
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective hydrogenation of 5-hydroxymethylfurfural and its acetal with 1,3-propanediol to 2,5-bis(hydroxymethyl)furan using supported rhenium-promoted nickel catalysts in water†
Jan J.
Wiesfeld
a,
Minjune
Kim
b,
Kiyotaka
Nakajima
 *bc and
Emiel J. M.
Hensen
*a
*bc and
Emiel J. M.
Hensen
*a
aLaboratory of Inorganic Materials and Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.j.m.hensen@tue.nl
bInstitute for Catalysis, Hokkaido University, Kita 21 Nishi 10, Kita-ku, Sapporo 0010021, Japan. E-mail: nakajima@cat.hokudai.ac.jp
cAdvanced Low Carbon Technology Research and Development Program (ALCA), Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi 332-0012, Japan
First published on 21st January 2020
Abstract
The high reactivity of the formyl group of 5-hydroxymethylfurfural (5-HMF) is problematic, because it leads to undesired oligomerization reactions. This is usually countered by working in dilute non-aqueous solutions. Here, we present a novel approach to convert concentrated aqueous solutions of 5-HMF to 2,5-bishydroxymethylfuran (BHMF), which is a prospective monomer for polyesters and self-healing polymers. Our approach is based on the protection of the formyl group of 5-HMF using acetalization with 1,3-propanediol. Hydrogenation is carried out using an optimized bimetallic Ni–Re catalyst supported on TiO2 at a carefully controlled pH, resulting in balanced rates of deprotection and hydrogenation and high BHMF yield. Under optimized conditions at a benign temperature of 40 °C, hydrogenation of concentrated solutions (10–20 wt%) of protected 5-HMF in water gave 81–89% yields of BHMF without having to resort to platinum-group metals such as palladium or platinum.
Introduction
Climate change is a main driver for the decarbonization of the energy industry. The chemical industry cannot decarbonize like the energy sector, as materials are mostly based on carbon. Therefore, there is a great need to substitute or supplement petroleum-based feedstock of the chemical industry by renewable sources.1–3 Lignocellulosic biomass is considered a treasure trove of industrially relevant chemicals, accessible through digestion to a selection of small compounds called platform molecules.4,5 Among a variety of platform molecules, 5-hydroxymethylfurfural (5-HMF) is especially important, as it can be obtained by acid-catalyzed dehydration of hexoses, which form a dominant part of lignocellulose.6,7From a chemical point of view, 5-HMF is interesting because it possesses aldehyde and alcohol functionalities as well as a rigid unsaturated furan ring. As a result, it can be converted to many high value-added compounds. Especially, oxidation to 2,5-furandicarboxylic acid (FDCA) is currently at the center of the attention of many researchers, because it can be used as a monomer for the production of polyethylene 2,5-furandicarboxylate, a biobased analogue of polyethylene terephthalate (PET).8–10 Two other prospective monomers that can be obtained from 5-HMF by hydrogenation are 2,5-bis(hydroxymethyl)furan (BHMF) and 2,5-bis (hydroxymethyl)tetrahydrofuran (BHMTHF). Both diol monomers can be used to produce polyesters, but have other uses as well. BHMTHF has received most attention, because it can be ring-opened. In this way, a biorenewable route to 1,6-hexanediol can be established to replace current fossil resource based pathways for the large scale production of plastics.11,12 BHMF is more difficult to obtain, but interesting as a monomer for thermosets and self-healing polymers because of its two conjugated double bonds.13,14
Recent literature has shown that rhenium-promoted iridium, palladium and platinum are excellent catalysts for the selective hydrogenation of aldehydes, carboxylic acids and esters, including the aldehyde moiety of HMF. Hydrogenation of 5-HMF has been studied intensively using heterogeneous catalysts, which are usually based on one or more platinum group metals (PGMs), yielding products such as BHMF, BHMTHF, and/or ring-opened, deoxygenated and decarbonylated products depending on the harshness of the conditions employed.15 A comparison of several catalytic systems for the hydrogenation of 5-HMF to BHMF and BHMTHF is listed in Table S1 of the ESI.† The oxophilic nature of the rhenium promoter helps to activate polar bonds and improves the reaction rate of the expensive PGM-based active phase.16–19 Ir-ReOx/TiO2 hydrogenated HMF to BHMF quantitatively (BHMF yield >99%), whereas bimetallic ReOx-modified Rh–Ir/SiO2 catalyst converted furfural to 1,5-pentanediol at high concentration (50 wt%).17,20 Despite the high activities, escalating prices of and increasing demand for PGMs inspired us to look for cheaper, non-critical metals capable of efficient hydrogenation. These scarcity aspects are less pressing for rhenium. Herein, we present an approach to convert 5-HMF in high yield to BHMF in water using a heterogeneous catalyst based on the earth-abundant metal nickel using rhenium as a promoter. To achieve practically interesting yields in water, we protect the formyl group of 5-HMF by acetalization with 1,3-propanediol, another biobased platform chemical. Direct oxidation of the acetal form of 5-HMF, propanediol-HMF (PDHMF), is possible and provides an attractive route to obtain FDCA and its carboxylates in high yield from concentrated solutions.8,21 Acetal protection of the highly reactive formyl group suppressed not only heavy by-product formation occurring predominantly during oxidation reactions in concentrated reaction mixtures, but also simple self-degradation of pure HMF itself during thermal treatment. The protection strategy, which supresses side reactions, can be potentially applicable to a variety of HMF-based reactions at commercially relevant concentrations, which includes BHMF formation. However, direct hydrogenation of an acetal is not possible and requires deprotection prior to hydrogenation.22
This work is organized in the following manner. Firstly, the activity of the catalysts and their selectivity towards different hydrogenated products is investigated using dilute and concentrated solutions of 5-HMF to study the effects of the rhenium promotor on conversion and selectivity of Ni/TiO2 catalyst towards hydrogenated products (Scheme 1a). The catalysts were then screened for highest BHMF yield from concentrated solutions of PDHMF, which liberates 5-HMF during the course of the reaction as acetals are water-labile (Scheme 1b). The innovation in this work is the balancing of the deprotection rate of PDHMF by control of the pH and the hydrogenation rate of the liberated 5-HMF to maximize the BHMF yield and avoid undesirable humin formation even in a concentrated solution. This approach is generic and can be applied to other catalytic reactions in which biobased platform molecules that often carry a reactive formyl moiety need to be converted.
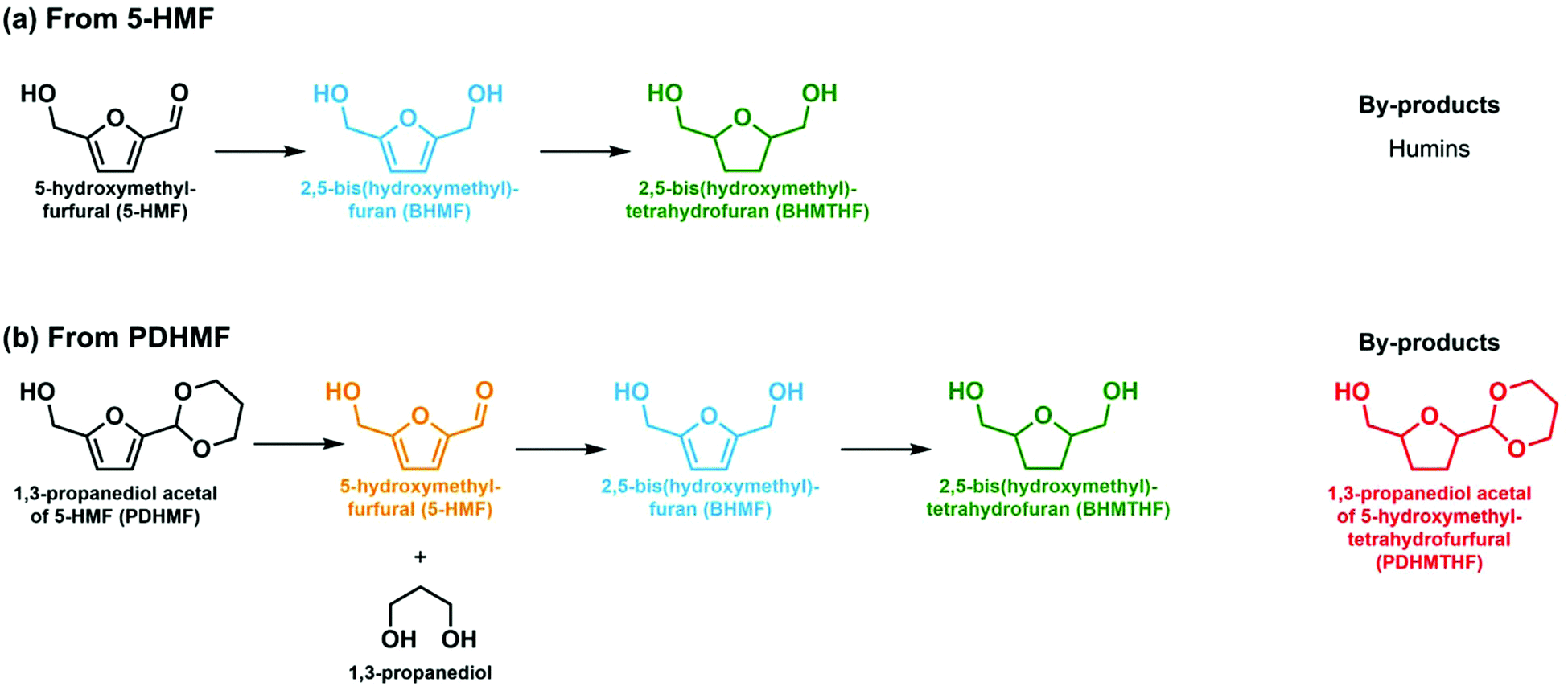 | ||
| Scheme 1 Reaction routes of hydrogenation of 5-HMF and PDHMF. | ||
Methods
Chemicals
TiO2 (P25) was procured from Degussa. Nickel nitrate hexahydrate (Ni(NO3)2·6H2O, ≥![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 98.5%), perrhenic acid (HReO4, 75–80 wt% aqueous), 5-hydroxymethylfurfural (5-HMF, ≥99%), 1-butanol (1-BuOH, 98%), 1,3-propanediol (PDO, 98%) and CDCl3 (99.96% deuterated, silver stabilized) were purchased from Sigma Aldrich. 2,5-bis(hydroxymethyl)-furan (BHMF, 95%) was acquired from Wako Pure Chemical Industries Ltd. 2,5-Bis(hydroxymethyl)-tetrahydrofuran (BHMTHF, 95%) was obtained from Carbosynth. All chemicals were used without further purification.
98.5%), perrhenic acid (HReO4, 75–80 wt% aqueous), 5-hydroxymethylfurfural (5-HMF, ≥99%), 1-butanol (1-BuOH, 98%), 1,3-propanediol (PDO, 98%) and CDCl3 (99.96% deuterated, silver stabilized) were purchased from Sigma Aldrich. 2,5-bis(hydroxymethyl)-furan (BHMF, 95%) was acquired from Wako Pure Chemical Industries Ltd. 2,5-Bis(hydroxymethyl)-tetrahydrofuran (BHMTHF, 95%) was obtained from Carbosynth. All chemicals were used without further purification.
All supported catalysts were prepared by wet-impregnation with quantities listed per gram of catalyst. Metal loading of Ni/TiO2 and Re/TiO2 was fixed at 0.5 mmol g−1 support, resulting in weight loadings of respectively 3 wt% and 9.5 wt%. Nickel loading of bimetallic NiRex catalysts was equally fixed at 3 wt%. x denotes the intended Ni:Re atomic ratios of 0.5, 1 and 2, which corresponds with Re weight loadings of 4.8 wt%, 9.5 wt% and 19 wt% respectively.
In a typical procedure HReO4 (40–159 μL) was added to a 5 mL aqueous solution of Ni(NO3)2·6H2O (148.6 mg). The appropriate amount of TiO2 was added slowly and the resulting paste was homogenized for three hours after which the water was evaporated at 85 °C. The catalysts were dried further at 110 °C under vacuum, thoroughly ground in an agate mortar and sieve fractioned (125–500 μm). The catalysts were reduced under a flow of 10% H2/He (100 mL min−1) whilst heating at ramp rate of 2 °C min−1 to final temperatures of 300 °C for Ni/TiO2 and NiRex/TiO2, and 250 °C for Re/TiO2 to prevent loss of volatile ReOx species. Final temperatures were maintained for 3 hours in all cases. Since all catalysts share the same support (TiO2), they will be referred to by the metals present and ratio when appropriate for the sake of brevity.
PDHMF was prepared by thermal treatment of HMF in pure 1,3-propanediol in the absence of any acid catalyst. Briefly, the mixture of HMF (0.53 g, 4.2 mmol) and 1,3-propanediol (2 mL, 28 mmol) was stirred at 333 K for 6 hours, which gave both high HMF conversion (76.7%) and PDHMF selectivity (94.1%). The catalyst-free environment necessitates the use of a considerable excess of 1,3-PDO as formation of an acetal is an equilibrium reaction. After conventional work-up procedures, PDHMF with 90–97% purity can be obtained, and we then used it as a substrate for hydrogenation reactions. 1H NMR spectra of PD-HMF were recorded on a Bruker Avance III HD.
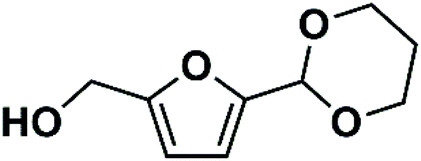
|
(5-(1,3-Dioxan-2-yl) furan-2-yl)methanol (PDHMF): 1H NMR (400 MHz, CDCl3): 6.39 (d, 1H), 6.27 (d, 1H), 5.56 (s, 1H), 4.60 (s, 1H), 4.25 (m, 2H), 3.95 (m, 2H), 2.24 (m, 1H), 1.45 (m, 1H) |
Characterization
Elemental analysis was performed on a Spectroblue EOP ICP optical emission spectrometer with axial plasma viewing, equipped with a free-running 27.12 MHz generator operating at 1400 W. Prior to the measurement, the samples were digested in mixtures of concentrated HNO3 and H2SO4 (2:5 parts by volume) and heated to 250 °C until full dissolution was observed.
X-ray photoelectron spectroscopy (XPS) was conducted on a Thermo Scientific K-alpha equipped with a monochromatic small-spot X-ray source and a 180° double focusing hemispherical analyzer with a 128-channel detector. Initial pressure was 8 × 10−8 mbar or less which increased to 2 × 10−7 mbar due to the active argon charge compensation dual beam source during measurement. For a typical sample preparation, freshly reduced catalyst was pressed down on carbon tape supported by an aluminium sample plate under inert atmosphere. The inert conditions were retained by using a vacuum transfer module. Spectra were recorded using an Al Kα X-ray source (1486.6 eV, 72 W) and a spot size of 400 μm. Survey scans were taken at a constant pass energy of 200 eV, 0.5 eV step size, region scans at 50 eV constant pass energy with a step size of 0.1 eV. XP spectra were calibrated to the C–C carbon signal (284.8 eV) obtained from adventitious carbon and deconvoluted with CasaXPS. The peak areas thus obtained were used to estimate surface chemical composition.
Temperature programmed reduction (TPR) plots were recorded on a Micromeritics Autochem II 2920. Powdered catalyst (100 mg–150 mg) was loaded between two quartz wool plugs in a quartz glass U-tube. Samples were pretreated in a 10 mL min−1 He flow at 130 °C for 2 hours to remove physisorbed water and other adsorbates. TPR was then performed in a 50 mL min−1 flow of 4% H2 in N2 in a temperature range of 50 °C to 750 °C, 10 °C min−1. The H2 signal was calibrated using a CuO/SiO2 reference material.
CO chemisorption data was obtained from a Micromeritics ASAP 2020 to determine the number of active sites. Powdered catalyst (100 mg–150 mg) was loaded between two quartz wool plugs in a quartz glass U-tube. Prior to measurement, the samples were reduced in situ at 300 °C for 4 hours, ramp rate 2 °C min−1 in a flow of pure H2. CO chemisorption was then performed at 35 °C. Active sites were determined by extrapolating the linear part of the isotherm to the ordinate. Particle size calculations were based on total metal content assuming complete reduction of the metals and using a stoichiometry factor of 1.5 CO/active site.
HAADF-STEM images were acquired on an aberration corrected JEOL ARM-200CF TEM/STEM operating at 200 kV. This microscope was also equipped with a Centurio silicon drift detector (SDD) system for X-ray energy dispersive spectroscopy (XEDS) analysis. Quantification of the XEDS spectra was carried out via the Thermo NORAN System SIX (NSS) software using the Cliff-Lorimer method assuming no X-ray absorption.23 The Ni Kα (peak range: 7331 eV–7623 eV) and Re Lα (peak range: 8496 eV–8806 eV) were used to quantify the relative proportions of the two elements in the areas analyzed. Standard-less Gaussian fitting was applied to the two characteristic peaks, with the k-factors pre-determined by the NSS software (kNi Kα = 1.217, kRe Lα = 2.652).
Catalytic activity measurements
All hydrogenation experiments were conducted in 10 mL autoclaves equipped with a gas delivery system, and Teflon insert and magnetic stirring bar. The autoclaves were loaded with appropriate amounts of substrate (20 mg, 200 mg or 400 mg) and catalyst (10 mg or 100 mg) in a glovebox. Premixed solutions of Na2CO3 in degassed water (2.0 mL) and internal standard 1-butanol (1-BuOH, 1 μL) was added externally through a septum in order to retain the inert atmosphere in the autoclave. The autoclaves were then flushed 6 times with H2 or N2 prior to pressurizing the vessel to the desired pressure (5 MPa). Stirring was provided by a magnetic stirring plate set to 750 rpm and the autoclaves were heated to the appropriate temperature with a band heater connected to a programmable controller (Eurotherm 2408). Heating and stirring were stopped after completion of the reaction and pressure was released carefully. Catalyst was separated by centrifugation and the reaction liquor was analyzed by an Interscience Focus GC-FID over a Stabilwax-DA column (30 m × 0.53 mm I.D., df = 1.00 μm). Product quantification was based on mass response factors determined by five-point calibration lines of commercial compounds, referenced to 1-BuOH as internal standard. PDHMF calibration was performed using the as-synthesized compound.Results and discussion
Effect of catalyst composition on 5-HMF hydrogenation in dilute solutions
To address PGM scarcity, we evaluated nickel catalysts for the hydrogenation of 5-HMF. Titania is a useful support in this kind of catalytic chemistry, as it can improve the hydrogenation performance of supported metals with respect to aldehydes, carboxylic acids and esters.24–26 We prepared a range of nickel–rhenium catalysts supported on titania by straightforward wetness impregnation followed by drying and reduction (see Experimental methods, Tables S2–S4 and Fig. S1–S3 in the ESI†). The resulting materials will be denoted by the supported metal(s) for the sake of brevity. Under benign conditions (5-HMF, 20 mg; H2O, 2 mL; catalyst, 10 mg catalyst, molar ratio 5-HMF/Ni = 31; temperature 40 °C; PH2 5 MPa; time 4 hours), the nickel-containing catalysts can almost completely convert 1 wt% 5-HMF in water into BHMF and BHMTHF. The product distribution depends on the atomic Re/Ni ratio (Fig. 1). Ni and NiRe0.5 predominantly lead to BHMF formation, while NiRe1 and NiRe2 are significantly more selective to BHMTHF. Re is poorly active in comparison to the other catalysts and only gives BHMF.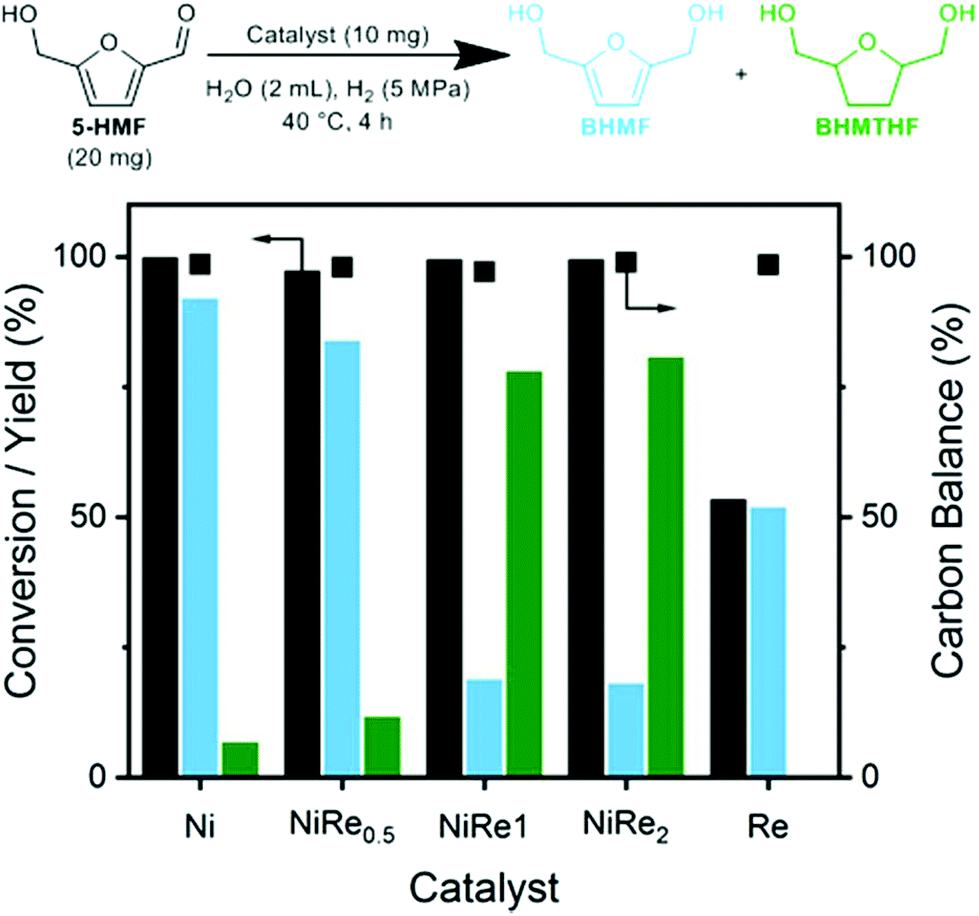 | ||
| Fig. 1 Evaluation of titania-supported Ni, NiRex and Re catalysts in the hydrogenation of aqueous 1 wt% 5-HMF solutions. | ||
From these results, we conclude that ring hydrogenation by nickel is greatly enhanced in the presence of rhenium promoter when the atomic Re to Ni ratio exceeds 0.5. The difference among Ni, Re and NiRex catalysts towards hydrogenation of the formyl group and the furan ring can be attributed to the activation behavior of 5-HMF and BHMF. Ni and Re sites in respectively pure Ni and Re catalysts only activate the formyl group of 5-HMF, which leads to selective BHMF formation. Considering the potential mechanism, the formyl group is activated through its association to nickel, forming an η2(C,O)-aldehyde complex (Fig. 2, blue box).27,28 The actual hydrogenation is proposed to proceed through sequential addition of a hydride and a proton. This infers that molecular hydrogen dissociates heterolytically, which has been postulated for these catalysts in several earlier works.29,30 The oxidic species present on the catalyst are expected to aid in the heterolytic dissociation of hydrogen This effect has been well-investigated by the group of Tomishige for a variety of metal oxide-Ir/SiO2 catalysts.31 Side-reactions arising from the nickel-formyl interaction include decarbonylation and hydrodeoxygenation. These respectively occur after forming a η1(C)-acyl species through cleavage of the formyl C–H bond (Fig. 2, top red box) or after breaking the formyl C–O bond succeeding its hydrogenation (Fig. 2, rightmost red box). Both side-reactions only take place at higher temperature and were not encountered in our test reactions.32–35 For rhenium, the exact binding mode is unknown. However, the group of Shimizu found a nearly quantitative hydrogenation of 3-phenylpropionic acid towards 3-phenylpropanol with titania-supported rhenium as catalyst and postulated the preferential adsorption of the carboxylic acid group over the phenyl moiety on oxophilic rhenium as one of the reasons for the high selectivity towards the aromatic alcohol.36,37 The mechanism assisted by oxophilic rhenium was fully supported by DFT studies.
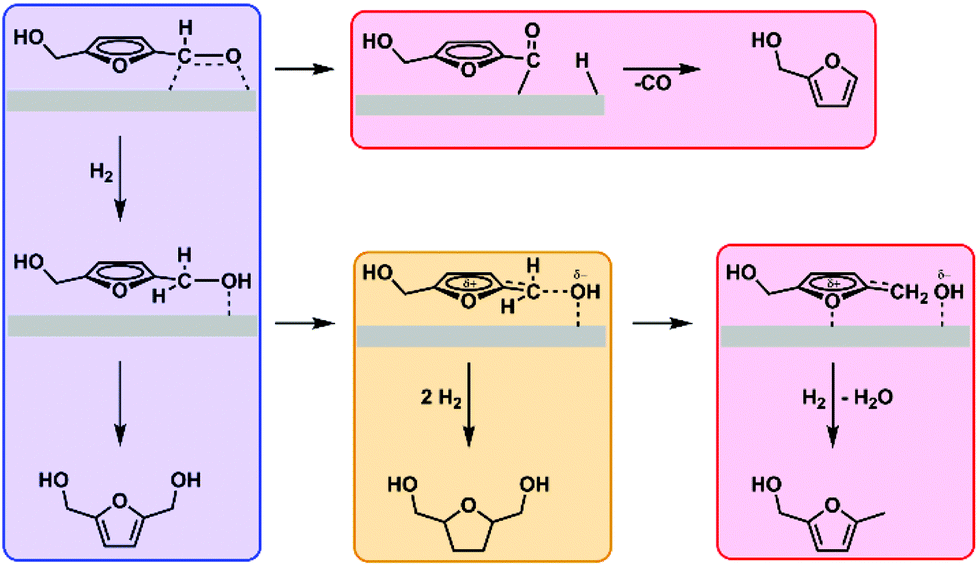 | ||
| Fig. 2 Proposed pathways for C–O hydrogenation (blue), furanic ring hydrogenation (yellow), and high-temperature decarbonylation and deoxygenation (red). | ||
The promoting effect of rhenium therefore lies in its oxophilic nature, readily forming complexes with the formyl oxygen and, potentially, the furanic oxygen.38 Unlike 3-phenylpropionic acid, the formyl and furan ring are conjugated. Strong polarization of the formyl group by rhenium can thus activate the furan ring, promoting ring hydrogenation by forcing the ring in the required orientation. This effect only occurs on the nickel-containing catalysts, however, and this underlines the promoting role rhenium displays under the employed conditions as rhenium by itself shows poor activity compared to the other materials and is unable to hydrogenate the furan ring. Studies performed by the group of Rosseinsky conclude that the formation from BHMTHF proceeds through BHMF.39 This is in line with our findings, as BHMTHF was never encountered in absence of BHMF in any of our reactions starting from 5-HMF. A similar pathway has been found by the group of Tomishige for hydrogenation of furfural to tetrahydrofurfuryl alcohol, which proceeds through furfuryl alcohol as intermediate.40 Both groups found that the selectivity towards the saturated alcohol depends on the conversion of the unsaturated aldehyde. With our materials we find that both the furan ring and the formyl group in 5-HMF can be stabilized and activated simultaneously only on bimetallic NiRex catalysts and with a high Re loading (Re/Ni atomic ratio >0.5), thus promoting the hydrogenation of both of these functional groups. As a consequence, BHMTHF is the dominant reaction product over NiRe1 and NiRe2 catalysts (Fig. 2, yellow box). This behavior is comparable to that of the Ir-ReOx/SiO2 catalysts investigated by Tomishige et al. In their work, which involves the selective hydrogenation of crotonaldehyde to crotyl alcohol among other α,β unsaturated alcohols, they propose that oxidic species activate the aldehyde group of the reactant and aid in the heterolytic dissociation of hydrogen into protons and hydrides.17,31 The hydrides formed in this manner are especially reactive towards polar unsaturated groups such as aldehydes, leading to the formation of alcohols. Their family of MOx-Ir/SiO2 catalysts, however, are not particularly active for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogenation in contrast to the materials studied in this work. We postulate that this is due to the presence of nickel instead of iridium, as nickel, palladium and platinum-based catalysts generally excel at CC hydrogenation. However, supported monometallic Ni, Pd and Pt catalysts are not overly active towards 5-HMF furan hydrogenation at moderate temperatures (Table S1,† entries 1, 3 and 5) and selectivity towards BHMTHF strongly benefits from synergistic effects resulting from bimetallic systems (Table S1,† entries 2 and 4). A combination of these effects likely explains our findings in Fig. 1.
C hydrogenation in contrast to the materials studied in this work. We postulate that this is due to the presence of nickel instead of iridium, as nickel, palladium and platinum-based catalysts generally excel at CC hydrogenation. However, supported monometallic Ni, Pd and Pt catalysts are not overly active towards 5-HMF furan hydrogenation at moderate temperatures (Table S1,† entries 1, 3 and 5) and selectivity towards BHMTHF strongly benefits from synergistic effects resulting from bimetallic systems (Table S1,† entries 2 and 4). A combination of these effects likely explains our findings in Fig. 1.
Control experiments using BHMF gave us an important insight into these reaction mechanisms (Table 1). Ni, Re and NiRe1 catalysts are inactive for ring-hydrogenation of BHMF at 40 °C in a dilute solution (1 wt%), whereas the NiRe1 catalyst can promote hydrogenation of both the furan ring and the formyl group in 5-HMF under the same reaction conditions, yielding BHMTHF (Fig. 1). Therefore, the interaction of the formyl group in 5-HMF with oxophilic Re site could induce additional interaction between the furan ring and active Ni site of NiRe1, which leads to simultaneous hydrogenation of these two functional groups. Due to a lower affinity of the furan ring with both Ni and Re species, the interaction between the nucleophilic oxygen atom in the formyl group and oxophilic Re promoter present in close proximity with active Ni, confirmed with HAADF-STEM and EDS (ESI Fig. S3†), can be identified as the main driving force for ring hydrogenation. One exception is noted during a high-temperature reaction: direct ring-hydrogenation of BHMF with NiRe1 can be achieved at 90 °C (Fig. 2, yellow box). In order to develop a more efficient process for BHMF production with high productivity, we next increased the initial 5-HMF concentration to 10 wt% and conducted the reactions without changing other parameters including reaction temperature, catalyst weight, hydrogen pressure, and so on. This means that the reactions were carried out at a ten-fold ratio of substrate to catalyst (5-HMF/Ni). Table 2 summarizes the activities in the hydrogenation of 10 wt% 5-HMF solution (5-HMF, 200 mg; H2O, 2 mL; catalyst, 10 mg catalyst, molar ratio 5-HMF/Ni = 310; temperature 40 °C; PH2 5 MPa; time 4 hours).
| Catalyst | Temp. (°C) | X BHMF (%) | Y BHMTHF (%) | Y unknown (%) | C.B.b (%) |
|---|---|---|---|---|---|
| a Reagents and conditions: Catalyst (10 mg), BHMF (20 mg, molar ratio BHMF/Ni = 31), H2O (2 mL), 1-butanol (1 μL), H2 (5 MPa), 40 °C, 4 hours, 750 rpm. b Carbon balance. | |||||
| Ni | 40 | 5.1 | n.d. | 5.1 | 94.9 |
| NiRe1 | 40 | 4.6 | 2.9 | 1.7 | 98.3 |
| NiRe1 | 90 | 67.8 | 57.3 | 10.5 | 89.5 |
| Re | 40 | 4.6 | n.d. | 4.6 | 95.4 |
| Entry | Catalyst | Cat. (mg) | X 5-HMF (%) | Y BHMF (%) | Y BHMTHF (%) | C.B.b (%) |
|---|---|---|---|---|---|---|
| a Reagents and conditions: 5-HMF (200 mg, molar ratio 5-HMF/Ni = 312 or 31), H2O (2 mL), 1-butanol (1 μL), H2 (5 MPa), 40 °C, 4 hours, 750 rpm. b Carbon balance. c Conditions as in footnote a, but reaction was extended for 8 hours and heated to 90 °C from 40 °C for this period. d Conditions as [a] but with addition of 0.5 eq. of Na2CO3. | ||||||
| 1 | Ni | 10 | 11.2 | 9.87 | <0.1 | 87.7 |
| 2 | NiRe0.5 | 10 | 21.5 | 18.1 | <0.1 | 84.3 |
| 3 | NiRe1 | 10 | 29.9 | 25.5 | <0.1 | 85.5 |
| 4 | NiRe1 | 10 | 70.5c | 2.1 | 34.5 | 66.3 |
| 5 | NiRe2 | 10 | 53.8 | 45.6 | <0.1 | 84.7 |
| 6 | NiRe2 | 10 | 98.2d | 72.1 | <0.1 | 73.9 |
| 7 | NiRe2 | 100 | 97.2 | 83.3 | 2.6 | 88.8 |
| 8 | Re | 10 | 5.3 | 4.5 | <0.1 | 85.5 |
Activity tests using concentrated 5-HMF solutions
A drastic increase in 5-HMF/Ni ratio to 312 resulted in a decrease in the performance of all studied catalysts (entries 1, 2, 3, 5, and 8) compared to those tested at a 5-HMF/Ni ratio of 31 in Fig. 1. Monometallic Ni and Re showed lower activity than bimetallic catalysts with respect to 5-HMF conversion and BHMF yield. NiRe2 catalyst showed the highest activity (46% of BHMF yield at 54% of 5-HMF conversion) among the catalysts tested. Interestingly, NiRe1 and NiRe2 gave only BHMF in concentrated 5-HMF solution (hydrogenation limited to the formyl group), while simultaneous hydrogenation of the furan ring and the formyl group leading to BHMTHF was predominant in diluted 5-HMF solution (Fig. 1). This tendency for no-BHMTHF formation did not change, even after the increase of the 5-HMF/Ni ratio to 31 in a NiRe2-catalyzed reaction. In this case an acceptable BHMF yield of 83% was obtained at nearly complete 5-HMF conversion (97%, entry 7). We hypothesize this change is due to the molecular orientation of adsorbed 5-HMF on the surface of NiRe1 and NiRe2 in concentrated solution. Interaction of the furan ring with the active Ni sites is limited due to steric hindrance, i.e. when the NiRex surface is fully covered with 5-HMF and/or BHMF in concentrated solution. This prevents the formation of BHMTHF by simultaneous hydrogenation of the furan ring and the formyl group (Fig. 3).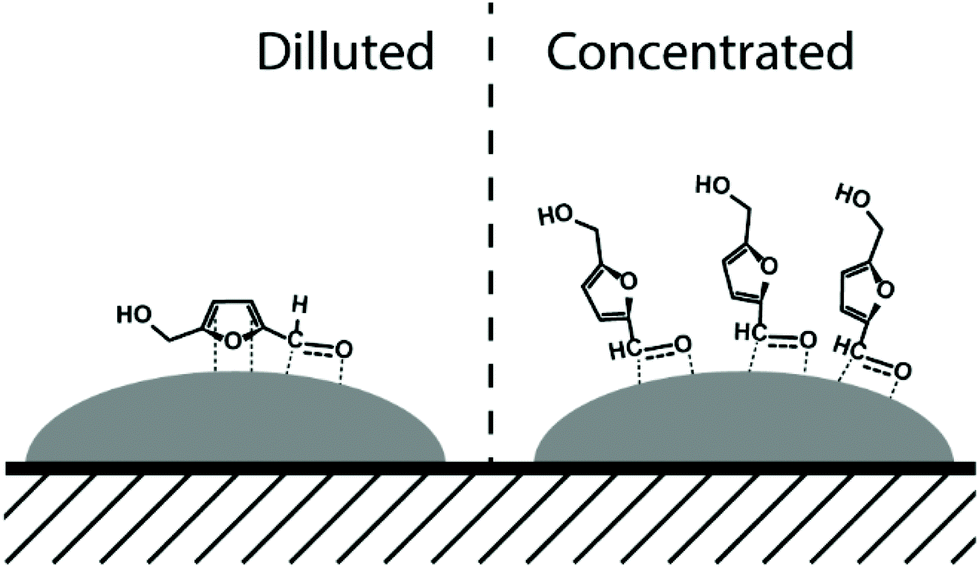 | ||
| Fig. 3 Proposed 5-HMF orientations in dilute (1 wt%) and concentrated (10 wt%) solutions over NiRe1 and NiRe2 bimetallic catalysts. | ||
The reaction time can be shortened by increasing the reaction temperature. However, a high temperature reaction at 90 °C using NiRe1 is not efficient for BHMF formation because of the simultaneous hydrogenation of furan and formyl moieties, resulting in a moderate BHMTHF yield (34.5%) (entry 4). It should be noted that carbon balances for entries 1, 2, 3, 5, 7, and 8 of Table 2 are in the 84–88% range, which is significantly lower than those in the 97–99% range obtained from the hydrogenation of 1 wt% 5-HMF solution (Fig. 1). Low carbon balances can be explained by the formation of undesired humin oligomers in concentrated aqueous solutions of 5-HMF, which was also evident from the yellow coloration of reaction mixtures and the formation of a brown solid residue (insoluble humins), even without catalyst or in the presence of the bare titania support. Presence of humins on the catalytic surface was verified with TGA (Fig. 4). Fresh materials showed practically no mass loss, while catalysts used in the hydrogenation of 1 wt% 5-HMF and 10 wt% 5-HMF lost roughly 10% and 20% of their mass, respectively. Therefore, we conclude that the hydrogenation of concentrated 5-HMF solution with NiRe bimetallic catalysts is not applicable for efficient BHMF production, because the catalysts most probably deactivate before reaching full 5-HMF conversion in these runs due to deposition of humins.
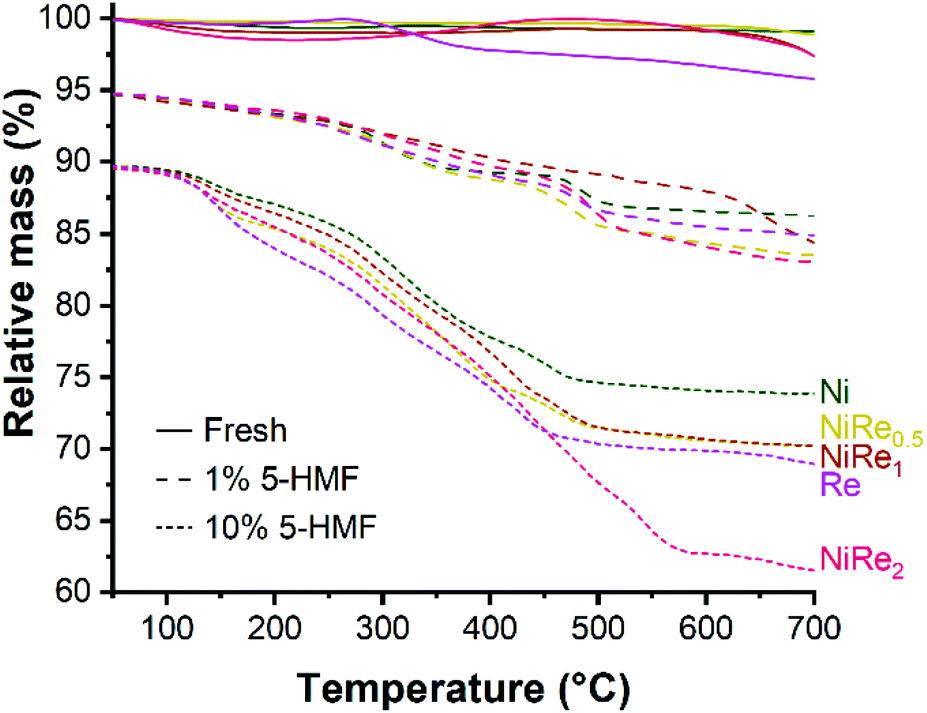 | ||
| Fig. 4 TG curves showing relative mass loss of fresh and spent Ni, NiRex and Re used for the hydrogenation of 1 wt% and 10 wt% 5-HMF. TG curve sets are offset by −5% (1 wt% 5-HMF) and −10% (10 wt% 5-HMF) for clarity. | ||
Catalyst screening using concentrated PDHMF solutions
We extended 5-HMF hydrogenation using NiRe bimetallic catalysts to PDHMF and aimed for a more sophisticated BHMF process. Initial screening using 10 wt% PDHMF was performed in pH-neutral environment in order to facilitate the required deprotection. The results are summarized in Table 3. The strategy worked very well as we were able to reach high conversion of both protected 5-HMF (as PDHMF) and 5-HMF itself, which is now considered an intermediate. The bimetallic catalysts outperform both Ni/TiO2 and Re/TiO2 monometallic catalysts, which produce only small amounts of hydrogenated products.| Catalyst | X PDHMF (%) | Y 5-HMF (%) | Y BHMF (%) | Y BHMTHF (%) | C.B.b (%) | PDO rec.c (%) |
|---|---|---|---|---|---|---|
| a Reagents and conditions: Catalyst (10 mg), PDHMF (200 mg, molar ratio PDHMF/Ni = 213), H2O (2 mL), 1-butanol (1 μL), H2 (5 MPa), 40 °C, 4 hours, 750 rpm. b Carbon balance. c PDO recovery presented as a sum of unreacted PDHMF and free 1,3-propanediol. PDO originates from deprotection of PDHMF during the course of the reaction. It is excluded from the carbon balance. | ||||||
| Ni | 98.3 | 57.5 | 18.7 | <0.1 | 78.0 | 93.8 |
| NiRe0.5 | 94.4 | 8.5 | 78.7 | 5.0 | 97.8 | 91.8 |
| NiRe1 | 94.7 | 4.5 | 77.1 | 4.7 | 91.7 | 83.0 |
| NiRe2 | 99.2 | 1.1 | 78.8 | 6.1 | 87.4 | 76.5 |
| Re | 98.2 | 66.5 | 5.87 | <0.1 | 74.7 | 92.3 |
The bimetallic catalysts are very selective to BHMF and only small amounts of BHMTHF are produced in all cases, and similar results were also obtained in the test reactions with concentrated 5-HMF solutions (Table 2). However, we do observe a decaying trend in both carbon balance and 1,3-propanediol (PDO) recovery. Peculiar here is that the selectivities to the hydrogenation products are very similar even with decaying carbon balances. However, a viable explanation is that rhenium oxides tend to be Brønsted acidic or, alternatively, Brønsted acid sites can be generated from metallic rhenium in contact with water, which yields acidic hydroxyl species due to the oxophilicity of rhenium.38,41,42 It is therefore possible that higher levels of rhenium will increase deprotection rates, as acetals are acid labile. This makes the deprotected 5-HMF vulnerable to degradation, a process which itself is enhanced by higher levels of Brønsted acidity. Additionally, PDO by itself does not degrade when in contact with NiRe2 catalyst, although up to 35% of the PDO deteriorates during hydrogenation of 5-HMF (ESI Table S5†). However, no strong PDO deterioration was detected when replacing 5-HMF with BHMF. This indicates that PDO can form side-products with 5-HMF when exposed to elevated levels of Brønsted acidity, but not with BHMF.
Optimization of acetal deprotection rate by varying pH
The screening above showed that NiRe0.5 is the preferred catalyst to produce BHMF, as it combines a decent BHMF selectivity with very good carbon balance and PDO recovery (Table 3). However, a fair amount of 5-HMF is also recovered, which means the rate of deprotection is too high. This is not optimal as this liberated 5-HMF can still engage in side-reactions. As acetal stability depends on the alkalinity of the solution, the rate of deprotection can be controlled by careful adjustment of the pH. We tested the addition of various amounts of Na2CO3 to the reaction liquor, expressed as equivalents (eq.) to PDHMF. The results are summarized in Fig. 5 and complete data is provided in ESI Table S6.† The influence of base is significant. At 0.1 eq. of Na2CO3, we were only able to reach a PDHMF conversion of 53%. No trace of 5-HMF was found, meaning that any liberated 5-HMF is immediately hydrogenated, but the yields of BHMF and BHMTHF were very low. Curiously, we discovered the emergence of an unexpected side-product at high Na2CO3 eq., which we identified as the 1,3-propanediol acetal of 5-hydroxymethyltetrahydrofuran (PDHMTHF) by NMR (ESI Table S6 and Fig. S4†). It appears that under these conditions, ring hydrogenation is possible but not preferred, surmised from the low conversion. This is in contrast to our verification experiments to hydrogenate pure BHMF (Table 1). This indicates that the activation barrier for PDHMF ring-hydrogenation might be lower than for BHMF hydrogenation, but is still difficult to overcome. A plausible mechanism for the PDHMF ring-hydrogenation is that the molecule can freely rotate over the furan-acetal bond, which might aid in finding an orientation where the ring can be hydrogenated. In contrast, 5-HMF cannot freely rotate over the furan-formyl bond as the formyl CO bond is conjugated to the furan ring, which forces the formyl group planar to the furan ring through resonance.
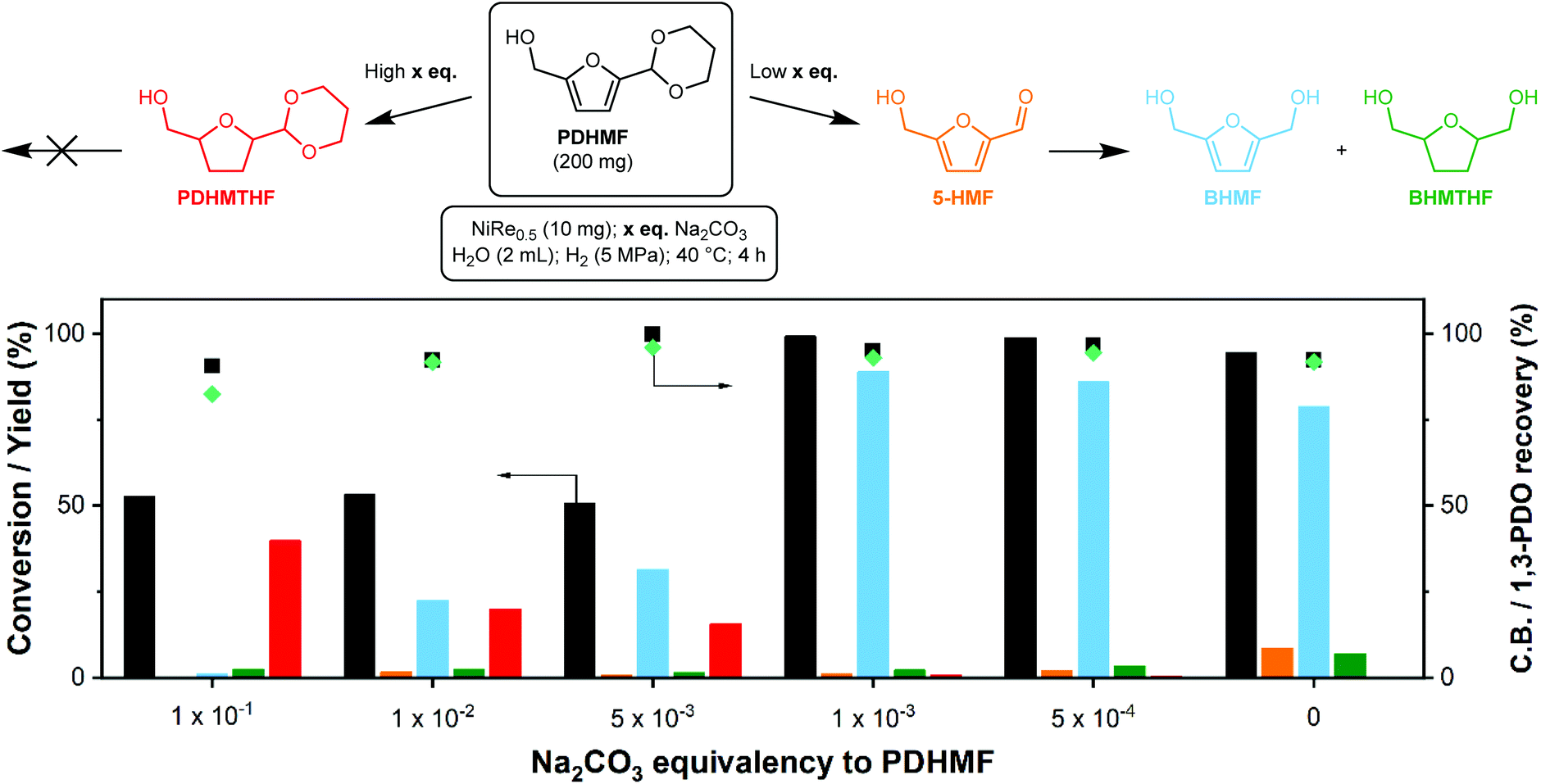 | ||
| Fig. 5 Conversion and product distribution as function of Na2CO3 equivalencies for the hydrogenation of aqueous 10 wt% PDHMF solutions using NiRe0.5 as catalyst. Carbon balances are marked with black squares. Teal diamond markers indicate 1,3-PDO recovery as a sum of unreacted PDHMF, PDHMTHF and free 1,3-propanediol. 1,3-PDO originates from deprotection of PDHMF during the course of the reaction. It is excluded from the carbon balance. | ||
The high stability of the acetal group towards deprotection at higher pH thus suppressed the deprotection step which causes the reactive formyl group to be unavailable for hydrogenation. Consequently, this resulted in gradual and direct ring-hydrogenation of PDHMF to PDHMTHF. We conducted the hydrogenation of 5-HMF in the presence of Na2CO3 (0.5 eq.) as a control experiment (Table 2, entry 6) in order to measure the effect of Na2CO3 on ring-hydrogenation of 5-HMF. Compared to the result without addition of Na2CO3 (entry 5), BHMTHF selectivity was not increased, although the BHMF yield was significantly improved (72%, Table 2, entries 5 and 6). The carbon balance was negatively affected, dropping from 85% to 74%, which indicates that addition of base did not suppress degradation of 5-HMF.
From 0.1 to 5 × 10−3 eq. Na2CO3, the conversion remained relatively stable, but formyl hydrogenation is gradually favored over furan ring-hydrogenation as we could observe increasing BHMF and decreasing PDHMTHF yields. This indicates how sensitive the acetal is to the pH, which only drops from 11.2 to 10.7 over this range. Near-full conversion was achieved (>99%) by lowering the amount of Na2CO3 even further to 1 × 10−3 eq., which also significantly increased the BHMF yield (89%). The small amount of base was, however, still able to mediate the deprotection rate, as far less 5-HMF was recovered compared to the uncontrolled reaction. This translates in a slightly higher carbon balance. The promoting role of rhenium under the optimized conditions was verified by replacing NiRe0.5 by Ni. Using the same conditions, we recovered 56% 5-HMF and obtained a BHMF yield of just 34%. There are several reports on selective BHMF formation with >98% yield at commercially relevant concentrations as summarized in Table S1,† but most of them requires Pd, Pt, and Ir-based catalysts. The use of PDHMF enables selective production of BHMF over a PGM-free catalyst (Ni-ReOx) in concentrated solutions, which is clearly distinct from other approaches reported in previous papers.
Tuning the acetal deprotection rate to the rate of hydrogenation is paramount to obtain high conversion and optimal BHMF yield and selectivity. To gain more insight in these rates, we conducted a kinetic study at 2 different Na2CO3 eq. The results are presented in Fig. 6(a) and (b). For the optimized amount of base (1 × 10−3 eq. Na2CO3, Fig. 6(a)), PDHMF is gradually deprotected over the entire duration of the reaction and liberated 5-HMF is almost immediately consumed. After 3 hours, the rate of hydrogenation starts to lag behind the deprotection rate as the amount of free 5-HMF approaches 8%. However, at this point nearly 80% of the PDHMF is consumed, allowing the hydrogenation rate to catch up again and, after 4 hours, PDHMF is fully consumed and about 1% of 5-HMF remains. BHMF hydrogenation under these conditions is very unfavorable, as already mentioned, and this explains the very high BHMF selectivity. Removal of the catalyst after 2 hours of reaction ceases production of BHMF, confirming the heterogeneous nature of the reaction. A higher base amount of Na2CO3 (0.05 eq.) significantly attenuates the PDHMF conversion rate (Fig. 6(b)). BHMF and BHMTHF yields are almost negligible and no 5-HMF is produced. Instead, the main product is PDHMTHF, formed by ring hydrogenation of PDHMF. Here, we can conclude that furan ring-hydrogenation can occur even when the acetal is still present. However, this reaction is not preferred and no acetal deprotection occurs, even after the ring has been hydrogenated as neither BHMTHF or 5-HMTHF were detected. In absence of H2, PDHMF conversion is very low and 5-HMF is in equilibrium with PDHMF (ESI Fig. S5†). Initially, the amount of 5-HMF recovered vs. the amount of converted PDHMF is fairly similar. However, after 4 hours, we can see the 5-HMF yield leveling off, suggesting formation of side-products.
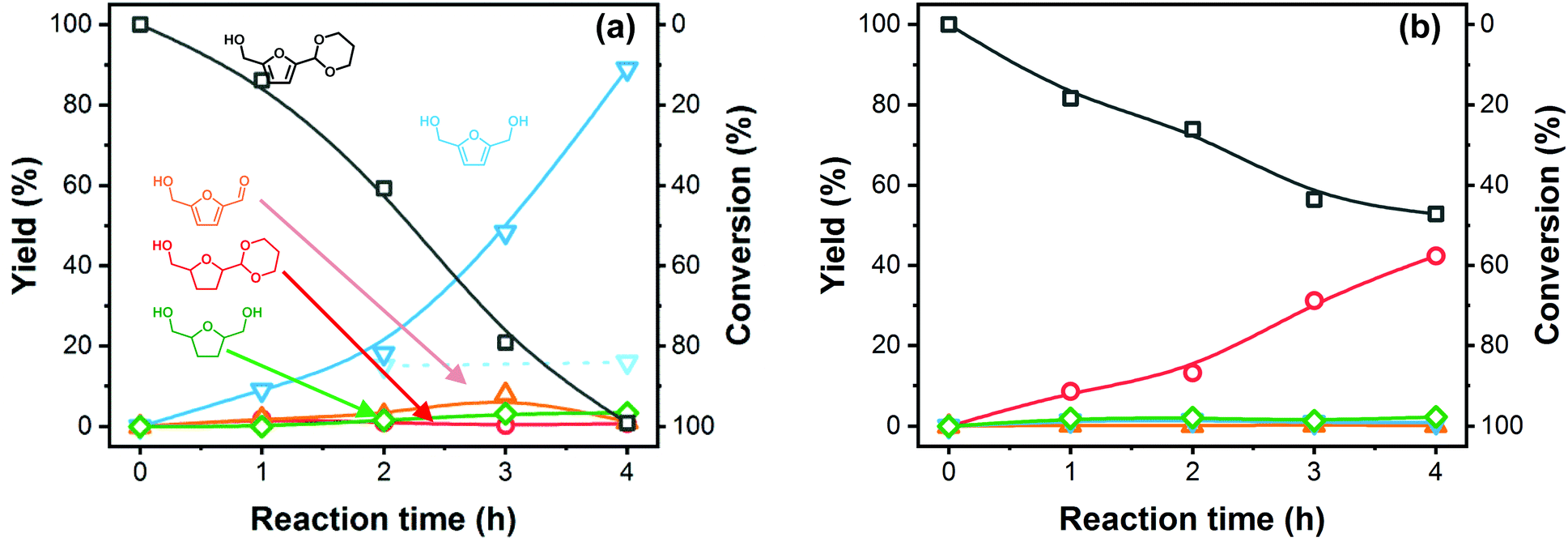 | ||
| Fig. 6 Time courses of BHMF formation from PDHMF with NiRe0.5 using (a) 1 × 10−3, and (b) 5 × 10−2 eq. Na2CO3. Lines provide guidance for the reader's eye. Reagents and conditions: Catalyst (10 mg), PDHMF (200 mg, molar ratio PDHMF/Ni = 214), H2O (2 mL), 1-butanol (1 μL), H2 (5 MPa), 40 °C, 750 rpm. Dashed line and transparent symbols (a) show BHMF yield after catalyst removal. | ||
Fig. 7 shows the proposed reaction pathway. The acetalization reaction is an equilibrium reaction. Deprotection of PDHMF proceeds gradually under optimized conditions as shown in Fig. 6(a). Although weakly Brønsted acidic hydroxyls and Lewis acid sites on the surface also contribute to deprotection steps, we expect that the rates for initial and second hydrolysis steps, giving hemiacetal of PDHMF and bare 5-HMF, are strongly dependent on the pH of the reaction mixture. There are three compounds (i.e. PDHMF, the hemi-acetal of PDHMF and bare 5-HMF) in the reaction mixture. The concentrations of these compounds approach an equilibrium under the reaction conditions as confirmed in ESI Fig. S5,† when no hydrogenation takes place in the absence of H2. However, the concentration of PDHMF decreases in the course of the hydrogenation reaction, because 5-HMF is now involved in the hydrogenation. In theory, an attack from a hydride species (depicted in gray) can break the C–O bond of the hemiacetal but (hemi)acetals are only very weakly electrophilic, so this reaction is very unlikely; therefore, hydrogenation of bare 5-HMF can be determined as a main path for efficient BHMF formation. Due to a short life time of bare 5-HMF and high stability towards hydrogenation with NiRe0.5 catalyst, most of 1,3-propanediol used as protecting reagent can be recovered after the reaction and used repeatedly for subsequent reactions.
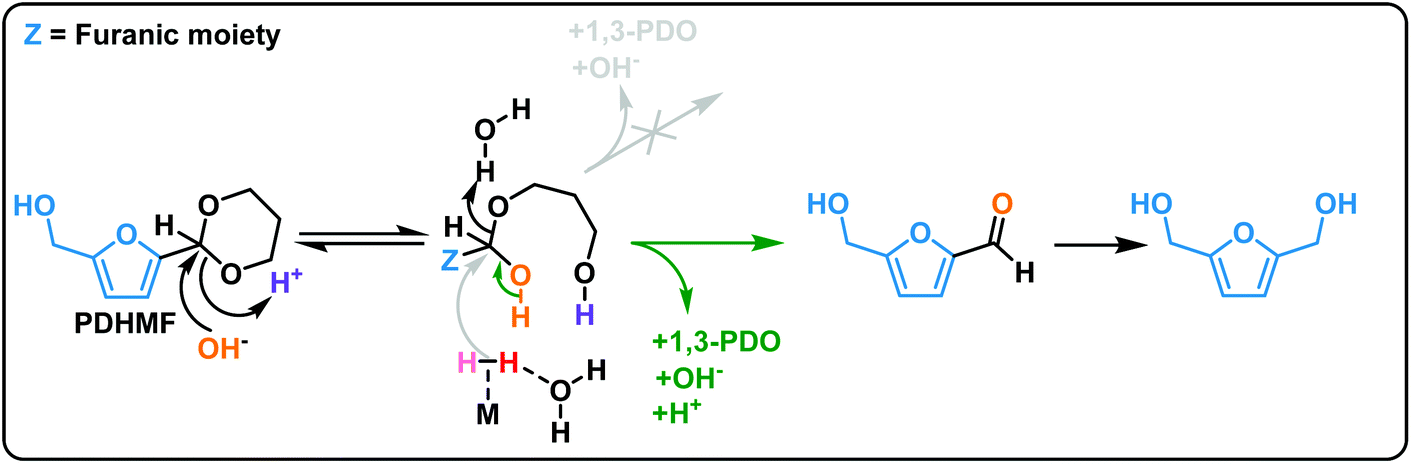 | ||
| Fig. 7 Proposed reaction pathway for hydrogenation of PDHMF via 5-HMF as intermediate. | ||
Inspired by these results, we decided to test the limits of the optimum catalyst and the protection method by using a loading of 20 wt% PDHMF and an optimized Na2CO3 (1 × 10−3 eq.). The results are presented in Table 4. After 4 hours of reaction we recovered 11% of unreacted PDHMF and a substantial amount of 5-HMF (21%) and BHMF (57%). However, nearly all PDHMF was converted (99%) and 5-HMF levels dropped to 5% after extending the reaction to a total of 6 hours. More importantly, the BHMF yield increased to 81% with satisfactorily low BHMTHF and PDHMTHF yields and a carbon balance of 91%.
| t (h) | X PDHMF (%) | Y 5-HMF (%) | Y BHMF (%) | Y BHMTHF (%) | Y PDHMTHF (%) | C.B.c (%) | PDO rec.d (%) |
|---|---|---|---|---|---|---|---|
| a Reagents and conditions: Catalyst (10 mg), PDHMF (400 mg, molar ratio PDHMF/Ni = 428), H2O (2 mL), 1-butanol (1 μL), H2 (5 MPa), 40 °C, 1 × 10−3 equivalence of Na2CO3 to PDHMF, 750 rpm. b 1,3-Propanediol acetal of 5-hydroxymethyltetrahydrofurfural. c Carbon balance. d PDO recovery presented as a sum of unreacted PDHMF, PDHMTHF and free 1,3-propanediol. PDO originates from deprotection of PDHMF during the course of the reaction. It is excluded from the carbon balance. | |||||||
| 4 | 88.8 | 20.9 | 57.4 | 1.3 | 2.6 | 93.4 | 95.6 |
| 6 | 98.7 | 5.2 | 80.9 | 2.1 | 2.0 | 91.4 | 90.2 |
Conclusions
We used titania-supported rhenium-promoted nickel catalysts for the selective hydrogenation of 5-HMF to BHMF and BHMTHF in water at low temperature. Rhenium adopts several promoting roles in this catalytic chemistry. In dilute 5-HMF solutions (1 wt%), rhenium promotes the hydrogenation of the furan ring and full conversion can be reached with all nickel-containing catalysts with carbon balances exceeding 95%. In more concentrated solutions of 5-HMF (10 wt%), higher loadings of rhenium increase the overall activity of the catalyst, although full conversion was never reached due to humin buildup on the catalyst surface. With NiRe2, the most active material, 54% conversion was obtained with a BHMF yield of 46% and a carbon balance of 85%. BHMTHF was not produced in this case, which we attribute to competitive adsorption between unreacted 5-HMF and BHMF still adsorbed to the catalytic surface and it is expected that this locally high concentration of 5-HMF also promotes the formation of humins. The benefit of using rhenium on the overall activity is more prominent when a concentrated solution (10 wt%) of PDHMF, the acetal of 5-HMF with 1,3-propanediol, is hydrogenated, as all bimetallic NiRe catalysts can fully convert the substrate unlike their monometallic counterparts. Although all rhenium-promoted catalysts produce BHMF and BHMTHF in nearly equal amounts in pH-neutral solutions, NiRe0.5 is identified as the optimal catalyst, as it produces the lowest amount of side-products. The carbon balance, although improved compared to 10 wt% 5-HMF (92% vs. 85%), is still suboptimal, which is caused by degradation of the relatively high amounts of unprotected 5-HMF (9%) formed during the reaction. The reason is that the deprotection rate was too high compared to the hydrogenation rate at the neutral pH. We discovered that the deprotection rate can be attenuated by increasing the pH. Too high pH leads to a too low deprotection rate, essentially blocking the formation of the formyl and, instead, the slow and undesired hydrogenation of the furan ring of PDHMF occurs. Careful balancing of the deprotection and hydrogenation rates by judicious choice of the pH proved to be the key in selectively producing BHMF in high yields (81–89%) using concentrated solutions of PDHMF (10–20 wt%) in water.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors would like to thank M.G.W.M. Verhoeven for support with XPS processing. This work was supported by The Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), an NWO Gravitation programme funded by the Ministry of Education, Culture and Science of the Government of The Netherlands.References
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- P. Daorattanachai, P. Khemthong, N. Viriya-Empikul, N. Laosiripojana and K. Faungnawakij, Carbohydr. Res., 2012, 363, 58–61 CrossRef CAS PubMed.
- J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- M. Kim, Y. Su, A. Fukuoka, E. J. M. Hensen and K. Nakajima, Angew. Chem., Int. Ed., 2018, 57, 8235–8239 CrossRef CAS PubMed.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS PubMed.
- Z. Zhang and K. Deng, ACS Catal., 2015, 5, 6529–6544 CrossRef CAS.
- J. He, K. Huang, K. J. Barnett, S. H. Krishna, D. M. Alonso, Z. J. Brentzel, S. P. Burt, T. Walker, W. F. Banholzer, C. T. Maravelias, I. Hermans, J. A. Dumesic and G. W. Huber, Faraday Discuss., 2017, 202, 247–267 RSC.
- J. He, S. P. Burt, M. Ball, D. Zhao, I. Hermans, J. A. Dumesic and G. W. Huber, ACS Catal., 2018, 8, 1427–1439 CrossRef CAS.
- J. Zhang, T. Wang, X. Tang, L. Peng, J. Wei and L. Lin, BioResources, 2018, 13, 7137–7154 Search PubMed.
- E. A. Baroncini, S. Kumar Yadav, G. R. Palmese and J. F. Stanzione, J. Appl. Polym. Sci., 2016, 133, 44103 CrossRef.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS PubMed.
- Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Catal. Sci. Technol., 2016, 6, 5668–5683 RSC.
- M. Tamura, K. Tokonami, Y. Nakagawa and K. Tomishige, Chem. Commun., 2013, 49, 7034–7036 RSC.
- E. J. M. Hensen, M. W. G. M. (Tiny) Verhoeven, A. Ciftci, J. Pritchard and E. A. Pidko, Catal. Today, 2016, 279, 10–18 Search PubMed.
- S. T. Thompson and H. H. Lamb, ACS Catal., 2016, 6, 7438–7447 CrossRef CAS.
- S. Liu, Y. Amada, M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2014, 4, 2535–2549 RSC.
- M. Kim, Y. Su, T. Aoshima, A. Fukuoka, E. J. M. Hensen and K. Nakajima, ACS Catal., 2019, 4277–4285 CrossRef CAS.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, 2006 Search PubMed.
- G. Cliff and G. W. Lorimer, J. Microsc., 1975, 103, 203–207 CrossRef.
- W. Rachmady, ProQuest Diss. Theses, 2001, 334, 322 Search PubMed.
- K. Liu, J. Pritchard, L. Lu, R. Van Putten, M. W. G. M. Verhoeven, M. Schmitkamp, X. Huang, L. Lefort, C. J. Kiely, E. J. M. Hensen and E. A. Pidko, Chem. Commun., 2017, 53, 9761–9764 RSC.
- M. Englisch, A. Jentys and J. A. Lercher, J. Catal., 1997, 166, 25–35 CrossRef CAS.
- J. R. Roy, M. A. Laliberté, S. Lavoie, M. Castonguay and P. H. McBreen, Surf. Sci., 2005, 578, 43–56 CrossRef CAS.
- D. P. Duarte, R. Martínez and L. J. Hoyos, Ind. Eng. Chem. Res., 2016, 55, 54–63 CrossRef CAS.
- G. Fan, Y. Zhou, H. Fu, X. Ye, R. Li, H. Chen and X. Li, Chin. J. Chem., 2011, 29, 229–236 CrossRef CAS.
- H. Hattori and T. Shishido, Catal. Surv. Jpn., 1997, 1, 205–213 CrossRef CAS.
- M. Tamura, K. Tokonami, Y. Nakagawa and K. Tomishige, ACS Catal., 2016, 6, 3600–3609 CrossRef CAS.
- M. Mavrikakis and M. A. Barteau, J. Mol. Catal. A: Chem., 1998, 131, 135–147 CrossRef CAS.
- R. Shekhar, M. A. Barteau, R. V. Plank and J. M. Vohs, J. Phys. Chem. B, 2002, 101, 7939–7951 CrossRef.
- C. P. Jiménez-Gómez, J. A. Cecilia, C. García-Sancho, R. Moreno-Tost and P. Maireles-Torres, ACS Sustainable Chem. Eng., 2019, 7, 7676–7685 CrossRef.
- A. Banerjee and S. H. Mushrif, J. Phys. Chem. C, 2018, 122, 18383–18394 CrossRef CAS.
- T. Toyao, S. M. A. H. Siddiki, Y. Morita, T. Kamachi, A. S. Touchy, W. Onodera, K. Kon, S. Furukawa, H. Ariga, K. Asakura, K. Yoshizawa and K. I. Shimizu, Chem. – Eur. J., 2017, 23, 14848–14859 CrossRef CAS PubMed.
- T. Toyao, S. M. A. H. Siddiki, A. S. Touchy, W. Onodera, K. Kon, Y. Morita, T. Kamachi, K. Yoshizawa and K. I. Shimizu, Chem. – Eur. J., 2017, 23, 980 CrossRef CAS.
- M. Chia, Y. J. Pagán-Torres, D. Hibbitts, Q. Tan, H. N. Pham, A. K. Datye, M. Neurock, R. J. Davis and J. A. Dumesic, J. Am. Chem. Soc., 2011, 133, 12675–12689 CrossRef CAS PubMed.
- N. Perret, A. Grigoropoulos, M. Zanella, T. D. Manning, J. B. Claridge and M. J. Rosseinsky, ChemSusChem, 2016, 9, 521–531 CrossRef CAS PubMed.
- Y. Nakagawa, H. Nakazawa, H. Watanabe and K. Tomishige, ChemCatChem, 2012, 4, 1791–1797 CrossRef CAS.
- D. L. King, L. Zhang, G. Xia, A. M. Karim, D. J. Heldebrant, X. Wang, T. Peterson and Y. Wang, Appl. Catal., B, 2010, 99, 206–213 CrossRef CAS.
- K. Tomishige, M. Tamura and Y. Nakagawa, Chem. Rec., 2014, 14, 1041–1054 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03856f |
| This journal is © The Royal Society of Chemistry 2020 |