NaPi (50 mM, pH 7.5), incubated at 30 °C, 250 rpm for 18 h. SrCAR = Segniliparus rugosus carboxylic acid reductase. Conversion values were determined from HPLC/GC-MS analyses. | |||
| 1 |
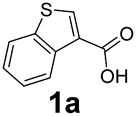
|
0 | >99 |
| 2 |
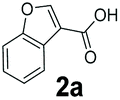
|
<1 | >98 |
| 3 |
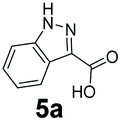
|
1 | 96 |
| 4 |
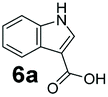
|
96 | 0 |
| 5 |
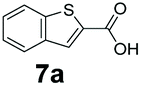
|
0 | >99 |
| 6 |
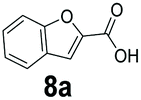
|
0 | 98 |
| 7 |
|
0 | 48 |
| 8 |
|
0 | >99 |
| 9 |
|
0 | 93 |
| 10 |
|
0 | 15 |
| 11 |
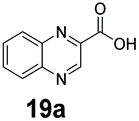
|
37 | 63 |
| 12 |
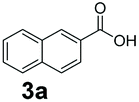
|
0 | >99 |
| 13 |
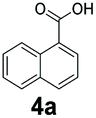
|
0 | 98 |
| 14 |
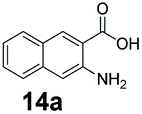
|
0 | 87 |
| 15 |
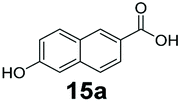
|
0 | 73 |
| 16 |
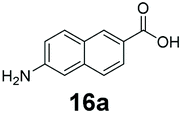
|
0 | 98 |
| 17 |
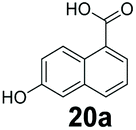
|
0 | 78 |
| 18 |
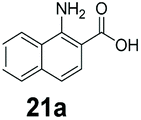
|
0 | 82 |
| 19 |
|
83 | 0 |
Despite the weak activity of SrCAR towards structurally challenging (hetero)aromatic carboxylic acids (e.g. substrates bearing steric bulk at adjacent carbon to the carboxylate) observed from our initial rate study (Fig. 2), moderate conversion values were achieved upon longer biotransformation incubation (18 h, Table 1) indicating potential for application of CAR enzymes for valorization of these substrates. In addition, there is also an opportunity to further improve the efficiency of CAR enzymes, for example via (muti)site saturation of crucial active site residues that have recently been identified from structural,22,34 computational35,37 and directed evolution36 studies.
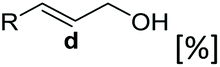
Scope for conversion of acrylic acids to corresponding alcohols using recombinant E. coli cells
Allylic alcohols or their saturated analogues are important building blocks for organic synthesis; hence we sought to explore the CAR-route for the conversion of acrylic acid derivatives which are abundant in biomass to the corresponding alcohols. Cinnamic acid 26a and derivatives bearing p-substituents on the aromatic ring such as weakly e−-withdrawing groups p-halogens (27a, 29a) and weakly e−-donating groups such as p-Me (28a), as well as strong e−-donating groups p-OH 48a were tolerated by SrCAR and endogenous EcADH, generating the corresponding alcohols from the parent carboxylates in good to excellent conversion values (60–92%, Table 2, entries 1–6). Interestingly, while we had anticipated the formation of the corresponding allylic alcohols (cinnamyl alcohol derivatives) as the final reduction product, GCMS analysis of the biotransformation reactions revealed the formation of corresponding saturated alcohols (hydrocinnamyl alcohols) as the major product in most cases, indicating a novel CC reducing activity of E. coli whole-cell biocatalysts, Table 2. Substrate 30a which carries two carboxyl groups was reduced to the corresponding diol with high conversion, again with the acrylic carboxylate moiety reduced to the corresponding hydrocinnamyl alcohol (72%).
|
|
|||
|---|---|---|---|
| Entry | Substrates | Conversions [%] | |
|
|
|
|
|
| Reaction contained 5 mM substrate, 20 mM D-glucose, 10 mM MgCl2, 2% v/v DMSO and fresh resting E. coli cells containing overexpressed SrCAR at OD600 = 30, all in NaPi (50 mM, pH 7.5), incubated at 30 °C, 250 rpm for 18 h. Conversion values were determined from HPLC/GC-MS analyses.a Enantiomeric excess (ee) and absolute configuration for chiral products not determined. NB: 3–15% of saturated acid analogues of the α,β-unsaturated acids were detected with substrates 27a, 28a, 29a, 35a, 36a, 42a, 48a and 49a. 4–8% of decarboxylated product detected with substrate 48a & 49a. <1–10% of α,β-unsaturated aldehyde was detected in most reactions and 28% of propanal derivative was detected with substrate 38a. | |||
| 1 |
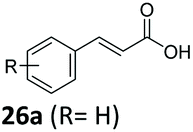
|
6 | 92 |
| 2 | 27a (R = p-F) | <1 | 60 |
| 3 | 28a (R = p-Me) | <1 | 71 |
| 4 | 29a (R = p-Br) | <1 | 82 |
| 5 | 31a (R = p-Ph) | 4 | 92 |
| 6 | 48a (R = p-OH) | <1 | 90 |
| 7 |
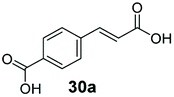
|
|
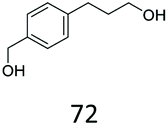
|
| 8 |
|
<1 | 87 |
| 9 |
|
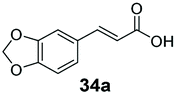
6 | 75 |
| 10 |
|
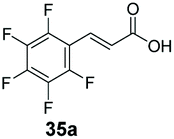
<1 | 68 |
| 11 |
|
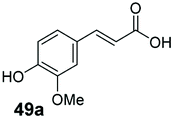
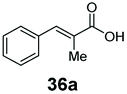
10 | 74a |
| 12 |
|
70 | 0 |
| 13 |
|
0 | 84a |
| 14 |
|
58 | 43a |
| 15 |
|
<1 | >99 |
| 16 |
|
<1 | 87 |
| 17 |
|
11 | 80 |
| 18 |
|
0 | 90 |
| 19 |
|
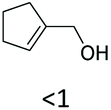
|

|
| 20 |
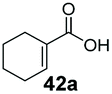
|
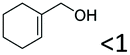
|
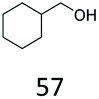
|
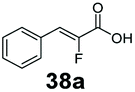
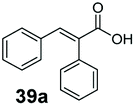
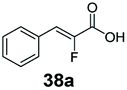
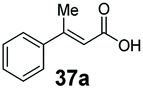
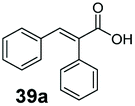
Bulky substituents on the aromatic moiety such as p-Ph group 31a were tolerated, affording high conversion, yielding predominantly the corresponding hydrocinnamyl alcohol (92%). Similarly, di-, and penta-functionalised cinnamic acid derivatives 34a, 35a, 49a were converted to the corresponding hydrocinnamyl alcohols as final products (Table 2, entries 8–10). Furthermore, cinnamic acid derivatives bearing small substituents at the α-carbon to the carboxylate (α-Me 36a) as well as bulky substituents (α-Ph 39a) were accepted, yielding the corresponding hydrocinnamyl alcohols as major products, while β-Me substitution (37a) generated a mixture of the corresponding allylic alcohol and the saturated analogue. Interestingly, reduction of α-F cinnamic acid 38a yielded the corresponding allylic alcohol (70%) as the sole alcohol product, while the saturated aldehyde, α-F hydrocinnamaldehyde 38e was also detected (28%).
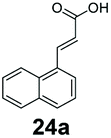
Bicyclic aromatic substituents at β-carbon such as naphthyl (24a, 25a) were excellent substrates, yielding the corresponding 1-propanol in up to >99% conversion. Similarly, acrylic acids bearing heteroaromatic systems at β-carbon (44a, 45a), as well as α,β-unsaturated cyclic carboxylic acids 41a, 42a were accepted, affording the corresponding 1-propanols and saturated cyclic methanols respectively (57–90% conversion).
The presence of intermediates suggest C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C reducing activity of E. coli whole-cells on α,β-unsaturated acids and aldehydes
C reducing activity of E. coli whole-cells on α,β-unsaturated acids and aldehydes
The CC reducing activity of E. coli whole-cells enabled an unexpected 3-step hydrogenation route for the conversion α,β-unsaturated carboxylic acids to the corresponding saturated alcohols. An in vitro system applying SrCAR as purified enzyme preparation and supplying stoichiometric amounts of NADPH and ATP for the reduction of α,β-unsaturated acid 26a or the corresponding aldehyde lacked CC reducing activity. This suggests the presence of (likely oxygen-stable) E. coli CC reducing enzymes, perhaps of the ene/enoate reductases (EREDs) or short chain dehydrogenases/reductases (SDRs) enzyme families.38,39 GCMS analysis of intermediates detected from biotransformation reactions applying SrCAR as whole-cell catalyst highlights two plausible routes to the saturated alcohol derivatives. In the first route, the α,β-unsaturated aldehyde intermediate generated from the CAR step can undergo CC hydrogenation to generate the corresponding propanal derivative prior the carbonyl reduction step. Evidence for this route is supported by detection of α-F hydrocinnamaldehyde 38e from analysis of biotransformation for reduction of α-F cinnamic acid 38a, ESI, Fig. S13.† In this case, the propanal derivative was unreactive with EcADH and therefore accumulates up to 28%. A complementary route to the saturated alcohol via CC reducing activity on α,β-unsaturated carboxylic acids can occur prior to carboxylate and carbonyl reduction steps (see reaction scheme, Table 2). Detection of the corresponding saturated carboxylic acid analogues from α,β-unsaturated carboxylates for substrates 27a, 28a, 29a, 36a, 42a and 48a supports the latter, ESI, Fig. S14–S16.†
Our whole-cell CAR system demonstrates for the first time (to the best of our knowledge) the use of E. coli expressing CAR as an efficient biocatalytic multi-step hydrogenation system enabling conversion of a wide range of acrylic acids to the corresponding saturated alcohols. This self-sufficient system provides the 6e− required for the three step reduction process (i.e. carboxylate reduction, α,β CC bond reduction and CO reduction) at the expense of exogenously added glucose. A similar trend has been previously observed with the fungus Mucor sp. A-73 cells, in this case the reduction of C6 α,β-unsaturated carboxylic acids (e.g. hexenoic acid, sorbic acid) to the corresponding α,β saturated alcohols was achieved40 suggesting that recombinant fungal cells containing CAR can further be developed for this multi-step hydrogenation process. In addition, a wide range benzo-fused bulky (hetero)aromatic alcohols can be accessed via this simple recombinant E. coli whole-cell system harboring CAR.
In vitro biotransformation to access benzofused hetero(aromatic) alcohols and allylic alcohols
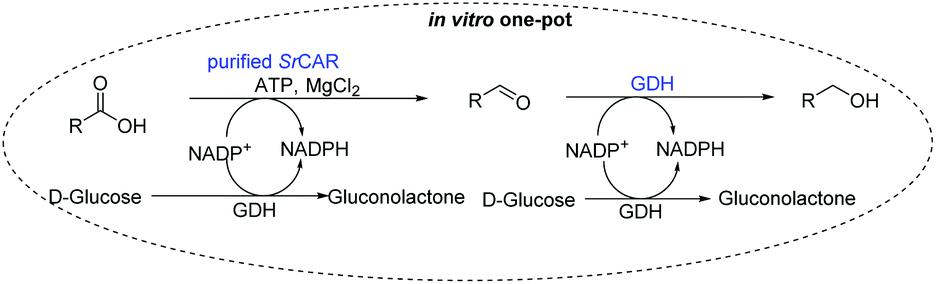
We sought an alternative route to access allylic alcohols from the α,β-unsaturated carboxylates as these are highly versatile building blocks for organic synthesis as they allow further functionalisation of the allylic moiety.41 Hence, we next aimed to develop a simple in vitro approach for the conversion of a variety of carboxylic acids to the corresponding alcohols; with selective access to allylic alcohols of particular interest. To circumvent the need to use a stoichiometric amount of NADPH in the two NADPH-dependent reduction steps, we employed glucose dehydrogenase (GDH)-based NADPH-recycling system. Next, we attempted to identify a carbonyl reductase for the second step. Inspired by recent report on the promiscuous carbonyl reductase activity of GDH on naphthoquinones,42,43 we envisioned a system where GDH is applied as a bifunctional catalyst; to recycle NADPH and as carbonyl reductase. In this system, GDH catalyses the oxidation of glucose to recycle NADPH while concurrently reducing the aldehyde intermediates generated from the CAR step to the corresponding alcohols.Bifunctional glucose dehydrogenase as promiscuous carbonyl reductase
We first examined the performance of isolated Bacillus subtilis GDH (BsGDH, GenBank reference: AFQ56330.1) as a bifunctional catalyst for glucose oxidation and promiscuous carbonyl reduction of benzo-fused (hetero)aromatics aldehydes to the corresponding alcohols. We attempted a one-pot biotransformation reaction containing a carboxylic acid substrate 8a, a stoichiometric amount of ATP, a catalytic amount of NADP+, D-glucose (4 equivalents), purified SrCAR and BsGDH. This system yielded the corresponding alcohol in up to 77% conversion. Having established the substrate scope for the CAR-catalysed carboxylate reduction, we next aimed to investigate the substrate scope of GDH-carbonyl reduction step. Surprisingly, GDH displayed a broad substrate scope and was capable of reducing a wide range of benzo-fused (hetero)aromatic aldehydes to generate the corresponding primary alcohols in moderate to excellent conversion values (37–98%), Table 3. N-heteroaromatic carboxylates 6a, 9a and sterically demanding naphthoic acid derivatives, 14a, 21a & 22a only underwent a one-step reduction to the corresponding aldehydes, indicating that the aldehydes formed were unreactive with BsGDH.|
|
|||
|---|---|---|---|
| Entry | Substrates | Conversion [%] | |
|
|
|
||
| In vitro reaction contained 5 mM carboxylic acid substrate, 20 mM D-glucose, 10 mM MgCl2, 2% v/v DMSO, purified SrCAR (0.5 mg mL−1), purified BsGDH (0.6 mg mL−1). Reaction performed in NaPi (50 mM, pH 7.5), incubated at 30 °C, 250 rpm for 18 h.a 1 mg mL−1 purified GDH was used. SrCAR = Segniliparus rugosus carboxylic acid reductase; BsGDH = Bacillus subtilis glucose dehydrogenase. Conversion values were determined from HPLC/GC-MS analyses. | |||
| 1 |
|
4 | 91a |
| 2 |
|
2 | 96a |
| 3 |
|
10 | 68 |
| 4 |
|
79 | 0 |
| 5 |
|
0 | >99 |
| 6 |
|
0 | 77 |
| 7 |
|
43 | 0 |
| 8 |
|
7 | >92 |
| 9 |
|
6 | >93 |
| 10 |
|
14 | >85 |
| 11 |
|
13 | 85 |
| 12 |
|
0 | >99 |
| 13 |
|
0 | 92 |
| 14 |
|
>99 | 0 |
| 15 |
|
0 | 92a |
| 16 |
|
0 | 96 |
| 17 |
|
18 | 37 |
| 18 |
|
55 | 0 |
| 19 |
|
40 | 0 |
|
|
|
|
| 20 | 26a (R = H) | 11 | 87a |
| 21 | 27a (R = p-F) | 66 | 14 |
| 22 | 29a (R = p-Br) | 25 | 22 |
| 23 | 28a (R = p-Me) | 56 | 41 |
| 24 | 31a (R = p-Ph) | 36 | 30 |
| 25 | 33a (R = p-NO2) | 48 | 16 |
| 26 | 48a (R = p-OH) | >99 | 0 |
| 27 |
|
>99 | 0 |
| 28 |
|
56 | 44 |
| 29 |
|
<1 | 99 |
| 30 |
|
7 | 94a |
| 31 |
|
83 | 7 |
| 32 |
|
0 | >99a |
| 33 |
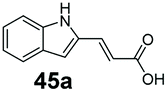
|
97 | 0 |
| 34 |
|
20 | 70 |
In vitro one-pot CAR–GDH cascade enables selective access to allylic alcohols
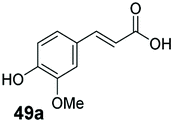
Finally, the CAR–GDH in vitro system was tested against α,β-unsaturated carboxylic acids to allow selective access to the corresponding allylic alcohols. Indeed, the one-pot CAR–GDH system was capable of converting α,β-unsaturated carboxylic acids to the corresponding allylic alcohols with 14 to >99% conversion. Saturated alcohol products were not detected with this system. Cinnamic acid derivatives bearing weak or strong e−-withdrawing p-substituents on the aromatic ring such as p-halogens and p-NO2 (27a, 29a, 33a) as well as weakly e−-donating groups such as p-Me (28a) and bulky p-Ph 30a yielded the corresponding allylic alcohols albeit in low conversion values (14–41%), Table 3, entries 21–25. BsGDH was inert towards p-hydroxycinnamaldehydes as p-hydroxycinnamic acid derivatives such as p-coumaric acid 48a and ferulic acid 49a only underwent a one-step reduction to the corresponding aldehydes with >99% conversion (Table 3, entries 26 & 27). Similarly, α,β-unsaturated carboxylates bearing heteroaromatic substituent at the β-carbon such as in 45a underwent a one-step reduction to the corresponding aldehydes (97% conversion, Table 3, entry 33) indicating that BsGDH lacked carbonyl reducing activity towards the corresponding aldehyde. However, cinnamic acid derivatives bearing small groups at the α- or β-Me carbon to the carboxylate moiety (α-Me 36a, β-Me 37a, and α-F 38a) were accepted by the CAR–GDH system, affording the corresponding allylic alcohols in up to 99% conversion, Table 3. Similarly, α,β-unsaturated carboxylates bearing bulky bicyclic aromatic substituents at the β-carbon such as naphthyl (24a) was converted to the allylic alcohols (>99% conversion) and the cyclic carboxylate afforded 70% of the corresponding α,β-unsaturated alcohol whereas cinnamic acid derivative bearing a bulky substituent at the α-carbon such as α-Ph (39a) only afforded 7% of the allylic alcohol with the aldehyde intermediate accumulating. Analysis of biotransformation reactions for selected substrates (8a, 26a, 38a) incubated for 1 h showed that the carbonyl reduction rate (vs. CAR-catalysed carboxylate reduction) is significantly slower, and differs depending on the intermediate aldehydes (ESI Fig. S2†). Hence, this leads to the observed differences in the amount of aldehyde intermediates accumulating for biotransformation of different carboxylic acids (Table 3). Biotransformation reactions performed for substrates (e.g.26a, 27a & 28a) under identical conditions, but supplying 2 equivalents of NADPH while excluding GDH and glucose, led to the accumulation of the corresponding aldehydes.The results from Table 3 clearly demonstrate that our simple novel in vitro system provides a green, potentially cheaper biocatalytic route for the synthesis of a wide variety of synthetically useful allylic alcohols.41 In particular, this system allows indirect screening of promiscuous carbonyl reductase activity of GDH against a wide range of hard-to-access aldehydes, uncovering the versatility of GDH as carbonyl reductase for structurally diverse carbonyl compounds. The bifuntionality of GDH (for NADPH regeneration and carbonyl reduction) ensured a significant reduction of reaction components and simplified optimisation process. We suggest that our indirect approach to monitor promiscuous enzymatic activity can be extended to explore catalytic promiscuity of SDRs.44,45 To avoid ambiguity in the indirect profiling of promiscuous carbonyl activity of GDH, we relied on the use of a stoichiometric amount of ATP to demonstrate the potential for the in vitro CAR–GDH system as a proof of concept. However, for future economically viable application, we suggest that a simple ATP co-factor regeneration system can be coupled to the CAR–GDH in vitro system. For example, a family-2 polyphosphate kinase (PPK2, e.g. CHU0107) which catalyses two-step phosphorylation of AMP to ATP (via ADP) to ensure a straight-forward ATP regeneration for CAR-based cascades22,46 is suitable for the CAR–GDH system.
Preparative scale reactions
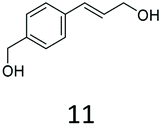
Our analytical whole-cell indirect substrate profiling of EcADH highlights aldehydes that are non-reactive with the EcADH, e.g. indole-3-carboxyaldehyde 6b. This can be advantageous if selective access to the aldehyde product is desired via a CAR-whole-cell process and in synthetic cascades for which the aldehyde serves as an intermediate for a subsequent step (e.g. reductive amination). Hence, we sought to demonstrate the applicability of CAR expressing recombinant E. coli cells for selective preparative synthesis of indole-3-carboxyaldehye 6b, an important precursor for the preparation of many biologically active compounds including indole alkaloids. We attempted a 100 mg scale whole-cell biotransformation at 10 mM substrate concentration, using 100 mM sodium phosphate buffer. However, we observed a steady drop in the reaction pH, from an initial pH of 8 to pH < 6 within 6 h of the incubation. Increasing buffer strength to 150 mM, or changing the buffer to Tris-HCl did not stabilise the reaction pH, neither did decreasing the substrate concentration to 5 mM. Hence, we resorted to hourly adjustment of the reaction pH during the incubation period. With this strategy, 100 mg indole-3-carboxylic acid 6a was exclusively converted to the corresponding aldehyde 6b by the recombinant resting E. coli cells, affording 95% conversion and an isolated yield of 85% after 7 h. Similarly, we explored the unexpected CC reduction activity observed with recombinant E. coli cells. Using the same reaction conditions, 100 mg of (E)-3-(naphthalen-1-yl)acrylic acid 24a was converted to 3-(naphthalen-1-yl)propan-1-ol 24f, in 75% conversion and 54% isolated yield, after 7 h.
Finally, by applying the in vitro one-pot two enzyme CAR–GDH system and using catalytic amount of NADP+, 100 mg cinnamic acid 1a at 10 mM substrate loading was converted to cinnamyl alcohol 1c, affording 75% conversion and 62% isolated yield.
Conclusion
In summary, we have developed simple and efficient in vivo and in vitro biocatalytic systems that enable access to a variety of benzo-fused (hetero)aromatic alcohols, allylic alcohols and their saturated analogues from the corresponding carboxylic acids. We reveal the broad substrate scope of E. coli endogenous ADH, as well as novel CC bond reducing activity of E. coli cells. Hence, recombinant E. coli cells expressing SrCAR can be applied as efficient multi-step hydrogenation catalysts to convert a variety of α,β-unsaturated carboxylic acids to the corresponding saturated primary alcohols. Where allylic alcohols are required, an alternative in vitro system can yield these compounds. The promiscuous carbonyl reductase activity of BsGDH towards a wide range of benzo-fused (hetero)aromatic aldehydes and α,β-unsaturated aldehydes supports a simple in vitro system coupling CAR-catalysed carboxylate reduction and GDH-catalysed carbonyl reduction to access a variety of (hetero)aromatic alcohols and allylic alcohols from the parent carboxylates. The application of GDH as a bifunctional enzyme in this system (i.e. as glucose oxidant and carbonyl reductase), enabled development of a two-enzyme system, which efficiently couples NADP(H) recycling system to the two hydrogenation steps from carboxylate to alcohol. The development of these simple whole-cell and in vitro systems provide alternative green hydrogenation catalytic routes that can potentially replace harsh abiotic reducing agents for the preparation industrial alcohols from biomass derived carboxylic acids. Altogether, we present novel biocatalytic routes that enable the conversion of a wide range of biomass-derived carboxylic acids to a variety of primary alcohols.
Experimental section
Chemicals
Commercially available chemicals and reagents of highest purity were purchased from Sigma-Aldrich (Poole, Dorset, UK), or Fluorochem (Hadfield, Derbyshire, UK) unless stated otherwise. HPLC solvents were obtained from Sigma-Aldrich (Poole, Dorset, UK) or ROMIL (Waterbeach, Cambridge, UK) and GC gases from BOC gases (Guildford, UK). Enzyme nicotinamide cofactors NADP+ and NADPH were purchased from Bio Basic (Markham, Ontario, Canada), and Adenosine triphosphate (ATP) was sourced from Sigma-Aldrich (Poole, Dorset, UK). LB auto induction media was purchased from Formedium (Hunstanton, UK).Preparation of CARSfp whole-cell biocatalyst
Plasmids of carboxylic acid reductases TpCAR, SrCAR, and plasmid of phosphopantetheine transferase (Sfp) from Bacillus subtilis were sourced from in-house plasmid collections and cloned as previously reported.12E. coli cells were transformed with CAR and Sfp plasmids. A single colony of recombinant E. coli BL21 (DE3) containing pET28-b-FL-SrCAR and pCDF1b-Sfp was inoculated into 10 mL lysogeny broth (LB) (1% tryptone, 0.5% yeast extract, 1% NaCl) and incubated overnight at 37 °C in an orbital shaker with 200 rpm shaking. This starter culture was used as inoculum. A 2 L flask containing 500 mL LB autoinduction media, supplemented with kanamycin (30 μg mL−1) and streptomycin (50 μg mL−1) was inoculated with 5 mL of the starter culture. Cultivation was performed at 37 °C in an orbital shaker with 200 rpm shaking, initially for 6 h. Incubation was continued at 24 °C and 200 rpm for a further 42 h. Cells from a 500 mL culture were harvested by centrifugation at 6000 rpm for 15 min, and washed in sodium phosphate buffer (sodium phosphate, 50 mM, pH 7.5). Harvested cells containing CARSfp were used as fresh resting whole cells or purified according to a previously reported method.12Analytical scale whole-cell biotransformation
Recombinant E. coli cells containing SrCARsfp were applied as fresh resting cells for the conversion of carboxylic acids to the corresponding alcohols via the aldehyde intermediate. Glucose was added to enable in situ regeneration of cofactors. Reactions were performed in 2 mL Eppendorf tubes. A typical 500 μL reaction mixture contained 5 mM carboxylic acid substrate, 2% v/v DMSO, 20 mM glucose, 10 mM MgCl2, E. coli resting whole-cells containing over-expressed CARsfp to the final OD600 of 30 in NaPi buffer (50 mM, pH 7.5). Reaction mixtures were incubated at 30 °C with 250 rpm shaking for 18 h, after which the samples were prepared for RP-HPLC or GC-MS analysis.Preparative whole-cell biotransformation
Preparative scale biotransformation reactions were performed in a 250 mL glass flask sealed with breathable membrane to ensure adequate aeration. Biotransformation reactions contained 4 mM MgCl2, 120 mM D-glucose, 100 mg of carboxylic acid 6a or 24a (at 5 mM or 10 mM) and 125 g L−1 of fresh resting recombinant E. coli cells expressing SrCAR in 100 mM sodium phosphate buffer supplemented with 50 mM NaCl, pH 8.0. Following addition of all components, reaction pH was adjusted to pH 8, incubated at 30 °C with 180 rpm shaking, and hourly pH adjustments to pH ∼8.0 was performed. After 7 h incubation, the reaction was basified to pH ∼12 with 10 M NaOH, extracted twice into EtOAc with centrifugation (4000 r.p.m, 5 min, 10 °C). The organic layers were combined, dried with anhydrous MgSO4 and solution was carefully concentrated.Indole-3-carboxyladehyde 6b was prepared from indole-3-carboxylic acid 6a, yellow solid was isolated, 85% yield. 1H NMR (400 MHz, DMSO-D6) δ 12.14 (s, 1H), 9.96 (s, 1H), 8.29 (s, 1H), 8.15–8.08 (dt, 1H), 7.53 (dt, J = 8.2, 0.9 Hz, 1H), 7.30–7.15 (m, 2H). 13C NMR (101 MHz, DMSO) δ 184.77, 137.55 (d, J = 137.6 Hz), 123.94, 123.27, 121.93, 120.65, 117.99, 112.23. HRMS calcd for C9H8NO+ for 146.06 [M + H]+, found 146.0602.
3-(Naphthalen-1-yl)propan-1-ol 24f was prepared from 3-(naphthalen-1-yl)acrylic acid 24a, brown solid was isolated, 54% yield. 1H NMR (400 MHz, DMSO) δ 8.09 (d, J = 8.1 Hz, 1H), 7.91 (d, J = 7.6, 1.7 Hz, 1H), 7.76 (d, J = 8.1 Hz, 1H), 7.57–7.49 (m, 2H), 7.45–7.35 (m, 2H), 4.57 (s, 1H), 3.51 (t, J = 6.3 Hz, 2H), 3.08 (t, 2H), 1.87–1.75 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 138.58, 133.68, 131.61, 128.78, 126.49, 126.08 (d, J = 2.6 Hz), 125.77 (d, J = 9.6 Hz), 123.97, 60.59, 34.05, 28.99. HRMS calcd for C13H15O+ for 187.11 [M + H]+, found 187.5821.
Analytical scale in vitro biotransformation
For the one-pot, two-step in vitro conversion of benzofused (hetero)aromatic acids or acrylic acids derivatives respectively to the corresponding (hetero)aromatic primary alcohols or allylic alcohols, purified enzymes were used. A typical 500 μL reaction contained 5 mM carboxylic acid substrate, 2% v/v DMSO, 7.5 mM ATP, 10 mM MgCl2, 20 mM D-glucose, 0.4 mM NADP+, purified SrCAR (0.4 mg mL−1), and purified BsGDH (0.5–1 mg mL−1) in 50 mM sodium phosphate buffer, pH 7.5. Reaction mixtures in 2 mL tightly closed Eppendorf's were incubated at 30 °C with 250 rpm shaking for 18 h, after which the samples were prepared for RP-HPLC or GC-MS analysis.Preparative in vitro biotransformation
In vitro preparative scale biotransformation reactions were performed in 250 mL glass flask. Purified enzyme biotransformation reactions contained 4 mM MgCl2, 30 mM D-glucose 1 mM NADP+, 12 mM ATP, 100 mg of 26a (10 mM), 2% v/v DMSO, 0.8 mg mL−1 purified SrCAR and 2 mg mL−1 purified GDH in 100 mM sodium phosphate buffer (supplemented with 50 mM NaCl, pH 8.0) to a total volume of 60 mL. The reaction was incubated at 30 °C with 180 rpm shaking. After 27 h of incubation, the reaction was basified to pH ∼12 with 10 M NaOH, extracted twice into EtOAc with intermediate centrifugation (4000 r.p.m, 5 min, 10 °C). The organic layers were combined, dried with anhydrous MgSO4 and solvent was carefully concentrated.Cinnamyl alcohol 26c was prepared from cinnamic acid 26a. Pale yellow liquid was isolated, 62% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.44–7.26 (m, 5H), 6.56 (d, J = 15.9, 2.0 Hz, 1H), 6.38 (dt, J = 16.0, 5.0 Hz, 1H), 4.87 (s, 1H), 4.12 (t, J = 5.3, 1.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 136.89, 130.80, 128.59, 128.39, 127.18, 126.11, 61.47.
Initial rate measurements
Specific activities where determined from initial rates of CAR-catalysed carboxylate reduction. Unless stated otherwise, measurements were performed in triplicate at 370 nm (ε = 2.216 mM−1 cm−1) using a Tecan infinite M200 microplate reader (Tecan Group, Switzerland). The samples were prepared in 96-well microtitre plates. A typical reaction mixture in a well contained 1–2 mM carboxylic acid substrate, 2 mM MgCl2, 1 mM ATP, 0.5 mM NADPH, 1% (v/v) DMSO and 20–100 μg of purified SrCAR in a total volume of 200 μl (50 mM sodium phosphate, pH 7.5). A unit of SrCAR was equal to the amount of the pure enzyme required to consume 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) μmol NADPH per min. Activity measurement for substrates 24a, 25a, 31a, 34a and 45a were determined from initial reaction rates measured by RP-HPLC using the same reaction conditions. In this case, reactions were stopped after 5–10 min incubation and the samples were prepared for analysis on RP-HPLC. Specific activities for these substrates were determined from HPLC activity measurements relative to reference activities for substrates 1a and 36a (which have been standardised using both microplate reader and RP-HPLC measurements) under the same reaction conditions.
μmol NADPH per min. Activity measurement for substrates 24a, 25a, 31a, 34a and 45a were determined from initial reaction rates measured by RP-HPLC using the same reaction conditions. In this case, reactions were stopped after 5–10 min incubation and the samples were prepared for analysis on RP-HPLC. Specific activities for these substrates were determined from HPLC activity measurements relative to reference activities for substrates 1a and 36a (which have been standardised using both microplate reader and RP-HPLC measurements) under the same reaction conditions.
Analysis: chromatography columns and conditions and sample preparation
Reverse phase HPLC was performed on an Agilent system (Santa Clara, CA, USA) equipped with a G1379A degasser, G1312A binary pump, a G1367A well plate autosampler unit, a G1316A temperature-controlled column compartment and a G1315C diode array detector. Syncronis C18 column, 250 mm length, 4.6 mm diameter, 5 μm particle size (Thermo Scientific; Waltham, MA USA) was used. Substrate standards and product markers, and the resulting biotransformation products were analysed by reverse phase HPLC using isocratic methods with different solvent ratios of acetonitrile and water, supplemented with 0.1% triflouroacetic acid. The flow rate was maintained at 1 mL min−1 and elutes were detected by the U.V. detector (λ = 245 nm). To account for the variation in UV response between the starting material and the product, relative response factors were experimentally determined. Correction factors were calculated from the ratio of the slopes of standard curves plotted for varying concentrations of both the acid and the corresponding products at the UV detection wavelength of 245 nm.GCMS analyses were performed on Agilent 5977A Series GC/MSD System with an Agilent 7890B Series GC coupled to Mass Selective Detector. Data analysis was performed using GC/MSD MassHunter Data Acquisition and ChemStation Data Analysis. A 30 m × 0.25 mm × 0.1 μm VF-5HT column (Agilent, Santa Clara, CA, USA) was used. The parameters of the method include: inlet temperature = 240 °C, detector temperature = 250 °C, MS source = 230 °C, helium flow = 1.2 mL min−1; oven temperature between 50–360 °C, 30 °C min−1.
Spectra from 1H and 13C NMR runs were recorded on a Bruker Avance 400 instrument (400 MHz for 1H and 100 MHz for 13C) in DMSO-d6, using residual protic solvent as an internal standard. Reported chemical shifts (δ) (in parts per million (ppm)) are relative to the residual protic solvent signal. High-resolution mass spectrometry (HRMS) was recorded using a Waters LCT time-of-flight mass spectrometer, connected to a Waters Alliance LC (Waters, Milford, MA, USA). Data were processed with Waters Masslynx software.
Where analysis of biotransformation reactions was performed on the reverse phase HPLC, the reaction was stopped by addition of 3 volumes of acetonitrile and vigorous mixing. The reaction mixture was centrifuged (15 °C, 13000 rpm, 10 min); the clear supernatant was collected and centrifuged further. The clarified solution was transferred to HPLC vials for analysis.
Where analysis of biotransformation was performed on the GC-MS, an equal volume of EtOAc (containing a known concentration of an internal standard where necessary) was added to biotransformation mixture, vigorously mixed, centrifuged (15 °C, 13000 rpm, 10 min) and the organic layer was extracted. The aqueous fraction was then acidified to a pH of ∼2 and further extracted into EtOAc. Where the substrates contained an amino group, a further extraction step was performed, the aqueous fraction was basified to pH ∼12 and extracted into EtOAc. The organic fractions were combined and dried over anhydrous MgSO4 and the samples were transferred to vials for analysis on GC-MS.
Conflicts of interest
The authors declare that they have no conflict of interest with the contents of this article.Acknowledgements
This work was supported by the grants BBSRC BB/K017802 and ERC pre-FAB 695013. The Authors wish to thank Rachel Heath (Manchester) for providing the GDH plasmid, Deepankar Gahloth and Sasha Derrington (Manchester) for useful discussion, and Reynard Spiess (Manchester) for assistance with high resolution mass spectroscopy. D. L. is a Royal Society Wolfson Merit Award holder.References
- S.-H. Kim, Y. Huang, C. Sawatdeenarunat, S. Sung and V. S.-Y. Lin, J. Mater. Chem., 2011, 21, 12103–12109 RSC.
- Q. Wu, X. Bao, W. Guo, B. Wang, Y. Li, H. Luo, H. Wang and N. Ren, Biotechnol. Adv., 2019, 37, 599–615 CrossRef CAS PubMed.
- X. Zhao, H. Zhou, V. S. Sikarwar, M. Zhao, A.-H. A. Park, P. S. Fennell, L. Shen and L.-S. Fan, Energy Environ. Sci., 2017, 10, 1885–1910 RSC.
- L. J. Goossen, N. Rodríguez and K. Goossen, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 CrossRef CAS PubMed.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409 CrossRef.
- G. A. Aleku, B. Nowicka and N. J. Turner, ChemCatChem, 2018, 10, 124–135 CrossRef CAS.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961–969 CrossRef CAS PubMed.
- F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS PubMed.
- J. Mangas-Sanchez, S. P. France, S. L. Montgomery, G. A. Aleku, H. Man, M. Sharma, J. I. Ramsden, G. Grogan and N. J. Turner, Curr. Opin. Chem. Biol., 2017, 37, 19–25 CrossRef CAS PubMed.
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12, 2859–2881 CrossRef CAS PubMed.
- G. A. Aleku, J. Mangas-Sanchez, J. Citoler, S. P. France, S. L. Montgomery, R. S. Heath, M. P. Thompson and N. J. Turner, ChemCatChem, 2018, 10, 515–519 CrossRef CAS.
- D. Gahloth, M. S. Dunstan, D. Quaglia, E. Klumbys, M. P. Lockhart-Cairns, A. M. Hill, S. R. Derrington, N. S. Scrutton, N. J. Turner and D. Leys, Nat. Chem. Biol., 2017, 13, 975–981 CrossRef CAS PubMed.
- G. A. Aleku, C. Prause, R. T. Bradshaw-Allen, K. Plasch, S. M. Glueck, S. S. Bailey, K. A. P. Payne, D. A. Parker, K. Faber and D. Leys, ChemCatChem, 2018, 10, 3736–3745 CrossRef CAS PubMed.
- A. J. L. Wood, N. J. Weise, J. D. Frampton, M. S. Dunstan, M. A. Hollas, S. R. Derrington, R. C. Lloyd, D. Quaglia, F. Parmeggiani, D. Leys, N. J. Turner and S. L. Flitsch, Angew. Chem., 2017, 129, 14690–14693 CrossRef.
- M. Winkler, Curr. Opin. Chem. Biol., 2018, 43, 23–29 CrossRef CAS PubMed.
- M. Petchey, A. Cuetos, B. Rowlinson, S. Dannevald, A. Frese, P. W. Sutton, S. Lovelock, R. C. Lloyd, I. J. S. Fairlamb and G. Grogan, Angew. Chem., 2018, 130, 11758–11762 CrossRef.
- H. K. Philpott, P. J. Thomas, D. Tew, D. E. Fuerst and S. L. Lovelock, Green Chem., 2018, 20, 3426–3431 RSC.
- D. Gahloth, G. A. Aleku and D. Leys, J. Biotechnol., 2020, 307, 107–113 CrossRef CAS PubMed.
- S. R. Derrington, N. J. Turner and S. P. France, J. Biotechnol., 2019, 304, 78–88 CrossRef CAS PubMed.
- G. A. Strohmeier, I. C. Eiteljörg, A. Schwarz and M. Winkler, Chem. – Eur. J., 2019, 25, 6119–6123 CrossRef CAS PubMed.
- T. P. Fedorchuk, A. N. Khusnutdinova, E. Evdokimova, R. Flick, R. Di Leo, P. Stogios, A. Savchenko and A. F. Yakunin, J. Am. Chem. Soc., 2020, 142, 1038–1048 CrossRef CAS PubMed.
- J. Citoler, S. R. Derrington, J. L. Galman, H. Bevinakatti and N. J. Turner, Green Chem., 2019, 21, 4932–4935 RSC.
- W. Finnigan, R. Cutlan, R. Snajdrova, J. P. Adams, J. A. Littlechild and N. J. Harmer, ChemCatChem, 2019, 11, 3474–3489 CrossRef CAS PubMed.
- M. Horvat, G. Fiume, S. Fritsche and M. Winkler, J. Biotechnol., 2019, 304, 44–51 CrossRef CAS PubMed.
- A. N. Khusnutdinova, R. Flick, A. Popovic, G. Brown, A. Tchigvintsev, B. Nocek, K. Correia, J. C. Joo, R. Mahadevan and A. F. Yakunin, Biotechnol. J., 2017, 12, 1600751 CrossRef PubMed.
- M. K. Akhtar, N. J. Turner and P. R. Jones, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 87–92 CrossRef CAS PubMed.
- L. Kramer, E. D. Hankore, Y. Liu, K. Liu, E. Jimenez, J. Guo and W. Niu, ChemBioChem, 2018, 19, 1452–1460 CrossRef CAS PubMed.
- R. C. Hoye, J. Chem. Educ., 1999, 76, 33 CrossRef CAS.
- M. Hudlicky, J. Chem. Educ., 1980, 57, A173 CrossRef.
- T. J. Korstanje, J. I. van der Vlugt, C. J. Elsevier and B. de Bruin, Science, 2015, 350, 298–302 CrossRef CAS PubMed.
- T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Hölscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed.
- W. Finnigan, A. Thomas, H. Cromar, B. Gough, R. Snajdrova, J. P. Adams, J. A. Littlechild and N. J. Harmer, ChemCatChem, 2017, 9, 1005–1017 CrossRef CAS PubMed.
- D. Gahloth, M. S. Dunstan, D. Quaglia, E. Klumbys, M. P. Lockhart-Cairns, A. M. Hill, S. R. Derrington, N. S. Scrutton, N. J. Turner and D. Leys, Nat. Chem. Biol., 2017, 13, 975–981 CrossRef CAS PubMed.
- G. Qu, B. Liu, K. Zhang, Y. Jiang, J. Guo, R. Wang, Y. Miao, C. Zhai and Z. Sun, J. Biotechnol., 2019, 306, 97–104 CrossRef CAS PubMed.
- D. Schwendenwein, A. K. Ressmann, M. Doerr, M. Höhne, U. T. Bornscheuer, M. D. Mihovilovic, F. Rudroff and M. Winkler, Adv. Synth. Catal., 2019, 361(11), 2371–2733 Search PubMed.
- G. Qu, M. Fu, L. Zhao, B. Liu, P. Liu, W. Fan, J.-A. Ma and Z. Sun, J. Chem. Inf. Model., 2019, 59, 832–841 CrossRef CAS PubMed.
- J. Chan Joo, A. N. Khusnutdinova, R. Flick, T. Kim, U. T. Bornscheuer, A. F. Yakunin and R. Mahadevan, Chem. Sci., 2017, 8, 1406–1413 RSC.
- H. S. Toogood and N. S. Scrutton, ACS Catal., 2018, 8, 3532–3549 CrossRef CAS PubMed.
- S. Kurogochi, S. Tahara and J. Mizutani, Agric. Biol. Chem., 1975, 39, 825–831 CAS.
- A. Lumbroso, M. L. Cooke and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 1890–1932 CrossRef CAS PubMed.
- S. M. Husain, M. A. Schätzle, S. Lüdeke and M. Müller, Angew. Chem., Int. Ed., 2014, 53, 9806–9811 CrossRef CAS PubMed.
- D. Conradt, M. A. Schätzle, S. M. Husain and M. Müller, ChemCatChem, 2015, 7, 3116–3120 CrossRef CAS.
- S. Roth, A. Präg, C. Wechsler, M. Marolt, S. Ferlaino, S. Lüdeke, N. Sandon, D. Wetzl, H. Iding, B. Wirz and M. Müller, ChemBioChem, 2017, 18, 1703–1706 CrossRef CAS PubMed.
- S. Roth, M. B. Kilgore, T. M. Kutchan and M. Müller, ChemBioChem, 2018, 19, 1849–1852 CrossRef CAS PubMed.
- B. P. Nocek, A. N. Khusnutdinova, M. Ruszkowski, R. Flick, M. Burda, K. Batyrova, G. Brown, A. Mucha, A. Joachimiak, Ł. Berlicki and A. F. Yakunin, ACS Catal., 2018, 8, 10746–10760 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Preliminary biotransformation data and sample chromatograms of biotransformation reactions. See DOI: 10.1039/d0gc00867b |
| This journal is © The Royal Society of Chemistry 2020 |