 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast room-temperature functionalization of silicon nanoparticles using alkyl silanols†
Alyssa F. J.
van den Boom
 a,
Sidharam P.
Pujari
a,
Fatma
Bannani
b,
Hafedh
Driss
c and
Han
Zuilhof
*acd
a,
Sidharam P.
Pujari
a,
Fatma
Bannani
b,
Hafedh
Driss
c and
Han
Zuilhof
*acd
aLaboratory of Organic Chemistry, Wageningen University, Stippeneng 4, 6708 WE Wageningen, The Netherlands. E-mail: han.zuilhof@wur.nl
bDepartment of Chemistry, King Abdulaziz University, Jeddah, Saudi Arabia
cChemical and Materials Engineering, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
dSchool of Pharmaceutical Science and Technology, Tianjin University, 92 Weijin Road, Tianjin, P. R. China
First published on 5th March 2020
Abstract
Silicon nanoparticles (Si NPs) are a good alternative to conventional heavy metal-containing quantum dots in many applications, due to their low toxicity, low cost, and the high natural abundance of the starting material. Recently, much synthetic progress has been made, and crystalline Si NPs can now be prepared in a matter of hours. However, the passivation of these particles is still a time-consuming and difficult process, usually requiring high temperatures and/or harsh reaction conditions. In this paper, we report an easy method for the room-temperature functionalization of hydrogen-terminated Si NPs. Using silanol compounds, a range of functionalized Si NPs could be produced in only 1 h reaction time at room temperature. The coated NPs were fully characterized to determine the efficiency of binding and the effects of coating on the optical properties of the NPs. It was found that Si NPs were effectively functionalized, and that coated NPs could be extracted from the reaction mixture in a straightforward manner. The silanol coating increases the quantum yield of fluorescence, decreases the spectral width and causes a small (∼50 nm) blue-shift in both the excitation and emission spectra of the Si NPs, compared to unfunctionalized particles.
Introduction
Semiconductor nanoparticles have proven to be useful for many different applications, including bio-imaging,1–3 LEDs,4–6 and solar cells.7–12 Silicon nanoparticles (Si NPs) are, in this regard, particularly interesting, due to their low toxicity and the high natural abundance of the starting material.13,14 However, several issues have so far stood in the way of their widespread use. First, synthesis of the NPs is time-consuming and difficult, often requiring high temperatures or harsh chemicals.15–25 Second, once the Si NPs are formed, they have to be rapidly passivated to prevent the loss of their original properties.26 This is especially the case for hydrogen-terminated Si NPs (H-Si NPs). Such particles are of significant interest due to their near-ideal electronic and electrical properties, yet their susceptibility to oxidation requires extensive protection. In the literature, passivation of H-Si NPs has been achieved by heating to 190 °C in the presence of a suitable ligand,27 etching using phosphorous pentachloride followed by modification with an amine,20,28 thermally-promoted thiolation reactions,29 reacting with bifunctional alkenes,26 halogenation followed by a Grignard reaction,24,30 platinum-catalyzed reactions,23,25,31 or reactions in the presence of radical initiators.32,33 These procedures generally suffer from long reaction times and/or the use of non-trivial reaction conditions, making them less attractive for application on a larger scale. Furthermore, the type of ligand that can be attached in such procedures is usually limited by the reaction conditions used. In most cases, the ligand may also contain only one functional group, as having multiple functional groups interferes with the reaction. This limits the possibility for further functionalization. Clearly, an easy, fast, and versatile passivation method for H-Si NPs is still needed.In this paper, we report the use of silanol compounds with a range of functional groups to passivate H-Si NPs (obtained via a microwave-based procedure developed in our group,34 building upon earlier protocols from He’s group for other types of Si NPs).35,36 This silanol reaction showed promise due to the discovery that flat Si surfaces could readily be functionalized in this manner,37 yielding densely packed, hydrolytically stable monolayers in <1 h at room temperature. As this reaction proceeded without the need for additional solvents or catalysts, this constitutes a significant advantage over previously mentioned routes. Here, only mono-hydroxylated silanols were considered, as silanols with two or three hydroxyl groups might yield multilayers on the surface of the Si NPs, as reported for flat surfaces.37 We show that passivation with silanols is an easy and fast method to obtain functionalized Si NPs. The silanol coating can easily be tuned by selecting silanols with different side groups, and due to the specificity of the reaction, even silanols with multiple functional groups can be used. Finally, we show it is possible to obtain pure, passivated Si NPs with good optical properties in less than 4 h of combined reaction (1 h) and purification (3 h) time.
Experimental
Synthetic procedures
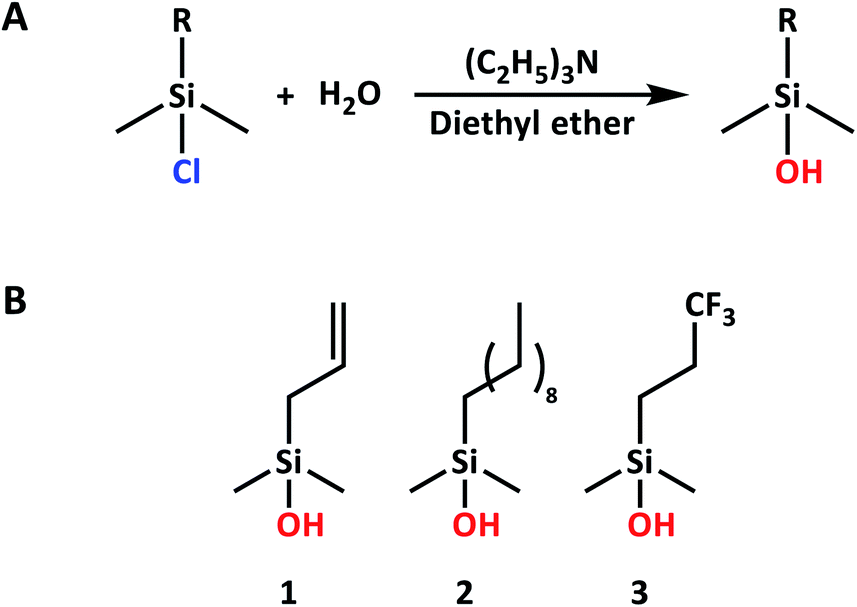 | ||
| Scheme 1 (A) Reaction scheme for silanol synthesis. (B) Overview of silanols used in this study. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 chlorobenzene and DMF was added inside a glovebox.38,39 The reaction mixture was stirred at room temperature for 3.5 h with 365 nm light irradiation. The distance between the lamp and the solution was ∼4 cm, so as not to heat the sample. The lamp was removed, and the solvents were evaporated in vacuo. A light brown gel formed, from which Si NPs and DMPA were extracted using chlorobenzene. The solvent was again removed in vacuo, and the functionalized Si NPs were extracted using diethyl ether.
:1 chlorobenzene and DMF was added inside a glovebox.38,39 The reaction mixture was stirred at room temperature for 3.5 h with 365 nm light irradiation. The distance between the lamp and the solution was ∼4 cm, so as not to heat the sample. The lamp was removed, and the solvents were evaporated in vacuo. A light brown gel formed, from which Si NPs and DMPA were extracted using chlorobenzene. The solvent was again removed in vacuo, and the functionalized Si NPs were extracted using diethyl ether.
To de-protect the acetate-capped sugar, the Si NPs were dispersed in a 0.015 M solution of sodium methoxide in methanol.40 The reaction mixture was stirred for 45 min, and sodium methoxide was precipitated out of the solution by the addition of anhydrous pentane. The solvents in the clear dispersion were removed in vacuo to obtain 27 mg of dry thioglucose-coated Si NPs for analysis.
Materials and methods
Results and discussion
Silanol-based functionalization of silicon nanoparticles
The passivation of H-Si NPs with silanol compounds (Fig. 1) proceeded readily at room temperature with a straightforward procedure: silanol was simply added to a H-Si NP suspension in DMSO (as obtained without further purification from Si NP synthesis), and the mixture was stirred for 1 h at room temperature. The ease of this reaction is in line with the calculated exothermicity, namely ΔH = −16.9 kcal mol−1 in DMSO, as obtained from quantum-chemical wB97XD/6-311+G(d,p) calculations using a dielectric SMD model mimicking DMSO. The reason for the extra 30 min of stirring time, compared to the analogous reaction on flat surfaces,37 was to ensure a high packing density on the Si NP surface, as this would minimize future oxidation. Escorihuela et al. further reported that for flat surfaces, the reaction speed could be increased by irradiation with UV light. However, this was not tested here, as the subsequent (relatively slow) evaporation of DMSO in vacuo meant the Si NPs would be exposed to silanol for at least 2 h anyway.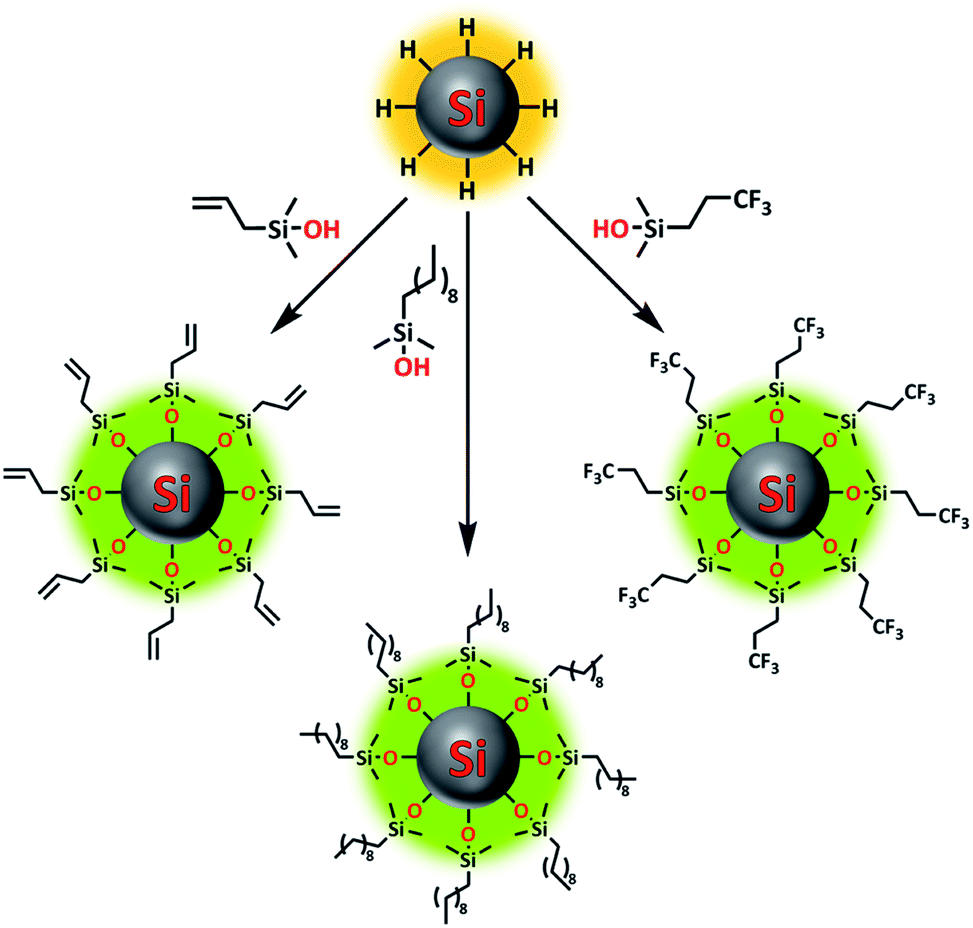 | ||
| Fig. 1 Overview image showing functionalization of silicon nanoparticles using various silanol compounds. | ||
For both silanols 1 and 3, excess silanol was removed simultaneously with DMSO, simplifying the purification procedure; only one extraction step with diethyl ether was necessary to remove the remaining traces of unbound silanol and obtain a dry powder. Meanwhile, 2 could not be removed by vacuum at the current temperature. Therefore, for this silanol, three extractions with diethyl ether were performed to obtain a dry powder. In all cases, an estimated yield of ∼45% was obtained, assuming all Si NP surface atoms had reacted with the silanol, and no Si–H bonds remained.
To confirm that the passivation process did not affect the size of the Si NPs, TEM imaging was performed. For both passivated and non-passivated Si NPs, the average size of the particles was around 6 ± 1 nm (Fig. S13†), slightly (∼1.5 nm) smaller than previously reported by Pujari et al. for H-Si NPs synthesized using similar conditions.34
High-resolution TEM of single Si NPs confirmed the crystallinity of the functionalized NPs, in line with data shown by Pujari et al.34 Additionally, the interplanar spacing of the crystalline particles was measured to be 0.31 nm in the (111) direction, and 0.19 nm in the (110) direction (Fig. 2D and E). This corresponds to literature values found for crystalline silicon wafers (3.13 and 1.92 Å, respectively).42
 | ||
| Fig. 2 High-resolution TEM images of 3-Si NPs. Scale bars are 200 nm (A) and 10 nm (B and C). The insert in (A) shows the size distribution. (D) shows the interplanar spacing of a Si(111) NP measured along the red line in (C), and (E) shows the spacing of a Si(110) NP measured in (B). | ||
Recently, a paper by Oliinyk et al. raised the question whether Si NPs could really be formed in a microwave reaction.43 Even though they specifically doubted their formation in an aqueous solution, and suggested the formation of fluorescent NPs of a carbon or hybrid composition, their investigation is also relevant here. Based on the interplanar crystal spacing found, and the silicon signal observed using XPS (vide infra), we would argue that crystalline Si NPs were indeed formed in this reaction, rather than hybrid organo-silicon or carbon NPs. Furthermore, Oliinyk et al. attributed the source of fluorescence in the paper by Zhong et al.36 to the formation of carbon nanoparticles and/or organic fluorophores. If this had been the case here, the organic fluorophores should have been visible in the NMR and IR spectra discussed below. However, in these spectra, only signals from the silanol coating could be seen, indicating that no other functional groups were present in the sample. Combined, these data imply the formation of well-defined, fluorescent, crystalline Si NPs, rather than non-crystalline and hybrid carbon nanodots, in the microwave reaction described above.
Characterization of silanol-functionalized particles
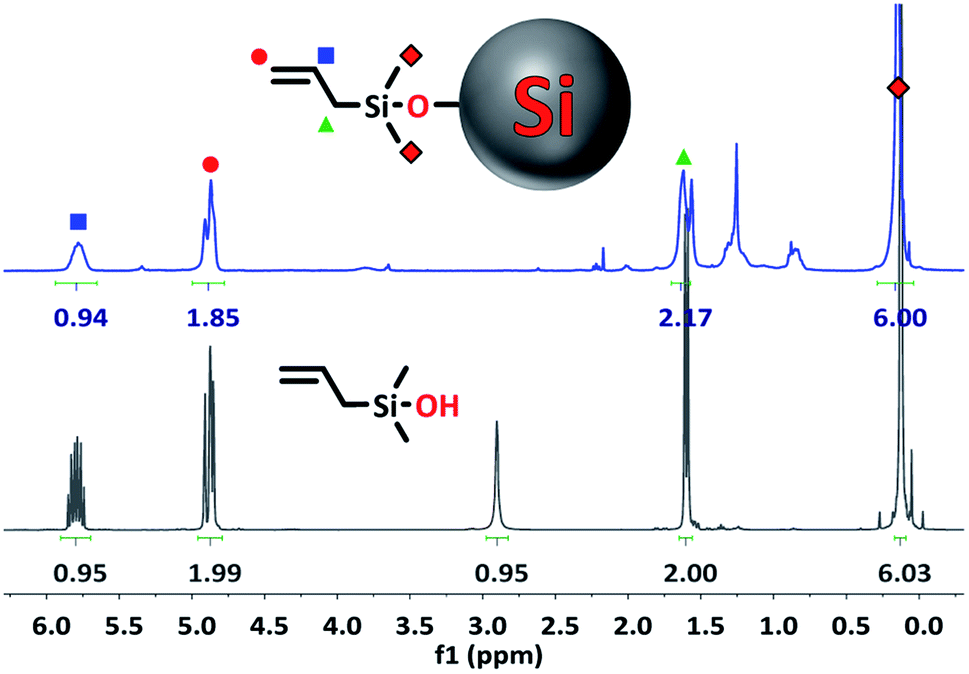 | ||
| Fig. 3 1H NMR spectrum of 1-Si NPs (top) and pure unbound 1 (bottom) in CDCl3. | ||
All signals belonging to the alkyl groups of the silanol can clearly be seen in the spectrum of the functionalized Si NPs, while the signal from the –OH group has disappeared (5.55 ppm in DMSO-d6, Fig. S1†). This indicates that the silanol has successfully reacted via the –OH group. In addition, the integration ratio of the peaks closely matches the ratio found for the unbound silanol, although some deviation is sometimes found due to the low signal of the functionalized NPs.
Binding is further confirmed by the broadening of the silanol peaks, caused by attachment of the organic compound to a slowly-rotating NP.19,25 Since rotation speed is related to size, more severe peak broadening is observed for larger particles. This is why the peak broadening here is more severe compared to the spectra of the 1–4 nm NPs reported in the literature.19,25 Meanwhile, 1H NMR of TiO2 NPs of ∼10.4 × 7.4 nm showed spectra with nearly unresolvable peaks.44,45
Final proof of successful binding is given by the slight change in chemical shift for hydrogen atoms belonging to the methyl groups of the silanol (Fig. 3, ∼0.2 ppm). This shift is upfield when measured in CDCl3, and downfield when measured in DMSO-d6. Hydrogen atoms further up the alkyl chain of the silanol seem to be unaffected by the Si NP surface.
In addition to this, only small contaminant peaks can be found in the spectrum, indicating that the purification was successful. Since NPs, due to their slow-tumbling nature, only give very small signals compared to organic compounds, any contaminants are clearly visible in the spectrum.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond vibrations at 1632, 990, 932, and 893 cm−1, and the C–H vibration at 3080 cm−1. Additionally, the strong band at 1059 cm−1 indicates the presence of a Si–O–Si bond, as expected after passivation, while the absence of a broad peak at ∼3270 cm−1 reflects the disappearance of the –OH group.
C bond vibrations at 1632, 990, 932, and 893 cm−1, and the C–H vibration at 3080 cm−1. Additionally, the strong band at 1059 cm−1 indicates the presence of a Si–O–Si bond, as expected after passivation, while the absence of a broad peak at ∼3270 cm−1 reflects the disappearance of the –OH group.
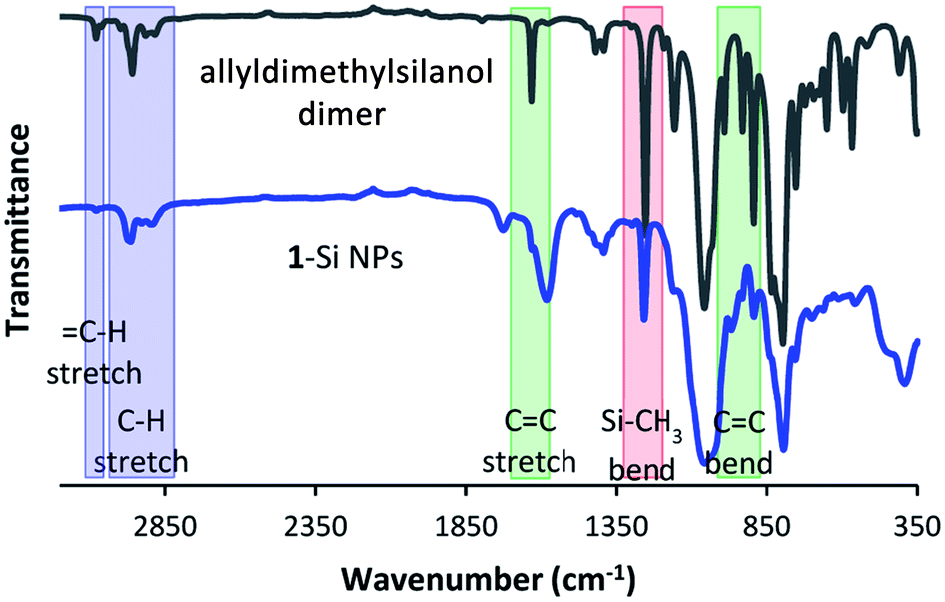 | ||
| Fig. 4 ATR-IR spectra of 12 and 1-Si NPs. Several notable peaks are highlighted. | ||
For this sample in particular, the IR spectrum is interesting, as it provides further evidence that the silanol has indeed reacted via the –OH group, rather than the vinyl group. Not only would binding via the vinyl group lead to the disappearance of the CC signals at 1632, 990, 932 and 893 cm−1, it would also lead to the formation of a new signal at around 1210 cm−1.46
The absence of this latter peak is not directly clear from the spectrum in Fig. 4. However, when the difference spectrum between passivated and non-passivated Si NPs is plotted, the lack of signal in this region becomes clear (Fig. S5†). In addition, further signals characteristic of 1 also show up in the difference spectrum, including several peaks in the fingerprint region. This is also observed for the other passivations: the difference spectra show characteristic peaks belonging to the silanol used during the reaction (Fig. S6 and S7†). Only for 2-Si NPs is the comparison less clear, due to the small number of characteristic peaks that can be uniquely attributed to 2.
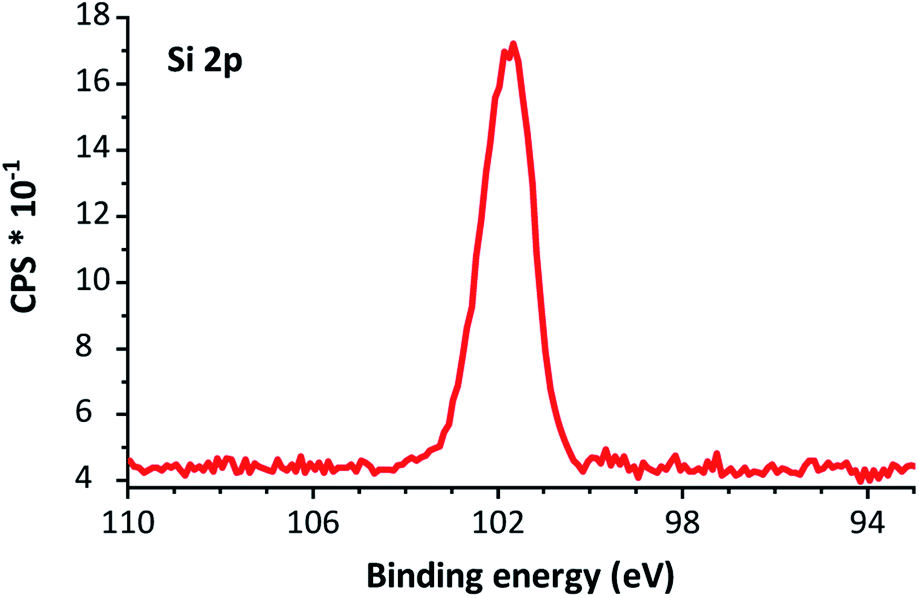 | ||
| Fig. 5 XPS silicon 2p narrow scan of 2-Si NPs. The peak is centered around 101.8 eV. | ||
As no peak at >103 eV is present in Fig. 5, we tentatively attribute the peak at 1060 cm−1, as observed in the IR spectrum, to the Si–O–Si bonds from the silanol functionalization, rather than oxidation by atmospheric oxygen.
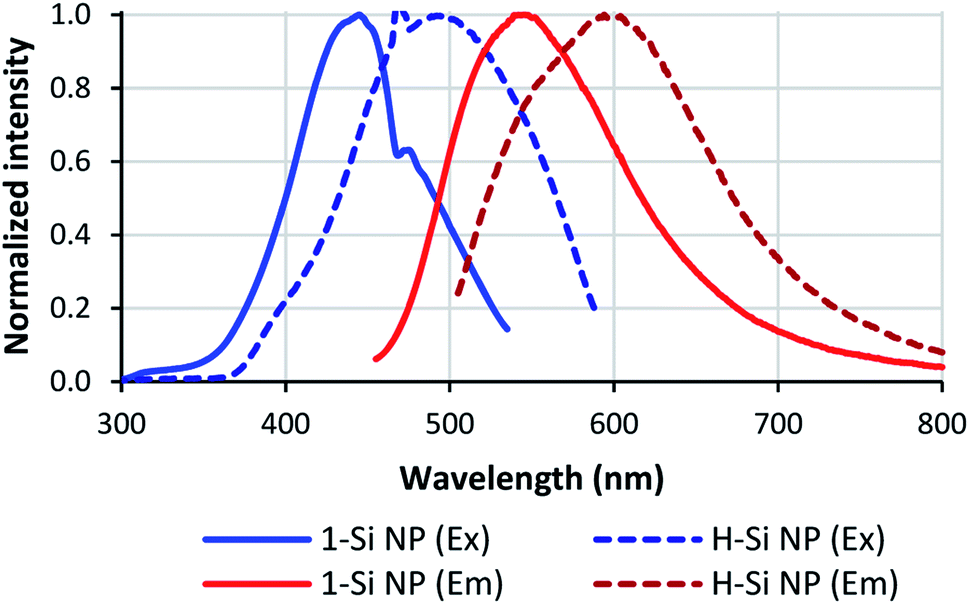 | ||
| Fig. 6 Optical properties of Si NPs. Full lines: Si NPs passivated with 1; dashed lines: non-passivated Si NPs. Excitation spectra are shown in blue, emission spectra in red. | ||
Besides shifting the positions of the excitation and emission maxima, the silanol coating seems to reduce the spectral width of the Si NPs. For non-passivated Si NPs, the spectral widths of the excitation and emission spectra were 0.68 and 0.53 eV, respectively. For 1-passivated Si NPs the excitation width decreased to 0.57 eV, and for 3-Si NPs the excitation and emission width decreased to 0.43 and 0.41 eV, respectively. Meanwhile, no change in spectral width was observed for 2-Si NPs; the excitation spectrum had a spectral width of 0.69 eV, and the emission spectrum a width of 0.50 eV.
Finally, the fluorescence lifetime and quantum yield were also affected by the passivation process. The average fluorescence lifetime of 3-Si NPs increased from 4.1 to 6.9 ns after passivation, while for 2-Si NPs the lifetime decreased to 3.3 ns. These short ns lifetimes are typical for Si NPs with alkyl termination, prepared using wet-chemical methods, as shown in previous studies on free-standing Si NPs.31,48–52 In addition, it has been shown that an electronegative cap (such as silanol) or an electronegative environment (such as DMSO) can also cause short ns lifetimes for Si NPs.53
The quantum yield of the Si NPs increased for all silanols, though the increase for 2-Si NPs was not as large as for the other silanols. This increase could be partially due to the purification procedure for the passivated Si NPs; as the H-Si NP sample was not purified before quantum yield determination (to minimize oxidation), this sample contains traces of non-fluorescent impurities that increase the optical absorption of the sample, and thereby slightly decrease the apparent quantum yield for the H-Si NPs compared to the purified samples. The quantum yield for these silanol-functionalized Si NPs compares favorably with other modes of functionalization.34
Reactivity of silanol-based Si NPs: water-soluble, glucose-coated Si NPs
As a proof of concept, to show that water-soluble, bio-functional Si NPs could be prepared in this manner, 1-thio-β-D-glucose tetraacetate was reacted with 1-Si NPs via a thiol-ene click reaction (Fig. 7 and 8). Previous research on thiol-ene click reactions used a reaction time of 1 h.47 Here, due to the size and bulkiness of the sugar ligand, a longer reaction time of 3.5 h was used, to ensure a higher packing density. Due to the extensive purification needed during synthesis of these NPs, and the small scale of the experiment (30 mg of H-Si NPs), this reaction yielded only 27 mg. Successful binding of the protected thiol-sugar was confirmed by IR, with the disappearance of the S–H stretch vibration after the click reaction (Fig. S15†).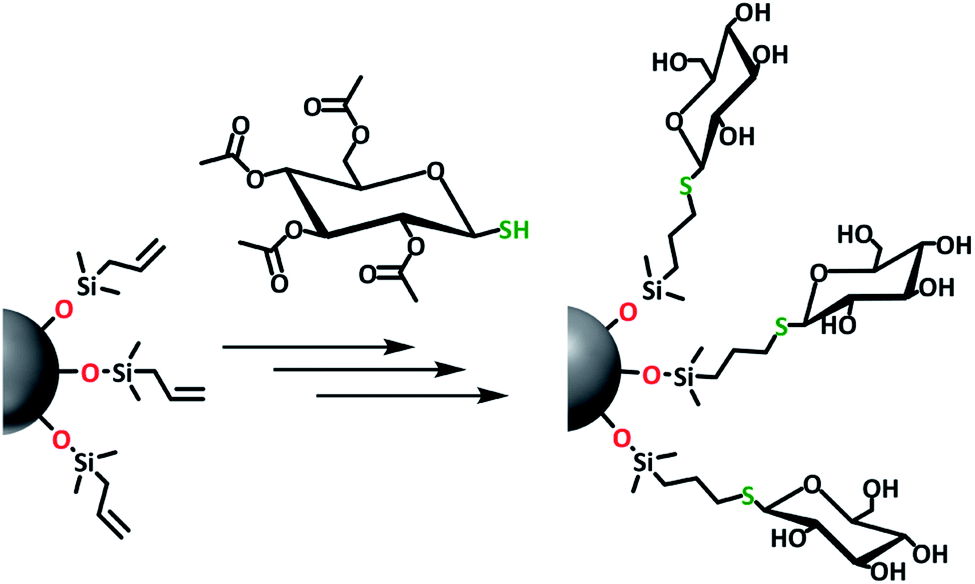 | ||
| Fig. 7 Schematic overview of the synthesis of water-soluble, glucose-coated Si NPs. | ||
 | ||
| Fig. 8 Glucose-coated Si NPs in water under normal light (left) and 365 nm irradiation (middle). The right picture shows the solubility of glucose-coated Si NPs in hexane (top layer) and water (bottom layer). | ||
After deprotection, the thioglucose-coated Si NPs were dispersed in water for fluorescence measurements. An excitation maximum of 370 nm and an emission maximum of 460 nm were found, with a quantum yield of 35% compared to quinine sulfate (Fig. S12†). This is a significant blue-shift compared to the Si NPs coated only with silanol, likely due to UV-induced oxidation.
Conclusions
We have demonstrated a fast, easy and efficient way to passivate hydrogen-terminated Si nanoparticles (H-Si NPs) using silanol compounds at room temperature. The reaction was specific enough to allow the presence of other functional groups in the silanol ligand, and both the passivation and purification of the Si NPs were simple procedures with opportunities for scale-up. The silanol-coated Si NPs produced here showed little oxidation and improved quantum yields. Finally, purified silanol-coated Si NPs could be produced and purified from hydrogen-terminated Si NPs in under 4 h, showing a significant improvement over previous methods for H-Si NP functionalization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Netherlands Organization for Scientific Research (NWO) in the framework of the Materials for Sustainability program. Low-resolution electron microscopy work was performed at the Wageningen Electron Microscopy Center (WEMC) of Wageningen University. Barend van Lagen is acknowledged for helpful instrumental assistance.Notes and references
- X. Cheng, S. B. Lowe, P. J. Reece and J. J. Gooding, Chem. Soc. Rev., 2014, 43, 2680 RSC.
- Y. Su, X. Ji and Y. He, Adv. Mater., 2016, 28, 10567 CrossRef CAS PubMed.
- S. Wu, Y. Zhong, Y. Zhou, B. Song, B. Chu, X. Ji, Y. Wu, Y. Su and Y. He, J. Am. Chem. Soc., 2015, 137, 14726 CrossRef CAS PubMed.
- B. Ghosh, Y. Masuda, Y. Wakayama, Y. Imanaka, J. Inoue, K. Hashi, K. Deguchi, H. Yamada, Y. Sakka and S. Ohki, et al. , Adv. Funct. Mater., 2014, 24, 7151 CAS.
- S. Coe, W.-K. Woo, M. Bawendi and V. Bulovic, Nature, 2002, 420, 800 CrossRef CAS PubMed.
- L. Yang, Y. Liu, Y.-L. Zhong, X.-X. Jiang, B. Song, X.-Y. Ji, Y.-Y. Su, L.-S. Liao and Y. He, Appl. Phys. Lett., 2015, 106, 173109 CrossRef.
- I. Robel, V. Subramanian, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2006, 128, 2385 CrossRef CAS PubMed.
- G. Y. Kwak, T. G. Kim, S. Hong, A. Kim, M. H. Ha and K. J. Kim, Sol. Energy, 2018, 164, 89 CrossRef CAS.
- B. Liu, J. Hu, L. Jia, J. Liu, X. Ren, X. Zhang, X. Guo and S. Liu, Sol. Energy, 2018, 167, 102 CrossRef CAS.
- T. Subramani, J. Chen, Y. L. Sun, W. Jevasuwan and N. Fukata, Nano Energy, 2017, 35, 154 CrossRef CAS.
- J. Duan, H. Zhang, Q. Tang, B. He and L. Yu, J. Mater. Chem. A, 2015, 3, 17497 RSC.
- B. Song and Y. He, Nano Today, 2019, 26, 149 CrossRef CAS.
- S. Bhattacharjee, I. M. C. M. Rietjens, M. P. Singh, T. M. Atkins, T. K. Purkait, Z. Xu, S. Regli, A. Shukaliak, R. J. Clark and B. S. Mitchell, et al. , Nanoscale, 2013, 5, 4870 RSC.
- L. Ruizendaal, S. Bhattacharjee, K. Pournazari, M. Rosso-Vasic, L. H. J. De Haan, G. M. Alink, A. T. M. Marcelis and H. Zuilhof, Nanotoxicology, 2009, 3, 339 CrossRef CAS.
- X. Li, Y. He, S. S. Talukdar and M. T. Swihart, Langmuir, 2003, 19, 8490 CrossRef CAS.
- F. Erogbogbo, K. T. Yong, R. Hu, W. C. Law, H. Ding, C. W. Chang, P. N. Prasad and M. T. Swihart, ACS Nano, 2010, 4, 5131 CrossRef CAS PubMed.
- W. Ren, Y. Wang, Q. Tan, J. Yu, U. J. Etim, Z. Zhong and F. Su, Electrochim. Acta, 2019, 320, 134625 CrossRef CAS.
- F. Erogbogbo, C. A. Tien, C. W. Chang, K. T. Yong, W. C. Law, H. Ding, I. Roy, M. T. Swihart and P. N. Prasad, Bioconjugate Chem., 2011, 22, 1081 CrossRef CAS.
- A. Shiohara, S. Hanada, S. Prabakar, K. Fujioka, T. H. Lim, K. Yamamoto, P. T. Northcote and R. D. Tilley, J. Am. Chem. Soc., 2010, 132, 248 CrossRef CAS PubMed.
- M. Dasog, G. B. De los Reyes, L. V. Titova, F. A. Hegmann and J. G. C. Veinot, ACS Nano, 2014, 8, 9636 CrossRef CAS PubMed.
- C. M. Hessel, E. J. Henderson and J. G. C. Veinot, Chem. Mater., 2006, 18, 6139 CrossRef CAS.
- Z. Kang, Y. Liu, C. H. A. Tsang, D. D. D. Ma, X. Fan, N.-B. Wong and S.-T. Lee, Adv. Mater., 2009, 21, 661 CrossRef CAS.
- M. Rosso-vasic, E. Spruijt, Z. Popovíc, K. Overgaag, B. van Lagen, B. Grandidier, D. Vanmaekelbergh, D. Domínguez-Gutiérrez, L. De Cola and H. Zuilhof, J. Mater. Chem., 2009, 19, 5926 RSC.
- C.-S. Yang, R. A. Bley, S. M. Kauzlarich, H. W. H. Lee and G. R. Delgado, J. Am. Chem. Soc., 1999, 121, 5191 CrossRef CAS.
- W. Biesta, B. van Lagen, V. S. Gevaert, A. T. M. Marcelis, J. M. J. Paulusse, M. W. F. Nielen and H. Zuilhof, Chem. Mater., 2012, 24, 4311 CrossRef CAS.
- Y. Yu, C. M. Hessel, T. D. Bogart, M. G. Panthani, M. R. Rasch and B. A. Korgel, Langmuir, 2013, 29, 1533 CrossRef CAS PubMed.
- J. A. Kelly, A. M. Shukaliak, M. D. Fleischauer and J. G. C. Veinot, J. Am. Chem. Soc., 2011, 133, 9564 CrossRef CAS PubMed.
- M. Dasog and J. G. C. Veinot, Phys. Status Solidi A, 2012, 209, 1844 CrossRef CAS.
- Y. Yu, C. E. Rowland, R. D. Schaller and B. A. Korgel, Langmuir, 2015, 31, 6886 CrossRef CAS PubMed.
- M. Dasog, K. Bader and J. G. C. Veinot, Chem. Mater., 2015, 27, 1153 CrossRef CAS.
- M. Rosso-vasic, E. Spruijt, B. Van Lagen, L. De Cola and H. Zuilhof, Small, 2008, 4, 1835 CrossRef CAS.
- I. M. D. Höhlein, J. Kehrle, T. K. Purkait, J. G. C. Veinot and B. Rieger, Nanoscale, 2015, 7, 914 RSC.
- J. M. Buriak, Chem. Mater., 2014, 26, 763 CrossRef CAS.
- S. P. Pujari, H. Driss, F. Bannani, B. van Lagen and H. Zuilhof, Chem. Mater., 2018, 30, 6503 CrossRef CAS PubMed.
- Y. Zhong, X. Sun, S. Wang, F. Peng, F. Bao, Y. Su, Y. Li, S.-T. Lee and Y. He, ACS Nano, 2015, 9, 5958 CrossRef CAS PubMed.
- Y. Zhong, F. Peng, F. Bao, S. Wang, X. Ji, L. Yang, Y. Su, S.-T. Lee and Y. He, J. Am. Chem. Soc., 2013, 135, 8350 CrossRef CAS PubMed.
- J. Escorihuela and H. Zuilhof, J. Am. Chem. Soc., 2017, 139, 5870 CrossRef CAS PubMed.
- N. S. Bhairamadgi, S. Gangarapu, M. A. Caipa Campos, J. M. J. Paulusse, C. J. M. Van Rijn and H. Zuilhof, Langmuir, 2013, 29, 4535 CrossRef CAS.
- M. A. C. Campos, J. M. J. Paulusse and H. Zuilhof, Chem. Commun., 2010, 46, 5512 RSC.
- K. Ágoston, A. Dobó, J. Rákó, J. Kerékgyárto and Z. Szurmai, Carbohydr. Res., 2001, 330, 183 CrossRef.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850 RSC.
- C. Xiao, J. Guo, P. Zhang, C. Chen, L. Chen and L. Qian, Sci. Rep., 2017, 7, 40750 CrossRef CAS PubMed.
- B. V. Oliinyk, D. Korytko, V. Lysenko and S. Alekseev, Chem. Mater., 2019, 31, 7167 CrossRef CAS.
- E. Schechtel, R. Dören, H. Frerichs, M. Panthöfer, M. Mondeshki and W. Tremel, Langmuir, 2019, 35, 12518 CrossRef CAS PubMed.
- Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211 CrossRef CAS.
- B. Arkles and G. Larson, Silicon Compounds: Silanes & Silicones, Gelest Inc., Morrisville, 3rd edn, 2013 Search PubMed.
- L. Ruizendaal, S. P. Pujari, V. Gevaerts, J. M. J. Paulusse and H. Zuilhof, Chem.–Asian J., 2011, 6, 2776 CrossRef CAS PubMed.
- K. Dohnalová, A. N. Poddubny, A. A. Prokofiev, W. D. De Boer, C. P. Umesh, J. M. Paulusse, H. Zuilhof and T. Gregorkiewicz, Light: Sci. Appl., 2013, 2, e47 CrossRef.
- K. Dohnalová, A. Fučíková, C. P. Umesh, J. Humpolíčková, J. M. J. Paulusse, J. Valenta, H. Zuilhof, M. Hof and T. Gregorkiewicz, Small, 2012, 8, 3185 CrossRef PubMed.
- D. S. English, L. E. Pell, Z. Yu, P. F. Barbara and B. A. Korgel, Nano Lett., 2002, 2, 681 CrossRef CAS.
- K. Kůsova, O. Cibulka, K. Dohnalová, I. Pelant, J. Valenta, A. Fučíková, K. Žídek, J. Lang, J. Englich and P. Matějka, et al. , ACS Nano, 2010, 4, 4495 CrossRef PubMed.
- J. H. Warner, A. Hoshino, K. Yamamoto and R. D. Tilley, Angew. Chem., Int. Ed., 2005, 44, 4550 CrossRef CAS PubMed.
- A. N. Poddubny and K. Dohnalová, Phys. Rev. B, 2014, 90, 245439 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fd00102f |
| This journal is © The Royal Society of Chemistry 2020 |