
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDissolving used rubber tires†
Sijia
Zheng
,
Mengchen
Liao
,
Yang
Chen
and
Michael A.
Brook
 *
*
Department of Chemistry and Chemical Biology, McMaster University, 1280 Main St W, Hamilton, ON, Canada L8S 4M1. E-mail: mabrook@mcmaster.ca
First published on 25th November 2019
Abstract
Used automobile tires present an enormous environmental burden. Efficient methods for degradation of the sulfur crosslinks in organic elastomers have proven elusive. We show that the reductive silylation of RS-SR bonds to silyl thio ethers RSSiR′′3 in up to 90% yield using a variety of hydrosilicones occurs in the presence of <1 mol% B(C6F5)3 for model compounds. Sulfur-cured automotive rubber required 10 wt% catalyst for efficient sulfide cleavage. At temperatures ranging from room temperature to 100 °C recoveries of organic polymers as oils from tires using this one step process ranged from 56% for complex mixtures of rubber crumb from ground tires to 93% for butyl rubber (bicycle inner tubes; 87% yield at 100 °C over 30 minutes). After removal of inorganic materials by simple filtration, the recovered polymeric oils were radically or oxidatively crosslinked to generate new elastomers that can be optionally reinforced with the solids recovered in the initial reduction procedure. This mild process constitutes a facile route to reutilize the organic polymers found in automobile and other sulfur-crosslinked rubbers.
Introduction
The radically induced1 vulcanization of alkene-containing hydrocarbon polymers, reported by Goodyear in 1844,2 is a technological advancement that remains an integral part of modern life; sales of automobile tires prepared using this process are expected to reach 3 billion units in 2019.3 The elastomer products, crosslinked with sulfur oligomers, are incredibly robust products, which pose a major challenge; they are far too stable to be readily recycled.Automobile tires exemplify polymers derived from fossil fuels that are destined for single use. Used tires constitute a significant environmental burden, particularly because of the scale of production.4 In large part, used tires are simply placed in stockpiles,5 from which leaching into the environment of their many (toxic) constituents occurs.6 Dangerous, highly polluting, difficult-to-arrest tire fires at such storage facilities are not uncommon.7
Some automotive rubber is exploited as fuel in the cement industry; capturing SO2 during combustion may be problematic. Some tires are turned into crumb and used as fillers,8 for example, in asphalt, cement or turf replacements from which, however, leaching of contaminants may still occur.9 There is a longstanding need to recover the organic materials from tires for sustainable reuse. Although the S–S bond strength is only ∼280 kJ mol−1,10 practicable processes for S–S cleavage in vulcanized tires have not been reported. Aggressive chemical approaches, for example, reactive reduction with LiAlH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11,12 or amines13 have not proven commercially viable. Reuse strategies therefore typically involve energetically intensive, relatively inefficient pyrolytic conversion into fuel gas, low grade carbon black and other low value materials.14 The world is in dire need of new efficient methods for recycling waste tire rubber, especially in ways that allow the recovery and reuse of the basic building blocks.
11,12 or amines13 have not proven commercially viable. Reuse strategies therefore typically involve energetically intensive, relatively inefficient pyrolytic conversion into fuel gas, low grade carbon black and other low value materials.14 The world is in dire need of new efficient methods for recycling waste tire rubber, especially in ways that allow the recovery and reuse of the basic building blocks.
The weak Si–H bond in hydrosilanes makes them excellent reducing agents, particularly when strong Si–heteroatom bonds are formed in the process.15 Thus, reductive hydrolysis/alcoholysis16 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydrosilylation,17 among others, are efficient processes. Key to the work described here is the Lewis acid-catalyzed (typically B(C6F5)3 = BCF) reduction of carbonyls (Fig. 1a), ethers, silanols, alkoxysilanes (the Piers–Rubinsztajn reaction18–20Fig. 1b), benzylic sulfides, and thioacetals (Fig. 1c) using HSi functional groups.21 These reactions are normally easy to control, often work at room temperature, and the main experimental issues are associated with managing the co-products when they are flammable gases, including hydrogen or alkanes.
O hydrosilylation,17 among others, are efficient processes. Key to the work described here is the Lewis acid-catalyzed (typically B(C6F5)3 = BCF) reduction of carbonyls (Fig. 1a), ethers, silanols, alkoxysilanes (the Piers–Rubinsztajn reaction18–20Fig. 1b), benzylic sulfides, and thioacetals (Fig. 1c) using HSi functional groups.21 These reactions are normally easy to control, often work at room temperature, and the main experimental issues are associated with managing the co-products when they are flammable gases, including hydrogen or alkanes.
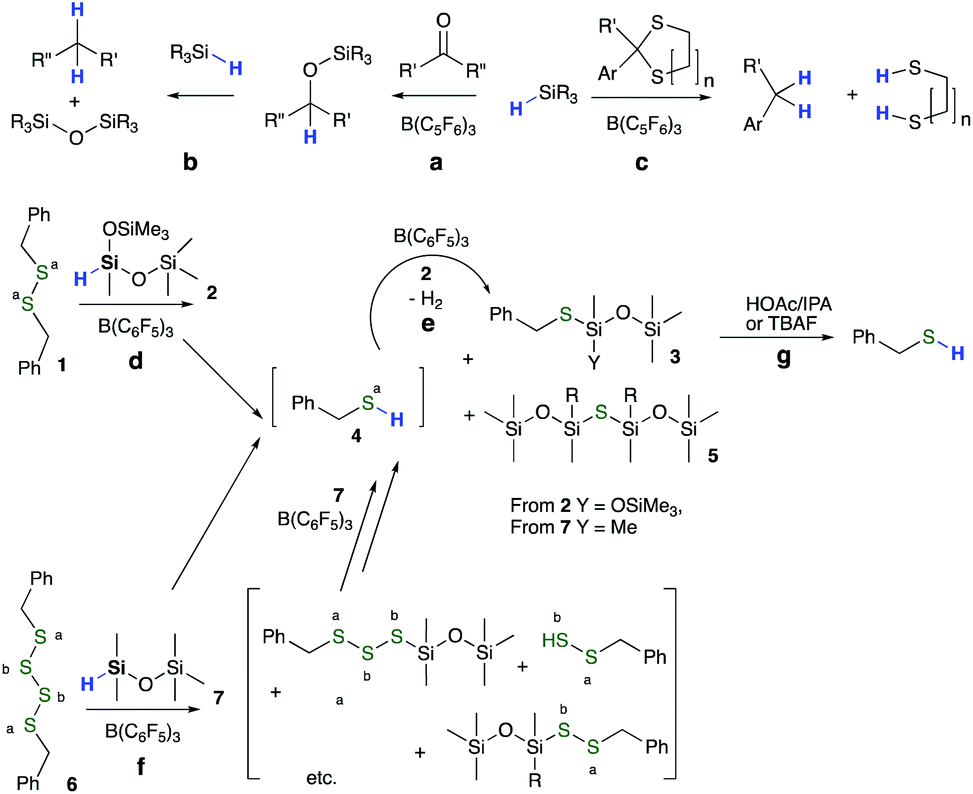 | ||
| Fig. 1 Selected examples of B(C6F5)3-catalyzed reduction by SiH groups. (a) Carbonyl reduction to silyl ethers; (b) silyl ether reduction to alkanes (Piers-Rubinsztajn); (c) aromatic thioacetal cleavage to alkanes; (d) disulfide cleavage to silyl thio ethers (this work); (e) reduction of thiols to silyl thio ethers; (f) silyl thiol ether formation from oligosulfides, and, (g) Si–S cleavage using acid or fluoride catalysis. | ||
We report that, catalyzed by B(C6F5)3, hydrosilanes effectively reduce S–S bonds of model organic disulfides, tetrasulfides and, more importantly, complex sulfur-crosslinked solid automotive rubbers in the forms of bicycle inner tubes, solid tires or tire crumb in good to excellent yield. The products are polymeric, silyl-protected thiolated organic oils that are readily separated from the accompanying, unreactive solids, such as fillers, fiber and metal reinforcements, pigments, etc. simply by filtration or centrifugation. The products, sulfur-containing polymeric oils, may be converted back into (reinforced) rubbers using simple oxidative or radical processes.
Results
Model reductions of dibenzyl disulfide and tetrasulfide
Model reductions of dibenzyl disulfide 1 (n = 1) were undertaken with bis(trimethylsiloxy)methylsilane 2 (HSiMe(OSiMe3)2, MDHM) (the common nomenclature used for silicones is Q: SiO4/2; T: MeSiO3/2; D: MeSiO2/2 and M: Me3SiO∼) as reducing agent in the presence of BCF (Fig. 1d, e, Fig. 2, Table S1, and Fig. S1, ESI†). With less than one equivalent of hydrosiloxane, residual starting material and only product 3 were recovered, demonstrating that the reaction of SiH with S–H, e.g., in compound 4, is faster than that with S–S bonds. Complete reduction of 1 → 3 required 2 equiv. of the hydrosiloxane and occurred in 90% yield using only 0.8 mol% BCF; the other sulfur-based product 5 was removed under reduced pressure. Reduction of the analogous tetrasulfide 6 to 3 using HSiMe2OSiMe37 required five equivalents of MDHM and demonstrated that both C–Sa–Sa and Sa–Sb–Sb linkages undergo efficient reductive silylation (Fig. 1f, 2 and Fig. S2, ESI†).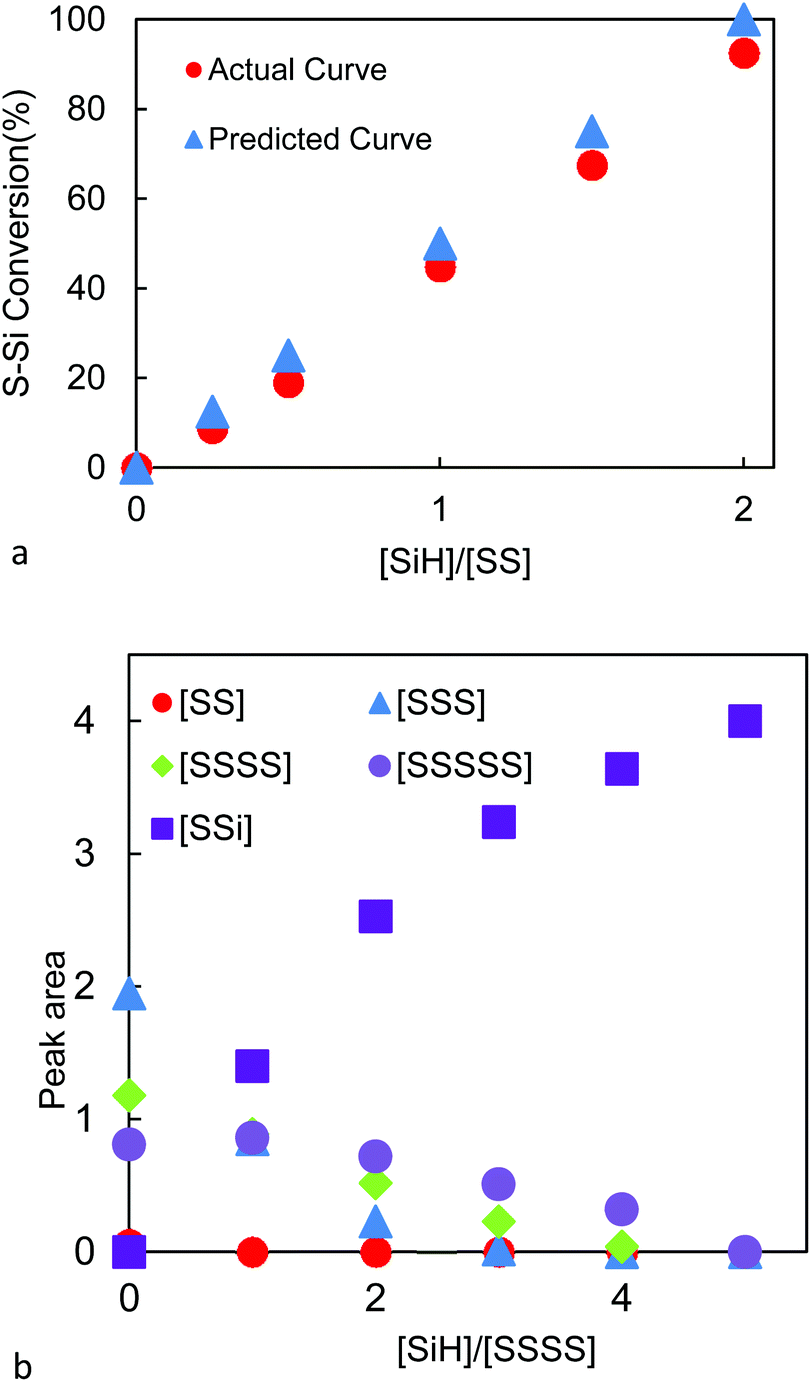 | ||
| Fig. 2 (a) Disulfide conversion in benzyl disulfide system versus [SiH]/[SS] using MDHM. (b) Polysulfide conversion in benzyl tetrasulfide system versus [SiH]/[SSSS] using MDHM. For integration data, see Table S2.† | ||
Reduction of used automotive rubbers
Automotive tires contain a complex variety of constituents, including (spent) catalysts for their formation, antioxidants, colorants, particulate reinforcing agents like carbon black and/or organosulfur-modified silica, and fibrous reinforcing agents including nylon cord and woven steel.14 Holding together this complex assortment of excipients is the sulfur-crosslinked elastomer. We reasoned that the (oligo)sulfide linkages in rubber tires could be reductively cleaved using hydrosilanes in analogy to the reactions with the oligosulfide model compounds (Fig. 1d–f).Scrap rubber from automobile tires is available in large quantity in the form of ‘rubber crumb’. It is formed by shredding tires from multiple sources to remove metal wires and polyester cord and grinding the resulting product to various crumb sizes. The typical organic constituents in crumb mixtures include isobutene isoprene (IIR), butadiene rubber (BR), styrene–butadiene rubber (SBR), isoprene rubber (IR) and natural rubber (NR). Thermal degradation profiles allow one to determine the constitution of sulfur-crosslinked elastomers; depending on their structure, the polymers degrade between 300–485 °C;22,23 (Fig. S5, and S6, Table S3†) while inorganic carbon (carbon black) thermally decomposes from 560–800 °C in oxygen.24 In the rubbers tested, the organic rubber content was approximately 60 wt% (Table 1, Fig. S11†).
| Rubber | Rubber constituentsa | Components | ||
|---|---|---|---|---|
| Organic (%) | Carbon black (%) | Other solids (%) | ||
| a EPDM terpolymer of ethylene, propylene and diene, IR/NR isoprene rubber/nature rubber, IIR isobutene isoprene rubber, BR butadiene rubber. Constituents were determined by a combination of TGA, which showed separate decomposition temperatures for IIR and IR/NR, and 1H NMR. | ||||
| EPDM | EPDM | 55.1 | 26.4 | 18.5 |
| Inner tube | 61.1 | 33.0 | 5.9 | |
| Truck tread -1 | IR/NR | 62.2 | 28.4 | 9.4 |
| Truck tread -2 | IR/NR | 65.0 | 23.0 | 12.0 |
| Tread (snow tire) | IR/NR + BR | 63.0 | 22.9 | 14.1 |
| Side wall (snow tire) | IIR + IR/NR + BR | 59.4 | 22.2 | 18.4 |
| Crumb-1 | BR + EPDM | 47.6 | 44.6 | 8.8 |
| Crumb-2 | IIR + PBD | 61.6 | 31.5 | 6.9 |
Two separate sources of commercial crumb were compared for their reactivity under the reducing silylation conditions. Much more BCF catalyst (10 wt% compared to the rubber starting material) was required to achieve reasonable yields of reduction with rubbers than with the model compounds (<1 mol%), which is not surprising given the complexity of the mixed rubber starting materials and the fact that they have been exposed to degradation and various environments during use.
With 7 as reducing agent, organic oils were recovered in 36% yield from one crumb rubber source and a moderate 56% yield from another (2 lots, Fig. 3g). Use of second and third reduction steps with fresh catalyst and hydrosilane significantly improved overall conversion of elastomer to oils (steps (1) 56% → (2) +29.6% → (3) +2.5% (total 88% organic recovery) – the inorganic constituents were removed by centrifugal separation (Tables 2 and 3, Fig. S14†)). In retrospect, in a practical sense, only the improvement in overall efficiency of the second step could be justified. The reduction process was readily visible by eye, as black dispersions were converted to yellow oils (Fig. 3b → f,g, Fig. S7 and S8, ESI†).
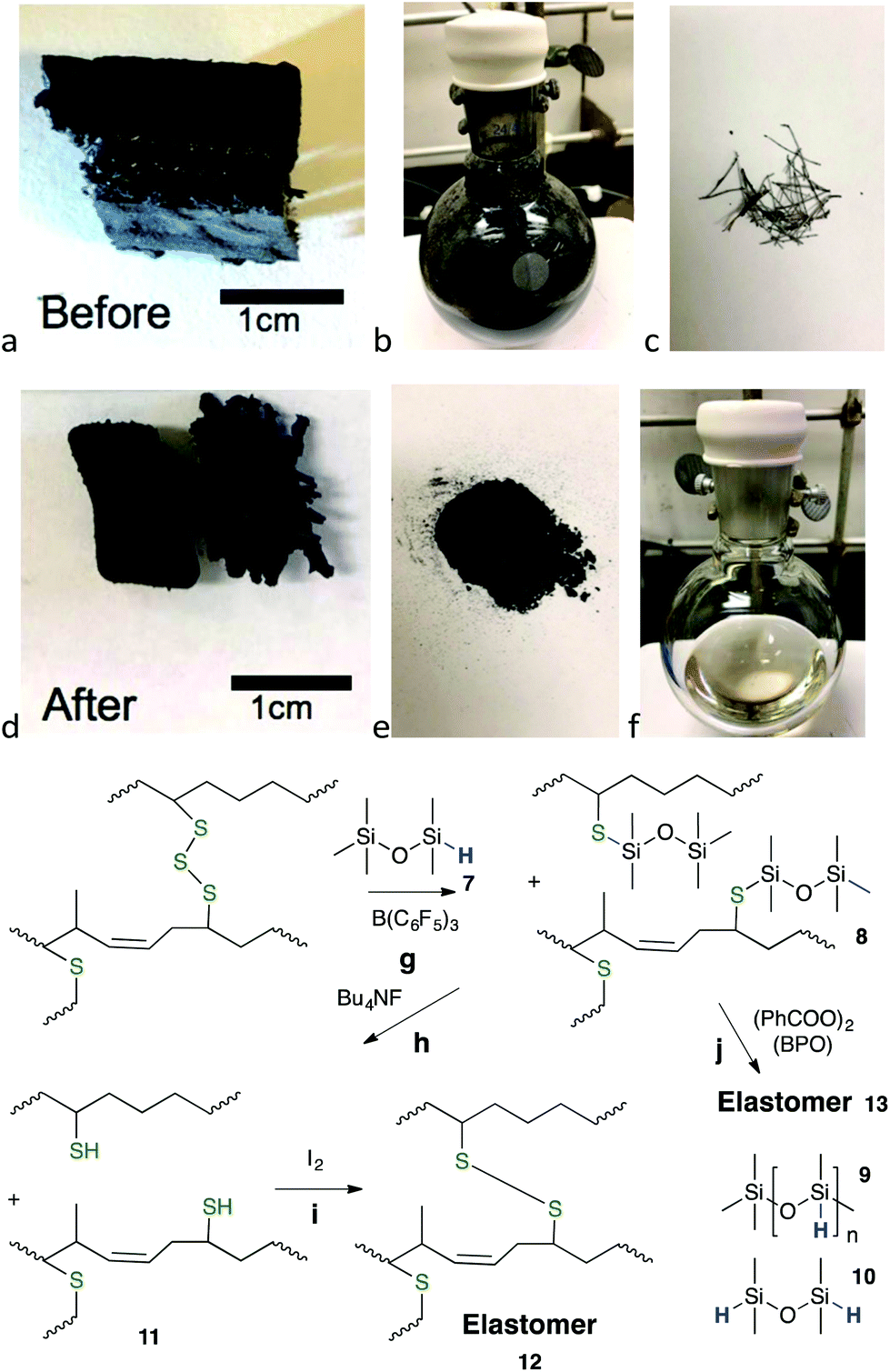 | ||
| Fig. 3 (a) Section of snow tire tread cross-section, unreacted. (b) Reductive dispersion using 7 and 10 wt% BCF after 6 days. (c) Metal wires that self-affixed to the magnetic stir bar. (d) Shrunken sections after reaction. (e) Inorganic and unreacted rubber constituents isolated by gravity filtration. (f) Product silyl-protected thiolated organic oils 8 in toluene (56% yield; note: the recovered oil may be a darker color, depending on starting material and the specific reaction, Fig. S7†). (g) Reductive silylation (a → d, e, f). (h) Cleavage of SiS bonds. (i) Oxidative coupling produces elastomers crosslinked by disulfides. (j) Radicals form elastomers from alkene-containing oils. Steps i and j can optionally use recovered inorganic constituents as reinforcing agents in the elastomers. | ||
| Rubber | Rubber constituentsb | Starting components | Yieldc (%) | ||
|---|---|---|---|---|---|
| Organic (%) | Carbon black (%) | Other solids (%) | Organic oil | ||
| a Experimental conditions: Rubber (300 mg); B(C6F5)3 (30 mg, 10 wt%), 12 mL toluene 7 (1.5 mL) at 60 °C for 48 h. b Constituents were determined by a combination of TGA, which showed separate decomposition temperatures for IIR and IR/NR, and 1H NMR. EPDM = terpolymer of ethylene, propylene and diene, IR/NR isoprene rubber or natural rubber, IIR isobutene isoprene rubber, BR butadiene rubber. c Organic yield = (total organic-recovered organic)/total organic × 100. Organic composition established using TGA (Fig. S6, S9, S11, Table S3,† and Table 1). d Cumulative yield for 3 process steps. e Process utilized hydrosilicone 10 (1.5 mL) at 100 °C for 30 min. f External road contacting component only. g Cross-section of entire tread from core to external surface. | |||||
| Crumb-1 | BR + IR/NR + EPDM | 47.6 | 44.6 | 8.8 | 35.8 |
| Crumb-2 | IIR + BR | 61.6 | 31.5 | 6.9 | 56.2 |
| Repeat process with 1st product | 85.8 | ||||
| Repeat process with 2nd product | 88.3d | ||||
| EPDM | EPDM | 55.1 | 26.4 | 18.5 | 60.4 |
| Inner tube | IIR | 61.1 | 33.0 | 5.9 | 92.9 |
| 100 °C 30 mine | 87.0 | ||||
| Truck treadf | IR/NR | 65.0 | 23.0 | 12.0 | 89.8 |
| Tread (snow tire)g | IR/NR + BR | 63.0 | 22.9 | 14.1 | 51.8 |
| Side wall (snow tire) | IIR + IR/NR + BR | 59.4 | 22.2 | 18.4 | 53.8 |
| Starting rubber componentsb | Parameters | Results | ||||||
|---|---|---|---|---|---|---|---|---|
| Sample (mg) | Form | Organic (mg) | Inorganicc (mg) | Hydrosilanes (mL) | Temperature (°C) & Time (h) | Organic yieldd (%) | Residual solide (mg) | |
| a Experimental conditions: B(C6F5)3/rubber = 10 wt%, 12 mL toluene. b Based on TGA. c Includes carbon black and thermally stable inorganic moieties. d Fraction of available elastomer converted into organic soluble oils. e Residual solid contains carbon black, inorganics and residual polymeric rubber. f 49.0 mg of residual powder and 188 mg of residual coupon still containing elastomer (Fig. S10†). g Process for degradation of Crumb-2 using multiple reduction cycles; 169.0 mg of starting material were used in the 2nd reduction and 65.0 mg in the 3rd (Fig. S14b†). h The rubber powder was allowed to swell in reaction mixture for 3 h before adding B(C6F5)3 catalyst. i Catalyst solution was added immediately after addition of hydrosilane. | ||||||||
| Crumb 1 (300) | Powder | 143 | 157 | 7 (1.5) | 60/48 | 35.8 | 206.7 | |
| Crumb 2 | Firstg | Powder | 185 | 115 | 7 (1.5) | 60/48 | 56.2 | 182.9 |
| Secondg | Powder | 80.9 | 102 | 7 (1.5) | 60/48 | 85.8 | 74.0 | |
| Thirdg | Powder | 26.3 | 47.7 | 7 (1.5) | 60/48 | 88.3 | 64.0 | |
| EPDM (300) | Powder | 165 | 135 | 7 (1.5) | 60/48 | 60.4 | 162.0 | |
| Inner Tube (300) | Coupon | 183 | 117 | 7 (1.5) | 60/48 | 62.4 | 49.0 + 118.0f | |
| Inner Tube (300) | Powder | 183 | 117 | 7 (1.5) | 60/48 | 92.9 | 112.7 | |
| Inner Tube (2000) | Powder | 1222 | 778 | 7 (10.0) | 60/48 | 89.1 | 821.0 | |
| Inner tube (300) | Powderh | 183 | 117 | 7 (1.5) | 100/0.5 | 79.0 | 150.0 | |
| Inner tube (300) | Powderh | 183 | 117 | 7 (1.5) | 100/2 | 80.3 | 143.4 | |
| Inner tube (300) | Powderh | 183 | 117 | 7 (1.5) | 100/4 | 81.5 | 142.3 | |
| Inner tube (300) | Powderh | 183 | 117 | 7 (1.5) | 100/10 | 85.0 | 122.0 | |
| Inner tube (300) | Powderh | 183 | 117 | MHMH (1.5) | 100/0.5 | 87.0 | 151.0 | |
| Inner tube (300) | Powderi | 183 | 117 | MHMH (1.5) | 100/0.5 | 85.0 | 151.0 | |
| Truck Tread 1 (300) | Powder | 187 | 113 | MMH (1.5) | 60/48 | 88.3 | 109.5 | |
| Truck Tread 1 (300) | Powder (Soxhlet) | 175 | 125 | MMH (1.5) | 60/48 | 87.6 | 115.1 | |
| Truck Tread 1(2000) | Powder | 1244 | 756 | MMH (10.0) | 60/48 | 84.6 | 861.0 | |
| Truck Tread 2 (300) | Powder | 195 | 105 | 7 (1.5) | 60/48 | 89.8 | 109.0 | |
| Tread (snow tire) (300) | Powder | 189 | 111 | 7 (1.5) | 60/48 | 51.8 | 180.0 | |
| Side wall (snow tire) (300) | Powder | 178 | 122 | 7 (1.5) | 60/48 | 53.8 | 183.0 | |
Improved recoveries of organic polymers were observed with single composition rubbers. For example, about 60% of an EPDM elastomer (ethylene propylene diene terpolymer, ‘pond liner’) was converted to a soluble oil using reductive silylation with 7 and B(C6F5)3 in toluene at 60 °C. Automotive rubbers were efficiently reduced to oils in one step in yields ranging from 52–93%. These included: IIR from a (used) bicycle inner tube; IR/NR from the outermost section of truck tire tread; a mixture of IIR, IR/NR and BR from a snow tire side wall; and IR/NR, BR from a snow tire tread (Tables 2 and 3). The process is easily seen from the reduction of a bulk section of snow tire tread (Fig. 3a). Shortly after the reduction reaction started, the reinforcing steel wires separated from the bulk rubber and were collected on the magnetic stir bar (Fig. 3c); accompanied by particulate formation to give a black dispersion (Fig. 3b) – the bulk rubber underwent shrinkage, but not complete disintegration (Fig. 3d, Fig. S19†). Filtration allowed separation of a black solid mass comprised primarily of inorganic excipients and a yellow solution of silylated organic oils in toluene (Fig. 3e, f, Fig. S7 and S18†).
Oligosulfides were converted into silyl thiol ethers during reduction (Fig. 3g and Fig. S7†). Therefore, the residual solids and recovered non-volatile liquids often exhibited a weight gain when compared to the starting rubber mass (Table S4†). The product oils 8 (Fig. 3, for 1H NMR data, see Fig. S15†) typically exhibited a bimodal distribution of molecular weight, with a low fraction centered near 10000 g mol−1 and a broad peak centered near 1 million g mol−1 (Table S5, Fig. S16†).
Several factors were manipulated to improve the efficiency of the reduction process. A variety of hydrosilicones are commercially available that vary in the density of SiH groups. Model studies on the reduction of the organic sulfides or automotive rubbers were undertaken with 2 or 7, respectively, because the use of small molecules facilitated characterization of the reaction products. Either compound is too expensive for practical use. Attempts to facilitate reduction of rubbers with the inexpensive, high SiH density polymer Me3Si(OSiMeH)nSiMe39 were unsuccessful (Fig. S17†) because the silicone product of the reduction is a network polymer, which led to the formation of intractable tars. By contrast, the use of inexpensive, high SiH density HMe2SiOSiMe2H 10 led to efficacious, rapid reduction of rubbers (Tables 2 and 3, Fig. S17†).
Unlike the model compounds above, relatively large quantities of the BCF catalyst, 10 wt% against the rubber, were required for the reduction of rubbers to occur efficiently. Pre-swelling the rubber in commonly used organic solvents like acetone,25 or an initial Soxhlet extraction using acetone to remove potential catalyst inhibitors, e.g., amines, free sulfur, acetone soluble colorants, antioxidants, processing rubber additives, etc., did not appreciably increase either the rate or yield of the reduction (Table 3, Fig. S14†). We continue to work on process optimization to reduce the quantities of catalyst required.
Several other factors were found to the affect the efficiency of the reduction of rubbers, including surface area, process temperature and reducing agent. Unsurprisingly, the reduction of bulk elastomers, including cylindrical sections of bicycle inner tube (diameter ∼16.9 mm, thickness ∼0.82 mm, IIR, Fig. S7†), and a cross-section of an automobile snow tire (IR/NR + BR, Fig. 3), were slow to occur and low yielding at 60 °C using 7 (Fig. S6†). In both cases, the objects underwent significant shrinkage (Fig. S19†), and increased crosslink density as shown by an increase in Young's modulus (Table S4†), but maintained their shape. Cryogenic grinding of the starting rubbers to increase rubber surface area (to particle size ∼330 μm, Fig. 4) led to significant enhancements in yield; an increase from 52 to 93% was observed in the case of the inner tube (IIR, Table 3).
 | ||
| Fig. 4 (a) The right front tire was removed from a toy car. (b) A silicone mold of the tire. (c) Organic oil 8 was prepared by reduction of truck tire tread (Sailun, IR/NR) with hydrosilane 7. (d) Silicone mold filled with 0.707 g 8 + 1 wt% BPO + 0.3010 g residual solid. (e) New tire after curing. (f) Tire replacement. (g) The residual solid during the preparation of 8 could be included in the pre-elastomer formulation. (h) Close up showing: (i) the original tire, (ii) tire made without additional inorganic excipients 13, and (iii) tire including inorganic excipients. | ||
The tire reductive silylation studies were initially undertaken using relatively mild temperatures because literature suggests that the B(C6F5)3 catalyst undergoes degradation at temperatures above 80 °C in the presence of moisture.26 Current studies with rubber reduction, however, showed this not to be problematic. A 93% yield of recovered organic polymer (IIR from inner tube) was achieved at 100 °C after 18 hours using 10, but an 87% yield had already been achieved in the first 30 minutes (Table 3). This result suggests that reduction processes at 100 °C or higher could be adapted to a continuous process.
Initial studies for reusing/re-crosslinking the recovered oil focused initially on the regeneration of thiols from silyl thio ethers. There are few reports of the reactivity of Si–S compounds. Me3Si–S–SiMe3 is very labile, undergoing rapid degradation simply in the presence of water (vapor) to form H2S and Me3SiOH.27 By contrast, the hydrolysis of silicone-based thio ethers was much less facile. Alcoholysis of 3 (Fig. 1g) yielded 12% product only once acetic acid was added to isopropanol solutions (the less sterically hindered thio ether PhCH2SSiMe2OSiMe3 underwent rapid, quantitative cleavage under the same conditions). The silylated polymeric oils derived from elastomers 8 were yet less reactive. It was necessary to use more aggressive nucleophiles for silicon, such as Bu4NF28 to regenerate the silyl free thiols 11 (Fig. 3h and Fig. S20†).
Once cleaved, the freed thiols on the organic polymers could be crosslinked into a new elastomer 12 by oxidative coupling using iodine in isopropyl alcohol29 (Fig. 3i and Fig. S21†). However, it was also discovered that, if silylated polymers 8 were derived from IR/NR or BR and possessed residual alkenes, re-crosslinking did not require removal of the silyl groups; simply adding a radical initiator such as benzoyl peroxide (BPO) and heating led to new elastomers 13 (Fig. 3j).
The ability to create new elastomers from the recovered polymeric oils was demonstrated by creating a new automotive tire (for a child's toy) using 8. A mold of the tire was made in silicone rubber (Fig. 4a and b). Silylated oil 8 derived from IR/NR (tire tread) was placed in the mold in the presence of BPO and heated to give a new, soft elastomer (Fig. 4b + c → e,f durometer Shore OO 68, Table S6†). Adding to 8 the inorganic excipients (recovered from the production of 8, Fig. 4h), and then curing oxidatively, led to harder, more brittle elastomers (Shore A 91; original rubber Shore A 60).
Reductive silylation processes have shown synthetic merit in many arenas. The relatively weak SiH bond,30 particularly in the in the presence of B(C6F5)3 and related Lewis acids, readily reduces a variety of bonds15 driven, in large part, by the thermodynamic benefit of forming Si–heteroatom bonds (heteroatomO, N, S, etc.).31 We have demonstrated that this type of process works effectively with S–S bonds to form thio silyl ethers. The key finding of this work is that, in addition to clean model compounds, the process works with complex and dirty samples – used automotive tires – to convert sulfur-cured elastomers into polymeric oils, in up to ∼90% yield. The process can be rendered practicable at temperatures as low as 100 °C and the product oils can find new utility in elastomeric objects using at least two different cure modes. We are currently establishing the quality of elastomers that can be produced from different used tire feedstocks. The recovered inorganic mass can also be repurposed as a filler in those new elastomers. Thus, reductive silylation provides a new opportunity to find commercial value in materials that are environmentally problematic. It is not possible to effectively ‘reuse’ automotive rubber, but reductive silylation is worthy of consideration as a strategy for recycling and reuse.
Experimental
Materials
Pentamethyldisiloxane 7, bis(trimethylsiloxy)methylsilane 2, (HMe2SiOSiMe2H, MDHM) 10, (Me3Si(OSiMeH)nOSiMe3, MDHnM) 9, were purchased from Gelest and used after drying over molecular sieves overnight. Dibenzyl disulfide, benzyl bromide, tetrabutylammonium fluoride trihydrate (TBAF), and iodine (I2) were obtained from Sigma Aldrich and used as received. Benzoyl peroxide (BPO) was purchased from BDH. Sodium tetrasulfide was purchased from Dojindo. B(C6F5)3 (BCF) was prepared by Grignard reaction following a literature procedure;32 we acknowledge with gratitude Prof. David Emslie, McMaster University, for providing this sample. Rubber samples: bicycle inner tube (Chaoyang 700 × 38/45C bicycle inner tube, China), EPDM (pond liner, purchased at a local garden centre, producer unknown), Crumb-1 (Als-RC, Amazon, Canada), Crumb-2 (Canadian Eco Rubber Ltd, Emterra, Canada), were used as received. Truck tread 1: a piece of truck tread, not part of a complete tire, was found at a local garbage dump (origin unknown). Truck tread 2: (Sailun 225/70R19.5). In both cases, samples were cut only from the external, road contacting tread part (Fig. S3†); both rubbers were based on polyisoprene. The tread and side wall samples – cross-sections – were cut from different parts of a used car tire (snow tire, Cooper 185/65R4, Fig. 5). Naphthalene (internal standard) was purchased from Fisher. Toluene (solvent) received from Caledon (HPLC grade) was dried over activated alumina before use. Deuterated NMR solvents were obtained from Cambridge Isotope Laboratories. Glass apparatus were dried overnight at 120 °C and cooled under a dry nitrogen atmosphere for 30 min prior to use. | ||
| Fig. 5 (a–c) A commercial snow tire was cut into (d) sidewall and tread sections using a reciprocating saw with blade designed for cutting metal. (e) Tread 1.200 × 1.519 × 1.125 cm. (f) Sidewall cut into 1.595 × 1.427 × 0.6096 cm. These samples were subjected to ‘bulk reduction’ of their as-cut shape. (g) For other reductions, the rubbers were chopped into fine pieces after cooling with liquid nitrogen and ground to powder using coffee grinder (KitchenAid), (h) sidewall. (i) Tread. | ||
Methods
The MS parameters (for LC-MS) chosen were as follows: ESI, gas temperature at 225 °C, drying gas at 13 L min−1, nebulizer pressure at 20 psi, sheath gas temperature at 400 °C, sheath gas flow at 12 L min−1, VCap at 3500 V, nozzle voltage at 1000 V, fragmentor at 375 V, and Oct 1 RF Vpp at 750 V. The data were acquired in electrospray positive mode from m/z 50 to 1000 at a scan rate of 1.5 Hz. The mass was auto recalibrated using reference lock mass from Agilent ESI-T Tuning Mix (for Ion Trap).
The polymer constituents of rubber samples were estimated from carbon high-resolution magic angle spinning (13C HR-MAS) NMR spectroscopy (Fig. S5†). TGA: Thermogravimetric analysis (TGA) according to ASTM D 6370-99 (American Society for Testing and Materials) was used to measure the organic carbon (polymer), carbon black content and inorganic residue of the component.24 A small amount of test sample (2 to 5 mg) was placed into the alumina pan of the calibrated Thermogravimetric Analyzer (Mettler Toledo TGA/DSC 3+). A 100 cm3 min−1 argon purge was applied and the furnace was heated from 50 °C to 560 °C at 10 °C min−1. Then, the furnace was cooled to 300 °C and the purge gas was changed to air at 100 cm3 min−1. The temperature was allowed to equilibrate for 2 min before the furnace was heated to 800 °C at 10 °C min−1.
The constituents of sulfur-cured rubbers have well-defined thermal decomposition profiles (Table S3†). Within the thermal decomposition range of the organic polymers, there are further distinctions. TGA and DTA data (Fig. S6†) show the constituents of the rubber samples tested (Table 1).
Preparation of dibenzyl tetrasulfide
To a pre-dried 200 mL round-bottomed flask purged with dry N2 were added sodium tetrasulfide (0.098 g, 0.562 mmol), benzyl bromide (0.209 g, 1.22 mmol) and dry THF (50 mL, 44.45 g, 0.616 mol) as solvent. The mixture was stirred for 23 d and collected by vacuum filtration. The mixture was purified using column chromatography; the low polarity impurity (S8) was removed using hexanes, after which the elution solvent was switched to dichloromethane, to give a yield of 77% (137 mg, based on the different sulfides found in the product. Note: it was not possible to detect tetrasulfide or higher polysulfide linkages in the GC-MS, which may be due to thermal degradation of polysulfide bond (when n > 3).33,341H NMR (600 MHz, acetone-d6): δ 3.72 (s, 0.11H), 4.11 (s, 2.02H), 4.24 (s, 1.20H), 4.30 (s, 0.67H), 7.28–7.40 (m, 9.21H) ppm. 13C NMR (600 MHz, acetone-d6): δ 29.85, 43.55, 43.99, 44.54, 128.36, 129.42, 130.33, 137.54, 137.80 ppm. GC-MS C14H14S2, MW: 246: [M+ − 1] = 65.2 (18), 77.2 (8), 91.1 (90), 121.1 (5), 181.2 (18), 246.1 (20); S8, MW: 256: [M+ − 1] = 64.1 (100), 77.1 (7), 91.1 (66), 127.9 (60), 160.0 (66), 191.9 (53), 207.1 (5), 223.9 (7), 255.8 (98), 315.1 (5). C14H14S3, MW: 278: [M+ − 1] = 65.2 (13), 77.1 (3), 91.2 (100), 123.1 (3), 181.2 (2), 213.1 (56), 278.1 (13).
Dibenzyl disulfide reduction using bis(trimethylsiloxy)methylsilane ([SiH]/[SS] = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1, molar ratio between hydrosilane and disulfide)
:1, molar ratio between hydrosilane and disulfide)
To a pre-dried 200 mL round-bottomed flask purged with dry N2 gas, was added dibenzyl disulfide (0.50 g, 2.04 mmol) and bis(trimethylsiloxy)methylsilane (0.91 g, 4.09 mmol) together with dry DCM (3.81 g, 44.86 mmol) as solvent. Freshly prepared B(C6F5)3 stock solution was added (0.167 mL, 0.0326 mmol) after 5 min stirring. The total reaction time was 3 h, then the reaction was quenched by adding neutral alumina. Each datapoint in Fig. 2 was obtained using the identical reaction protocol, only changing the ratio of hydrosilane to disulfide [SiH]/[SS] (Table S1†).
Titration of disulfides to establish relative reactivity of functional groups
Conversion in the reaction was shown by peak area of the hydrogens on the carbon adjacent to the disulfide bond (–C![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif) 2SS) in 1H NMR, which was plotted against different ratios of hydrosilane (Si) to disulphide. Analogous techniques were used to follow the reduction of the tetrasulfide (Fig. 2).
2SS) in 1H NMR, which was plotted against different ratios of hydrosilane (Si) to disulphide. Analogous techniques were used to follow the reduction of the tetrasulfide (Fig. 2).
Reduction of rubbers: general procedure
The reaction conditions for the reduction of rubber powder are given in Tables 2 and 3. The general experimental procedure is as follows. The cryogenically ground rubber powder was allowed to swell in dry toluene (12 mL) for 30 min.Pentamethyldisiloxane 7 was added to the reaction mixture. The stock catalyst solution was added to initiate the reaction. The suspension was heated in a 60 °C oil bath for 48 h. The residual undissolved rubber powder was washed with toluene and separated by centrifugation (Eppendorf, Centrifuge 5424, at 12000 rpm for 20 min). The extraction process was repeated twice to completely remove soluble compounds. The supernatants were mixed, and the solvents were removed by rotary evaporation. Any residual (organic) volatiles were removed under a stream of N2 over 24 h. The recovered organic liquid was characterized by NMR. The recovered rubber powders were dried at 100 °C overnight and then examined by TGA. The bulk sample were reduced by the similar protocol and more details were provided in ESI.†
Reduction with inexpensive tetramethyldisiloxane MHMH
Cryogenically ground inner tube powder (300 mg) was placed in dry toluene (12 mL), followed by tetramethyldisiloxane MHMH (1.5 mL, 8.5 mmol). The stock catalyst solution (600 μL, stock catalyst concentration: 50 mg mL−1 in toluene, catalyst concentration in reaction: 10 wt%/inner tube) was added immediately afterwards to initiate the reaction. The suspension was heated in a preheated 100 °C oil bath for 30 min. The reaction flask was put into a room temperature water bath to quench the reaction and followed by a separation process using the same protocol stated in the former paragraph. The organic yield was 87%.Desilylation of product polymeric oils to give 11
The silylated organic oil 8 (based tire tread, derived from IR/NR, 0.50 g) was desilylated by treatment with TBAF solution (0.5 g TBAF, 1.92 mmol TBAF, dissolved in 10 mL THF containing 0.1 mL methanol) for 24 h at 80 °C. The solvent and siloxane fragments were removed by using a rotary evaporator, followed by kugelrohr distillation; loss of silicone was clearly seen in the 1H NMR (200 °C, 3 h, Fig. S20†).Crosslinking of recovered oils
Preparation of toy tire using a peroxide initiator (Fig. 4)
A silicone mode was prepared with a two-part liquid component kit (Sylgard 184). Two components were mixed at the recommended ratio of 10 parts (10.0 g) base to 1 part curing agent (1.0 g). The mixing process was performed using a planetary centrifugal mixer (FlackTek Inc.) with a duration of 5 min at a speed of 3000 rpm. In order to fabricate bubble free elastomer, the mixed uncured PDMS was thoroughly degassed in a vacuum desiccator at low pressure for 30 min.The right front tire of a toy car was removed from the toy and placed in the degassed, uncured mixture. The mixture was cured in an 80 °C oven overnight. The tire was removed from the cured mold.
Used rubber powder from the truck tread (Sailun) (2.0 g) was reduced by pentamethyldisiloxane 7 in a 100 °C oil bath for 18 h to give a polymeric oil 8 (81.5% yield). Solvents in the supernatants, after centrifugation, were removed by rotary evaporation; any residual volatiles and silicone by-products were removed using a stream of N2 over 24 h. The oil was accompanied by residual undissolved rubber powder that was washed with toluene and separated by centrifugation (Eppendorf, Centrifuge 5424, at 12000 rpm for 20 min), dried an 80 °C oven for 18 h, and ground into powder using a mortar and pestle (Fig. 4d).
The silylated organic oil 8 from the former step (comprised of IR/NR derivatives, 0.707 g) was dissolved in hexanes (10 mL), benzoyl peroxide (BPO, 0.01 g, 1 wt%, 0.0413 mmol) and, optionally, ground residual inorganic solids (from the preparation of 8, 0.3010 g), were added sequentially and mixed to give a homogeneous dispersion. After the solvent was removed by rotary evaporation, the mixture was placed in the silicone mold and degassed under vacuum in a desiccator for 30 min. The curing process was performed at 100 °C for 18 h. The formulations for rubber with different residual solid are listed in Table S6.†
Conclusions
Utilization of the reactions described here to create polymeric oils by the reductive silylation of used automotive rubbers, and subsequent oxidation to new elastomers, permits the completion of a full cycle of use for these organic materials. Mild, efficient reduction of S–S bonds of organic disulfides, including those found in used, sulfur-cured rubbers permits the formation of silylated polymers 8 – already in 87% yield at 100 °C within 30 minutes – that can be separated from the inorganic constituents. Radical cure of the oils if alkenes are present, or following desilylation, oxidative cure of thiolated polymers leads to new elastomers 12, 13 that, optionally, can be reinforced by the inorganic constituents recovered during the reduction process. The practice of this recycling process has the potential to reduce the environmental impact of used, sulfur-crosslinked elastomers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge with gratitude the financial support of the Natural Sciences and Engineering Research Council of Canada. We also appreciate provision of real rubber samples from Emterra Canada (rubber tire crumb); Active Green and Ross (used Cooper automobile tire); and, Marshall Tirecraft (Sailun truck tire – neither positive nor negative inference should be associated with the tires obtained from these specific manufacturers). SZ also thanks the Chinese Scholarship Council for a scholarship.Notes and references
- A. Y. Coran, in The Science and Technology of Rubber (Fourth Edition), ed. J. E. Mark, B. Erman and C. M. Roland, Academic Press, Boston, 2013, pp. 337–381, DOI:10.1016/B978-0-12-394584-6.00007-8.
- C. Goodyear, US Patent3633, 1844 Search PubMed.
- Freedonia Group, 2015, Industry Study 3357.
- J. J. Evans, Rev. Environ. Contam. Toxicol., 1997, 151, 67–115 CrossRef CAS PubMed.
- K. Stevenson, B. Stallwood and A. G. Hart, Biorem. J., 2008, 12, 1–11 CrossRef CAS.
- X. Liu, J. Wang, A. Gheni and M. A. ElGawady, Waste Manage., 2018, 81, 53–60 CrossRef CAS.
- A. Singh, S. N. Spak, E. A. Stone, J. Downard, R. L. Bullard, M. Pooley, P. A. Kostle, M. W. Mainprize, M. D. Wichman, T. M. Peters, D. Beardsley and C. O. Stanier, Atmos. Environ., 2015, 104, 273–283 CrossRef CAS.
- Rubber Recycling: Challenges and Developments, ed. J. K. Kim, P. Saha, S. Thomas, J. T. Haponiuk and M. K. Aswathi, RSC, 2019 Search PubMed.
- H. Cheng, Y. Hu and M. Reinhard, Environ. Sci. Technol., 2014, 48, 2114–2129 CrossRef CAS.
- M. Ochmann, A. Hussain, I. von Ahnen, A. A. Cordones, K. Hong, J. H. Lee, R. Ma, K. Adamczyk, T. K. Kim, R. W. Schoenlein, O. Vendrell and N. Huse, J. Am. Chem. Soc., 2018, 140, 6554–6561 CrossRef CAS.
- M. L. Studebaker and L. G. Nabors, Rubber Chem. Technol., 1959, 32, 941–961 CrossRef CAS.
- M. L. Studebaker, Rubber Chem. Technol., 1970, 43, 624–650 CrossRef CAS.
- R. Walvekar, Z. M. Afiq, S. Ramarad and S. Khalid, MATEC Web Conf., 2018, 152, 01007 CrossRef.
- D. Czajczyńska, R. Krzyżyńska, H. Jouhara and N. Spencer, Energy, 2017, 134, 1121–1131 CrossRef.
- M. A. Brook, in Silicon in Organic, Organometallic and Polymer Chemistry, Wiley, New York, 2000, ch. 7, pp. 171–188 Search PubMed.
- E. Lukevics and M. Dzintara, J. Organomet. Chem., 1985, 295, 265–315 CrossRef CAS.
- B. MarcIniec, J. Gulinski, W. Urbaniak and Z. W. Kornetka, Comprehensive Handbook on Hydrosilylation Chemistry, Pergamon, Oxford, 1992 Search PubMed.
- M. A. Brook, Chem. – Eur. J., 2018, 24, 8458–8469 CrossRef CAS.
- V. Gevorgyan, M. Rubin, J. X. Liu and Y. Yamamoto, J. Org. Chem., 2001, 66, 1672–1675 CrossRef CAS.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- K. Saito, K. Kondo and T. Akiyama, Org. Lett., 2015, 17, 3366–3369 CrossRef CAS.
- E. Jakab and M. Omastová, J. Anal. Appl. Pyrolysis, 2005, 74, 204–214 CrossRef CAS.
- D. W. Brazier and G. H. Nickel, Rubber Chem. Technol., 1975, 48, 661–677 CrossRef.
- ASTM, 2014, D6370-99.
- ASTM, 2002, D 297–93.
- W. E. Piers, Adv. Organomet. Chem., 2005, 52, 1–77 CAS.
- J.-H. So, P. Boudjouk, H. H. Hong and W. P. Weber, Inorg. Synth., 1992, 29, 30–32 CAS.
- R. D. Crouch, Synth. Commun., 2013, 43, 2265–2279 CrossRef CAS.
- B. Zeynizadeh, J. Chem. Res., 2002, 2002, 564–566 CrossRef.
- M. Oestreich, J. Hermeke and J. Mohr, Chem. Soc. Rev., 2015, 44, 2202–2220 RSC.
- F. J. Fernández-Alvarez, A. M. Aitani and L. A. Oro, Catal. Sci. Technol., 2014, 4, 611–624 RSC.
- M. A. Beckett, D. S. Brassington, S. J. Coles and M. B. Hursthouse, Inorg. Chem. Commun., 2000, 3, 530–533 CrossRef CAS.
- H. Akbulut, S. Yamada and T. Endo, RSC Adv., 2016, 6, 108689–108696 RSC.
- K. L. Stensaas, A. S. Brownell, S. Ahuja, J. K. Harriss and S. R. Herman, J. Sulfur Chem., 2008, 29, 433–443 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of reactivity of di- and tetrasulfides, rubber components, efficiency of repeated reduction and GPC data of produced oils. Figures showing NMR data of starting rubbers, product oils, TGA data or starting rubbers, and products oils, photographs showing the process from rubber to oils to new rubbers. See DOI: 10.1039/c9gc03545a |
| This journal is © The Royal Society of Chemistry 2020 |