
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRigid biobased polycarbonates with good processability based on a spirocyclic diol derived from citric acid†
Olivier
Bonjour
 a,
Ilme
Liblikas
b,
Tõnis
Pehk
c,
Truong
Khai-Nghi
d,
Kari
Rissanen
d,
Lauri
Vares
*b and
Patric
Jannasch
*ab
a,
Ilme
Liblikas
b,
Tõnis
Pehk
c,
Truong
Khai-Nghi
d,
Kari
Rissanen
d,
Lauri
Vares
*b and
Patric
Jannasch
*ab
aDepartment of Chemistry, Lund University, Box 124, Lund 221 00, Sweden. E-mail: patric.jannasch@chem.lu.se
bInstitute of Technology, University of Tartu, Nooruse 1, Tartu 50411, Estonia. E-mail: lauri.vares@ut.ee
cLaboratory of Chemical Physics, National Institute of Chemical Physics and Biophysics, Akadeemia tee 23, Tallinn 12618, Estonia
dUniversity of Jyvaskyla, Department of Chemistry, P.O. Box 35, Survontie 9 B, Jyväskylä 40014, Finland
First published on 28th May 2020
Abstract
Introducing biobased polymers from renewable sources for use as high-performance thermoplastics with high demands on mechanical rigidity, transparency, thermal stability, as well as good processability, is a significant challenge. In the present work we have designed and prepared a rigid biobased bis-spirocylic diol by di-cycloketalization of a bicyclic diketone (cis-bicyclo[3.3.0]octane-3,7-dione, obtained from citric acid) using trimethylolpropane. This spiro-diol monomer has two reactive primary hydroxyl groups and the synthesis from inexpensive biobased starting materials is straightforward and readily upscalable, involving no chromatographic purification. In order to explore the usefulness of the new monomer, it was employed in melt polycondensations with diphenylcarbonate at up to 280 °C to form rigid fully amorphous polycarbonates (PCs). Molecular weights (MWs) up to Mn = 28 kg mol−1 were achieved, and thermal and dynamic mechanical measurements showed glass transitions up to Tg = 100 °C, with no thermal decomposition until Td ∼350 °C. Solvent cast films had excellent mechanical flexibility and strength, as well as a high transparency with only slight coloration. Results by dynamic melt rheology implied that the high-MW PCs had a good processability at 170 °C, with a stable shear modulus over time, but started to degrade via chain scission reactions when the temperature approached 200 °C. In conclusion, the present work demonstrates the straightforward preparation of the citric acid-based spiro-diol, and indicates that it is an efficient building block for the preparation of rigid biobased PCs and other condensation polymers.
Introduction
The development of sustainable biobased alternatives to fossil-based monomers and polymers has become increasingly important in recent years due to growing environmental, social and economic concern.1,2 However, it is a considerable challenge to chemically design economically viable biobased polymers which meet all the strict requirements from high-end applications on high-performance plastics with good processability.3 For example, it is crucial that plastics intended for a wide range of automotive, construction, coating and packaging applications have a high degree of transparency and adequate thermal and mechanical properties to maintain dimensions and mechanical strength at elevated temperatures. This requires rigid polymers with a high glass transition temperature (Tg), high molecular weight (MW), as well as high thermal stability. Consequently, there is currently a need for biobased building blocks and monomers containing rigid aromatic and alicyclic rings to produce polymers with inflexible macromolecular chain structures.3Lignin is the most accessible source of rigid aromatic building blocks in the form of various phenolic compounds.4 However, the depolymerization and purification of lignin is a complex and difficult process and so far vanillin is the only lignin-based compound that is commercially available. Despite these limitations, several high-Tg lignin-based polymers5 in the form of, e.g., polymethacrylates6,7 and polyesters,8–10 have been reported. When it comes to alicyclic monomers for polycondensation processes, isosorbide is probably the most studied and exploited alternative.11 Isosorbide is a nonotoxic and chiral bicylic diol produced from sorbitol, which, in turn, is a reduction product of glucose.12,13 Despite the apparent benefits of incorporating isosorbide into polymers, the limited reactivity and stability are obvious drawbacks.14 The low reactivity of the two non-equivalent secondary hydroxyl groups15 impedes the formation of high MW polymers via polycondensation, and low MW polymers have insufficient mechanical properties. Increasing the polycondensation temperature (>230 °C) or reaction time to favor polymer chain growth typically leads to discoloration, cross-linking and degradation due to the limited stability towards thermooxidation.14 Methods to improve the reactivity of isosorbide and other isohexides have been investigated and the strategies include the conversion of the secondary hydroxyl groups into primary ones via carbon chain extension,16,17 and the development of more efficient catalyst systems.18 Still, the intrinsic instability of the fused furan ring system needs attention. Gao, et al. have recently reported on octahydro-2,5-pentalenediol (OPD, Scheme 1), derived from citric acid.19,20 Just like isosorbide, it has a rigid V-shaped structure, but instead of furan rings it has two cis-fused carbocycles. This alicyclic diol yielded PCs with good thermal stability (thermal decomposition at 276 °C) and Tgs up to 80 °C, demonstrating that the mimicking of the isosorbide bicyclic structure is an efficient approach to increase the chain rigidity of polymers. However, similarly to isosorbide, the secondary hydroxyl groups of OPD have low reactivity in polycondensation reactions and are likely to lead to low and medium MW polymers.
 | ||
| Scheme 1 Outline of the compounds based on diketone 1 reported by McKeown and Gao, and in the present work (inside box). Spiro-diol 3 (mixture of cis and trans) was synthesized in a ketalization reaction of diketone 1 and a slight excess of TMP 2 (yields of 74 and 91% were obtained when the product was isolated by crystallization and chromatography, respectively). | ||
The introduction of cyclic ketal and acetal units into polymers is a further attractive strategy to increase the Tg and improve mechanical properties of polymers.21 When an aldehyde or a ketone reacts with a suitable diol, a cyclic acetal or ketal, respectively, is formed. From this follows that the use of cylic ketones will lead to spiro-cyclic products, which are especially rigid.21 The reactions proceed via acid catalyzed equilibria and the removal of water is necessary to reach high conversion. Acetals and ketals are typically stable towards bases but are susceptible to hydrolysis under acidic conditions.22 Du Prez and coworkers prepared spirocyclic diol (“spiro-diol”) monomers in ketalization reactions of glycerol and two equivalents of 1,4-cyclohexanedione and 4,4′-dicyclohexanone, respectively.23 Both monomers were successfully used to prepare PCs of high MW, but the synthesis of polyesters proved difficult and only low MW products were obtained. Recently, the biobased diketones camphorquinone and 1,4-cyclohexanedione were combined with glycerol to obtain stiff spiro-diols.24 The corresponding PCs showed good mechanical properties, high thermal stability and high Tg values. However, the isolation of these diol monomers required chromatographic purification, which makes them less attractive on larger scales. In another piece of work, Mankar et al. synthesized a spiro-diol by acetal formation in a reaction of pentaerythritol with two equivalents of vanillin.25 Incorporation of this rigid spiro-cyclic diol in copolyesters produced from 1,6-hexanediol and dimethyl terephthalate resulted in improved Tg and thermal stability.25 Hence, this gave materials similar to Akestra™, a commercial fossil-based high-performance polyester produced from a spiro-diol, ethylene glycol and terephthalic acid.26 Using a similar strategy, Warlin and coworkers reported the synthesis of a rigid spiro-diol from pentaerythritol and two equivalents of fructose-based 5-hydroxymethylfurfural.27 This monomer was employed to produce polyesters and poly(urethane-urea)s with increased Tgs. However, the investigated spiro-diol was found to be heat-sensitive, causing coloration and branching during the polyester synthesis. Besides the synthesis of various cyclic monomers, polycycloacetals and polycycloketals have been formed in direct polycondensations of tetraols and dialdehydes or diketones, respectively.13 For example, McKeown et al. has reacted 1,5-diethylbicyclo[3.3.0]octane-3,7-dione with pentaerythritol in a polycondensation and achieved a spiro-polymer with a very high Tg.28 However, the degree of polymerization remained mediocre at ∼30, and the polymer was rather easily hydrolyzed in solution unless special precautions were taken (Scheme 1).
In the present work we have designed and prepared a novel and readily accessible biobased tetracyclic aliphatic spiro-diol 3 which combines the thermal stability of the rigid V-shaped alicyclic rings with the high reactivity of primary hydroxyl groups installed via the spiro rings (Scheme 1). The stiff bicyclic diketone cis-bicyclo[3.3.0]octane-3,7-dione 1 was employed as a starting material. This diketone is derived via a Weiss–Cook condensation between glyoxal and dimethyl-1,3-acetonedicarboxylate (DAC).29 Aqueous glyoxal can be prepared via oxidation of ethylene glycol.30 A plant-based ethylene glycol production is currently under development by Avantium N.V.31 DAC is readily derived from citric acid after decarboxylation and esterification.32–34 Citric acid is an extensively used biobased platform chemical, produced via fermentation of glucose in more than 1.6 million tons annually.35 Thus, the synthesis route of 1 from citric acid is well-developed and involves only inexpensive acids (cat. H2SO4 with H2O2 or ClSO3H, aq. HCl, AcOH), base (NaOH) and MeOH. For the ketalization, we employed trimethylolpropane (TMP, 2), a triol widely used in the polymer industry that is available from Perstorp with partly renewable content (Evyron™).36 The ketalization reaction between diketone 1 and two equivalents of TMP 2 then afforded the stiff spiro-diol 3, which bears two six-membered spiro rings connected through a bicyclo[3.3.0]octane entity. Depending on the origin of the TMP (0, 20 or 50% renewable grade), spiro-diol 3 has a 40, 50 or 70% content from renewable sources. Consequently, the overall synthesis route to spiro-diol 3 is well-developed, readily up-scalable, and this monomer can be envisioned as a valuable biobased building block in various applications, including the preparation of different condensation polymers. In order to investigate the usefulness spiro-diol 3, we have in the present work studied the preparation of PCs in polycondensations with diphenylcarbonate, and subsequently investigated the thermal and mechanical properties, as well as the processability of the resulting materials.
Results and discussion
Monomer synthesis and structure elucidation
A roughly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two isomers of spiro-diol 3 was formed in the ketalization reaction of diketone 1 with two equivalents of TMP 2 in the presence of an acid catalyst in toluene/cyclohexane (Fig. 1a and Fig. S1–S6†). We speculated that these isomers are diastereomers that differ by mutual orientation of the terminal ethyl and hydroxymethyl groups (Scheme 1). The C2 symmetric isomer was conveniently named as 3-trans and the Cs symmetric as 3-cis. We separated these two isomers with the aim to unambiguously confirm the diastereomers. This separation turned out to be a routine task by standard flash chromatography on silica gel. The different symmetry of 3-trans and 3-cis is evident from their NMR spectra (Fig. 1 and Table S1†).
:1 mixture of two isomers of spiro-diol 3 was formed in the ketalization reaction of diketone 1 with two equivalents of TMP 2 in the presence of an acid catalyst in toluene/cyclohexane (Fig. 1a and Fig. S1–S6†). We speculated that these isomers are diastereomers that differ by mutual orientation of the terminal ethyl and hydroxymethyl groups (Scheme 1). The C2 symmetric isomer was conveniently named as 3-trans and the Cs symmetric as 3-cis. We separated these two isomers with the aim to unambiguously confirm the diastereomers. This separation turned out to be a routine task by standard flash chromatography on silica gel. The different symmetry of 3-trans and 3-cis is evident from their NMR spectra (Fig. 1 and Table S1†).
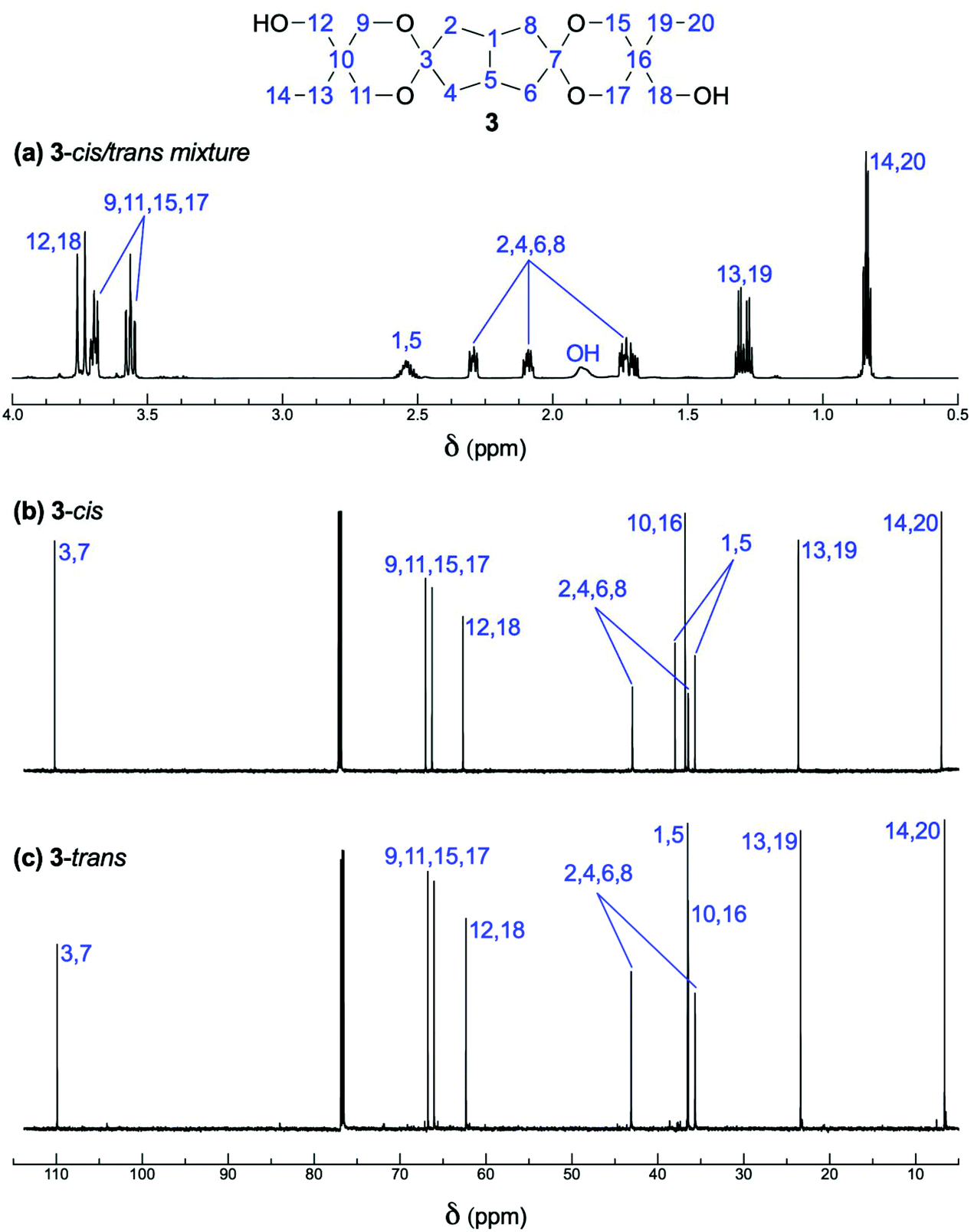 | ||
| Fig. 1 (a) 1H spectrum of the mixture of 3-trans and 3-cis after isolation via double crystallization, and 13C spectra of the (b) 3-cis and (c) 3-trans isomers after separation. | ||
In the bicyclo[3.3.0]octane subunit, the trans isomer has equivalent C-1 and C-5 and H-1 and H-5 chemical shifts, while the cis isomer shows different ones, separated in the 13C and 1H NMR spectra by 2.3 and 0.03 ppm, respectively. Further confirmation of the assignment of stereoisomers was obtained after single crystal X-ray analysis of the trans isomer. The single crystals of the compound 3-trans were obtained by slow evaporation of chloroform at ambient temperature. Fig. 2 presents the structure of 3-trans. In the solid-state the ethyl substituents of 3-trans are equatorial, while the hydroxymethyl groups are axial. The crystal lattice is stabilized via hydrogen bonds between the hydroxyl groups of 3-trans with its neighboring molecules. Crystal data, data collection and refinement details are given in the ESI.† Despite many attempts, no suitable single crystals from 3-cis could be obtained and thus its X-ray structure could not be determined. In the obtained conformation, the methylene carbons of the dioxane rings have both one trans and one gauche orientation to the bicycle methylene carbon atoms. However, from two bicycle methylene carbons, only one is having two gauche orientations. This is the main reason for the significant differences in the methylene carbon chemical shifts in the bicycle (7.4 in trans vs. 6.5 ppm in cis isomer) and the quite close values of the methylene carbon chemical shifts from the dioxane rings (for more comprehensive data, see Table S1†).
 | ||
| Fig. 2 The X-ray structure of 3-trans (thermal displacement ellipsoids are shown at the 50% probability level). | ||
During the course of the work we optimized the ketalization reaction, work-up and product isolation procedure to increase the efficiency for large scale synthesis of spiro-diol 3. In a preparative scale we chose to work with the diastereomeric mixture for practical reasons. This is also most relevant from an industrial point of view. We evaluated various solvents (mixtures of cyclohexane/DMF and cyclohexane/toluene, pure toluene), varied the stoichiometry of the reagents (1.95–3.5 eq. of TMP) and the amount of catalyst (1–20 mol% p-toluenesulfonic acid, [p-TsOH]). The use of toluene as a reaction medium resulted in some darkening during the ketalization, probably due to the relatively high temperature needed for reflux. The lower boiling cyclohexane did not fully dissolve the starting materials. However, the addition of a small amount of DMF (10%) resulted in complete dissolution and a clean reaction. The removal of DMF is nevertheless tedious and its use is not recommended due to toxicity issues.37 Finally, a 3:2 mixture of cyclohexane/toluene was identified as the best solvent system, affording a clean conversion without any noticeable by-product formation. The amount of acid catalyst had only a minor impact on the reaction outcome, and we found 1 mol% of p-TsOH to be a suitable concentration. An excess of TMP corresponding to 3.5 equiv. in relation to the diketone resulted in a somewhat faster ketalization reaction. Still, a smaller excess (2.2 equiv., i.e. 1.1 equiv. in relation to one carbonyl group) was enough to achieve a high conversion during 16 h. Detailed data from the optimization is presented in Table S2.†
After cooling the reaction mixture and removal of the solvents by evaporation, three different work-up/product isolation methods were evaluated: (a) direct crystallization of 3 in EtOAc/hex, 1:1, (b) dissolution of the directly crystallized product in EtOAc, washing with water/brine, followed by a second crystallization, and (c) flash chromatography. The direct crystallization (method a) afforded the crude product in 85% yield. However, by careful analysis of 13C NMR spectra, we detected a small amount of unreacted TMP present in the product. The aqueous wash (method b) removed the traces of TMP and the second crystallization afforded the pure product 3 in 74% yield. Finally, the purification of the crude reaction mixture by flash chromatography (method c) afforded 3 in 91% yield. Still, this method is less attractive from practical and economical points of view.
The hydrolytic stability of spiro-diol 3 in acidic environment was evaluated by using conditions reported by Liu and Thayumanavan38 after modification: 1% aq. KH2PO4 (pH = 4.5) in CD3CN at 20 and 60 °C, respectively, and 10 mM aq. trifluoroacetic acid (TFA) in CD3CN at 20 °C. The experiments were carried out directly in NMR tubes and monitored by recording 1H NMR spectra at regular intervals. In the phosphate buffer at 20 °C, the diol was stable and no degradation was observed even after 7 days. However, after raising to 60 °C traces (<5%) of new signals that correspond to the partially hydrolyzed diol were observed after 24 h. In 10 mM aqueous TFA, the hydrolysis occurred faster. After 4 h at 20 °C roughly 50% of spiro-diol 3 was left, and after 24 h all the diol had been cleanly converted into diketone 1 and TMP 2 (see Fig. S11† for 1H NMR spectra at different time intervals).
Polycarbonate preparation and structure characterization
The diastereomeric mixture of spiro-diol 3 was employed in the synthesis of PCs using diphenylcarbonate (DPC) as the carbonyl source (Scheme 2). Polymerizations were carried out as solvent-free melt polycondensations and we initially evaluated two different catalysts [NaHCO3, Li(Acac)], varied the ratio 3:DPC, and experimented with various heating/vacuum profiles (see Table S3† for detailed descriptions). A procedure adapted from Du Prez et al. was used as a starting point.23 After this optimization to control MW and avoid degraded products, we prepared two PC samples using NaHCO3 as catalyst and keeping the molar ratio of diol 3 and DPC at 1.0:1.1. The MW of the first sample was purposely kept low and the second one high, to give the samples denoted PC-l and PC-h, respectively (Scheme 2). In the preparation of PC-h, melt polycondensation temperatures up to 280 °C were applied, whereas for PC-l the maximum temperature was limited to 210 °C. In the final stage when the temperature was raised to 280 °C, PC-h became highly viscous and started slowly to darken, after which the polymerization was terminated. In contrast, PC-l remained a viscous flowing melt throughout the polycondensation at 210 °C with no apparent color change. Both PC-l and PC-h were purified by precipitation in methanol from chloroform solutions. Further details on their synthesis are given in the Experimental section.
 | ||
| Scheme 2 Synthesis of PCs by melt polycondensations of diol 3 with DPC using the mixture of the 3-trans and 3-cis isomers. | ||
Characterization by size-exclusion chromatography (SEC) using poly(ethylene oxide) standards showed Mn = 7.5 and 27.7 kg mol−1 for PC-l and PC-h, respectively (Table 1). As anticipated, PC-h reached a high MW, while that of PC-l was significantly lower. Moreover, the analysis revealed polydispersities of Đ = 3.55 and 3.40, respectively. Under normal (ideal) conditions a value of Đ close to 2 is expected for polycondensations driven to high monomer conversions.39 Hence, the higher values obtained in the present case may imply limited degradation via ring opening of the ketal rings during the polymerizations, which would result in branching and a broadening of the chain length distribution. This was also hinted by the shape of the SEC curves, which show high-MW shoulders (Fig. S12†). Similar findings have previously been reported by Du Prez et al. who found Đ in the range 2.8–3.2 for PCs (Mn = 24–33 kg mol−1) prepared by melt polycondensation of DPC and ketal diol monomers.23 For polyesters based on the same ketal monomers, the range in Đ was 2.3–4.0. In addition, Suh and coworkers synthesized a PC by polycondensation of DPC and the bis(glycerol) ketal of camphorquinone, and reported Đ = 2.25 (Mn = 8.3 kg mol−1).24
| Sample | Isolated yield (%) | M n (SEC) (kg mol−1) | Đ (SEC) | M n (NMR) (kg mol−1) | T g (DSC) (°C) | T g (DMA) (°C) | T d,95 (TGA) (°C) |
|---|---|---|---|---|---|---|---|
| PC-l | 86 | 7.5 | 3.55 | 7 | 85 | 85 | 352 |
| PC-h | 90 | 27.7 | 3.40 | 22 | 100 | 99 | 360 |
Characterization by 1H and 13C NMR spectroscopy confirmed the structure of the PCs (see Fig. 3 and Fig. S7 and S8†). Detailed analysis of the spectra of PC-h indicated that the cis and trans isomers of 3 preserved the shift differences. The carbonyl region at 155 ppm revealed the dyad level sequences of the polymer, showing clearly the presence of cis–cis, cis–trans and trans–trans dyads in roughly 1:2:1 ratios (Fig. 3). This indicates that the reactivity of each hydroxyl group is independent, and is hence not influenced by the relative orientation of the other hydroxyl group in diol 3. Close inspection of the 1H and 13C NMR spectra also indicated the complete lack of any aromatic signals potentially originating from DPC residues at the chain ends. On the other hand, small signals from terminal –CH2OH groups were clearly present at 62.7 ppm in the 13C NMR spectra (Fig. S9 and S10†). Assuming linear chains with exclusively –CH2OH end groups, the intensity of these signals in the spectra of both PC-l and PC-h were in level with that expected from their respective degree of polymerization measured by SEC. Hence, the end group analysis based on the signals from terminal –CH2OH groups indicated Mn = 7.0 and 22 kg mol−1 for PC-l and PC-h, respectively. The slightly lower values obtained in the end group analysis may indicate limited branching, which is in agreement with the finding that Đ > 2. Still, we cannot rule out that the observed differences in the Mn values are explained simply because SEC analysis using poly(ethylene oxide) standards and calculations based on small NMR signals give different results.
 | ||
| Fig. 3 (a) 1H and (b) 13C spectra of PC-h. 13C signals indicating the 1:2:1 ratios of cis–cis, cis–trans and trans–trans dyads are shown separately in the carbonyl region. | ||
After drying, the precipitates of both PC-l and PC-h were completely soluble in CHCl3 and THF, partially soluble in 1,4-dioxane, EtOAc and toluene, and insoluble in water, MeOH, iPrOH, acetone, hexanes, diethyl ether and CH3CN. The PC-h precipitate had a “cotton-like” appearance with clearly visible fibers, reflecting its high MW, while the PC-l precipitate was a fine powder as expected from its low MW. Both PC samples were cast from solution to study their ability to form films. The PC-h films were only slightly colored (Fig. 4) with high transparency (Fig. S13†) and gloss (between 75 and 118 gloss units [GU]), combined with an excellent mechanical flexibility (ESI Movie†) and strength (Fig. S14†). The films formed by PC-l were less colored, equally transparent, but showed brittleness due to the lower MW.
 | ||
| Fig. 4 Photograph of a 45 μm thick film of PC-h cast from CHCl3 solution. | ||
Thermal characterization
The Tg and the thermal decomposition temperature (Td,95) of the PCs were evaluated by DSC and TGA measurements, respectively. At the Tg the polymer starts to soften and lose its mechanical rigidity, and hence this value normally sets the upper use temperature in a given application. As seen in Fig. 5a, PC-h showed Tg = 100 °C, which is in range with polystyrene.40 As expected, the DSC traces did not show any crystallization or melt transitions. Hence, PC-h is a fully amorphous polymer that will retain mechanical rigidity at temperatures approaching 100 °C. Poly(trimethylene carbonate) and poly(propylene carbonate) have Tg values at approximately −17 °C41 and 30 °C,42 respectively, which demonstrates that the incorporation of the tetracyclic spiro-diol 3 has the potential to significantly increase the Tg of aliphatic PCs. The high Tg of PC-h may open up for, e.g., “hot-fill” applications in food packaging and handling. The value may further be compared to the semicrystalline PET (Tg = 75 °C),43 as well as with PC based on bisphenol A (Tg ∼150 °C),44 and the Akestra™ family of polyesters (Tg in the range 90–110 °C, depending on monomer content).26 The DSC analysis of PC-l showed a Tg value of 85 °C, i.e., 15 °C below that of PC-h. This was anticipated because of the significantly lower Mn value of the former polymer (7.5 vs. 27.7 kg mol−1). The lower Tg and Mw values suggest potential applications as, e.g., specialty polyols in the preparation of polyurethanes.
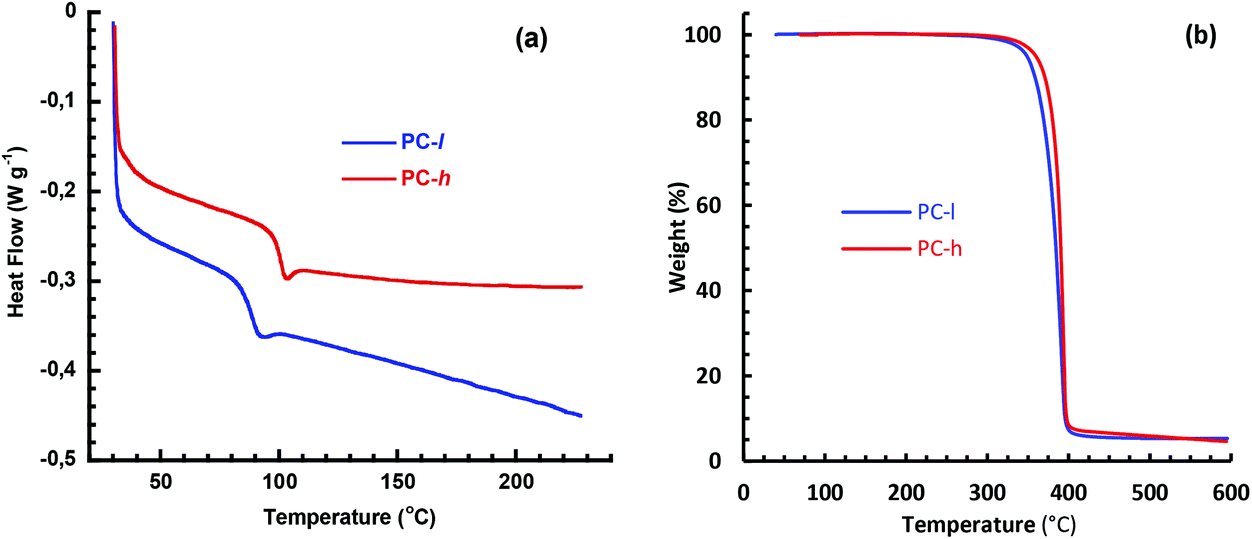 | ||
| Fig. 5 DSC (a) and TGA (b) traces of PC-h and -l, respectively. | ||
The Td,95 value measured by TGA analysis typically determines the maximum melt processing temperature of a polymer. Both PC-h and -l show thermal decomposition in a single step at Td,95 ∼350 °C (Fig. 5b). This indicated that the thermal stability of these materials is sufficiently high to allow melt processing which (depending on method, MW, etc.) is typically performed ca. 50–100 °C above Tg. In order to study the long-term stability, isothermal TGA measurements were carried out at 170 and 200 °C, respectively, during 8 h with the samples in the melt state. As seen in Fig. 6, the two PC samples showed different behavior under these conditions. At 170 °C, PC-l showed an initial weight loss of just above 2 wt%, before reaching a plateau. At 200 °C the initial weight loss had the same magnitude, but was followed by a constant weight loss (decomposition) rate of ∼0.30 wt% per h. The initial weight loss may be due to the presence residual solvent or monomer. For PC-h, the weight slowly increased by ∼0.8 wt% and then seemed to reach a plateau value. This increase was most probably due to limited oxidation of the sample. In contrast, the same sample displayed a steady weight loss rate of ∼0.15 wt% per h at 200 °C, i.e., half that of PC-l.
 | ||
| Fig. 6 Isothermal TGA measurements of PC-l and -h at 170 and 200 °C. | ||
Dynamic mechanical characterization
The dynamic mechanical properties of the PCs were studied by DMA analysis of hot-pressed sample bars at 1 Hz in the linear viscoelastic region (tensile strain 0.02%). Fig. 7 shows the tensile storage (E′) and loss (E′′) moduli of the two PC samples during heating at a rate of 2 °C min−1. As can be seen, the E′ value on the glassy plateau was higher for PC-h because of its higher Mn value compared to PC-l. Consequently, PC-h had an E′ value of 0.90 GPa at 90 °C, while PC-l had an E′ value of 0.42 GPa at 60 °C. The decrease in E′ marked the start of the glass transitions of the two PCs. Notably, the transition occurred in a broader temperature range for PC-l than for PC-h, which may be a consequence of the broader Đ value and hence larger heterogeneity of the former sample. After the transition region, i.e., just before entering the rubbery plateau, the samples deformed too much to continue the measurements. The Tg was indicated by the maximum in E′′ and showed Tg = 85 and 100 °C for PC-l and PC-h, respectively. These values were essentially the same as those evaluated by DSC, which demonstrated a very good correlation between the two methods.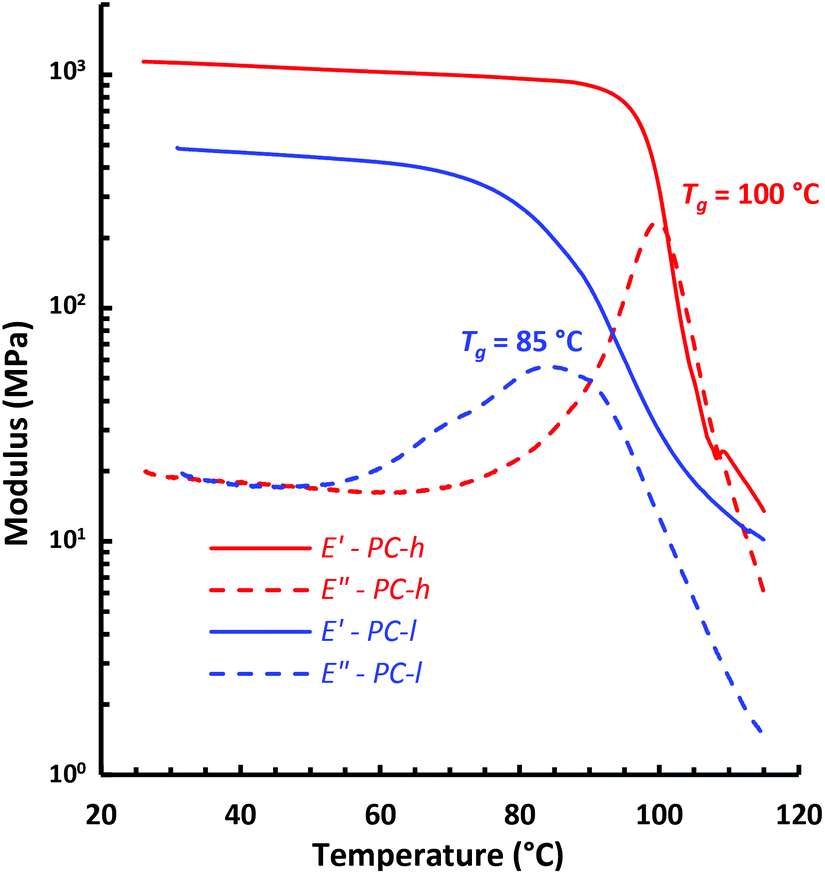 | ||
| Fig. 7 Tensile storage (E′) and loss (E′′) moduli of PC-l and -h, as measured by DMA at 1 Hz, 0.02% strain. | ||
Dynamic melt rheology
Dynamic rheology measurements were primarily carried out to assess the stability and processability of the PCs in the melt state. Rheological properties are related to MW and structure, and changes in the modulus may thus indicate degradation by, e.g., polymer chain scission or crosslinking reactions.45 The data of the present polymers was measured by using hot-pressed sample discs in a parallel plate arrangement. Time sweeps were carried out during 20 min at a frequency of 1 Hz at 0.2% strain, which was within the linear viscoelastic region.Fig. 8a shows the melt shear storage modulus (G′) and phase shift (δ) of PC-h as a function of time at 170 and 200 °C, respectively. As can be seen, the data at 170 °C were constant over time with G′ and δ equal to 42 kPa and 43°, respectively. This indicated a high stability of the sample without apparent signs of any polymer chain degradation. Initially, the measurement at 200 °C showed a decrease in G′ and an increase in δ, in relation to the data at 170 °C. This was consistent with the expected decrease in viscosity and less elastic response of the melt as T − Tg increased. In sharp contrast to the data at 170 °C, there was a clear decrease in G′ and a corresponding increase in δ observed over time at 200 °C. These changes towards a less elastic material are consistent with a reduction in MW over time. Hence, the data in Fig. 8a implies that PC-h is processable in the melt at 170 °C, but starts to degrade by chain scission reactions when the temperature approaches 200 °C.
 | ||
| Fig. 8 Variation of the melt shear storage modulus (G′) and phase shift (δ) during time sweeps of PC-h at 170 and 200 °C (a) and PC-l at 170 °C (b), as measured by rheology at 1 Hz, 0.2% strain (note the different scales for G′). | ||
Fig. 8b shows the corresponding values of G′ and δ of PC-l as a function of time at 170 °C. Compared to the data of PC-h, the initially measured value of G′ was lower and the δ value higher. This was consistent with the lower MW of PC-l, resulting in a less elastic response. Over time, the data of PC-l showed a clear increase in G′ and a corresponding decrease in δ. These trends towards a more elastic material are consistent with the occurrence of crosslinking reactions. In conclusion, chain scission dominated the degradation of the high-MW PC-h at high temperature, while the lower MW PC-l (with a higher concentration of chain ends) instead degraded by crosslinking. However, at 170 °C the PC-h melt showed a very stable rheological behavior without any signs of degradation. This temperature was 70 °C above the Tg and 180 °C below the Td,95, which indicates a good processability of PC-h. Fig. 9 shows a frequency sweep in the range 0.01–100 Hz on PC-h at 170 °C. At low frequency, the shear loss modulus (G′′) dominated in the viscous melt region. However, at approx. 0.6 Hz there was a crossover, and G′ instead started to dominate as the sample approached the rubbery plateau.
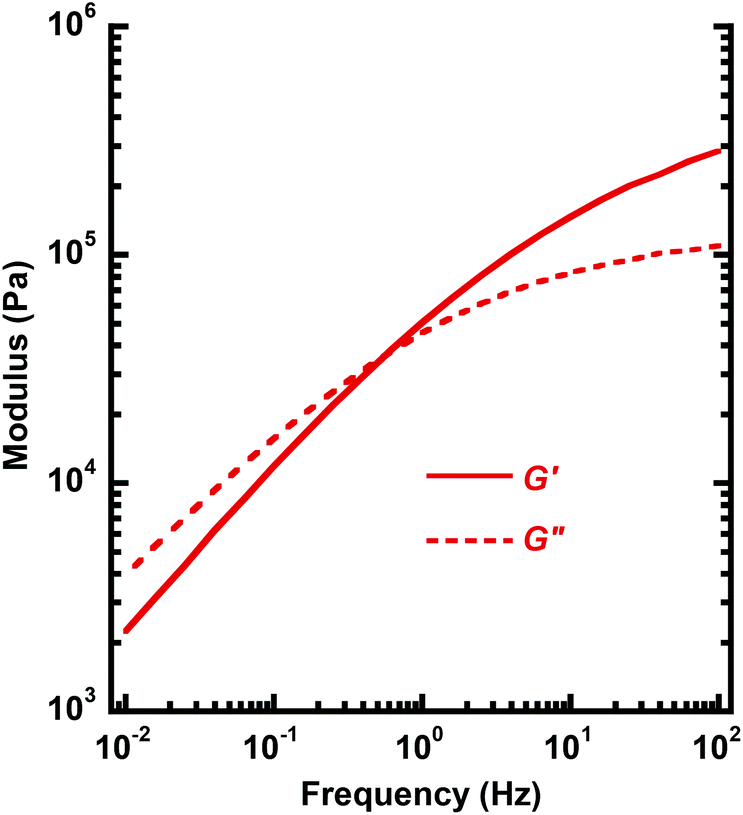 | ||
| Fig. 9 Melt shear storage (G′) and loss (G′′) modulus of PC-h as a function of frequency at 170 °C (0.2% strain). | ||
Hydrolytic stability study
The current PCs contain potentially sensitive ketal groups which motivated a preliminary investigation of the hydrolytic stability. Ketal groups are typically quite stable under basic conditions but tend to degrade at low pH.22 Sample pieces of PC-h were immersed in aqueous solutions with pH = 3 and pH = 8, respectively, at 37 °C during 18 days. After the immersion, the samples were dried, weighed, and analyzed by SEC to evaluate any decrease in MW. However, no changes in sample weight, Mn or Đ were detected. Consequently, the ketal-containing PC-h was seemingly stable under these conditions, at least partly because of its high Tg. The susceptibility of ketal and acetal groups in polymers to hydrolyze varies and depends on the precise molecular structure.22Experimental section
Materials
All reagents and solvents were obtained from commercial sources and used without further purification. Diketone 1 was synthesized from commercial DAC (purity 97%) according to the published procedure.46 TMP and DPC were obtained from Acros and had 98% and 99% purity grades, respectively. Reactions were monitored by thin-layer chromatography (TLC, Alugram Xtra SIL G/UV254), and TLC plates were visualized by staining with vanilla or phosphomolybdic acid solution. Silica gel 60 (0.040–0.063 mm, 230–400 mesh) was used in the flash chromatography.Synthesis of spiro-diol 3
A stirred mixture of bicyclo[3.3.0]octane-3,7-dione 1 (5.39 g, 39.1 mmol), trimethylolpropane (11.53 g, 85.9 mmol, 2.2 eq.), p-toluenesulfonic acid monohydrate (74 mg, 0.39 mmol), toluene (50 mL), and cyclohexane (75 mL) was refluxed for 16 h in a 250 mL round-bottom flask equipped with a Dean-Stark water trap. After cooling the reaction mixture to room temperature, the solvent was removed using a rotary evaporator. The crude product was then crystallized from the mixture of EtOAc and hexane to yield 12.26 g (85%) of diol 3 containing some unreacted TMP. This sample was dissolved in EtOAc (200 mL), washed with water (2 × 50 mL) and brine (30 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. The second crystallization yielded 10.64 g (74%) of spirodiol 3 as a white crystalline compound.Following an alternative procedure, diol 3 was isolated by chromatography. A stirred mixture of bicyclo[3.3.0]octane-3,7-dione 1 (3.70 g, 26.8 mmol), TMP (12.57 g, 93.8 mmol, 3.5 eq.), p-toluenesulfonic acid monohydrate (0.05 g), DMF (15 mL), and cyclohexane (110 mL) was refluxed for 8 h in a 250 mL round-bottom flask equipped with a Dean-Stark water trap. After cooling the reaction mixture to room temperature, NaHCO3 (1.0 g) was added and the solvent was removed by using a rotary evaporator. Next, toluene (100 mL) was added and the solid material was filtered off. The mixture was concentrated once more to remove DMF together with toluene. The residue was dissolved in small amount of DCM before loading on the top of the flash chromatography column. A gradual elution system (20–100% EtOAc in petrol ether) was used to afford 9.05 g (91%) of diol 3 (fractions of 3-trans and 3-cis were combined). The same elution system was used to separate the isomers. 3-Trans eluted faster [Rf = 0.56 (70% EtOAc in petrol ether)] than 3-cis [Rf = 0.39 (70% EtOAc in petrol ether)]. The assigned NMR data on the 3-trans and 3-cis isomers is presented in Table S1.†
For the HRMS, IR and Mp measurements, a mixture of cis and trans isomers were used.
HRMS (ESI): calcd for C20H34O6 [M + Na]+ 393.2248, found 393.2249.
IR (ATR) νmax (cm−1): 3433, 2966, 2862, 1471, 1313, 1134, 1112, 1043, 879, 758.
Mp = 102–103 °C.
Polycondensations
The PCs were synthesized by melt polycondensations using a Büchi Kugelrohr device with Glass Oven B-585 using a round-bottom tube (25 × 140 mm) as a reaction vessel. Representative procedures for the preparation of PC-h and PC-l are given below.PC -h: Spiro-diol 3 (2.017 g, 5.44 mmol), diphenylcarbonate (1.283 g, 5.99 mmol, 1.1 eq.) and NaHCO3 (11 mg, 0.6 mol%) were weighed and placed in the reaction tube. The reaction tube was placed into the Büchi apparatus and the mixture was melted at 115 °C at a rotation rate of 30 rpm. Subsequently the flask was degassed under vacuum (100 mbar) and purged with argon three times before heating the reaction mixture. First, the tube was kept at 160 °C for 1.5 h at atmospheric pressure followed by 20 min at 200 mbar. Then the temperature was increased at constant speed to 200 °C during 1.5 h and kept for 45 min. Next, the pressure was reduced to 100 mbar and after 30 min the temperature was increased to 220 °C. During the next 2 h, the temperature was gradually increased to 260 °C, and the pressure reduced to 0.36 mbar. The rotation rate of the reaction vessel was reduced to 6 rpm as the reaction mixture became very viscous. In the final stage, the temperature was raised to 280 °C during 1 h. The product was then cooled down to room temperature, dissolved in chloroform (30 mL), and precipitated in ice cold methanol (350 mL). Finally, the precipitate was filtered and dried under vacuum during 40 h to afford 1.944 g of PC-h. The assigned NMR data of PC-h is presented in Table S1.†
PC -l: Spiro-diol 3 (2.205 g, 5.95 mmol), diphenylcarbonate (1.402 g, 6.55 mmol, 1.1 eq.) and NaHCO3 (8 mg) were weighed into the reaction vessel and placed into a Büchi oven. The reaction mixture was melted, degassed and kept under a blanket of argon at 120 °C. After that the reaction mixture was heated to 160 °C and kept for 2 h. During the second hour at 160 °C, moderate vacuum (50 mbar) was applied. Next, the temperature was gradually raised to 200 °C over 30 min while keeping the vacuum constant (50 mbar). Over the next hour, the temperature was increased to 210 °C and the pressure was gradually reduced to 0.3 mbar. After 15 min under these conditions (210 °C, 0.3 mbar), the reaction mixture was cooled, dissolved in chloroform (30 mL), whereafter the product was precipitated in cold methanol (350 mL). After filtration and drying 2.05 g of PC-l was obtained.
Structural characterization
The structure of monomers and polymers was determined by NMR spectroscopy using a Bruker 800 or 400 MHz spectrometer with the samples dissolved in chloroform-d. The 1H NMR and 13C NMR spectra were recorded at 800 or 400 MHz and 201 or 101 MHz, respectively. The chemical shifts were calibrated using residual solvent signals (for 1H, CDCl3: δ = 7.26 ppm, for 13C, CDCl3: δ = 77.0 ppm) and are given in ppm. The HRMS analysis of 3 was performed using a Thermo Electron LTQ Orbitrap XL analyzer and FTIR was recorded using Shimadzu IRAffinity-1 spectrophotometer. The MWs of the PCs were determined by size-exclusion chromatography (SEC) in CHCl3. The SEC setup included three Shodex columns coupled in series (KF-805, -804, and -802.5) placed in a Shimadzu CTO-20A prominence column oven, a Shimadzu RID-20A refractive index detector, with Shimadzu LabSolution software. All samples were run at 40 °C at an elution rate of 1 mL min−1. Calibration was done by using poly(ethylene oxide) standards (Mn = 3.86, 12.60, 49.64 and 96.1 kg mol−1).Thermogravimetry and calorimetry
Thermogravimetric analysis (TGA) was performed on a TA Instruments TGA Q500. Samples of 1–5 mg were heated to 600 °C at a rate of 10 °C min−1 under nitrogen flux (60 mL min−1). The thermal decomposition temperature (Td,95) was determined at 5% loss of the total weight.Differential scanning calorimetry (DSC) measurements were performed on TA Instruments DSC Q2000. Samples from 2–7 mg were first heated up to 150–250 °C (depending on their respective thermal decomposition temperature) at a rate of 10 °C min−1. They were kept at this temperature for 2 min, then cooled to −50 °C and kept isothermal for 2 min before being heated up again to the previous temperature, at the same heating rate.
Dynamic mechanical measurements
Dynamic mechanical analysis (DMA) was carried out on TA Instruments DMA Q800. Rectangular bars of PC-h and PC-l (dimensions: 35 × 5 × 1 mm3) were hot-pressed between two Teflon plates using a hydraulic press (Specac, GS15011) at 150 °C during 2 min, then cooled to 100 °C during 5 min, and finally cooled to room temperature. The samples were analyzed at a frequency of 1 Hz, over the temperature range between 25 to 150 °C at a heating rate of 2 °C min−1. The measurements were carried out in the linear viscoelastic region at a strain of 0.02%. The Tg value was determined at the maximum peak value of the loss modulus.Melt rheology
Dynamic rheology measurements were performed with TA Instruments Advanced Rheometer AR2000 ETC. The experiments were made using parallel plates (∅ = 25 mm). Discs of PC-h and PC-l (∅ = 25 mm, t = 1 mm) were hot-pressed as described above. A time sweep was carried out during 20 min at 170 and 200 °C, at a frequency of 1 Hz at 0.2% strain, which was within the linear viscoelastic region. Moreover, a frequency sweep was performed on PC-h at 170 °C, under a strain of 0.2% over the range 0.01–100 Hz.Measurements of optical transmission and gloss
Polymer optical transmission measurements were performed on a Perkin Elmer Lambda 40 UV/VIS Specrometer using 32 μm thick film of PC-h cast from CHCl3 solution. The gloss was measured using a Byk Gardner Spectro-guide at 60° angle. Two PC-h films of different thicknesses (cast from CHCl3) were used and the measurements were repeated three times.Conclusions
We have successfully developed a straightforward and readily upscalable synthetic pathway to a biobased alicyclic monomer using inexpensive starting materials. Hence, a bicyclic ketone derived from citric acid was reacted with two equivalents of trimethylolpropane in a ketalization reaction to form the spirocyclic diol in high yields. This product carried two primary hydroxyl groups and was obtained as an equimolar mixture of cis and trans isomers. In an initial evaluation of this spiro-diol, we developed and carried out melt polycondensations of the new monomer and diphenylcarbonate to produce rigid PCs. The results showed that products with high MWs and Tg values up to 100 °C were achieved. Still, further analysis indicated that the products were slightly branched. This hinted that a limited ring opening of the ketal units occurred during the polycondensations, which were carried out at temperatures up to 280 °C. After hot-pressing and solvent casting, the PC formed flexible and mechanically strong samples with high transparency and only slight coloration. Melt rheology implied that the polymer had good processability up to at least 170 °C, but started to degrade as the temperature approached 200 °C. Overall, the results indicate that the citric acid-based spiro-diol is an efficient building block for the preparation of high-Tg condensation polymers. We are currently investigating the use of the spiro-diol monomer to prepare polyesters, polyurethanes and polyacrylates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Regional Development Fund (project no. MOBTT21) and the Estonia-Russia Cross Border Cooperation Programme (ER30). O. B. is grateful to the Swedish Research Council Formas for financial support (diarienr 2016-00468), and P. J. acknowledges funding by the Swedish Foundation for Strategic Environmental Research through the “STEPS” project (no. 2016/1489). T. P. thanks The Estonian Ministry of Education and Research (grant no. IUT23-7) and the European Regional Development Fund (grant no. TK134). We are grateful to Jevgenija Martõnova and Prof. Riina Aav for obtaining single crystals for X-ray and this was supported by the Estonian Research Council (grant PRG339). Dr Janek Peterson (Akzo Nobel Baltics) is acknowledged for the gloss measurements. Sergo Kasvandik and Merilin Saarma are thanked for HRMS analyses, Dr Koit Herodses for the UV/Vis measurements and Livia Matt for preparing the SEC and NMR graphs.References
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- H. T. H. Nguyen, P. Qi, M. Rostagno, A. Feteha and S. A. Miller, J. Mater. Chem. A, 2018, 6, 9298–9331 RSC.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116(4), 2275–2306 CrossRef CAS PubMed.
- A. L. Holmberg, K. H. Reno, N. A. Nguyen, R. P. Wool and T. H. Epps III, ACS Macro Lett., 2016, 5(5), 574–578 CrossRef CAS PubMed.
- A. L. Holmberg, N. A. Nguyen, M. G. Karavolias, K. H. Reno, R. P. Wool and T. H. Epps III, Macromolecules, 2016, 49(4), 1286–1295 CrossRef CAS.
- L. Mialon, A. G. Pemba and S. A. Miller, Green Chem., 2010, 12, 1704–1706 RSC.
- J. Zhang, C. C. Pang and G. L. Wu, RSC Adv., 2016, 6, 11848 RSC.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Polym. Chem., 2015, 6, 6058–6066 RSC.
- D. J. Saxon, A. M. Luke, H. Sajjad, W. B. Tolman and T. M. Reineke, Prog. Polym. Sci., 2020, 101, 101196 CrossRef CAS.
- C. Dussenne, T. Delaunay, V. Wiatz, H. Wyart, I. Suisse and M. Sauthier, Green Chem., 2017, 19, 5332–5344 RSC.
- F. Delbecq, M. R. Khodadadi, D. Rodriguez Padron, R. Varma and C. Len, Mol. Catal., 2020, 482, 110648 CrossRef CAS.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J. P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS.
- B. A. J. Noordover, V. G. van Staalduinen, R. Duchateau, C. E. Koning, B. van, M. Mak, A. Heise, A. E. Frissen and J. van Haveren, Biomacromolecules, 2006, 7, 3406–3416 CrossRef CAS PubMed.
- J. Wu, S. Thiyagarajan, C. Fonseca Guerra, P. Eduard, M. Lutz, B. A. J. Noordover, C. E. Koning and D. S. van Es, ChemSusChem, 2017, 10, 3202–3211 CrossRef CAS PubMed.
- P. Villo, L. Matt, L. Toom, I. Liblikas, T. Pehk and L. Vares, J. Org. Chem., 2016, 81, 7510–7517 CrossRef CAS PubMed.
- W. Qian, X. Tan, Q. Su, W. Cheng, F. Xu, L. Dong and S. Zhang, ChemSusChem, 2019, 12, 1169–1178 CrossRef CAS PubMed.
- X. Liu, C. Pang, J. Ma and H. Gao, Macromolecules, 2017, 50, 7949–7958 CrossRef CAS.
- C. Pang, X. Jiang, Y. Yu, X. Liu, J. Lian, J. Ma and H. Gao, ACS Sustainable Chem. Eng., 2018, 6, 17059–17067 CrossRef CAS.
- A. Hufendiek, S. Lingier and F. E. Du Prez, Polym. Chem., 2019, 10, 9–33 RSC.
- E. H. Cordes and H. G. Bull, Chem. Rev., 1974, 74, 581–603 CrossRef CAS.
- S. Lingier, Y. Spiesschaert, B. Dhanis, S. De Wildeman and F. E. Du Prez, Macromolecules, 2017, 50, 5346–5352 CrossRef CAS.
- G.-H. Choi, D. Y. Hwang and D. H. Suh, Macromolecules, 2015, 48, 6839–6845 CrossRef CAS.
- S. V. Mankar, M. N. Garcia Gonzalez, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 19090–19103 CrossRef CAS.
- Akestra™ by Perstorp AB, CAS number: 102070-64-4, https://www.perstorp.com/en/products/akestra_100.
- N. Warlin, M. N. Garcia Gonzalez, S. Mankar, N. G. Valsange, M. Sayed, S.-H. Pyo, N. Rehnberg, S. Lundmark, R. Hatti-Kaul, P. Jannasch and B. Zhang, Green Chem., 2019, 21, 6667–6684 RSC.
- S. Makhseed and N. B. McKeown, Chem. Commun., 1999, 255–256 RSC.
- S. H. Bertz, J. M. Cook, A. Gawish and U. Weiss, Org. Synth., 1986, 64, 27–38 CrossRef CAS.
- G. Mattioda and A. Blanc, Ullmann's Encyclopedia of Industrial Chemistry, 2011, DOI:10.1002/14356007.a12_491.pub2.
- https://www.avantium.com/technologies/ray/ .
- R. Adams, H. M. Chiles and C. F. Rassweiler, Org. Synth., 1925, 5, 5 CrossRef.
- E. Jakfalvi, B. L. Gregorne, T. Morasz, G. Toth, A. Szabo, J. Toereki and S. Olah, WO2004089867A2, 2004.
- Y. Chen, J. Chen and L. Heng, CN103288629A, 2013.
- C. R. Soccol, L. P. S. Vandenberghe, C. Rodrigues and A. Pandey, Food Technol. Biotechnol., 2006, 44, 141–149 CAS.
- Perstorp AB offers TMP in 20 and 50% renewable grade (Evyron™). ( https://www.perstorp.com/en/products/proenvironment_polyols/evyron/).
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- B. Liu and S. Thayumanavan, J. Am. Chem. Soc., 2017, 139, 2306–2317 CrossRef CAS PubMed.
- P. J. Flory, Principles of polymer chemistry, Cornell University Press, San Diego, 1953 Search PubMed.
- J. Maul, B. G. Frushour, J. R. Kontoff, H. Eichenauer, K.-H. Ott and C. Schade, Ullmann's Encyclopedia of Industrial Chemistry, 2007, DOI:10.1002/14356007.a21_615.pub2.
- A. P. Pêgo, A. A. Poot, D. W. Grijpma and J. Feijen, J. Mater. Sci.: Mater. Med., 2003, 14, 767–773 CrossRef PubMed.
- A. Yoshida, S. Honda, H. Goto and H. Sugimoto, Polym. Chem., 2014, 5, 1883–1890 RSC.
- E. Gubbels, T. Heitz, M. Yamamoto, V. Chilekar, S. Zarbakhsh, M. Gepraegs, H. Köpnick, M. Schmidt, W. Brügging, J. Rüter and W. Kaminsky, Ullmann's Encyclopedia of Industrial Chemistry, 2018, DOI:10.1002/14356007.a21_227.pub2.
- G. Abts, T. Eckel and R. Wehrmann, Ullmann's Encyclopedia of Industrial Chemistry, 2014, DOI:10.1002/14356007.a21_207.pub2.
- G. Filippone, S. C. Carroccio, R. Mendichi, L. Gioiella, N. Tz. Dintcheva and C. Gambarotti, Polymer, 2015, 72, 134–141 CrossRef CAS.
- J. A. Cadieux, D. J. Buller and P. D. Wilson, Org. Lett., 2003, 5, 3983–3986 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1986990. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0gc00849d |
| This journal is © The Royal Society of Chemistry 2020 |