
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic transfer hydrogenation of maleic acid with stoichiometric amounts of formic acid in aqueous phase: paving the way for more sustainable succinic acid production†
M.
López Granados
 *a,
J.
Moreno
b,
A. C.
Alba-Rubio
c,
J.
Iglesias
b,
D.
Martín Alonso
de and
R.
Mariscal
a
*a,
J.
Moreno
b,
A. C.
Alba-Rubio
c,
J.
Iglesias
b,
D.
Martín Alonso
de and
R.
Mariscal
a
aEQS Group (Sustainable Energy and Chemistry Group), Institute of Catalysis and Petrochemistry (CSIC), C/Marie Curie, 2, 28049 Madrid, Spain. E-mail: mlgranados@icp.csic.es
bChemical & Environmental Engineering Group, Universidad Rey Juan Carlos, C/Tulipan, s/n, Mostoles, Madrid 28933, Spain
cDepartment of Chemical Engineering, University of Toledo, Toledo, OH 43606, USA
dGlucan Biorenewables LLC, Madison, WI 53719, USA
eDepartment of Chemical and Biological Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, WI 53706, USA
First published on 25th February 2020
Abstract
The aqueous phase hydrogenation of maleic acid (MAc) to succinic acid (SAc) is demonstrated in the absence of any organic solvent and using stoichiometric amount of formic acid (FAc) as source of H2. Among the different noble metals (Pd, Au, Ru, Pt and Rh) and supports investigated (γ-Al2O3, TiO2, CeO2, ZrO2, WO3, CeZrO4, carbon, nicanite, SiO2 and TS-1), Pd/C was identified as the best catalyst. We observe that the undesirable formation of malic acid (MalAc) by hydration of MAc must be prevented. The transformation of MAc to SAc with negligible formation of MalAc is possible by using relatively mild temperature (140–150 °C) and a high catalyst to MAc ratio (i.e. fixed bed continuous flow reactor). Using the carboxylate forms (disodium maleate and sodium formate) instead of the acids results in an increase of the reaction rate. In a fixed bed reactor under a continuous flow of 15 wt% of MAc at a WHSV = 12 h−1 (contact time = 5 min), at 150 °C, 10 bar of N2 and using a formic acid/maleic acid molar ratio = 1, a yield of SAc close to 98% was obtained, equivalent to a productivity of 1.87 g SAc per gcat per·h. Leaching of Pd was below 0.02 ppm. No deactivation was observed in long term experiments at 150 °C (ca. 730 h), although the characterization of the used catalyst by CO chemisorption and TEM and XPS studies showed certain sintering of Pd particles. Regarding the mechanism of the reaction, kinetic isotopic experiments using deuterated DCOOH indicated that the reaction must essentially proceed via catalytic transfer hydrogenation, formyl H of formic acid is involved in the rate determining step of the reaction. When using maleate and formate sodium salts, the second H needed for the reaction is supplied by the solvent (H2O molecules). A preliminary environmental assessment (Life Cycle Analysis, LCA) of this CTH approach indicates that for relevant environmental categories of the LCA (such as climate change and consumption of fossil resources) the CTH process is greener than conventional hydrogenation process; the benefits are even larger if biomass-derived FAc is involved.
Introduction
Succinic acid (SAc) and its dehydrated form, succinic anhydride, have multiple applications in the pharmaceutical, food, detergent, cosmetic, textile, photography, agrochemical and chemical industries.1,2 Polymerization of SAc with diamines or with diols renders polyamides or polyesters with interesting properties, such as commercially available Bionolle® (a polybutylene succinate-based polymer). Moreover SAc is an intermediate in the production of γ-butyrolactone (GBL), 1,4-butanediol (1,4BDO), and tetrahydrofuran (THF) from maleic acid (MAc).3,4 These latter compounds currently find also numerous applications as solvents and as intermediates in the synthesis of pharmaceuticals, agrochemicals, and polymers.Until few years ago, SAc was only produced from petrochemical maleic anhydride. The process requires first the liquid phase hydrogenation of maleic anhydride (120–180 °C, 0.5–4.0 MPa, Ni or Pd catalysts) rendering succinic anhydride that is subsequently hydrated with hot water to produce SAc.2,5 Another alternative route is the production of succinic anhydride as a by-product of the gas phase hydrogenation of maleic anhydride to GBL and/or 1,4-butanediol (1,4-BDO). The succinic anhydride can be recycled back to reactor for complete hydrogenation or separated and purified.2
More recently, fermentation of sugars seems to gain competitiveness versus the petrochemical route.6–9 The production of SAc from biomass feedstocks (glucose, starch, glycerin, etc.) requires the utilization of succinate producers microrganisms.6 However, the fermentation process also presents drawbacks that threatens its viability, such as, the handling of microorganisms, the precise control of the pH, its low productivity (fermentation requires long reaction times) and the complex and costly separation–purification steps of the succinic acid from the fermentation broth.10
The liquid phase oxidation of furfural to SAc using H2O2 is another alternative and high yields have been reported (in some cases close to 90%4,11). However, the current high price of H2O2 makes the production of C4 dicarboxylic acids like maleic and succinic unaffordable through this H2O2-driven oxidation, when compared with the petrochemical and fermentation routes.12
In this work we investigate a different route to produce SAc from MAc: the aqueous phase hydrogenation using stoichiometric formic acid (FAc) as source of H2. This approach presents features aligned with the principles of green chemistry. First, instead of using organic solvents, we used water, the greener solvent. Second, the utilization of formic acid prevents storing and handling of H2 at high pressure. Third, when conducting the reaction with H2, an inefficient excess of stoichiometric H2 pressure is needed versus the stoichiometric amount of formic acid demonstrated in this work. Finally, as it will be also shown along this article, the catalytic transfer hydrogenation with formic acid allows reducing some of the environmental impact caused by the conventional hydrogenation pathway.
An additional very interesting feature of this process is that SAc is easily separated from the downstream liquid by precipitation due to its relatively low solubility in H2O (2.65% at 5 °C, 4.26% at 10 °C).13 This means that when feeding relatively concentrated MAc, which is much more soluble than SAc (31.62% at 5 °C),14 most of the SAc is precipitated downstream by simple cooling: the lower the temperature, the more SAc precipitates. The remaining solution of diluted SAc can be recycled back to the reactor. This is a much straightforward and simpler protocol than that required for the separation and purification SAc obtained by fermentation.
Interestingly, although both FAc and MAc are currently derived from fossil feedstocks, their potential production from lignocellulosic feedstocks has already been technically demonstrated. Renewable FAc can be produced by for instance the so-called OxFA process15–17 or as a by-product in the production of levulinic acid from lignocellulose.18 Renewable MAc can be produced by oxidation of different lignocellulose derived platforms, i.e. butanol,19 levulinic acid,20 hydroxymethylfurfural,21–24 and furfural.25–29,30–33 Consequently, using bio-based FAc and MAc will turn the SAc produced into a renewable chemical. In this work we take the first step to make this possible: to demonstrate that the formic acid-driven hydrogenation of MAc in aqueous phase is efficient and technically and environmentally viable.
FAc is a well-known hydrogenating agent of a variety of organic functionalities like C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO and C–O groups.34 However, to the best of our knowledge, the FAc mediated hydrogenation of olefins has been conducted in organic solvents and with an excess of FAc. There is a lack of investigations regarding the use of water as solvent and of a stoichiometric amount of FAc.
C, CO and C–O groups.34 However, to the best of our knowledge, the FAc mediated hydrogenation of olefins has been conducted in organic solvents and with an excess of FAc. There is a lack of investigations regarding the use of water as solvent and of a stoichiometric amount of FAc.
Recently Zeng et al. have reported the aqueous phase FAc-mediated production of succinic acid, but from the trans isomer of MAc, fumaric acid, and using twice the stoichiometric amount of formic acid.35 As we will show later, this isomer is more easily reduced with FAc than MAc. Besides, a homogeneous Pd(AcO)2 catalyst was utilized, which is difficult to separate from the reaction mixture and consequently problematic to reuse and, more important, the incorporation of Xantphos (4,5-bis(diphenylphosphino)-9,9-dimethylxanthene) as a ligand was required to reach 94% SAc yield after 20 h of reaction. No results regarding the stability of this catalyst was reported although long term stability is required for an industrial application. Within this context, solid catalysts are a much better option when considering a practical application because they can readily be used in a fixed bed reactor under continuous operation.
A much simpler, more active and robust solid catalyst (carbon supported Pd catalyst) was identified in our work. We show that this catalyst conducts the reaction at similar temperatures and pressures to those used in the H2-industrial petrochemical process. We also demonstrate that the process can be conducted in water and using a stoichiometric amount of formic acid. The catalyst we report here shows very efficient atom economy: 100% of the H atoms of formic acid ended in the succinic acid molecule and close to 100% of MAc ended up as SAc (formation of malic and fumaric acid is negligible). We have also proved the long-term stability and robustness of the catalyst in a fixed bed reactor feeding relatively concentrated solutions of MAc for over 730 h.
Experimental
Reagents and catalysts
Maleic acid (99 wt%), formic acid (98 wt%), malic acid (99 wt%), fumaric acid (99 wt%) and levulinic acid (99 wt%) were supplied by Sigma Aldrich. Commercial catalysts based on palladium, platinum, ruthenium and rhodium (5 wt%) supported on activated charcoal were supplied by Sigma Aldrich. Carbon supported gold catalyst was prepared by a deposition–precipitation protocol described more in detail in the ESI.† The preparation of Pd supported on different supports is also described in the ESI.†Characterization of the catalysts
The Pd content of the fresh and used catalysts was determined by ICP-AES at Galbraith Laboratories, Inc. using a sodium peroxide fusion protocol for pretreating the sample. In summary samples were fussed with sodium peroxide over a Bunsen burner and subsequently the fused sample was dissolved in water. The Pd leached into the reaction mixtures was analyzed without previous digestion using an ICP-OES spectrophotometer (PQ9000 Analytik Jena).The average surface size of Pd particles was determined by HRTEM and CO chemisorption. HRTEM microscopy pictures were taken using a JEOL JEM-2100 F model microscope, working with an acceleration voltage of 300 kV. Samples were prepared by ultrasonically dispersing some powder sample in absolute ethanol and putting a droplet of the suspension on a Cu grid. The diameter of more than 250 Pd particles was measured to calculate the average surface diameter.
The amount of CO chemisorbed over Pd particles was determined by the breakthrough curves obtained by switching the feed from only Ar to a flow containing Ar, He and CO. Helium is used to trace the dead volume of the reactor and to set time = 0 for the breakthrough curves. A careful description of the experiment is described in the ESI,† in short, the protocol is as follows. The samples (50 mg) were loaded in a U-tube reactor which exit is connected to a Mass Spectrometer in order to analyze in real time the composition of the gases exiting the reactor. Two cycles were conducted, in the first cycle the CO breakthrough curve is delayed with respect to t = 0 due to irreversible and reversible CO chemisorption. Once CO signal reaches a steady value (30 min) the feed switches back to only Ar. Once CO is fully removed from the flow (no CO signal is detected) a second cycle was done. The second CO breakthrough curve is delayed only because of the reversible CO chemisorption (see an example in ESI Fig. 1†). The difference between these two breakthrough curves is the irreversible CO chemisorption. The number of atoms of Pd at the surface of the Pd particles (Pdsur) was calculated assuming a CO/Pdsur = 1. The dispersion of Pd was Pdsur/Pdtotal and the average particle size was estimated following the equation d(nm) = 1.12/dispersion.36
Catalytic activity experiments
The reactor was then immersed in a silicone bath set at the selected temperature (t0 of the experiment) and stirred at 1000 rpm for a given reaction time. To stop the reaction, the reactor was taken out from the silicone bath and let it cool down to room temperature. The reactor was depressurized and a known amount (around 0.5 g) of an aqueous levulinic acid (LAc) solution (around 0.15–0.20 g of LAc per g of solution) was incorporated to the reaction mixture as an internal standard. This solution was filtered through a 0.22 μm syringe filter and analyzed in an Agilent 1200 HPLC chromatograph equipped with a refraction index detector, and a Bio-Rad Aminex HPX-87H column (300 mm × 7.8 mm). A 0.005 M H2SO4 mobile phase was employed at 368 K and a 0.8 mL min−1 flow rate. MAc conversion and product yields were calculated according to the following formulas:
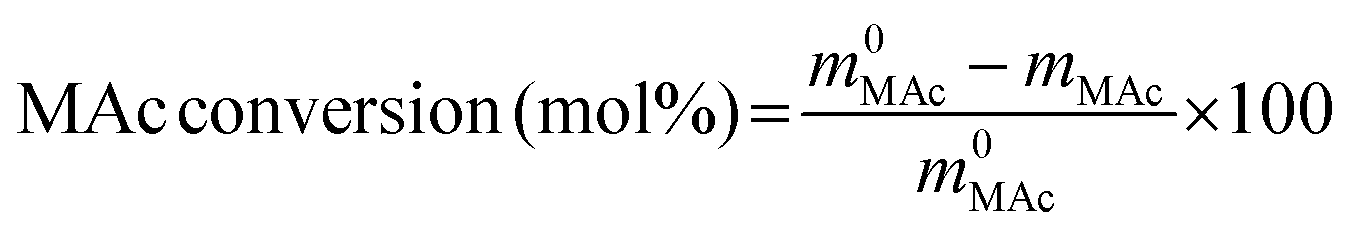
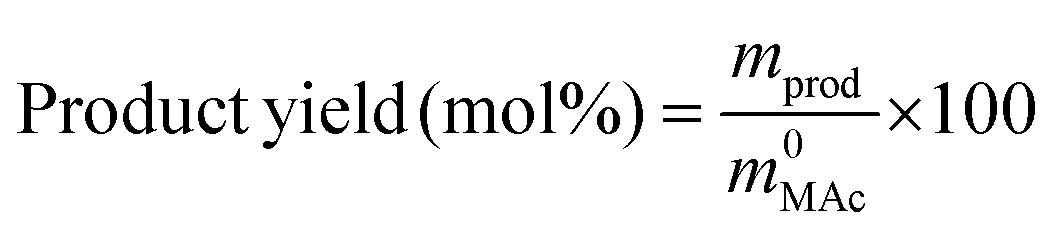
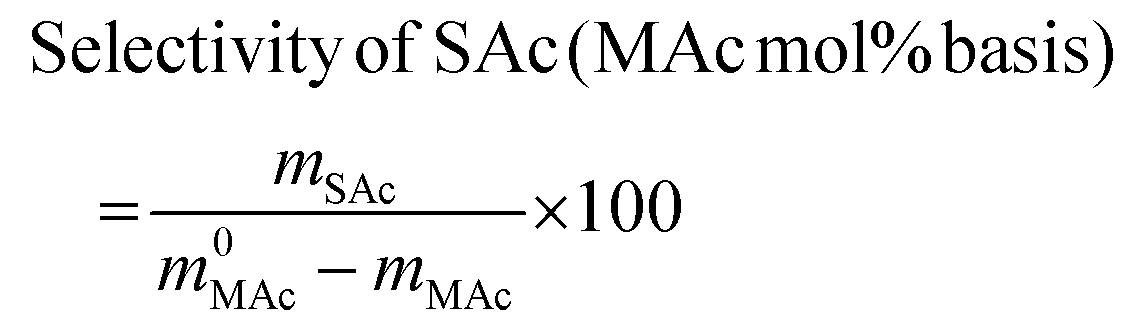
where m0MAc and m0FAc refers to the mole quantities initially loaded into the reactor, and mMAc, mFAc, mSAc and mprod refer to the number of moles of the MAc, FAc, SAc and, in general, of any product of the reaction, respectively, in the mixture after reaction. The HPLC chromatographic factor of the organic products was calculated by analyzing solutions with known concentrations of the different organic products.
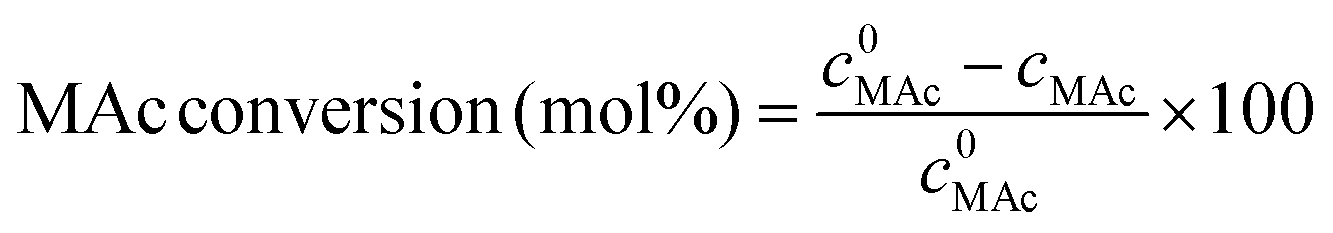
where c0MAc refers to the mole per g of aliquot fed into the reactor, and cMAc and cprod refer to the number of mole per g of aliquot of MAc and products, respectively, in the reaction mixture sampled at a given time.10 The carbon balance was calculated on MAc mol basis and for these experiments was always 100% (±5%).
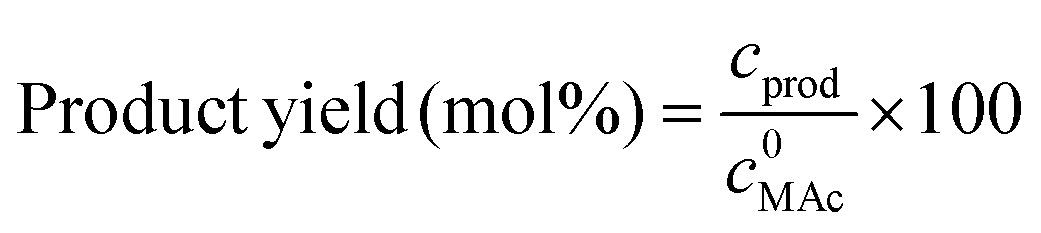
Results and discussion
Study of the catalytic properties of different catalysts and optimization of the reaction parameters
| Catalyst | X MAc (%) | X FAc (%) | Y SAc (%) | Y MalAc (%) | Y FumAc (%) | Sel.SAc (%) (based on MAc) | Sel.SAc (%) (based on FAc) |
|---|---|---|---|---|---|---|---|
| X: conversion, Y: yield, Sel.: selectivity. | |||||||
| Pd/C | 99 | 24 | 96 | 1 | 3 | 97 | 100 |
| Ru/C | 92 | 6 | 24 | 2 | 18 | 26 | 100 |
| Au/C | 98 | 15 | 24 | 1 | 24 | 25 | 40 |
| Pt/C | 61 | 4 | 16 | 1 | 18 | 26 | 100 |
| Rh/C | 67 | 2 | 7 | 4 | 18 | 11 | 87 |
It is clear that Pd/C is the most active and selective among of the different catalyst tested: it showed the highest activity both in MAc and FAc conversion (99% and 24%, respectively) and the highest yield of SAc (96%). It is also worth stressing that SAc selectivity in MAc and FAc mol basis are also the highest (96% and 100%, respectively). Other diacids like malic acid (MalAc) and fumaric acid (FumAc) were also formed respectively by hydration of the double bond and isomerization of MAc, respectively, but in minor amounts.
The rest of the metals present a much lower yield of SA, visibly low in the case of Pt and Rh. The main reason seems to be the worse capacity of these metals to transfer the hydrogen from FAc to MAc: the FAc conversion for Ru, Au, Pt and Rh is visibly smaller than that of Pd. In the case of Au the conversion of formic acid is quite high but its effective capacity is very low (only around 40% of converted FAc is used in the hydrogenation). Moreover, in all these metals there is a lack of carbon balance with respect to MAc. Thus, in the case of Ru catalyst around 44% of the converted MAc was not identified by HPLC. A white precipitate, that turned out to be FumAc when dissolved with NaOH and analyzed, explains this lack of carbon balance (solubility of FumAc in water is around 6–8 mg g−1 of solution10). Therefore, FumAc yields must be higher than that tabulated. In practice if MAc is not rapidly hydrogenated, isomerizes under the reaction conditions to scarcely soluble FumAc. This is a problem when considering a practical application under continuous operation mode because FumAc precipitation may result in the plugging of the reactor. In summary, the following order can be established between the metals in terms of SAc yield Pd ≫ Ru ≈ Au > Pt > Rh. Consequently, Pd/C catalyst was selected for further studies.
Study of the effect of FAc/MAc mol ratio, pressure and temperature of reaction on Pd/C catalyst
Table 2 summarises the results obtained with the Pd/C catalyst when changing the FAc/MAc mol ratio, initial pressures of N2 and reaction temperatures. The rest of the reaction conditions were those of Table 1 experiments. First three rows refer to the effect of the FAc/MAc mol ratio. Using a FAc/MAc ratio close to 1 is the best option because of the saving in FAc. The results clearly showed that the conversion of MAc and the yield of SAc decrease upon lowering the FAc concentration. Thus, conversion of MAc was 96, 84 and 71% and the yield of SAc was 96, 70 and 40% for FAc/MAc ratio of 4, 2 and 1, respectively, clearly indicating that the reaction rate of transformation of MAc into SAc also decreases. However, a positive result is that the selectivity of SAc in FAc basis for ratios of 1 and 2, was also quite high (89 and 100%, respectively), indicating that the H2 contained in the converted FAc also ends up essentially in SAc. In other words, FAc utilization was very effective regardless the FAc/MAc ratio.| Exp | FAc/Mac (mol ratio) | T (°C) | P in. N2 (bar)a | X MAc (%) | X FAc (%) | Y SAc (%) (SelSAc)c | Y MalAc (%) | Y FumAc (%) |
|---|---|---|---|---|---|---|---|---|
| a Initial pressure of N2. b atm.: Atmospheric pressure. c Selectivity of SAc based on FAc. d Using sodium formate as H2 source. Same conditions as entry 9, except 2 h of reaction time. | ||||||||
| 1 | 4 | 110 | atm.b | 99 | 24 | 96 (100) | 1 | 3 |
| 2 | 2 | 110 | atm. | 84 | 35 | 70 (100) | 1 | 12 |
| 3 | 1 | 110 | atm. | 71 | 45 | 40 (89) | 1 | 11 |
| 4 | 1 | 110 | 10 | 73 | 60 | 49 (82) | 1 | 11 |
| 5 | 1 | 110 | 30 | 75 | 61 | 47 (77) | 2 | 11 |
| 6 | 1 | 110 | 50 | 69 | 52 | 44 (85) | 1 | 12 |
| 7 | 1 | 130 | 10 | 91 | 79 | 67 (85) | 3 | 12 |
| 8 | 1 | 140 | 10 | 97 | 87 | 87 (100) | 6 | 4 |
| 9 | 1 | 150 | 10 | 99 | 90 | 83 (92) | 10 | 4 |
| 10d | 1 | 150 | 10 | 100 | 98 | 98 (100) | 0 | 2 |
It is also worth noticing that the yield of FumAc also increases upon decreasing the ratio (from 3 for a ratio of 4 to 11–12 for lower ratios). So it seems that when working with low FAc/MAc ratio, MAc is not rapidly hydrogenated to SAc and is isomerized to FumAc. The same can also occurs, in a less extension, with MalAc. Consequently, when using FAc/MAc close to 1, the reaction conditions must be changed to increase the hydrogenation rate of MAc to SAc, and minimize the hydration to MalAc and, especially, the isomerization to FumAc. FumAc is very insoluble and can cause plugging issues in a hypothetical application in continuous mode (MalAc is much more soluble).
Next, the effect of N2 pressure and reaction temperature was explored. Entries 3, 4, 5 and 6 summarizes the results conducted at atmospheric pressure, 10, 30 and 50 bar. Stainless steel batch reactor was used for experiments at high pressure. The FAc/MAc was kept = 1 because our intention is to use the stoichiometric ratio. Increasing the pressure results in a small but visible effect on the FAc conversion and the SAc yield, the latter went through a maximum at 10 bar (49 and smoothly decreases for larger pressure. At 50 bar the SAc yield was 44%. It is important to notice that the SAc selectivity in FAc basis were very high in all cases (above 75%). Summarizing, pressure has a limited effect beyond N2 pressure of 10 bar.
The effect of the reaction temperature is discussed next (entries 4 and 7–9). As expected, the reaction temperature has a positive effect on the conversion of reactants because the reaction rates become faster. Thus, the yield of SAc increases with temperature and reached values well above 80% for 140 and 150 °C. Interestingly, the yield of FumAc decreases also with temperature. However, the increase of the reaction temperature comes with more formation of MalAc and actually its yield reaches 10% at 150 °C.
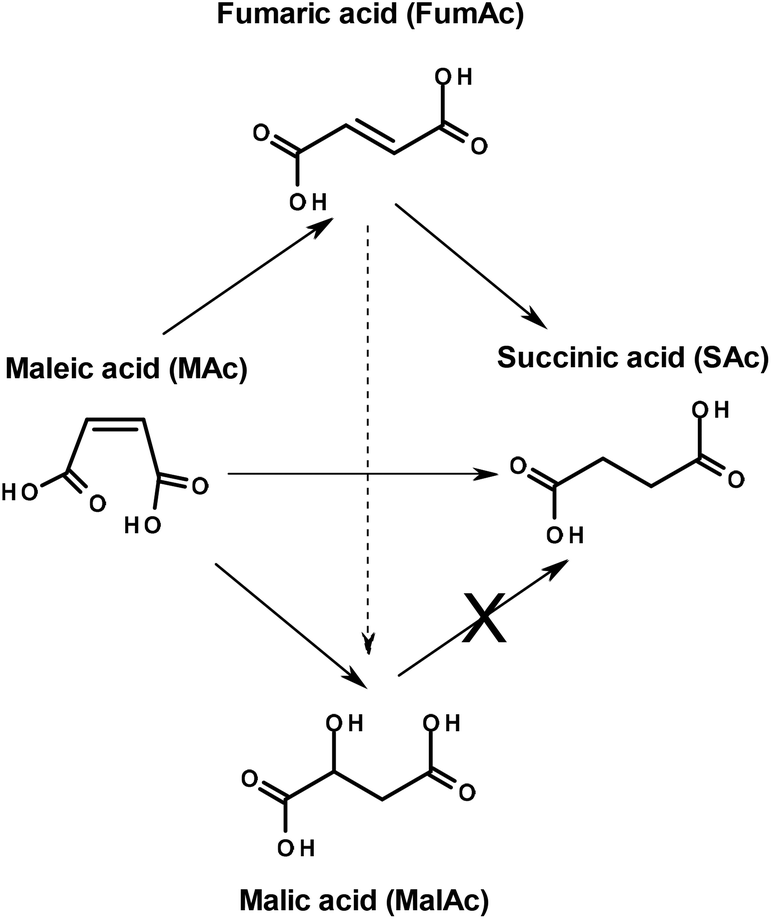
In an attempt of gaining information of the formation of MalAc and FumAc and consequently of the reaction network, several blank experiments were conducted at atmospheric pressure (not tabulated). In a blank experiment conducted under the reaction conditions of entry 3 but in the absence of FAc, the conversion of MAc is low and limited to FumAc (yield of only 2%) and MalAc (4%). In contrast, in entry 3 FumAc and MalAc yield were 11% and 1%, respectively. In another blank experiment contacting MAc with only FAc (no catalyst) the conversion of MAc is also very limited to FumAc (yield of 1%) and to MalAc (1%). This indicates that the formation of MalAc from MAc is also very limited and seems to be driven either thermally or catalyzed by formic acid. In the case of the isomerization of MAc to FumAc, the reaction is also very limited and enhanced by the simultaneous presence of both the Pd/C catalyst and formic acid (acid catalysis).
Interestingly, when conducting the hydrogenation of FumAc in the presence of both catalyst and FAc under the reaction conditions of entry 3, FumAc was almost quantitatively hydrogenated to SAc (yield was 100%, results in ESI Table 1†). FumAc is scarcely soluble in water at room temperature but its solubility rises to around 10% at 110 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 facilitating the hydrogenation reaction. Very low yields of MAc and MalAc (<0.1 and <0.2%, respectively) were observed. This may suggest that in our reaction (FAc-driven hydrogenation of MAc), FumAc is the intermediate of the reaction (MAc is isomerised to FumAc) which is subsequently and rapidly hydrogenated to SAc. However, in an additional experiment conducted with both FumAc and MAc initially present in the reactor (see ESI Table 1† and discussion there in), it was found that SAc basically derives from MAc. In this experiment it was also found that conversion of FAc is lower than when with only FumAc is present. All these data indicate that FumAc hydrogenation to SAc, although very rapid when no MAc is present, is significantly inhibited by MAc.
13 facilitating the hydrogenation reaction. Very low yields of MAc and MalAc (<0.1 and <0.2%, respectively) were observed. This may suggest that in our reaction (FAc-driven hydrogenation of MAc), FumAc is the intermediate of the reaction (MAc is isomerised to FumAc) which is subsequently and rapidly hydrogenated to SAc. However, in an additional experiment conducted with both FumAc and MAc initially present in the reactor (see ESI Table 1† and discussion there in), it was found that SAc basically derives from MAc. In this experiment it was also found that conversion of FAc is lower than when with only FumAc is present. All these data indicate that FumAc hydrogenation to SAc, although very rapid when no MAc is present, is significantly inhibited by MAc.
The possibility that FumAc is the intermediate of the reaction (MAc is isomerised to FumAc which is subsequently and effectively hydrogenated) can be ruled out. FumAc is an undesired by-product which hydrogenation is inhibited until MAc concentration is low. We will see below in practice that FumAc is hydrogenated once MAc concentration is depleted (high MAc conversion).
Finally, the hydrogenolysis of MalAc under reaction conditions of entry 1 (not tabulated) rendered a negligible amount of SAc, below 1%. This means that MalAc was very stable and cannot be transformed to SAc under the reaction conditions explored in this study. The network of reactions depicted in Scheme 1 can be proposed considering the results so far obtained. Under the reaction conditions here explored, MAc can be directly hydrogenated to SAc, isomerized to FumAc or hydrated to MalAc. The isomerization of FumAc to MalAc cannot be ruled out. The FumAc can also be subsequently hydrogenated to SAc but MalAc cannot be transformed to SAc.
 | ||
| Scheme 1 | ||
Study of other supports for Pd particles
The effect of the support was also studied by testing different materials (γ-Al2O3, SiO2, TiO2, WO3, ZrO2, CeZrO4, TS-1 and nicanite) as supports of Pd nanoparticles. The same Pd loading of the Pd/C commercial catalyst was used (close to 5 wt%). For the sake of summarizing, the preparation protocol and the results are thoroughly described in the ESI.† The commercial Pd/C showed the best SAc yield among all supported catalysts studied (ESI Fig. 2†). Pd/γ-Al2O3 also displayed quite similar SAc yield than Pd/C but also presented a high yield of MalAc and besides long term operation studies showed that Pd/γ-Al2O3 rapidly deactivates (see ESI Fig. 3†).Study of the effect of time on catalytic properties of Pd/C catalyst
Fig. 1 shows the reaction progress vs. time of Pd/C catalyst at 150 °C and 10 bar of initial N2 pressure (rest of conditions as entry 9). The data shows that MAc is unavoidably converted to MalAc as the reaction progresses but at a much slower reaction rate than that of SAc formation. At 4 h the yield of MalAc is close to 10%. The figure also shows that, initially, FumAc yield slightly increases upon reaction time and only decreases for reaction times longer than 1 h, once the conversion of MAc is quite high and MAc concentration has extensively been depleted. As suggested above, the presence of MAc inhibits the otherwise very fast hydrogenation of FumAc to SAc. Once MAc concentration approaches zero, FumAc can be definitively hydrogenated. Unfortunately, under these reaction conditions the full conversion of the initial MAc to SAc is not possible due to the formation of MalAc that cannot be hydrogenated with the Pd/C catalyst. It must also be stressed that FAc conversion and SAc yield are essentially (within the experimental error) the same, confirming the high efficiency in the use of H atoms of FAc (in some of the experiments SAc yield is slightly above the FAc conversion but taking into account that the experimental error in data deduced from HPLC is within ±2% and that the weighing of FAc and MAc also results in actual variations of the nominal FAc/MAc of ±5%, we can conclude that SAc yield and FAc conversion are essentially the same). This means that the SAc formation is intimately linked to the conversion of FAc and that the decomposition of FAc into CO and H2O can be considered negligible.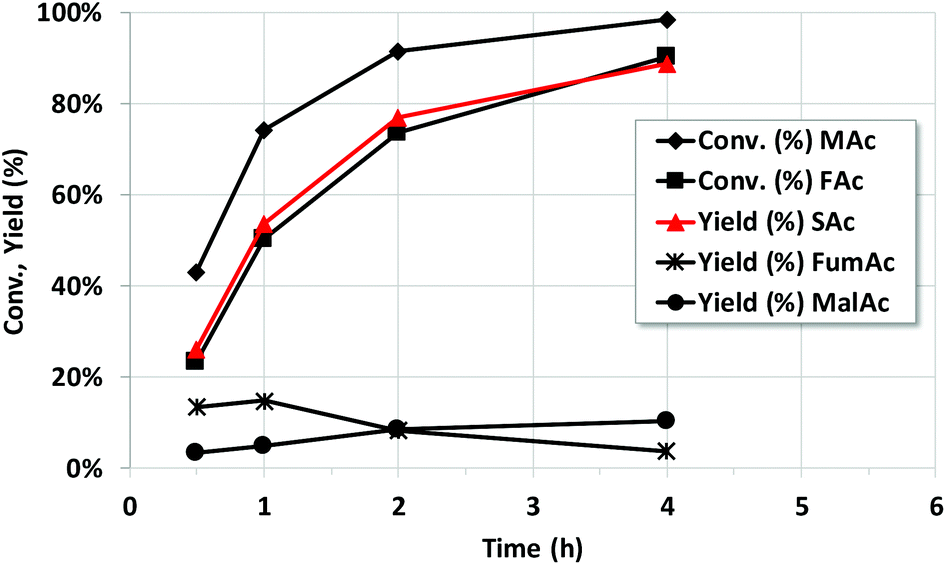 | ||
| Fig. 1 Catalytic properties of Pd/C catalyst as a function of reaction time. Reaction conditions: 5 g of reaction mixture, 5 wt% MAc, 1 wt% of catalyst, mol FAc/MAc = 1, 150 °C, initial N2 pressure = 10 bar. | ||
Study of the catalyst loading
The effect of the catalyst loading (from 0 to 2 wt%) was also studied. The reaction time was 4 h and the rest of the reaction conditions are those of Fig. 1. As expected, (see Fig. 2) the yield of SAc increases upon increasing the catalyst loading. Special attention must also be given to the yield of MalAc. When no catalyst is incorporated, SAc is not formed, and MAc is hydrated to MalAc up to a yield of 21%. When the catalyst loading increases, SAc is more rapidly formed and the MalAc yield decreases. At the maximum wt% studied (2 wt%, equivalent to a catalyst to substrate ratio of 0.4) the MalAc yield was 7.5%. Therefore, the faster the formation of SAc at higher catalyst loading, the faster the consumption of MAc; the latter prevents the formation of MalAc. In practice, a much higher effective catalyst to substrate ratio can be easily feasible by continuously feeding the liquid into a fixed bed.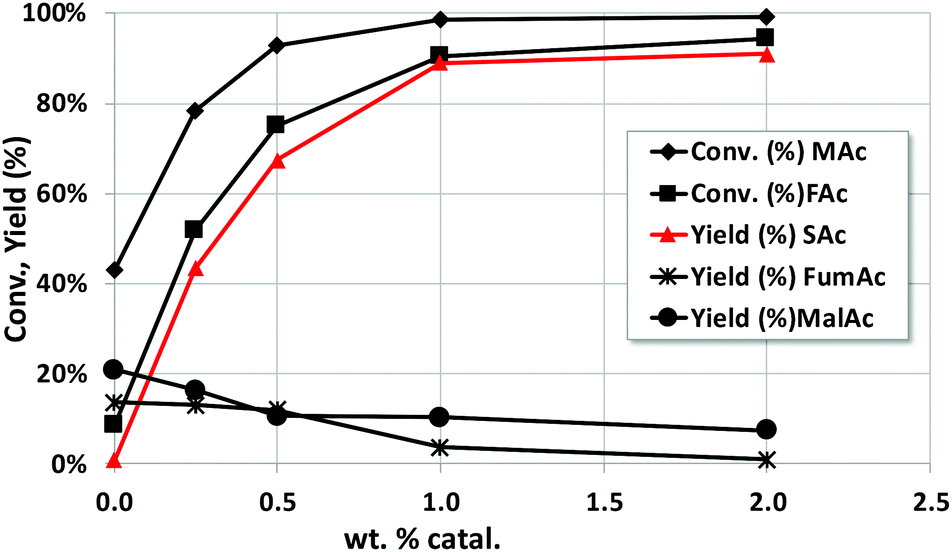 | ||
| Fig. 2 Effect of wt% of catalyst on the catalytic properties of Pd/C catalyst. Reaction conditions: 5 g of reaction mixture, 5 wt% MAc, mol FAc/MAc = 1, 150 °C, initial N2 pressure = 10 bar, time of reaction = 4 h. | ||
Study of the effect of concentration of MAc on Pd/C catalyst activity
Fig. 3 presents the results obtained with concentrations of MAc up to 40 wt%. The concentration of the catalyst was increased accordingly in order to maintain the catalyst/MAc wt ratio of 0.2. When conducting the experiments at higher MAc concentration, special attention must be paid to the solubility of SAc in water.10,13,37 | ||
| Fig. 3 Effect of wt% of MAc on the catalytic properties of Pd/C catalyst. Reaction conditions: 5 g of reaction mixture, wtcatalyst/MAc = 0.20, mol FAc/MAc = 1, 150 °C, initial N2 pressure = 10 bar, time of reaction = 2 h. | ||
The solubility of SAc in water at room temperature is around 7–8 g per 100 g of solution and rises up to around 57 g per 100 g at 100 °C. Consequently, when using up to 40% of MAc there should not be any SAc precipitate while conducting the reaction at 150 °C. But precipitation occurs when halting the reaction and cooling down the reactor for sampling and analysis. To conduct the analysis few tenths of mL of a 50 wt% solution of NaOH was added to the reaction mixture to dissolve the SAc as disodium succinate. Disodium succinate has a much higher solubility in water at room temperature than the acid (above 35 g per 100 g of water). The addition was done before incorporating the internal standard.
The results showed that, when using 10 and 20 wt% of MAc, the yield of SAc is even slightly better than with 5 wt% of MAc. After reaching 30 wt% of MAc, the yield dramatically declines but these results indicate that solutions up to 30 wt% of MAc can be processed without compromising the yield of SAc.
Effect of neutralizing the protons of MAc and FAc
The utilization of sodium formate as source of H2 instead of formic acid was also explored (entry 10 of Table 2). The reaction conditions were those of entry 9 in Table 2 but at a reaction time of only 2 h. MAc was then largely converted to SAc (yield was 98%), better than that in entry 9 of Table 2 (83%). Besides, no MalAc was observed that compares very favorably to the case of using formic acid (entry 9, 10%). It is well known that the formate counterion is key when defining the hydrogen-donating capacity of formate moiety and that Na+ is one of the best among the different counterions tested.34,38,39 It is worth realizing that the use of formate instead of formic acid poses the question of the origin of the second hydrogen required to fully hydrogenate the double bond. This issue will be examined later when discussing the mechanism of reaction.Taking into account the results using Na formate, the effect of neutralizing the different protons of the acids was also studied by incorporating increasingly larger amounts of NaOH to the reaction mixture. Thus, Fig. 4 shows the results after incorporating a NaOH/total acid proton mol ratio of 0.33, 0.66, 1 and 1.5. Taking into account that the pKa1 and pKa2 of maleic acid are 1.90 and 6.09 and pKa of formic acid is 3.77, the incorporation of these amounts of NaOH roughly corresponds, respectively, to the neutralisation of the first proton of maleic acid, that of formic acid, that of the second acid proton of MAc and finally the incorporation of an excess of NaOH. Neutralisation of protons results in such increase of the reaction rate that the concentration of MAc used for these experiments had to be increased up to 20% and the temperature decreased to 100 °C in order to see differences between the different experiments. The rest of the reaction conditions were kept constant. The results indicate that the yield of SAc reaches a maximum when all the protons have been neutralized (ratio of 1). MalAc yield was also negligible for the neutral pH experiment. For NaOH/acid protons of 1.5 the yield of SAc worsens again.
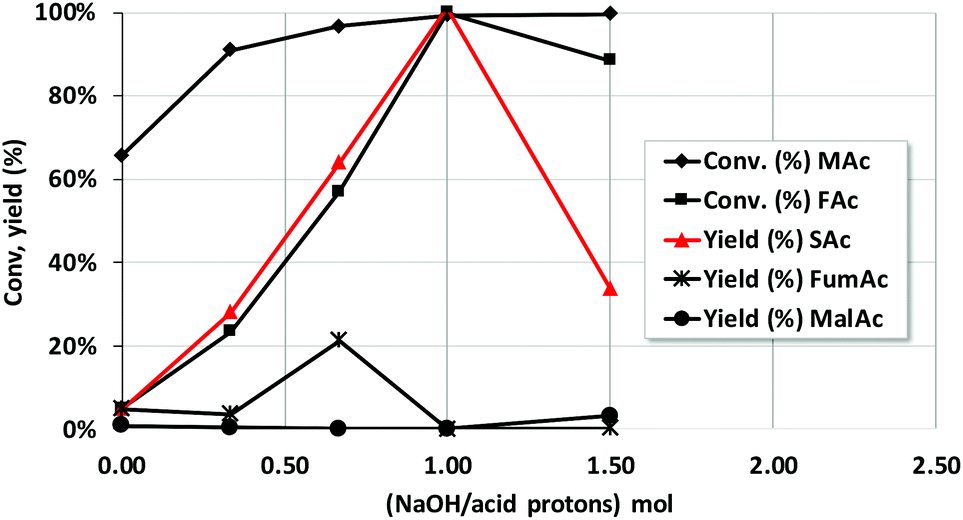 | ||
| Fig. 4 Effect of the incorporation of NaOH to the reaction mixture on the catalytic properties of Pd/C catalyst. Reaction conditions: 5 g of reaction mixture conc. of MAc solution = 20 wt%, wt catalyst/MAc = 0.025, mol FAc/MAc = 1, 100 °C, 10 bar, reaction time = 2 h. | ||
The reaction rate is faster when carboxylate anions, instead of acids, are present. Reaction rate increases according to the following trend: maleic acid < hydrogen maleate < formate < maleate. It could be possible that anions can be adsorbed on the surface more strongly than the acids, following the same trend described above, increasing their surface concentration accordingly and consequently accelerating the reaction. At very high OH− concentration, higher than that needed to have fully neutralised maleic acid to maleate, the formation of SAc is inhibited. The possibility that the concentration of OH− can determine the reaction rate should be ruled out because when the protons are sequentially neutralised, the concentration of OH− increases more than two orders of magnitude and the reaction rate does not (see ESI† for the data and a more detailed discussion). In any case, more research is needed to understand why reaction rate changes so clearly when acid protons are sequentially neutralised.
Catalyst stability tests of Pd/C catalyst: continuous operation in a fixed bed reactor
The robustness of the Pd/C catalysts was studied both with the fully neutralized acids and with non-neutralized acids. The former case is presented first. Previous results (see Fig. 4) showed us that the fastest reaction rate for SAc formation is obtained for (NaOH/acid protons) = 1. Interestingly, neutralising both MAc and FAc also increases the pH of the solution what may prevent corrosion issues in the reactor. Fig. 5 shows the catalytic properties of the Pd/C catalyst when operating in a fixed bed reactor under a continuous flow and using a solution of 2.5 wt% of MAc. The catalyst was first tested at 130 °C, after an initial transient period, no deactivation was observed for 140 h on stream and the yield was above 99.0% throughout the experiment. Temperature was then decreased to 110 °C in order to lower the SAc yield and assess on causes of deactivation with low to medium impact that may not be detected working at conditions well above those needed for a complete conversion. The catalyst was stable for other 310 h on stream: the SAc yield was above 85% for the time on stream investigated (except for the initial transient period). In practice, the catalyst could have been operating for ca. 450 h on stream without noticeable deactivation at a space time yield (STY) of above 0.30 g SAc per gcat per h what actually means that in this 450 h of operation 136 g of SAc per gcat would have been obtained. | ||
| Fig. 5 Catalytic properties of Pd/C catalyst under continuous operation incorporating NaOH to fully neutralize MAc and FAc. Reaction conditions: conc, of MAc solution = 2.5 wt%, mol FAc/MAc = 1, 10 bar, catalyst loading = 0.2 g, flow rate = 0.04 mL min−1, WHSV = 12 h−1, initial temperature = 130 °C, after 140 h on stream temperature was decreased to 110 °C. | ||
Fig. 6 shows the stability study using initially 4.5 wt% (first 80 h) and later 10 wt% of MAc. Both MAc and FAc were neutralized. The rest of the reaction conditions were those of Fig. 5. No sign of deactivation was observed for the 150 h the catalyst was under operation. The space time yield (STY) of SAc when using 4.5 wt% and 10 wt% of MAc were approximately 0.57 and 1.27 g SAc per gcat per h, respectively. In the overall experiment the catalyst reached to produce ca. 134 g SAc per gcat or, in terms of Pd, 2704 g SAc per gPd.
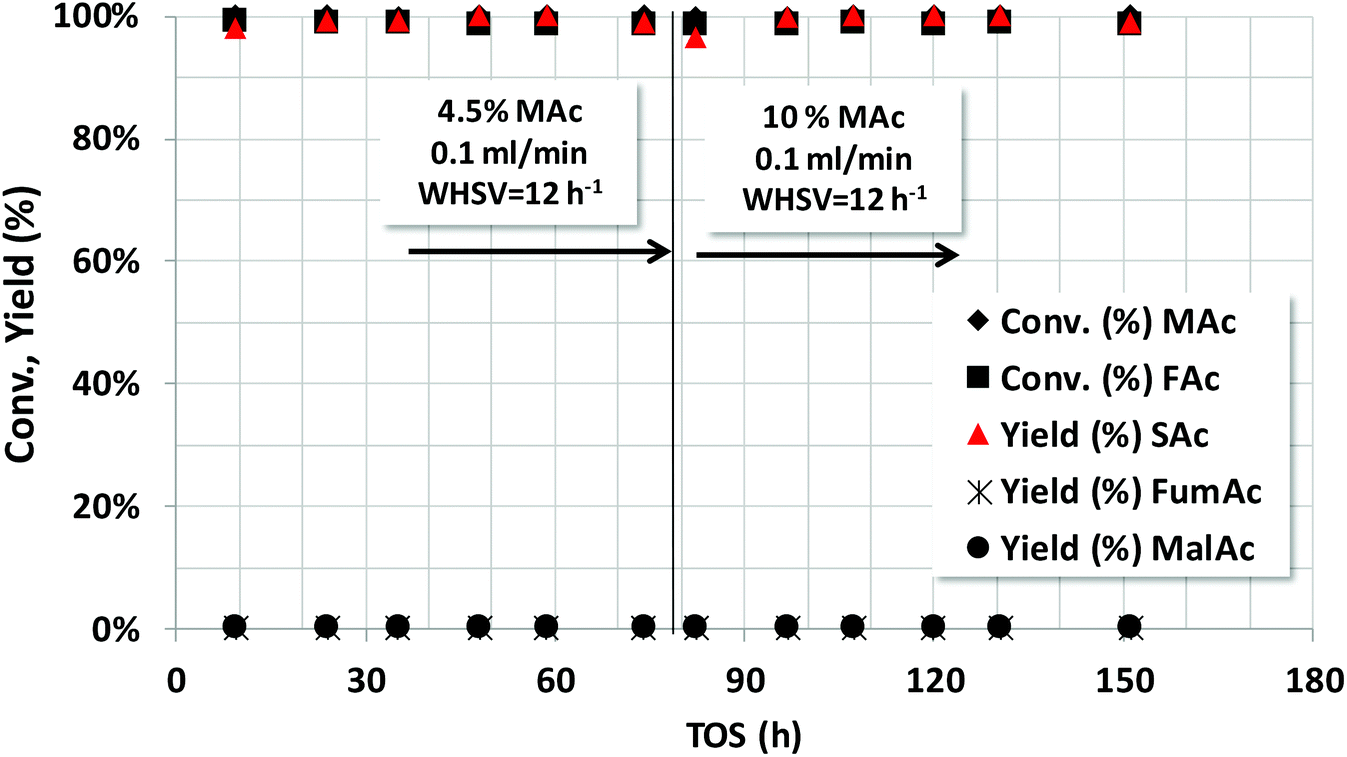 | ||
| Fig. 6 Catalytic properties of Pd/C catalyst under continuous operation using 4.5 and 10 wt% of MAc. NaOH was incorporated in order to fully neutralize MAc and FAc. Reaction conditions: mol FAc/MAc = 1, 10 bar, catalyst loading = 0.5 g, WHSV = 12 h−1, initial temperature = 150 °C. | ||
The long-term stability was also studied with the acids. Although using neutralized acids may prevent corrosion problems in the reactor and present the advantage of faster reaction rates, conducting the reaction with acids facilitates the separation of the SAc by spontaneous precipitation. Fig. 7 represents the catalytic properties of the Pd/C catalyst operating in continuous flow using a solution of 5, 10 and 15 wt% MAc and an equivalent mol FAc/MAc = 1. The rest of the reaction conditions are in the caption. Except for the transient initial results, the SAc yield was close to 98% for the duration of the experiments (ca. 730 h on stream, more than 30 days). The STY obtained using 5, 10 and 15 wt% of MAc was 0.60, 1.23 and 1.87 g SAc per gcat per h, respectively. In the overall experiment the catalyst reached to produce ca. 950 g SAc per gcat or, in terms of Pd, more than 19000 g SAc per gPd.
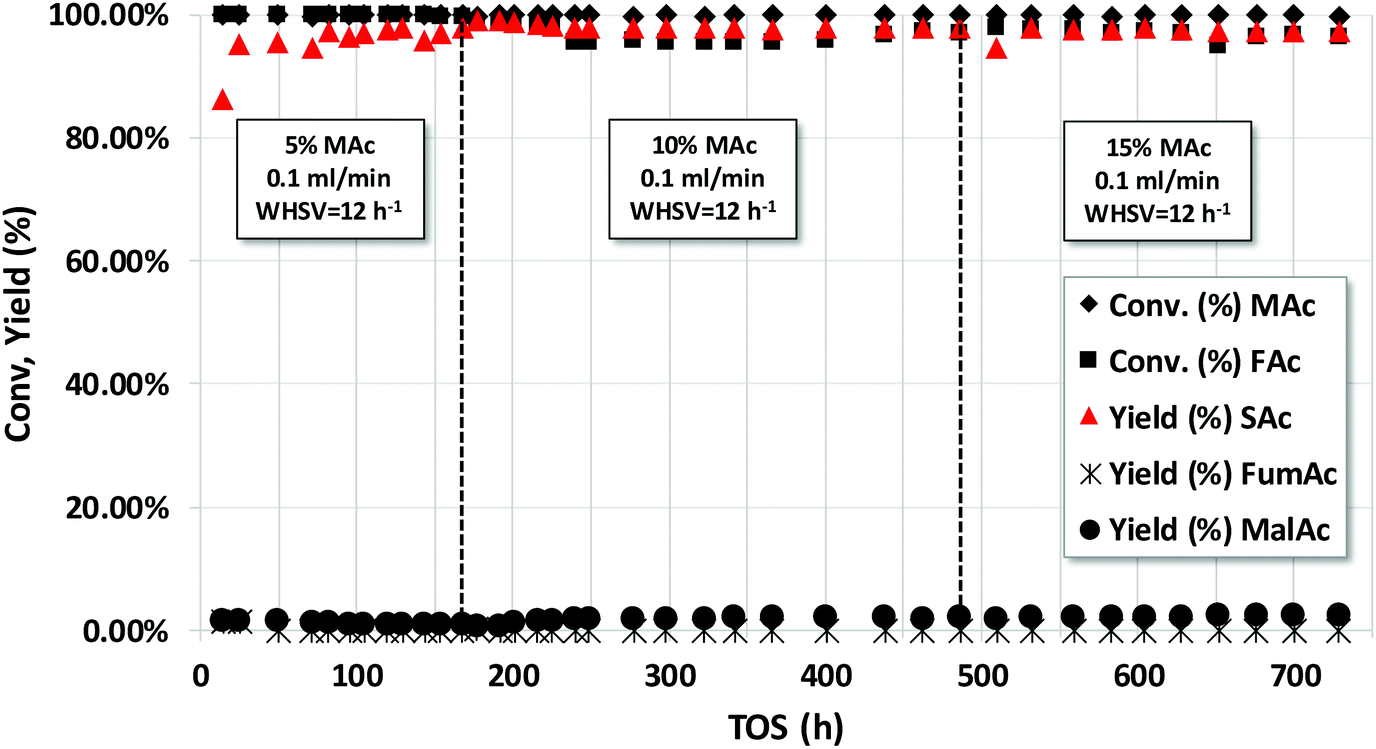 | ||
| Fig. 7 Catalytic properties of Pd/C catalyst under continuous operation using 5, 10 and 15 wt% of MAc. Reaction conditions: mol FAc/MAc = 1, 150 °C, 10 bar, catalyst loading = 0.50 g, 0.1 mL min−1 (WHSV ca. 12 h−1). | ||
These results visibly contrast with those obtained with a more diluted 2.5 wt% of MAc using a Au/C catalyst (see ESI Fig. 4,† the initial yield of SAc was 100% but slightly and continuously deactivated, reaching a yield of 83% after 73 h on stream) or Pd/γ-Al2O3 (ESI Fig. 3,† with an initial SAc yield of ca. 70% that deactivates more rapidly and reaches ca. a 30% in less than 80 h).
Characterization of the fresh and used Pd/C catalysts
The Pd content (Pd wt%) of the fresh and used catalyst (downloaded from the reactor after the experiments of Fig. 7) was 4.97 ± 0.45 and 4.74 ± 0.42, respectively. Both values are similar within the experimental error. The Pd concentration of the aqueous solutions sampled during the study of ESI Fig. 6† was in all cases below 0.02 mg L−1, the detection limit of the technique. Assuming that the Pd concentration in the samples was 0.02 mg L−1, less than 0.36% of the total Pd initially loaded in the fresh catalyst would have been leached during the 370 h on stream. Thus, Pd leaching can be considered irrelevant.The dispersion and the average diameter of the Pd particles in the fresh and used Pd/C catalysts are summarized in Table 3. The used Pd/C sample corresponds to that of Fig. 7. Table 3 gives the information obtained from two different techniques: CO chemisorption and TEM studies (see ESI Fig. 5† for representative TEM pictures). Both techniques confirm that the Pd particles undertake a sintering during the catalyst utilization.
| CO chemisorption | TEM studies | |||
|---|---|---|---|---|
| Dispersiona (%) | Size (nm) | Dispersion (%) | Sizeb (nm) | |
| a Dispersion and particle size (diameter in nm) in Pd are related by equation size d = 1.12/dispersion.36 b Particle size estimated by TEM was calculated as the mean surface diameter (∑nidi3/∑nidi2) obtained from the surface distribution of particles (nidi2vs. di).36 See ESI† section for representative pictures and further details of TEM studies. | ||||
| Fresh | 51.6 ± 2.3 | 2.2 ± 0.1 | 33.4 ± 0.5 | 3.35 ± 0.05 |
| Used | 31.1 ± 2.8 | 3.6 ± 0.3 | 18.7 ± 0.4 | 6.00 ± 0.12 |
CO chemisorption shows that the average particle size increases from 2.2 to 3.6 nm and TEM from 3.35 to 6.00, whereas the dispersion estimated from CO chemisorption changes from 51.6 to 31.1% and that of TEM studies from 33.4 to 18.7%. In general, particle sizes estimated by TEM studies are always larger than that obtained from CO chemisorption because of the difficulty to account for particles with sizes in the range of 1 nm and smaller. Besides, some of larger particles were the result of the agglomeration of several Pd particles and can inadvertently be accounted as only one particle. On the contrary CO chemisorption can differentiate these individual particles because it titrates almost all the surface atoms except those few in the contacting region of the individual particles of the agglomerate. In any case both techniques have observed a 1.6–1.8 fold increase of the particles size and a 0.6 fold reduction in the dispersion of the particles.
Experimental evidences of CTH mechanism
In principle, two routes can be proposed when FAc is used as the reduction agent for the hydrogenation reaction. A first option is that the reaction proceeds through a concerted CTH mechanism in which H atoms in the formic acid are directly transferred to the double bond of MAc. Another option is that formic acid decomposes over the surface of the catalyst yielding gas CO2 and the H atoms (either as gas H2 or as adsorbed on the Pd surface) needed for the MAc hydrogenation.When the hydrogenation was conducted using gas H2 at a pressure equal to the pressure that would be generated by the decomposition of the formic acid it was found that the H2-gas reaction rate is slower than that using formic acid (see ESI Fig. 6† and discussion therein). This result suggests that the reaction does not completely proceed via H2 released from the decomposition of formic acid. This experiment is not conclusive. More compelling evidences for unveiling the mechanism are deduced from isotopic experiments.
Table 4 summarizes the isotopic kinetic experiments. The rates of conversion of FA (rFAc) in the absence of MAc, using formic acid (HCOOH) and deuterated formic acid in the formyl position (DCOOH) are included. This reaction represents the direct decomposition of FAc, into gas CO2 and H atoms (or gas H2). These rates are compared with the FAc conversion in the presence of MA (rFAcMAc) and with the rate of formation of SAc (rSAcMAc).
| In the absence of MAca | In the presence of MAcb | ||
|---|---|---|---|
| rFAc | rFAcMAc | rSAcMAc | |
| a No MAc was incorporated for the determination of the rate of direct decomposition of FAc. b Reaction conditions: 5 g of reaction mixture, 5 wt% MAc, 0.5 wt% of catalyst, mol FAc/MAc = 1, 150 °C, initial N2 pressure = 10 bar, time of reaction 1 h. | |||
| HCOOH | 1.06 ± 0.01 | 0.57 ± 0.01 | 0.59 ± 0.01 |
| DCOOH | 0.91 ± 0.02 | 0.38 ± 0.01 | 0.38 ± 0.02 |
First, we compare rSAcMAc using either formic acid (HCOOH) or deuterated DCOOH (last column of the Table 4). The rate rSAcMAc with HCOOH is faster than that with DCOOH (0.59 vs. 0.38 mmol SAc per gcat per min, respectively). The ratio between these rates is 1.55 ± 0.07 evidencing that there is a kinetic isotope effect in the formation of SAc. The same effect is found when comparing rFAcMAc using HCOOH and DCOOH (0.57 vs. 0.38 mmol FAc per gcat per min). In contrast, the ratio between the rates of direct decomposition (rFAc) of HCOOH and DCOOH is 1.16 ± 0.02 (1.06 divided by 0.91). This ratio is lower than the ratio for rFAcMAc and rSAcMAc (1.55 vs. 1.16). These results indicate, first, that formic acid is involved in the rate determining step of the SAc formation, more specifically, formyl H is involved. Second, that the direct decomposition of formic acid to H atoms is not involved in the rate determining step of the hydrogenation reaction, otherwise the same isotopic effect would have been observed for rFAcc and rFAcMAc. Consequently, the rate determining step has to involve the concerted and direct transference of H atoms from FAc to maleic acid.
Additional support to these conclusions comes from the comparison between the results from the same row. Interestingly, when comparing the rates within the same first row of experiments (experiments using HCOOH) we found that rFAc is almost two times faster than rFAcMAc (1.06 vs. 0.57 mmol FAc per gcat per min) and rSAcMAc (0.59 ± 0.01 mmol SAc per gcat per min). The same occurs when comparing the rates of the second row when the reaction is conducted with DCOOH (0.91 ± 0.02 vs. 0.38 ± 0.01). It is also worth noticing that rFAcMAc is similar to rSAcMAc, what means that FAc decomposition and SAc formation are intimately coupled and that when MAc is present the rate of formic acid consumption is limited by the formation of SAc. In other words, FAc is only converted once SAc is formed. These results are in agreement with a rate determining step involving a CTH mechanism.
Spencer et al.40 have also concluded that the hydrogenation of unsaturated C–C bonds using FAc and Pd/C as catalyst operates via a CTH mechanism. In their case an alkyne (methyl phenylpropiolate) was hydrogenated to the corresponding cis-alkene using trimethylamine as solvent. Besides, Spencer et al.40 also demonstrated that the hydrogen transfer occurs via two palladium-diformate species from two different FAc molecules: the two formyl hydrogens from the two diformate species concertedly hydrogenate the double bond. The acid proton was not involved in the hydrogenation. Spencer et al. conducted their experiments with a FAc/MAc mol ratio of 10. Pd-diformate species cannot be involved in our case, otherwise the yield of SAc would have been maximum 50%: we have used a stoichiometric FAc/MAc ratio and consequently only one formic molecule can be involved in the hydrogenation. In our case the second hydrogen to hydrogenate the double bond is not a formyl H from a second FAc. It must come from another source.
A first possibility is the acid protons. However, the results of Fig. 4 describing the effect of neutralizing the protons of FAc and MAc showed that the hydrogenation is very fast once all the protons of formic acid and maleic acid are neutralized. Consequently, in (close to) neutral conditions the participation of H+ in the hydrogenation can be ruled out. This left H2O as the only possible source hydrogen. The presence of H+ in the medium (partial or no neutralisation of the acids) clearly slowed down the reaction rate that means that if protons are involved in the hydrogenation, they result in an inhibition of the faster mechanism involving H2O (ESI Scheme 1† summarized the CTH mechanism proposed). Actually, when we conducted the reaction with aprotic solvents (DMSO or acetonitrile) under the conditions of Fig. 1, the SAc yield after 1 h of reaction was negligible (below 2%, the results are not tabulated), whereas in contrast a SAc yield of 50% is obtained in water (see Fig. 1). This fact gives additional support to the hypothesis that water is directly involved in the donation of the second H needed for the hydrogenation and that the presence of H+ is actually inhibiting the reaction. Within this context, very recently the important role and the direct participation of H2O in the mechanism of hydrogenation of CC and CO bonds in liquid phase on Pd catalysts have already been demonstrated.41
A preliminary assessment of the environmental impact: CHT from formic acid vs. H2 gas hydrogenation
The CTH approach involves some features that could significantly improve the sustainability of the SAc production as compared to the conventional petrochemical way. In particular, using FAc as a reduction agent avoids storing and handling H2 at high pressure, and thus this probably constitutes one of the most promising advantages to achieve a greener process. However, an evaluation of the environmental impact caused by these two alternatives (using FAc or high-pressure H2) is mandatory to achieve a closer understanding of the improvements in the sustainability of SAc production that may be accomplished by implementing a CTH transformation path.Life Cycle Assessment (LCA) is a well-known, widely used methodology for quantifying the environmental impacts ascribed to industrial systems, including the steps beyond the factory gate, that is, understanding the full life cycle of a product or process.42 Completing an LCA study is a complex task because of the great amount of information that must be collected. A first approach can be addressed by applying simplifications and using LCA databases. In this case, we present a simplified comparative assessment of the production of 1 kg of SAc by conventional hydrogenation and catalytic transfer hydrogenation using FAc as a reduction agent, respectively. ESI Fig. 7† shows the boundaries of the evaluated systems. In our approximation, these exclusively include the production of the reduction agents (H2 or FAc) and their conditioning to reach the reaction pressure. We assume that both have been obtained from fossil feedstocks. This approach is needed because of the great differences required to drive both types of reductions, mainly focused on the pressure conditions, quite high in the case of conventional hydrogenation. Assumptions for data collection and for LCA calculations methodology have been detailed in the ESI.†
Fig. 8 shows the relative contribution of the evaluated systems to the different environmental impacts categories. The catalytic hydrogen transfer pathway using FAc as hydrogen donor leads to lower impacts, compared to conventional hydrogenation, in categories such as climate change (CC), human toxicity (HT), water use and cumulative energy demand (CED). Differences in data corresponding to climate change (CC) and cumulative energy demand (CED) are especially remarkable indicating that CTH involves a saving of around 50% of the environmental impacts caused by conventional hydrogenation. This fact is mainly related to two factors. First, the production of hydrogen by steam reforming of natural gas (the most common industrial process) implies large carbon dioxide emissions per kg of H2.43
 | ||
| Fig. 8 Relative contribution to different environmental impact categories of FAc production from MAc by conventional hydrogenation (blue bars) and catalytic hydrogen transfer from FAc (green bars). CC: climate change; RD: resource depletion, FW: fresh water demand; HT: total human toxicity; POF: photochemical ozone formation; CED: cumulative energy demand. | ||
A significant part of this is inherent to the process since CO2 is a co-product of the reaction scheme (CH4 + 2H2O ↔ CO2 + 4H2). Thus, just the stoichiometry in the production of hydrogen involves a direct emission of 5.5 kg of CO2 per kg of H2. Adding the rest of the required steps increases the CO2 emissions to more than 10 kg CO2-eq per kg of H2.43,44 On the other hand, the production of 1 kg of FAc by the methyl formate route (the most usual one) leads to 3 kg CO2-eq per kg of FAc,45 which is an enormous difference compared to hydrogen production-related emissions. Despite FAc consumption during MAc reduction also leads to a direct emission of CO2 (0.38 kg of CO2 per kg SAc), this quantity is much lower than the stoichiometrically emitted by the production of the required hydrogen to produce an equivalent amount of SAc: 5.5 kg of CO2 per kg H2 × 0.254 kg H2 consumed (see ESI†) = 1.4 kg of direct emission of CO2.
As for the CED category, using FAc instead of H2 saves primary energy consumption. This is mainly due to the different processes and raw materials used for the manufacture of both reducing agent. Whereas the methyl formate route, based on methanol carbonylation, consumes a primary energy demand of 73 MJ kg−1 FAc, steam reforming of natural gas requires 209 MJ of primary energy per kg of H2, almost exclusively associated to the consumption of this fossil fuel as both, reactant and heating agent.
In the case of human toxicity (HT), it is remarkable that the impact produced by the conventional hydrogenation is not due to the production of hydrogen, but mainly to the consumption of electricity required for gas compression up to the hydrogenation pressure (4.0 MPa). ESI Table 4† shows the relative contribution of the hydrogen manufacture and gas compression steps to each environmental impact category. As shown, the use of electricity is the key contributor to human toxicity (HT) and freshwater eutrophication (FE) categories and it is responsible for half of other environmental indicators, such as terrestrial acidification (TA) and blue water consumption.
Regarding terrestrial acidification (TA) and freshwater eutrophication (FE), these are the weakest environmental points associated to the use of FAc as hydrogen donor. This is ascribed to the production of FAc from methyl formate, a process that involves higher emissions of nitrogen oxides and phosphates than those corresponding to the steam reforming process for hydrogen manufacture.
Despite the simplified assessment of the environmental performance of the SAc production by formic acid CTH, the results make evident the environmental benefits associated with this technology as compared to conventional hydrogenation of MAc. LCA indicates a clear reduction in the impacts associated to climate change and non-renewable resources depletion by using this new approach.
Recently, LCA of the production of biomass-derived FAc has been conducted in comparison with the conventional petrochemical route.46 This analysis has showed that, depending on the type of biomass, renewable FAc may present further improvements in some of the environmental impact categories like CC, HT and water use (see ESI† for a more detailed discussion). Consequently, these improvements will benefit likewise the CTH of MAc to SAc that uses biomass derived FAc.
Conclusions
Among the different noble metal and support studied, Pd/C is the most efficient and robust catalyst for the catalytic transfer hydrogenation of maleic acid to succinic acid using formic acid as H2 source. Interestingly the reaction was conducted in water medium and with a stoichiometric amount of formic acid.In a fixed bed reactor and under a continuous flow of MAc–FAc solutions no sign of deactivation was observed either using neutralized (up tp 10 wt% of equivalent MAc) or non-neutralized maleic and formic acids (up to 15 wt% of MAc). In the case of acid solutions, the catalyst was working for ca. 730 h.
A space time yield (STY) of above 1.27 g SAc per gcat per h was achieved with disodium maleate and sodium formate solutions working with a 10 wt% of equivalent MAc, at 150 °C, 10 bar of N2 and a WHSV = 12 h−1 (contact time = 5 min). In the case of using 15 wt% of non-neutralized MAc, the STY was 1.87 g SAc per gcat per h. A total of 950 g SAc per gcat were produced during the long-term experiments with non-neutralised MAc-FAc experiments, that in terms of Pd represents more than 19000 g SAc per gPd.
The reaction really proceeds via catalytic transfer hydrogenation in which formyl H of formic acid provides one of the H needed for the hydrogenation. In close to neutral pH conditions the reaction is faster than in acidic pH and water supplies the second H needed for the reaction. In acidic pH the involvement of acid protons cannot be ruled out with the data so far available, but the presence of protons slows down the reaction rate.
A preliminary environmental assessment indicates that the FAc-based CTH technology results in a significant reductions of relevant environmental indicators (such as climate change and consumption of fossil resources) as compared to those of conventional hydrogenation route, that can be even more favorable if biomass-derived FAc is used instead of conventional petrochemical FAc.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Research, Innovation and University (MICINN) (projects CTQ2015-64226-C3-1-R, RTI2018-094918-B-C41, and RTI2018-094918-B-C42) and from CSIC (i-link1048 project) is gratefully acknowledged. Y. Rodenas is also thanked for the preparation of some of the catalysts and some of the measurements of the catalytic activity. A. C. A. R. also thanks the University of Toledo for her start-up package.References
- I. Bechthold, K. Bretz, S. Kabasci, R. Kopitzky and A. Springer, Chem. Eng. Technol., 2008, 34, 647–654 CrossRef.
- C. Fumagalli, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2006 Search PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- J. M. Pinazo, M. E. Domine, V. Parvulescu and F. Petru, Catal. Today, 2015, 239, 17–24 CrossRef CAS.
- R. K. Saxena, S. Saran, J. Isar and R. Kaushik, in Current Developments in Biotechnology and Bioengineering: Production, Isolation and Purification of Industrial Products, 2016, pp. 601–629 Search PubMed.
- B. C. Klein, J. F. L. Silva, T. L. Junqueira, S. C. Rabelo, P. V. Arruda, J. L. Ienczak, P. E. Mantelatto, J. G. C. Pradella, S. V. Junior and A. Bonomi, Biofuels, Bioprod. Biorefin., 2017, 11, 1051–1064 CrossRef CAS.
- 2013 BioConSepT_Market-potential-for-selected-platform-chemicals_report1, http://www.bioconsept.eu/wp-content/uploads/BioConSepT_Market-potential-for-selected-platform-chemicals_report1.pdf.
- A. Carlson, B. Coggio, K. Lau, C. Mercogliano and J. Millis, in Chemicals and Fuels from Bio-Based Building Blocks, ed. F. Cavani, S. Albonetti, F. Basile and A. Gandini, Wiley-VCH Verlag, 2016, pp. 173–190 Search PubMed.
- C. S. López-Garzón and A. J. J. Straathof, Biotechnol. Adv., 2014, 32, 873–904 CrossRef PubMed.
- W. Zhu, F. Tao, S. Chen, M. Li, Y. Yang and G. Lv, ACS Sustainable Chem. Eng., 2019, 7, 296–305 CrossRef CAS.
- I. Agirre, I. Gandarias, P. L. Arias and M. López Granados, Biomass Convers. Biorefin. DOI:10.1007/s13399-019-00462-w.
- S. Black and F. L. Muller, Org. Process Res. Dev., 2010, 14, 661–665 CrossRef CAS.
- Q. Yu, S. Black and H. Wei, J. Chem. Eng. Data, 2009, 54, 2123–2125 CrossRef CAS.
- J. Albert and P. Wasserscheid, Green Chem., 2015, 17, 5164–5171 RSC.
- J. Albert, R. Wölfel, A. Bösmann and P. Wasserscheid, Energy Environ. Sci., 2012, 5, 7956–7962 RSC.
- J. Reichert, B. Brunner, A. Jess, P. Wasserscheid and J. Albert, Energy Environ. Sci., 2015, 8, 2985–2990 RSC.
- S. G. Wettstein, D. M. Alonso, Y. Chong and J. A. Dumesic, Energy Environ. Sci., 2012, 5, 8199–8203 RSC.
- G. Pavarelli, J. Velasquez Ochoa, A. Caldarelli, F. Puzzo, F. Cavani and J. L. Dubois, ChemSusChem, 2015, 8, 2250–2259 CrossRef CAS PubMed.
- A. Chatzidimitriou and J. Q. Bond, Green Chem., 2015, 17, 4367–4376 RSC.
- G. Lv, C. Chen, B. Lu, J. Li, Y. Yang, C. Chen, T. Deng, Y. Zhu and X. Hou, RSC Adv., 2016, 6, 101277–101282 RSC.
- J. Lan, J. Lin, Z. Chen and G. Yin, ACS Catal., 2015, 5, 2035–2041 CrossRef CAS.
- Z. Du, J. Ma, F. Wang, J. Liu and J. Xu, Green Chem., 2011, 13, 554–557 RSC.
- X. Li and Y. Zhang, Green Chem., 2016, 18, 643–647 RSC.
- V. Laserna, A. Heras, N. Alonso-Fagúndez, A. C. Alba-Rubio, R. Mariscal, M. López Granados and M. Mengibar, Catal. Today, 2014, 234, 285–294 CrossRef.
- N. Alonso-Fagúndez, I. Agirrezabal-Telleria, P. L. Arias, J. L. G. Fierro, R. Mariscal and M. López Granados, RSC Adv., 2014, 4, 54960–54972 RSC.
- A. C. Alba-Rubio, J. L. G. Fierro, L. León-Reina, R. Mariscal, J. A. Dumesic and M. López Granados, Appl. Catal., B, 2017, 202, 269–280 CrossRef CAS.
- Y. Rodenas, J. L. G. Fierro, R. Mariscal, M. Retuerto and M. López Granados, Top. Catal., 2019, 62, 560–569 CrossRef CAS.
- Y. Rodenas, R. Mariscal, J. L. G. Fierro, D. Martín Alonso, J. A. Dumesic and M. López Granados, Green Chem., 2018, 20, 2845–2856 RSC.
- S. Shi, H. Guo and G. Yin, Catal. Commun., 2011, 12, 731–733 CrossRef CAS.
- H. Guo and G. Yin, J. Phys. Chem. C, 2011, 115, 17516–17522 CrossRef CAS.
- J. Lan, Z. Chen, J. Lin and G. Yin, Green Chem., 2014, 16, 4351–4358 RSC.
- Y. Huang, C. Wu, W. Yuan, Y. Xia, X. Liu, H. Yang and H. Wang, J. Chin. Chem. Soc., 2017, 64, 786–794 CrossRef CAS.
- M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS.
- T. Liu, Y. Zeng, H. Zhang, T. Wei, X. Wu and N. Li, Tetrahedron Lett., 2016, 57, 4845–4849 CrossRef CAS.
- G. Bergeret and P. Gallezot, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, Wiley-VCH Verlag, 2008, pp. 439–467 Search PubMed.
- Q. Li, D. Wang, Y. Wu, W. Li, Y. Zhang, J. Xing and Z. Su, Sep. Purif. Technol., 2010, 72, 294–300 CrossRef CAS.
- S. Rajagopal and A. F. Spatola, Appl. Catal., A, 1997, 152, 69–81 CrossRef CAS.
- S. Rajagopal and A. F. Spatola, J. Org. Chem., 1995, 60, 1347–1355 CrossRef CAS.
- J. Yu and J. B. Spencer, Chem. Commun., 1998, 1935–1936 RSC.
- Z. Zhao, R. Bababrik, W. Xue, Y. Li, N. M. Briggs, D.-T. Nguyen, U. Nguyen, S. P. Crossley, S. Wang, B. Wang and D. E. Resasco, Nat. Catal., 2019, 5, 431–436 CrossRef.
- J. Moreno, C. Pablos and J. Marugán, in Quantitative Methods for Food Safety and Quality in the Vegetable Industry, ed. F. Pérez-Rodríguez, P. Skandamis and V. Valdramidis, Springer International Publishing, Cham, 2018, pp. 255–293 Search PubMed.
- J. Dufour, D. P. Serrano, J. L. Gálvez, J. Moreno and C. García, Int. J. Hydrogen Energy, 2009, 34, 1370–1376 CrossRef CAS.
- N. Z. Muradov and T. N. Veziroǧlu, Int. J. Hydrogen Energy, 2005, 30, 225–237 CrossRef CAS.
- J. Sutter, Ecoinvent Rep., 2007, 1–402 Search PubMed.
- H. H. Khoo, W. L. Ee and V. Isoni, Green Chem., 2016, 18, 1912–1922 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc04221k |
| This journal is © The Royal Society of Chemistry 2020 |