DOI:
10.1039/D0GC00820F
(Paper)
Green Chem., 2020,
22, 3809-3819
Continuous hydroformylation of 1-decene in an aqueous biphasic system enabled by methylated cyclodextrins†
Received
6th March 2020
, Accepted 9th April 2020
First published on 14th April 2020
Abstract
For the first time, randomly methylated β-cyclodextrin was applied as the mass transfer agent in a continuous process. Considering the example of the Rh-catalyzed hydroformylation of 1-decene, process development was shown, where cyclodextrin was used together with a catalyst system that was continuously recovered and recycled using an aqueous biphasic system. In initial experiments, water-soluble and commercially available Rh/TPPTS and Rh/sulfoxantphos catalyst systems were scaled up from 50 ml into 1000 ml high-pressure autoclave systems to demonstrate their scalability. Both these systems were compared, and they afforded excellent chemoselectivity (>99%) toward the desired linear aldehyde product. In particular, higher regioselectivity (up to 31) was achieved for the Rh/sulfoxantphos system. Investigations regarding the long-term stability of the mass transfer agent and both catalyst systems were carried out in a continuously operated miniplant process. It was shown that the process could be successfully operated under the steady state for over 200 h with chemoselectivity of >97% toward the desired aldehyde product. Simultaneously, extremely low Rh leaching (total: 0.59%) was observed over the entire period of 200 h.
1. Introduction
Hydroformylation is widely used in industry to manufacture high-value-added aldehydes and alcohols, and it is considered to be one of the largest homogenously catalyzed reactions in industry. A major issue persisting with the use of homogenous catalysts is their recovery and separation from the product mixture, particularly for higher olefins; this is important due to the cost of the employed transition metals. These days, distillation has become the most commonly used separation method in the hydroformylation of alkenes and therefore the most well-known method.1 However, distillation conditions that are required to separate high-boiling products may lead to catalyst decomposition along with concomitant loss of metal. Hence, cheaper cobalt catalysts are used, ensuring that the processes remain economically viable.2 However, cobalt catalysts are less active than rhodium catalysts, requiring higher reaction temperatures and pressures.3 Therefore, the challenge involved in formulating techniques to convert higher olefins in hydroformylation persist, particularly in a manner that can address the separation problem and minimize environmental impact, while not compromising on catalyst selectivity and activity. Some concepts focused on this involve the intensification of the monophasic flow process for hydroformylation.4 Other approaches have been developed in both industry and academia to tackle the main issue of catalyst separation from the product by using rhodium catalysts. Examples of these are the use of supercritical CO2 (scCO2),5 CO2-expanded liquid (CXL),6,7 supported ionic liquid phase (SILP),7 organic solvent nanofiltration (OSN),8 thermomorphic multiphase systems (TMS),9 microemulsions,10 or biphasic systems.11 Some of them use abundant resources (such as carbon dioxide and the environmentally friendly solvent, water), but others do not. The use of water as a solvent for process applications is particularly beneficial from the economic and environmental impact viewpoints since water is fairly accessible, nontoxic, nonflammable, odorless, and has a high heat capacity and heat of vaporization.12 Nevertheless, these benefits have to compete with its intrinsic limitation of low organic substrate solubility in the aqueous catalyst phase, which have also been confirmed by early kinetic studies.13,14 Therefore, several approaches have been developed to tackle the issue of low space–time yield in biphasic aqueous–organic reaction systems involving poorly water-soluble substrates, namely, by using surfactants, cosolvents, thermoregulated ligands, and cyclodextrins (CDs) to achieve improved hydroformylation, as summarized in the review article by Matsinha et al.14
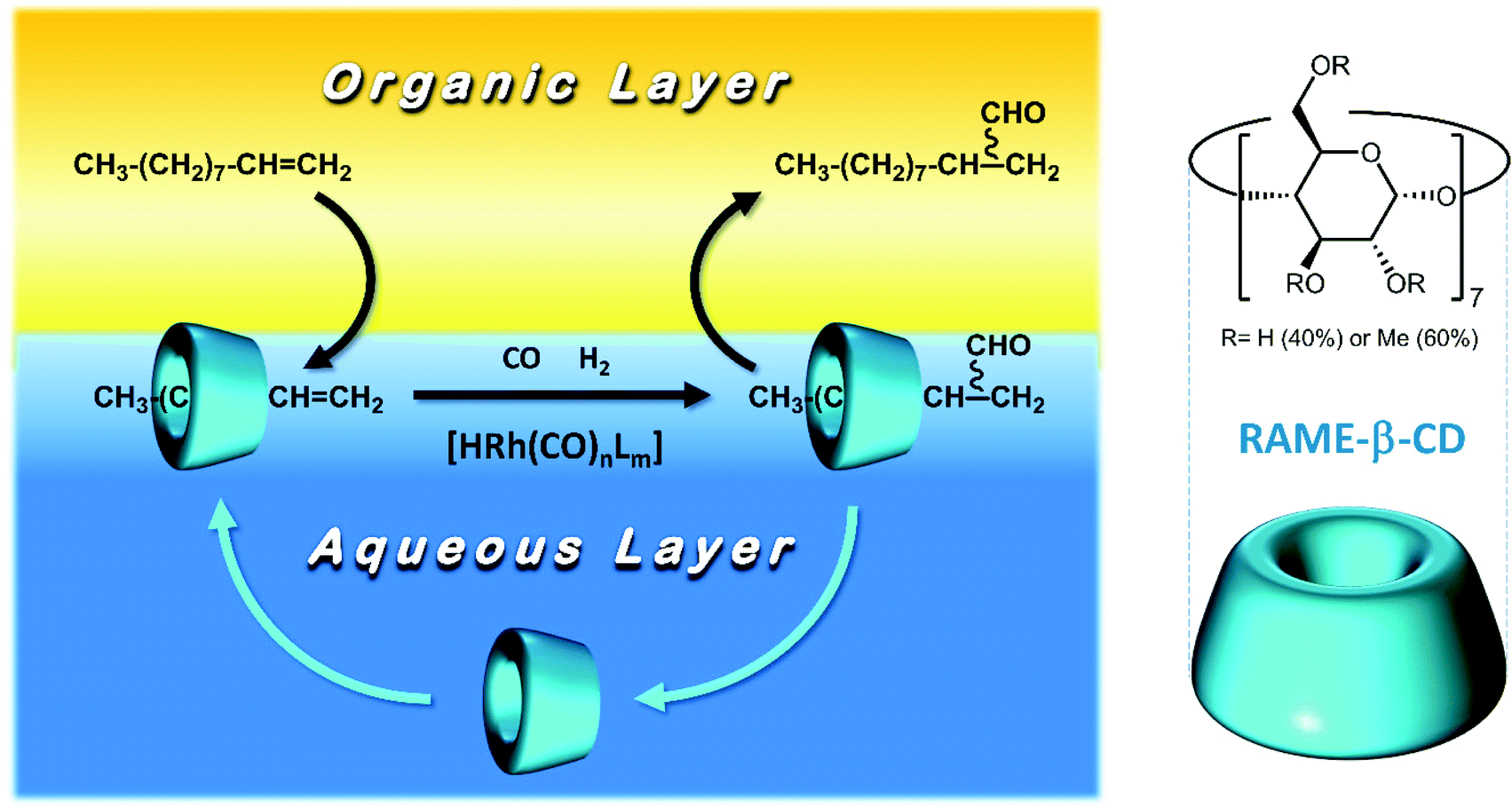
From this perspective, this work further focuses on the use of CDs as the mass transfer agent to overcome mass transfer limitations in the biphasic hydroformylation of 1-decene. CDs are cyclic oligosaccharides comprising six (α-CD), seven (β-CD), or eight (γ-CD) α-D-glucopyranose units.15 They form conical cylinders with the outer hydrophilic surface and inner hydrophobic surface. The inner surface forms a cavity and allows for the formation of an inclusion complex when it binds with a hydrophobic substrate (Fig. 1).16 To vary their size and shape, native CDs can be modified by substituting their hydroxyl groups with various other functional groups. These are referred to as mono- or polysubstituted CDs. Due to their ability to form inclusion complexes with a wide range of compounds, native and modified CDs have applications in various fields.17 In the field of aqueous biphasic organometallic catalysis, CDs have mainly been employed as mass transfer agents16,18 in order to increase the solubility of hydrophobic substrates in water. For example, in 1995, Monflier et al.16,19–22 described rhodium-catalyzed hydroformylation in an aqueous/organic two-phase system of 1-decene in the presence of randomly methylated β-CD (RAME-β-CD; Fig. 1).21,23 The phase separation between the organic and aqueous phases was described to be fast; further, the rhodium and phosphorus contents in the organic phase were found to be less than 0.5 and 1.2 ppm, respectively. Moreover, in 1999, Tilloy et al. proposed the reusability of the Rh/TPPTS/RAME-β-CD catalytic system by performing the rhodium-catalyzed hydroformylation of 1-decene with 5 catalytic recycle runs without any loss of activity.24 Later, in 2004, Leclercq et al. investigated the same reaction using the sulfoxantphos ligand, which led to improved performances in terms of chemoselectivity and regioselectivity.20 Although CDs are highly promising as mass transfer agents in aqueous biphasic systems for the selective hydroformylation of higher olefins, they have never been applied in a continuously operated process. As our research group has gained expertise in various continuously operated recycling strategies for homogeneous catalysts with regard to hydroformylation25,26 in earlier years, we combined our knowledge with the group of Monflier to understand if CDs can be employed for such applications. Therefore, in this work, we transfer two catalytic systems (Rh/TPPTS/RAME-β-CD and Rh/sulfoxantphos/RAME-β-CD) into one of our continuously operated miniplants for the purpose of aqueous biphasic hydroformylation. Since this has never been attempted before, both these systems were first scaled up in differently sized batch autoclaves in order to understand their performance in terms of chemo-/regioselectivity and conversion. Further, their reaction profile was determined to be the benchmark for the residence time in the subsequent continuous process. During continuous operations, the influence of various parameters (such as CD concentration and stirrer speed) on the catalytic activity was determined. Eventually, the effective recycling of the catalytic systems (supported by leaching data) with the optimized parameters was conducted via a long-term experiment, revealing the potential of CDs for such applications.
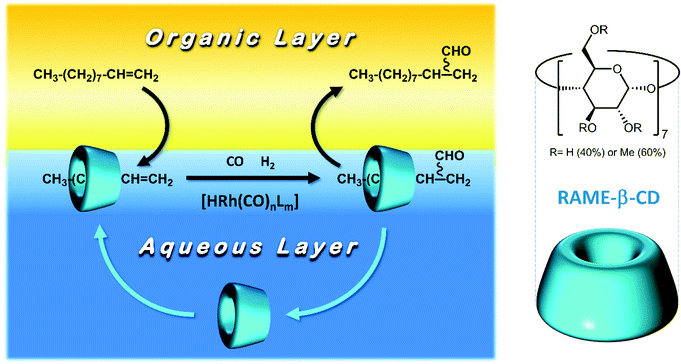 |
| | Fig. 1 Principle of aqueous biphasic organometallic catalysis mediated by RAME-β-CD. | |
2. Results and discussion
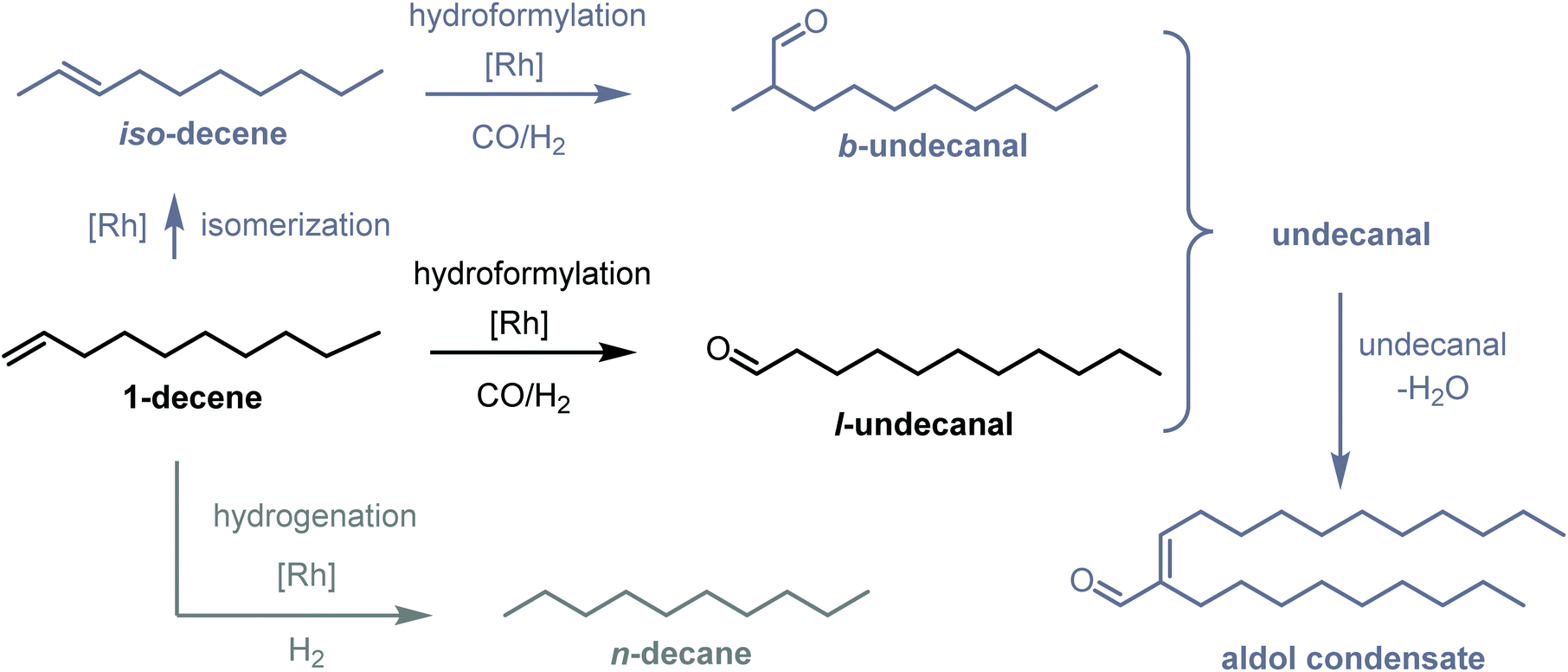
Based on the studies by Monflier et al.,16,19–22 1-decene was chosen as the model substrate for CD-based hydroformylation. The reaction network is shown in Fig. 2. The main product from the hydroformylation is undecanal, which is referred to as l-undecanal (linear undecanal). 1-Decene isomers may occur as byproducts and are referred to as iso-decene (isomerized decene). The other byproducts are n-decane (which results from the hydrogenation of 1-decene or iso-decene) and the branched hydroformylation product, namely, 2-methyldecanal (which can arise from the hydroformylation of 1-decene or iso-decene). Further, 2-methyldecanal is the representative of all the branched aldehydes since it is formed as a byproduct in the highest amount. These branched aldehydes are categorized as b-undecanal (branched undecanal). All the undecanal isomers are categorized into undecanal (l-undecanal + b-undecanal). The remaining byproducts are aldol condensates, which can result from the condensation reaction of two aldehydes. All these products are summarized, and 2-nonyl-tridec-2-enal is considered to be the representative of the aldol condensates.
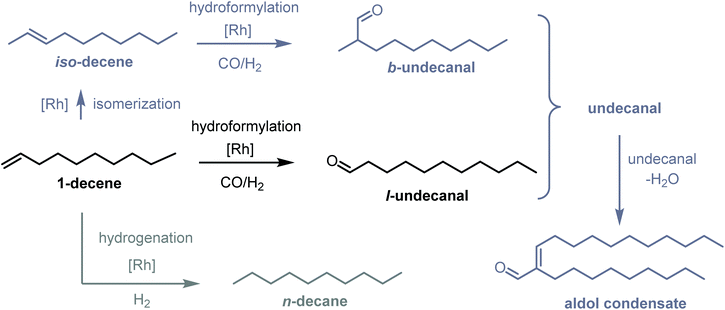 |
| | Fig. 2 Reaction network of 1-decene hydroformylation and typical byproducts. | |
In this work, two catalyst systems have been investigated, since they yielded worthwhile results in earlier publications with regard to high product chemo-/regioselectivity. The already published results are listed in Table 2.18,20,24 Both these systems use water as the solvent, Rh(acac)(CO)2 as the precursor, and RAME-β-CDs (Fig. 1) as the mass transfer agent. The only difference is the choice of phosphine ligand, namely, monodentate TPPTS and bidentate sulfoxantphos.
2.1. Scaled up batch experiments
Scaled up experiments were first performed since the original reactions were carried out in 50 ml high-pressure autoclaves. Therefore, two types of high-pressure autoclaves were used for the experiments (Fig. 3). A high-pressure autoclave with a total volume of 300 ml was used as the first reference as well as for the scaling up tests. For a facilitating comparability between the experiments, the gas-to-liquid volume ratio was maintained as per the original experiments performed by Monflier et al.19 and Leclercq et al.,20 yielding a volume of 100 ml for the reaction solution. In this work, this reactor is also referred to as the 300 ml high-pressure autoclave. Another reactor is built into the continuous miniplant process and was used in the batch or continuous mode. This reactor had a total volume of 1000 ml and is correspondingly referred to subsequently. To maintain the gas-to-liquid volume ratio constant, the liquid volume was set to 330 ml.
 |
| | Fig. 3 Scaled up reactors used in this work. | |
2.1.1. Scaled up Rh/TPPTS catalyst systems.
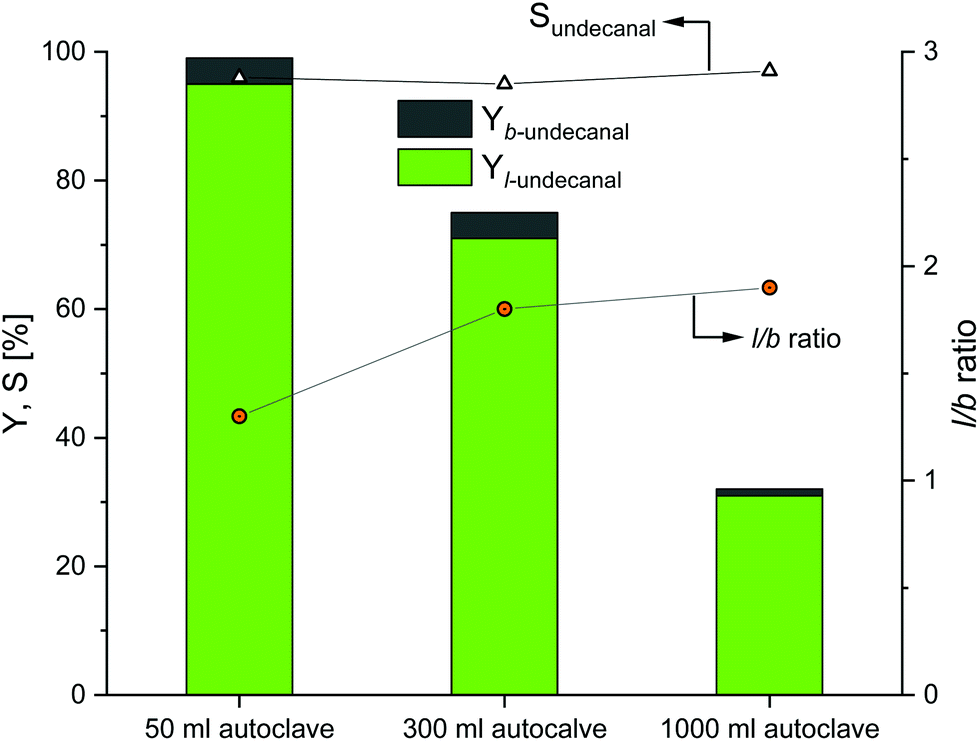
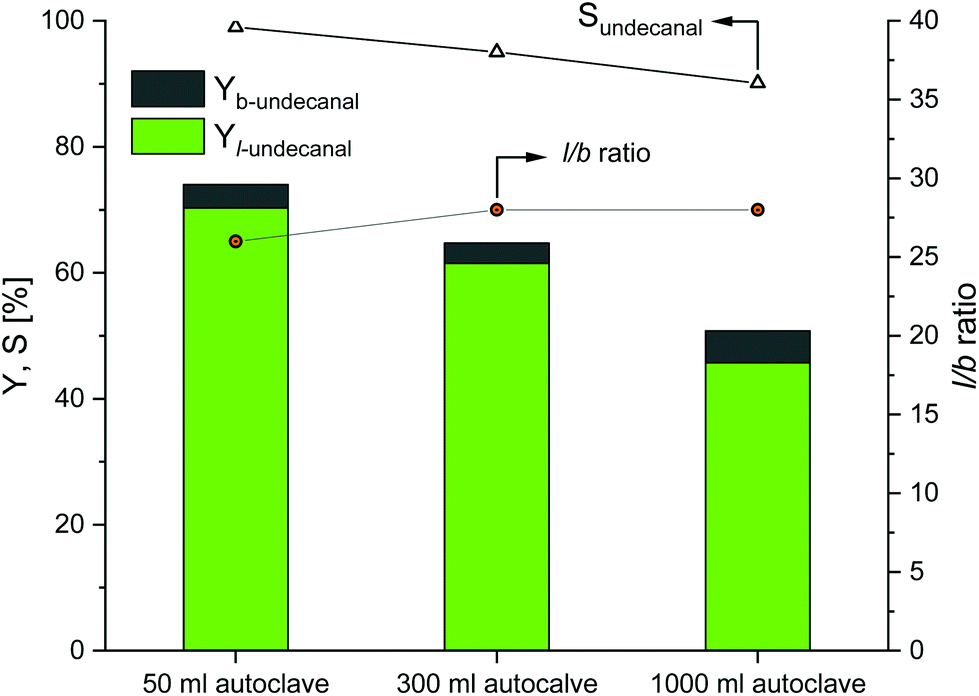
The first scaling up experiments were conducted for the Rh/TPPTS catalyst system. Based on the reaction data from the original experiments (Table 1), the Rh(acac)(CO)2 precursor and TPPTS ligand were used to convert 1-decene into undecanal. The reaction temperature was set to 80 °C at a phosphorus-to-rhodium ratio (nP/nRh) of 5 and CD-to-rhodium ratio (nCD/nRh) of 12. The reaction time was set to 6 h, since this is the maximum possible residence time in the reactor for the continuous process at the rhodium-to-substrate ratio of 496. Before the start of the reaction, preforming was conducted for 2 h. In the original experiments, pressure of 50 bar was used for the reactions to ensure sufficient gas solubility and to preclude the limitation of the reaction. Due to apparatus technology, only pressures of 40 bar could be used for the 300 and 1000 ml autoclaves. However, Mathivet et al. mentioned that reaction pressure of 10 bar was sufficient; any further increase in pressure only had a minor impact.27 The stirrer speed was set to 800 min−1 for the scaling up experiment. The results are shown in Fig. 4.
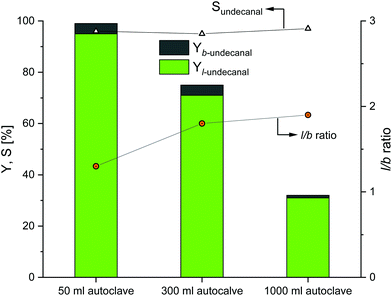 |
| | Fig. 4 Results of the CD-mediated aqueous biphasic hydroformylation of 1-decene using TPPTS depending on the reactor size. Here, the 50 ml autoclave data were adapted from Hapiot et al.21 Conditions: 1. 50 ml autoclave:21 Rh(acac)(CO)2: 0.041 mmol, TPPTS: 0.21 mmol, CDs: 0.48 mmol, water: 11.5 ml, 1-decene: 20.35 mmol, T = 80 °C, p(CO/H2) = 50 bar, n = 1500 min−1, no preformation. 2. 300 and 1000 ml autoclaves: preforming: p(CO/H2) = 20 bar, nCO/nH2 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, T = 80 °C, t = 2 h; reaction: p(CO/H2) = 40 bar, nCO/nH2 = 1:1, T = 80 °C, t = 6 h, mwater/msubstrate = 4:1, nsubstrate/nRh = 496, nCD/nRh = 12, nP/nRh = 5, n = 800 min−1. :1, T = 80 °C, t = 2 h; reaction: p(CO/H2) = 40 bar, nCO/nH2 = 1:1, T = 80 °C, t = 6 h, mwater/msubstrate = 4:1, nsubstrate/nRh = 496, nCD/nRh = 12, nP/nRh = 5, n = 800 min−1. | |
Table 1 Comparison of aldehydes (chemo-)selectivity and linear-to-branched aldehyde (l/b) ratio (regioselectivity) for the rhodium-catalyzed hydroformylation of higher olefins by Rh/TPPTS and Rh/sulfoxantphos systems (adapted from Hapiot et al.21 and Leclercq et al.20)
| Entry |
Olefin |
CD |
Ligand |
Selectivityc [%] |
l/b |
| Experimental conditions: Rh(acac)(CO)2: 0.04 mmol; TPPTS/sulfoxantphos: 0.21 mmol; CD: 0.48 mmol; water: 11.5 ml; olefin: 20.35 mmol; n-undecanal (internal standard): 2.03 mmol; p(CO/H2, 1:1): 50 bar. T = 80 °C, t = 6 h. T = 120 °C, t = 24 h. Chemoselectivity toward undecanal. |
| 1a |
1-Decene |
RAME-β-CD |
TPPTS |
98 |
1.7 |
| 2b |
1-Decene |
RAME-β-CD |
Sulfoxantphos |
>99 |
26 |
Evidently, as the reactor size increased, the catalytic activity as compared to the original 50 ml experiments decreased. Nearly full conversion could be achieved in the 50 ml autoclaves as compared to the 300 ml autoclaves; the conversion decreases to 75% and eventually to 33% when compared to the 1000 ml autoclaves. The chemoselectivity of all the three experiments are in the same range (over 95%), which indicates that the Rh/TPPTS catalyst system works as expected for all the experiments. Further, with respect to regioselectivity, the l/b ratio reached a value close to 1.8 for all the experiments.
As these experiments were conducted to demonstrate the influences of different parameters as barriers for the potential transfer into a continuous process, the obtained results revealed that a transfer is generally possible when the selectivities remain constant. However, in a continuous process, lower conversions have to be expected due to the potential mass transfer limitations due to an increase in the size of the autoclaves. Since this is a three-phase gas–liquid–liquid system, there may be limitations at the gas–liquid phase interface and/or at the liquid–liquid phase interface, which is supported by the investigations of Sieffert et al.; they suggested that the reaction does not take place in the bulk water phase, but it occurs at the aqueous phase/interface layer.28 Therefore, there would be stronger dependence between the phase interface and reaction rate and consequently the conversion rate. With scaling up and an increase in volume, the amount of substance gets proportionally scaled, but the phase interface does not. The reaction profiles for the 300 and 1000 ml systems are provided in the ESI,† revealing the potential undecanal yield for different residence times in the miniplant reactor.
2.1.2. Scaled up Rh/sulfoxantphos catalyst system.
Scaled up experiments performed for Rh/sulfoxantphos catalyst systems are shown in Fig. 5. Reaction conditions do not drastically differ from the previous experiments: only the reaction temperature was increased from 80 °C to 120 °C and the preforming time was extended from 2 to 12 h. Similar to the results obtained from the Rh/TPPTS catalyst system, the conversion of 1-decene and undecanal yield generally decrease with an increase in the reactor size. The reaction profiles of the 300 and 1000 ml systems are provided in the ESI,† revealing the potential undecanal yield for different residence times in the miniplant reactor for continuous operations. However, the decrease in conversion is less as compared to that for the TPPTS system because of the surface properties of sulfoxantphos. Here, the conversion decreases from 71% (50 ml autoclave) to 51% (1000 ml autoclave). The undecanal yield analogously decreases with the conversion from 70% to 46% as the chemoselectivity values for all the experiments remain over 90%. Further, the regioselectivity (l/b ratio) of around 27 for all the experiments shows the high regioselectivity toward the linear product, i.e., aldehyde.
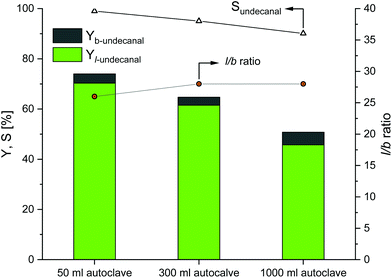 |
| | Fig. 5 Results of the CD-mediated aqueous biphasic hydroformylation of 1-decene using sulfoxantphos depending on the reactor size. Here, the 50 ml autoclave data were adapted from earlier studies.20 Conditions: 1. 50 ml autoclave:20 Rh(acac)(CO)2: 0.04 mmol, sulfoxantphos: 0.21 mmol, CD: 0.48 mmol, water: 11.5 ml, olefin: 20.35 mmol, n-undecanal (internal standard): 2.03 mmol, p(CO/H2) = 50 bar, T = 120 °C. 2. 300 ml autoclave: preforming: p(CO/H2) = 20 bar, nCO/nH2 = 1:1, T = 120 °C, t = 12 h; reaction: p(CO/H2) = 40 bar, nCO/nH2 = 1:1, T = 120 °C, t = 6 h, mw/mS = 4:1, nS/ncat = 496, nCD/ncat = 12, nP/ncat = 5, n = 800 min−1. 3. 1000 ml autoclave: preforming: p(CO/H2) = 20 bar, nCO/nH2 = 1:1, T = 120 °C, t = 12 h; reaction: p(CO/H2) = 40 bar, nCO/nH2 = 1:1, T = 120 °C, τ = 6 h, mw/mS = 4:1, cCat = 0.006 mol%, nCD/nCat = 12, n = 800 min−1. | |
2.2. Continuous miniplant experiments
The proofs of concept for the successful scaling up of the Rh/TPPTS and Rh/sulfoxantphos catalyst systems have been shown in earlier batch experiments. As regio- and chemoselectivity are nearly constant for these experiments, the catalyst systems are transferred for the first time into a continuous process (Fig. 10). In this study, the long-term stability of catalytic systems in combination with CD, as well as influences of parameters such as CD concentration and stirrer speed in the reactor, were investigated.
2.2.1. Variations in CD concentrations with TPPTS.
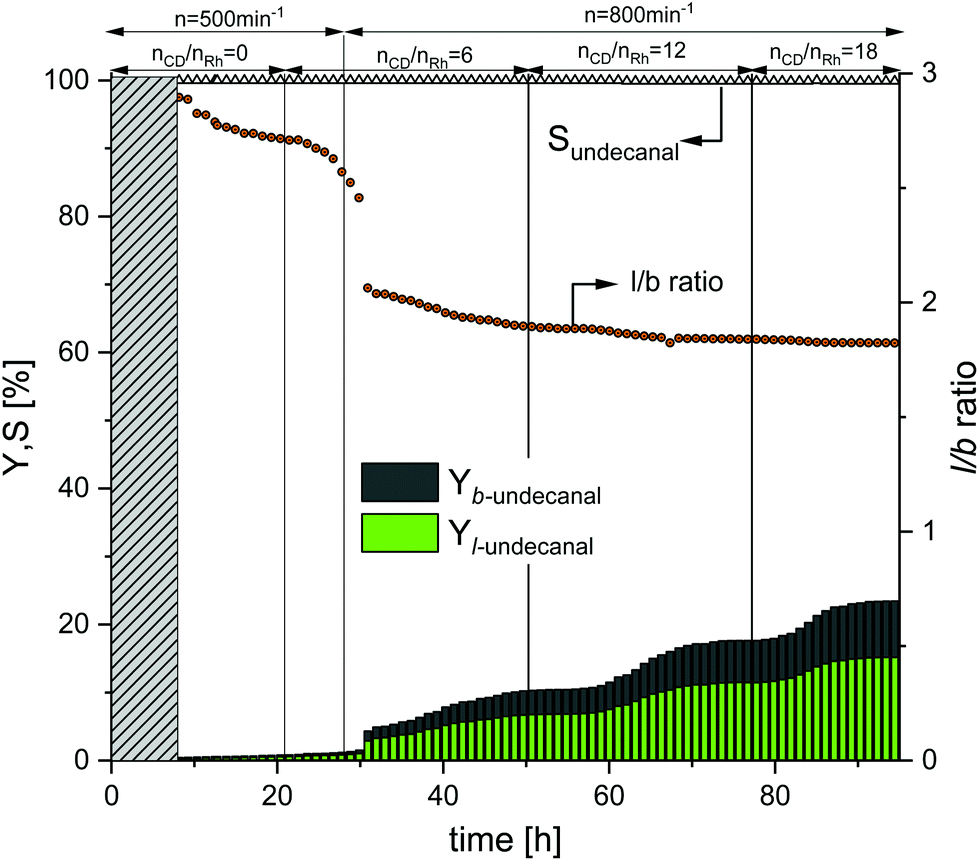
In the first continuous experiment, the Rh/TPPTS catalyst system was used to show the effect of CDs as a mass transfer agent during the reaction under biphasic conditions. The obtained results are shown in Fig. 6. The first 8 h are the startup time of the plant (shown with the grey bar). Initially, no CDs were added to the system that proved the poor reaction activity, similar to that with the aqueous biphasic mixture. As expected from hydroformylation experiments in aqueous biphasic systems as reported in the literature,29 the observable yield of undecanal (linear and branched) was low, under 0.5% after 21 h at a stirrer speed of 500 min−1. To increase the reaction performance, CDs were added to the system at a molar ratio of 6 with respect to the rhodium catalyst. After 29 h, the yield of undecanal increased only to 1%. Consequently, the stirrer speed was increased from 500 min−1 to 800 min−1 to increase the mixing in the reactor and potentially lowering the mass transfer limitations. Under these conditions, the yield of undecanal reached a steady value of 11.5% after 47 h. After 52 h, more CDs were added to the reactor. The molar ratio of CDs to rhodium increased to 12, which was used earlier in the batch experiments (Fig. 4 and 5). The yield increased after some induction time, which roughly equaled to the residence time; therefore, after 65 h, the undecanal yield was about 20%. As compared to the batch experiment in the 1000 ml autoclave (ESI†) under the same conditions, the undecanal yield was determined to be 10% less. This could be attributed to the recycling of the catalyst. To further increase the conversion rate, more CDs were added after 76 h, resulting in a molar ratio of CDs to rhodium of 18. The conversion increased further, reaching a steady state value of 32%.
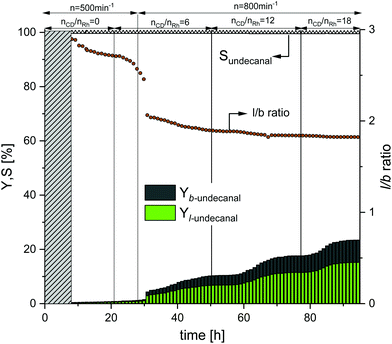 |
| | Fig. 6 Yield and selectivity over time for a continuous operation with variations in the CD concentration and stirrer speed when using TPPTS as the ligand. Conditions: Preforming: p(CO/H2) = 20 bar, nCO/nH2 = 1:1, T = 80 °C, t = 2 h; reaction: p(CO/H2) = 40 bar, nCO/nH2 = 1:1, T = 80 °C, τ = 6 h, mw/mS = 4:1, cCat = 0.0064 mol%, nCD/nCat = 12, nP/nCat = 5, ṁ1-decene = 10.2 g h−1, ṁRecycle = 41.1 g h−1. | |
Initially, the regioselectivity (l/b ratio) was ∼2.8. After the first addition of CDs, the ratio rapidly dropped to 2.0 and then slowly reached a value within 1.8–1.9, which was the ratio determined for TPPTS in the batch experiments. The further addition of CDs had no influence on the ratio. The chemoselectivity (∼99%) was calculated over the entire duration of the experiment and therefore was higher than that in the batch experiments (Fig. 4). In Table 2, the catalyst leaching results for this experiment are listed. The samples taken from the product stream showed the total leaching (L) of the ligand of 1.2% and L value of rhodium of 0.8% after 94 h.
Table 2 Leaching results with respect to the catalyst and ligand for continuous experiments when using TPPTS as the ligand
|
t [h] |
P [ppm] |
L
ligand [%] |
L
ligand [% h−1] |
Rh [ppm] |
L
Rh [%] |
L
Rh [% h−1] |
| 0–39 |
3 |
0.64 |
0.0165 |
1 |
0.22 |
0.0057 |
| 39–87 |
2 |
0.48 |
0.0102 |
2 |
0.50 |
0.0106 |
|
L(94 h) |
— |
1.20 |
0.0128 |
— |
0.80 |
0.0086 |
2.2.2. Variations in stirrer speeds with sulfoxantphos.
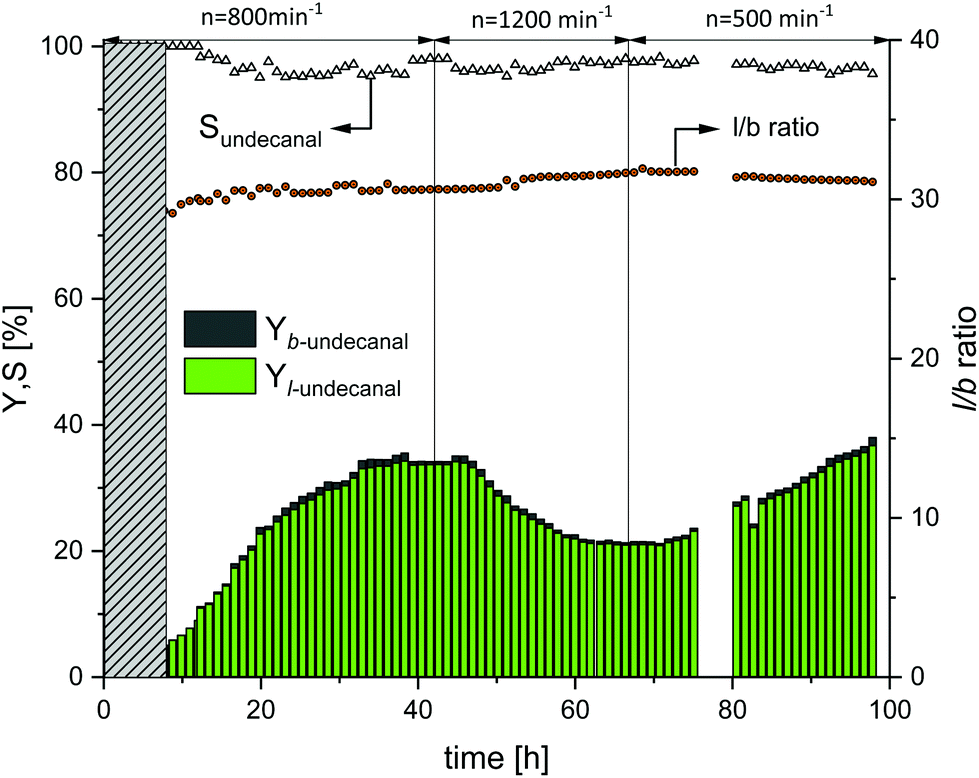
With an increase in the CD concentration in the reactor, the mass transfer increased and hence the product yield for the Rh/TPPTS catalyst system also increased: the same was expected for the Rh/sulfoxantphos system. Therefore, the subsequent continuous experiment does not only investigate the feasibility of the Rh/sulfoxantphos catalyst system in a continuous process, but also reveals the mixing behavior of the phase system in a more comprehensive manner. For this, the molar ratio of CD to rhodium was maintained constant at 12 and the stirrer speed at the beginning was set to 800 min−1. The obtained results are shown in Fig. 7. The first 8 h represent the startup time of the plant (denoted by the grey bar). Thereafter, the yield of undecanal steadily increased to 32% until the 34th hour, which is marginally lower than the expected yield from the batch reaction profile experiment (38%) (ESI†). The miniplant operation was considered to be stable at this point. To investigate if the mass transfer limitation limited the conversion, the stirrer speed was increased from 800 to 1200 min−1 to mechanically enhance mass transfer between both the phases and consequently resulting in higher conversion. However, the undecanal yield decreased to 20%. However, at the same time, foam formation increased in the reactor, which is considered to be the potential reason for the decreased yield since mass transfer between the liquid–liquid phases can be inhibited by foam. After the plant was steady again, the stirrer speed was reduced from 1200 to 500 min−1 at about 66 h. The undecanal yield slowly increased after 6 h (equal to the residence time). At the end of the experiment, the yield was 34%. Furthermore, it should be noted that the foam phase reduced considerably at 500 min−1. However, changes in the stirrer speed and therefore in the mass transfer did not influence the chemoselectivity at all, which was maintained at >95% for the aldehyde product. The regioselectivity (l/b ratio) started at 27.5 and slowly increased to a steady-state value of 30.5 after 20 h, which are similar to the ratios achieved in the batch tests. Because of the differences in the range of measurement as well as continuous operation fluctuations, no direct influences of stirrer speed and l/b ratio are evident.
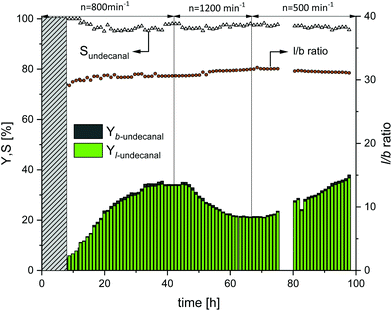 |
| | Fig. 7 Yield and selectivity over time in continuous operation with variations in stirrer speed using sulfoxantphos as the ligand. Conditions: Preforming: p(CO/H2) = 20 bar, nCO/nH2 = 1:1, T = 120 °C, t = 12 h; reaction: p(CO/H2) = 40 bar, nCO/nH2 = 1:1, T = 120 °C, τ = 6 h, mw/mS = 4:1, cCat = 0.0064 mol%, nCD/nCat = 12, nP/nCat = 5, ṁ1-decene = 10.2 g h−1, ṁrecycle = 41.1 g h−1. | |
In Table 3, the catalyst leaching results are represented. In the beginning, the leaching values of rhodium and ligand were slightly higher. This is most likely due to the startup procedure and lower average residence time in the decanter. Further loss corresponded approximately to the value determined for the entire operating time. Over the operating time of 93 h and with an average leaching of the ligand of 0.0122% h−1, the total ligand leaching added to 1.14%. The average loss of rhodium was 0.0059% h−1, and therefore, the total rhodium leaching added to 0.55%.
Table 3 Leaching results of the catalyst and ligand for continuous variations in the stirrer speed using sulfoxantphos as the ligand
|
t [h] |
P [ppm] |
L
ligand [%] |
L
ligand [% h−1] |
Rh [ppm] |
L
Rh [%] |
L
Rh [% h−1] |
| 0–5 |
4 |
0.09 |
0.0170 |
2 |
0.07 |
0.0128 |
| 5–22 |
4 |
0.24 |
0.0147 |
1 |
0.09 |
0.0055 |
| 22–45 |
3 |
0.30 |
0.0129 |
1 |
0.13 |
0.0056 |
| 45–93 |
3 |
0.50 |
0.0105 |
1 |
0.26 |
0.0055 |
|
L(93 h) |
— |
1.14 |
0.0122 |
— |
0.55 |
0.0059 |
2.2.3. Long-term experiment with sulfoxantphos.
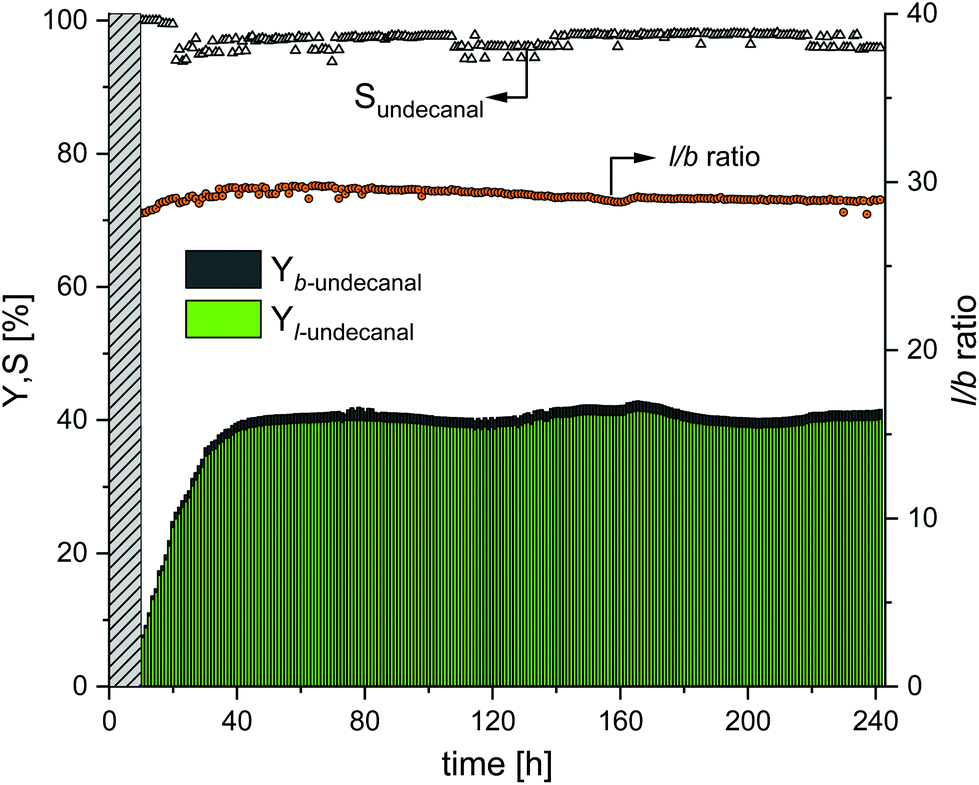
In this experiment, the earlier results from the miniplant (which afforded the highest yields and selectivities) were used to show the long-term effects when the plant was running in the steady state. Therefore, the experiment was carried out with a sulfoxantphos catalyst system as it generally afforded higher yields and higher regioselectivities as compared to the Rh/TPPTS catalyst system. The amount of CD was set to nCD/nRh = 18 for the experiment shown in Fig. 6 and the stirrer speed was set to 500 min−1 for the experiment shown in Fig. 7. The results of this experiment are shown in Fig. 8. At the beginning of the startup phase, the undecanal yield was ∼6%. This steadily increased until reaching a stationary value of 39% after 38 h, which was also expected from the batch reaction profile experiments (ESI†). This value was maintained stable in a steady-state operation over 200 h with maximum deviations of 1.5% until the end of the test after 241 h. The chemoselectivity of undecanal was initially >99%. Until the stationary point was reached, the chemoselectivity dropped to 97%, but retained this value with maximum deviations of 1% over the entire operating time. This behavior of chemoselectivity was also observed in the earlier continuous experiment with sulfoxantphos (Fig. 7). The regioselectivity (l/b ratio) was 27, which increased until reaching the stationary value of 29.6 after 40 h of operation. This value was maintained with minimum deviation until the end of the experiment.
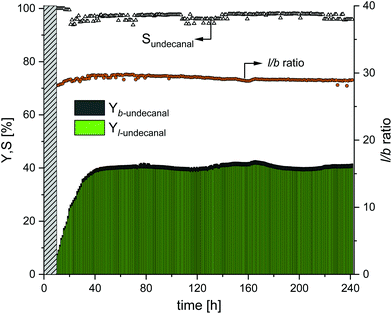 |
| | Fig. 8 Yield and selectivity over time in long-term experiments using sulfoxantphos as the ligand. Conditions: Preforming: p(CO/H2) = 20 bar, nCO/nH2 = 1:1, T = 120 °C, t = 12 h; reaction: p(CO/H2) = 40 bar, nCO/nH2 = 1:1, T = 120 °C, τ = 6 h, mw/mS = 4:1, cCat = 0.0064 mol%, nCD/nCat = 18, nP/nCat = 5, n = 500 min−1, ṁ1-decene = 12.7 g h−1, ṁrecycle = 51.4 g h−1. | |
In Table 4, the catalyst leaching results are presented for the entire time of operation. In general, no significant deviation in leaching was observed, which supports the evidence that CDs are very efficient as mass transfer agents. Over the operating time of 241 h, the average leaching of ligand was 0.0098% h−1; therefore, the total leaching of the ligand added to 2.17%. The average loss of rhodium was 0.0027% h−1, and therefore, the total rhodium leaching added to 0.59%. The long-term stability of the system could, therefore, be demonstrated in this experiment. No makeup stream was needed to maintain the catalytic activity over the entire course of the experiment. During the experiment, a white solid deposit was observed in the decanter through a viewing window. This was noticed for the first time after ∼70 h of operation, and this deposit increased over the test period. The formation of this solid could be attributed to an increase in the CD concentration at the beginning in the sulfoxantphos system (nCD/nRh = 18 instead of nCD/nCat = 12 as compared to the batch experiments) and not to the long operating time itself. Solubility problems, therefore, appear to occur, which further exacerbate with an increase in the CD concentration, which could become problematic during continuous operation and should not be considered.
Table 4 Leaching results of the catalyst and ligand for continuous experimentation for 240 h using sulfoxantphos as the ligand
|
t [h] |
P [ppm] |
L
ligand [%] |
L
ligand [% h−1] |
Rh [ppm] |
L
Rh [%] |
L
Rh [% h−1] |
| 0–8 |
2 |
0.07 |
0.0098 |
0.3 |
0.01 |
0.0022 |
| 8–19 |
2 |
0.10 |
0.0098 |
0.3 |
0.02 |
0.0026 |
| 19–30 |
2 |
0.10 |
0.0098 |
0.3 |
0.03 |
0.0027 |
| 30–43 |
2 |
0.12 |
0.0098 |
0.4 |
0.03 |
0.0031 |
| 43–64 |
2 |
0.20 |
0.0098 |
0.3 |
0.05 |
0.0026 |
| 64–95 |
2 |
0.30 |
0.0098 |
0.3 |
0.07 |
0.0025 |
| 95–122 |
2 |
0.26 |
0.0098 |
0.3 |
0.06 |
0.0023 |
| 122–168 |
2 |
0.45 |
0.0098 |
0.4 |
0.14 |
0.0032 |
| 168–184 |
2 |
0.15 |
0.0098 |
0.4 |
0.05 |
0.0033 |
| 184–221 |
2 |
0.36 |
0.0098 |
0.3 |
0.08 |
0.0023 |
|
L(240 h) |
— |
2.17 |
0.0098 |
— |
0.59 |
0.0027 |
2.2.4. Concept of distillation and substrate recycling.
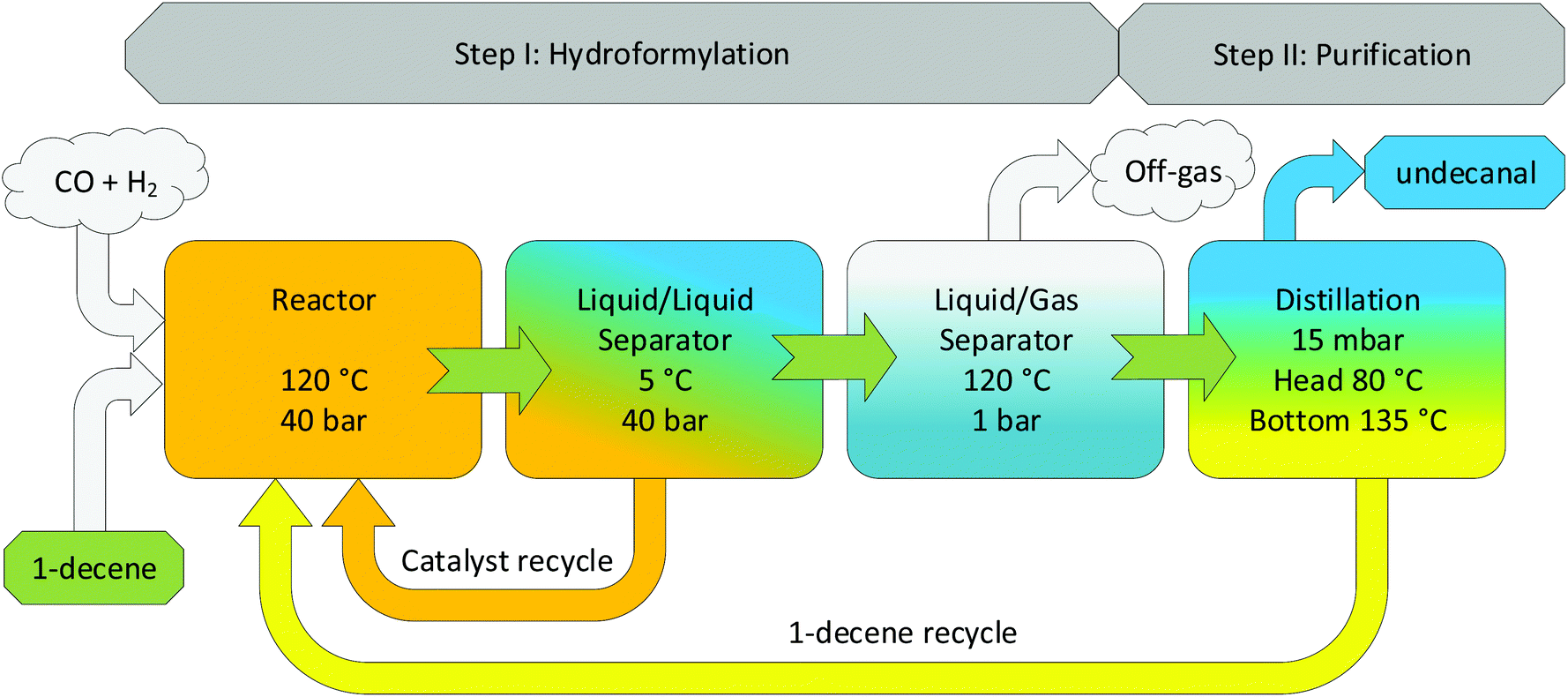
The aqueous biphasic hydroformylation of 1-decene using CDs as the mass transfer agent exhibited high stability, low catalyst leaching, and high chemo- and regioselectivity in the continuous experiments under consideration; however, it lacked high turnover rates. To make it a more attractive process for commercial applications, process intensification is imperative. Consequently, several techniques have been formulated, e.g., increasing the residence time of 1-decene in the reactor by increasing the reactor volume or decreasing the feed and recycle flow rates to the reactor in order to increase conversion. Further, the increased mixing behavior can yield higher turnover, which can be achieved by changing the stirrer geometry and stirrer speed, which has already been shown earlier (Fig. 7). Further investigations in this particular field are currently ongoing. The third way to improve the efficiency of this process is to recover and recycle the nonconverted substrate after the reaction. Fig. 8 shows that up to 58% of the nonconverted substrate leaves the process after phase separation. A huge improvement could be made if the substrate could be recovered and recycled. Therefore, a potential concept of this process has been proposed, as shown in Fig. 9, by integrating (step I: hydroformylation) a second purification step into the already existing system by using a distillation column to separate the substrate from the product.
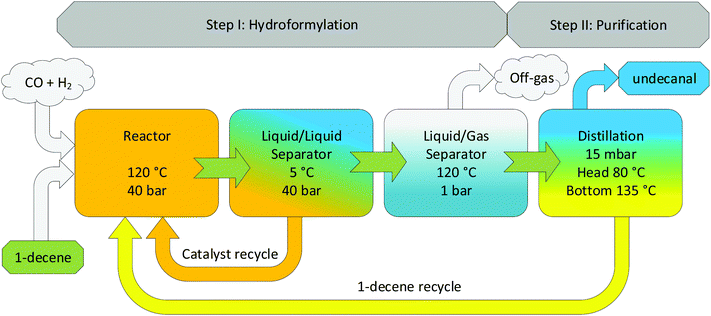 |
| | Fig. 9 Process concept for an intensified process for CD-based hydroformylation. | |
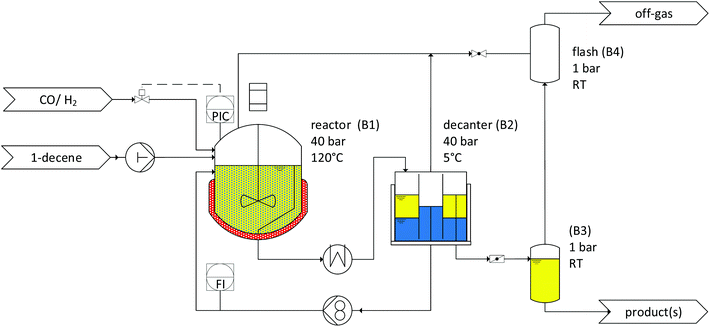 |
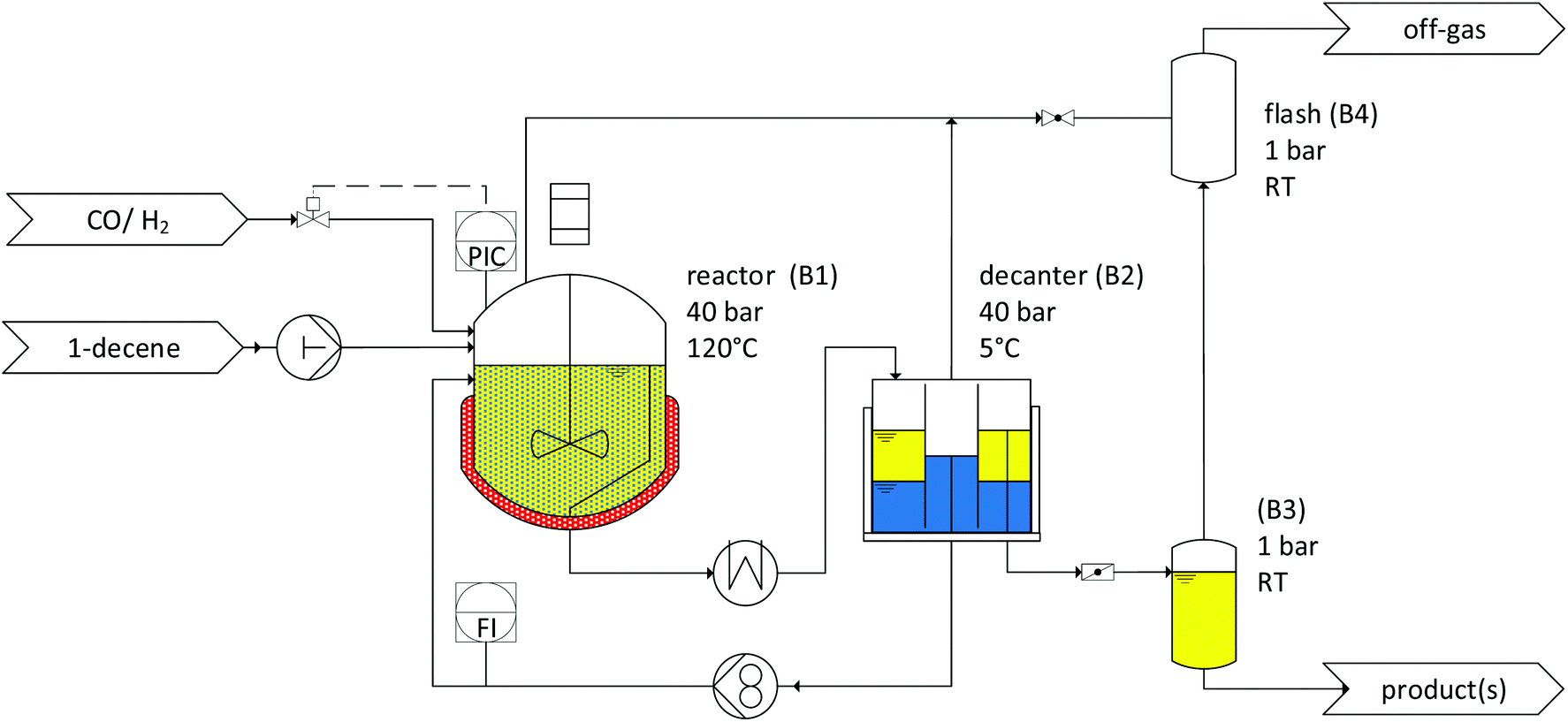
| | Fig. 10 Flowchart of the continuously operated miniplant. | |
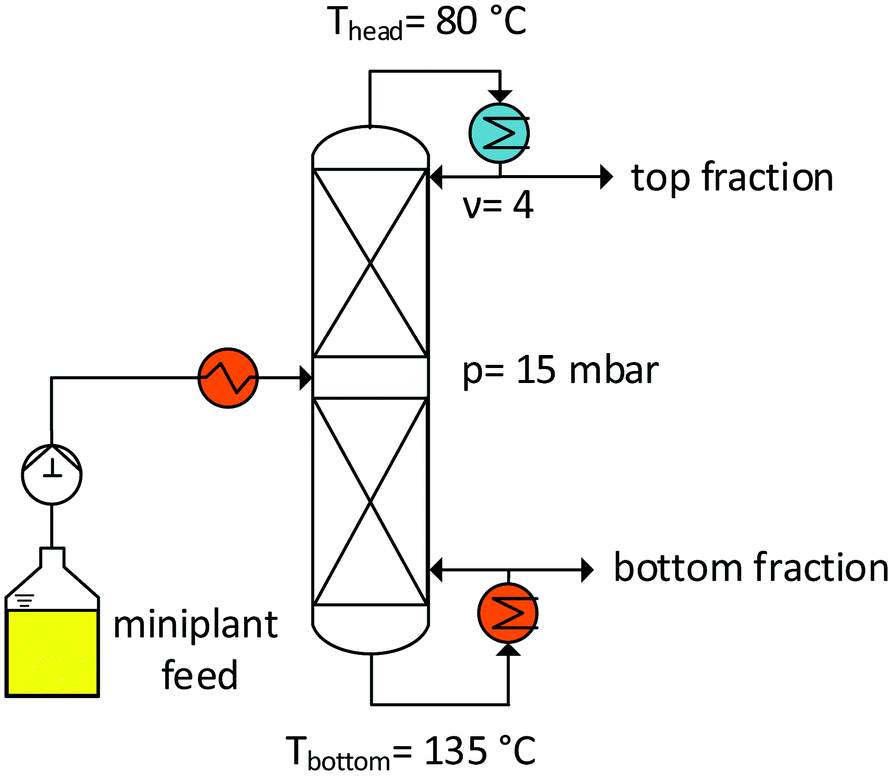
To prove that this concept is feasible, a distillation column (Fig. 11) was setup, as described in the Experimental section. In particular, vacuum distillation (temperature: <150 °C) was chosen to suppress the isomerization reactions of the substrate, but also for efficient product and substrate separation. The results of continuous distillation over 8 h are shown in the ESI.† A distillate stream (top fraction) with a composition of ∼94.2 wt% 1-decene and 5.8 wt% decane/decene isomers can be formulated. The regioselectivity (l/b ratio) of decene in the feed and at the top of the column remains constant at a value of ∼6 wt%. Therefore, the isomer portion of the feed, when completely separated, is decisive for determining the quality of the distillate stream. Therefore, the general feasibility of the process concept is evident.
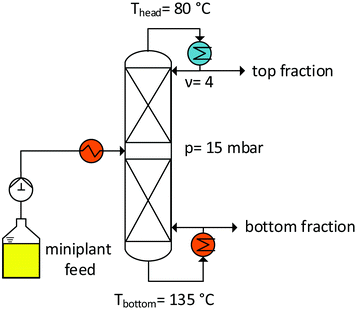 |
| | Fig. 11 Distillation column for substrate recycling. | |
3. Experimental section
3.1. Chemicals
In Table 5, all the chemicals used in this work are listed. All the solvents and substrates were degassed before the reaction, but not purified further. The purity of each component was tested via gas chromatography.
Table 5 Chemicals used in this work
| Chemicals |
Manufacture |
Purity [%] |
| 1-Decene |
Acros organics |
99 |
| Argon |
Messer Industriegase |
— |
| Rh(acac)(CO)2 |
Umicore |
— |
| Triphenylphosphine-3,3′,3′′-trisulfonic acid trisodium salt |
Oxea GmbH |
— |
| Sulfoxantphos |
MOLISA |
— |
| Di-n-butylether |
Acros organics |
99.5 |
| Isopropanol |
VWR international |
98 |
| CO/H2 |
Messer Industriegase |
99.9 |
| Bi-distilled water |
— |
— |
3.2. Analytics
A gas chromatograph from Agilent Technologies (Type 7890A) with an HP-5 column (30 m × 0.32 mm × 0.25 m) and a flame ionization detector (FID) was used for gas chromatographic measurements of the offline samples from the batch and continuous experiments. n-Dodecane was used as the standard and isopropanol, the solvent. For measuring the online samples for continuous experiments, a gas chromatograph (Type 7890A, Agilent Technologies) was used for the gas chromatographic measurements. This system was equipped with a FID. An HP-5 (30 m × 0.25 mm × 0.25 m) was used as the separation column. A sample volume of 1 μl was injected with a split ratio of 1:50 with a constant supply of helium as the carrier gas.
The contents of rhodium and phosphorus in the liquid phases were quantified via inductively coupled plasma emission spectroscopy (ICP-OES, Thermo Elemental Iris Intrepid). The catalyst leaching rates for rhodium and phosphorus were calculated on the basis of the total amount of the related product fraction, the appropriate mass fraction (ppm) contained, and the initial mass of rhodium and phosphorus at the start of the miniplant. For example, 1% h−1 of leaching represents a loss of 1% of the initial catalyst amount via product flow in 1 h.
3.3. Experimental setup
Batch experiments.
A 300 ml high-pressure autoclave (Parr Instrument Company) was used to perform the batch experiments. The autoclave was pressure-stable up to 250 bar and was equipped with a blade stirrer. The reactor was used for creating the time curves and performing kinetic experiments for scaling up the reaction. After weighing the catalyst, ligand, polar solvent, and CDs, the autoclave was closed and installed in the reaction equipment. The substrates and solvents were degassed with argon via frit for 30 min. The 1-decene substrate was subsequently transferred to a dropping funnel via a cannula. The dropping funnel, when filled, was pressurized with the system pressure from the reactor. When the predetermined time for preforming was reached, the substrate was transferred into the reactor; thereafter, it was continuously fed with syngas. The Schlenk technique was used for the preparation of all the components and the inert transfer of all the substances into the reactor at all times.
A 1000 ml high-pressure autoclave (Büchi) was used to perform further scaling up experiments. The reactor was stable up to 60 bar, including a blade stirrer and baffles. The procedure for carrying out the experiments is the same as that for the 300 ml high-pressure autoclave. The reactor was also used as a continuous stirred-tank reactor (CSTR) for performing continuous miniplant experiments.
Continuous experiments.
The miniplant comprised a 1000 mL CSTR, which was pressure-stable up to 60 bar. In addition, the reactor was designed with a blade stirrer and baffles. The reactor was connected to a decanter (B2) unit, where the reaction mixture was separated into polar (blue) and nonpolar (yellow) phases. The catalyst phase (blue) was then fed back by a gear pump to the reactor, while the nonpolar phase was collected on a scale. The miniplant was automated and equipped with an online gas chromatograph (Fig. 10). The initial mixtures containing catalyst, ligand, water, and CDs were prepared for all the miniplant apparatuses (reactor and decanter) at once. Therefore, the substances were weighed into several bottles and flushed with argon. The miniplant was tempered and evacuated, and the reactor (B1) was filled with the mixture via a pressure-stable dropping funnel. Then, syngas was added in a stepwise manner in each apparatus and pipe section. At the beginning of the operation, the pump for 1-decene was activated. Simultaneously, continuous syngas feeding and pressure control were started. During the miniplant operation, samples from the product phase were periodically analyzed with an online gas chromatograph from the decanter. Further, offline samples were taken from the product and catalyst phases for ICP analysis, but also for the validation of the results obtained from the online gas chromatograph.
Distillation setup.
Fig. 11 shows the general setup of the distillation column used for the recovery of the nonconverted substrate from the product stream. The column was operated at a pressure of 15 mbar by means of a diaphragm vacuum pump. The product solution from the miniplant (Fig. 10, B2) was conveyed into the column via a storage tank with a solenoid-driven diaphragm metering pump. The feed solution was heated in the column by using a heating rod; the formed steam rose and passed through 15 separation stages that were prepared using Sulzer DX (30 mm) laboratory packs (each pack was 5.5 cm high and had a HETP value of 0.085 m). At the top of the column, the distillate stream passed through a water-cooled condenser in which a split was installed in order to set the reflux. The distillate stream was collected in a container. The bottom of the column was designed as a natural circulation evaporator to ensure free mixing. In addition, there is an overflow at the bottom from which the bottom product reaches another container. To protect the bottom of the column from drying out due to the condensed gas, the overflow was equipped with a gas trap.
4. Conclusion
The Rh-catalyzed aqueous biphasic hydroformylation of 1-decene was successfully conducted only using water as the environmentally friendly solvent and RAME-β-CD as the green mass transfer agent. Initially, two catalyst systems comprising of Rh/TPPTS and Rh/sulfoxantphos were scaled up from a 50 ml autoclave to a 1000 ml high-pressure autoclave to evaluate the feasibility of these phase systems for a potential continuous process application. Herein, both these systems performed efficiently in terms of chemoselectivity (>95%) toward the aldehyde product. However, these experiments reveal that for both these catalyst systems, the reaction rates slowed down in the larger autoclaves; therefore, conversion decreased from 99% to 30% (Rh/TPPTS) and from 70% to 50% (Rh/sulfoxantphos). However, as the chemoselectivity remained high for both these systems, they were tested in two continuous experiments (each over 90 h). These experiments showed that the reaction activity could be drastically influenced by the stirrer speed and nCD/nRh values inside the reactor. The Rh/sulfoxantphos catalyst system not only performed better in terms of aldehyde yield (45% as compared to that the Rh/TPPTS system, i.e., 30%), but also afforded higher regioselectivities (l/b ratios), i.e., 27, as compared to the value of 1.7; therefore, this system was chosen for the long-term experiment. For a total operation time of 240 h in a continuous miniplant and that of more than 200 h for steady-state operation, the leaching of catalyst was determined to be extremely low, affording ligand loss of 0.0098% h−1 and rhodium loss of 0.0027% h−1. As compared to a commercialized hydroformylation process (Shell) and other recently proposed processes (SILP/CXL and Aqueous TMS) in the literature, a CD-based system does not yield the highest turnover rates in continuous operations, but one of the highest chemo- and regioselectivities and lowest rhodium leaching rates (Table 6). Further, the CD process necessitates very mild conditions (120 °C and 40 bar) as compared to other processes.
Table 6 Comparison of different concepts for the continuous hydroformylation of higher olefins
| Parameter |
CDsa (Rh) |
SILP/CXLb (Rh)30 |
Aqueous TMS (Rh)26 |
Shell (Co)31 |
|
Referring to the experiment shown in Fig. 8.
SILP/CXL: supported ionic liquid phase + carbon-dioxide-expanded liquids; TOS: time on stream.
|
|
T [°C] |
120 |
100 |
140 |
200 |
|
p [bar] |
40 |
100 |
21 |
80 |
|
S
aldehyde [%] |
97 |
98 |
93 |
80 |
|
l/b ratio [—] |
27 |
3 |
19 |
— |
| TOF [h−1] |
19 |
500 |
1500 |
20 |
| TOS [h] |
240 |
40 |
21 |
— |
| Rh loss [ppm h−1] |
0.01 |
0.005 |
0.7 |
— |
Reaction rate improvements such as stirrer speed, stirrer type, phase ratios, and CD concentrations are still under investigation and will be discussed in an additional publication in more detail. On the other hand, process optimization was successfully described by the continuous distillation of the product mixture (undecanal, 1-decene, and iso-decene) and recovering nonconverted 1-decene substrate with 94% purity. With the possibility of continuous distillation of the nonconverted substrate, a new process concept was proposed by implementing a second recycling loop into the process. Therefore, combining an aqueous biphasic hydroformylation process using methylated β-CDs for higher olefins with extremely low catalyst leaching and high chemo- and regioselectivities toward the corresponding aldehyde product can be achieved.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) – TRR 63 “Integrierte chemische Prozesse in flüssigen Mehrphasensystemen” (Teilprojekt D3) – 56091768. Authors would like to thank Umicore AG &Co. KG for donating the rhodium precursor Rh(acac)(CO2) and Oxea GmbH for donating the ligand NaTPPTS.
References
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- F. Hebrard and P. Kalck, Chem. Rev., 2009, 109, 4272–4282 CrossRef CAS PubMed.
-
P. W. N. M. Leeuwen, Homogeneous Catalysis. Understanding the Art, Kluwer Academic Publishers, Dordrecht, 2004 Search PubMed.
-
(a) H. Masui, E. Honda, S. Niitsu, M. Shoji and T. Takahashi, Int. J. Org. Chem., 2018, 08, 135–141 CrossRef CAS;
(b) S. Wang, J. Zhang, F. Peng, Z. Tang and Y. Sun, Ind. Eng. Chem. Res., 2020, 59, 88–98 CrossRef CAS;
(c) M. L. Abrams, J. Y. Buser, J. R. Calvin, M. D. Johnson, B. R. Jones, G. Lambertus, C. R. Landis, J. R. Martinelli, S. A. May, A. D. McFarland and J. R. Stout, Org. Process Res. Dev., 2016, 20, 901–910 CrossRef CAS;
(d) S. A. May, J. Flow Chem., 2017, 7, 137–145 CrossRef CAS.
- U. Hintermair, G. Franciò and W. Leitner, Chem. Commun., 2011, 47, 3691–3701 RSC.
- Z. Xie and B. Subramaniam, ACS Sustainable Chem. Eng., 2014, 2, 2748–2757 CrossRef CAS.
- J. M. Marinkovic, A. Riisager, R. Franke, P. Wasserscheid and M. Haumann, Ind. Eng. Chem. Res., 2019, 58, 2409–2420 CrossRef CAS.
- M. Janssen, C. Müller and D. Vogt, Dalton Trans., 2010, 39, 8403–8411 RSC.
- J. Bianga, K. U. Künnemann, T. Gaide, A. J. Vorholt, T. Seidensticker, J. M. Dreimann and D. Vogt, Chem. – Eur. J., 2019, 11586–11608 CrossRef CAS PubMed.
- M. Schwarze, T. Pogrzeba, K. Seifert, T. Hamerla and R. Schomäcker, Catal. Today, 2015, 247, 55–63 CrossRef CAS.
- B. Cornils and E. G. Kuntz, J. Organomet. Chem., 1995, 502, 177–186 CrossRef CAS.
-
D. J. Adams, P. J. Dyson and S. J. Tavener, Chemistry in alternative reaction media, Wiley, Chichester, West Sussex, 2005 Search PubMed.
-
(a) S. K. Sharma and R. V. Jasra, Catal. Today, 2015, 247, 70–81 CrossRef CAS;
(b) L. Obrecht, P. C. J. Kamer and W. Laan, Catal. Sci. Technol., 2013, 3, 541–551 RSC;
(c) Y. Zhang, Z.-S. Mao and J. Chen, Ind. Eng. Chem. Res., 2001, 40, 4496–4505 CrossRef CAS.
- L. C. Matsinha, S. Siangwata, G. S. Smith and B. C. E. Makhubela, Catal. Rev. Sci. Eng., 2019, 61, 111–133 CrossRef CAS.
-
(a)
Cyclodextrin Fundamentals, Reactivity and Analysis, ed. S. Fourmentin, G. Crini and E. Lichtfouse, Springer International Publishing, Cham, 2018, vol. 16 Search PubMed;
(b) J. Szejtli, Chem. Rev., 1998, 98, 1743–1754 CrossRef CAS.
- H. Bricout, F. Hapiot, A. Ponchel, S. Tilloy and E. Monflier, Sustainability, 2009, 1, 924–945 CrossRef CAS.
-
(a) J. Yi, W. Liang, X. Wei, J. Yao, Z. Yan, D. Su, Z. Zhong, G. Gao, W. Wu and C. Yang, Chin. Chem. Lett., 2018, 29, 87–90 CrossRef CAS;
(b) A. Cocq, H. Bricout, F. Djedaïni-Pilard, S. Tilloy and E. Monflier, Catalysts, 2020, 10, 56 CrossRef CAS;
(c) X. Wei, W. Wu, R. Matsushita, Z. Yan, D. Zhou, J. J. Chruma, M. Nishijima, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, J. Am. Chem. Soc., 2018, 140, 3959–3974 CrossRef CAS PubMed.
- F. Hapiot, S. Menuel, H. Bricout, S. Tilloy and E. Monflier, Appl. Organomet. Chem., 2015, 29, 580–587 CrossRef CAS.
- E. Monflier, S. Tilloy, G. Fremy, Y. Castanet and A. Mortreux, Tetrahedron Lett., 1995, 36, 9481–9484 CrossRef CAS
http://www.sciencedirect.com/science/article/pii/004040399502073X
.
- L. Leclercq, F. Hapiot, S. Tilloy, K. Ramkisoensing, J. N. H. Reek, P. W. N. M. van Leeuwen and E. Monflier, Organometallics, 2005, 24, 2070–2075 CrossRef CAS.
- F. Hapiot, L. Leclercq, N. Azaroual, S. Fourmentin, S. Tilloy and E. Monflier, Curr. Org. Synth., 2008, 5, 162–172 CrossRef CAS.
- F. Hapiot, S. Tilloy and E. Monflier, Chem. Rev., 2006, 106, 767–781 CrossRef CAS PubMed.
-
(a) F. Hapiot, H. Bricout, S. Tilloy and E. Monflier, Top. Curr. Chem., 2013, 342, 49–78 CrossRef CAS PubMed;
(b) H. Bricout, F. Hapiot, A. Ponchel, S. Tilloy and E. Monflier, Curr. Org. Chem., 2010, 14, 1296–1307 CrossRef CAS.
- S. Tilloy, F. Bertoux, A. Mortreux and E. Monflier, Catal. Today, 1999, 48, 245–253 CrossRef CAS.
-
(a) A. Behr, A. Kämper, R. Kuhlmann, A. J. Vorholt and R. Franke, Catal. Sci. Technol., 2016, 6, 208–214 RSC;
(b) M. Zagajewski, J. M. Dreimann, M. Thönes and A. Behr, Chem. Eng. Process., 2016, 99, 115–123 CrossRef CAS;
(c) K. U. Künnemann, J. Bianga, R. Scheel, T. Seidensticker, J. M. Dreimann and D. Vogt, Org. Process Res. Dev., 2020, 24, 41–49 CrossRef;
(d) H. Warmeling, A. Behr and A. J. Vorholt, Chem. Eng. Sci., 2016, 149, 229–248 CrossRef CAS.
- T. Gaide, J. M. Dreimann, A. Behr and A. J. Vorholt, Angew. Chem., Int. Ed., 2016, 55, 2924–2928 CrossRef CAS PubMed.
- T. Mathivet, C. Méliet, Y. Castanet, A. Mortreux, L. Caron, S. Tilloy and E. Monflier, J. Mol. Catal. A: Chem., 2001, 176, 105–116 CrossRef CAS
http://www.sciencedirect.com/science/article/pii/S1381116901002291
.
- N. Sieffert and G. Wipff, Chem. – Eur. J., 2007, 13, 1978–1990 CrossRef CAS PubMed.
-
(a) H. W. F. Warmeling, T. Hafki, T. von Söhnen and A. J. Vorholt, Chem. Eng. J., 2017, 326, 298–307 CrossRef CAS;
(b) H. W. F. Warmeling, D. Janz, M. Peters and A. J. Vorholt, Chem. Eng. J., 2017, 330, 585–595 CrossRef CAS.
- U. Hintermair, Z. Gong, A. Serbanovic, M. J. Muldoon, C. C. Santini and D. J. Cole-Hamilton, Dalton Trans., 2010, 39, 8501–8510 RSC.
- P. B. Webb, T. E. Kunene and D. J. Cole-Hamilton, Green Chem., 2005, 7, 373 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Reaction profiles for the Rh/TPPTS and Rh/sulfoxantphos systems are shown for both 300 and 1000 ml autoclaves. Further, the composition of the distillate stream for the continuously operated distillation is shown. See DOI: 10.1039/d0gc00820f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
L.
Schurm
a,
L.
Schurm