
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Separation and purification of biomacromolecules based on microfluidics
Filipa A.
Vicente
 a,
Igor
Plazl
bc,
Sónia P. M.
Ventura
*a and
Polona
Žnidaršič-Plazl
*bc
a,
Igor
Plazl
bc,
Sónia P. M.
Ventura
*a and
Polona
Žnidaršič-Plazl
*bc
aCICECO – Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal. E-mail: spventura@ua.pt
bUniversity of Ljubljana, Faculty of Chemistry and Chemical Technology, Večna pot 113, SI-1000 Ljubljana, Slovenia. E-mail: polona.znidarsic@fkkt.uni-lj.si
cUniversity of Ljubljana, Chair of Microprocess Engineering and Technology – COMPETE, Večna pot 113, SI-1000 Ljubljana, Slovenia
First published on 19th March 2020
Abstract
Separation and purification of biomacromolecules either in biopharmaceuticals and fine chemicals manufacturing, or in diagnostics and biological characterization, can substantially benefit from application of microfluidic devices. Small volumes of equipment, very efficient mass and heat transfer together with high process control result in process intensification, high throughputs, low energy consumption and reduced waste production as compared to conventional processing. This review highlights microfluidics-based separation and purification of proteins and nucleic acids with the focus on liquid–liquid extractions, particularly with biocompatible aqueous two-phase systems, which represent a cost-effective and green alternative. A variety of microflow set-ups are shown to enable sustainable and efficient isolation of target biomolecules both for preparative, as well as for analytical purposes.
Introduction
Over the past two decades, microfluidic devices have been the focus of numerous studies due to their ability to process fluids either for analyses, reactions or separations in a very efficient and controllable way. The benefits of these devices, typically having at least one characteristic dimension in the range of micrometres and thus, high surface to volume ratio, comprise small amounts of sample and reagents needed, very efficient mass and heat transfer and controlled process conditions.1,2 Microfluidics have shown outstanding breakthroughs in several fields comprising chemistry, biotechnology, biomedicine and process engineering.Tremendous improvement in high-throughput bioprocess development and the productivity of biotransformations and fermentations, as well as recent trends towards continuous production in pharma and fine chemicals production, exposed the downstream processing as a manufacturing bottleneck. This is particularly evident in the production of biopharmaceuticals such as monoclonal antibodies and therapeutic enzymes, where purification could reach up to 90% of total production costs. Novel protein-based drugs for treatment of previously untreatable diseases boosted unprecedented growth of their market, which is currently hindered by the lack of cost-efficient and controlled product isolation.3 Furthermore, protein extraction from marine organisms (e.g. phycobiliproteins),4 plants5 (e.g. recombinant pharmaceutical proteins, such as human growth hormone, recombinant human intrinsic factor, hepatitis B virus antigen, etc.) and wastes (e.g. lactoferrin from whey) have gain an increased attention as they can be used in various applications comprising medicine, food industry and cosmetics. On the other hand, high-throughput and high-efficiency separation and purification processes to be applied on proteins and nucleic acids are needed in diagnosis, or for biochemical characterization of cells and biological material. Sample volumes used in general biology and medical research are becoming smaller and concentrations are dramatically decreasing.6
In all cases, accomplishment of high purities and yields of (bio)macromolecules in a short time strongly rely on innovative technological solutions. Miniaturization along with a reduced number of unit operations through process integration leads to intensification, both on the laboratory/analytical scale, as well as at the production level. Microchips have been in the past two decades successfully applied in many bioseparation techniques comprising cells separation (reviewed in ref. 7), cell lysis and/or extraction (reviewed in ref. 1, 8 and 9), blood fractionation (reviewed in ref. 10), integrated isolation of products of biocatalytic processes (partly reviewed in ref. 11 and 12), steroid extraction,13 antibiotics purification,14 in capillary electrophoresis for protein separation (reviewed in ref. 15), as well as in chromatography (reviewed in ref. 16–18). Several of these applications focused on the use of microfluidic devices for process intensification and reduction of sample volumes. Therefore, their adoption typically resulted in a decrease of processing costs along with the reduction of the reagents/solvents required and time needed to pursue separation. Thereby, the energy costs and environmental impact of the processes were also reduced, as previously reviewed.3,19
This review aims at discussing some basic phenomena underlying benefits of microfluidic devices and their use in extraction and separation of proteins and nucleic acids with the emphasis on liquid–liquid extraction (LLE). The use of more biocompatible and non-destructive solvent systems such as aqueous two-phase systems (ATPS) applied on the sustainable processing of biomacromolecules is also highlighted. A short overview of other microflow-based separation techniques for selected biomacromolecules is given along with some outlook and current trends in the field.
Process intensification via miniaturization
The emergence of microreactor technology and process intensification through miniaturization has provided a new platform for accelerating the development of the next generation of chemical and biochemical process technologies. The process intensification provides insights into different scales and can be defined as the development of novel and sustainable equipment that, compared to the state-of-the-art, results in dramatic process improvements related to equipment size, waste production, energy consumption and other factors.20 The application of microreactor technology in (bio)chemical processes meets these criteria, with known reduction of the equipment size. However, besides spatial benefits, microfluidic devices also provide enhanced heat and mass transport, safety, environmental impact, and others.6 An obvious effect of shrinking a system to the micrometre scale is the large increase in surface area relative to volume, often by several orders of magnitude. Specific surface areas of microstructured devices lie between 1 × 104 and 5 × 104 m2 m−3, while those of traditional reactors are generally about 100 m2 m−3. Decreasing in volume, which typically amount to a few microliters, replaces batch with continuous flow processes and process parameters such as pressure, temperature, residence time, and flow rate are more easily controlled for processes that take place in small volumes.21 A key advantage that miniaturization brings is the knowledge and ability for building microscale systems in a controlled and repeatable manner.22Fluid flow at the microscale
The microfluidics concept was firstly proposed in 1969 by Lew and Fung23 without being completely aware of the microfluidic phenomenon per se. These authors demonstrated that the (micro)circulation flow within the blood vessels and the air flow within the bronchioles and alveolar ducts and sacs of the lungs were subjected to a change upon the entry (inlet) of a new vessel or branch, respectively, and that this flow was determined by the low Reynolds numbers (Re). At this point, it was established the main phenomenon dictating the flow pattern in natural microscale conditions, though, only later researchers became aware of the benefits of working in microscale and start to understand it.Nowadays, microfluidics is a research field that develops methods and devices to control, manipulate, and analyse flows on nano- to microliter scales.21 The main features of microscale systems are reflected in fluid dynamics therefore, the understanding of fundamental mechanisms involved in fluid flow characteristics at the microscale is essential since their behaviour affects the transport phenomena and microfluidic applications. Fluid behaviour at the microscale is increasingly influenced by viscosity rather than inertia. Viscosity, the internal friction of a fluid, produces a resistance to shear and a tendency for the fluid to move in parallel layers known as laminar flow, while the inertia, tendency of a body in motion to retain its initial motion, counters laminar flow and can ultimately result in turbulent flow.21,22,24 The laminar flows cause velocity profiles in the microchannel to appear typically parabolic in shape, which can lead to a relatively broad residence-time distribution. While the ratio of inertial to viscous forces is related with the Re, the capillary number (Ca) represents the relative effect of viscous drag forces versus surface tension forces acting across an interface between a liquid and a gas, or between two immiscible liquids. While the dynamics of single-phase flow in microchannels is very similar to that in large diameter channels, this is not the case for multiphase flow. A deeper understanding is needed to describe multiphase flow, where the modelling-based study of multiphase flow fundamentals at the microscale plays a key role.
Flow pattern, such as parallel flow, droplets, segmented or slugs and annular flow, depends upon the interaction amongst the gravitational, interfacial, inertial and viscous forces. Therefore, flow pattern, together with the pressure drop, represents the most important characteristics of multiphase flow in micro channels. In the microfluidics, the surface forces that govern the physical phenomena, like surface tension, frictional or viscous force, wall adhesion and wall wettability etc., become significant in multiphase flow at the microscale. Thus, the surface forces in the microfluidic devices are dominant compared with body forces.25 The microchannel geometry and its inner surface properties (roughness, hydrophobicity) also contribute for the establishment of a stable flow pattern of multiphase flow.
The most typical flow patterns of two-phase flow used for bioprocess extraction, separation and purification in microfluidic devices are parallel, droplet or slug flow. In slug flow of two immiscible liquids, the continuous phase of a liquid is segmented by discrete droplets of the distributed liquid phase. Slug flow is known by several other names, such as Taylor flow, plug flow and most often, segmented flow.26 Segmented flow is increasingly being used in various industrial processes due to its unique hydrodynamic characteristics. Mass transfer between the two phases is enhanced by internal recirculation within the liquid slug and droplet of the distributed liquid phase, the large interfacial area and the small diffusion paths.24,26,27 To describe the extraction process in microfluidics based on segmented flow, the convective transport and diffusion in all three directions within the droplet and slug must be taken into account, while the mass transport of extraction component through the interphase surface is defined by diffusion and partition coefficients.28
Y-shaped inlet channels (Fig. 1A–C) allow the typical formation of parallel laminar flow, with the two immiscible liquids being introduced concurrently through both inlets and moving alongside until the exit. However, all the previous parameters need to be properly addressed to maintain this microflow stable along the full microchip length. This will be also crucial to achieve a solute concentration equilibrium and allow a complete phase separation at the chip outlets. A third inlet/outlet can be introduced as well in the chip (ψ-shaped microchannel; Fig. 1D–F), inducing the formation of a second interface in the system. In turn, an increase on the interfacial area occurs, allowing a more efficient mass transfer. From the extraction and/or purification point of view, the major advantage of this fluid flow system is that there is the phase separation at the channel exit, allowing the recovery of the phase containing the compound of interest to be further processed, while the contaminants-based phase can be discarded. As a downside, parallel laminar flow only allows mass transfer across one or two parallel interfacial areas by diffusion, demanding long residence times and thus, long micro-channels are required to achieve a complete separation.
 | ||
| Fig. 1 Different examples of microfluidic devices applied for parallel (A to F) and segmented flows (VII to XI). (A) to (C) and (D) to (F) are a representation of a laminar flow within a microchip with two and three inlets/outlets with a straight and serpentine main channel, respectively. (G) to (K) evidence the two most common conformations originating a segmented flow, namely T-shape/branch conformation (G and H) and flow focusing geometry (I to K). Segmented flow can display distinct regimes, for instance slugs (I) and droplets (G, H, J, K). | ||
Parallel flow pattern can be shifted to a segmented microflow following some changes in the merging conformation of the inlets, namely by creating a perpendicular conformation, also known as T-shape conformation or T-branch (Fig. 1G and H), or through the so-called flow focusing geometries (Fig. 1I–K). In the first scenario, the organic and aqueous phases are put in contact through a 90° angle, leading to the formation of small droplets. In the second case, there is the creation of a three inlets microchip with two immiscible liquids flowing simultaneously: one in the central/inner channel and the other in the outer channels, preferably contacting at a 90° angle. After merging but not mixing, the fluids pass through a small orifice, leading to the droplet formation. In both cases, the formation of droplets/slugs is originated due to the generation and competition of shear forces and surface tensions between the aqueous and the organic phases at this junction. At this point, there is some flow instability and non-linearity, thus leading to droplet formation (spherical droplets not touching the channel walls, Fig. 1G–K), slug/plug-flow (elongated droplets touching the channel walls, Fig. 1I) or annular-flow (thinner and longer slugs do not touching the walls), as reviewed in ref. 29 and 30. When parallel to segmented flow transitions occur, there is a considerable increase in the surface to volume ratio, leading to a higher mass transfer and a shorter residence time. These account for the major advantages of this flow pattern. In contrast, the greatest drawback might be the continuous/disperse phases separation at the outlets.31–34
Integration of microfluidic units
Another key feature of microflow devices is their ability to combine different unit operations either in a consecutive streamline leading to end-to-end processing,35 or on a single chip (Lab on a chip). The latter approach has been widely exploited in analytics by developing a variety of micro total analysis systems (μTAS).Recent trends towards continuous manufacturing systems in pharma and fine chemicals production, supported also by FDA's recommendation, clearly opens the space for integrated processing and control. The use of highly adaptable smaller equipment with real-time monitoring could result in lower production costs, improved product quality, increased safety and shorter processing times.11,35 Furthermore, such systems allow distributed and on-demand manufacturing, preventing shortages of drugs and chemicals, as well as reduced formulation complexity relative to tablets needing yearlong stability.35
Development of a flexible, plug-and-play platform capable of complex multistep synthesis, multiple in-line purifications, post-synthesis work-up and handling, semi-batch crystallization, real-time process monitoring, and ultimately, formulation of high-purity drug products has recently opened up a new era in continuous flow pharmaceuticals production.35 Purification of biomacromolecules like therapeutic proteins, antibodies, enzymes and nucleic acids mostly requires several steps of extraction and polishing, which are typically comprising filtration, centrifugation, membrane technologies, and various types of chromatography, hence yielding high downstream processing costs.36 Liquid–liquid extractions using non-denaturing solvent systems are gaining increased attention as a cost-effective alternative.36,37
Liquid–liquid microextraction
LLE comprises the mass transfer of a solute from the feed, i.e. a liquid containing the molecule of interest, towards the extraction in a solvent immiscible with the feeding phase. The mass transfer of the solute across the interface takes place until the thermodynamic equilibrium is reached. After the separation is completed, the extract comprises the solvent containing the solute, while the raffinate consists of the feeding phase and the remaining solute. As batch extraction is limited by thermodynamic equilibrium, multi-stage continuous operation, usually performed in a counter-current flow, is often applied. For the industrial-scale extractions, mixer-settlers, extraction columns, and centrifugal extractors are commonly used.Typically, LLE is accomplished using an organic solvent to extract the solute from the aqueous phase. However, most organic solvents are hazardous for the biomolecules and the environment.38 As an attractive alternative, aqueous two-phase systems, ATPSs, (also called as aqueous biphasic systems, ABS) emerged as a more benign type of LLE since they are mainly composed of water (65–90%) and do not require the use of organic solvents in the whole process. Mild operation conditions that allow the biomolecules to keep their native conformations and biological activities are thus provided.39 These systems consist of two aqueous solutions of immiscible compounds, for instance two polymers, a polymer and an inorganic salt or an IL, among others.39–41 The ATPSs present highly flexible separation systems, since a vast array of compounds can be used in extractions and purifications providing a good selectivity and yield, as reviewed in ref. 39 and 40.
Due to the previously stated benefits of microflow systems as compared to conventional apparatuses, there has been an increased interest in applying these devices for LLEs. This is evident from the constant increase in the number of publications published per year containing the keywords “liquid–liquid extraction” and “microfluidic devices” or “microfluidics” or “microchips”, shown in Fig. 2. Both co-current and counter-current stratified (parallel) flow patterns have a common advantage over droplet-based flow patterns as they allow the simultaneous phase separation at the exit of the microfluidic device during the extraction process. However, both have as well significant limitations due to the instability of flow patterns and consequently, the low capacity and productivity. Some solutions have been proposed in order to stabilize the co-flowing immiscible streams, like to introduce the membrane, or a series of micro-pillars placed in the extraction channel, which in turn reduces the extraction efficiency. On the other hand, the development of a multistage countercurrent extraction with high effectiveness indicated that it is still challenging to balance the pressure loss with micropumps after every stage. The continuously operated system consisted of integrated devices for highly efficient droplet-based microfluidic liquid–liquid extraction and phase separation is currently a very promising option to replace the conventional macroscale systems in terms of process intensification and to meet high industry expectations.
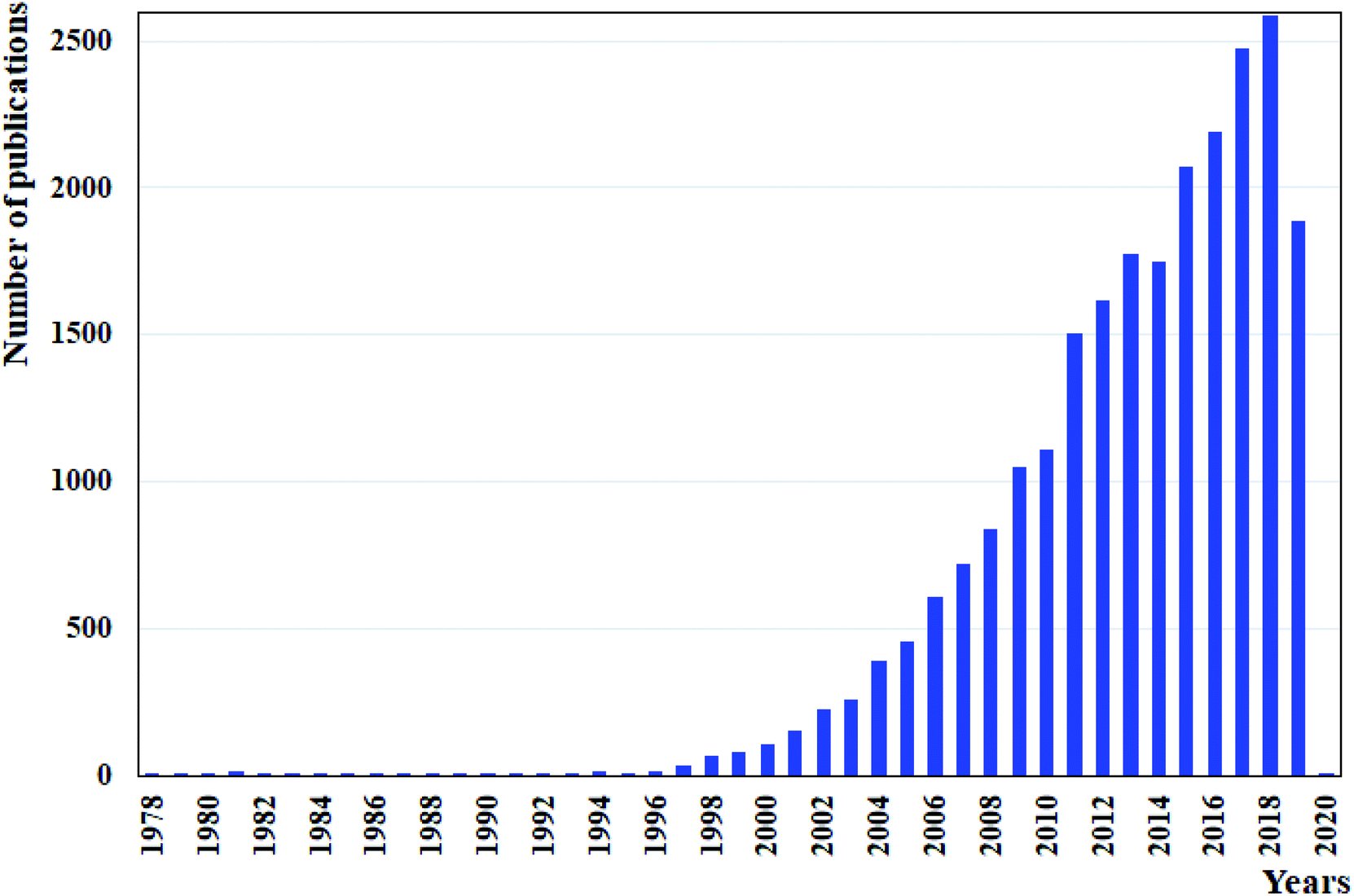 | ||
| Fig. 2 Number of articles published per year on the use microfluidic devices for LLE by using “liquid–liquid extraction” and “microfluidic devices” or “microfluidics” or “microchips” as keywords in Web of Science. Data assessed in October 2019. | ||
Miniaturization of mixing and reaction procedures was already well optimized. However, extraction and separation of compounds within a microfluidic device are still the limiting step to accomplish the entire process in a single microchip. Assmann and co-workers42 have focused their attention on reviewing the strategies to stabilize and promote phase separation while using different types of flow. Instead, we focused on the combining benefits of microfluidic devices and their use in conventional and more sustainable LLE on the separation of proteins and nucleic acids.
Conventional LLE
Organic solvent-based LLE is most often used for the extraction and/or purification of molecules that are stable in these solvents, e.g. steroids13 or dyes.43–45 Since biomacromolecules easily denaturate in organic solvents, there are only a few works reporting the microextraction of proteins and nucleic acids using this methodology, as summarized in Table 1. Zhang et al.46 developed a rapid and high efficient approach for bacterial pathogen identification and quantification, while applying a laminar flow. The authors designed a microfluidic device composed of microwell arrays to selectively extract and purify deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) of both Gram positive and negative bacteria, such as Staphylococcus aureus and Pseudomonas aeruginosa. To evaluate the device efficiency, purified nucleic acids were added to the aqueous phase alongside labelled bovine serum albumin (BSA), used as a model protein to access the amount of protein that might also be retained in the microwells with the nucleic acids. The DNA purification results showed that 92.9% of protein and 93.2% of RNA partitioned to the organic phase (phenol/chloroform/isoamyl alcohol), at a flow rate of 0.45 mL min−1 and pH 8. This result was improved by increasing the flow rate to 0.65 mL min−1, resulting in an almost pure DNA recovery in the aqueous phase. The DNA recovery was also proved to be dependent upon the pH of the organic phase. By reducing the phase pH to 4.6, and only at a flow rate of 0.65 mL min−1, a complete DNA recovery (>99.9%) was obtained in the organic phase, alongside with more than 95% of BSA. Yet, this allows the recovery of 94.2% of a purer RNA in the aqueous phase. In general, these results have evidenced the possibility to manipulate the nucleic acid partition between both organic and aqueous phases by the fine tuning of the flow rates as well as the organic phase pH. Therefore, such results turn this approach into a very appealing option to be applied in real matrices. The real nucleic acid purification was attempted from bacterial lysates, using a cell suspension from 5000 to 5 CFU (colony forming units) and the results were then compared to a column-based solid phase extraction. Herein, the chip presented recoveries between 85% and 100% for both DNA and RNA covering all CFU range, with exception of the Staphylococcus aureus RNA displaying only a recovery between 70 and 80%. On the other hand, recoveries from the alternative method decreased with cell density and attained only recoveries of nucleic acids between 15 and 20%. This study showed the great potential of a microfluidic device to be applied in the biomolecule's extraction field, even in presence of a real and complex matrix, such as a bacterial lysate. Meanwhile, Morales and Zahn47 have analysed the extraction of rhodamine labelled BSA and DNA using both laminar and segmented flow patterns. Herein, the authors used a typical two inlets/outlets chip with a main serpentine channel usually applied for laminar flow to also perform a segmented regime. They realized that, by manipulating the flow rates ratio, both laminar and segmented flows were possible to induce. In laminar regime, the authors found a much higher flow rate for the aqueous phase than for the organic phase, which lead to a capillary number of 0.72. For segmented flow, these were drastically reduced until the flow rates ratio allowed a capillary number of 0.07. At this point, interfacial forces have dominated the viscous forces and slugs were formed. Once this condition was established, the biomolecules extraction occurred allowing to recover 78% of BSA in the organic (phenol/chloroform/isoamyl alcohol) phase, while 8% of DNA was collected in the aqueous phase, for the laminar flow. By replacing this flow pattern for a droplet regime, the biomolecules extraction was enhanced due to the additional convective process. As a result, purities around 96% and 97% for the protein and DNA, respectively, were found. Following these results, the authors adopted segmented flow, shortened the main channel length, and constructed a new microfluidic device by replacing the inlet region for a normal T-shape conformation. This new device was used to study the extraction of an Escherichia coli plasmid by introducing directly the bacterial cell lysate into the microchip. The results were very good, showing recoveries of the genetic material higher than 90%. However, replacing the laminar by the segmented flow within the same device without taking additional measurements such as the outlets coating, the physical separation of both phases at the outlets was not achieved, and thus, the authors have carried the phase separation “off-chip”, representing the main disadvantage of the approach. This study clearly evidenced that segmented flow could be much more advantageous from the mass transfer point of view owing to its higher interfacial area, thus presenting a considerable higher extraction efficiency. However, some of the drawbacks persist, namely the difficulty of phase separation at the outlet, and the absence of a stable profile and a well characterized flow regime.| Molecules extracted | Microfluidic device | Solvents | Ref. |
|---|---|---|---|
| Bovine serum albumin (BSA), DNA and RNA | A polydimethylsiloxane microfluidic device with one inlet/outlet and a main channel in contact with several micro-wells | Organic phase: phenol/chloroform/isoamyl alcohol, aqueous phase: bacterial lysate | 46 |
| Rhodamine labelled BSA, DNA and bacterial plasmid | Two PDMS microchip with two inlets/outlets and a serpentine main channel: one with a Y-shape inlet and the other with a T-shape (cf.Fig. 1) | Organic phase: phenol/chloroform/isoamyl alcohol, aqueous phase: phosphate-buffered saline (PBS) + DNA and BSA | 47 |
| Rhodamine labelled BSA | Two glass microfluidic devices: one with two inlets while the other has three, both converging into a straight main channel with one outlet (cf.Fig. 3A) | Organic phase: phenol/chloroform/isoamyl alcohol, aqueous phase: water + SDS | 48 |
The previous study was a strong indication that the inlets conformation of microchips is not the only factor influencing the type of flow. For example, by changing the flow rates ratio, the flow regime can be shifted. Reddy and Zahn48 corroborated this accomplishment and have shown the importance of using a co-solvent to maintain a stable laminar microflow. Here, two distinct apparatus in a Y-shape conformation with two and three inlets, respectively, were used. The results showed the need of adding an anionic surfactant (sodium dodecyl sulphate, SDS) to the aqueous phase to achieve a stratified flow. Without the surfactant, the interfacial tension between the organic (phenol/chloroform/isoamyl alcohol) and aqueous phases was much higher, preventing a laminar flow. Besides, in its absence, the capillary number was so low that a slug profile was obtained. Thus, at flow rates of 2.5, 1.25, and 2.5 μL min−1 for the aqueous–organic–aqueous phases, respectively, a laminar flow was achieved (Fig. 3A). Though, if this flow rate was reduced to 1, 0.5, 1 μL min−1, the laminar flow became thinner and gave rise to a tortuous jet with droplet ejection (Fig. 3B–D), even in the presence of a surfactant. Afterwards, this microplatform was applied to the BSA extraction from the aqueous phase. The protein diffused towards the organic phase, precipitating in the aqueous–organic interface. However, the extraction of BSA was not complete due to the limited surface area of the interface created. The publications in this direction are summarized in Table 1.
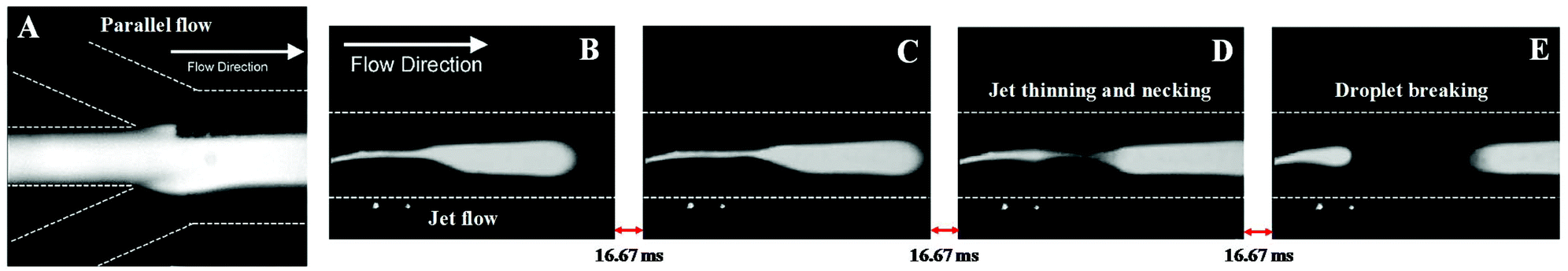 | ||
| Fig. 3 (A) Microchip inlets with laminar flow at flow rates of 2.5, 1.25, and 2.5 μL min−1 for the aqueous–organic–aqueous phases. (B–E) By reducing these flow rates from 2.5, 1.25 and 2.5 to 1, 0.5, 1 μL min−1, respectively, the laminar flow gives rise to a tortuous jet with droplet ejection. Reprinted from Interfacial stabilization of organic–aqueous two-phase microflows for a miniaturized DNA extraction module, Vol. 286, Issue 1, Varun Reddy and Jeffrey D. Zahn, Pages 158–165, Copyright (2005), with permission from Elsevier. | ||
Aqueous two-phase microextraction
As aforementioned, ATPS can be formed by combining different compounds. The studies applying aqueous two-phase microextraction range from the conventional polymer–polymer-49–51 and polymer–salt-52–56 based ATPS to the recent surfactant–salt,57,58 polymer–surfactant,59 protein–polymer,60 IL–water,61 IL–salt53 or even the IL–sugar62 for the extraction of different proteins, as detailed in Table 2. This type of LLE is, by itself, a great improvement for the solvent extraction field as well reviewed in ref. 39 and 41 and as such, in this review, the focus will be on the combined advantages of ATPS and microfluidic devices.| Protein | Molecular weight (kDa) | pI |
|---|---|---|
| Bovine serum albumin, BSA | ∼69.3 | 5.82 |
| (Escherichia coli) β-galactosidase | ∼116.7 | 5.30 |
| (Aequoera victoria) green fluorescent protein, GFP | ∼26.9 | 5.67 |
| (Schistosoma japonicum) glutathione S-transferase | 23.4–25.5 | 6.09–6.73 |
| (Human) Immunoglobulin G, IgG | ∼150 | 6.60–7.20 |
| (Staphylococcus aureus) Protein A | ∼42 | 4.85–5.10 |
| (Bovine) insulin | ∼11.4 | 7.60 |
| (Bacillus licheniformis) α-amylase | ∼58.5 | 6.33 |
| (Halobacterium salinarium) bacteriorhodopsin | ∼28.3 | 4.58 |
| (Bovine) trypsin | ∼25.8 | 8.40 |
In order to further accelerate separation of electrically charged species within microfluidic devices, an externally imposed electric field has been often introduced. This not only allows the extraction and separation of biomolecules between both phases but also the separation between the fastest molecules.60 Typically, the partition is carried accordingly to the biomolecule affinity/preferential interactions towards each phase; yet, here there is also the possibility of further separating the charged molecules according to their surface charge, upon the use of an external electric field.60 More recently, in 2017, Vobecká and co-workers have searched the possibility of controlling the droplet motion in ATPS, again by applying a DC electric field. The authors have shown the possibility to control electrically the motion of the salt droplets (phosphate, sulphate and carbonate species) in PEG/salt-based ATPS, by a DC electric field.63
Polymer–polymer-based ATPS
Polymer–polymer-based were the first ATPS studied, which explains its extensive characterization and use in literature.40 These systems have been applied to the microextraction of trypsin,50 BSA,49,51 protein A, insulin, immunoglobulin G (IgG) and green fluorescent protein (GFP).51 Herein, besides the diversity of analytes studied, several microfluidic devices were also proposed, as summarized in Table 3.| Cells or (bio)molecule extracted | ATPS components | Microfluidic device | Ref. |
|---|---|---|---|
| Trypsin | PEG 35000, dextran 10000 and 500000 | Two PDMS microfluidic devices: one with three inlets and the other with seven inlets, both converging into a straight main channel and one outlet. | 50 |
| BSA | PEG 8000, dextran 500000 | A PMMA microdevice with three inlets converging into a straight main channel and one outlet. The main channel has several gel bridges connecting it to two reservoirs at which an electric field is applied | 49 |
| BSA, GFP, immunoglobulin G (IgG), protein A and insulin | PEG 1000, dextran 20 | A PDMS microfluidic device with three inlets, converging into a straight main channel and one outlet | 51 |
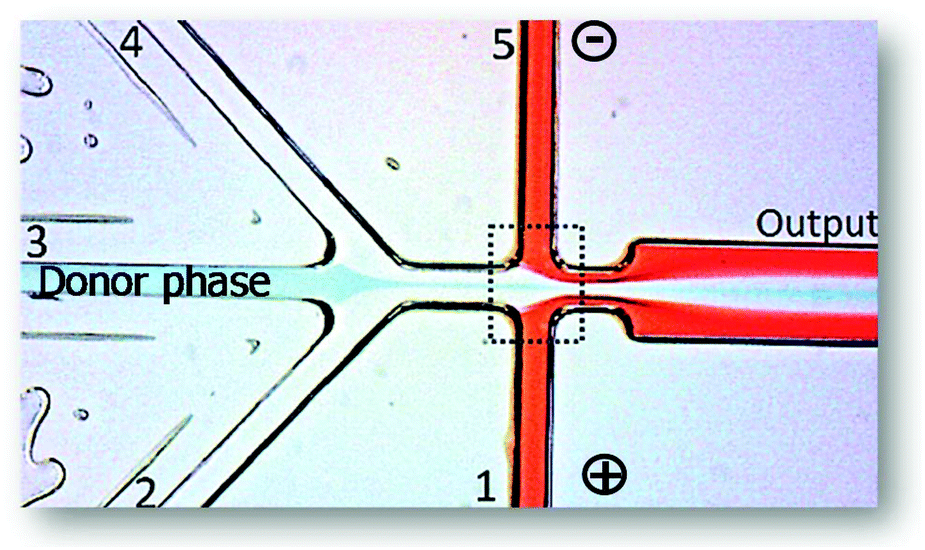
Münchow and co-workers49 used the charge of proteins to develop a microfluidic device able to promote the protein partition by an electric field applied to the chip, thus promoting an electrophoresis at microscale, while simultaneously crossing its principles with those from the ATPS. For this purpose, the authors created a three inlets chip that converged into a straight main channel and one outlet, in which the main channel was connected to side reservoirs by gel bridges (Fig. 4). The electric field was applied through the main channels to allow the mobilization of proteins according to their electric mobility. Herein, BSA was dissolved in the dextran (Dex)-rich phase, thus remaining in this phase upon voltages up to 2.5 V. Nevertheless, by augmenting the voltage, the authors reported that BSA has migrated to the opposite phase rich in PEG, thus following the electric potential imposed. In contrast, when BSA was introduced in the PEG outer phases, it migrated preferentially to the middle Dex-rich phase, a result exclusively controlled by the BSA preferential interactions with the dextran, independently of the electric field path imposed. Moreover, the migration of BSA for dextran was proven to be pH-independent.
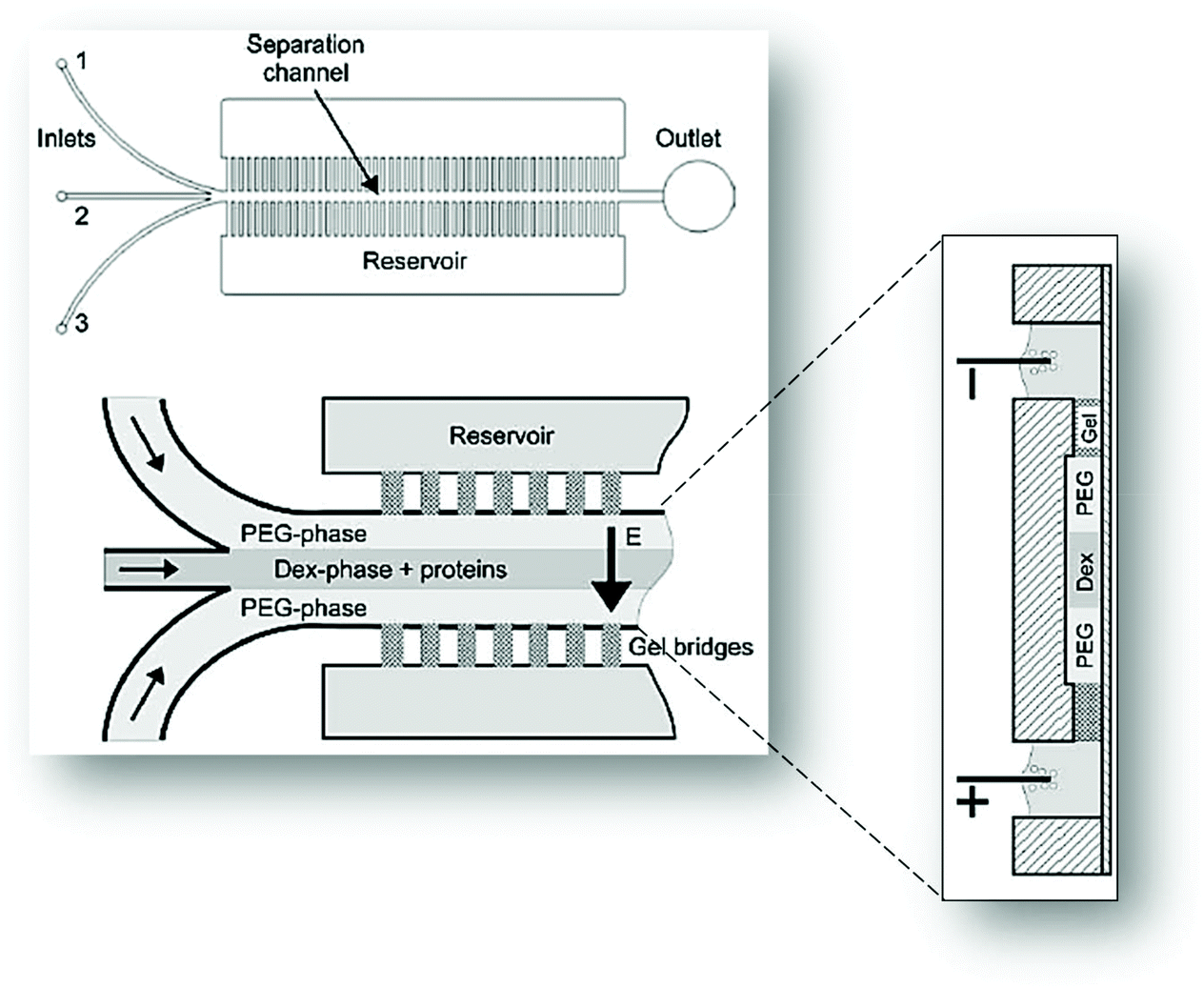 | ||
| Fig. 4 Schematic representation of the three inlets/one outlet microfluidic device created by Münchow and co-workers49 for the electrophoretic partition of proteins. Adapted from ref. 49 with permission from The Royal Society of Chemistry. | ||
Polymer–polymer-based ATPS were also used to selectively deliver chemicals to cells as demonstrated by Frampton and co-workers.50 In this work, the delivery of trypsin to immobilized cells was studied. Trypsin is a cationic enzyme used to detach cells from the supports they are adhered. The authors have measured the number of cells detached and accumulated in the polymers flowing in the microchip. Herein, a negatively charged dextran was applied to clearly evidence the selective migration of trypsin positively charged to dextran layer. The authors have demonstrated the highest performance of the ATPS against the poor results obtained for the conventional aqueous medium to precisely deliver protein molecules to cells.
Aires-Barros et al.51 studied the partition coefficient of several labelled proteins, including protein A, insulin, immunoglobulin G (IgG), and Green Fluorescent Protein (GFP), with a microchip with three inlets, converging into a straight main channel and one outlet. The authors aimed to quickly determine the partition coefficient of the biomolecules, by achieving it without the final phase separation. All proteins selected were fluorescent or previously labelled with a fluorescent marker, making possible their fast “on-chip” detection and quantification at the end of the main channel. With this, the authors avoided the need for the phases’ separation at the outlets, allowing thus the quantification of the biomolecules “off-chip”.
With the microfluidic chip developed, the authors reduced from several hours (macroscale) to 30 minutes (microscale), the time of analysis of the ATPS compositions, phase separation and biomolecules quantification. While the time of analysis was decreased, the partition coefficient values obtained were similar for both micro and macroscale.
Summing up, these works showed a similar microfluidic approach or microchip device used on the partition study of distinct proteins. However, during the development of these studies some disadvantages emerged, e.g. the high viscosity of polymer–polymer-based ATPS which negatively interfered with the flow rates, or the similar polarities between phases, making the success of the separation a more difficult task.51
Polymer–salt-based ATPS
One of the alternatives found in literature to surpass the polarity problems highlighted for the polymer–polymer-based ATPS were those composed of polymers + salts, which were already reported on the extraction of a wide range of biomolecules.41,65 The authors pointed out several advantages for these to be used within a microfluidic device, namely their faster thermodynamic equilibrium, higher polarity difference between the phases and their lower viscosity. Polymer–salt ATPS have been applied on the extraction of BSA,51,52,55,62 β-galactosidase,52 GFP,36,51,52 glutathione S-transferase (GST),52 genomic DNA,52 IgG,36,51,54,66 protein A,51 insulin,51 α-amylase67 and bacteriorhodopsin53 (summary of all reports available in Table 4). Singh and collaborators52 were the pioneers to apply polymer–salt ATPS into a microfluidic device. In this work, a reusable glass microchip with three inlets, converging into a serpentine main channel later diverging into two outlets was investigated. This device was initially tested using fluorescent labelled BSA and β-galactosidase to allow the visual identification of the preferential affinity of both proteins for the saline or polymeric phases, respectively. The authors also studied the partition of both GFP and GST in their native variants and after being genetically tagged with two different sequences each. In this way, they intended the manipulation of their partition towards the polymeric phase, thus increasing the purity and recoveries of the extracted proteins from 16 to ∼50% for the native and tagged GFP, respectively, and from nearly zero to around 40% for the wild-type and tagged GST, respectively. They also used a cell lysate of Escherichia coli to study the partition of all the components. The recombinant tagged proteins partitioned almost equally between both phases, though 75–90% of total proteins were collected in the salt-rich phase owing to its higher flow rate. Meanwhile, the genomic DNA was adhered to the walls or stayed near the interface migrating to the saline phase. The β-galactosidase, GFP and GST, showed a selectivity between 3 and 5, when their partition coefficients were correlated with the data found for total proteins. During the experiments, some reproducibility issues were identified and justified by the incomplete phase separation in the outlets and dead volumes in the chip. Moreover, the small amounts of materials tested were also pointed out as source of experimental flaws, these causing some interferences with the “off-chip” detections.| Biomolecule extracted | ATPS components | Microfluidic device | Ref. |
|---|---|---|---|
| BSA, GFP, immunoglobulin G (IgG), protein A and insulin | PEG 1000, dextran 20; PEG 1000, phosphate buffer (K2HPO4/KH2PO4) | A PDMS microfluidic device with three inlets, converging into a straight main channel and one outlet | 51 |
| BSA | PEG 4000, (NH4)2SO4 | A coaxial capillary microfluidic device composed of tubular and square glass tubes, creating two outer phases and an inner phase | 55 |
| BSA, β-galactosidase, green fluorescent protein (GFP), glutathione S-transferase, genomic DNA | PEG 4000, potassium phosphate buffer (K2HPO4/KH2PO4) | A glass microfluidic device with three inlets which merged into a single serpentine main channel and then diverged into two outlets | 52 |
| IgG | PEG 3350, phosphate buffer (K2HPO4/NaH2PO4), NaCl | A PDMS microfluidic devices with three inlets/outlets and a serpentine main channel | 54 |
| PEG 400, 1000, 3350; potassium phosphate salts (K2HPO4/KH2PO4) | A PDMS microfluidic devices with three inlets/outlets and a serpentine main channel | 66 | |
| α-Amylase | PEG 4000, K2HPO4 | A glass microchip with two and three inlets/outlets | 67 |
| GFP, LYTAG-GFP and IgG | PEG 3350, 4000, 6000, 8000: potassium phosphate salts (K2HPO4/KH2PO4) | 8 microfluidic devices in a single PDMS chip with two inlets converging into a serpentine main channel and a common outlet at the center of the chip | 36 |
| BSA | PEG 4000, K2HPO4 | Glass microchannel systems with two inlets/outlets and a serpentine main channel | 62 |
| Bacteriorhodopsin | PEG 8000, KH2PO4/K2HPO4 | A PDMS microfluidic device with a stable three-phase stream in the microchannel | 53 |
Tong et al.55 proposed an innovative microfluidic device composed of capillary glass tubes. Two outer phases and one inner phase were created by introducing two square capillaries near the inlet and outlet. This coaxial capillary device allowed the formation of two interfaces after phase separation. The operation parameters, i.e. the flow, mass transfer and contact time conditions, were evaluated through the partition of rhodamine B. In this work, the BSA partition was tested as well as the impact of the flow rate, several cycles of extraction and different BSA concentrations. The recovery of BSA on the outer phase increased with the number of ATPS cycles, from 34.2% to 71.1% for the first and third cycles, respectively. Moreover, when the flow rate of the outer phase augmented, the BSA recovery rate declined due to mass transfer issues, because of the reduced contact time.
The purification of antibodies is nowadays a hot topic in the field of microfluidics, especially regarding IgG.36,51,54,66 Aires-Barros et al.54 were the first to attempt the IgG purification using a polymer–salt ATPS with a two inlets/outlets chip. However, the authors have replaced it by a modified microchip with three inlets/outlets, which allowed the complete phase separation at the outlets. For that, the middle outlet width was decreased, while the outer outlets were increased. Once in the main channel, the labelled IgG diffused from the salt-rich phase towards the polymeric phases until its concentration reached a plateau at a 10 cm length from the inlets of a total of a 16.8 cm microchannel length. Nonetheless, the extraction required all the channel length so that the IgG remaining in the interface could completely migrate to the PEG-rich phase. These data was refereed as being in agreement with both simulation and experimental results. Moreover, the authors found out that the ATPS size reduction from macro to microscale was not considerably affecting the antibody partition, but have reduced the operation time.54 Rito-Palomares and collaborators66 used a similar chip to study the partition of IgG to distinct polymeric phases. In this study, the authors observed that, by increasing the polymer molecular weight it was possible to manipulate the affinity of the antibody towards any phase of the ATPS. When PEG 400 was used, IgG partitioned completely to the polymeric phase, whereas PEG 3350 led to the recovery of most of the antibody in the salt-rich phase. PEG 1000 lies in the between, showing an equal distribution of IgG among both phases. Again, the authors have demonstrated that similar results were obtained for the micro and macroscales. More recently, Aires-Barros and collaborators36,51 proposed two new chips. In these, the typical device with two or three inlets/outlets with a final phase separation to determine the biomolecule partition coefficient was not used. Firstly, a microchip with three inlets converging into a main channel and one outlet coupled with fluorescent microscopy to detect the target molecule was proposed.51 Several other labelled or fluorescent proteins apart from the labelled IgG, namely BSA, protein A, insulin and GFP were tested. The results showed a preferable migration of all proteins for the PEG-rich phase and an increased salting-out effect from the salt for high saline concentrations, similarly to what was reported for polymer-dextran-based ATPS. The authors justify the results obtained by the reduction on viscosity when a dextran-rich phase is replaced by a phosphate-rich phase, consequently increasing the K. Then, a second chip was developed, containing 8 identical microfluidic devices with two inlets converging into a serpentine main channel and a central outlet common to all devices (Fig. 5).36 The outlet was driven by a negative pressure, which means that it is connected to a syringe pump in a pulling mode, at a constant flow rate of 2 μL min−1. As a result, each phase flow rate was controlled by its viscosity and density. Initially, the chip performance was evaluated by the fractionation of GFP and LYTAG-GFP. Both tagged and untagged proteins partitioned preferably for the PEG-rich phase, with the LYTAG-GFP displaying a 2.5 to 6-fold increase in the partition due to the LYTAG affinity for the PEG molecules. For this tagged protein, the PEG molecular weight had no effect on the protein partition, whereas the untagged GFP suffered a slight decrease in the partition when the PEG molecular weight was increased, which can be a result of the higher viscosity of the polymer, as detailed by the authors.
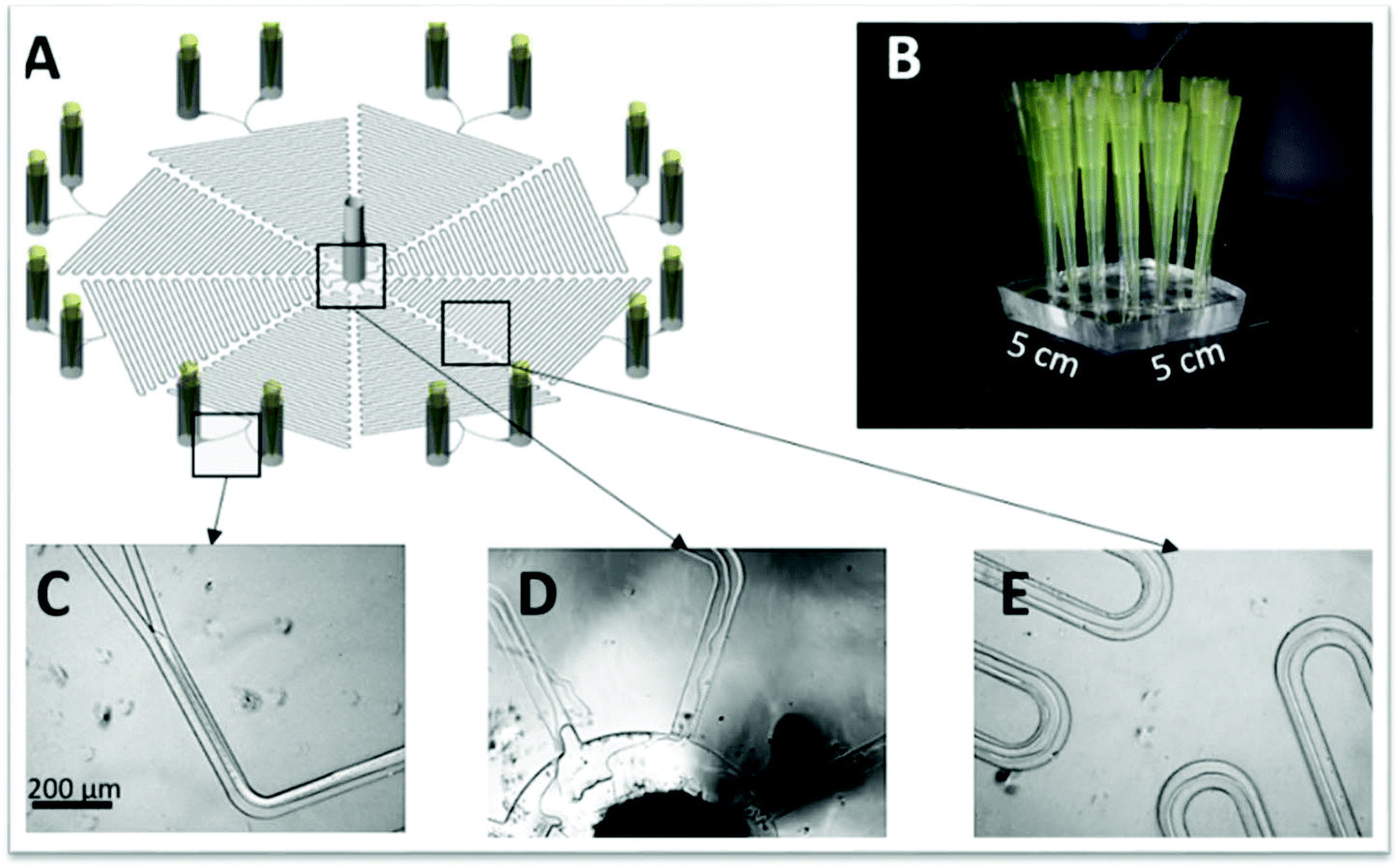 | ||
| Fig. 5 (A) Schematic representation of the PDMS microchip developed by Aires-Barros and collaborators36 with 8 microfluidic devices in a single chip (B). (C–D) Pictures of the ATPS phases inside distinct locations of the device. Reprinted from A multiplexed microfluidic toolbox for the rapid optimization of affinity-driven partition in aqueous two phase systems, Vol. 1515, Issue 15, E. J. S. Bras, R. R. G. Soares, A. M. Azevedo, P. Fernandes, M. Arévalo-Rodríguez, V. Chu, J. P. Conde and M. R. Aires-Barros, Pages 252–259, Copyright (2017), with permission from Elsevier. | ||
This microfluidic device was then applied for the IgG extraction with the aid of LYTAG-Z fusion proteins owing to the ability of the antibody to bind to the Z-domain, while the LYTAG had more affinity to PEG. The results obtained for the ATPS with PEG 8000 showed that in the LYTAG-Z absence, there was no significant difference between the systems with lower tie-line length (TLL). On the other hand, in its presence, the IgG partition increased ∼2-fold for the lower TLL and the highest K was obtained for the highest TLL due to the higher PEG concentration on the PEG-rich phase. A different behaviour was obtained for systems with PEG 3350. Here, the IgG partition to the PEG-rich phase was evident in both the LYTAG-Z presence and absence. Nevertheless, a different trend is observed regarding the partition and the system’ TLL. In the LYTAG-Z absence, the partition seems to be independent of the TLL, while in its presence the antibody partition increases with the decrease of the TLL. This behaviour was discussed by authors as being related with the steric hindrance effects and/or exclusion volume effects. In general, with the PEG 3350-based ATPS, it is possible to achieve K values around 59%, these higher than the ones obtained with the PEG 8000-based system. The data was compared with the macroscale results showing the same tendency. However, for some cases, there was an underestimation of K at extreme values, which was attributed to light dispersion inside the chip. After optimizing the IgG extraction, the authors focused on the back-extraction of the antibody to a phosphate buffer phase spiked with cholinium. For that, a second chip was developed, this including three inlets, a serpentine main channel and two outlets. Although the similarity between devices, the latest suffered some modifications, since this is the combination of two chips into one, as shown in Fig. 6. This device was divided in two sections; the first used to extract the IgG and the second to carry the back-extraction. The first section is described by the authors as composed of two inlets converging into a main channel and this diverging again in two channels; one is the first outlet for the salt-rich phase, whereas the second one is the PEG-rich phase that is going to merge with the third inlet containing the new salt phase spiked with cholinium. At the end, both phases left the chip through the second outlet. This back-extraction was possible due to the use of LYTAG-Protein A added to the first salt phase having a higher affinity to cholinium than to PEG. During the extraction step, the IgG bound to the LYTAG-Protein A partitioned towards the PEG-rich phase as there was no cholinium present. Later, on the back-extraction section, the IgG bound to the LYTAG-Protein A migrated towards the new saline phase owing to its higher affinity to cholinium. It is noteworthy to mention that this was carried in a sample spiked with bovine serum to mimic a real sample. The results showed that the presence of impurities did not affect the partition profile. Bacteriorhodopsin and α-amylase extractions were investigated by Park et al.53 and Novak and co-workers,67 respectively.
 | ||
| Fig. 6 Schematic representation of the microchip developed by Aires-Barros and collaborators36 to perform the extraction and back-extraction of IgG. Reprinted from A multiplexed microfluidic toolbox for the rapid optimization of affinity-driven partition in aqueous two phase systems, Vol. 1515, Issue 15, E. J. S. Bras, R. R. G. Soares, A. M. Azevedo, P. Fernandes, M. Arévalo-Rodríguez, V. Chu, J. P. Conde and M. R. Aires-Barros, Pages 252–259, Copyright (2017), with permission from Elsevier. | ||
For the extraction of the bacteriorhodopsin (integral membrane protein found in “purple membrane”, the Archaea cell membrane, mainly in Halobacteria species) from a pre-treated cell lysate, two microfluidic devices were investigated. One of these devices was used for the protein microextraction and the other for the micro-dialysis allowing the sucrose removal (used for the sample pre-treatment) and a higher protein purification (Fig. 7). In this work, the pH effect and the influence of the number of ATPS cycles on the protein purity and recovery were studied, as well as the effect of the buffer flow rate and the pH on the sucrose removal during the micro-dialysis. The results showed that the bacteriorhodopsin recovery increased with the pH rise to 7.0 and decreased with the number of ATPS cycles. However, its purity increased with the number of ATPS cycles and with the pH decrease, an opposite behaviour to the sucrose, which slightly decreased with the pH increase.
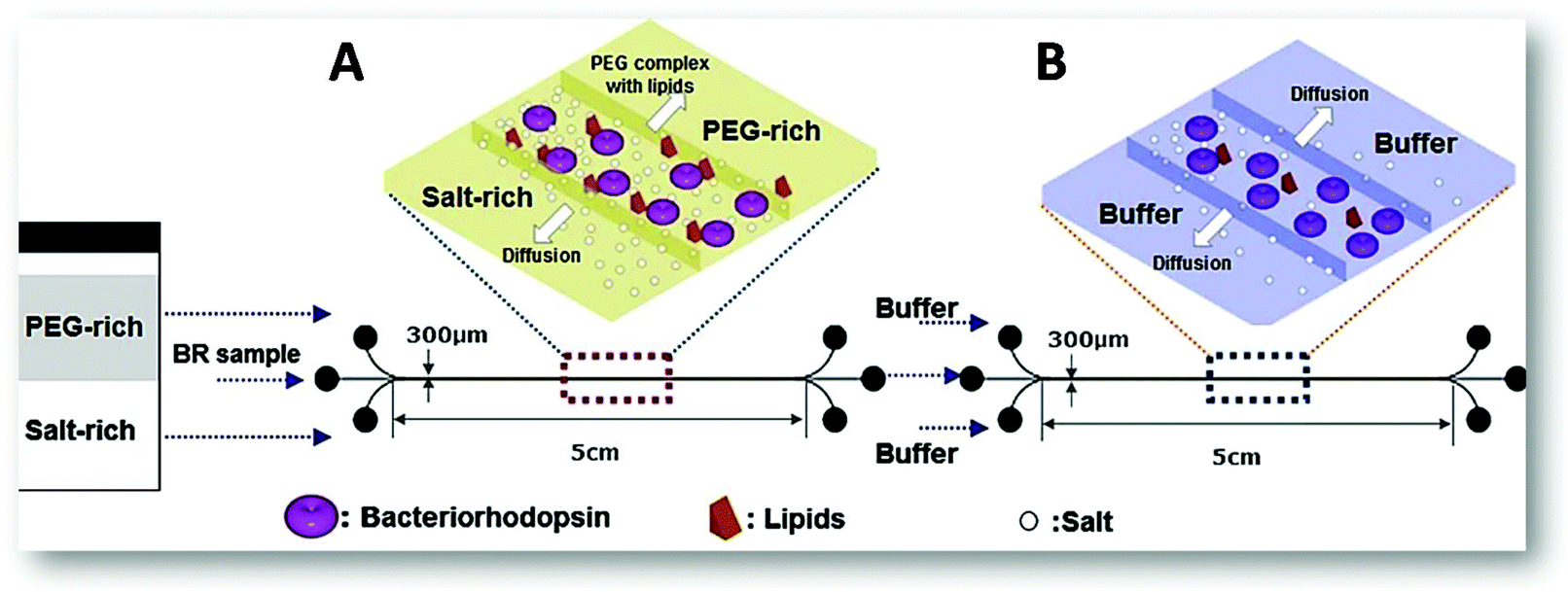 | ||
| Fig. 7 Schematic representation of the two microfluidic devices applied on the purification of bacteriorhodopsin.53 (A) Represents the extraction process using ATPS to purify the protein from the cell lysate sample; and (B) corresponds to three-flow desalting micro-dialysis applied on the removal of contaminant proteins and excess of sucrose after fractionation of the sample stream from the laminar-flow extraction process. Reprinted from Y. S. Huh, C.-M. Jeong, H. N. Chang, S. Y. Lee, W. H. Hong and T. J. Park, Biomicrofluidics, 2010, 4, 14103, with the permission of AIP Publishing. | ||
The authors concluded that when only the ATPS was applied, the recovery rate obtained was around 90%, the sucrose removal was about 17.4% and the total purity corresponded to 0.435, which represents a 1.16 purification-fold. However, after the micro-dialysis (Fig. 7B), the protein recovery rate decreased to 79%, although the sucrose removal and purity increased to 65.3% and 0.503 (1.55 purification-fold), respectively. Concerning the α-amylase extraction,67 the authors compared the influence of two vs. three inlets/outlets microchips and the advantages of the latest. In the first chip, the diffusion time was considerable higher than in the second due to the longer diffusion path needed, respectively 40.6 seconds and 8.2 seconds. The extraction efficiency of the two inlets chip was only 29% compared to the 52% of the microfluidic device with three inlets, as a result of the two interfaces present. Both results are lower than the obtained in the batch system (74%). Nonetheless, the latest approach required 2.5 hours just to allow the phases to reach the thermodynamic equilibrium not to mention the additional timing for the phase separation and enzyme quantification. Thus, the three inlets/outlets microchip seemed a better option since within a few seconds it was possible to achieve a reasonable extraction efficiency.
Other ATPSs
As mentioned before, the possible combinations to form ATPS extend far beyond the polymer–polymer and polymer–salt mixtures discussed above. Polymer–surfactant,59 IL–salt,53 IL–sugar62 and protein–polymer60 based ATPS have been reported and many of them applied on the microextraction of IgG and membrane proteins,59 bacteriorhodopsin,53 BSA62 and amino acids (lysine, glutamic acid and tryptophan).60 The specifications of each ATPS components and microchip characteristics are gathered in Table 5 in addition to the respective biomolecule being extracted.| Cells or (bio)molecule extracted | Type of ATPS | ATPS components | Microfluidic device | Ref. |
|---|---|---|---|---|
| BSA | IL–sugar | [C4mim][BF4], D-Fructose | Glass microchannel systems with two inlets/outlets and a serpentine main channel | 62 |
| Bacteriorhodopsin | IL–salt | 1-Hexyl-3-methylimidazolium hexafluorophosphate ([C6mim][PF6], KH2PO4/K2HPO4 | A PDMS microfluidic device with a stable three-phase stream in the microchannel | 53 |
| Lysine, glutamic acid and tryptophan | Protein–polymer | Sodium caseinate, PEG 6000 | A polycarbonate chip with 5 inlets, a straight main channel and one outlet. An electric field is applied in two of the inlets | 60 |
| IgG and membrane proteins | Polymer–surfactant | PEG 6000, Zwittergent 3-10 + SDS + Triton X-114 | A PDMS microchip with three inlets/outlets and serpentine microchannels | 59 |
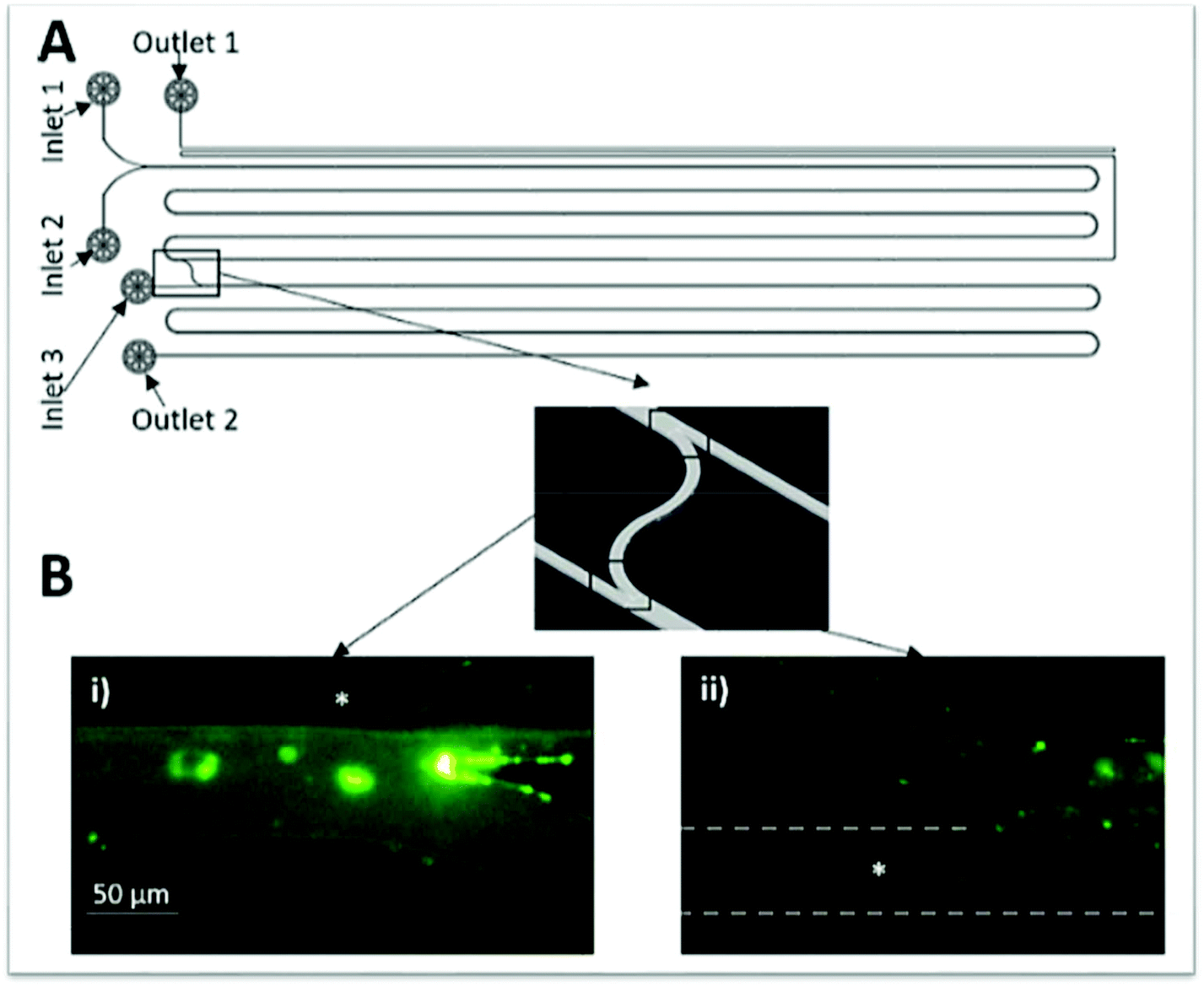
Starting with a less complex biomolecule, Campos and collaborators60 created a device for an electrophoretic extraction of amino acids. This chip displayed 5 inlets converging into a straight main channel and one outlet (Fig. 8), being the electric field applied in the perpendicular inlets (inlets 1 and 5 from Fig. 8). The authors used sodium caseinate in buffer as the donor phase of the amino acids, namely lysine, glutamic acid, tryptophan, and PEG as the acceptor phase. When the electric field was applied, the amino acids migrated accordingly to their charge. After applying an electric field, the PEG phases introduced in inlets 2 and 4 acted as a virtual membrane for the selective extraction of compounds with distinct mobility. This means that the amino acids with higher mobility will cross this boundary whereas, the ones with lower mobility will remain in the donor phase.
 | ||
| Fig. 8 Photograph of the microfluidic device used in literature.60 It is visible the application of an electric field in inlets 1 and 5. The donor phase contains the amino acids being extracted to the acceptor phases (1, 2, 4 and 5). The extraction occurs inside the dashed line. Adapted from ref. 60 with permission from The Royal Society of Chemistry. | ||
Results showed that, when no electric field was applied, or its strength was 7.4 kV m−1, no migration of the amino acids from the donor phase was observed. Only when the electric field was ≥14.7 kV m−1, the amino acids with higher mobility, particularly lysine, crossed the phase boundary. This means that ∼70% of glutamic acid and tryptophan were recovered in the donor phase while only ∼47% of lysine remained in the same phase. To improve the amino acids selectivity, authors functionalized lysine and glutamic acid with fluorescein isothiocyanate to create a higher difference in their mobility. Consequently, at an electric field of 14.7 kV m−1, the glutamic acid and lysine ratio was 87% higher than with no electric field and by further increase this field to 22.1 kV m−1, both amino acids could be separated almost completely, although promoting the loss of glutamic acid to the collector channel. This showed that the chip had not been well planned since the outlet region could present more outlets, resulting in the complete, or at least, more efficient separation of the phases containing the different amino acids. Even though the authors used laminar flow, they did not make use of its easier way to separate the phases at the outlet, besides they never mentioned how the phases were separated, only that they were analysed by HPLC.
Liu et al.59 reported the separation of membrane proteins (liposoluble) from the water soluble ones using a polymer–surfactant-based ATPS inside a three inlets/outlets microfluidic device. This approach is known to purify membrane proteins without leading to their denaturation, which is only possible due to the different affinities of the lipo- and water-soluble proteins towards the surfactant- and polymeric-rich phases, respectively. Initially, a labelled IgG was used as a model biomolecule to evaluate the microchip performance considering the channel width, diffusion time and interface area. The results revealed a good antibody partition from the surfactant phase (zwittergent 3-10 + SDS + Triton X-114) towards the PEG-rich phase with a recovery of 90.8%. The extraction and detection of the membrane proteins then proceeded using capillary electrophoresis, SDS-PAGE and nano-HPLC-MS/MS. As expected, the water-soluble proteins diffused towards the outer polymeric phases while the membrane proteins stayed in the surfactant-rich phase. The results showed a 90% purification of membrane proteins within 5 to 7 seconds, which corresponds to the highest purification found so far.
The extraction of bacteriorhodopsin was performed also using an IL–salt-based ATPS53 in a three inlets/outlets chip. The authors opted to replace the polymer by a hydrophobic IL (1-hexyl-3-methylimidazolium hexafluorophosphate, [C6mim][PF6]) owing to its ability to remove more easily the contaminant lipids and hydrophobic proteins, besides its immiscibility with water. The results for this ATPS were identical to those obtained using a polymer–salt ATPS, however, the protein recovery decrease with the pH from 9.023 of the previous systems to 8.432. Nevertheless, when an additional step of micro-dialysis was incorporated in the system (Fig. 7B), the sucrose removal increased from 35.6 to 75.5%, which is a 10% increase compared to the conventional ATPS. The total purity was also improved with the dialysis incorporation from 0.493 to 0.508, corresponding to a 1.41 and 1.55 purification fold, respectively. These values were higher than those obtained for the polymer–salt ATPS. Although this system was not as good as the traditional in terms of the protein recovery, it was much better as far as purity was concerned.53 This study showed the importance of coupling microchips to enhance the molecule purification and shows how crucial it is to carefully select the ATPS.
Another interesting example is BSA extraction using an IL-sugar and PEG-phosphate ATPSs in a two inlets/outlets microfluidic device with a parallel flow and a liquid–liquid interface in the middle of a microchannel enabling the efficient phase separation at the exit of the Y-shaped channel.62 It was found that changes in the IL (1-butyl-3-methylimidazolium tetrafluoroborate, [C4mim][BF4]) concentration and pH of a D-fructose-rich phase highly affected the BSA partition coefficient. Furthermore, the decrease of viscosity of almost 10 times was obtained when using IL-based ATPS as compared to the PEG-phosphate ATPS, resulting in much more favourable flow ratio of both phases and thereby several times more efficient extraction. This microfluidics-based approach using IL-based ATPS appeared as a very promising tool for protein extraction.62 An overview of all the research done in this field up to this date, as well as each type of microfluidic device used for the extractions and the solvents applied is detailed in Tables 3–5. Unlike for the conventional LLE, it is here possible to easily compare distinct works since almost all use the common two or three inlets/outlets microchips. Additionally, within the same type of ATPS, the system phase formers are the same or belong to the same family. For instance, the polymer–polymer-based ATPS are always composed of PEG and dextran, which often display the same molecular weight in different works. These similarities between different works help drawing some general conclusions, namely regarding the best system and conditions for a class of compounds. For the separation of the biomolecules using polymer–polymer-based ATPS, it was seen that some molecules migrate preferably for the PEG-rich phase whereas others migrate towards the dextran-rich phase due to their natural affinity for these phases. Nevertheless, it was also shown that this affinity can be manipulated, if necessary, for the success of the separation. On the other hand, when the polymer–salt results for the protein extraction are compared with those obtained with alternative ATPS, it seems that these innovative systems enhance the proteins extraction/purification.
However, more studies are required in microscale to confirm this, especially with the adequate choice of the systems components. Proved only in macroscale,39,41 ILs-based ATPS seem to be a good choice, since they can be designed to meet the requirements of a specific application, due to their design solvent nature. Despite some of the good results already described for microscale, their application to process real matrices is still scarcely approached. Thereby, and in our opinion, future works starting by the initial optimization should then investigate the applicability of the processes optimized but in the real systems or extracts. Moreover, most proteins under study were coupled with fluorescent dyes to facilitate their detection and quantification. However, when a real sample is processed aiming at a biomolecule purification, the proteins will not always be tagged. This means that other detection/quantification techniques should be considered and investigated.
Overview of other (non-)conventional separation techniques
It is well known that chromatography and (capillary) electrophoresis are high resolution separation and analytical techniques commonly used for the macroscale separation and quantification of macromolecules, such as proteins and nucleic acids. The first can separate all types of molecules by the proper choice of the stationary phase and/or presence of specific ligands, whereas the second offers the separation of charged species according to the molecule charge/size ratio upon the application of an electric field. Both techniques offer the possibility to distinguish small differences in the biomolecule structure and properties, however they have also great costs associated, time-consuming protocols, and sometimes require skilled personnel and laboratory infrastructures. By miniaturizing both techniques, such drawbacks can be overcome while developing portable devices applicable to a wide range of applications, namely for diagnosis and for monitoring the presence of specific proteins in human samples68 and food.69Table 6 summarizes the works approaching the proteins and nucleic acids separation by different techniques within a microfluidic device. More precisely, electrophoresis can be differentiated in capillary zone electrophoresis, capillary gel electrophoresis, isotachophoresis, micellar electrokinetic chromatography and isoelectric focusing, though they are not discriminated in the table. All of these have been extensively applied for protein and DNA analysis as demonstrated by the several reviews published on the subject, with the emphasis on the development of the technique at microscale and improvements on the materials used.15,68–75 Microfluidics also appear as an attractive approach to revolutionize point-of-site detection for distinct areas, for instance medical diagnosis and research, food analysis and environmental monitoring, by assisting in the creation of label-free DNA biosensing devices, as recently reviewed by Dutta and co-workers.76 Interestingly, Nazzaro et al.69 summarized the cost associated with the equipment and reagents as well as the time required for the different steps used in routine food protein analysis through different techniques, including SDS-PAGE, reverse-phase HPLC, conventional capillary electrophoresis and miniaturized capillary electrophoresis. Microfluidics allow a reliable, reproducible and sensitive analysis within a few minutes and with much lower costs than all the remaining alternatives.| Microfluidics cross-linked with | Number of publications | Ref. |
|---|---|---|
| a At least 39 papers reporting proteins and DNA electrophoresis within a microfluidic device. There might be more papers regarding simply the DNA separation for analysis. | ||
| Conventional liquid–liquid extraction | 3 | 46–48 |
| ATPS | 13 | 36, 49–55, 59, 60, 62, 66, 67 |
| Electrophoresis | 39a | 49,60 and 78–115 |
| Chromatography | 22 | 116–137 |
| Protein crystallization | 36 | 28 and 138–173 |
The chromatographic separation of proteins can be accomplished through different approaches, namely size exclusion chromatography, ion exchange chromatography, hydrophobic interaction chromatography and affinity chromatography depending on the properties of the target biomolecule, as reviewed in distinct works.16,17,77
Herein, it is much more difficult to introduce all the operation steps in a single device and still maintaining a good performance. Yet, Yuan and Oleschuk18 have just reported the late advances in liquid chromatography within a microfluidic device in terms of the stationary phase and detection of the molecule being separated. Tetala and Vijayalakshmi17 have also overviewed the stationary phase as well as its surface modification through the addition of functional groups and ligands, and have summarized some applications for the separation of nucleic acids and proteins. Some exceptions for the protein extraction and separation through a combined process using electrophoresis and ATPS were discussed earlier in detail since they fit the review scope.
Miniaturization of protein crystallization has been another cutting-edge subject over the last years since it requires small volumes of sample and crystallization reagents, offer a high-throughput screening and allow the monitorization of the protein crystallization, which, in turn, facilitates the crystal structure analysis. Usually, protein crystallization is known to be the bottleneck of the protein structure analysis owing to the immense time required until a good crystallization of the protein is obtained, in addition to the several failed attempts in trying. Therefore, having a fine tuning of the crystallization process smooths and accelerates the entire process while resulting in a high-quality protein study. The advances made over the years in this field as well as the different approaches to obtain a good protein crystallization have been reviewed by distinct authors.174–178 Gavira174 paid a special attention to the protein nature and its crystallization process while also reviewing the different methods being applied to achieve a good protein crystallization. Furthermore, the protein crystallization can be achieved using different approaches, which should be selected accordingly to the final purpose of the study. If the intention is to analyse the protein within the microchip by X-ray analysis, a droplet-based crystallization is the best option since it allows the formation of a single crystal, facilitating the protein structure analysis. In contrast, if the protein crystallization is intended for other purposes than the protein structure analysis, a well-based protein crystallization might be the best option.175 Looking at protein crystallization from a different perspective, such as a process step and its scalability, it is required a minimal, yet efficient mixing and good mass transfer that lead to significant enhancement in crystal characteristics and reduction of operation time. This can be achieved using meso oscillatory flow reactor (meso-OFR), in which protein crystals are subjected to fluid shear forces induced by oscillatory flow mixing and solid–liquid interfaces. As a result, there is the occurrence of a strong nucleation by attrition at low supersaturation that lead to the formation of a high number of small crystals with different sizes and shapes from tetragonal, orthorhombic and needle-shaped crystals, to microcrystals and precipitates. Meso-OFRs offer also the reduction of the metastable zone and by controlling the oscillation amplitude and frequency it is possible to influence the induction time and size of the crystals.179,180 These results open the potential to exploit meso-OFRs to control protein nucleation for the design of protein crystallization. Therefore, there is a strong need for the development of micro- and mesoscale devices with integrated in situ analytic techniques to improve the current knowledge in protein crystallization, for both structural determination and downstream processing purposes.
Among the most promising strategies for the successful protein manufacturing is a holistic approach to develop and design processes. It aims to control critical quality attributes through a concerted optimization of both upstream and downstream process parameters. By this approach, implementation of miniaturized system of downstream steps is used during e.g. the screening of mutants producing specific target proteins, where problems associated with their isolation and purification (e.g. agglomeration, hardly removed side products), could significantly improve the overall process performance and scale-up.
In the field of protein analysis, Kitamori's group has shown a tremendous breakthrough by implementation of micro and nanoscale channels. Recent demonstration of a single IgG molecule detection using enzyme-linked immunosorbent assay (ELISA) within the sophisticated miniaturized device confirms the amazing potential of such systems for various applications.
Critical analysis and perspectives
From the analysis of the published results here discussed, the use of the microfluidic devices for LLE is on its infancy, still trying to learn the basics that may eventually lead to its widespread use and application. Nevertheless, the preliminary results obtained so far show that this innovative field can improve extraction/separation processes with faster, more efficient and selective purifications. However, to increase the reliability of the results obtained so far, the study of real matrices is required, since these may affect the flow regime, the surface properties of the system, as well as the partition behaviour of target molecules, due to the matrix higher complexity that can be a residue, biomass or a biomaterial. Often, the target compounds are present in very low concentration and it is here that microfluidics could be a major advantage given its well-known process intensification ability. This could be further enhanced by the ability to use microchips in series, helping on the clarification, pre-purification, purification and dialysis in a single run, as it is done today at macroscale. Park et al.53 have shown the possibility to pursue this approach by coupling two microchips for the purification of bacteriorhodopsin, in which the first chip was mainly for a pre-purification and the second allowed the micro-dialysis of sugars, resulting in a final product with higher purification. The feasibility of microfluidics in series was here demonstrated but it is still seldom explored in literature. It should be highlighted that microfluidics are an excellent approach for the purification intensification of compounds present in crude extracts at low concentration that are difficult to achieve by other approaches, namely antibodies from plasma or serum, growth factors, among others included in the concept of “high-value, low volume”.Process intensification at microscale can be further improved by counter-current, cross-current or fractional extraction arrangements similarly to what happens in macroscale.181 However, different features need to be addressed as in microfluidics, viscosity and surface wetting are more effective at controlling flow than gravity and inertia.182 On the other hand, multistage counter-current extractions require a good phase separation, hence limiting more the type of flow used to laminar flow,181–183 with few studies of segmented pattern.184 The bottleneck of this type of extraction at lower scale resides in the difficulty to maintain a stable interface and balance the pressure between the two inlets and outlets. By selectively modifying the surface of each inlet/outlet as well as each half of the microchannel it is possible to achieve a successful counter-current flow. Nevertheless, this has only been reported for simpler molecules as dyes, evidencing a focus for future studies.181,182 In the same line of scarce application and experimental considerations is the use of cross-current flow. It has been studied in microfluidic scale in cells, and particularly in blood cells separation. Despite the fact that separation of cells is out of the focus of this review, it helps us to emphasize the need for new studies to address the advantages and/or disadvantages of the microfluidic technique for different flows, applied to the separation of biomacromolecules.
Over the last decade, almost all research was carried by applying conventional liquid–liquid microextraction, however there are several other fractionation approaches that could be implemented and were not deeply studied so far. As previously discussed, ATPS were used, more recurrently the ones based in conventional solvents, and much less using unconventional solvents like ILs. In this sense, we believe that much more could be investigated regarding the use of ILs and other alternative solvents, considering the set of valuable properties these solvents present.39,41 These reviews39,41 have already summarized and discussed all the works done up to date regarding the ATPS extraction and purification of (bio)molecules and guided towards the best strategy of different families of compounds. Thus, a previous selection of the system applied should be carried out prior to its application in microfluidics. Nevertheless, it arises here as an easy and feasible opportunity. Additionally, these two works39,41 also considered the use of real matrices and the main problems that could arise during the extraction, so it might also be facilitating when applying these systems in microscale. Microfluidics and alternative-based ATPS emerged in the scientific community approximately at the same time, though the ATPS-based microextraction is clearly behind in the ATPS evolution, and therefore, a major opportunity is here displayed. Different proteins have been reported in the studies contemplating the use of microfluidics, however, some considerations need to be stated. Microfluidics can open the door for the purification of globular proteins, since their potential denaturation may be avoided easily. Jaspe et al.,185 have demonstrated that shear rates up to ∼2 × 105 s−1 do not seem to destabilize the folded of the globular horse cytochrome c protein. Furthermore, it was shown that it would be necessary an extremely high shear rate to destabilize a small protein (∼100 amino-acids) in water. Such shear rates are very difficult to achieve using laminar flow, hence the probability of protein denaturation inside a microfluidic device is very low.16 One of the latest researches reported on the use of alternative purification processes applying ATPS to purify proteins was the use of aqueous micellar two-phase systems (AMTPS). Briefly, AMTPS are a case of ATPS where the phase separation is mainly dictated by changing temperature. These have been studied on the purification of several biomolecules, being particularly interesting to be used in the purification of labile molecules, like proteins4,186,187 and antibodies.188,189 By applying these systems, the processes of separation can gain with the use of unconventional solvents like surfactants, deep eutectic solvents, and copolymers, regarding selectivity, extraction efficiency and purity of the final product.
Finally, it is true that in the last decade microfluidics have suffered a tremendous improvement and advancement, having an estimated market projection of $27.91 billion by 2023.190 However, it is well recognized the economic demands are still significative. As recently argued “If the microfluidics solution is not faster by at least one order of magnitude or offers other significant improvements in performance, the cost of the existing conventional solutions will define the maximum price for a microfluidic system”.191 As previously discussed, the manufacturing of microfluidics is the major economical drawback to be surpassed.191 In this sense, several strategies have been investigated,192 namely their 3D-printing,193 as reported last year.
We believe that the full potential of these devices will only be assessed when integrated with different units like reaction, sensing, mixing, pumping, injection, detection, diagnosis56 or alternative separations194 into a single chip. Actually, despite the efforts on this review to present the main developments on the dual function of miniaturization and separation of proteins and antibodies, much more could be investigated, since some other strategies of separation like chromatography, electrophoresis, and ultrafiltration195 can be integrated with ATPS to improve process conditions and to achieve better selectivity and purity parameters.195 In the end, and contrarily to what some (young) scientists have been arguing in different conferences, these approaches can process as much as the application requires, with high mass transfer, with the schematics we want and need e.g. in parallel, in series, several devices connected, with and without temperature shock, and most important, integrating different steps in the same microfluidic unit.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 & UIDP/50011/2020, financed by national funds through the Portuguese Foundation for Science and Technology/MCTES. S. P. M. Ventura acknowledges FCT for the contract IF/00402/2015 under the Investigador FCT 2015. I. P. and P. Ž.-P. were financially supported by the Slovenian Research Agency through Grants P2-0191 and N2-0067, as well as through the European Union's H2020 project COMPETE (Grant 811040), which is gratefully acknowledged.References
- S. Hardt and T. Hahn, Lab Chip, 2012, 12, 434–442 RSC.
- A. Pohar and I. Plazl, Chem. Biochem. Eng. Q., 2009, 23, 537–544 CAS.
- R. dos Santos, A. L. Carvalho and A. C. A. Roque, Biotechnol. Adv., 2017, 35, 41–50 CrossRef CAS.
- F. A. Vicente, I. S. Cardoso, M. Martins, C. V. M. Gonçalves, A. C. R. V. Dias, P. Domingues, J. A. P. Coutinho and S. P. M. Ventura, Green Chem., 2019, 21, 3816–3826 RSC.
- J. F. Buyel and R. Fischer, Bioengineered, 2014, 5, 138–142 CrossRef PubMed.
- K. Shirai, K. Mawatari, R. Ohta, H. Shimizu and T. Kitamori, Analyst, 2018, 143, 943–948 RSC.
- S. Tripathi, Y. V. B. Varun Kumar, A. Prabhakar, S. S. Joshi and A. Agrawal, J. Micromech. Microeng., 2015, 25, 83001 CrossRef.
- Q. Huang, S. Mao, M. Khan and J.-M. Lin, Analyst, 2019, 144, 808–823 RSC.
- M. Shehadul Islam, A. Aryasomayajula and P. R. Selvaganapathy, Micromachines, 2017, 8, 83 CrossRef.
- H. W. Hou, A. A. S. Bhagat, W. C. Lee, S. Huang, J. Han and C. T. Lim, Micromachines, 2011, 2, 319–343 CrossRef.
- R. Wohlgemuth, I. Plazl, P. Žnidaršič-Plazl, K. V. Gernaey and J. M. Woodley, Trends Biotechnol., 2015, 33, 302–314 CrossRef CAS PubMed.
- P. Žnidaršič-Plazl, Biotechnol. J., 2019, 14, 1800580 CrossRef.
- P. Znidarsic-Plazl and I. Plazl, Lab Chip, 2007, 7, 883–889 RSC.
- L. Vobecká, A. Romanov, Z. Slouka, P. Hasal and M. Přibyl, Nat. Biotechnol., 2018, 47, 73–79 Search PubMed.
- M. Dawod, N. E. Arvin and R. T. Kennedy, Analyst, 2017, 142, 1847–1866 RSC.
- I. Rodríguez-Ruiz, V. Babenko, S. Martínez-Rodríguez and J. A. Gavira, Analyst, 2018, 143, 606–619 RSC.
- K. K. R. Tetala and M. A. Vijayalakshmi, Anal. Chim. Acta, 2016, 906, 7–21 CrossRef CAS.
- X. Yuan and R. D. Oleschuk, Anal. Chem., 2018, 90, 283–301 CrossRef CAS PubMed.
- M. P. C. Marques and P. Fernandes, Molecules, 2011, 16, 8368–8401 CrossRef CAS PubMed.
- A. I. Stankiewicz and J. A. Moulijn, Chem. Eng. Prog., 2000, 96, 22–34 CAS.
- P. Žnidaršič-Plazl and I. Plazl, in Comprehensive Biotechnology, ed. M. Moo-Young, Academic Press, Burlington, 2nd edn, 2011, pp. 289–301 Search PubMed.
- J. Atencia and D. J. Beebe, Nature, 2005, 437, 648–655 CrossRef CAS PubMed.
- H. S. Lew and Y. C. Fung, J. Biomech., 1969, 2, 105–119 CrossRef CAS PubMed.
- J. P. Brody and P. Yager, Sens. Actuators, A, 1997, 58, 13–18 CrossRef CAS.
- R. Gupta, D. F. Fletcher and B. S. Haynes, J. Comput. Multiphase Flows, 2010, 2, 1–31 CrossRef CAS.
- R. Gupta, D. F. Fletcher and B. S. Haynes, J. Comput. Multiphase Flows, 2010, 2, 1–31 CrossRef CAS.
- J. P. Brody, P. Yager, R. E. Goldstein and R. H. Austin, Biophys. J., 1996, 71, 3430–3441 CrossRef CAS.
- Y. Yu, X. Wang, D. Oberthür, A. Meyer, M. Perbandt, L. Duan and Q. Kang, J. Appl. Crystallogr., 2012, 45, 53–60 CrossRef CAS.
- T. Thorsen, R. W. Roberts, F. H. Arnold and S. R. Quake, Phys. Rev. Lett., 2001, 86, 4163–4166 CrossRef CAS PubMed.
- A. Huebner, S. Sharma, M. Srisa-Art, F. Hollfelder and J. B. Edel, Lab Chip, 2008, 8, 1244–1254 RSC.
- N. Wang, S. Mao, W. Liu, J. Wu, H. Li and J.-M. Lin, RSC Adv., 2014, 4, 11919–11926 RSC.
- K. Wang, K. Qin, T. Wang and G. Luo, RSC Adv., 2015, 5, 6470–6474 RSC.
- W.-T. Wang, F.-N. Sang, J.-H. Xu, Y.-D. Wang and G.-S. Luo, RSC Adv., 2015, 5, 82056–82064 RSC.
- C. E. Poulsen, R. C. R. Wootton, A. Wolff, A. J. deMello and K. S. Elvira, Anal. Chem., 2015, 87, 6265–6270 CrossRef CAS PubMed.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. M. Revalor and D. R. Snead, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- E. J. S. Bras, R. R. G. Soares, A. M. Azevedo, P. Fernandes, M. Arévalo-Rodríguez, V. Chu, J. P. Conde and M. R. Aires-Barros, J. Chromatogr. A, 2017, 1515, 252–259 CrossRef CAS PubMed.
- D. Platis and N. E. Labrou, J. Chromatogr. A, 2006, 1128, 114–124 CrossRef CAS PubMed.
- P. G. Mazzola, A. M. Lopes, F. A. Hasmann, A. F. Jozala, T. C. V. Penna, P. O. Magalhaes, C. O. Rangel-Yagui and A. Pessoa Jr., J. Chem. Technol. Biotechnol., 2008, 83, 143–157 CrossRef CAS.
- M. G. Freire, A. F. M. Cláudio, J. M. M. Araújo, J. A. P. Coutinho, I. M. Marrucho, J. N. Canongia Lopes and L. P. N. Rebelo, Chem. Soc. Rev., 2012, 41, 4966–4995 RSC.
- R. R. G. Soares, A. M. Azevedo, J. M. Van Alstine and M. R. Aires-Barros, Biotechnol. J., 2015, 10, 1158–1169 CrossRef CAS PubMed.
- S. P. M. Ventura, F. A. e Silva, M. V. Quental, D. Mondal, M. G. Freire and J. A. P. Coutinho, Chem. Rev., 2017, 117, 6984–7052 CrossRef CAS PubMed.
- N. Assmann, A. Ładosz and P. Rudolf von Rohr, Chem. Eng. Technol., 2013, 36, 921–936 CrossRef CAS.
- M. Surmeian, A. Hibara, M. Slyadnev, K. Uchiyama, H. Hisamoto and T. Kitamori, Anal. Lett., 2001, 34, 1421–1429 CrossRef CAS.
- M. Surmeian, M. N. Slyadnev, H. Hisamoto, A. Hibara, K. Uchiyama and T. Kitamori, Anal. Chem., 2002, 74, 2014–2020 CrossRef CAS PubMed.
- Y. Kikutani, K. Mawatari, A. Hibara and T. Kitamori, Microchim. Acta, 2009, 164, 241–247 CrossRef CAS.
- R. Zhang, H.-Q. Gong, X. Zeng, C. Lou and C. Sze, Anal. Chem., 2012, 85, 1484–1491 CrossRef PubMed.
- M. C. Morales and J. D. Zahn, Microfluid. Nanofluid., 2010, 9, 1041–1049 CrossRef CAS.
- V. Reddy and J. D. Zahn, J. Colloid Interface Sci., 2005, 286, 158–165 CrossRef CAS PubMed.
- G. Münchow, S. Hardt, J. P. Kutter and K. S. Drese, Lab Chip, 2007, 7, 98–102 RSC.
- J. Frampton, D. Lai, H. Sriram and S. Takayama, Biomed. Microdevices, 2011, 13, 1043–1051 CrossRef CAS PubMed.
- D. F. C. Silva, A. M. Azevedo, P. Fernandes, V. Chu, J. P. Conde and M. R. Aires-Barros, J. Chromatogr. A, 2017, 1487, 242–247 CrossRef CAS PubMed.
- R. J. Meagher, Y. K. Light and A. K. Singh, Lab Chip, 2008, 8, 527–532 RSC.
- Y. S. Huh, C.-M. Jeong, H. N. Chang, S. Y. Lee, W. H. Hong and T. J. Park, Biomicrofluidics, 2010, 4, 14103 CrossRef PubMed.
- D. F. C. Silva, A. M. Azevedo, P. Fernandes, V. Chu, J. P. Conde and M. R. Aires-Barros, J. Chromatogr. A, 2012, 1249, 1–7 CrossRef CAS PubMed.
- Y. Huang, T. Meng, T. Guo, W. Li, W. Yan, X. Li, S. Wang and Z. Tong, Microfluid. Nanofluid., 2014, 16, 483–491 CrossRef CAS.
- R. R. G. Soares, P. Novo, A. M. Azevedo, P. Fernandes, M. R. Aires-Barros, V. Chu and J. P. Conde, Lab Chip, 2014, 14, 4284–4294 RSC.
- Y. S. Song, Y. H. Choi and D. H. Kim, J. Chromatogr. A, 2007, 1162, 180–186 CrossRef CAS PubMed.
- Y. H. Choi, Y. S. Song and D. H. Kim, J. Chromatogr. A, 2010, 1217, 3723–3728 CrossRef CAS PubMed.
- R. Hu, X. Feng, P. Chen, M. Fu, H. Chen, L. Guo and B.-F. Liu, J. Chromatogr. A, 2011, 1218, 171–177 CrossRef CAS PubMed.
- C. D. M. Campos, J. K. Park, P. Neuzil, J. A. F. da Silva and A. Manz, RSC Adv., 2014, 4, 49485–49490 RSC.
- L. Qi, Y. Wang, Y. Li, G. Zheng, C. Li and H. Su, Anal. Bioanal. Chem., 2015, 407, 3617–3625 CrossRef CAS PubMed.
- U. Novak, A. Pohar, I. Plazl and P. Žnidaršič-Plazl, Sep. Purif. Technol., 2012, 97, 172–178 CrossRef CAS.
- L. Vobecká, E. Khafizova, T. Stragier, Z. Slouka and M. Přibyl, Microfluid. Nanofluid., 2017, 21, 51 CrossRef.
- R. Apweiler, A. Bairoch, C. H. Wu, W. C. Barker, B. Boeckmann, S. Ferro, E. Gasteiger, H. Huang, R. Lopez, M. Magrane, M. J. Martin, D. A. Natale, C. O'Donovan, N. Redaschi and L.-S. L. Yeh, Nucleic Acids Res., 2004, 32, D115–D119 CrossRef CAS PubMed.
- M. G. Freire, A. F. M. Cláudio, J. M. M. Araújo, J. A. P. Coutinho, I. M. Marrucho, J. N. Canongia Lopes and L. P. N. Rebelo, Chem. Soc. Rev., 2012, 41, 4966–4995 RSC.
- E. Espitia-Saloma, P. Vâzquez-Villegas, M. Rito-Palomares and O. Aguilar, Biotechnol. J., 2016, 11, 708–716 CrossRef CAS PubMed.
- U. Novak, M. Lakner, I. Plazl and P. Žnidaršič-Plazl, Microfluid. Nanofluid., 2015, 19, 75–83 CrossRef CAS.
- W. Ruige and Y. S. Fung, Bioanalysis, 2015, 7, 907–922 CrossRef PubMed.
- F. Nazzaro, P. Orlando, F. Fratianni, A. Di Luccia and R. Coppola, Nutrients, 2012, 4, 1475–1489 CrossRef CAS PubMed.
- M. Gong, N. Zhang and N. Maddukuri, Anal. Methods, 2018, 10, 3131–3143 RSC.
- T. Tran, I. Ayed, A. Pallandre and M. Taverna, Recent innovations in protein separation on microchips by electrophoretic methods: An update, 2010, vol. 31 Search PubMed.
- H. Chen and Z. H. Fan, Electrophoresis, 2009, 30, 758–765 CrossRef CAS PubMed.
- R. Ashton, C. Padala and R. S. Kane, Curr. Opin. Biotechnol., 2003, 14, 497–504 CrossRef CAS PubMed.
- S. Liu and A. Guttman, TrAC, Trends Anal. Chem., 2004, 23, 422–431 CrossRef CAS.
- S. K. Kailasa and S. H. Kang, Sep. Purif. Rev., 2009, 38, 242–288 CrossRef CAS.
- G. Dutta, J. Rainbow, U. Zupancic, S. Papamatthaiou, P. Estrela and D. Moschou, Chemosensers, 2018, 6 Search PubMed.
- F. A. Gomez, in Protein Chromatography: Methods and Protocols, ed. D. Walls and S. T. Loughran, Humana Press, Totowa, NJ, 2011, pp. 137–150 Search PubMed.
- H. Ranchon, R. Malbec, V. Picot, A. Boutonnet, P. Terrapanich, P. Joseph, T. Leïchlé and A. Bancaud, Lab Chip, 2016, 16, 1243–1253 RSC.
- M. He and A. E. Herr, Anal. Chem., 2009, 81, 8177–8184 CrossRef CAS PubMed.
- Y. Liu, R. S. Foote, S. C. Jacobson, R. S. Ramsey and J. M. Ramsey, Anal. Chem., 2000, 72, 4608–4613 CrossRef CAS PubMed.
- B. Gumuscu, J. G. Bomer, H. L. de Boer, A. van den Berg and J. C. T. Eijkel, Microsyst. Nanoeng., 2017, 3, 17001 CrossRef CAS PubMed.
- P. Novo, M. Jender, M. Dell'Aica, R. P. Zahedi and D. Janasek, Procedia Eng., 2016, 168, 1382–1385 CrossRef CAS.
- Y.-A. Song, M. Chan, C. Celio, S. R. Tannenbaum, J. S. Wishnok and J. Han, Anal. Chem., 2010, 82, 2317–2325 CrossRef CAS PubMed.
- A. V. Hatch, A. E. Herr, D. J. Throckmorton, J. S. Brennan and A. K. Singh, Anal. Chem., 2006, 78, 4976–4984 CrossRef CAS PubMed.
- S. Saedinia, K. L. Nastiuk, J. J. Krolewski, G. P. Li and M. Bachman, Biomicrofluidics, 2014, 8, 14107 CrossRef PubMed.
- R. C. Lo and V. M. Ugaz, Lab Chip, 2008, 8, 2135–2145 RSC.
- H. Nagata, M. Tabuchi, K. Hirano and Y. Baba, Electrophoresis, 2005, 26, 2247–2253 CrossRef CAS PubMed.
- B. L. Thompson, C. Birch, D. A. Nelson, J. Li, J. A. DuVall, D. Le Roux, A.-C. Tsuei, D. L. Mills, B. E. Root and J. P. Landers, Lab Chip, 2016, 16, 4569–4580 RSC.
- D. Oh, M. Lee, J. Park and S. H. Kang, J. Sep. Sci., 2010, 33, 1109–1114 CAS.
- C. L. Colyer, S. D. Mangru and D. J. Harrison, J. Chromatogr. A, 1997, 781, 271–276 CrossRef CAS PubMed.
- Y. Cong, L. Zhang, D. Tao, Y. Liang, W. Zhang and Y. Zhang, J. Sep. Sci., 2008, 31, 588–594 CrossRef CAS PubMed.
- R. Wu, Z. Wang, W. Zhao, W. Shu-Biu Yeung and Y. Fung, Multi-dimension microchip-capillary electrophoresis device for determination of functional proteins in infant milk formula, 2013, vol. 1274 Search PubMed.
- H. Shadpour and S. A. Soper, in 2004 AIChE Annual Meeting, Conference Proceedings, Austin, TX, United States, 2004, pp. 1933–1939.
- R. G. Wu, Z. P. Wang and D. Y. P. Seah, in 17th International Conference on Miniaturized Systems for Chemistry and Life Sciences, Freiburg, Germany, 2013.
- L. Gutzweiler, T. Gleichmann, L. Tanguy, P. Koltay, R. Zengerle and L. Riegger, Electrophoresis, 2017, 38, 1764–1770 CrossRef CAS PubMed.
- A. Han, K. Hosokawa and M. Maeda, Electrophoresis, 2009, 30, 3507–3513 CrossRef CAS PubMed.
- M. Yu, H.-Y. Wang and A. Woolley, Electrophoresis, 2009, 30, 4230–4236 CrossRef CAS PubMed.
- A. Wainright, U. T. Nguyen, T. Bjornson and T. D. Boone, Electrophoresis, 2003, 24, 3784–3792 CrossRef CAS PubMed.
- J. J. Lu, S. Wang, G. Li, W. Wang, Q. Pu and S. Liu, Anal. Chem., 2012, 84, 7001–7007 CrossRef CAS PubMed.
- R. S. Foote, J. Khandurina, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 2005, 77, 57–63 CrossRef CAS PubMed.
- N. Minc, C. Fütterer, K. D. Dorfman, A. Bancaud, C. Gosse, C. Goubault and J.-L. Viovy, Anal. Chem., 2004, 76, 3770–3776 CrossRef CAS PubMed.
- E. Ollikainen, A. Bonabi, N. Nordman, V. Jokinen, T. Kotiaho, R. Kostiainen and T. Sikanen, J. Chromatogr. A, 2016, 1440, 249–254 CrossRef CAS PubMed.
- W. Hellmich, C. Pelargus, K. Leffhalm, A. Ros and D. Anselmetti, Electrophoresis, 2005, 26, 3689–3696 CrossRef CAS PubMed.
- J. Liu, T. Pan, A. T. Woolley and M. L. Lee, Anal. Chem., 2004, 76, 6948–6955 CrossRef CAS PubMed.
- S. Akashi, K. Suzuki, A. Arai, N. Yamada, E.-I. Suzuki, K. Hirayama, S. Nakamura and Y. Nishimura, Rapid Commun. Mass Spectrom., 2006, 20, 1932–1938 CrossRef CAS PubMed.
- H. Shadpour and S. A. Soper, Anal. Chem., 2006, 78, 3519–3527 CrossRef CAS PubMed.
- S. Wang, S. K. Njoroge, K. Battle, C. Zhang, B. C. Hollins, S. A. Soper and J. Feng, Lab Chip, 2012, 12, 3362–3369 RSC.
- Y.-C. Wang, M. H. Choi and J. Han, Anal. Chem., 2004, 76, 4426–4431 CrossRef CAS PubMed.
- J. K. Osiri, H. Shadpour and S. A. Soper, Anal. Bioanal. Chem., 2010, 398, 489–498 CrossRef CAS PubMed.
- J.-Z. Pan, P. Fang, X.-X. Fang, T.-T. Hu, J. Fang and Q. Fang, Sci. Rep., 2018, 8, 1791 CrossRef PubMed.
- F.-C. Huang, C.-S. Liao and G.-B. Lee, Electrophoresis, 2006, 27, 3297–3305 CrossRef CAS PubMed.
- V. Dolnik and S. Liu, Applications of capillary electrophoresis on microchip, 2005, vol. 28 Search PubMed.
- H. Nagata, M. Tabuchi, K. Hirano and Y. Baba, Chromatogr. J. Sep. Detect. Sci., 2005, 26, 23–28 CAS.
- K. Macounová, C. R. Cabrera and P. Yager, Anal. Chem., 2001, 73, 1627–1633 CrossRef PubMed.
- R. Walczak, K. Adamski and W. Kubicki, Multidisciplinary Digital Publishing Institute Proceedings, 2017, p. 1 Search PubMed.
- K. Killeen, H. Yin, D. Sobek, R. Brennen and T. Van de Goor, in Proc MicroTAS, 2003, vol. 2003, pp. 481–484.
- M. S. Shapiro, S. J. Haswell, G. J. Lye and D. G. Bracewell, Biotechnol. Prog., 2009, 25, 277–285 CrossRef CAS PubMed.
- L. J. Millet, J. D. Lucheon, R. F. Standaert, S. T. Retterer and M. J. Doktycz, Lab Chip, 2015, 15, 1799–1811 RSC.
- C. Yu, M. H. Davey, F. Svec and J. M. J. Fréchet, Anal. Chem., 2001, 73, 5088–5096 CrossRef CAS PubMed.
- H. Zhong and Z. El Rassi, J. Sep. Sci., 2009, 32, 1642–1653 CrossRef CAS PubMed.
- Y. Li, H. D. Tolley and M. L. Lee, Anal. Chem., 2009, 81, 9416–9424 CrossRef CAS PubMed.
- J. Liu, C.-F. Chen, C.-W. Tsao, C.-C. Chang, C.-C. Chu and D. L. DeVoe, Anal. Chem., 2009, 81, 2545–2554 CrossRef CAS PubMed.
- K. Deshpande, T. Ahamed, L. A. M. van der Wielen, J. H. ter Horst, P. J. Jansens and M. Ottens, Lab Chip, 2009, 9, 600–605 RSC.
- C. Martin and A. M. Lenhoff, Biochem. Eng. J., 2011, 53, 216–222 CrossRef CAS PubMed.
- G. Chirica, J. Lachmann and J. Chan, Anal. Chem., 2006, 78, 5362–5368 CrossRef CAS PubMed.
- M. C. Peoples, T. M. Phillips and H. T. Karnes, J. Chromatogr. B, 2007, 848, 200–207 CrossRef CAS PubMed.
- A. S. Chan, M. K. Danquah, D. Agyei, P. G. Hartley and Y. Zhu, Sep. Sci. Technol., 2014, 49, 854–860 CrossRef CAS.
- C. Gao, X. Sun and A. T. Woolley, J. Chromatogr. A, 2013, 1291, 92–96 CrossRef CAS PubMed.
- A. S. Chan, M. K. Danquah, D. Agyei, P. G. Hartley and Y. Zhu, J. Anal. Methods Chem., 2014, 2014, 175457 Search PubMed.
- D. S. Reichmuth, T. J. Shepodd and B. J. Kirby, Anal. Chem., 2005, 77, 2997–3000 CrossRef CAS PubMed.
- A. G. Deshpande, N. J. Darton, K. Yunus, A. C. Fisher and N. K. H. Slater, Nat. Biotechnol., 2012, 29, 494–501 CAS.
- M. E. Sandison, K. T. Jensen, F. Gesellchen, J. M. Cooper and A. R. Pitt, Analyst, 2014, 139, 4974–4981 RSC.
- C. D. García, D. J. Hadley, W. W. Wilson and C. S. Henry, Biotechnol. Prog., 2003, 19, 1006–1010 CrossRef PubMed.
- B. Teste, F. Malloggi, J.-M. Siaugue, A. Varenne, F. Kanoufi and S. Descroix, Lab Chip, 2011, 11, 4207–4213 RSC.
- M. C. Peoples and H. T. Karnes, Anal. Chem., 2008, 80, 3853–3858 CrossRef CAS PubMed.
- H. Yin, K. Killeen, R. Brennen, D. Sobek, M. Werlich and T. van de Goor, Anal. Chem., 2005, 77, 527–533 CrossRef CAS PubMed.
- I. M. Lazar, P. Trisiripisal and H. A. Sarvaiya, Anal. Chem., 2006, 78, 5513–5524 CrossRef CAS PubMed.
- L. Wang, K. Sun, X. Hu, G. Li, Q. Jin and J. Zhao, Sens. Actuators, B, 2015, 219, 105–111 CrossRef CAS.
- C. L. Hansen, E. Skordalakes, J. M. Berger and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16531LP–1616536 CrossRef PubMed.
- M. Maeki, Y. Teshima, S. Yosizuka, H. Yamaguchi, K. Yamashita, H. Maeda and M. Miyazaki, in 16th International Conference on Miniaturized Systems for Chemistry and Life Sciences, Okinawa, Japan, 2012.
- M. Maeki, Y. Teshima, S. Yoshizuka, H. Yamaguchi, K. Yamashita and M. Miyazaki, Chem. – Eur. J., 2013, 20, 1049–1056 CrossRef PubMed.
- T. Xu, K. Chakrabarty and V. K. Pamula, IEEE Trans. Comput. Des. Integr. Circuits Syst., 2010, 29, 552–565 Search PubMed.
- S. Guha, S. L. Perry, A. S. Pawate and P. J. A. Kenis, Sens. Actuators, B, 2012, 174, 1–9 CrossRef CAS PubMed.
- J. D. Ng, P. J. Clark, R. C. Stevens and P. Kuhn, Acta Crystallogr. Sect. D: Biol. Crystallogr., 2008, 64, 189–197 CrossRef CAS PubMed.
- F. Li and R. Lakerveld, Cryst. Growth Des., 2017, 17, 3062–3070 CrossRef CAS.
- J. E. Kreutz, L. Li, L. S. Roach, T. Hatakeyama and R. F. Ismagilov, J. Am. Chem. Soc., 2009, 131, 6042–6043 CrossRef CAS PubMed.
- M. Lounaci, P. Rigolet, C. Abraham, M. Le Berre and Y. Chen, Microelectron. Eng., 2007, 84, 1758–1761 CrossRef CAS.
- H.-J. Wu, T. Basta, M. Morphew, D. C. Rees, M. H. B. Stowell and Y. C. Lee, in The 8th Annual IEEE International Conference on Nano/Micro Engineered and Molecular Systems, 2013, pp. 84–87.
- M. Ildefonso, N. Candoni and S. Veesler, Org. Process Res. Dev., 2012, 16, 556–560 CrossRef CAS.
- S. Zhang, C. J. J. Gerard, A. Ikni, G. Ferry, L. M. Vuillard, J. A. Boutin, N. Ferte, R. Grossier, N. Candoni and S. Veesler, J. Cryst. Growth, 2017, 472, 18–28 CrossRef CAS.
- X. Zhou, L. Lau, W. W. L. Lam, S. W. N. Au and B. Zheng, Anal. Chem., 2007, 79, 4924–4930 CrossRef CAS PubMed.
- L. Li, D. Mustafi, Q. Fu, V. Tereshko, D. L. Chen, J. D. Tice and R. F. Ismagilov, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19243LP–1919248 CrossRef PubMed.
- Y. Zhu, L.-N. Zhu, R. Guo, H.-J. Cui, S. Ye and Q. Fang, Sci. Rep., 2014, 4, 5046 CrossRef CAS PubMed.
- J. Ferreira, F. Castro, F. Rocha and S. Kuhn, Chem. Eng. Sci., 2018, 191, 232–244 CrossRef CAS.
- B. G. Abdallah, S. Roy-Chowdhury, R. Fromme, P. Fromme and A. Ros, Cryst. Growth Des., 2016, 16, 2074–2082 CrossRef CAS PubMed.
- M. Heymann, A. Opthalage, J. L. Wierman, S. Akella, D. M. E. Szebenyi, S. M. Gruner and S. Fraden, IUCrJ, 2014, 1, 349–360 CrossRef CAS PubMed.
- B. Zheng, L. S. Roach and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 11170–11171 CrossRef CAS PubMed.
- C. Longuet, A. Yamada, Y. Chen, D. Baigl and J. Fattaccioli, J. Cryst. Growth, 2014, 386, 179–182 CrossRef CAS.
- C. J. Gerdts, V. Tereshko, M. K. Yadav, I. Dementieva, F. Collart, A. Joachimiak, R. C. Stevens, P. Kuhn, A. Kossiakoff and R. F. Ismagilov, Angew. Chem., Int. Ed., 2006, 45, 8156–8160 CrossRef CAS PubMed.
- G. Li, Q. Chen, J. Li, X. Hu and J. Zhao, Anal. Chem., 2010, 82, 4362–4369 CrossRef CAS PubMed.
- Y. Zang, B. Kammerer, M. Eisenkolb, K. Lohr and H. Kiefer, PLoS One, 2011, 6, e25282 CrossRef CAS PubMed.
- A. S. Pawate, V. Šrajer, J. Schieferstein, S. Guha, R. Henning, I. Kosheleva, M. Schmidt, Z. Ren, P. J. A. Kenis and S. L. Perry, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2015, 71, 823–830 CrossRef CAS PubMed.
- B. Zheng, C. J. Gerdts and R. F. Ismagilov, Curr. Opin. Struct. Biol., 2005, 15, 548–555 CrossRef CAS PubMed.
- D. L. Chen, L. Li, S. Reyes, D. N. Adamson and R. F. Ismagilov, Langmuir, 2007, 23, 2255–2260 CrossRef CAS PubMed.
- G. Kisselman, W. Qiu, V. Romanov, C. M. Thompson, R. Lam, K. P. Battaile, E. F. Pai and N. Y. Chirgadze, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 533–539 CrossRef CAS PubMed.
- D. S. Khvostichenko, J. M. Schieferstein, A. S. Pawate, P. D. Laible and P. J. A. Kenis, Cryst. Growth Des., 2014, 14, 4886–4890 CrossRef CAS PubMed.
- J. M. Schieferstein, A. S. Pawate, C. Sun, F. Wan, P. N. Sheraden, J. Broecker, O. P. Ernst, R. B. Gennis and P. J. A. Kenis, Biomicrofluidics, 2017, 11, 24118 CrossRef PubMed.
- B. T. C. Lau, C. A. Baitz, X. P. Dong and C. L. Hansen, J. Am. Chem. Soc., 2007, 129, 454–455 CrossRef CAS PubMed.
- D. Zhu, X. Zhou and B. Zheng, Micromachines, 2015, 6 Search PubMed.
- B. Zheng, J. D. Tice, L. S. Roach and R. F. Ismagilov, Angew. Chem., Int. Ed., 2004, 43, 2508–2511 CrossRef CAS PubMed.
- C. L. Hansen, S. Classen, J. M. Berger and S. R. Quake, J. Am. Chem. Soc., 2006, 128, 3142–3143 CrossRef CAS PubMed.
- C. W. Carruthers Jr., C. Gerdts, M. D. Johnson and P. Webb, PLoS One, 2013, 8, e82298 CrossRef PubMed.
- M. Maeki, S. Yamazaki, A. S. Pawate, A. Ishida, H. Tani, K. Yamashita, M. Sugishima, K. Watanabe, M. Tokeshi, P. J. A. Kenis and M. Miyazaki, CrystEngComm, 2016, 18, 7722–7727 RSC.
- J. A. Gavira, Arch. Biochem. Biophys., 2016, 602, 3–11 CrossRef CAS PubMed.
- M. Maeki, H. Yamaguchi, M. Tokeshi and M. Miyazaki, Anal. Sci., 2016, 32, 3–9 CrossRef CAS PubMed.
- S. Sui and S. L. Perry, Struct. Dyn., 2017, 4, 32202 CrossRef.
- J. Wang, J. Zhang and J. Han, Front. Chem. Eng. China, 2010, 4, 26–36 CrossRef CAS.
- J. Leng and J.-B. Salmon, Lab Chip, 2009, 9, 24–34 RSC.
- F. Castro, A. Ferreira, J. A. Teixeira and F. Rocha, Cryst. Growth Des., 2016, 16, 3748–3755 CrossRef CAS.
- F. Castro, A. Ferreira, J. A. Teixeira and F. Rocha, Cryst. Growth Des., 2018, 18, 5940–5946 CrossRef CAS.
- D. Jaritsch, A. Holbach and N. Kockmann, J. Fluids Eng., 2014, 9, 136 Search PubMed.
- A. Aota, M. Nonaka, A. Hibara and T. Kitamori, Angew. Chem., Int. Ed., 2007, 46, 878–880 CrossRef CAS PubMed.
- M. Watanabe, Analyst, 2011, 136, 1420–1424 RSC.
- A. Holbach and N. Kockmann, Green Process. Synth., 2013, 2, 157–167 CAS.
- J. Jaspe and S. J. Hagen, Biophys. J., 2006, 91, 3415–3424 CrossRef CAS PubMed.
- F. A. Vicente, L. P. Malpiedi, F. A. e Silva, A. Pessoa Jr., J. A. P. Coutinho and S. P. M. Ventura, Sep. Purif. Technol., 2014, 135, 259–267 CrossRef CAS.
- F. A. Vicente, L. D. Lario, A. Pessoa and S. P. M. Ventura, Process Biochem., 2016, 51, 528–534 CrossRef CAS.
- L. P. Malpiedi, B. B. Nerli, D. S. P. Abdala, P. de A. Pessôa-Filho and A. Pessoa, Sep. Purif. Technol., 2014, 132, 295–301 CrossRef CAS.
- F. A. Vicente, J. Bairos, M. Roque, J. A. P. Coutinho, S. P. M. Ventura and M. G. Freire, ACS Sustainable Chem. Eng., 2019, 7, 15102–15113 CrossRef CAS.
- R. and Markets, Microfluidics Market by Application, Component and Material - Global Forecast to 2023, https://www.researchandmarkets.com/reports/4495405/microfluidics-market-by-application-component#rela0–4895469, (accessed 30 January 2020).
- H. Becker, Lab Chip, 2009, 9, 2759–2762 RSC.
- N. Bhattacharjee, A. Urrios, S. Kang and A. Folch, Lab Chip, 2016, 16, 1720–1742 RSC.
- T. Ching, Y. Li, R. Karyappa, A. Ohno, Y.-C. Toh and M. Hashimoto, Sens. Actuators, B, 2019, 297, 126609 CrossRef CAS.
- J. Huft, C. A. Haynes and C. L. Hansen, Anal. Chem., 2013, 85, 2999–3005 CrossRef CAS PubMed.
- J. De Jong, R. G. H. Lammertink and M. Wessling, Lab Chip, 2006, 6, 1125–1139 RSC.
| This journal is © The Royal Society of Chemistry 2020 |