
Structural characterization and osteogenic bioactivities of a novel Humulus lupulus polysaccharide†
Xiaoxia
Chen
a,
Tianyu
Li
b,
Degang
Qing
c,
Jun
Chen
d,
Qian
Zhang
*e and
Chunyan
Yan
 *ab
*ab
aCenter for Clinical Precision Medication, The First Affiliated Hospital of Guangdong Pharmaceutical University, Guangzhou 510006, China
bSchool of Clinical Pharmacy, Guangdong Pharmaceutical University, Guangzhou 510006, China. E-mail: ycybridge@gdpu.edu.cn
cXinjiang Institute of Traditional Chinese Medicine and Ethnodrug, Urumqi 830002, China
dSchool of Bioscience and Biopharmaceutics, Guangdong Pharmaceutical University, Guangzhou 510006, China
eSchool of Pharmacy, Guangdong Pharmaceutical University, Guangzhou 510006, China. E-mail: zqdrug@gdpu.edu.cn
First published on 16th December 2019
Abstract
Humulus lupulus is a perennial climbing plant of the subfamily Cannabioideae native to the Northern Hemisphere. The primary use of H. lupulus is in the brewing industry, where it is an essential ingredient for imparting a unique flavor (bitterness and aroma) to beer. The female flowers of H. lupulus are also used in traditional Chinese medicine, but the biologically active ingredients underlying its benefits remain unclear. China is the largest producer and consumer of H. lupulus in Asia. Using the waste from the beer-brewing process of H. lupulus as raw materials, the biologically active polysaccharides can be screened. This is useful for the full utilization of H. lupulus, potentially leading to disease prevention and treatment. In this study, we isolated a homogeneous polysaccharide (HLP50-1) with a molecular weight of 49.13 kDa from female flowers of H. lupulus via a DEAE-Cellulose 52 anion exchange column and a Sephadex G-75 gel filtration column. Methylation, GC-MS, and NMR analyses revealed that the HLP50-1 comprised →4)-α-D-Glcp-(1→, →6)-α-D-Manp-(1→, →3)-α-L-Rhap-(1→, β-D-Glcp-(1→, α-L-Araf-(1→, →4,6)-2-OAc-β-D-Galp-(1→, β-D-Galp-(1→, →3,6)-β-D-Glcp-(1→, →2,3,4)-α-D-Xylp-(1→, →6)-α-D-Glcp-(1→, →3)-α-D-Galp-(1→, →4)-α-D-Galp-(1→. Advanced structural analysis showed that the HLP50-1 contained irregular fragments of different sizes and shapes with a smooth surface. The aggregates appeared be composed of accumulated crystals. Furthermore, the osteogenic activities of the HLP50-1 were evaluated via MC3T3-E1 cells in vitro. The results showed that 0.13 μM HLP50-1 led to outstanding proliferation, differentiation, and mineralization of the MC3T3-E1 cells. Therefore, HLP50-1 has osteogenic effects, and it may be a candidate for the treatment of osteoporosis. It has broad application prospects in functional foods, health-care products, and pharmaceuticals.
1. Introduction
Osteoporosis is a systemic bone disease characterized by reduced bone mass and impaired bone microstructure.1 It has an increasing rate of incidence due to the aging population.2 Bone metabolic homeostasis is maintained by the balance between osteoblast-mediated bone formation and osteoclast-mediated bone resorption. Osteoporosis is triggered when bone resorption exceeds bone formation.1 Most of the drugs currently used to treat osteoporosis, including bisphosphonates, calcitonins, and estrogens, have varying degrees of toxic side effects;3 thus, safe and effective alternative osteoporosis drugs are needed urgently.In recent years, an increasing body of evidence has suggested that plant-derived bioactive compounds including polysaccharides can lower the risks of chronic diseases. For example, polysaccharides from Morinda officinalis can promote osteoblast differentiation by up-regulating markers related to osteogenic differentiation, combatting the effects of osteoporosis.4Polygonatum sibiricum polysaccharides inhibit osteoporosis by promoting osteoblast formation and blocking osteoclastogenesis via the Wnt/β-catenin signaling pathway.5
Humulus lupulus—a perennial, dioecious plant of the Cannabioideae subfamily that is native to the Northern Hemisphere, i.e., hops—is widely grown for brewing beer. It is an essential ingredient and offers a unique flavor (aroma and refreshing bitterness).6,7H. lupulus is also an important economic crop in the Xinjiang Uygur Autonomous Region and Gansu Province of China. In 2015, the acreage of H. lupulus was 2320 hectares, and the production was 5954 metric tons in China.8 During the beer-brewing process, only the fat-soluble aroma and bitter components are utilized. The remaining components, which include polysaccharides, are usually biologically active;9,10 however, these are often treated as waste. Therefore, using these waste components as raw materials to screen out active polysaccharides is not only beneficial to more fully utilize H. lupulus but to the prevent and treat diseases as well.
Several studies have found that polysaccharides are one of the most important components of H. lupulus, which have estrogen-like effects6,10 (estrogen maintains bone strength and stability).11,12 In our previous study,13 we isolated a novel heteropolysaccharide, HLP50-2, from female H. lupulus flowers, identified its structure and assessed its osteogenic activities. Experimental results indicated that HLP50-2 can promote the osteogenic mineralization of MC3T3-E1 cells. Thus, we deduced that H. lupulus polysaccharides may be effective in the prevention and treatment of osteoporosis.
In the study, the purification and structural characterization of a novel polysaccharide (HLP50-1) derived from H. lupulus is reported via column chromatography (DEAE-Cellulose 52 anion exchange column and Sephadex G-75 gel filtration column), chemical analyses (monosaccharide composition and methylation), and instrumental analyses (infrared spectroscopy, gas chromatography-mass spectrometry (GC-MS), nuclear magnetic resonance (NMR), and scanning electron microscopy). In addition, the in vitro osteogenic bioactivities of HLP50-1 were assessed using MC3T3-E1 cells.
2. Materials and methods
2.1. Reagents and materials
Female H. lupulus flowers were obtained from the Xinjiang Uygur Autonomous Region. The voucher specimen was identified by Professor Degang Qing from the Institute of Traditional Chinese Medicine, Xinjiang, China. Diethylaminoethyl cellulose (DEAE-52) and Sephadex G-75 were purchased from Whatman Co. (Maidstone, Kent, UK) and GE Healthcare (Chicago, IL, USA), respectively. Trifluoroacetic acid (TFA) and 17β-estradiol (E2) were supplied by Aladdin Industrial Co. (Shanghai, China). Standard monosaccharides, T-series dextran, and Alizarin Red S were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). The MC3T3-E1 cells were supplied by the American Type Culture Collection (ATCC, Manassas, VA, USA). Fetal bovine serum (FBS), trypsin, and α-modified minimum essential medium (α-MEM) were obtained from Gibco (New York, NY, USA). The alkaline phosphatase (ALP) assay kit was purchased from Beyotime (Shanghai, China). All other chemicals and solvents were of analytical grade.2.2. Extraction of crude polysaccharide
The dried female H. lupulus flowers (27 kg) were extracted with hot water 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (w/v, kg L−1) three times at 90 °C for 3 h each. The water extracts were combined, concentrated (60 °C), centrifuged at 1768g for 10 min, and precipitated by adding 90% ethanol to a final concentration of 50%, after which it was maintained at room temperature for 24 h to obtain the precipitate.14 The precipitate was then dissolved in an appropriate amount of deionized water and filtered. The filtrate and Sevag reagent15 (chloroform:n-butanol = 4:1, v/v) were mixed at a ratio of 5:1, shaken vigorously, and allowed to stand to remove the protein layer. This process was repeated three times to remove the proteins completely. Finally, the deproteinized solution was dialyzed against distilled water (cut-off Mw: 1000 Da) for 48 h and then freeze-dried to obtain a crude polysaccharide, denoted as HL50.
:10 (w/v, kg L−1) three times at 90 °C for 3 h each. The water extracts were combined, concentrated (60 °C), centrifuged at 1768g for 10 min, and precipitated by adding 90% ethanol to a final concentration of 50%, after which it was maintained at room temperature for 24 h to obtain the precipitate.14 The precipitate was then dissolved in an appropriate amount of deionized water and filtered. The filtrate and Sevag reagent15 (chloroform:n-butanol = 4:1, v/v) were mixed at a ratio of 5:1, shaken vigorously, and allowed to stand to remove the protein layer. This process was repeated three times to remove the proteins completely. Finally, the deproteinized solution was dialyzed against distilled water (cut-off Mw: 1000 Da) for 48 h and then freeze-dried to obtain a crude polysaccharide, denoted as HL50.
2.3. Isolation and purification of HLP50-1
The HL50 (2 g) was dissolved in 100 mL of distilled water and centrifuged at 4029g for 5 min. The supernatant (10 mL) was then fractionated by a DEAE-Cellulose 52 anion exchange column (∅ 2.5 × 40 cm) with 0.05 M sodium chloride at a flow rate of 1 mL min−1. Meanwhile, the content of the polysaccharide was determined by a phenol-sulfuric acid colorimetric method.16 The fraction (HL50-1) was collected, dialyzed against distilled water for 48 h (cut-off Mw: 100 Da), and lyophilized. The HL50-1 (20 mg) was then re-dissolved in 1 mL of distilled water, filtered with a 0.45 μm membrane, and further purified by a Sephadex G-75 gel filtration column (∅ 1.6 × 100 cm) eluted with distilled water (1 mL per 3 min) to obtain a purified polysaccharide, denoted as HLP50-1. The structural identification and osteogenic activities analysis of HLP50-1 were subsequently carried out.2.4. Homogeneity verification and molecular weight detection of HLP50-1
Specific optical rotation analysis was conducted to determinate the homogeneity of HLP50-1 (Micro-Optical Rotator AUTOPOL I; Rudolph Technologies, Wilmington, MA, USA) based on a previous method.17–19 HLP50-1 (80 mg) was dissolved in distilled water (1 mL), and anhydrous ethanol was added until the final concentration reached 50%. After standing at 4 °C for 24 h, the solution was centrifuged at 4029g for 5 min to obtain precipitate I. Anhydrous ethanol was then continuously added to the supernatant to a final concentration of 70%, and precipitate II was collected after standing at 4 °C for 24 h and centrifugation at 4029g for 5 min. These two precipitates were lyophilized and dissolved in distilled water (2 mg mL−1) for specific optical rotation analysis. The temperature and wavelength were 20 °C ± 0.3 °C and 589 nm, respectively, and an optical tube with a length of 100 mm was used.The homogeneity verification and molecular weight detection of HLP50-1 were performed using a classical high-performance gel permeation chromatography (HPGPC) method.18,20,21 A Waters 2695 high-performance liquid chromatograph (Massachusetts, USA) was equipped with two serially linked columns: a TSK-GEL G-5000 PWXL and a G-3000 PWXL gel column. The mobile phase was a 0.02 M KH2PO4 solution, the flow rate was 0.6 mL min−1, and the sample injection volume was 10 μL. The column temperature was 35 °C, and the detector was a Waters 2414 refractive index detector.
The standard curve was obtained by plotting the logarithm of the relative molecular weights (logMW) of Dextran T-series standards (Dextran T1000, T500, T70, T40, T10, and T5) as the ordinate and the elution volume (V) as the abscissa. HLP50-1 (5 mg mL−1) and the standards (2 mg mL−1) were dissolved in 0.02 M KH2PO4. The relative molecular weight and distribution of HLP50-1 could subsequently be obtained based on the standard curve using the Breeze data processing software.18
2.5. Analysis of infrared spectrum
HLP50-1 (2 mg) was milled with KBr powder (150 mg) and pressed into a tablet to record the Fourier-transform infrared (FT-IR) spectrum from 4000 to 400 cm−1.222.6. Analysis of monosaccharide composition
The monosaccharide composition of HLP50-1 was determined by high-performance liquid chromatography via the pre-column derivatization of 1-phenyl-3-methyl-5-pyrazolone (PMP).23,24 The HLP50-1 (5 mg) was hydrolyzed using 3 M trifluoroacetic acid (TFA, 2 mL) in a sealed tube at 120 °C for 6 h. After cooling, methanol was added to the hydrolysate and rotary evaporated to dryness to remove excess TFA. This was repeated three times, after which the product was re-dissolved with 1 mL of distilled water.The hydrolysate (100 μL) reacted with 0.3 M NaOH (100 μL) and 0.5 M PMP in methanol (100 μL) at 70 °C for 30 min, after which it was neutralized with 0.3 M HCl (105 μL). The derivative was subsequently extracted with an equal amount of chloroform to remove excess PMP, which was repeated three times. Ultimately, the aqueous layer was filtered through a 0.45 μm membrane and analyzed by HPLC. Meanwhile, the derivative of the mixed monosaccharide standards (mannose, rhamnose, glucuronic acid, galacturonic acid, glucose, galactose, xylose, arabinose, and fucose) was prepared according to the above steps for HPLC analysis.
The chromatography was conducted using an Agilent 1260 HPLC system (Santa Clara, CA, USA) equipped with a ZORBAX Eclipse XDB-C18 column (5 μm, 4.6 × 250 mm) and an ultraviolet detector with a detection wavelength of 250 nm. The mobile phase was a 0.05 M NaH2PO4–Na2HPO4 buffer solution (pH 6.7) and acetonitrile (83:17, v/v), the flow rate was 1 mL min−1, and the sample injection volume was 10 μL.
2.7. Analysis of absolute configuration of monosaccharide
The hydrolysis of HLP50-1 (5 mg) began with the method of monosaccharide composition analysis presented in section 2.6. L-Cysteine methyl ester (2.5 mg) and pyridine (1 mL) were subsequently added to the hydrolysate and reacted in a water bath at 60 °C (1 h). Next, 5 μL of O-tolylisothiocyanate was added, and the water bath reaction continued at 60 °C for 1 h. Meanwhile, standard monosaccharides (1 mg) were directly and similarly derivatized. Finally, the derivatized products were filtered through a 0.45 μm filter and analyzed with HPLC equipped with an Agilent ZORBAX Eclipse XDB-C18 column (5 μm, 4.6 × 250 mm) and an ultraviolet detector. The analysis conditions were as follows: the mobile phase was 25% CH3CN–H2O (0.1% acetic acid); the flow rate was 0.8 mL min−1; the column temperature was 35 °C; the sample injection volume was 10 μL; the detection wavelength was 250 nm; and the detection time was 35 min.2.8. Analysis of methylation and GC-MS
We used the method of Needs and Selvendran25 with minor adjustments to methylate the HLP50-1. Briefly, 8 mg of dried HLP50-1 were dissolved in 5 mL of anhydrous dimethylsulfoxide (DMSO) and sonicated for 1 h. Next, 600 mg of dried NaOH was ground with a glass rod and added quickly, followed by 1 h of sonication. Next, methyl iodide (500 μL) was added in the dark and sonicated for 10 min. The process was repeated three times, and the final sonication continued for 1 h. Subsequently, distilled water (2 mL) and chloroform (3 mL) were added to the reaction product followed by centrifugation at 4029g for 5 min after intense shaking. Finally, the chloroform layer was washed with distilled water twice and analyzed using FT-IR. In the FT-IR spectrum, the O–H absorption band (3200–3600 cm−1) disappeared, which proved that the HLP50-1 was completely methylated.The hydrolysis of the HLP50-1 methylation product was consistent with the method of monosaccharide composition analysis described in section 2.6. The hydrolysate was reduced with NaBH4 (20 mg) and kept at 40 °C for 30 min. Next, 100 μL of glacial acetic acid was added to terminate the reduction. The reaction product was dried via rotary evaporation with methanol (three times), after which it was acetylated by adding 2 mL of acetic anhydride and 2 mL of pyridine at 95 °C for 1 h. Finally, the acetylated derivative was concentrated to dryness, dissolved in chloroform (3 mL), washed with an equivalent volume of deionized water three times, and analyzed by GC-MS.
The GC-MS analysis was conducted using a TRACE 1300 gas chromatograph equipped with a TG-SQC column (0.25 μm, 15 m × 0.25 mm) from Thermo Fisher Scientific Inc. (Waltham, MA, USA). The following temperature program was adopted. The initial column temperature was 80 °C (held for 1 min), after which the temperature was increased to 280 °C at 5 °C min−1 and held for 1 min. The split injector temperature was 250 °C, the ion source temperature was 280 °C, helium (purity of 99.999%) was the carrier gas, and the flow rate was 1.5 mL min−1.
2.9. Analysis of nuclear magnetic resonance
HLP50-1 (80 mg) was dissolved in D2O. The 1H-NMR, 13C-NMR, heteronuclear single quantum correlation (HSQC), heteronuclear multiple bond correlation (HMBC), and 1H–1H correlation spectroscopy (1H–1H COSY) spectra were recorded with an NMR spectrometer (AV-500, Bruker, Dresden, Germany).2.10. Field emission scanning electronic microscopy (SEM) analysis
The microstructure of HLP50-1 was observed using SEM (JSM-6330F, Jeol, Tokyo, Japan).26 Briefly, the HLP50-1 (1 mg) was ground onto a double-sided adhesive and sputter-coated with gold using a vacuum spray-coating apparatus. Finally, images of the HLP50-1 under 500-, 1500-, 3000-, and 5000-fold magnification were obtained.2.11. Effect of HLP50-1 on proliferation of MC3T3-E1 cells
The MC3T3-E1 cells were maintained in α-MEM medium supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin (complete medium) in humidified air with 5% CO2 at 37 °C. The MC3T3-E1 cells were added to a 96-well culture plate at a density of 5 × 103 cells per well and incubated with complete medium in the presence or absence of different concentrations of HLP50-1 (200 μL; concentrations of 0.065, 0.13, 0.26, 0.52, 1.04, and 2.08 μM). The normal group was cultured with only the complete medium, and the positive control group (E2) was cultured with complete medium containing 0.1 μM of E2 in parallel (six replicate wells).After treatment for 48 h, 200 μL of complete medium was added, and then 20 μL of 3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2-H-tetrazolium bromide (MTT) solution (5 mg mL−1) was pipetted into each well and held for 4 h at 37 °C. Next, the medium was discarded, and the formazan crystal was dissolved in 150 μL of DMSO. Finally, the solution was rocked for 10 min, and the optical density (OD) at 490 nm was measured with a plate-reader (Multiskan GO, Thermo Fisher Scientific, Massachusetts, USA). The proliferation rate (R%) was calculated as follows:
| R% = (ODsample − ODnormal)/ODnormal × 100%. |
2.12. Effect of HLP50-1 on differentiation of MC3T3-E1 cells
Based on the MTT data, 0.065, 0.13, and 0.26 μM concentrations of the HLP50-1 were selected to analyze the effect of the HLP50-1 on the differentiation of the MC3T3-E1 cells. The MC3T3-E1 cells were added to a 24-well culture plate at a density of 3 × 104 cells per well and incubated with complete medium for 72 h. The complete medium was then changed to an osteogenic induction medium (10 mM β-glycerophosphate and 100 μg mL−1 vitamin C in complete medium) in the presence or absence of different concentrations of HLP50-1 (500 μL; concentrations of 0.065, 0.13, or 0.26 μM). A control group was cultured with osteogenic induction medium, and the normal group was cultured with only complete medium. The positive control group (E2) was cultured with osteogenic induction medium containing 0.1 μM E2; this was performed in parallel (three replicate wells). After treatment for 10 d, the cells were lysed using a 100 μL radioimmunoprecipitation assay (RIPA) buffer per well at 4 °C for 30 min, after which they were centrifuged at 12000g for 20 min (4 °C). The concentrations of the protein supernatant were determined using a bicinchoninic acid (BCA) protein concentration quantitation kit (Thermo Fisher Scientific, Massachusetts, USA), and the ALP activity was measured using an ALP assay kit (Beyotime, Shanghai, China).27
2.13. Effect of HLP50-1 on mineralization of MC3T3-E1 cells
The MC3T3-E1 cells were added to a 12-well culture plate at a density of 6 × 104 cells per well and incubated with complete medium for 72 h. The complete medium was then changed to an osteogenic induction medium in the presence or absence of different concentrations of HLP50-1 (1 mL; concentrations of 0.065, 0.13, or 0.26 μM). The control group was cultured with the osteogenic induction medium, the normal group was cultured with only complete medium, and the positive control group (E2) was cultured with osteogenic induction medium containing 0.1 μM E2, which was conducted in parallel (three replicate wells). After 18 d of culturing, the cells were fixed with 10% neutral formalin at room temperature for 30 min, washed three times with phosphate buffer (PBS), stained with 0.1% Alizarin Red S for 30 min in the dark, washed 3–5 times with PBS, and observed with an inverted biological microscope to image the mineralized nodule (OLYMPOS CKX41, Olympus Corporation, Takachiho, Japan). The mineralization rate (M%) was quantitatively analyzed by adding 400 μL of 10% cetylpyridinium per well in the dark for 1 h and calculated according to the following formula:28–30| M% = (ODsample − ODnormal)/ODnormal × 100%. |
2.14. Statistical analysis
The statistical analysis was performed using a GraphPad Prism 5. The data were expressed as the mean ± standard error of the mean (SEM) from at least three independent experiments. Statistical significance was determined using one-way analysis of variance (ANOVA). Probabilities (P) below 0.05 were considered to be statistically significant.3. Results and discussion
3.1. Separation and purification of HLP50-1
Ion exchange chromatography is the most classic and common method of polysaccharide separation. It provides a sample loading area with a wide specific surface area. Generally, crude polysaccharides are first separated and enriched by an anion exchange column followed by gel molecular sieve column chromatography to obtain homogeneous polysaccharides.31 In this study, the lyophilizate of the crude polysaccharides, HL50, was extracted with hot water followed by ethanol precipitation, deproteinization, and dialysis with a yield of 4.68% from H. lupulus. HL50 was separated using a DEAE-Cellulose 52 column using 0.05 M NaCl as the eluent to obtain HL50-1 with a yield of 0.57% (Fig. S1A†). Next, HL50-1 was further purified using a Sephadex G-75 column to obtain HLP50-1 with a yield of 17.8% (Fig. S1B†).3.2. Homogeneity verification and molecular weight determination of HLP50-1
HPGPC was used to detect the polysaccharide homogeneity and molecular weight. It is widely recognized as the most authoritative test to date.32 Fig. S2† shows that the HLP50-1 exhibited a narrow and symmetrical peak in the HPGPC with a relative molecular weight of 49.13 kDa. In addition, the difference in the specific optical rotation between the two precipitation fractions of the HLP50-1 was less than 5°, indicating that it was a homogeneous fraction. Thus, the HPGPC and specific optical rotation analysis suggested that HLP50-1 was a homogeneous polysaccharide.3.3. FT-IR spectral analysis of HLP50-1
The infrared spectrum of HLP50-1 is shown in Fig. S3.† It is a typical polysaccharide spectrum with a broad and intense band at 3390 cm−1 (O–H stretching vibrations) and a weak absorption band at 2930 cm−1 (C–H stretching vibrations).33 In addition, the asymmetrical and symmetrical stretching bands of the HLP50-1 that appeared around 1610 and 1412 cm−1 indicate the presence of COO− in the HLP50-1.18,34,35 The bands at 1044 cm−1 indicate the bending vibrational modes of C–O stretching in the pyranose form, and the band at 617 cm−1 revealed the existence of a pyranose ring in the HLP50-1.35,36 Moreover, the weak absorption at 868 cm−1 indicated the presence of the α-configuration in HLP50-1.36,373.4. Monosaccharide composition analysis of HLP50-1
The monosaccharide composition analysis of the PMP pre-column HPLC results indicated that HLP50-1 was composed of mannose, rhamnose, glucose, galactose, xylose, and arabinose (Fig. 1). The results further suggested that HLP50-1 was a heteropolysaccharide. Furthermore, the result showed that glucose, galactose, and arabinose were the most abundant monosaccharides.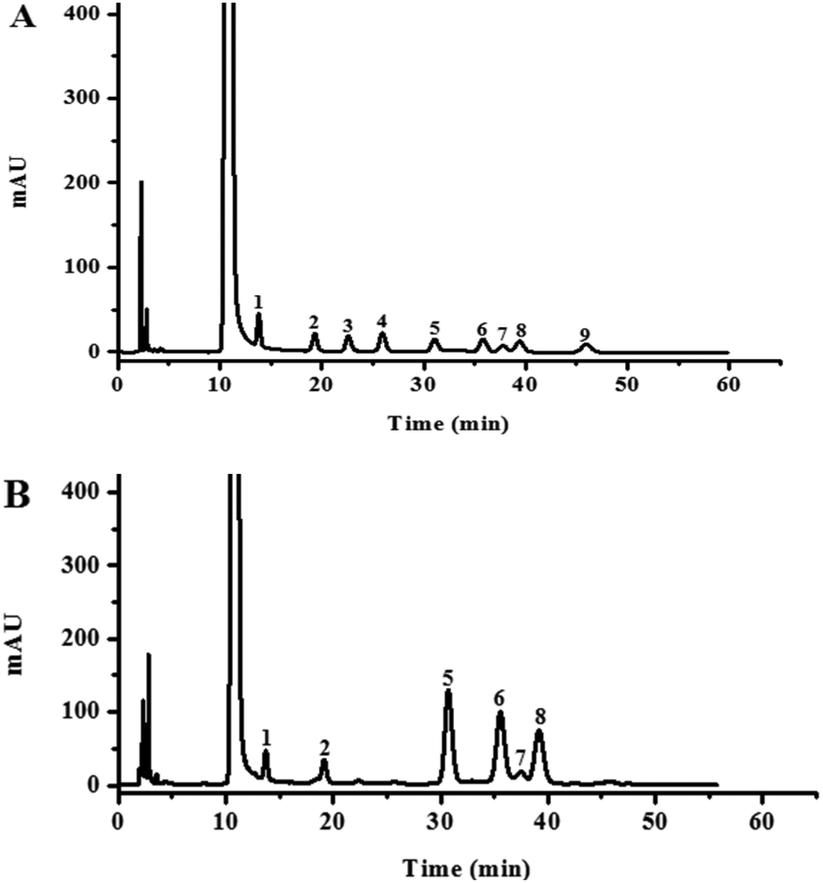 | ||
| Fig. 1 HPLC analysis of PMP pre-column derivatives of (A) nine standard monosaccharides (1. mannose; 2. rhamnose; 3. glucuronic acid; 4. galacturonic acid; 5. glucose; 6. galactose; 7. xylose; 8. arabinose; and 9. fucose) and (B) HLP50-1 hydrolysate. | ||
3.5. Absolute configuration analysis of monosaccharide of HLP50-1
The HPLC chromatograms of the HLP50-1 (Fig. 2B) after complete acid hydrolysis and derivatization were compared with the chromatograms of the derivatives of standard monosaccharides (Fig. 2A). The results indicated that HLP50-1 was composed of D-mannose (D-Man), D-galactose (D-Gal), D-glucose (D-Glc), L-arabinose (L-Ara), D-xylose (D-Xyl), and L-rhamnose (L-Rha). The D-Man, D-Gal, D-Glc, L-Ara, D-Xyl, and L-Rha standards were added to the derivative of HLP50-1 hydrolysate, and the results further indicated that HLP50-1 contained D-Man, D-Gal, D-Glc, L-Ara, D-Xyl, and L-Rha. | ||
| Fig. 2 HPLC chromatograms of (A) derivatives of standard monosaccharides and (B) HLP50-1 hydrolysate derivatives: HLP50-1 + standards with D-Man, D-Gal, D-Glc, L-Ara, D-Xyl, and L-Rha standards added to the derivative of the HLP50-1 hydrolysate (1. D-Man; 2. D-Gal; 3. D-Glc; 4. L-Ara; 5. D-Xyl; and 6. L-Rha). | ||
3.6. Methylation and GC-MS analysis of HLP50-1
Methylation and GC-MS analysis of the HLP50-1 were performed to determine the types and proportions of the sugar residues. After methylation, the hydroxyl band at 3200–3600 cm−1 basically disappeared, whereas the size of the methyl band at 2930 cm−1 increased in the infrared spectrum, demonstrating that HLP50-1 was completely methylated. Table 1 shows the results of the linkage types of the glycosidic bond according to the standard data of partially methylated alditol acetates (PMAAs) from the Complex Carbohydrate Research Centre (CCRC).38 The results implied that HLP50-1 was composed of →4)-D-Glcp-(1→, →6)-D-Manp-(1→, →3)-L-Rhap-(1→, D-Glcp-(1→, L-Araf-(1→, →4,6)-D-Galp-(1→, D-Galp-(1→, →3,6)-D-Glcp-(1→, →2,3,4,)-D-Xylp-(1→, →6)-D-Glcp-(1→, →3)-D-Galp-(1→, →4)-D-Galp-(1→ at a ratio of 4.9:5.0:8.7:5.1:25.9:9.2:3.6:14.6:5.0:4.3:2.7:11.0.
| PMAA | Type of linkage | Molar ratios | Mass fragments (m/z) |
|---|---|---|---|
| 1,4,5-Tri-O-acetyl-2,3,6-tri-O-methyl-D-glucitol | →4)-D-Glcp-(1→ | 4.9 | 43, 55, 71, 87, 99, 101, 117, 129, 131, 161, 173, 233 |
| 1,5,6-Tri-O-acetyl-2,3,4-tri-O-methyl-D-mannitol | →6)-D-Manp-(1→ | 5.0 | 43, 57, 71, 87, 99, 101, 117, 129, 161, 189, 233 |
| 1,3,5-Tri-O-acetyl-2,4-di-O-methyl-6-deoxy-L-mannitol | →3)-L-Rhap-(1→ | 8.7 | 43, 59, 71, 87, 99, 101, 113, 117, 131, 159, 173, 203, 233 |
| 1,5-Di-O-acetyl-2,3,4,6-tetra-O-methyl-D-glucitol | D-Glcp-(1→ | 5.1 | 43, 59, 71, 87, 101, 117, 129, 145, 161, 205 |
| 1,4-Di-O-acetyl-2,3,5-tri-O-methyl-L-arabitol | L-Araf-(1→ | 25.9 | 43, 59, 71, 87, 101, 117, 129, 145, 161 |
| 1,4,5,6-Tetra-O-acetyl-2,3-di-O-methyl-D-galactitol | →4,6)-D-Galp-(1→ | 9.2 | 43, 59, 71, 85, 101, 117, 127, 159, 201, 261 |
| 1,5-Di-O-acetyl-2,3,4,6-tetra-O-methyl-D-galactitol | D-Galp-(1→ | 3.6 | 43, 59, 71, 87, 101, 117, 129, 145, 161, 205 |
| 1,3,5,6-Tetra-O-acetyl-2,4-di-O-methyl-D-glucitol | →3,6)-D-Glcp-(1→ | 14.6 | 43, 59, 73, 87, 101, 117, 129, 159, 173, 189, 233, 305 |
| 1,2,3,4,5-Penta-O-acetyl-D-xylitol | →2,3,4)-D-Xylp-(1→ | 5.0 | 43, 73, 85, 103, 115, 139, 145, 157, 187, 217, 289 |
| 1,5,6-Tri-O-acetyl-2,3,4-tri-O-methyl-D-glucitol | →6)-D-Glcp-(1→ | 4.3 | 43, 71, 87, 99, 101, 117, 129, 161, 189, 233 |
| 1,3,5-Tri-O-acetyl-2,4,6-tri-O-methyl-D-galactitol | →3)-D-Galp-(1→ | 2.7 | 43, 71, 85, 87, 99, 101, 117, 129, 161, 233, 277 |
| 1,4,5-Tri-O-acetyl-2,3,6-tri-O-methyl-D-galactitol | →4)-D-Galp-(1→ | 11.0 | 43, 71, 87, 99, 101, 113, 117, 129, 131, 161, 173, 203, 233 |
3.7. NMR spectroscopy analysis of HLP50-1
To further interpret the structure of the HLP50-1, NMR analysis, including 1D-NMR (1H-NMR, 13C-NMR) and 2D-NMR (HSQC, HMBC and 1H–1H COSY), was performed. First, the amount of sugar residue and the chemical shifts of H1/C1 were determined by GC-MS combined with 1H-NMR, 13C-NMR, and HSQC analysis. The chemical shifts of the adjacent carbons and protons were then assigned using the HMBC and 1H–1H COSY data. Finally, the linkage sites and sequences were predicted by the HMBC spectrum. In addition, the α/β configurations of the sugar residues were decided by the chemical shift of the anomeric carbon and proton and the coupling constant of the anomeric proton.39The 1H-NMR and 13C-NMR (Fig. 3) spectra of HLP50-1 show that the proton and carbon signals were mainly distributed in the range of 3.0–5.5 and 60–110 ppm, respectively.40 The anomeric signals are highlighted in the figures. Importantly, the carbon signals at 171.1 and 23.3 ppm and the proton signal at 1.92 ppm indicate the presence of O-acetyl groups in HLP50-1.41,42 In addition, the 1.92/23.3 and 4.13/83.8 ppm signals in the HSQC (Fig. 3C) spectrum and the cross peaks at 4.13/171.1 and 1.92/171.1 ppm in the HMBC (Fig. 3D) spectrum revealed that the O-acetyl group was attached to the C-2 position of residue G. Based on the HSQC spectrum, the cross peaks at 5.08/100.6, 4.92/100.0, 4.94/107.8, 4.56/104.2, 5.16/109.4, 5.35/108.2, 5.00/107.5, 5.11/106.8, 4.41/103.1, 4.90/97.7, 4.65/100.7, and 5.26/99.7 ppm appeared, indicating that HLP50-1 contained twelve sugar residues (designated as A, B, D, E, F, G, I, J, K, L, M, and N). This finding was consistent with the GC-MS results.
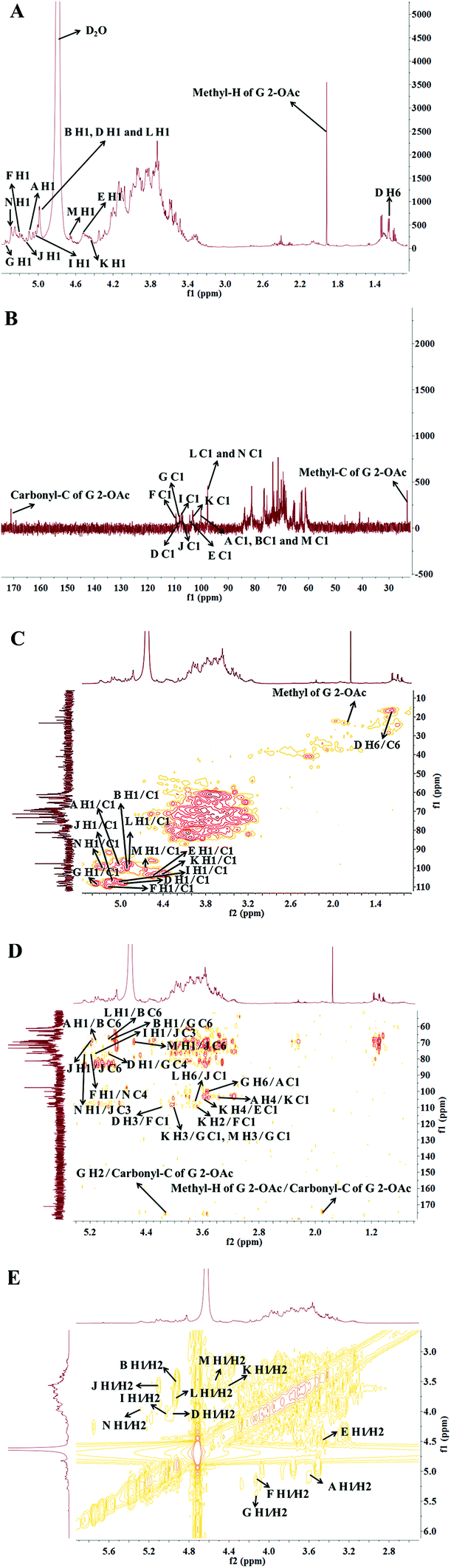 | ||
| Fig. 3 (A) 1H-NMR, (B) 13C-NMR, (C) HSQC, (D) HMBC, and (E) 1H–1H COSY spectra of HLP50-1. | ||
Fig. 3C shows distinct correlation peaks at 1.17/16.5 ppm. These peaks were identified as the H6/C6 signal of the rhamnose residue via the literature38 and monosaccharide composition analysis. The HMBC suggested the adjacent chemical shifts of H/C of this residue. The combination of the downfield shifts of C-3 (76.7 ppm) confirmed that the residue was the (1 → 3) linked rhamnose. Combined with literature data, GC-MS and NMR spectroscopy analyses were used to assign the chemical shifts of the 12 sugar residues of HLP50-1 (Table 2).37–45
| Chemical shifts δ (ppm) | |||||||
|---|---|---|---|---|---|---|---|
| Sugar residue | C1 | C2 | C3 | C4 | C5 | C6 | –Ac |
| H1 | H2 | H3 | H4 | H5 | H6 | ||
| →4)-α-D-Glcp-(1→ | 100.6 | 72.3 | 73.0 | 75.3 | 70.4 | 61.4 | |
| A | 5.08 | 3.58 | 3.13 | 3.37 | 3.76 | 3.64/3.68 | |
| →6)-α-D-Manp-(1→ | 100.0 | 76.1 | 73.5 | 71.9 | 73.7 | 68.7 | |
| B | 4.92 | 3.50 | 3.64 | 3.42 | 4.11 | 3.84/3.86 | |
| →3)-α-L-Rhap-(1→ | 107.8 | 73.9 | 76.7 | 70.4 | 69.0 | 16.5 | |
| D | 4.94 | 3.99 | 4.11 | 3.76 | 3.93 | 1.17 | |
| β-D-Glcp-(1→ | 104.2 | 74.2 | 73.7 | 73.5 | 76.6 | 60.8 | |
| E | 4.56 | 3.45 | 3.49 | 3.87 | 3.51 | 3.40/3.57 | |
| α-L-Araf-(1→ | 109.4 | 81.4 | 76.7 | 81.2 | 61.0 | — | |
| F | 5.16 | 4.14 | 3.99 | 4.04 | 3.76/3.82 | — | |
| →4,6)-2-OAc-β-D-Galp-(1→ | 108.2 | 83.8 | 73.4 | 76.5 | 72.3 | 69.6 | 171.1, 23.3 |
| G | 5.35 | 4.13 | 3.86 | 3.98 | 3.57 | 3.39/3.43 | 1.92 |
| β-D-Galp-(1→ | 107.5 | 74.0 | 72.3 | 73.4 | 76.6 | 62.3 | |
| I | 5.00 | 4.04 | 3.58 | 3.85 | 3.99 | 3.26/3.30 | |
| →3,6)-β-D-Glcp-(1→ | 106.8 | 76.2 | 76.8 | 72.8 | 75.6 | 69.3 | |
| J | 5.11 | 3.58 | 3.56 | 3.69 | 3.30 | 3.15/3.16 | |
| →2,3,4)-α-D-Xylp-(1→ | 103.1 | 80.0 | 76.8 | 81.3 | 60.6 | — | |
| K | 4.41 | 3.68 | 3.93 | 3.56 | 3.82/3.87 | — | |
| →6)-α-D-Glcp-(1→ | 97.7 | 72.8 | 74.2 | 70.4 | 73.9 | 69.1 | |
| L | 4.90 | 3.74 | 4.03 | 3.76 | 3.92 | 3.65/3.68 | |
| →3)-α-D-Galp-(1→ | 100.7 | 72.3 | 76.6 | 72.0 | 73.5 | 60.9 | |
| M | 4.65 | 3.55 | 4.02 | 3.40 | 3.87 | 3.39/3.51 | |
| →4)-α-D-Galp-(1→ | 99.7 | 76.5 | 72.8 | 76.8 | 72.4 | 60.6 | |
| N | 5.26 | 3.97 | 3.70 | 3.30 | 3.56 | 3.11/3.13 | |
The linkage sites and sequences of the sugar residues were further determined by HMBC. The cross peak at 4.11/109.4 ppm (D H3/F C1) in the HMBC spectrum confirmed that the O-3 of residue D was linked to the C-1 of residue F. Similarly, the signals at 4.94/76.5 ppm (D H1/G C4), 3.56/104.2 ppm (K H4/E C1), 3.93/108.2 ppm (K H3/G C1), 3.68/109.4 ppm (K H2/F C1), 4.92/69.6 ppm (B H1/G C6), 3.37/103.1 ppm (A H4/K C1), 5.08/68.7 ppm (A H1/B C6), 5.26/76.8 ppm (N H1/J C3), 5.16/76.8 ppm (F H1/N C4), 3.68/106.8 ppm (L H6/J C1), 4.02/108.2 ppm (M H3/G C1), 5.00/76.8 ppm (I H1/J C3), 4.65/69.3 ppm (M H1/J C6), 3.43/100.6 ppm (G H6/A C1), and 4.90/68.7 ppm (L H1/B C6) suggested that the O-1 of residue D was linked to the C-4 of residue G, the O-4 of residue K was linked to the C-1 of residue E, the O-3 of residue K was linked to the C-1 of residue G, the O-2 of residue K was linked to the C-1 of residue F, the O-1 of residue B was linked to the C-6 of residue G, the O-4 of residue A was linked to the C-1 of residue K, the O-1 of residue A was linked to the C-6 of residue B, the O-1 of residue N was linked to the C-3 of residue J, the O-1 of residue F was linked to the C-4 of residue N, the O-6 of residue L was linked to the C-1 of residue J, the O-3 of residue M was linked to the C-1 of residue G, the O-1 of residue I was linked to the C-3 of residue J, the O-1 of residue M was linked to the C-6 of residue J, the O-6 of residue G was linked to the C-1 of residue A, and the O-1 of residue L was linked to the C-6 of residue B, respectively. Additionally, the cross peak at 5.11/69.3 ppm (J H1/J C6) indicated that the O-1 of residue J was linked to the C-6 of residue J, which implied that there was a repetitive residue J in the HLP50-1. In general, the predicted structure of the HLP50-1 was inferred and presented in Fig. 4. Up to now, the structure of HLP50-1 has not been reported in the literature.
 | ||
| Fig. 4 Predicted structure of HLP50-1. | ||
3.8. Morphological analysis
SEM is commonly used to investigate the surface microstructures and aggregation morphologies of polysaccharides. It is an important tool for studying the microscopic information of polysaccharides. Fig. 5 shows that the HLP50-1 was in the form of irregular fragments of different sizes and shapes, and the surface of the polysaccharide was smooth. In addition, the aggregate of HLP50-1 appeared as an accumulation of crystals. | ||
| Fig. 5 SEM images of HLP50-1 (A: 500×, B: 1500×, C: 3000×, and D: 5000×). | ||
3.9. Effects of HLP50-1 on proliferation of MC3T3-E1 cells
The proliferation rate of the MC3T3-E1 cells first increased and then decreased with increasing concentrations of HLP50-1 (Fig. 6). Moreover, 0.065 μM (P < 0.001), 0.13 μM (P < 0.001), 0.26 μM (P < 0.001), 0.52 μM (P < 0.001), and 1.04 μM (P < 0.05) concentrations of HLP50-1 significantly promoted the proliferation of MC3T3-E1 cells versus the normal group; the proliferation rate reached a maximum (8.1%) for the 0.13 μM group. Thus, the effects of HLP50-1 on the differentiation and mineralization of the MC3T3-E1 cells were studied by selecting HLP50-1 concentrations of 0.065, 0.13, and 0.26 μM.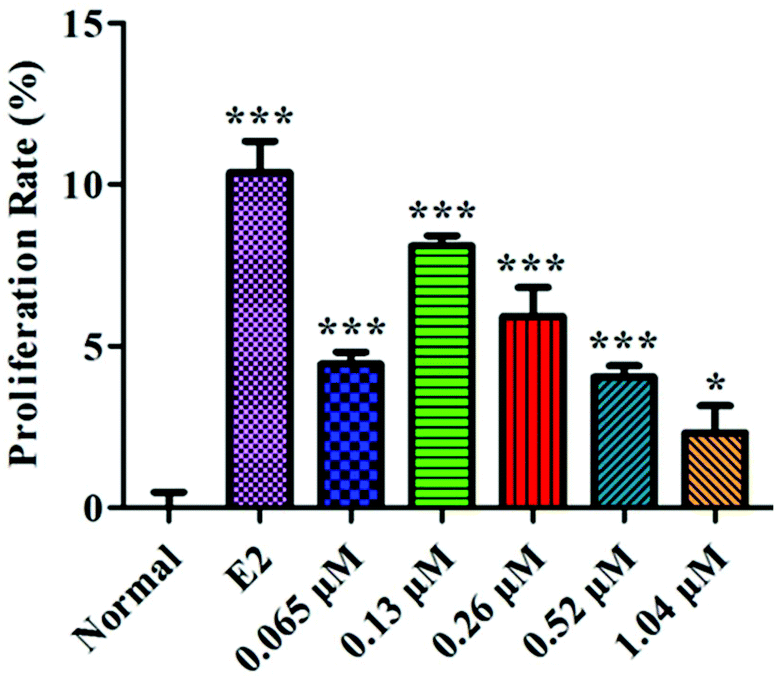 | ||
| Fig. 6 Effects of HLP50-1 on the proliferation of MC3T3-E1 cells. Data are the mean ± SEM. *P < 0.05, ***P < 0.001 versus normal. | ||
3.10. Effects of HLP50-1 on differentiation of MC3T3-E1 cells
ALP is a marker of the early differentiation of osteoblasts. As shown in Fig. 7, the ALP activity of the MC3T3-E1 cells in the control group was significantly higher than that of the normal group (P < 0.001), indicating that the osteoblast-induced cell model was successfully prepared. After 10 d of administration, the 0.13 μM HLP50-1 significantly promoted the ALP activity versus the control group (P < 0.001), suggesting that the effect of the HLP50-1 on the differentiation of the MC3T3-E1 cells was related to its concentration. | ||
| Fig. 7 Effects of HLP50-1 on the ALP activity of MC3T3-E1 cells. Data represent the mean ± SEM. ###P < 0.001 versus normal, ***P < 0.001 versus control. | ||
3.11. Effects of HLP50-1 on mineralization of MC3T3-E1 cells
Alizarin Red S complexed with calcium ions in the matrix to form red deposits. These calcium nodules could be used to qualitatively and quantitively evaluate the mineralization of osteoblasts. The MC3T3-E1 cells in each group with the osteogenic induction medium showed different degrees of mineralization after 18 d of administration (Fig. 8). Many calcium nodules were formed in the E2 and HLP50-1 groups, especially for the 0.13 μM HLP50-1. The mineralization rate of the 0.13 μM HLP50-1 was significantly higher than that of the control group (P < 0.001). However, the low and high concentrations of HLP50-1 had no effect on the mineralization of the MC3T3-E1 cells versus the control group. These results indicated that the 0.13 μM HLP50-1 significantly promoted the mineralization of the MC3T3-E1 cells.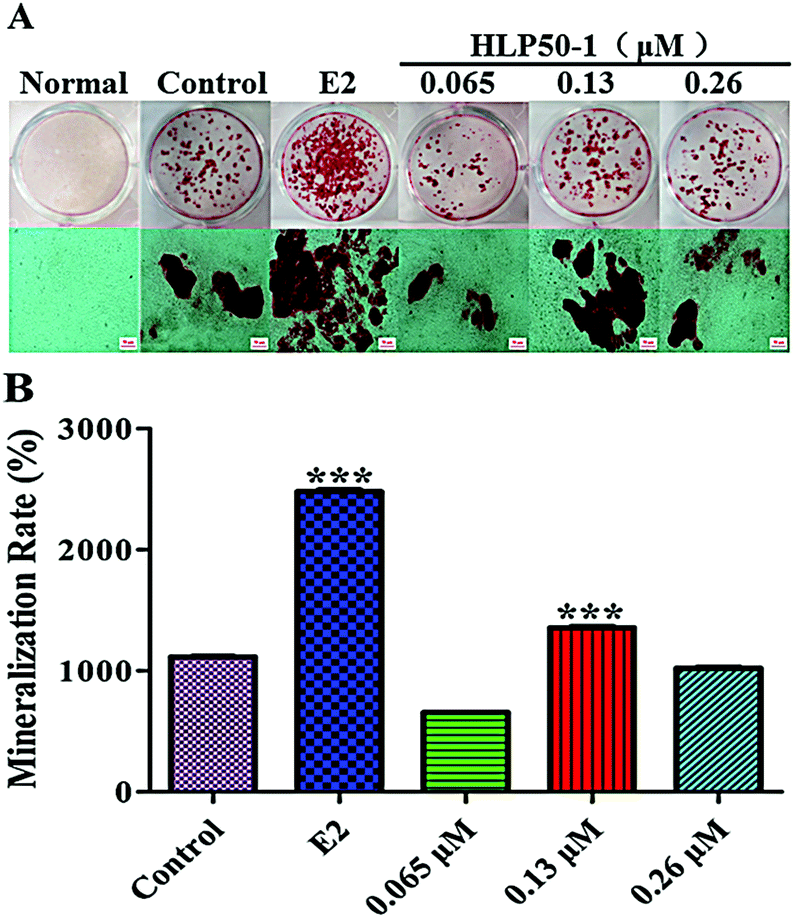 | ||
| Fig. 8 (A) Images of Alizarin Red S staining of calcium nodules. The scale bar represents 50 μm. (B) Quantitation of Alizarin Red S staining of MC3T3-E1 cells treated with HLP50-1 or E2. Data are the mean ± SEM. ***P < 0.001 versus control. | ||
In conclusion, 0.13 μM HLP50-1 had the highest proliferation rate (8.1%), ALP activity (130.7 U L−1), and mineralization rate (1353.6%). It indicated that the HLP50-1 promoted bone formation by promoting osteoblast differentiation and mineralization. But it is worth noting that HLP50-1 differs from HLP50-2, which was isolated and purified in our previous study.13 HLP50-2 was a homogeneous polysaccharide with better osteogenic activities, and comprised glucuronic acid, which may be a crucial structural feature of its anti-osteoporosis activity.46,47 Now, we are conducting transcriptome sequencing of MC3T3-E1 cells treated with H. lupulus polysaccharides, as well as corresponding RT-PCR and western blot detection. We will further study the relationship between the structure and osteogenic activities of H. lupulus polysaccharides based on these results.
4. Conclusions
In summary, a novel homogeneous heteropolysaccharide (HLP50-1; 49.13 kDa) was obtained from the female flowers of H. lupulus. The HLP50-1 was composed of →4)-α-D-Glcp-(1→, →6)-α-D-Manp-(1→, →3)-α-L-Rhap-(1→, β-D-Glcp-(1→, α-L-Araf-(1→, →4,6)-2-OAc-β-D-Galp-(1→, β-D-Galp-(1→, →3,6)-β-D-Glcp-(1→, →2,3,4,)-α-D-Xylp-(1→, →6)-α-D-Glcp-(1→, →3)-α-D-Galp-(1→, →4)-α-D-Galp-(1→, and the structure of the HLP50-1 was inferred for the first time. HLP50-1 took the form of irregular fragments of different sizes and shapes with a smooth surface, and its aggregates were accumulated crystals. Moreover, 0.13 μM HLP50-1 had the highest proliferation rate, ALP activity, and mineralization rate. In conclusion, the HLP50-1 promoted bone formation by promoting osteoblast differentiation and mineralization. Therefore, it may be a candidate for the treatment of osteoporosis. It has broad utility in functional foods, health-care products, and pharmaceuticals. Further study on the mechanism of HLP50-1 against osteoporosis in vitro is underway.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was funded by the National Natural Science Foundation of China (No. 81673557, U1703110 and 81973454) and the Innovative and Upgrading Schools Project of Guangdong Province (No. 2017KZDXM048).References
- X. Li, M. S. Ominsky, K. S. Warmington, S. Morony, J. Gong, J. Cao, Y. Gao, V. Shalhoub, B. Tipton, R. Haldankar, Q. Chen, A. Winters, T. Boone, Z. Geng, Q.-T. Niu, H. Z. Ke, P. J. Kostenuik, W. S. Simonet, D. L. Lacey and C. Paszty, Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis, J. Bone Miner. Res., 2009, 24, 578–588 CrossRef CAS.
- B. L. Riggs, S. Khosla and L. J. Melton, A unitary model for involutional osteoporosis: estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men, J. Bone Miner. Res., 1998, 13, 763–773 CrossRef CAS.
- W. Zhao, G. Shen, H. Ren, T. Qiu, Z. Zhang, J. Tang, K. Chen, D. Liang, Z. Yao, Z. Yang and X. Jiang, Research progress of the active monomer composition of rhizoma drynariae in the regulation of osteoporosis-related signaling pathways, Chin. J. Osteoporosis, 2017, 23, 122–140 Search PubMed.
- K. Jiang, D. Huang, D. Zhang, X. Wang, H. Cao, Q. Zhang and C. Yan, Investigation of inulins from the roots of Morinda officinalis for potential therapeutic application as anti-osteoporosis agent, Int. J. Biol. Macromol., 2018, 120, 170–179 CrossRef CAS.
- L. Du, M.-N. Nong, J.-M. Zhao, X.-M. Peng, S.-H. Zong and G.-F. Zeng, Polygonatum sibiricum polysaccharide inhibits osteoporosis by promoting osteoblast formation and blocking osteoclastogenesis through Wnt/β-catenin signalling pathway, Sci. Rep., 2016, 6, 1–12 CrossRef CAS.
- S. R. Milligan, J. C. Kalita, A. Heyerick, H. Rong, L. D. Cooman and D. D. Keukeleire, Identification of a potent phytoestrogen in hops (Humulus lupulus L.) and beer, J. Clin. Endocrinol. Metab., 1999, 83, 2249–2252 CrossRef PubMed.
- D. D. Yan, Y. F. Wong, R. A. Shellie, P. J. Marriott, S. P. Whittock and A. Koutoulis, Assessment of the phytochemical profiles of novel hop (Humulus lupulus L.) cultivars: A potential route to beer crafting, Food Chem., 2019, 275, 15–23 CrossRef CAS.
- Z. Liu, L. Wang and Y. Liu, Rapid differentiation of Chinese hop varieties (Humulus lupulus) using volatile fingerprinting by HS-SPME-GC-MS combined with multivariate statistical analysis, J. Sci. Food Agric., 2018, 98, 3758–3766 CrossRef CAS.
- E. Zhu, New type of hop product applied in beer production, China Brew., 2006, 10, 53–56 Search PubMed.
- L. Lin, Y. Jiang, Q. Zhang, L. Qin and H. Xin, Research progress of chemical constituents and pharmacological activities in Humulus lupulus, China J. Chin. Mater. Med., 2017, 42, 1830–1836 Search PubMed.
- L. Lin, T. Xia, Y. Jiang, C. Li, L. Qin and H. Xin, Anti-osteoporotic study on xanthohumol, an active ingredient in Humulus lupulus L, J. Pharm. Pract., 2018, 36, 219–223 Search PubMed.
- R. Pacifici, Estrogen, cytokines, and pathogenesis of postmenopausal osteoporosis, J. Bone Miner. Res., 1996, 11, 1043–1051 CrossRef CAS.
- X. Chen, X. Hou, D. Qing, Q. Zhang and C. Yan, Structural identification and osteogenic activity of a novel heteropolysaccharide obtained from female flowers of Humulus lupulus, Food Funct., 2019, 10, 824–835 RSC.
- C. Wang, D. Hua and C. Yan, Structural characterization and antioxidant activities of a novel fructan from Achyranthes bidentata Blume, a famous medicinal plant in China, Ind. Crops Prod., 2015, 70, 427–434 CrossRef CAS.
- M. G. Sevag, D. B. Lackman and J. Smolens, The isolation of the components of streptococcal nucleoproteins in serologically active form, J. Biol. Chem., 1938, 124, 425–436 CAS.
- M. Dubois, K. A. Gilles, J. K. Hamilton, P. A. Rebers and F. Smith, Colorimetric method for determination of sugars and related substances, Anal. Chem., 1956, 28, 350–356 CrossRef CAS.
- Y. Zhang, Z. Liang, W. Zhao, X. Huo, L. Zhang and X. Zhang, Separation, purification and compositional analysis of water soluble polysaccharide GPPS3 from Codonpsis pilosula, Chin. Pharm. J., 2005, 40, 1107–1109 CAS.
- Y. Jiao, D. Hua, D. Huang, Q. Zhang and C. Yan, Characterization of a new heteropolysaccharide from green guava and its application as an α-glucosidase inhibitor for the treatment of type II diabetes, Food Funct., 2018, 9, 3997–4007 RSC.
- N. Li, C. Yan, D. Hua and D. Zhang, Isolation, purification, and structural characterization of a novel polysaccharide from Ganoderma capense, Int. J. Biol. Macromol., 2013, 57, 285–290 CrossRef CAS.
- L. Sun, K. Feng, R. Jiang, J. Chen, Y. Zhao, R. Ma and H. Tong, Water-soluble polysaccharide from Bupleurum chinense DC: Isolation, structural features and antioxidant activity, Carbohydr. Polym., 2010, 79, 180–183 CrossRef CAS.
- K. Jahanbin, Structural characterization of a new water-soluble polysaccharide isolated from Acanthophyllum acerosum roots and its antioxidant activity, Int. J. Biol. Macromol., 2017, 107, 1227–1234 CrossRef.
- Y. Wang, Y. Xu, X. Ma, X. Liu, M. Yang, W. Fan, H. Ren, N. Efehi, X. Wang and X. Zhu, Extraction, purification, characterization and antioxidant activities of polysaccharides from Zizyphus jujuba cv. Linzexiaozao, Int. J. Biol. Macromol., 2018, 118, 2138–2148 CrossRef CAS.
- D. J. Strydom, Chromatographic separation of 1-phenyl-3-methyl-5-pyrazolone- derivatized neutral, acidic and basic aldoses, J. Chromatogr. A, 1994, 678, 17–23 CrossRef CAS.
- J. Yang, B. Zhu, J. Zheng, L. Sun, D. Zhou, X. Dong and C. Yu, Stimulation of lymphocyte proliferation byoyster glycogen sulfated at C-6 position, Carbohydr. Polym., 2013, 94, 301–308 CrossRef CAS.
- P. W. Needs and R. R. Selvendran, Avoiding oxidative degradation during sodium hydroxide/methyl iodide-mediated carbohydrate methylation in dimethyl sulfoxide, Carbohydr. Res., 1993, 245, 1–10 CrossRef CAS.
- A.-P. Chi, J.-P. Chen, Z.-Z. Wang, Z.-Y. Xiong and Q.-X. Li, Morphological and structural characterization of a polysaccharide from Gynostemma pentaphyllum, Makino and its anti-exercise fatigue activity, Carbohydr. Polym., 2008, 74, 868–874 CrossRef CAS.
- S. Song, B. Zhang, S. Wu, L. Huang, C. Ai, J. Pan, Y.-C. Su, Z. Wang and C. Wen, Structural characterization and osteogenic bioactivity of a sulfated polysaccharide from pacific abalone (Haliotis discus hannai Ino), Carbohydr. Polym., 2018, 182, 207–214 CrossRef CAS.
- X. Wang, M. Zhang, D. Zhang, X. Wang, H. Cao, Q. Zhang and C. Yan, Structural elucidation and anti-osteoporosis activities of polysaccharides obtained from Curculigo orchioides, Carbohydr. Polym., 2019, 203, 292–301 CrossRef CAS.
- E. J. Jeon, D.-H. Lee, Y.-J. Kim, J. Ahn, M. J. Kim, J.-T. Hwang, J. Hur, M. Kim, Y.-J. Jang, T.-Y. Ha, D.-H. Seo, J. S. Lee, M. J. Sung and C. H. Jung, Effects of yuja peel extract and its flavanones on osteopenia in ovariectomized rats and osteoblast differentiation, Mol. Nutr. Food Res., 2016, 60, 2587–2601 CrossRef CAS.
- E.-J. Kim, S.-Y. Bu, M.-K. Sung and M.-K. Choi, Effects of silicon on osteoblast activity and bone mineralization of MC3T3-E1 cells, Biol. Trace Elem. Res., 2013, 152, 105–112 CrossRef CAS.
- S. Lei, Bioactivities, isolation and purification methods of polysaccharides from natural products: A review, Int. J. Biol. Macromol., 2016, 92, 37–48 CrossRef.
- J. Xu, S.-L. Li, R.-Q. Yue, C.-H. Ko, J.-M. Hu, J. Liu, H.-M. Ho, T. Yi, Z.-Z. Zhao, J. Zhou, P.-C. Leung, H.-B. Chen and Q.-B. Han, A novel and rapid HPGPC-based strategy for quality control of saccharide-dominant herbal materials: Dendrobium officinale, a case study, Anal. Bioanal. Chem., 2014, 406, 6409–6417 CrossRef CAS.
- K. Jahanbin, A. Abbasian and M. Ahang, Isolation, purification and structural characterization of a new water-soluble polysaccharide from Eremurus stenophyllus (boiss. & buhse) baker roots, Carbohydr. Polym., 2017, 178, 386–393 CrossRef CAS.
- X. Zhai, C. Zhu, Y. Zhang, J. Sun, A. Alim and X. Yang, Chemical characteristics, antioxidant capacities and hepatoprotection of polysaccharides from pomegranate peel, Carbohydr. Polym., 2018, 202, 461–469 CrossRef CAS.
- X. Ji, F. Zhang, R. Zhang, F. Liu, Q. Peng and M. Wang, An acidic polysaccharide from Ziziphus Jujuba cv. Muzao: Purification and structural characterization, Food Chem., 2019, 274, 494–499 CrossRef CAS.
- M. Ka
![[c with combining breve]](https://www.rsc.org/images/entities/char_0063_0306.gif) uráková, P. Capek, V. Sasinková, N. Wellner and A. Ebringerová, FT-IR study of plant cell wall model compounds: pectic polysaccharides and hemicelluloses, Carbohydr. Polym., 2000, 43, 195–203 CrossRef.
uráková, P. Capek, V. Sasinková, N. Wellner and A. Ebringerová, FT-IR study of plant cell wall model compounds: pectic polysaccharides and hemicelluloses, Carbohydr. Polym., 2000, 43, 195–203 CrossRef. - P. B. S. Albuquerque, W. Barros Jr., G. R. C. Santos, M. T. S. Correia, P. A. S. Mourão, J. A. Teixeira and M. G. Carneiro-da-Cunha, Characterization and rheological study of the galactomannan extracted from seeds of Cassia grandis, Carbohydr. Polym., 2014, 104, 127–134 CrossRef CAS.
- N. D. Sanandiya and A. K. Siddhanta, Chemical studies on the polysaccharides of Salicornia brachiate, Carbohydr. Polym., 2014, 112, 300–307 CrossRef CAS.
- Y. Gong, J. Zhang, F. Gao, J. Zhou, Z. Xiang, C. Zhou, L. Wan and J. Chen, Structure Features and in vitro hypoglycemic activities of polysaccharides from different species of Maidong, Carbohydr. Polym., 2017, 173, 215–222 CrossRef CAS PubMed.
- J. M. Hazendonk, E. J. M. Reinerik, P. Waard and J. E. G. Dam, Structural analysis of acetylated hemicellulose polysaccharides from fibre flax (Linum usitatissimum, L.), Carbohydr. Res., 1996, 291, 141–154 CrossRef.
- S. V. Popov, R. G. Ovodova, V. V. Golovchenko, D. S. Khramova, P. A. Markov, V. V. Smirnov, A. S. Shashkov and Y. S. Ovodov, Pectic polysaccharides of the fresh plum Prunus domestica L. isolated with a simulated gastric fluid and their anti-inflammatory and antioxidant activities, Food Chem., 2014, 143, 106–113 CrossRef CAS.
- J. Yang, L. Wen, Y. Zhao, Y. Jiang, M. Tian, H. Liu, J. Liu and B. Yang, Structure identification of an arabinogalacturonan in Citrus reticulata, Blanco ‘Chachiensis’ peel, Food Hydrocolloids, 2018, 84, 481–488 CrossRef CAS.
- J. Yang, J. Tu, H. Liu, L. Wen, Y. Jiang and B. Yang, Identification of an immunostimulatory polysaccharide in banana, Food Chem., 2018, 277, 46–53 CrossRef.
- J. Yang, M. Yi, J. Pan, J. Zhao, L. Sun, X. Lin, Y. Cao, L. Huang, B. Zhu and C. Yu, Sea urchin (Strongylocentrotus intermedius) polysaccharide enhanced BMP-2 induced osteogenic differentiation and its structural analysis, J. Funct. Foods, 2015, 14, 519–528 CrossRef CAS.
- Q. Zhu, Y. Jiang, S. Lin, L. Wen, D. Wu, M. Zhao, F. Chen, Y. Jia and B. Yang, Structural identification of (1→6)-α-D-Glucan, a key responsible for the health benefits of longan, and evaluation of anticancer activity, Biomacromolecules, 2013, 14, 1999–2003 CrossRef CAS PubMed.
- X. Xie, M. Liu and Q. Meng, Angelica polysaccharide promotes proliferation and osteoblast differentiation of mesenchymal stem cells by regulation of long non-coding RNA H19, Bone Joint Res., 2019, 8, 323–333 CrossRef.
- S. K. Tat, J.-P. Pelletier, J. Vergés, D. Lajeunesse, E. Montell, H. Fahmi, M. Lavigne and J. Martel-Pelletier, Chondroitin and glucosamine sulfate in combination decrease the pro-resorptive properties of human osteoarthritis subchondral bone osteoblasts: a basic science study, Arthritis Res. Ther., 2007, 9, R117 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fo01918a |
| This journal is © The Royal Society of Chemistry 2020 |