
Novel heterogeneous ruthenium racemization catalyst for dynamic kinetic resolution of chiral aliphatic amines†
Koen
Adriaensen
 a,
Jannick
Vercammen
a,
Cédric
Van Goethem
a,
Samuel
Eyley
b,
Ivo
Vankelecom
a,
Wim
Thielemans
b and
Dirk
De Vos
*a
a,
Jannick
Vercammen
a,
Cédric
Van Goethem
a,
Samuel
Eyley
b,
Ivo
Vankelecom
a,
Wim
Thielemans
b and
Dirk
De Vos
*a
aCentre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions, Department of Microbial and Molecular Systems, KU Leuven – University of Leuven, Leuven Chem&Tech, Celestijnenlaan 200F, Post Box 2461, 3001 Heverlee, Belgium. E-mail: dirk.devos@kuleuven.be
bRenewable Materials and Nanotechnology Research Group, Department of Chemical Engineering, KU Leuven – University of Leuven, Campus Kulak Kortrijk, Etienne Sabbelaan 53, 8500 Kortrijk, Belgium
First published on 21st November 2019
Abstract
Only few dynamic kinetic resolution (DKR) systems are known for chiral aliphatic amines due to the difficult racemization of these amines. In this work, each aspect of the DKR of aliphatic amines is investigated. Various ruthenium catalysts were evaluated to increase their applicability in racemization as an alternative to established heterogeneous palladium catalysts. A heterogeneous Ru(III) on zeolite catalyst showed good activity for racemization in aprotic polar media. Next, kinetic resolution was evaluated; excellent yields (50%) and selectivities (>99%) were obtained in apolar solvents when employing isopentyl propionate as resolving agent. After evaluation of both components, the complete dynamic kinetic resolution of an aliphatic amine was established with good selectivity (97%), enantiomeric excess (96%) and a yield exceeding the kinetic resolution limit of 50%.
Introduction
The amount of waste generated in the production of active pharmaceutical ingredients (API) compared to other chemical sectors is significantly higher and remains a challenge to sustainability.1 One of the reasons for this relatively high waste generation is the complexity of the APIs, which often require a multi-step synthesis, stoichiometric or excess reagents, and extreme conditions leading to a poor atom-economy. Furthermore, many APIs contain chiral centres; as a result, one of the isomers may be active while the other is not or even potentially toxic.2 When a direct asymmetric synthesis is not possible, separation of enantiomers is required. With the exception of scenarios in which the undesirable enantiomer can be repurposed, a substantial amount of waste is generated. Moreover, the separation of the reaction mixture is often arduous since the enantiomers, in most aspects, have identical chemical and physical properties.To minimize waste generation, recycling processes have been developed. However, isolation of the desired enantiomer is often first required.3,4 Enantiomers can be separated by kinetic resolution, which utilizes an enantioselective catalyst (often an enzyme) to selectively convert one enantiomer. The resulting mixture can then be separated by conventional methods. An oxidation–reduction process is often used for recycling the undesirable enantiomer and yields again a racemic mixture, leading to a theoretically infinite number of cycles of kinetic resolution and oxidation–reduction.3 However, when these processes are combined into one step, an efficient system is created that is ideally able to provide the desired enantiomer with 100% yield and 100% enantiomeric excess. This strategy, called dynamic kinetic resolution, is therefore a valuable tool for the reduction of waste and to increase the efficiency of the pharmaceutical industry.
Many dynamic kinetic resolution (DKR) systems varying in substrate scope have been described.5–7 Despite the cost of amines, which are often produced from ketones and ammonia, relatively few DKR systems have been reported when compared to alcohols, especially for aliphatic amines. While research has led to efficient racemization systems for benzylic amines,8–11 aliphatic amines are less suitable substrates for these systems. Currently, reported racemization systems suffer from high catalyst loading, long reaction times, or severe incompatibility with enzymes.9,11,12 Heterogeneous RANEY metal (Co/Ni) racemization catalysts are excellent for the racemization but combine poorly with the enzyme in the one-pot DKR, leading to absence of product (RANEY Co) or long reaction times (RANEY Ni; 96 h).12 Homogeneous racemization catalysts are mainly based on Shvo's ruthenium transfer hydrogenation catalyst,9,13–15 while heterogeneous catalysts mostly consist of palladium nanoparticles supported on alkaline supports.8,16 In this contribution to the DKR of aliphatic amines, we first identify a novel heterogeneous ruthenium catalyst for amine racemization, look for appropriate kinetic resolution conditions, and finally evaluate the combination of both constituents into a functioning dynamic kinetic resolution for aliphatic amines (Fig. 1).
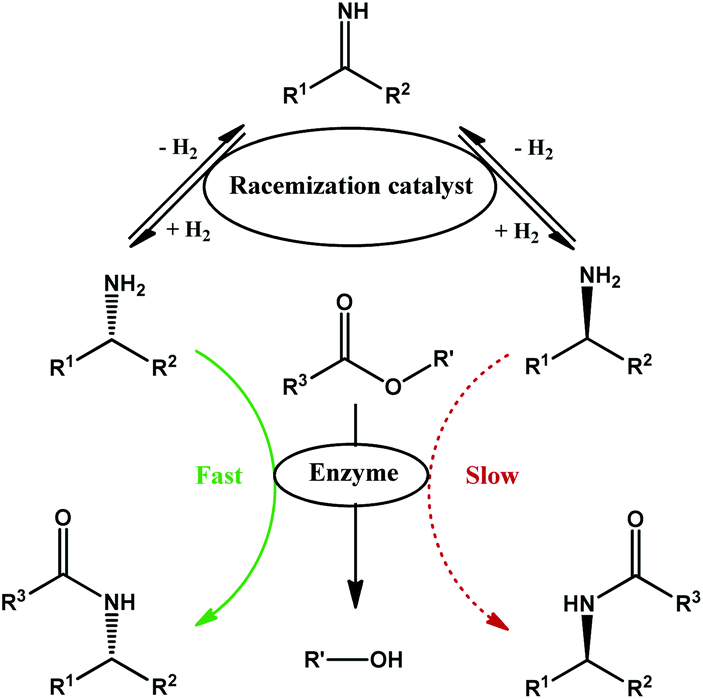 | ||
| Fig. 1 Schematic representation of dynamic kinetic resolution of chiral amines with esters as resolving agents. | ||
Results and discussion
Racemization of aliphatic amine
Racemization of aliphatic amines can be achieved via radical intermediates,17,18 but is generally approached by oxidation towards an imine intermediate, followed by a non-enantioselective reduction. Therefore, a suitable racemization catalyst should be able to perform dehydrogenation (oxidation) as well as hydrogenation (reduction) of the reactant multiple times in order to obtain a racemate. Transfer hydrogenation catalysts are known to perform both processes well; for such catalysts, ruthenium is among the most used elements.19,20| Catalyst | Conv. (%) | Sel. R-amine (%) | ee (%) |
|---|---|---|---|
| a Hydroxyapatite (HAP). b High-surface area (HSA) spinel support.22 | |||
| Ideal catalyst | 50 | 100 | 0 |
| Ru(0)/C | 14.8 | 100 | 71 |
| Ru(0)/C (red.) | 9.7 | 100 | 81 |
| Ru(0)/Al2O3 | 11.4 | 99.6 | 77 |
| Ru(0)/CaCO3 | 2.3 | 100 | 95 |
| Ru(0)/HAPa | 15.3 | 98.8 | 70 |
| Ru(0)/HSA-MgAl2O4b | 15.3 | 91.6 | 72 |
| Ru(0)/HSA-CaAl2O4b | 5.4 | 98.4 | 90 |
| Ru(0)/HSA-BaAl2O4b | 10.6 | 89.5 | 81 |
Some racemization activity was observed for reactions where ruthenium seemed to leach out of the support to yield a dark brown-yellow colour (entries 1 and 5–8).23 Since zerovalent Ru is very well retained on these supports, this observation could suggest that Ru is a better racemization catalyst in an ionic state. After this observation, some widely used homogeneous catalysts were screened alongside ionic ruthenium exchanged onto a zeolite support (Table 2). Homogeneous catalysts show very limited conversion and selectivity under these mild conditions (entries 1–6) while the zeolite catalyst leads to a significant drop in enantiomeric excess, coupled with a high selectivity for R-2-aminooctane (entries 7 and 8). Indeed, when homogeneous aliphatic amine racemization is reported, more severe reaction conditions seem to be required than those in Table 2 (70 °C).14 Paetzold et al. were able to perform a complete dynamic kinetic resolution of 2-aminooctane with 4.0 mol% of a modified Shvo-type racemization catalyst at an elevated temperature of 90 °C after 3 days to yield 85% (93% selectivity).9 Later, the same Bäckvall group reported that high temperatures are probably necessary to avoid coordination saturation of the amine substrate on the catalyst.14 However, elevated temperatures significantly decrease the number of suitable enzymes that can be used in a DKR system with the racemization system. Furthermore, the increase in temperature also promotes side reactions with the imine intermediate. Overall, racemization at lower temperature is highly preferable in the pursuit of this DKR strategy.
| Catalyst | Catalyst loading (mol%) | Conv. (%) | Sel. R-amine (%) | Sel. sec. amine (%) | ee (%) |
|---|---|---|---|---|---|
| a 1-Hydroxytetraphenylcyclopentadienyl(tetraphenyl-2,4-cyclopentadien-1-one)-μ-hydrotetracarbonyl-diruthenium(II). b 2.0 wt% ruthenium on zeolite Y. c 1.0 wt% ruthenium on zeolite Y. | |||||
| Ideal catalyst | Low | 50.0 | 100.0 | 0.0 | 0 |
| [Ru(p-cymene)Cl2]2 | 5 | 1.9 | 60.9 | 28.1 | 98 |
| [Ru(SS-TsDPEN)(p-cymene)(Py)](BF4) | 5 | 0.3 | 25.8 | 74.2 | >99 |
| Shvo's catalysta | 5 | 5.7 | 69.0 | 31.0 | 92 |
| Ru(acac)3 | 5 | trace | 100 | 0 | >99 |
| RuCl3·xH2O | 5 | trace | 100 | 0 | >99 |
| RuBr3·xH2O | 5 | trace | 100 | 0 | >99 |
| Ru(III)/Yb | 5 | 15.9 | 94.7 | 5.3 | 70 |
| Ru(III)/Yc | 2.5 | 15.3 | 100 | 0.0 | 69 |
Even though the Ru(III)/Y zeolite catalyst also displayed some leaching, racemization performance at the relatively low temperature of 70 °C is much higher than for any homogeneous ruthenium catalyst tested and reported so far. Furthermore, the catalyst loading was also fairly low (2.5 mol%), especially when compared to the Pd racemization catalyst of Kim et al. (12.0 mol%).11
| Support | Topology | Cation | SAR (in oxides) | Conv. (%) | Sel. R-amine (%) | Sel. sec. amine (%) | ee (%) |
|---|---|---|---|---|---|---|---|
| a Obtained from template-free zeolite synthesis. b pH-Control (∼7) during exchange of Ru(III) onto zeolite support. c Protic zeolite exchanged to ammonium form followed by an exchange to the Na form. | |||||||
| ZSM-5 | MFI | Na+ | 40–48 | 9.0 | 92.5 | 7.5 | 83 |
| MCM-22 | MWW | Na+ | 28 | 5.2 | 100 | 0.0 | 90 |
| Beta | *BEA | Na+ | 65 | 2.0 | 89.2 | 10.8 | 97 |
| Beta | *BEA | Na+ | 25 | 7.1 | 97.1 | 2.9 | 86 |
| Betaa | *BEA | Na+ | 9.2 | 11.0 | 96.4 | 3.6 | 79 |
| Mordenite | MOR | Na+ | 6.5 | 20.0 | 95.6 | 4.4 | 61 |
| Zeolite L | LTL | K+ | 6 | 24.4 | 99.0 | 1.0 | 52 |
| Y | FAU | Na+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
60 | 1.4 | 100 | 0.0 | 97 |
| Y | FAu | Na+c |
30 | 0.0 | — | — | >99 |
| Y | FAU | Na+ | 5.1 | 15.3 | 100 | 0.0 | 69 |
| Yb | FAU | Na+ | 5.1 | 26.1 | 100 | 0.0 | 48 |
| Yb | FAU | Cs+ | 5.1 | 21.4 | 100 | 0.0 | 57 |
Table 3 shows a trend where lower SARs result in higher activity; this is especially clear for the case of Beta zeolites where, for the same Ru content, the conversion increased as SAR decreased. A lower SAR corresponds to an increased framework negative charge, which can more easily compensate the charge of multiple charged ruthenium guests (e.g. Ru3+ ions) inside the channels. A similar trend is observed with the Y zeolites, where only the catalyst based on an Al rich faujasite (SAR = 5.1) appeared to give significant racemization. Note that the dealumination of Al-rich zeolites like NaY has profound impact on the ion exchange capacity and on the texture; this clarifies why the relation between SAR and catalytic performance is not necessarily a simple one.
Zeolites with 10-membered ring pore systems, like ZSM-5 and MCM-22 in general are less effective than zeolites with wider, 12-membered rings, like Beta zeolites, zeolite L, Mordenite or zeolite Y. For most zeolites, the tendency for formation of undesired secondary amines is satisfactorily low. This side reaction is usually associated with the acidity needed for the condensation of an amine with an intermediate imine.24 Note that negligible Brønsted acidity is expected for zeolites in their alkaline earth forms, with exchanged Na+, K+ or Cs+. Ru(III)/Y was chosen as racemization catalyst for further investigation due to the excellent commercial availability of the zeolite and the activity of the racemization catalyst as a whole.
According to Fig. 2, the optimal partial hydrogen pressure for the Ru(III)/Y catalyst system is around 10 bar, where a satisfactory low ee is achieved (17%), while preserving excellent R-amine selectivity (98.5%). Furthermore, racemization is also dependent on the choice of solvent (Table 4). Racemization activity generally seems better in more polar solvents; however, using alcohols as a solvent could create selectivity issues. Alcohols can be dehydrogenated towards carbonyl compounds, with which the amine will form imine products, leading to N-alkylated amines. This is the case for isopropyl alcohol (iPrOH) and ethanol (EtOH). When a tertiary alcohol is used as solvent (tBuOH), racemization performance does not improve over reaction with THF as the solvent. This may be due to the steric hindrance; alternatively, the beneficial effect of polarity may reach its optimum around a relative polarity of 0.2. Polar solvent molecules could increase reactivity by competition in coordination to the ruthenium moiety, since amines are known to saturate coordination spheres under mild reaction conditions.
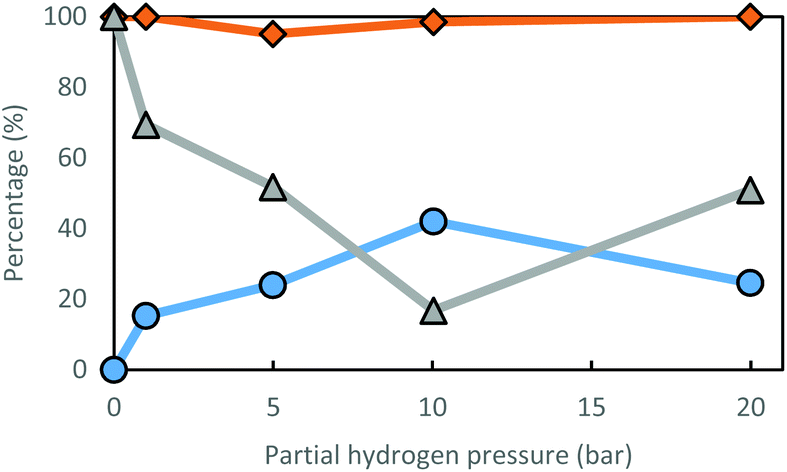 | ||
| Fig. 2 Influence of hydrogen pressure on conversion (○), selectivity for R-amine (◊) and enantiomeric excess (Δ) for the racemization of S-2-aminooctane with Ru3+/Y. Conditions: 85 mM substrate, 2.5 mol% catalyst, 2.0 mL THF, 70 °C, 24 h. | ||
| Solvent | Conv. (%) | Sel. R-amine (%) | ee (%) | Rel. polaritya |
|---|---|---|---|---|
| a Relative polarity as compared to H2O.25 b Methylcyclohexane (MeCy). c Side product is alkylated amine from a side reaction with the solvent. d Relative polarity of MTBE (data on CPME and TAME were not available). e Conversion may be low due to consumption of H2 by anisole in the hydrogenation towards cyclohexyl–methyl ether. | ||||
| MeCyb | 12.2 | 94.9 | 77 | 0.006 |
| iPr2O | 30.5 | 90.7c | 43 | 0.117 |
| CPME | 17.5 | 100 | 65 | ±0.124d |
| TAME | 17.5 | 89.2 | 68 | ±0.124d |
| 1,4-Dioxane | 16.6 | 100 | 67 | 0.164 |
| Anisolee | 4.9 | 100 | 90 | 0.198 |
| THF | 42.0 | 98.5 | 17 | 0.207 |
| t BuOH | 23.8 | 99.5 | 53 | 0.389 |
| iPrOH | 21.5 | 53.6 | 74 | 0.546 |
| EtOH | 15.7 | 23.0 | 92 | 0.654 |
Hydrogenation or hydrogen transfer reactions, catalysed by ionic Ru catalysts, may require an internal or external base for H2 activation as is the case for Shvo's and Noyori-Ikaryia catalysts, among many others.26 Therefore, we also tested the effect of different bases on the racemization reaction in THF (5 equivalents with respect to the substrate) (Table 5). Carbonate bases resulted in a dramatic loss of activity, of more than 90% for Na2CO3 and K2CO3, and 80% for lithium carbonate. Sodium and potassium hydroxide displayed the same trend as the carbonate bases. Addition of LiOH, however, did not lead to a decrease in activity; rather conversion and selectivity remained similar. Next to the quantitative effect on activity, all reactions with addition of inorganic base displayed a clear change in the colour of the solution. Without addition of base, the solution had a dark brown-yellow colour, indicating significant leaching of the Ru into solution. With addition of LiOH, the solution became colourless (see ESI† for visual representation). When analysed with ICP-OES, any leaching of ruthenium species in the absence of base (±44%) was confirmed to be prevented by the addition of LiOH (less than 0.2%).
| Additive | Conv. (%) | Sel. R-amine (%) | ee (%) |
|---|---|---|---|
| — | 42.0 | 98.5 | 17 |
| LiOH | 41.6 | 100 | 17 |
| NaOH | 2.8 | 100 | 94 |
| KOH | 1.0 | 100 | 98 |
| Li2CO3 | 8.8 | 100 | 82 |
| Na2CO3 | 1.1 | 100 | 98 |
| K2CO3 | 4.2 | 100 | 92 |
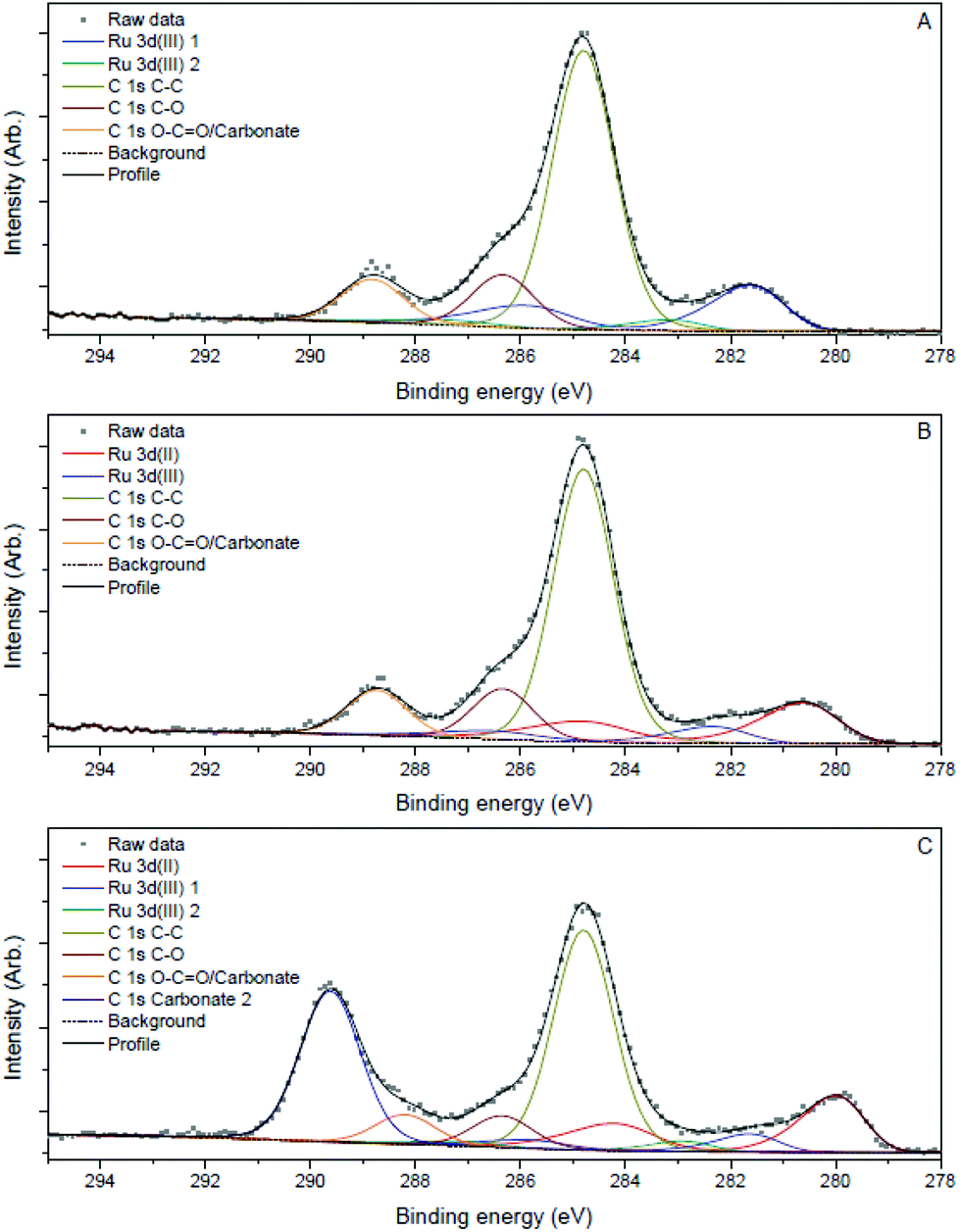 | ||
| Fig. 3 Ru 3d, C 1s XPS spectra of Ru3+/Y catalyst before reaction (A), after reaction (B) and after reaction with addition of LiOH (C). | ||
 | ||
| Fig. 4 TEM images of Ru3+/Y catalyst before reaction (left), after reaction without LiOH (middle) and with 5.0 equiv. of LiOH (right). | ||
Samples of the catalyst after reaction did not display major differences with the catalyst before reaction according to the TEM images; the nanoparticles had slightly increased in size to 2.0 nm and 2.3 nm after reaction without and with LiOH, respectively. However, XPS analysis showed that reduced ruthenium species were present next to Ru(III) after reaction, in contrast with only Ru(III) before reaction (Fig. 3B and C). The signals for ruthenium 3d species shift to lower eV, corresponding with a decrease in oxidation state best attributed to Ru(II) species according to literature.27 The signal for Ru(III) species remains and is visible as a shoulder on the larger Ru(II) signal. This suggests that both ruthenium species are present after reaction.
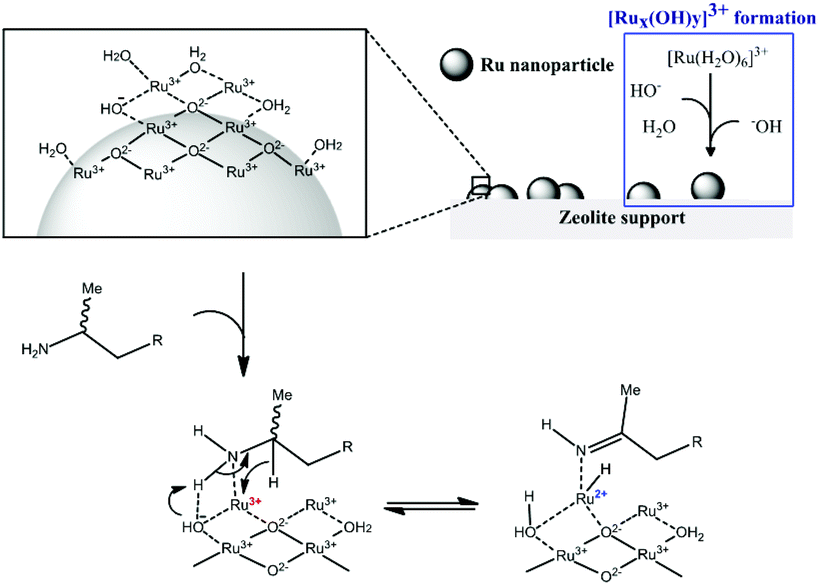 | ||
| Scheme 1 Proposed reaction mechanism for racemization of aliphatic amine with Ru3+/Y catalyst. | ||
The strong interaction between ionic Ru and the amine is indirectly proven by the observation that leaching only occured in the presence of the amine. In order to explain the beneficial effect of added hydroxide on the heterogeneity, one can assume that μ-bridging OH− anions provide the strongest possible bonds between neighbouring Ru centres in the oxyhydroxide clusters; hence extra OH− can firmly fix the Ru ions into the cluster, without affecting their activity.
Kinetic resolution of aliphatic amines
The kinetic resolution of amines is often reported with the Candida Antarctica lipase B enzyme (CalB).31–33 CalB is a robust, thermostable enzyme (R-acylase) with a broad substrate scope; it can be immobilized for continuous operation or extended reuse. Here, we test the applicability of acrylic resin immobilised CalB, also commercially known as Novozyme 435, for the kinetic resolution of the aliphatic amine 2-aminooctane.The stability of CalB in different organic solvents has been investigated before and it was found to decrease with increasing polarity of the solvent.34 Disruption of one of two important hydrogen bonds in the active site results in lowered activity. As shown in Table 6, using iPrOAc as the acylating agent in combination with an apolar solvent, like toluene, results in quantitative amide formation for the kinetic resolution of aliphatic amines, while the yield for resolution in a more polar solvent, like 2-MeTHF, drops to less than half (Table 6). There seems to be no difference between protic and aprotic polar solvent here; polarity seems sufficient to disrupt a critical hydrogen bond or induce denaturation of the enzyme.
| Solvent | Conv. (%) | Sel. R-amide (%) | Sel. N-alkyl amine (%) | ee (%) | Yield R-amide (%) |
|---|---|---|---|---|---|
| Ideal | 50.0 | 100.0 | 0.0 | 100 | 50 |
| THF | 58.1 | 56.1 | 42.5 | 96 | 33 |
| 2-MeTHF | 47.9 | 45.8 | 52.4 | 92 | 22 |
| MeCN | 47.6 | 45.6 | 52.6 | 93 | 22 |
| DCM | 16.3 | 38.6 | 59.7 | 92 | 6 |
| t BuOH | 27.9 | 95.5 | 0.0 | 91 | 27 |
| n-Octane | 74.0 | 71.6 | 21.2 | 82 | 49 |
| Toluene | 57.8 | 94.4 | 0.0 | 89 | 50 |
| MeCy | 72.1 | 75.6 | 18.5 | 86 | 50 |
Another important aspect in kinetic resolution is the resolving agent. Both esters and carbonates have been described in various DKRs,35–37 leading to amide or carbamate products respectively. When choosing a suitable resolving agent, two important factors have to be looked at. First of all, the resolving agent should only react selectively with the reactant as facilitated by the enzyme; an uncatalyzed, aselective reaction is not desired. Secondly, the corresponding leaving group of the resolving agent should be inert after it departs; its further reaction with the product, residual substrate or with the enzyme is not desirable.
In the case of aliphatic amines, carbonate resolving agents resulted in an uncatalyzed, aselective reaction; hence esters were examined more closely as the resolving agent of choice (Fig. 5). With commonly used acylating esters, like vinyl acetate and isopropyl acetate (iPrOAc), a significant alkylation side reaction resulted in N-alkylated-2-aminooctane formation. This alkylation can be prevented when switching to 2-alkoxyacetate esters as acyl donors, which react faster35 than iPrOAc, enabling us to suppress the alkylation. However, both with ethyl 2-methoxyacetate and with isopropyl 2-ethoxyacetate, some aselective acylation was still observed. Finally, employing larger resolving esters, near ideal kinetic resolution conditions were achieved, with selective, enzyme controlled acylation of the substrate amine, especially for isopentyl propionate as the ester of choice.
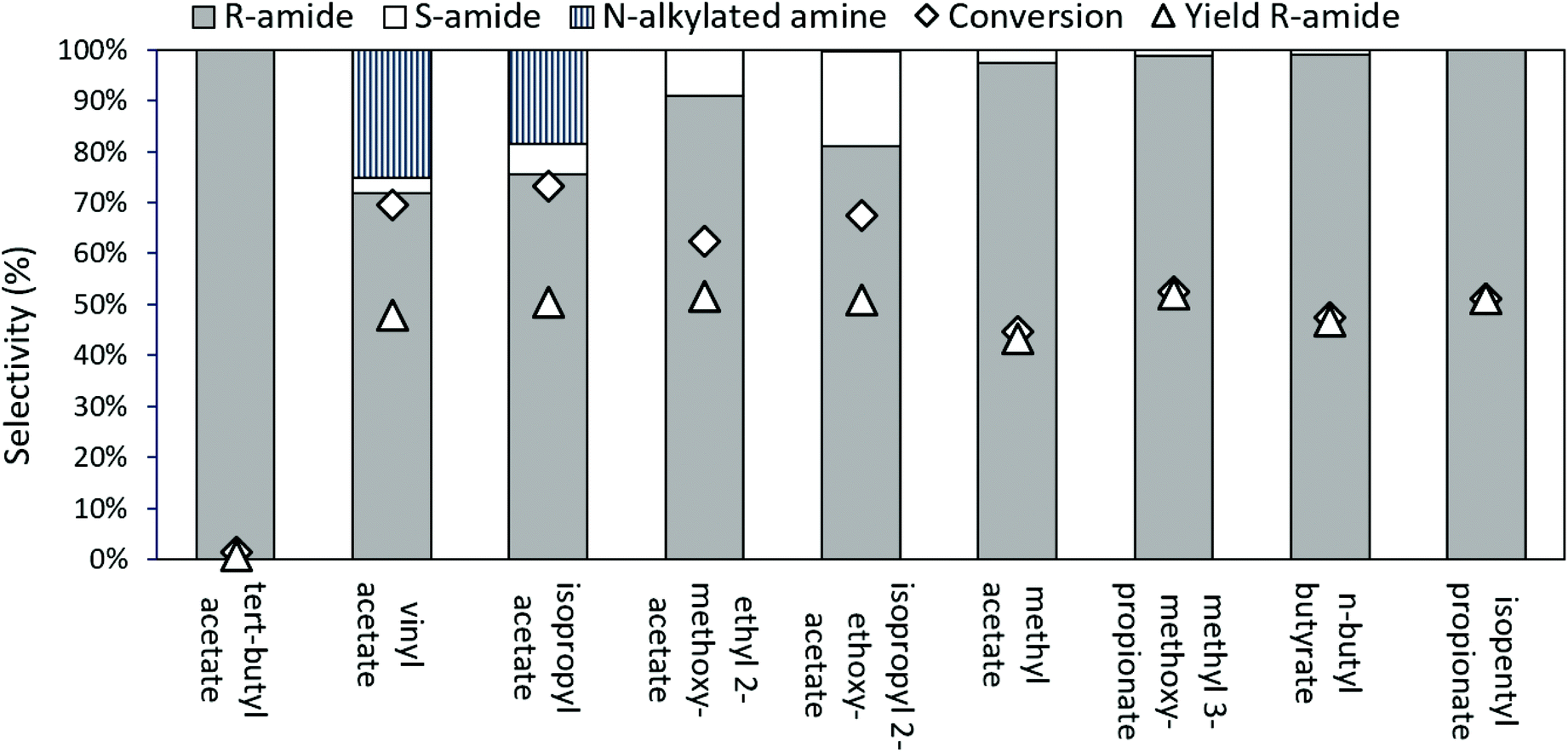 | ||
| Fig. 5 Selectivity, conversion (Δ) and yield of R-amide (◊) for different acylating agents in the kinetic resolution of rac-2-aminooctane with Novozyme 435. Conditions: 85 mM substrate, 50.0 mg Novozyme 435, 0.6 equiv. acylating agent, 2.0 mL cyclohexane, 70 °C, 24 h, 5.0 bar N2. | ||
Since CalB is a lipase, structures that mimic lipids are expected to have a better interaction with the active centre of the enzyme. The larger, more aliphatic acylating esters (such as methyl 3-methoxypropionate, butyl butyrate, or isopentyl propionate) probably fit better at the enzyme's active site than the smaller, more polar ones (such as isopropyl acetate, ethyl 2-methoxyacetate, methyl acetate, etc.). A small but sterically encumbered ester, like tert-butyl acetate may not fit well into the active site of the enzyme, while the small methyl ester might have too much space and consequently lose some activity.
Dynamic kinetic resolution of aliphatic amine
The dynamic kinetic resolution of aliphatic amines can be achieved by combining the findings in this work of both racemization and kinetic resolution. Since the racemization with Ru(III)/Y works best in aprotic polar solvents and kinetic resolution prefers apolar solvents (vide supra), a compromise needs to be found. Therefore, the dynamic kinetic resolution of aliphatic amines was evaluated for different solvents (Table 7). For polar solvents, like THF, the DKR is struggling to even reach the kinetic resolution limit of 50% yield. Much better results are observed when using apolar solvents, like methylcyclohexane (MeCy), exceeding the kinetic resolution limit with good selectivity (>97%) and enantiomeric excess (>95%).| Solvent | Conv. (%) | Sel. R-amide (%) | ee (%) | Yield R-amide (%) |
|---|---|---|---|---|
| Ideal | 100.0 | 100.0 | 100 | 100 |
| tBuOH | 5.9 | 98.8 | 98 | 5.8 |
| THF | 11.0 | 96.4 | 93 | 11 |
| 2-MeTHF | 33.0 | 97.1 | 97 | 32 |
| n-Octane | 62.2 | 94.8 | 94 | 59 |
| iPr2O | 82.0 | 73.6 | 96 | 60 |
| TAME | 63.0 | 96.3 | 95 | 61 |
| CPME | 64.7 | 97.0 | 96 | 63 |
| MeCy | 68.2 | 96.9 | 96 | 66 |
The remaining, unreacted amine is enantio-enriched with the S-enantiomer, suggesting inefficient racemization by the ruthenium catalyst under these conditions. Even with a compromise solvent, like diisopropyl ether (iPr2O) or cyclopentylmethyl ether (CPME), the DKR does not exceed 63% yield. Addition of base to the reaction media is harmful for the enzyme stability; product yield is far below kinetic resolution limit in case extra base is added.
Nevertheless, we have shown the unprecedented applicability of a new heterogeneous ruthenium catalyst for racemization of aliphatic amines at mild temperature and low catalyst loadings. We exceeded the kinetic resolution limit when the catalyst is combined with an enzyme in a dynamic kinetic resolution. Improvements to enzyme stability or catalyst immobilisation may further benefit this DKR system in the future.
Conclusion
In conclusion, we have found a novel heterogeneous ruthenium based catalyst for the racemization of aliphatic amines. The Ru(III)/Y catalyst can selectively racemise aliphatic amines with simple addition of 10 bars of hydrogen, at the comparatively low temperature of 70 °C. Any residual Ru leaching can be controlled by the addition of 5 equivalents of LiOH. Racemization has been shown to be best performed in aprotic polar solvents.Furthermore, a suitable resolving agent, viz. isopentyl propionate, was identified for kinetic resolution of the aliphatic model amine, 2-aminooctane, in order to convert one enantiomer selectively to an enantiopure amide. The enzyme performance of CalB in the kinetic resolution was studied and confirmed to be decreasing with increasing polarity of solvent.
Combination of the findings in racemization and kinetic resolution resulted in a novel dynamic kinetic resolution of aliphatic amines, exceeding the kinetic resolution limit of 50% yield with high selectivity (97%) and enantiomeric excess (96%).
Experimental
All reagents and solvents were purchased from commercial sources and used as delivered unless specified.Supports
Hydroxyapatite and high surface area spinels were synthesized according to literature.22,38Y zeolites, CBV-100 (SAR 5.1), CBV-720 (SAR 30) and CBV-760 (SAR 60), was obtained from Zeolyst and treated with an aqueous 1 M NaNO3 solution for 1 day. After stirring for 1 day at room temperature, the zeolite suspension is centrifuged and washed with deionized water (3×). CBV-720 and CBV-760 zeolites were exchanged to the ammonium form before treatment with NaNO3 by stirring the protic zeolite with 5.0 equivalent (in respect to the protic sites) of 7 M NH3 in methanol for 2 h at room temperature.
All other zeolite supports were treated in a similar manner as CBV-100 unless specified. Mordenite, Beta, ZSM-5 and MCM-22 zeolites were purchased from Zeocat, Sudchemie, Alsi-Penta Zeolithe GmbH and China Catalyst corporation respectively. Template-free Beta (SAR 9.2) and zeolite L synthesis was adapted from literature.39,40
Catalysts
Heterogeneous Ru(0) catalysts were synthesized when not commercially available. Ru(0)/HAP and Ru(0)/spinel were synthesized by wet impregnation of an aqueous RuCl3 solution for 5.0 wt% of ruthenium on support. Water was removed by stirring at room temperature until dry, after which the catalyst was heated to 60 °C in an oven and left overnight. Ruthenium was reduced under hydrogen flow (150 mL H2 min−1) with the following temperature program: 450 °C max. temperature; 2.5 °C min−1 ramp; 5 h at max. temperature.Ru(III)/zeolite catalysts were obtained by adding the zeolite support to an aqueous ruthenium solution with pH correction. RuCl3 hydrate (amount according to desired weight fraction) was dissolved in demineralized water by stirring. After complete dissolution and while stirring, the corresponding amount of zeolite support for the calculated weight fraction was added. After stirring for 15 min, pH levels of the suspension were checked and adjusted to around 7 with a 0.1 M aqueous solution of NaOH. pH levels were checked every 10 min and adjusted until constant. Next, the suspension was left stirring at room temperature for 24 h, after which it was centrifuged and washed thoroughly with demineralized water. The remaining solid was then dried in an oven at 60 °C.
Racemization of S-2-aminooctane
A glass liner is loaded with a stirring bar, 2.5 mol% catalyst, 28.5 μL amine (0.17 mmol; 85 mM), 2.0 mL solvent and 26.1 μL tetradecane (0.10 mmol; 50 mM) as internal standard. The glass liner is capped with a hollow Teflon cap and placed into a stainless steel custom autoclave. Specified gas loading is applied after purging by pressurizing/depressurizing with N2. The loaded autoclave is placed in an aluminium heating block set to the desired temperature and stirred at 500 rpm. The reaction is left stirring for 24 h, after which the autoclave is removed from the heating block and cooled in water. The autoclave is then depressurized and opened for sample recovery. Samples are taken by a syringe and filtered over a 0.20 μm PTFE filter before collection in a GC vial (1.0 mL). A few drops of triethylamine and acetic anhydride are added to the GC samples for better peak separation on GC.Kinetic resolution of rac-2-aminooctane
A glass liner is loaded with a stirring bar, 50.0 mg Novozyme 435 (CalB immobilized on acrylic resin), 28.5 μL amine (0.17 mmol; 85 mM), 0.6 equivalents (0.102 mmol) resolving agent (ester), 2.0 mL solvent and 26.1 μL tetradecane (0.10 mmol; 50 mM) as internal standard. The glass liner is capped with a hollow Teflon cap and placed into a stainless steel custom autoclave. Specified gas loading is applied after purging by pressurizing/depressurizing with N2. The loaded autoclave is placed in an aluminium heating block set to the desired temperature and stirred at 100 rpm. Reaction is left stirring for 24 h, after which the autoclave is removed from the heating block and cooled in water. The autoclave is then depressurized and opened for sample recovery. Samples are taken by syringe and filtered over a 0.20 μm PTFE filter before collection in a GC vial (1.0 mL).Dynamic kinetic resolution of rac-2-aminooctane
A glass liner is loaded with a stirring bar, 2.5 mol% catalyst, 50.0 mg novozyme 435 (CalB immobilized on acrylic resin, >5000 U g−1), 28.5 μL amine (0.17 mmol; 85 mM), 1.2 equivalents (0.204 mmol) resolving agent (ester), 2.0 mL solvent and 26.1 μL tetradecane (0.10 mmol; 50 mM) as internal standard. The glass liner is capped with a hollow Teflon cap and placed into a stainless steel custom autoclave. Specified gas loading is applied after purging by pressurizing/depressurizing with N2. The loaded autoclave is placed in an aluminium heating block set to the desired temperature and stirred at 100 rpm. Further reaction workup was performed as for the kinetic resolution (vide supra).Methods and machines
Conversion, selectivities, enantiomeric excesses and yields were calculated from GC data obtained with a CP-CHIRASIL-DEX CB chiral column (25 m), equipped with FID detector. Tetradecane was used as internal standard and effective carbon numbers were applied. Samples were run in parallel on a GC-MS from Agilent 6890-N with a 30 m HP-5MS column and Agilent 5973 mass-spectrometer.Leaching of ruthenium catalysts was quantified with ICP-OES using a Varian 720-ES machine equipped with a double-pass glass cyclonic spray chamber, a Sea Spray concentric glass nebulizer and a high solids torch. Samples were prepared by collection of the filtrate of reaction, removal of organics by rotary evaporation and subsequent digestion in a 3:1 solution of concentrated HCl and HNO3 (aqua regia).
Experimental information of XRD and XPS can be found in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Simon Smolders and Carlos Marquez for assistance with XRD and ICP-OES measurements. K. A. thanks Rik Verschueren for proofreading the manuscript. D. D. V. acknowledges the FWO for research project funding, the Flemish Government for long-term structural funding through the Methusalem program, and EoS (FWO-FNRS) for financial support. J. V. thanks the FWO for funding. C. V. G. and I. V. thank the Hercules fund for financial aid of project AKUL/13/19. W. T. and S. E. acknowledge financial support through the Accelerate3 project from the Interreg Vlaanderen-Nederland program and Flanders Innovation & Entrepreneurship and from KU Leuven internal grant 3E180424. W. T. also acknowledges support from the province West-Vlaanderen for his Provincial Chair in Advanced Materials.References
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- L. A. Nguyen, H. He and C. Pham-Huy, Int. J. Biomed. Sci., 2006, 2, 85–100 CAS.
- C. Moberg, Acc. Chem. Res., 2016, 49, 2736–2745 CrossRef CAS PubMed.
- P. O. Carvalho, Q. B. Cass, S. A. Calafatti, F. J. Contesini and R. Bizaco, Braz. J. Chem. Eng., 2006, 23, 291–300 CrossRef CAS.
- A. Parvulescu, J. Janssens, J. Vanderleyden and D. De Vos, Top. Catal., 2010, 53, 931–941 CrossRef CAS.
- M. Rachwalski, N. Vermue and F. P. J. T. Rutjes, Chem. Soc. Rev., 2013, 42, 9268–9282 RSC.
- F. F. Huerta, A. B. E. Minidis and J. E. Bäckvall, Chem. Soc. Rev., 2001, 30, 321–331 RSC.
- A. N. Parvulescu, P. A. Jacobs and D. E. De Vos, Chem. – Eur. J., 2007, 13, 2034–2043 CrossRef CAS.
- J. Paetzold and J. E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 17620–17621 CrossRef CAS.
- K. Engström, E. V. Johnston, O. Verho, K. P. J. Gustafson, M. Shakeri, C. W. Tai and J. E. Bäckvall, Angew. Chem., Int. Ed., 2013, 52, 14006–14010 CrossRef.
- M.-J. Kim, W.-H. Kim, K. Han, Y. K. Choi and J. Park, Org. Lett., 2007, 9, 1157–1159 CrossRef CAS.
- A. N. Parvulescu, P. A. Jacobs and D. E. De Vos, Adv. Synth. Catal., 2008, 350, 113–121 CrossRef CAS.
- L. K. Thalén, D. Zhao, J.-B. Sortais, J. Paetzold, C. Hoben and J.-E. Bäckvall, Chem. – Eur. J., 2009, 15, 3403–3410 CrossRef PubMed.
- O. Verho and J. E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 3996–4009 CrossRef CAS PubMed.
- Y. Shvo, D. Czarkie, Y. Rahamim and D. F. Chodosh, J. Am. Chem. Soc., 1986, 108(23), 7400–7402 CrossRef CAS.
- A. N. Parvulescu, P. A. Jacobs and D. E. De Vos, Appl. Catal., A, 2009, 368, 9–16 CrossRef CAS.
- Q. Yang, F. Zhao, N. Zhang, M. Liu, H. Hu, J. Zhang and S. Zhou, Chem. Commun., 2018, 54, 14065–14068 RSC.
- S. Gastaldi, S. Escoubet, N. Vanthuyne, G. Gil and M. P. Bertrand, Org. Lett., 2007, 9, 837–839 CrossRef CAS PubMed.
- C. Wang, X. Wu and J. Xiao, Chem. – Asian J., 2008, 3, 1750–1770 CrossRef CAS.
- C. Saluzzo and M. Lemaire, Adv. Synth. Catal., 2002, 344, 915–928 CrossRef CAS.
- Y. Ahn, S. B. Ko, M. J. Kim and J. Park, Coord. Chem. Rev., 2008, 252, 647–658 CrossRef CAS.
- J. Guo, H. Lou, H. Zhao, X. Wang and X. Zheng, Mater. Lett., 2004, 58, 1920–1923 CrossRef CAS.
- S. Agarwal and J. N. Ganguli, RSC Adv., 2014, 4(23), 11893 RSC.
- A. I. Biaglow, J. Šepa, R. J. Gorte and D. White, J. Catal., 1995, 151(2), 373–384 CrossRef CAS.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley, Weinheim, 2002 Search PubMed.
- C. P. Casey and J. B. Johnson, J. Am. Chem. Soc., 2005, 127, 1883–1894 CrossRef CAS PubMed.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- C. P. Casey, S. W. Singer, D. R. Powell, R. K. Hayashi and M. Kavana, J. Am. Chem. Soc., 2001, 123(6), 1090–1100 CrossRef CAS.
- M. Wills, Mod. Reduct. Methods, 2008, 271–296 CAS.
- K. Kaneda, K. Mori, T. Hara, T. Mizugaki and K. Ebitani, Catal. Surv. Asia, 2004, 8(4), 231–239 CrossRef CAS.
- C. Ortiz, Ma. L. Ferreira, O. Barbosa, J. C. S. dos Santos, R. C. Rodrigues, Á. Berenguer-Murcia, L. E. Briand and R. Fernandez-Lafuente, Catal. Sci. Technol., 2019, 9, 2380–2420 RSC.
- A. Tomin, G. Hornyánszky, K. Kupai, Z. Dorkó, L. Ürge, F. Darvas and L. Poppe, Process Biochem., 2010, 45, 859–865 CrossRef CAS.
- Z. Boros, D. Weiser, M. Márkus, E. Abaháziová, Á. Magyar, A. Tomin, B. Koczka, P. Kovács and L. Poppe, Process Biochem., 2013, 48, 1039–1047 CrossRef CAS.
- C. Li, T. Tan, H. Zhang and W. Feng, J. Biol. Chem., 2010, 285, 28434–28441 CrossRef CAS PubMed.
- M. Oláh, Z. Boros, G. Hornyánszky and L. Poppe, Tetrahedron, 2016, 72, 7249–7255 CrossRef.
- C. E. Hoben, L. Kanupp and J. E. Bäckvall, Tetrahedron Lett., 2008, 49, 977–979 CrossRef CAS.
- A. Parvulescu, D. De Vos and P. Jacobs, Chem. Commun., 2005, 5307 RSC.
- A. Peeters, L. Claes, I. Geukens, I. Stassen and D. De Vos, Appl. Catal., A, 2014, 469, 191–197 CrossRef CAS.
- B. Yilmaz, U. Müller, M. Feyen, S. Maurer, H. Zhang, X. Meng, F.-S. Xiao, X. Bao, W. Zhang, H. Imai, T. Yokoi, T. Tatsumi, H. Gies, T. De Baerdemaeker and D. De Vos, Catal. Sci. Technol., 2013, 3, 2580 RSC.
- J. Verduijn, U.S., Patent5242, 1993, 675 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc02615k |
| This journal is © The Royal Society of Chemistry 2020 |