DOI:
10.1039/D0GC00363H
(Paper)
Green Chem., 2020,
22, 2459-2467
Sustainability by design: automated nanoscale 2,3,4-trisubstituted quinazoline diversity†
Received
29th January 2020
, Accepted 21st February 2020
First published on 25th February 2020
Abstract
Small molecule synthesis is equally important for materials and pharmaceuticals. However, the traditional approach performed in pharmaceutical R&D including maintenance of million-sized libraries and optimization synthesis of hundreds or even thousands of chemicals on a mmol or larger scale lacks sustainability. Here, we exemplify the synthetic execution of a newly designed quinoxaline reaction towards a transformative sustainability in chemistry. This includes nanoscale synthesis, deep chemical space exploration, scalability over 6 orders of magnitude from milligram up to 10-gram resynthesis of quinazolines enabled by the simultaneous variation of four classes of building blocks. Benefits of our approach include a simple to perform, one-step procedure, mild reaction conditions and access to a very large chemical space through accessing many available building blocks. More than thousand derivatives were produced in an automated fashion on a nanoscale using positive pressure facilitated dispensing. Along with these advantages, there is a considerable reduction in synthetic effort, reagents, solvent, glass and plastic consumables and power consumption to decrease the footprint of synthetic chemistry.
Introduction
Chemistry is the central science that ameliorates human existence by producing compounds of great use in energy, agriculture, electronics, pharmaceuticals, personal care, cosmetics, and flavors and fragrances. However, with an ever-growing number of planetary inhabitants, sustainability is the only guaranty of a livable future. Achieving a sustainable future in chemistry is the major goal of green chemistry.1 Often chemists use the 12 Principles of Green Chemistry as a blueprint for chemical design for sustainability throughout the life cycle of chemicals.2 Efforts to increase the green footprint of organic synthetic chemistry (e.g. non chlorinated solvents, catalysis, non-toxic reagents) are therefore major transformative drivers of a sustainable future in industry and society.3 Despite these honorable ideas, the reality of contemporary chemistry in wide areas looks quite different: energy intensive processes, toxic reagents and solvents, expensive, wasteful and time-consuming sequential target syntheses resulting in overall low yields, clearly far from sustainability. Particularly, medicinal chemistry can be highlighted as a chemistry area where inclusion of sustainable methods and processes would have a tremendous impact.4 Pharmaceutical R&D is a highly compound intensive chemistry area. Large and medium sized pharma companies keep millions of compounds in screening libraries, traditionally on a gram scale, more recently on a multi milligram scale. These have to be regularly renewed to stay competitive and cope with ‘novelty erosion’.5 These screening compounds are mostly produced via multistep synthetic routes that are neither optimal nor green. Next, the hits from high throughput screening of the million compounds have to be optimized for ‘drug-like’ properties in tedious, lengthy and expensive processes involving the synthesis of thousands of intermediates and target compounds. Despite the exploratory scale, waste generation in early drug discovery is considerable. Conservatively, it has been estimated that up to 2 million kilograms of waste are produced per year in drug discovery with up to 1.5 million kilograms of additional waste produced per year in preclinical studies.6 Inevitably, drug compound optimization involves multiple low-yielding steps, often requires several solvent-intensive chromatographic purifications, and generates a large amount of plastic, glass and paper consumable waste. Key to sustainability in synthetic chemistry is waste reduction. The principle of prevention has been recently highlighted to overshadow all other principles of green chemistry.7 The emerging field of reaction miniaturization is therefore offering an outstanding opportunity in the ultra-conservation of advanced pharmaceutical intermediates and additionally in the overall reduction of waste achieved by performing reactions on a smaller scale.4 Moreover, multicomponent reaction chemistry is based on the principles of atom economy, step prevention, convergence and has been recognized since a long time to come close to ‘perfect’ green chemistry and has also been recently reviewed in this context.8 It has been repeatedly shown that by introducing a multicomponent reaction (MCR) into a target synthesis, the overall number of steps can be considerably reduced while yields are increased.9–11 A perfect example is the Teleprevir HCV inhibitor synthesis by Orru et al.10 Introducing two MCR steps in the synthesis of this complex peptidomimetic compound reduced the lengthy >20 step industrial synthesis by ∼50%. Here we exemplary showcase the discovery and scope and limitation study of a new reaction leading to the small molecule quinazoline scaffold based on several major sustainability principles.
Design – synthesis
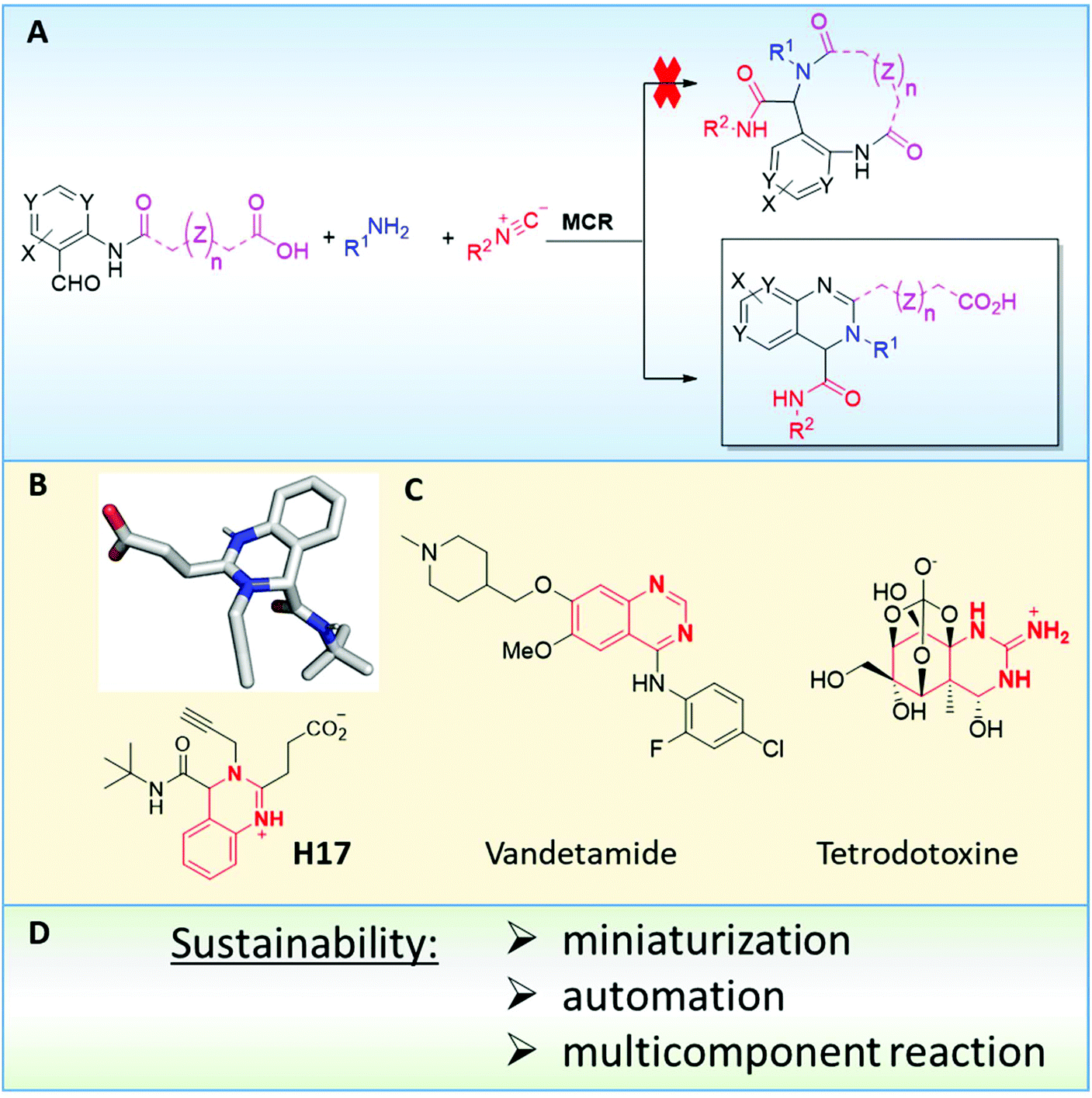
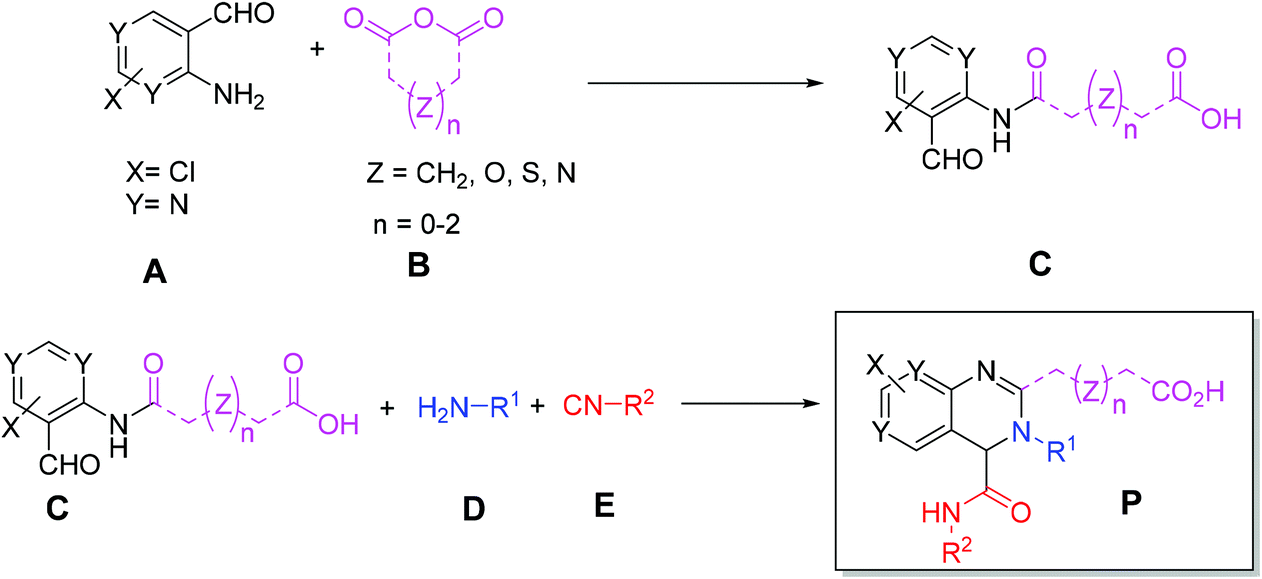
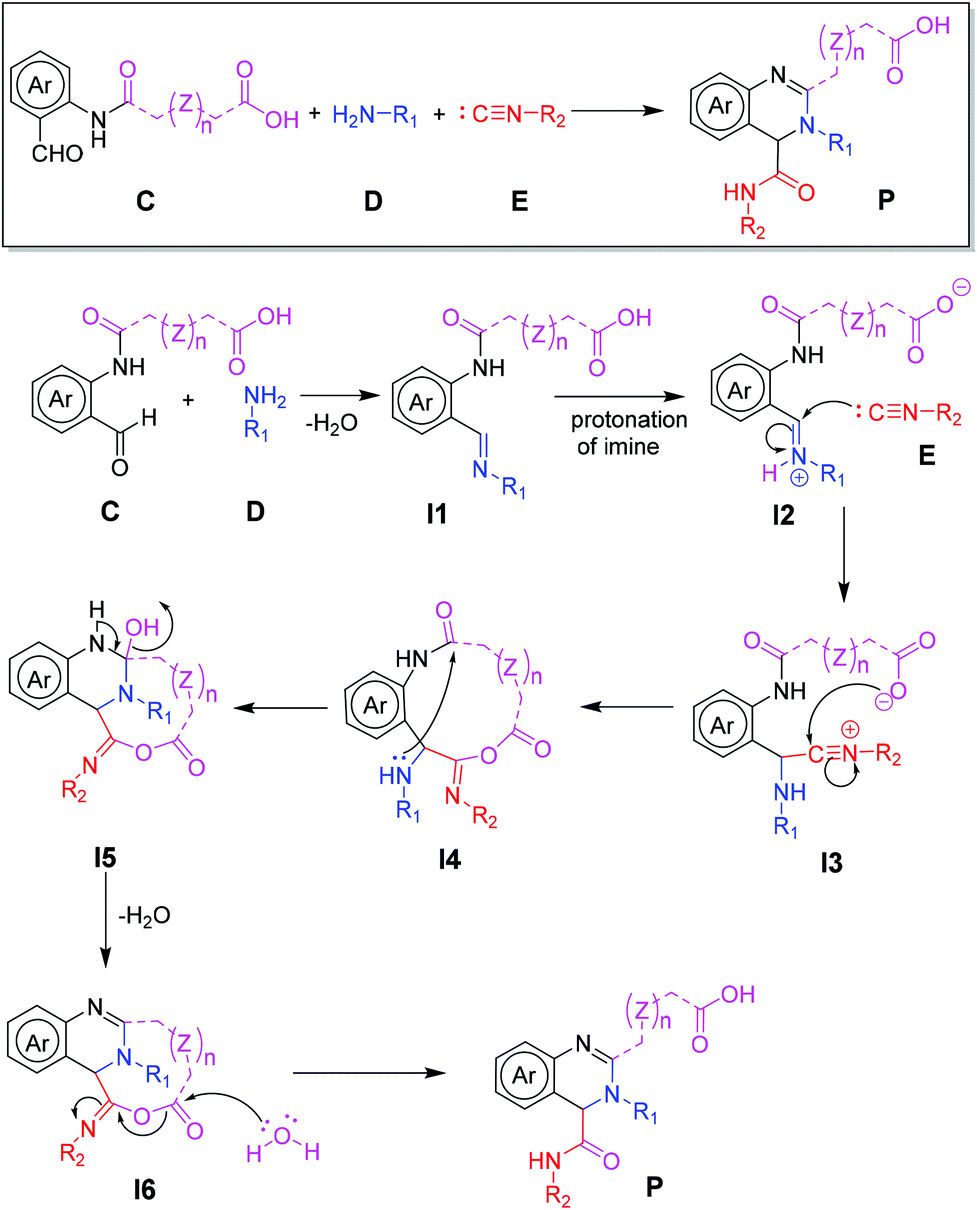
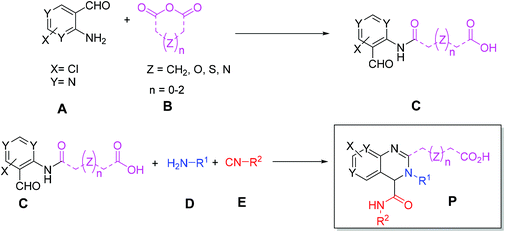
Initially, we designed a one-step general synthesis of medium cycles from readily accessible starting materials employing the archetypical Ugi-4CR.12–14 We hypothesized that suitable α-formyl-ω-carboxylic acids (C) will react with a medium cycle ring upon addition of a primary amine (D) and an isocyanide (E) (Fig. 1A). To gain access to a diversity of α-formyl-ω-carboxylic acids (C) we planned to react o-formyl anilines and heteroaromates (A) with cyclic carboxylic acid anhydrides (B). Both building blocks are commercially available in larger numbers with different substituents and ring sizes. Surprisingly and proven by a small molecule crystal structure, we found that a 2,3,4-trisubstituted quinazoline (P) is formed instead of the expected medium cycle (Fig. 1A and B and 2). Opportunistically we figured that the quinazoline scaffold shares key pharmacophoric and structural features of a large number of biologically relevant compounds (Fig. 1C).15
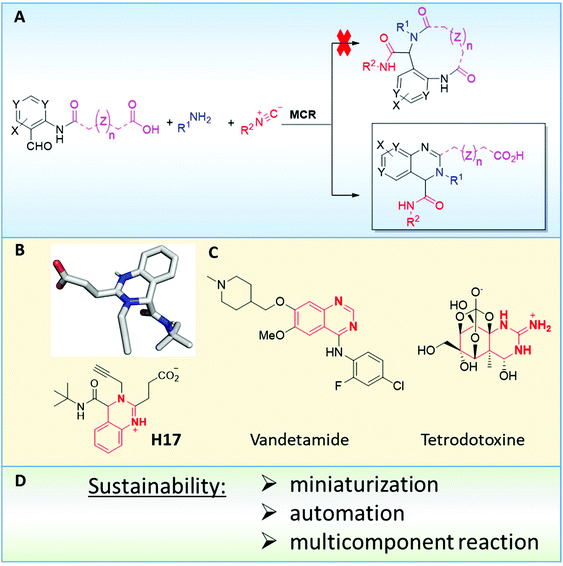 |
| | Fig. 1 Overall research design. (A) Design of medium cycle synthesis and surprising formation of highly substituted quinazolines instead; (B) crystal structure of compound H17; (C) the quinazoline moiety in drugs and natural products; (D) sustainability principles used in the design of the present research. | |
 |
| | Fig. 2 New 2-step quinazoline synthesis. | |
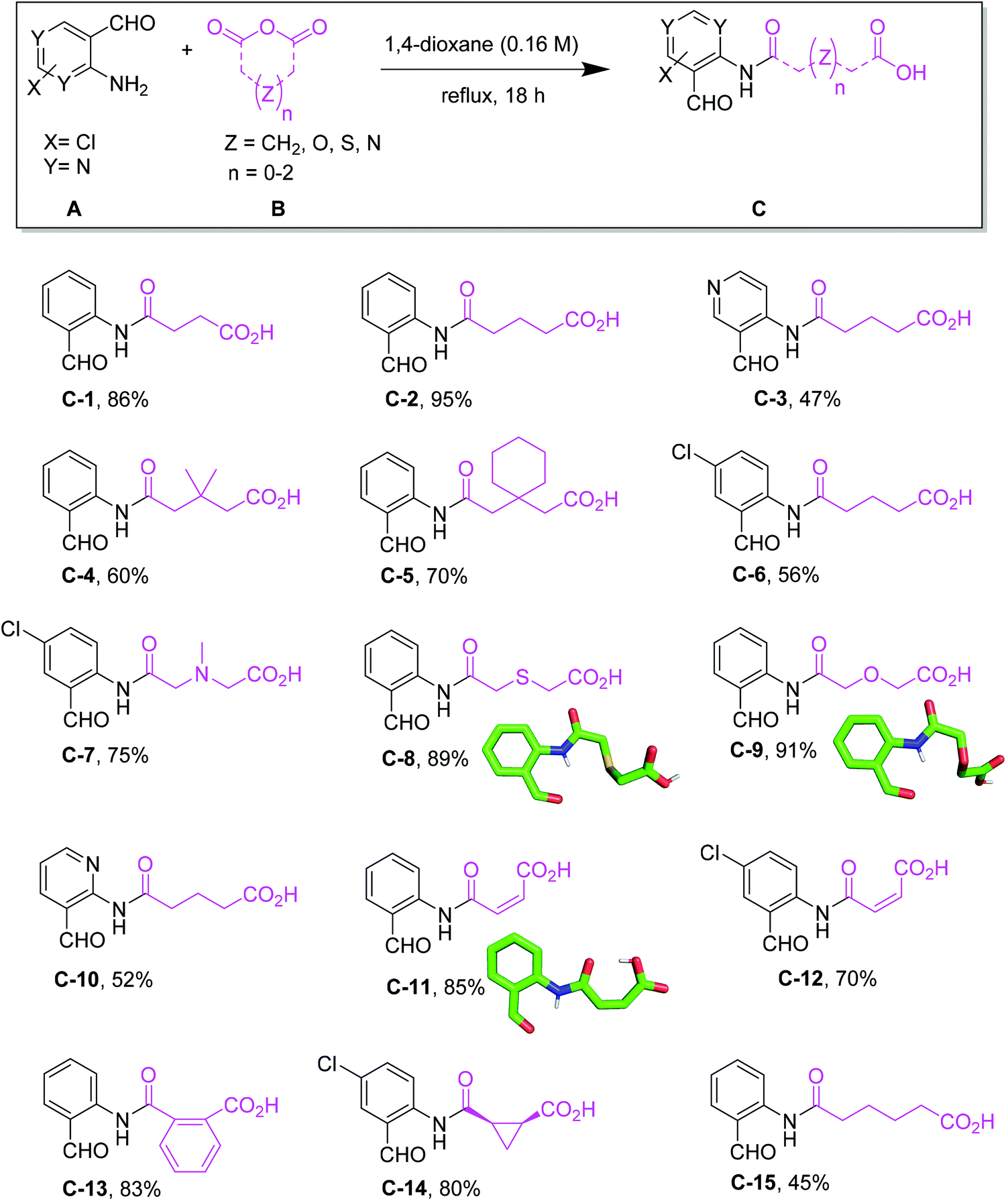
We commenced our study by the synthesis of α-formyl-ω-carboxylic acids as building blocks via a simple ring opening reaction of cyclic anhydride with various derivatives of 2-aminobenzaldehyde, both commercially available in great numbers. To optimize the reaction conditions of the model system using 2-aminobenzaldehyde/glutaric anhydride, we tried different solvents and mixtures, temperatures and ratios of reactants (ESI Table S1†). The best conditions were the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 ratio of 2-aminobenzaldehyde/glutaric anhydride and refluxing in dioxane to afford the desired product in 95% yield. Fifteen α-formyl-ω-carboxylic acids of different lengths were synthesized in good purity and yields, each on a gram scale (Fig. 3). This included benzene as well as pyridine derivatives (C-3, C-10). The diversity of the included anhydrides range from 5 to 7-membered simple aliphatic chains (C-1, C-2, C-15) to 3,3-dimethyl substituents (C-4), to cis-alkene (C-11, C-12), phenyl (C-13), and spiro cycle (C-5) and to a conformationally constrained cyclopropyl annulated ring (C-14). It also includes basic nitrogen (C-7), oxygen (C-9) or sulfur (C-8). The aminobenzaldehyde component showed variation in halogen (C-6, C-7, C-12, C-14) as well as two positional pyridine isomers (C-3, C-10).
:1.1 ratio of 2-aminobenzaldehyde/glutaric anhydride and refluxing in dioxane to afford the desired product in 95% yield. Fifteen α-formyl-ω-carboxylic acids of different lengths were synthesized in good purity and yields, each on a gram scale (Fig. 3). This included benzene as well as pyridine derivatives (C-3, C-10). The diversity of the included anhydrides range from 5 to 7-membered simple aliphatic chains (C-1, C-2, C-15) to 3,3-dimethyl substituents (C-4), to cis-alkene (C-11, C-12), phenyl (C-13), and spiro cycle (C-5) and to a conformationally constrained cyclopropyl annulated ring (C-14). It also includes basic nitrogen (C-7), oxygen (C-9) or sulfur (C-8). The aminobenzaldehyde component showed variation in halogen (C-6, C-7, C-12, C-14) as well as two positional pyridine isomers (C-3, C-10).
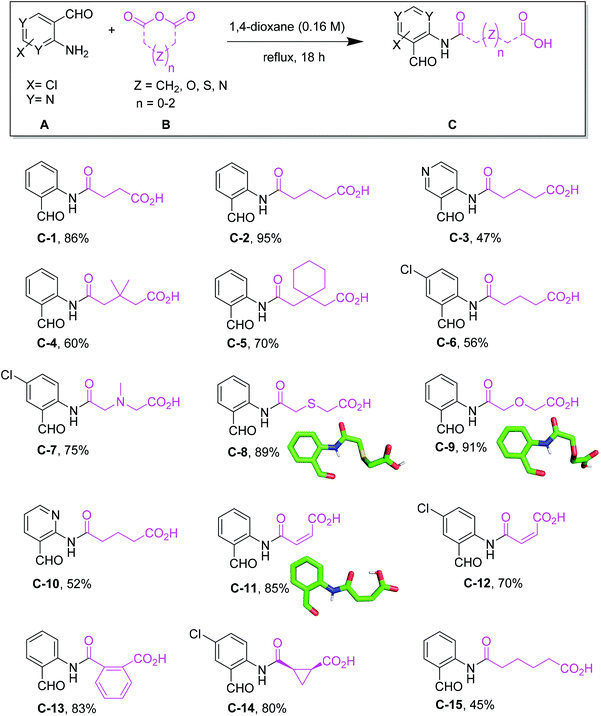 |
| | Fig. 3 Synthesis of α-formyl-ω-carboxyl building blocks C with isolated yields and selected crystal structures. | |
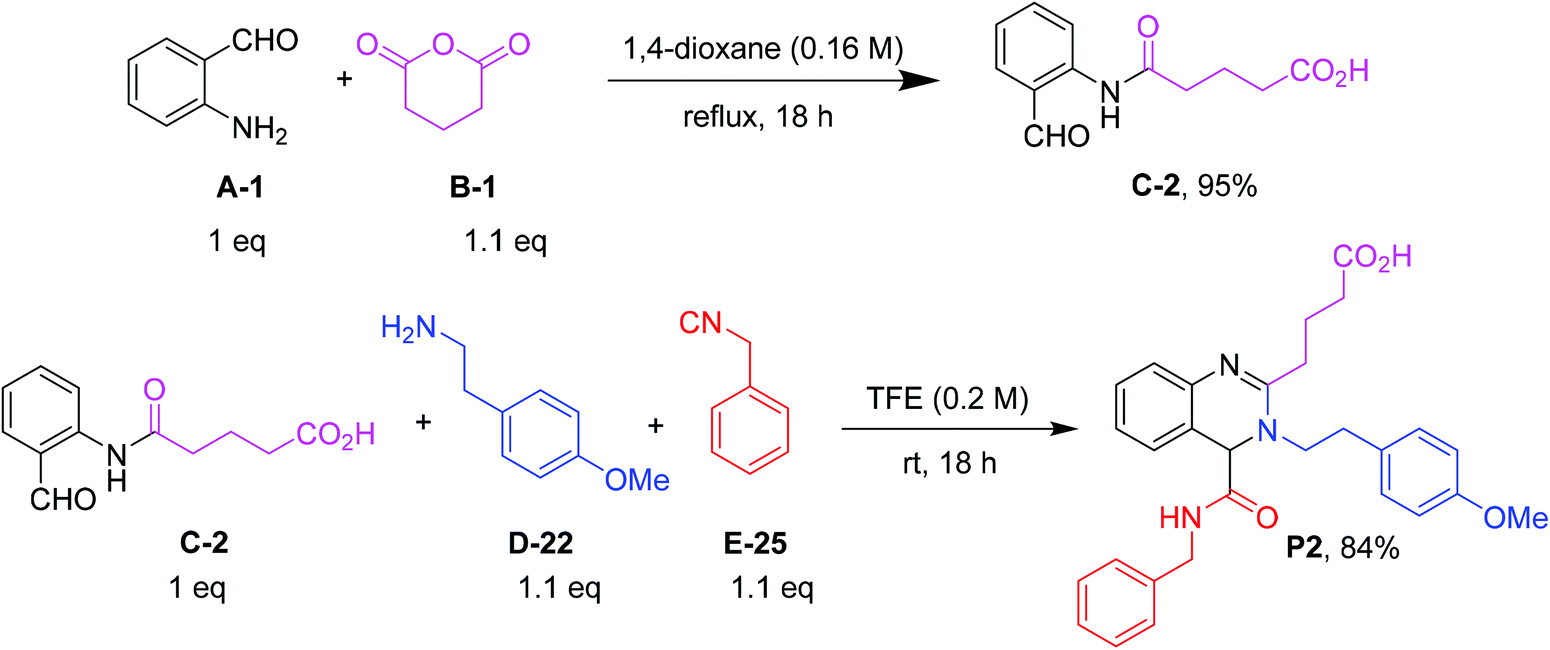
In the next step, we optimized the conditions of the quinazoline ring closure via a Ugi reaction (Fig. 4). The optimization was performed by using 5-((2-formylphenyl)amino)-5-oxopentanoic acid (C-2), 2-(4-methoxyphenyl)ethan-1-amine (D-22), and benzyl isocyanide (E-25) as a model reaction (ESI Table S2†). We probed various parameters including solvents, acid additives, ratios of reactants, microwaves, and sonication. Polar aprotic solvents such as THF and CH3CN gave the product in moderate yields of 30% and 22%, respectively, at room temperature, but methanol and trifluoroethanol showed better results. As trifluoroethanol (TFE, 76% yield) was slightly superior to MeOH (71% yield), we chose TFE for further scope and limitation studies. Next, different acids such as ZnCl2, Sc(OTf)3, Al(OTf)3, and p-TSA were screened. It was found that the addition of Lewis or Brønsted acids does not make a great difference in the percentage yield of products. Additionally, we investigated different stoichiometries of α-formyl-ω-carboxylic acids, amines and isocyanides. Using less than 2 equivalents of amine and isocyanide does not have any effect on the percentage yield, but it reduces the amount of unreacted starting materials. Moreover, we investigated sonication and microwave conditions, however the reaction works best at room temperature.
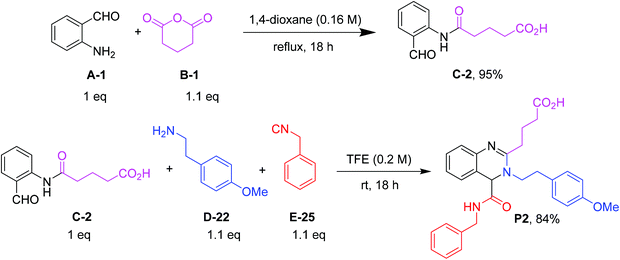 |
| | Fig. 4 Optimized conditions for the anhydride ring opening (6 mmol scale) and U-4CR (1 mmol scale). | |
With the optimized reaction conditions in hand, the scope and limitations of the Ugi-quinazoline reaction were further investigated by synthesizing large libraries of compounds using nanoscale automated chemistry (Fig. 5).
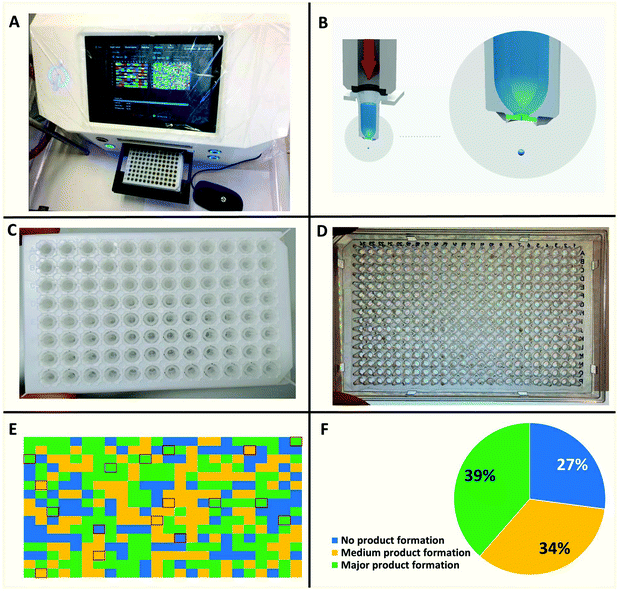 |
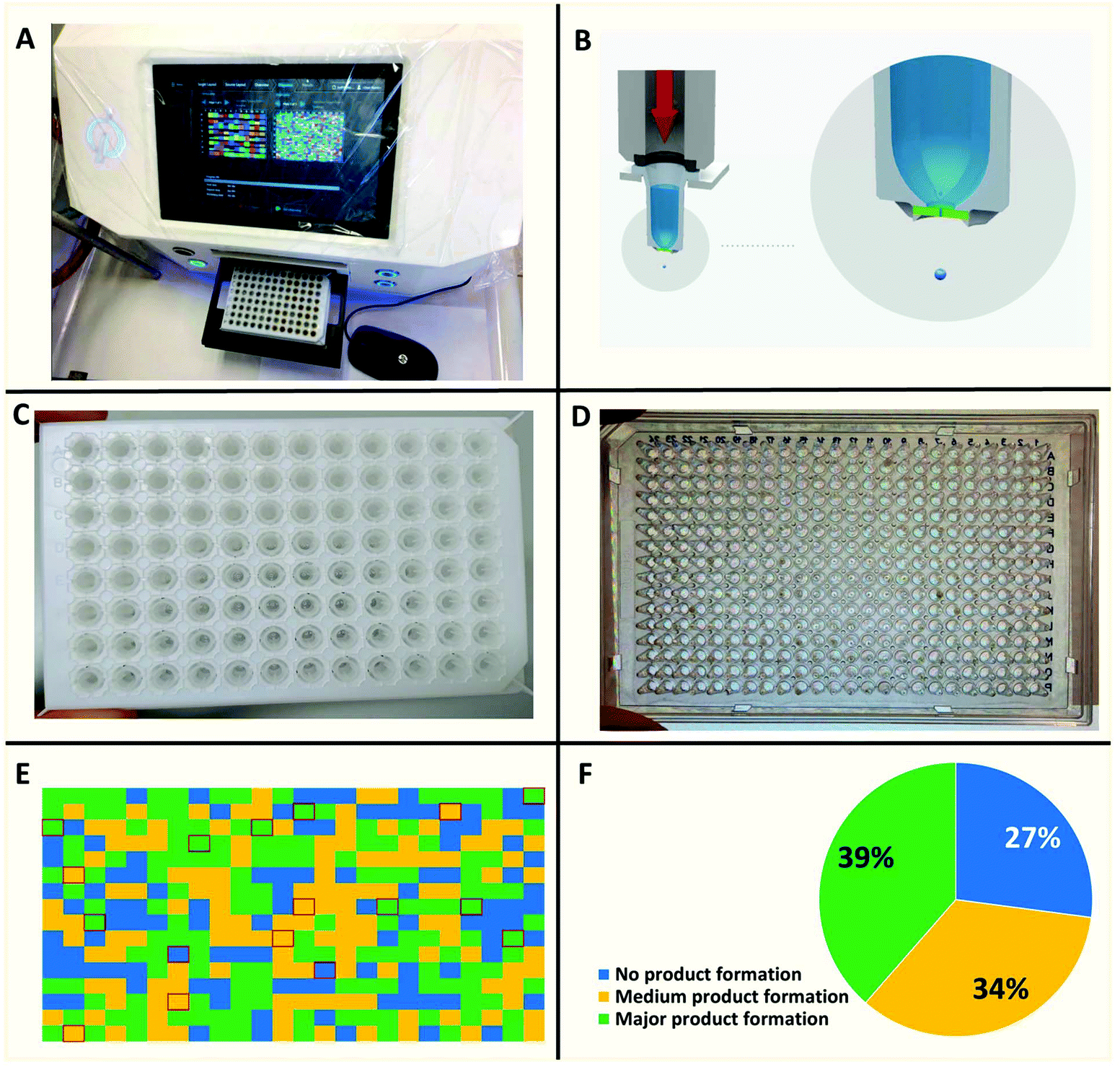
| | Fig. 5 Nanoscale synthesis of a library of quinazolines. (A) I-DOT nano dispenser instrument. (B) The “Immediate Drop on Demand Technology” (I-DOT) uses a non-contact, pressure-based dispensing technology. Applying a well-defined pressure pulse on top of a 96-well microliter plate with holes in the bottom of each well forms a highly precise nanoliter droplet that is released into any target plate. (C) A source 96-well plate with isocyanide and α-formyl-ω-carboxylic acid building blocks. (D) A 384-well polypropylene target plate. (E) A heat plot of a representative 384-well plate based on MS analysis. (F) A pie chart of the overall synthesis success of the 384-well plate. | |
Nanoscale synthesis
Recently we reported nanoscale synthesis of libraries of different scaffolds using acoustic dispensing ejection technology (ADE).16–18 A disadvantage of ADE is the limited solvent compatibility which somehow restricts organic chemical reactions. Currently, only solvents are compatible with a rather high boiling point and a low volatility such as DMSO, water, ethylene glycol and similar solvents. However, Ugi reactions prefer to be performed in low boiling alcohols and in the current reaction we found trifluoroethanol to be favored over other solvents. Therefore, we were evaluating a range of other nano-dispensing technologies which are compatible with a broader range of volatile solvents. The “Immediate Drop on Demand Technology” (I-DOT) is such a nano-dispensing technology and has been mostly applied to analytical and biological questions (Fig. 5A–D).19–22 A well-defined pressure pulse on top of a 96-well microliter plate housing the building block stock solutions with holes in the bottom of each well forms a highly precise nanoliter droplet that is released into any target plate (96-, 384-well or higher formats). The I-DOT source plate is located above the target plate that moves underneath. Each individual source well can dispense into each well in the target plate with a different and precise volume. Up to 96 different liquids can be used within one dispensing run. This approach enables highly efficient combinatorial dispensing useful in chemistry. Larger volumes are achieved by applying up to 400 pulses per second. As the ADE system, the platform works contact-less and is sustainable as it avoids the use of plastic tips. Using in-house written software, we synthesized non-combinatorial libraries in 384-well plates by combining three building blocks in each target plate well.
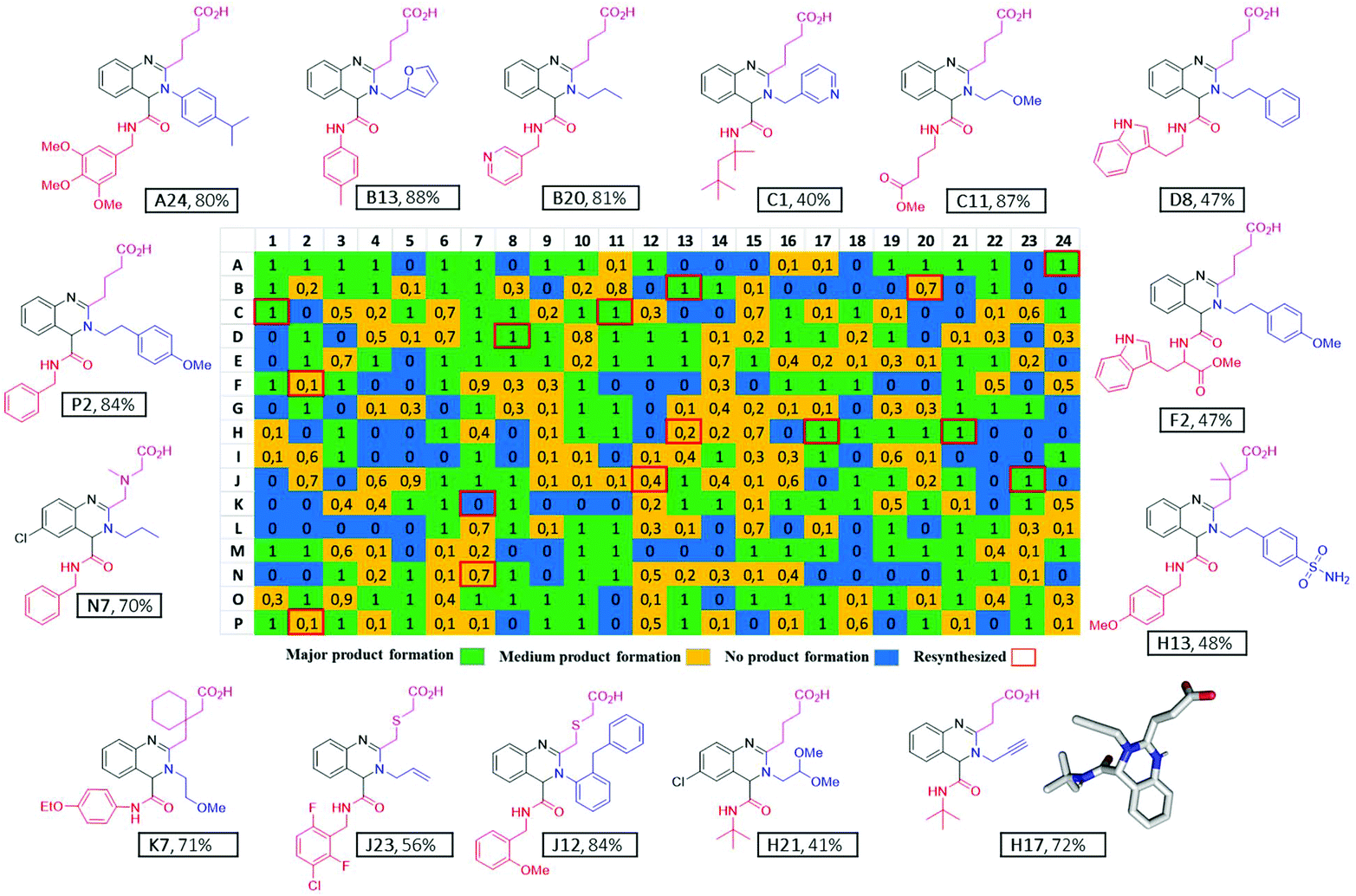
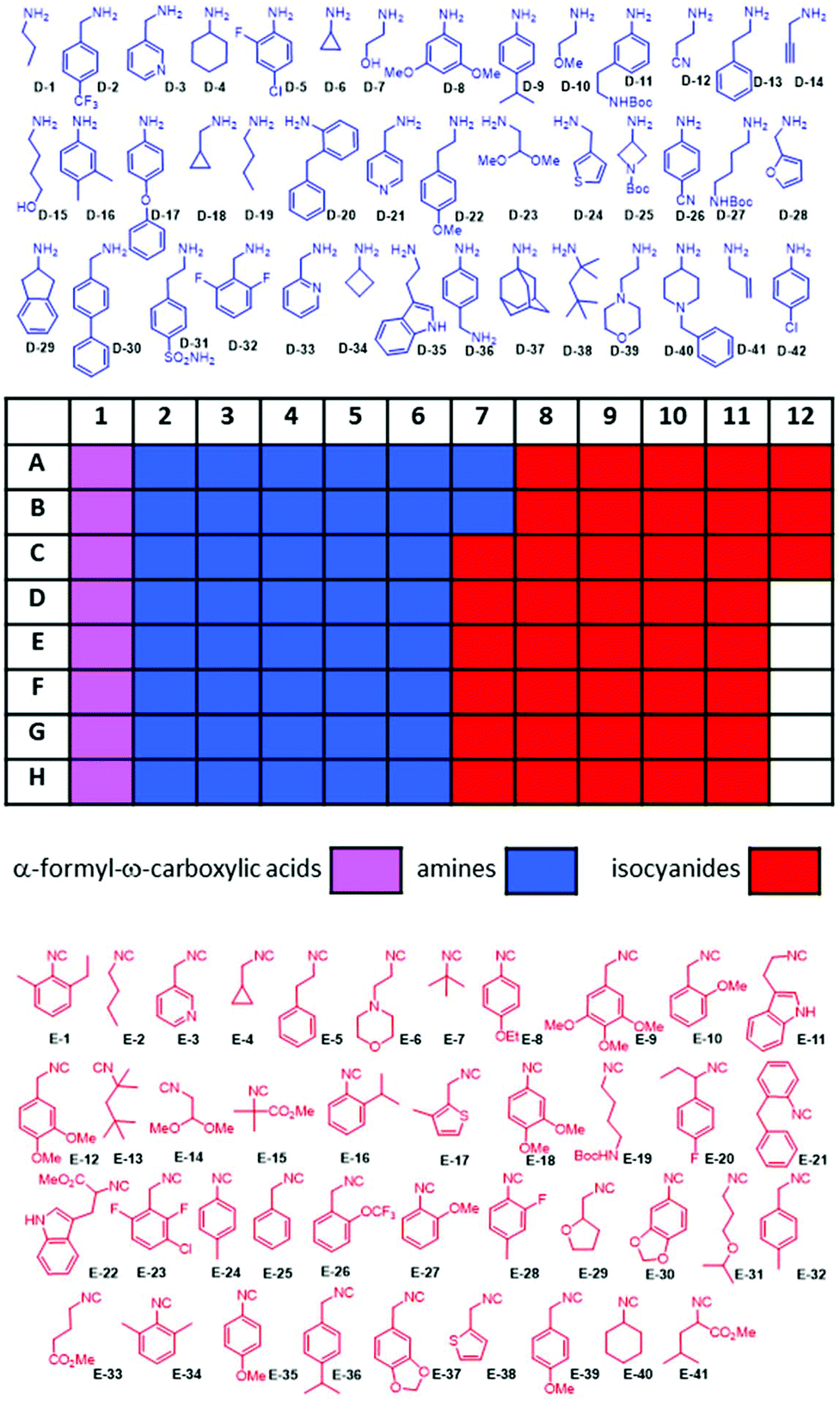
The reactions were performed at 0.16 M concentration with a total volume of 500 nL per well by dispensing TFE stock solutions of the building blocks into the target plate wells. 42 Primary amines and 41 isocyanides were used to build the libraries (Fig. 7). The time to fill a 384-well plate with the three starting material classes of building blocks was less than 20 min. The reactions were run at room temperature for 24 h by sealing the plates and leaving them on a shaker. Then the plates were desealed and analysis of the wells was performed by injecting the crude mixtures into a mass spectrometer. Using in-house written software, the raw MS data were analyzed for the presence of reaction products. A qualitative three-color classifier was used: blue for no reaction product, yellow for minor product formation and green for major product formation. A typical analysis outcome shown in Fig. 6 resulted in 39% major product and 34% minor product formation and 27% failed reactions (Fig. 5F). A total of three 384-well plates were filled resulting in 1.152 reactions. The used building blocks go much beyond what is usually employed during the report of a new reaction and scaffold. However, it is important to challenge new reactions with many different steric and electronic building block variants to gain a ‘real-world’ insight into the scope and limitations.
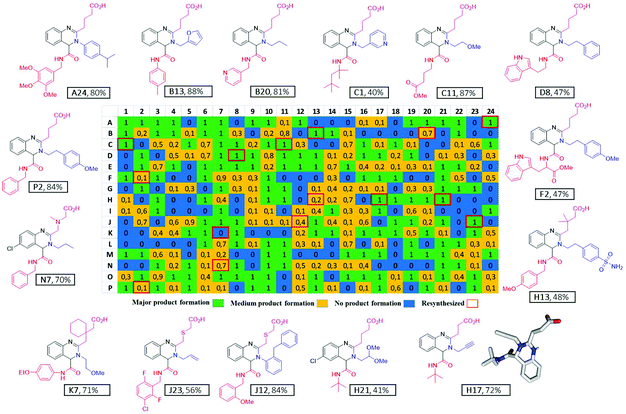 |
| | Fig. 6 Representative 384-well plate with compounds resynthesized on a mmol scale with isolated yields and an exemplary X-ray structure. (Note: K7 is shown blue because of I-DOT reagent transfer failure, but their synthesis worked nicely.) | |
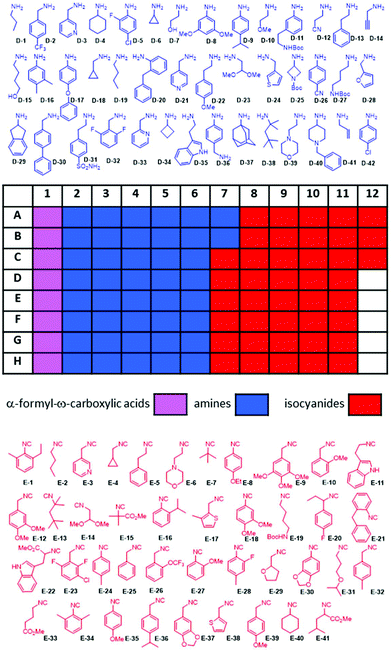 |
| | Fig. 7 96-Well source plate and the color-coded structures of the amine (blue) and isocyanide (red) building blocks used. The α-formyl-ω-carboxyl building blocks are shown in Fig. 3. | |
Of note is the great functional group tolerance of the new quinazoline synthesis: dimethoxyacetale (D-23, E-14), heterocycles such as tetrahydrofuran (E-29), indole (D-35, E-22), thiophene (D-24, E-38), morpholine (D-39, E-6), piperidine (D-40), azetidine (D-25), pyridine (D-3, D-21, D-33, E-3), furan (D-28), alkyne (D-14), alkene (D-41), primary alcohol (D-7, D-15), ether (D-10, D-17, E-31), Boc-protected amine (D-11, D-25, D-27, E-19), ester (E-15, E-22, E-41), nitrile (D-12), sulfonamide (D-31), and tertiary amine (D-40); aliphatic cycles from 3–6 membered (D-6, D-34, D-29, D-4), highly substituted benzene (E-23) and a very bulky adamantly group (D-37).
Mmol and gram scale synthesis
Scalability of a given reaction is important. For example, during the optimization process of a lead compound, a gram amount of high-quality material is needed for performing many different biological assays including pharmacokinetic studies in animals. Scalability is more complex than just making larger quantities of a particular compound. It is well known that many reactions are difficult to reproduce (yield, impurity profile, reaction setup, course, and workup), pose safety concerns (exotherms, decomposition profiles), and exhibit issues in waste handling (limited number of acceptable solvents), cost (yields, expensive stoichiometric reagents), sourcing (suppliers), and ease of operation (avoiding chromatography, fractional vacuum distillation, etc.). Thus, we investigated the scalability of our novel quinazoline synthesis from the nano scale to gram scale. Resynthesis of several compounds on a mmol scale confirmed the excellent performance of the new MCR quinazoline synthesis. 15 compounds could be isolated in 40 to 88% yields with an average of 66% and fully characterized by NMR, HRMS, IR and single-crystal X-ray crystallography (ESI page 76†), (Fig. 6).
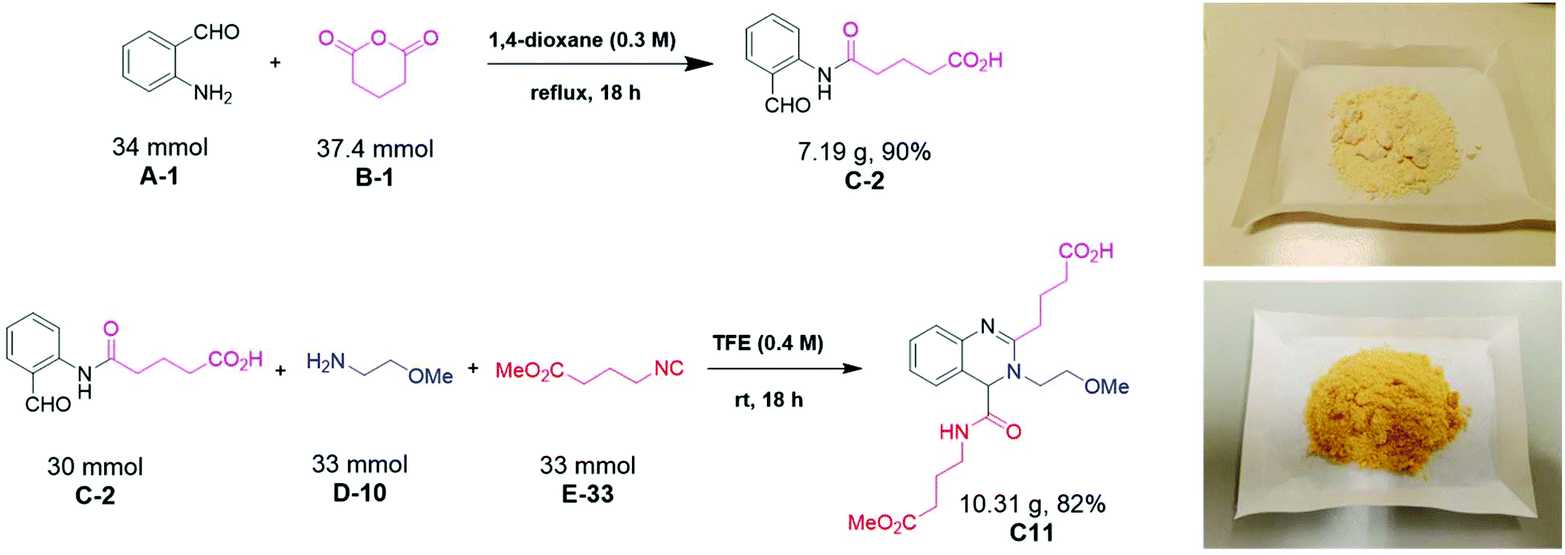
Next, we investigated the scalability of the reaction beyond 1 mmol (Fig. 8). The first acylation step was performed in refluxing dioxane to isolate 90% of α-formyl-ω-carboxylic acid (C-2) on a 7 g scale as a pale-yellow powder. The Ugi reaction in trifluoroethanol as a solvent at room temperature gave C11 in >80% yield and >10 g of a yellow solid. Thus, we could show that our newly discovered quinazoline synthesis is easily scalable over 6 orders of magnitude from nmol (81 nmol) to mmol (30 mmol).
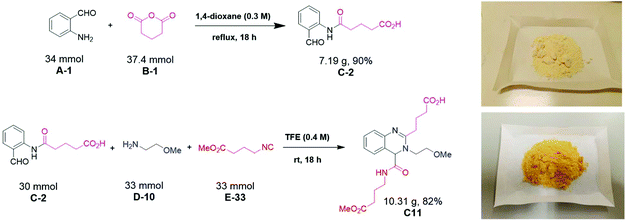 |
| | Fig. 8 Multicomponent quinazoline synthesis on a 10 g scale. | |
A mechanism of this unusual Ugi reaction is proposed in Fig. 9. Accordingly, a Schiff base (I1) is formed from the primary amine (D) and the α-formyl-ω-carboxylic acid (C) which is protonated (I2) and thereby activated towards the nucleophilic attack of the isocyanide forming the nitrilium ion (I3). Nucleophilic attack of the carboxylate to the nitrilium carbon (I4), followed by a transannular attack of the secondary amine to the benzamide carbonyl (I5) followed by dehydration yields the tricyclic intermediate (I6). Unusual in this mechanistic proposal is the avoidance of the common Mumm rearrangement (I4) which would give a strained medium cycle intermediate (ring size between 9 and 11, dependent on the ring size of the anhydride). We hypothesize that the medium cycle is very unstable based on the considerable antiperiplanar and transannular ring strains. The ring strain in medium sized carbocycles has been intensively investigated and described.23,24 Water instead hydrolyzes the N-heteroanhydride structure (I6) and the tricyclic bridgehead structure forms a favorable 6-membered bicyclic product (P).
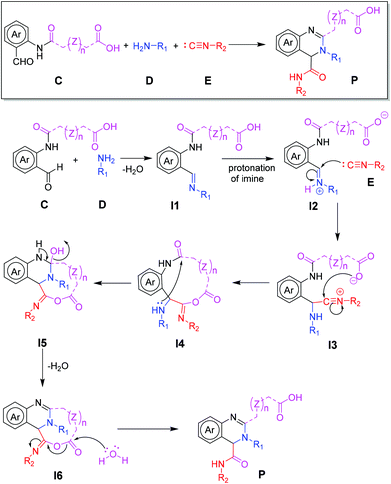 |
| | Fig. 9 Hypothesized reaction mechanism of this Ugi MCR. | |
Chemoinformatics
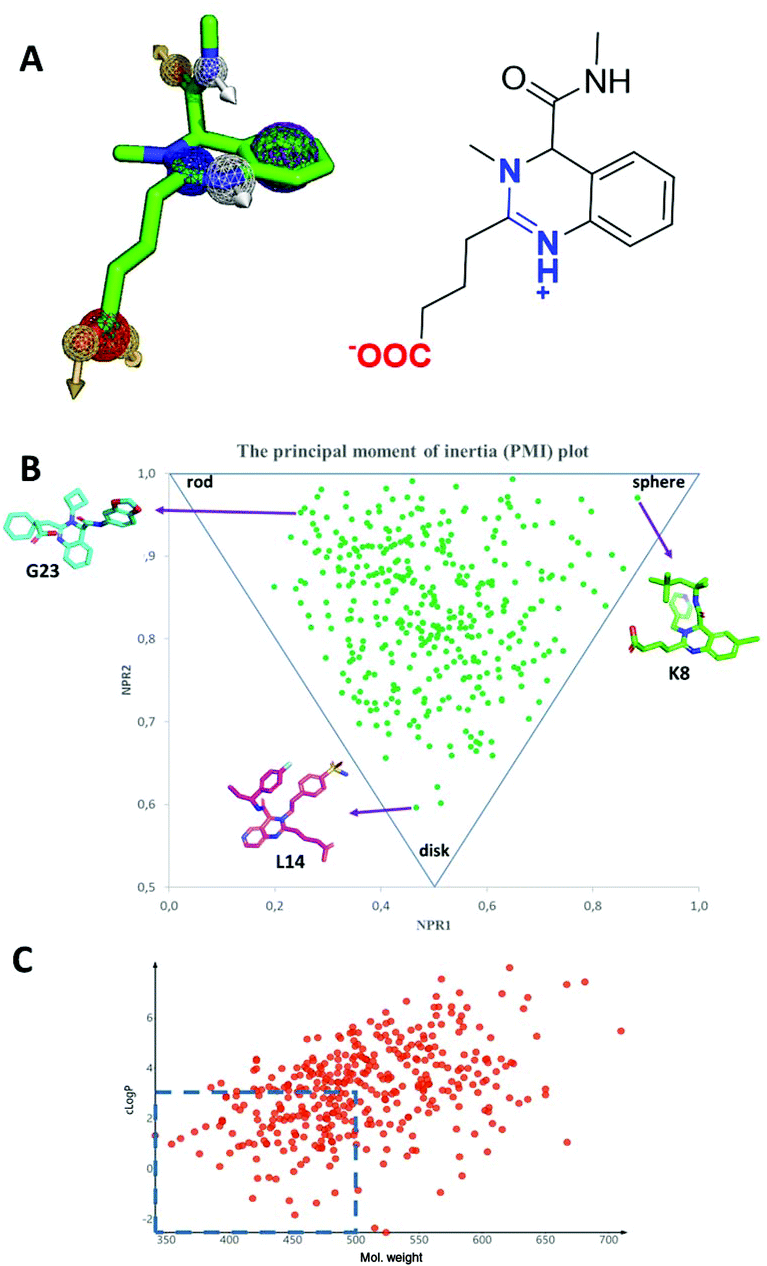
A simplified pharmacophore model and some calculated properties for the novel quinazoline scaffold are shown in Fig. 10. The compounds are zwitterions with a positive and negative charge area. The isocyanide-derived amide group lies orthogonal to the quinazoline ring plane, thus featuring a strong 3D character of the scaffold. The density and orientation of the scaffold-intrinsic pharmacophores provide ample opportunities to interact with biological receptors. The ‘escape from flatness’ 3D property (Fsp3) has recently been proposed to contribute to the drug-likeliness of small molecules (Fig. 10B).25 Calculation of some physicochemical parameters features additionally drug-like behavior. The lipophilicity and molecular weight are two other important determinants. The molecular weight and the lipophilicity of a considerable part of the synthesized library are in a ‘preferred’ region (Fig. 10C). Due to the scaffold intrinsic charge it can be expected that the molecules will not easily pass the cell membrane. However, many known charged bioactive compounds can pass the membrane if a transporter mechanism applies.
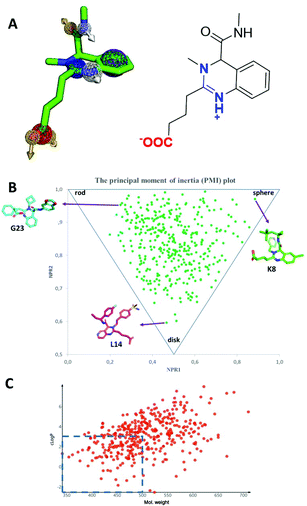 |
| | Fig. 10 Some chemoinformatic features of the quinazoline scaffold. (A) Pharmacophore model featuring positive and negative charges, an extended aromatic pi system, and an exocyclic secondary amide group with a hydrogen bond donor and an acceptor. (B) Moment of inertia analysis of the synthesized library as a measure of complexity (Fsp3) with three explicit compounds (G23, K8, L14) shown. (C) Lipophilicity/molecular weight analysis indicating a considerable portion of the quinazoline chemical space in the ‘preferred’ region MW < 500 and clogP < 3. | |
The accessibility and availability of small organic molecules with a high functional group density as seen in the structural complexity of pharmaceuticals presents a significant challenge to modern synthetic reactions. Many published methods that work well on simple substrates often fail when attempts are made to apply them to complex drug intermediates. Although the number of compounds synthesized in the industry is steadily increasing over the years, a heavy population bias towards only a few chemical reactions can be observed. It was noted that a limited arsenal of medicinal chemistry reactions may narrow the available chemistry space to structurally similar compounds which spill over to perhaps (unwanted) biased screening collections. Therefore, novel synthetic methods, e.g. multicomponent reactions, may not only unlock access to previously unattainable chemical matter, but also inspire new concepts as to how to design and build chemical matter.26 Our herein introduced quinazoline synthesis comprises an example of high throughput experimentation on a nano scale allowing for a rapid screening of the scope and limitations of a unique reaction towards a novel scaffold.27 The two-step synthesis involves in the first step a macroscopic gram-scale acylation of o-amino benzaldehydes or corresponding heterocycles with cyclic carboxylic acid anhydride to afford α-formyl-ω-carboxylic acid building blocks, followed by a microscopic nano-scale Ugi reaction to form highly substituted quinazolines according to an unprecedented mechanism. The minute reagent and solvent consumption of ∼50 mg of building blocks and ∼0.6 ml of solvent is a feature of this work contributing to the sustainability of the nano synthesis approach described herein. The nanoscale synthesis was facilitated by a positive pressure dispenser (I-DOT) and >1000 reactions could be processed in several 384-well plates in a short time and in an automated fashion. The nanoscale synthesis and the tip-less feature of I-DOT additionally contribute to lowering the synthetic chemistry footprint. MS analysis of the plates revealed that 1/3 of the expected products were formed well, while 1/3 where not formed at all. The automation aspect increases safety as chemists are less in contact with hazardous chemicals. The scalability of the nano reactions was shown by the resynthesis of 15 quinazolines on a mmol scale in generally good to very good yields. One quinazoline was even prepared on a 10 g scale in 73% overall yield (over two steps). Moreover, the scope of the building blocks was shown to be great and many functional groups were tolerated. The compounds of the library were found to have all over benign properties (MW, clogP and Fsp3) and the compounds were found to be stable at room temperature and in aqueous buffer at 37 °C. We are currently testing the compound libraries against therapeutic targets and screening results will be reported in due course.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research has been supported by ITN “Accelerated Early stage drug dIScovery” (AEGIS, grant agreement No 675555). Moreover, funding was received from the National Institute of Health (NIH) (2R01GM097082-05), the European Lead Factory (IMI) (grant agreement number 115489), the Qatar National Research Foundation (NPRP6-065-3-012) and COFUNDs ALERT (grant agreement no. 665250) and Prominent (grant agreement no. 754425). SS is supported by a KWF Kankerbestrijding grant (grant agreement no. 10504).
References
- P. T. Anastas and J. B. Zimmerman, Curr. Opin. Green Sustainable Chem., 2018, 13, 150–153 CrossRef.
- P. T. Anastas and J. B. Zimmerman, Chem, 2016, 1, 10–12 CAS.
- P. T. Anastas, ChemSusChem, 2009, 2, 391–392 CrossRef CAS PubMed.
- H. Wong and T. Cernak, Curr. Opin. Green Sustainable Chem., 2018, 11, 91–98 CrossRef.
- M. Follmann, H. Briem, A. Steinmeyer, A. Hillisch, M. H. Schmitt, H. Haning and H. Meier, Drug Discovery Today, 2019, 24, 668–672 CrossRef CAS PubMed.
- I. Aliagas, R. Berger, K. Goldberg, R. T. Nishimura, J. Reilly, P. Richardson, D. Richter, E. C. Sherer, B. A. Sparling and M. C. Bryan, J. Med. Chem., 2017, 60, 5955–5968 CrossRef CAS PubMed.
- L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC.
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
- T. Zarganes-Tzitzikas, C. G. Neochoritis and A. Dömling, ACS Med. Chem. Lett., 2019, 10, 389–392 CrossRef CAS PubMed.
- A. Znabet, M. M. Polak, E. Janssen, F. J. de Kanter, N. J. Turner, R. V. Orru and E. Ruijter, Chem. Commun., 2010, 46, 7918–7920 RSC.
- H. Wehlan, J. Oehme, A. Schäfer and K. Rossen, Org. Process Res. Dev., 2015, 19, 1980–1986 CrossRef CAS.
- A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef.
- A. Hameed, M. Al-Rashida, M. Uroos, S. A. Ali, Arshia, M. Ishtiaq and K. M. Khan, Expert Opin. Ther. Pat., 2018, 28, 281–297 CrossRef CAS PubMed.
- S. Shaabani, R. Xu, M. Ahmadianmoghaddam, L. Gao, M. Stahorsky, J. Olechno, R. Ellson, M. Kossenjans, V. Helan and A. Dömling, Green Chem., 2019, 21, 225–232 RSC.
- C. G. Neochoritis, S. Shaabani, M. Ahmadianmoghaddam, T. Zarganes-Tzitzikas, L. Gao, M. Novotná, T. Mitríková, A. R. Romero, M. I. Irianti, R. Xu, J. Olechno, R. Ellson, V. Helan, M. Kossenjans, M. R. Groves and A. Dömling, Sci. Adv., 2019, 5, eaaw4607 CrossRef PubMed.
- Y. Wang, S. Shaabani, M. Ahmadianmoghaddam, L. Gao, R. Xu, K. Kurpiewska, J. Kalinowska-Tluscik, J. Olechno, R. Ellson, M. Kossenjans, V. Helan, M. Groves and A. Dömling, ACS Cent. Sci., 2019, 5, 451–457 CrossRef CAS PubMed.
- M. Benz, M. R. Molla, A. Böser, A. Rosenfeld and P. A. Levkin, Nat. Commun., 2019, 10, 2879 CrossRef PubMed.
- L. F. Arenas da Silva, L. Schober, M. Sloff, A. Traube, M. L. Hart, W. F. Feitz and A. Stenzl, Cytotherapy, 2015, 17, 1655–1661 CrossRef CAS PubMed.
- G. J. Van Berkel, V. Kertesz and H. Boeltz, Bioanalysis, 2017, 9, 1667–1679 CrossRef PubMed.
- X. Zhang, S. Garnerone, M. Simonetti, L. Harbers, M. Nicos, R. Mirzazadeh, T. Venesio, A. Sapino, J. Hartman, C. Marchio, M. Bienko and N. Crosetto, Nat. Commun., 2019, 10, 4732 CrossRef PubMed.
- S. J. Brooks, S. E. Garcia-Garrido, M. E. Light, P. A. Cole and P. A. Gale, Chem. – Eur. J., 2007, 13, 3320–3329 CrossRef CAS PubMed.
- S. D. Appavoo, S. Huh, D. B. Diaz and A. K. Yudin, Chem. Rev., 2019, 119, 9724–9752 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed.
- S. M. Mennen, C. Alhambra, C. L. Allen, M. Barberis, S. Berritt, T. A. Brandt, A. D. Campbell, J. Castañón, A. H. Cherney, M. Christensen, D. B. Damon, J. Eugenio de Diego, S. García-Cerrada, P. García-Losada, R. Haro, J. Janey, D. C. Leitch, L. Li, F. Liu, P. C. Lobben, D. W. C. MacMillan, J. Magano, E. McInturff, S. Monfette, R. J. Post, D. Schultz, B. J. Sitter, J. M. Stevens, I. I. Strambeanu, J. Twilton, K. Wang and M. A. Zajac, Org. Process Res. Dev., 2019, 23, 1213–1242 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1968957–1968959 and 1972493. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0gc00363h |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Shabnam
Shaabani‡
a,
Shabnam
Shaabani‡