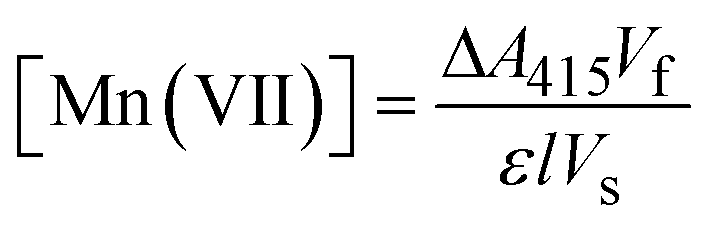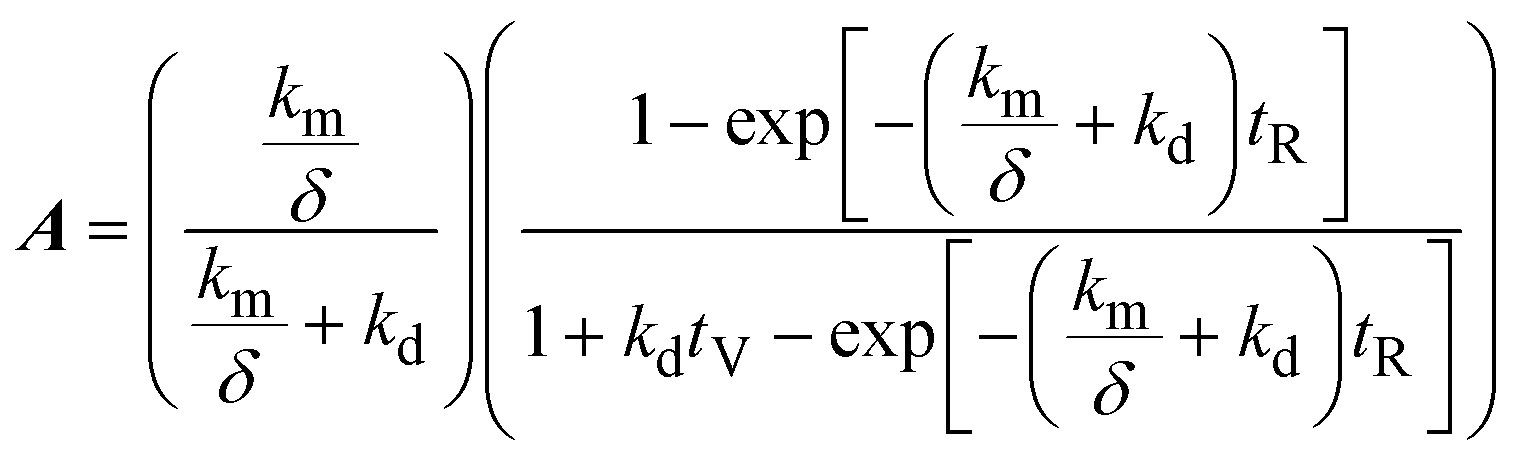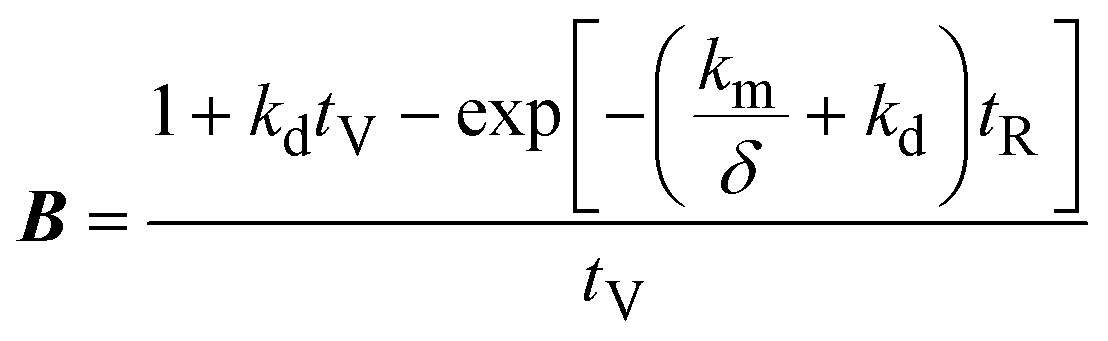Advanced electrochemical oxidation for the simultaneous removal of manganese and generation of permanganate oxidant†
Received
22nd March 2020
, Accepted 9th July 2020
First published on 10th July 2020
Abstract
Emerging electrochemical systems, such as advanced electro-oxidation, provide a potentially powerful alternative to conventional oxidation processes which can often be unsuitable for small, remote and decentralised system applications. The one electro-oxidation process, which may be well suited for these applications, is the use of high oxygen overpotential boron-doped diamond (BDD) electrodes, as a pre-oxidation step for the removal of various raw water contaminants. While BDD electro-oxidation has been studied extensively for the abatement of organic micropollutants, its application as a pre-oxidation technology for the removal of soluble manganese (Mn2+) in source waters for drinking water supply, has not been reported to-date. In this study, we summarise the results of tests using a bench-scale electro-oxidation system and synthetic Mn2+ solutions in order to consider the simultaneous removal of manganese and the generation of permanganate. The results showed that total manganese was reduced by 9.1, 38.7 and 57.4% at current densities of 10, 40 and 80 mA cm−2, respectively, with an initial Mn2+ concentration of 39 μM. Increased Mn removal at higher current density was attributed to increased generation of, and reaction with, hydroxyl radicals, indicated by a direct proportional relationship between pseudo-first order reaction rate constants for methanol (˙OH radical scavenger) and current density. A mathematical model was developed to describe Mn removal under mass transport limitations, and was found to correlate well with experimental data. Finally, a completely novel synthesis pathway for the generation of permanganate species (Mn7+) is presented, whereby concentrations up to 0.9 μM were synthesised from Mn2+ in circumneutral conditions.
Water impact
Electrochemical water treatment technologies present a promising alternative to conventional processes, particularly for small, remote and/or decentralised applications. Among these, advanced electro-oxidation processes using high oxygen overpotential electrodes like boron-doped diamond, already established for the efficient removal of various organic micropollutants, may also be applied advantageously as a pre-oxidation technique for the abatement of inorganic contaminants such as manganese.
|
1. Introduction
Electrochemical water treatment technologies are garnering much attention at present, as they provide a promising alternative to conventional processes for various water and wastewater applications. Much of this attention is in part due to their potential suitability for applications in small, remote and/or point-of-entry systems, as they can eliminate the chemical supply chain associated with conventional treatment processes through the electrochemical generation of process chemicals, on-site and on-demand. While technologies like electrocoagulation have been shown to be promising for remote drinking water system applications for both organic and inorganic contaminants,1,2 the process of electrochemical oxidation (electro-oxidation) has not yet received the same degree of attention. Furthermore, with the advent of advanced materials which decrease the parasitic oxygen evolution reaction (OER) potential, more sophisticated electrochemical oxidation processes such as advanced oxidation can proceed, as opposed to oxidation being limited directly on the electrode surface. One particular electrode material, which has been widely observed to treat effectively a number of common raw water pollutants through electro-oxidation, is boron-doped diamond (BDD).3
BDD electro-oxidation can proceed via a number of mechanisms including direct oxidation at the electrode surface, although most published research has indicated that the predominating mechanism is mediated through the generation of hydroxyl radicals (R1):4,5
| | | BDD + H2O → BDD(˙OH) + H+ + e− | (R1) |
At a lesser rate, it has also been shown that BDD electrodes are capable of generating other reactive oxygen species such as hydrogen peroxide
(R2)5,6 and ozone
(R3) and (R4):
6–8The most common and widely researched mechanism is the ˙OH radical generation by the BDD anode
(R1), which has been shown to be an efficient process for the treatment of a number of organic micro-pollutants.
3 Although effective pollutant degradation and mineralization has been demonstrated, the process is mass transfer limited due to the highly unstable and non-selective nature of the ˙OH radicals, which restricts the oxidation reactions to the proximity of the electrode surface,
9 with no detectable residual oxidant concentration in the bulk water solution.
One area that has not been thoroughly investigated is the application of BDD electro-oxidation for the abatement of inorganic contaminants, particularly that of manganese. Manganese, a naturally occurring metal found in rocks and mineral deposits, has a provisional maximum recommended concentration of 0.1 mg L−1 as defined by the World Health Organization (WHO) guidelines for drinking water quality.10 At circumneutral pH, Mn2+ can have a high solubility and is therefore frequently required to be converted to the higher valence state of Mn4+ by pre-oxidation, which has a significantly decreased solubility. Several studies have reported the use of conventional chemical oxidants for this purpose, such as chlorine,11–13 ozone,14,15 permanganate,11,13,16 ferrate17 and by biological processes such as manganese-oxidising bacteria.18 In addition to the aforementioned abatement methods, various manganese dioxide coated filter media, such as GreensandPlus™ (Inversand Company) and AD26 (AdEdge), are commercially available and used for removal of manganese, in addition to other contaminant species such as iron, hydrogen sulfide and radium from groundwater supplies. It is generally accepted that manganese dioxide deposits on sand media surfaces act as a catalyst in the oxidation–reduction reaction of manganese and require constant regeneration. While some electrochemical methods of manganese removal have been reported previously, these were limited to electrocoagulation.19–21 Although effective for conventional applications, these treatment options can be impractical and uneconomic for small, remote and decentralised treatment systems, as they all require the constant supply of process chemicals. The use of electro-oxidation for manganese removal may present a more favourable option that eliminates the chemical supply requirement associated with the conventional treatment processes.
While advanced electrochemical oxidation of manganese may be a novel technique for the removal of total manganese, through the conversion of soluble Mn2+ to insoluble Mn4+ (MnO2), an additional complementary phenomena may occur, this being the electro-synthesis of highly oxidative manganese species, specifically permanganate (Mn7+/MnO4−). Permanganate is conventionally synthesised in a two stage process: (i) the oxidation of manganese dioxide to potassium manganate (Mn6+) by oxygen in highly concentrated potassium hydroxide solutions, and (ii) electrochemical oxidation of potassium manganate to potassium permanganate in a highly alkaline electrolyte.22 Permanganate, dosed primarily in the salt form of potassium permanganate (KMnO4), is a widely used chemical in the water treatment industry for several applications including the oxidation of various contaminants, such as taste and odour compounds,23 phenolic compounds,24 pesticide and antibiotic organic micropollutants,25–27 algal toxins28 and heavy metals.29 Permanganate is also widely used as a pre-oxidant for the management and removal of soluble manganese(II)11,13,16 and iron(II).30 Because of its non-toxic reduction product (insoluble manganese dioxide), permanganate is known to be a “green oxidant”.22 Unlike highly transient hydroxyl radicals (self-decay kOH = 5 × 109 M−1 s−1 (ref. 31)), which are confined to the electrode surface,9 synthesised permanganate is sufficiently stable to diffuse from the electrode surface into the bulk solution to interact with contaminant species (including Mn2+), while still possessing a high redox potential (E0 = +1.51 VSHE). This characteristic differentiates permanganate from advanced electrochemical oxidation, as MnO4− concentrations can potentially function as a residual oxidant species in the water treatment-distribution system.
This paper summarises an investigation of the advanced electro-oxidation of solutions containing Mn2+ ions, whereby hydroxyl radicals generated at the electrode surface facilitate the oxidation of the manganese to higher valence state species for two purposes: (i) the formation of insoluble Mn oxides such as MnO2 (Mn4+), Mn2O3 (Mn3+) and Mn3O4(Mn2,3+), for Mn removal; (ii) the synthesis of highly oxidative permanganate species for contaminant degradation in the bulk water solution and/or distribution system. The research highlights a novel permanganate synthesis pathway and the first example of in situ permanganate production in circumneutral pH conditions, as well as a novel electrochemical pre-oxidation process for total manganese removal. In addition to providing a comprehensive experimental study of BDD advanced electrochemical oxidation for simultaneous total manganese removal and permanganate oxidant synthesis, the paper presents a mathematical model that describes the manganese removal process and supports the experimental findings.
2. Methods and materials
2.1 Electrochemical reactor setup
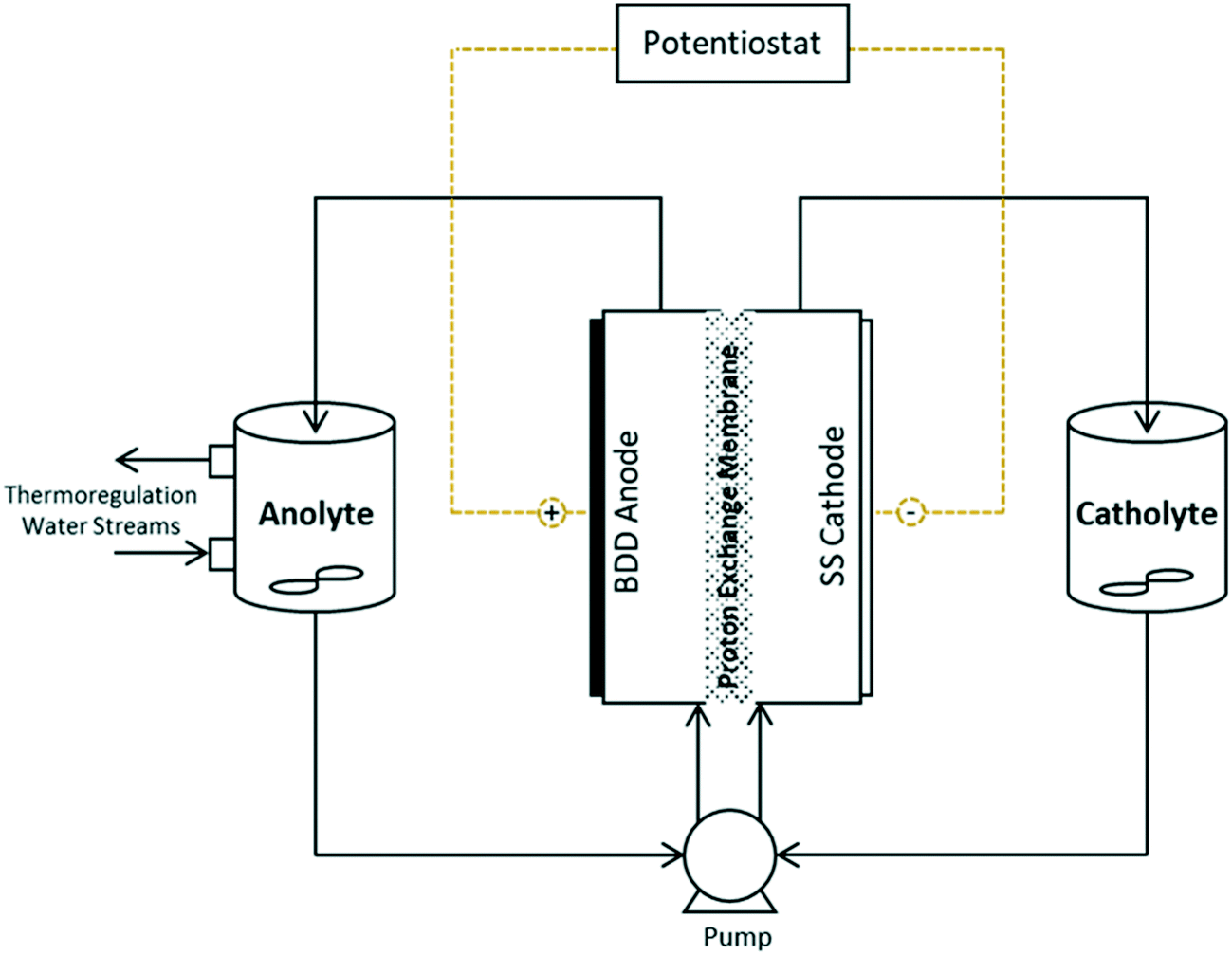
Experiments were conducted in a batch-recycle configuration, with the anolyte and catholyte separated by a Nafion-324 perfluorinated proton exchange membrane (Sigma-Aldrich) (Fig. 1). Water temperature was held constant using a thermo-regulated glass beaker and an applied thermal control (ATC) Kt recirculating chiller at 21.8 ± 0.8 °C. A single-drive Watson-Marlow 505S peristaltic pump, outfitted with two heads was used for the anolyte and catholyte flow, which was held constant at 355 mL min−1. The anolyte and catholyte solutions were stirred using magnetic stir bars. The thin-film (2–3 μm) monocrystalline BDD anode was commercially purchased (NeoCoat®) and prepared through the chemical vapour deposition method, on a 1 mm silicon substrate. The cathode material was austenitic, face centred cubic crystal stainless steel 304 (SS304) alloy. Both the anode and cathode had dimensions of 50 × 50 × 1 mm, and were separated by a 10 mm inter-electrode gap. The custom-made electrochemical cell was fabricated with electrically inert polyvinyl chloride and stainless steel hardware and braces for assembly. Inert rubber was used for the o-ring and gaskets in the assembled cell. All experiments were performed using a Keithley 2460-EC electrochemistry lab system potentiostat.
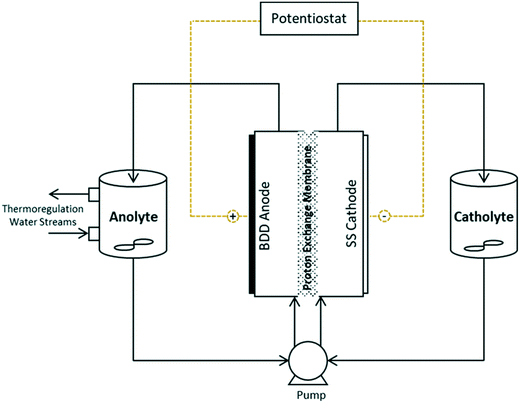 |
| | Fig. 1 Schematic diagram of electrochemical reactor apparatus for Mn reactions and permanganate generation. | |
2.2 Preparation of synthetic water (Mn2+ solution)
The water matrix used for all experiments was a phosphate buffer (pH = 7.1, 0.1 M), composed of ultrapure reverse osmosis water, NaH2PO4 and Na2HPO4 (Fisher Scientific). The desired Mn2+ concentration was attained by addition of MnCl2 (Acros Organics). Initial manganese concentrations of 175 and 39 μM (anolyte solution) were used. For all manganese removal and permanganate generation experiments, 900 mL of the synthetic water (phosphate buffer and Mn) and phosphate buffer were used for the anolyte and catholyte, respectively. Methanol degradation experiments (for hydroxyl radical quantification) used the same conditions, with the anolyte solutions containing methanol instead of manganese. During hydroxyl radical scavenger tests, HPLC grade (≥99.9) methanol (Sigma-Aldrich) was used at a concentration of either 2 mg mL−1 or 6 mg mL−1.
2.3 Analytical methods
2.3.1 Permanganate quantification.
The ABTS (Sigma-Aldrich) indirect method of analysis previously used for ferrate quantification32,33 was chosen because of its sensitivity and accuracy in determining low concentrations of the highly oxidative permanganate ion (MnO4−). This method for permanganate quantification has been described in detail elsewhere.34 In a 25 mL volumetric flask, 5 mL of acetate buffer (pH = 4.1) and 1 mL of ABTS reagent (1 g L−1) were added, in preparation for the addition of test samples, which were added to each flask throughout the electrolysis experiments. The indirect colorimetric method utilized a Shimadzu UV-4201PC spectrophotometer. Permanganate standards were prepared using potassium permanganate (>99%) (Alfa Aesar) in a concentration range of 0–4 mg L−1. During spectrophotometric analysis (380–500 nm), a visible maxima was yielded at 415 nm, characteristic of the generated ABTS˙+ species. Permanganate concentrations were determined from experimental measurements as follows:| | 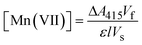 | (1) |
In eqn (1), ΔA415, Vf, ε, l and Vs represent the UV-absorbance at 415 nm (corrected with a blank), the final sample volume, the absorption coefficient as determined by the standards (140![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 030 M−1 cm−1), the cell path length (1 cm), and the volume of the sample extracted from the anolyte (5–15 mL), respectively.
030 M−1 cm−1), the cell path length (1 cm), and the volume of the sample extracted from the anolyte (5–15 mL), respectively.
New ABTS solutions were made prior to each electrolysis experiment with pure (reverse osmosis treated) water and stored in the refrigerator at 4 °C, to avoid degradation due to increased temperature and light exposure. Samples taken during permanganate generation experiments were added to the ABTS solution immediately, to avoid both the degradation of permanganate (prior to a subsequent addition to ABTS) and self-decay of ABTS. Immediately after sampling and addition to the ABTS solution, spectrophotometric analysis was conducted, once again to avoid the decay of ABTS˙+ which could contribute to ultraviolet absorbance.
2.3.2 Inductively coupled plasma atomic emission spectroscopy (ICP-OES).
Total dissolved manganese was quantified using ICP-OES (PerkinElmer – Avio 500). Samples were filtered prior to analysis using 0.45 μm syringe filter papers (GE Whatman cellulose nitrate membrane filter) and acidified with nitric acid (Sigma-Aldrich) to a 1 M final concentration. Manganese calibration was performed using an ICP 23 multi-element standard solution diluted in nitric acid (Merck) and all samples were measured in both the axial and radial direction. Quality control checks were performed every ten or fifteen samples. All glassware and syringe filters were soaked in 10% v/v nitric acid for at least 24 hours prior to being used and rinsed with ultrapure reverse osmosis water.
2.3.3 Cyclic voltammetry.
Cyclic voltammetry (CV) analysis was conducted using a Solartron 1470E potentiostat in the aforementioned water matrix, with a 45 × 24 mm BDD thin film monocrystalline anode on a silicon substrate (NeoCoat®), a Ag/AgCl reference electrode (=0.197 VSHE) and a Pt cathode, to inhibit electrochemical current limitations (decreasing the flow of charge) to the working electrode by minimising the overpotential of the hydrogen evolution reaction. A scan rate of 10 mV s−1 was used to yield well-defined voltammograms and was determined through a trial and error process. CVs were conducted over an oxidation potential range of 1.4–2.8 VAg/AgCl.
3. Results and discussion
3.1 Manganese removal
3.1.1 Effect of current density.
Prior to manganese electro-oxidation experiments, computational fluid dynamic (CFD) modelling and Raman micro-spectroscopy were conducted to understand the electrochemical cell water velocity distribution and evaluate anode quality, respectively. The CFD models demonstrated evenly distributed water flow across the electrode surfaces, consequently minimising any current density distribution variations that may exist due to local velocity variations.35 This evaluation was important in order to understand and ensure that the calculated current densities (absolute current over the entire electrode surface area) were achieved during the electrolysis experiments. Variations in current distribution on the electrode surface would result in reaction kinetic variations and consequently uneven hydroxyl radical formation (or direct oxidation variations at the electrode surface), which could significantly impact both soluble manganese removal and permanganate synthesis. Raman micro-spectroscopic analysis yielded a distinct Raman shift peak at 1332 cm−1, which is characteristic of diamond.36 Although high quality BDD electrodes have been found to have one of the greatest electrical potential ranges of water stability (−1.25 to 2.3 VSHE),37 any graphite impurities can narrow this potential range, resulting in the occurrence of the OER at lower operating potentials. If any graphite impurities existed, a peak would have been observed at 1580 cm−1, however, due the absence of this peak, it can be concluded that the anode has a high sp3/sp2 (diamond:graphite) carbon structure ratio and consequently a high diamond purity. Further analysis and description of the electrochemical cell's transport phenomena and BDD electrode quality have been described elsewhere38 and included in the supplemental materials.
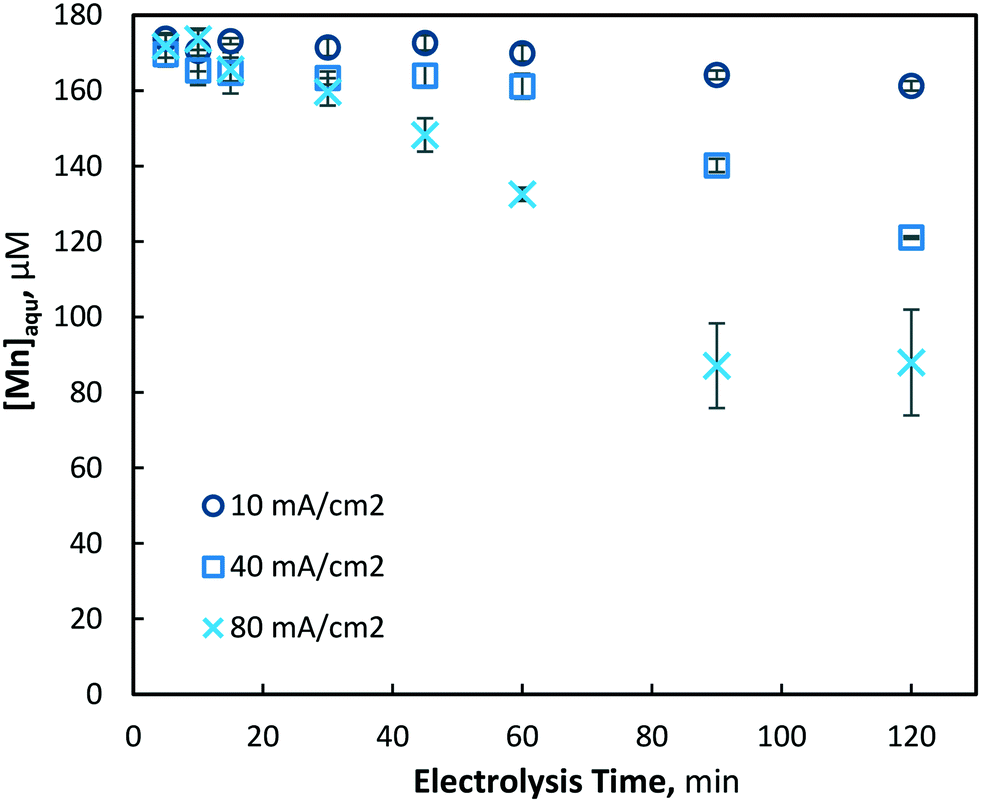
The electrochemical pre-oxidation and removal of total manganese were initially investigated at three current densities (10, 40 and 80 mA cm−2) and an initial Mn2+ concentration of 175 ± 4 μM (9.6 mg L−1); the higher than typical Mn concentration was used in order to facilitate greater accuracy in the analysis of reactions. Total manganese concentration was monitored throughout 120 minutes of electrolysis at regular intervals (5, 10, 15, 30, 45, 60, 90 and 120 min) for all current densities. In general, aqueous manganese (Mn2+) concentrations were observed to decrease with electrolysis time, as well as with the increase of current density (see Fig. 2). At 10, 40 and 80 mA cm−2 operations, final (120 min) manganese concentrations of 161, 121 and 87 μM were achieved, respectively. Operating cell voltages were relatively steady throughout electrolysis, typically observed in ranges of 6.0–6.6, 14.1–14.7 and 18.0–18.6 V. Operating voltages are highly dependent on several parameters, including solution conductivity, inter-electrode gap and electrode surface area, all of which affect the electrochemical cell resistance.
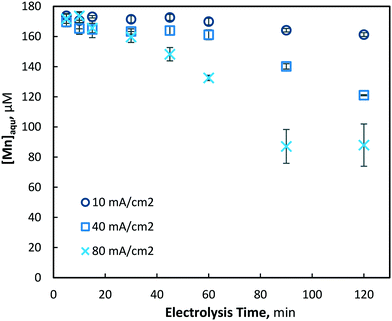 |
| | Fig. 2 Removal of aqueous manganese (Mn2+) with electrolysis time and current densities of 10, 40 and 80 mA cm−2 (pH = 7, [Mn2+]0 = 175 μM, T = 21.0 ± 0.8 °C). | |
In general, pollutant degradation via BDD electro-oxidation is a mass transport limited process, whereby the transport of pollutant species through the Nernst diffusion layer from the bulk water solution to the electrode surface (where either direct oxidation or hydroxyl radical mediated oxidation is limited) is the rate limiting step.3 While in the case of iron, the oxidation of Fe2+ to the higher oxidation state species of ferrate under similar conditions was found also to be diffusion limited, with no differences in ferrate generation observed over the same current density range,38 the generation of non-aqueous manganese species (MnO2/Mn4+, Mn2O3/Mn3+) was highly dependent on current density. The varying removal of manganese with respect to current density is likely due to one or both of the following mechanisms:
(1) Direct oxidation at the electrode surface:
| | | BDD(Mn2+) + 2H2O → MnO2 + 4H+ + 2e− | (R5) |
| | | BDD(4Mn2+) + 6H2O → 2Mn2O3 + 12H+ + 4e− | (R6) |
(2) Advanced oxidation at the electrode surface:
| | | BDD(2˙OH) + Mn2+ → MnO2 + 2H+ | (R7) |
| | | BDD(2˙OH) + 2Mn2+ + H2O → Mn2O3 + 4H+ | (R8) |
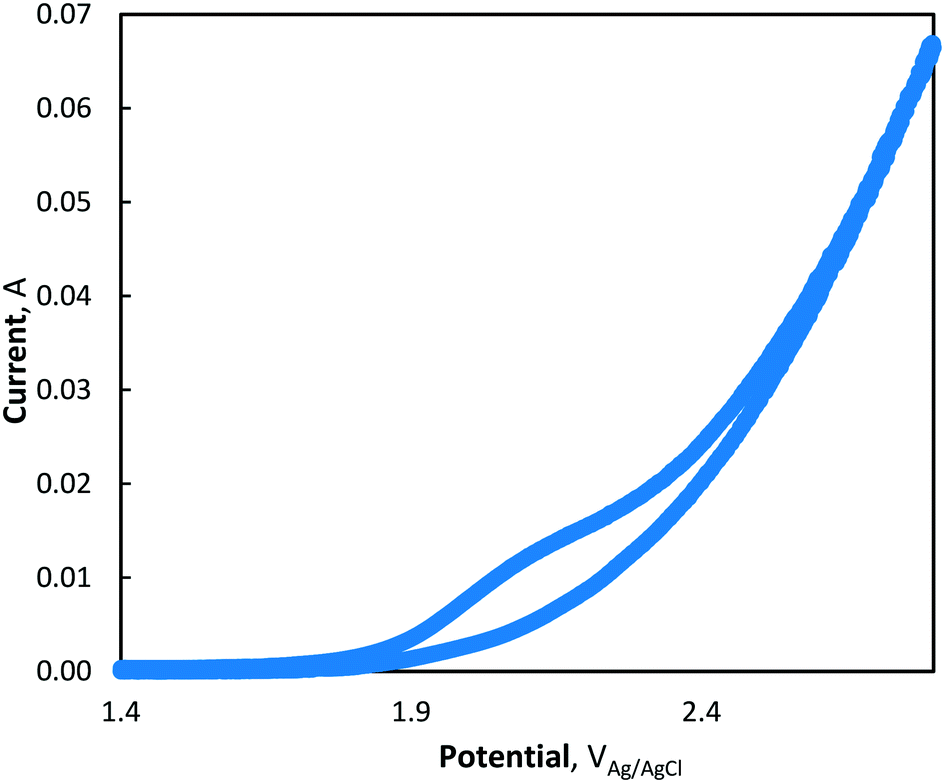
Using cyclic voltammetry, the presence of direct electron transfer mechanisms
(R5) and
(R6) was observed by the presence of a small shoulder in the voltammogram between 2.0–2.2 V
Ag/AgCl (see
Fig. 3), highlighting an oxidation mechanism by both direct and advanced oxidation processes. The steep increase in the voltammogram as the CV approaches 2.8 V
Ag/AgCl is characteristic of the OER.
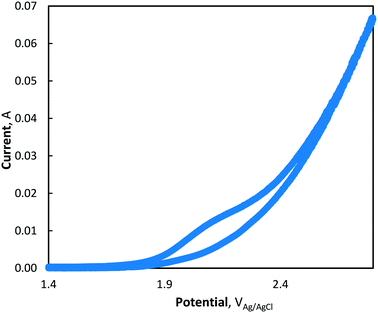 |
| | Fig. 3 Cyclic voltammogram of MnCl2 at pH = 7 (phosphate buffer) and 10 mV s−1 scan rate. | |
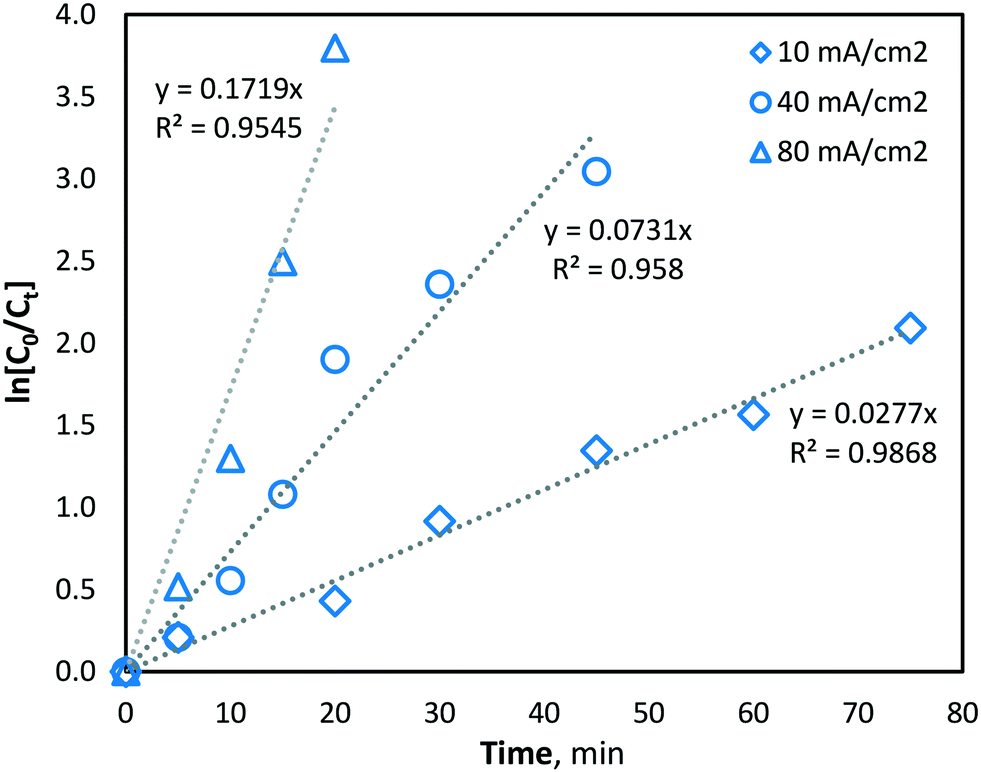
At higher current density operations, a greater rate of hydroxyl radical production is expected and therefore a greater rate of manganese removal. The determination of hydroxyl radical production was conducted indirectly using a strong ˙OH scavenger, namely, methanol (MeOH). MeOH was chosen as a suitable hydroxyl radical scavenger as it has been previously demonstrated to be oxidized solely via hydroxyl radicals (kOH ∼ 9.7 × 108 M−1 s−1 (ref. 31)) and not by direct oxidation during BDD electro-oxidation.39,40 The pseudo-steady-state MeOH degradation was first determined using an initial concentration of 62.5 mM in the same phosphate buffer water matrix used during manganese experiments. MeOH was quantified at regular intervals throughout electrolysis, in the same batch-recycle configuration used for all other experiments, using the nitroprusside indirect spectrophotometric method.41 From the results, the apparent pseudo-first-order reaction rate constant (k′) for MeOH degradation by hydroxyl radical reaction was yielded, giving the following values of k′ = 2.77 × 10−2, 7.31 × 10−2 and 1.72 × 10−1 s−1, for current densities of 10, 40 and 80 mA cm−2, respectively (Fig. 4). The values indicated significantly greater hydroxyl radical formation at higher current densities (as would be predicted by Faraday's law).
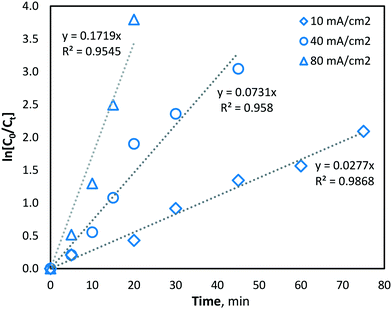 |
| | Fig. 4 Indirect quantification of hydroxyl radical formation (methanol degradation) with current densities of 10, 40 and 80 mA cm−2 (pH = 7, T = 21.0 ± 0.8 °C). | |
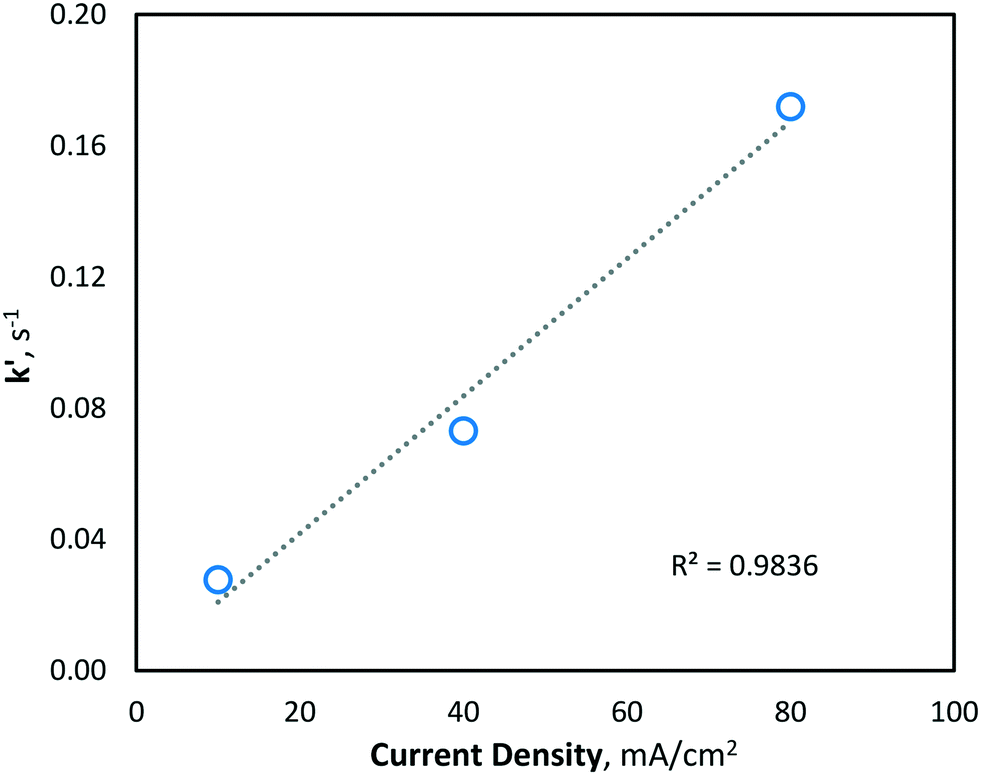
Since hydroxyl radicals are continuously generated at the electrode surface, MeOH degradation is described well by pseudo-first order kinetics (Fig. 4), as the concentration of hydroxyl radicals remains relatively unchanged and is continuously replenished throughout electrolysis. Since the second-order rate constant (kOH) of hydroxyl radicals and MeOH is known to be ∼9.7 × 108 M−1 s−1,31 the concentration of hydroxyl radicals at the various current density conditions can be calculated using the observed pseudo-first-order reaction rate constants (k′). As expected, the yielded k′ values (and corresponding hydroxyl radical concentrations) increase linearly with current density (Fig. 5).
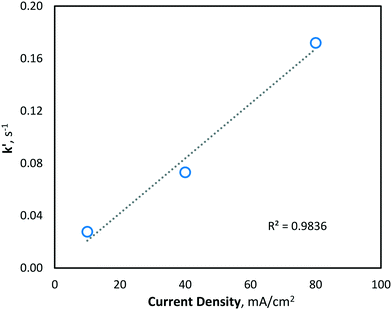 |
| | Fig. 5 Pseudo-first-order rate constant (k′) of MeOH degradation via hydroxyl radical oxidation at various current density conditions (10, 40 and 80 mA cm−2). | |
3.1.2 Effect of initial [Mn2+].
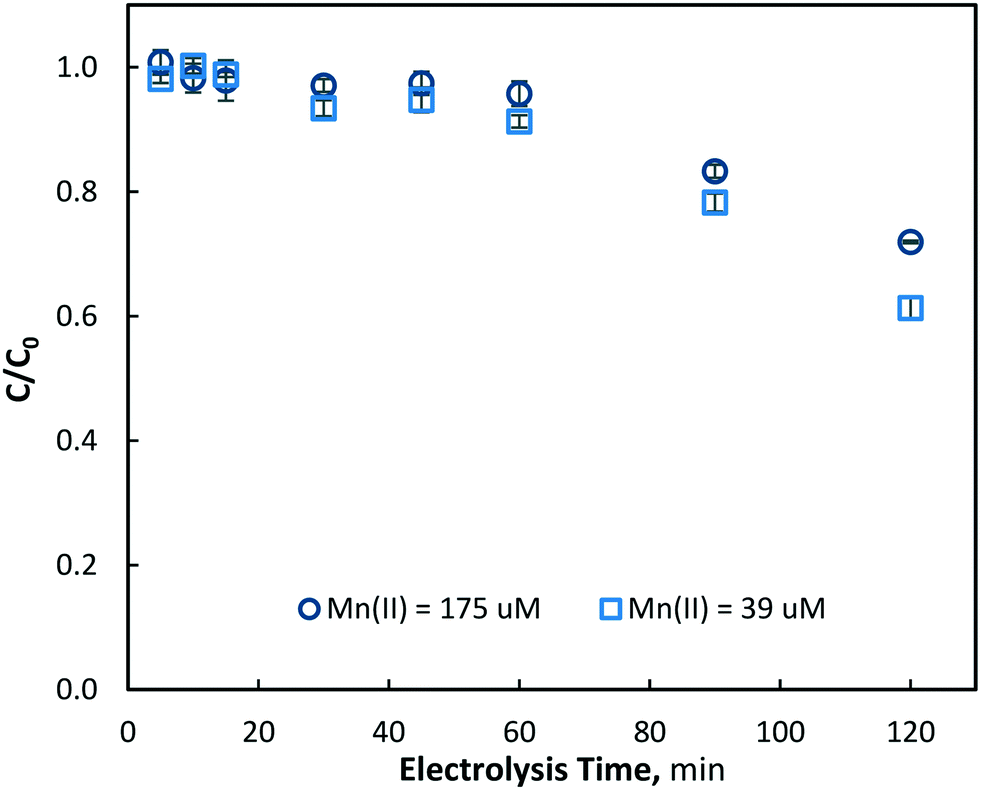
+The electrolysis experiments were conducted at a lower initial manganese concentration of Mn2+ = 39 ± 2 μM (2.2 mg L−1) under the same conditions as those for the Mn2+ = 175 μM tests. As expected, very similar results were observed, with the manganese removal increasing with both electrolysis time and current density. Since the hydroxyl radical formation rate is constant at each current density, the reduction in Mn2+ concentration was expected to be independent of its initial concentration, in accordance with a pseudo-first order reaction (k′ = kOH [˙OH]); confirmation of this for the initial period of electrolysis (<60 min) is shown in Fig. 6 for a current density of 40 mA cm−2. However, some small differences in the removal of manganese (Mn2+) were evident at the end of the electrolysis time (120 min) for the different initial concentrations. Thus, for the current densities of 10, 40 and 80 mA cm−2, the corresponding Mn2+ reduction was 7.2, 28.6 and 49.3% for Mn2+0 = 175 μM, and 9.0, 38.7 and 57.4% for Mn2+0 = 39 μM, respectively (Fig. 6). While it would be expected that manganese diffusion to the electrode surface at a higher concentration would be increased, only very little differences in removal were observed, indicating that the limited role of diffusion on oxidation process. However, these differences in removal may be due to hydroxyl radical consumption by chloride, the counter ion of manganese (dosed as MnCl2), which will be present at a much higher concentration during Mn2+ = 175 μM tests. Some researchers have previously found chloride to be readily reacted at the BDD electrode surface via advanced oxidation.42,43 Furthermore, at a constant current density and therefore constant hydroxyl radical generation, there will be a higher ˙OH to Mn2+ ratio at lower initial manganese concentrations, which could also explain the greater total manganese removal observed in Fig. 6.
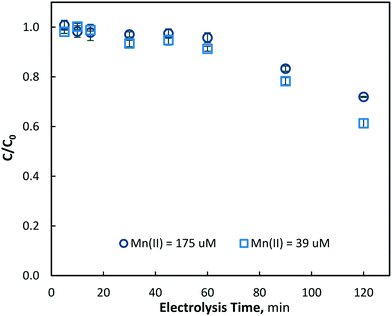 |
| | Fig. 6 Removal of aqueous manganese (Mn2+) with electrolysis time at 40 mA cm−2 with [Mn2+]0 = 175 and 39 μM, (pH = 7, T = 21.0 ± 0.8 °C). | |
3.1.3 Mathematical model.
A mathematical model was developed to understand and compare the experimentally observed results for electro-oxidation and removal of manganese, to those predicted by the theory. The simple model describes the oxidation of Mn2+ to higher valence state Mn species, under idealised conditions and assumptions. The model considers the time variation of manganese in two forms: dissolved manganese (Mn2+) and oxidised insoluble forms of manganese (Mn3,4,5+), as indicated by the subscripts “SOL” and “INS”, respectively. The model was derived through a mole balance of the electrochemical reactor and the anolyte vessel (Fig. 1). Two important assumptions made during the model development were: (i) plug flow mass transport through the electro-active volume of the reactor (i.e. no water flow velocity variations and perfect mixing), and (ii) the system and kinetics are completely mass transfer limited. The complete model development is described in the accompanying ESI.† The final expressions (eqn (2)–(4)) describe the generated concentration of insoluble manganese species (CINS) exiting the electrochemical reactor (and/or entering the anolyte reservoir), as a function of time during electrolysis (t), the mass transport coefficient (km) and degradation coefficient (kd). Other variables included in the final model expression are the inter-electrode gap (δ), the reactor residence time (tR) and anolyte reservoir residence time (tV).| | | CINS(IN,t) = A(1 − exp(−Bt))CSOL,0 | (2) |
where,| | 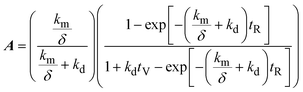 | (3) |
| | 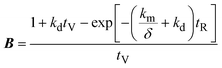 | (4) |
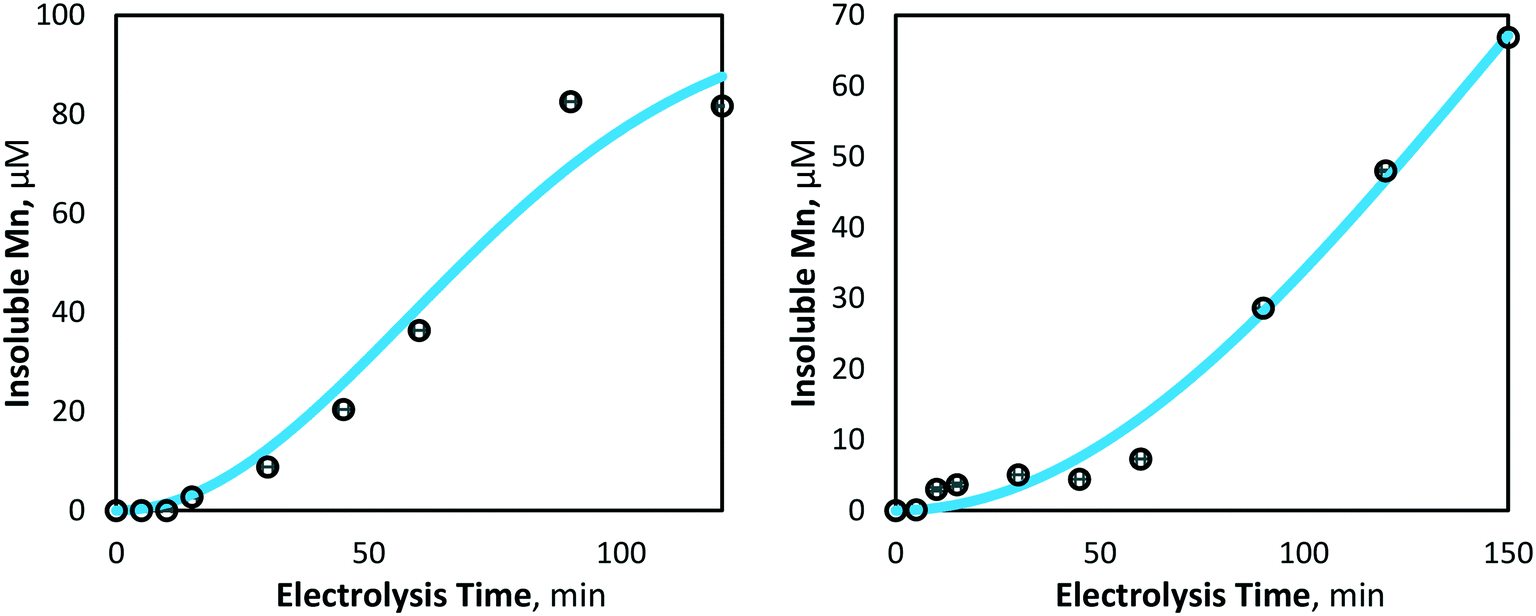
Using the curve fitting tool (MATLAB 2019b), the mass transfer coefficient (km, m s−1) and degradation coefficient (kd, s−1) for 80 and 40 mA cm−2 electrolysis were determined to be 0.532 × 10−9 and 2.445 × 10−9 m s−1, and 1.687 × 10−8 and 1.939 × 10−11 s−1, respectively. The model and experimental data for manganese removal were found to be in good agreement, with coefficients of determination of R2 = 0.97 and 0.98, for 80 and 40 mA cm−2 operations, respectively (Fig. 7), indicating that experimentally observed insoluble manganese generation results behave in a manner predictable by theory.
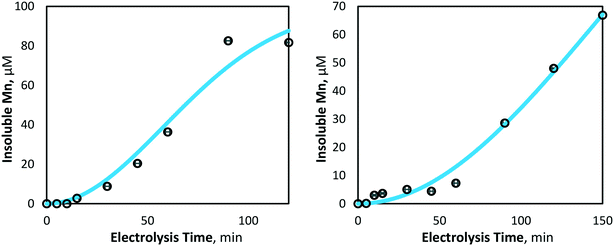 |
| | Fig. 7 Model and experimentally yielded results of insoluble manganese (Mn3+,4+) generation at 80 mA cm−2 (left) and 40 mA cm−2 (right) (pH = 7, [Mn2+]0 = 175 μM, T = 21.0 ± 0.8 °C). | |
In addition to confirming the validity of the experimental results compared to those predicted by theory, the model may also be useful in predicting and describing manganese removal in similar systems. The mass transport and reactor design variables governing the model, such as inter-electrode gap and electrode dimensions (and the resulting reactor residence time, tR), can be inputted to predict the resulting manganese removal. For example, if the current BDD electrode dimensions were changed from 50 × 50 mm with a 10 mm inter-electrode gap, to 100 × 300 mm and a 5 mm inter-electrode gap, the model predicted manganese removal would increase by two, in half of the time.
3.2 Permanganate synthesis
3.2.1 Effect of current density.
While the oxidation of Mn2+ was observed to progress principally to an intermediate oxidation state in the form of insoluble manganese (Mn3,4,5+), such as thermodynamically stable species MnO2, Mn2O3 and/or Mn3O4, there exists in theory an additional manganese species of higher redox potential that can be synthesised, this being MnO4− (permanganate). Under the high potential conditions investigated, the generation of permanganate can occur provided two phenomena are minimised: (i) the parasitic OER, which increases as electrical potential increases, and (ii) the parasitic reaction of insoluble manganese species formation. Although it was evident from the CV and ICP-OES analyses described in section 3.1 that both of these reactions were present, it was still considered likely that permanganate would be produced.
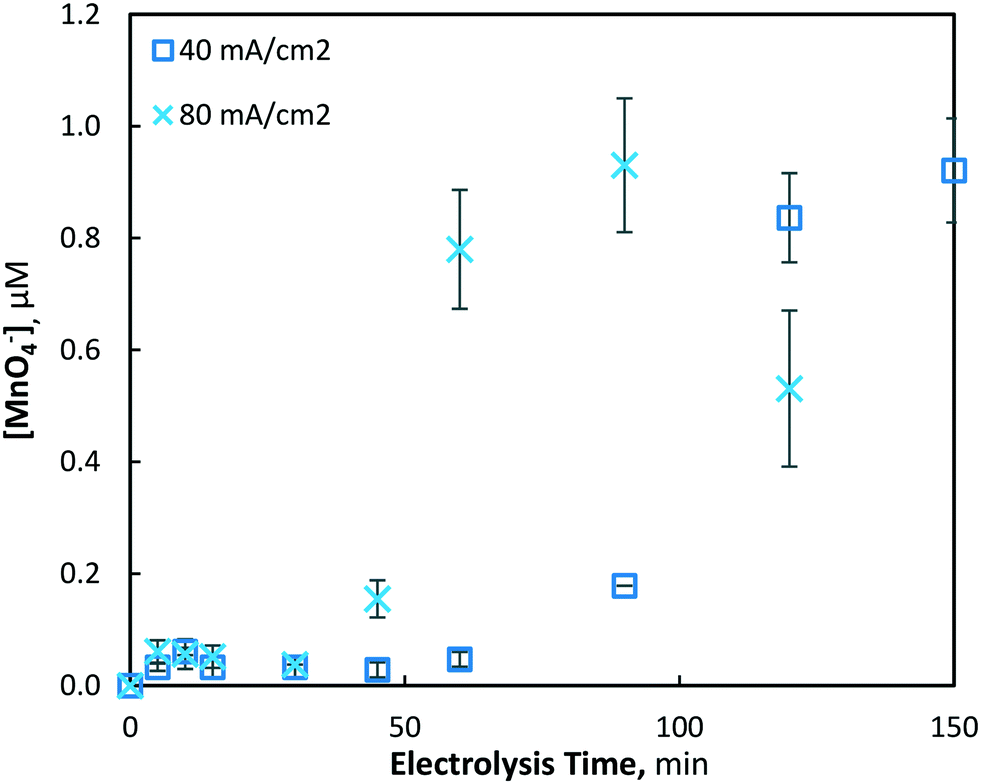
The electrochemical generation of permanganate was investigated under the same conditions used for the Mn2+ pre-oxidation experiments (10, 40 and 80 mA cm−2) with an initial Mn2+ concentration of 175(±4) μM (9.8 mg L−1). As was found in the investigation of manganese removal, only minor effects were evident at the lowest current density investigated (10 mA cm−2), and a maximum permanganate concentration of 0.05(±0.01) μM was yielded. Under the same conditions, the total manganese concentration decreased by only 7.2%, with 161.22 μM of Mn2+ remaining unreacted in solution. When the current density was increased to 40 and 80 mA cm−2, maximum permanganate concentrations of 0.921(±0.09) and 0.930(±0.12) μM were generated after 150 and 90 min of electrolysis, respectively (Fig. 8). In the latter case (80 mA cm−2), permanganate levels were observed to decrease after reaching a maximum, likely due to chemical reduction via oxidation reactions with other manganese species (Mn2–5+), as permanganate is an efficient chemical used for the oxidation and removal of dissolved manganese.11,13,16
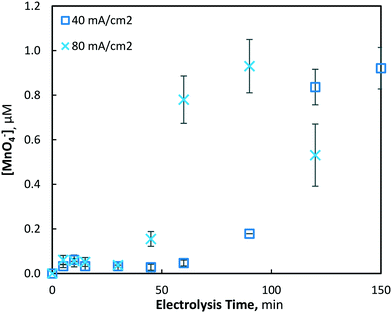 |
| | Fig. 8 Production of permanganate at 40 and 80 mA cm−2 over 120 min of electrolysis (pH = 7, [Mn2+]0 = 175 μM, T = 21.0 ± 0.8 °C). | |
After an initial lag period of minimal permanganate generation, a sharp increase in synthesis occurs after 30 and 60 min of electrolysis for the 80 and 40 mA cm−2 conditions, respectively. The same lag period prior to significant reductions of soluble manganese was also observed (Fig. 2), indicating the possibility of a similar mechanism responsible for the production of both permanganate and insoluble manganese species. This may be explained by the initial production of manganate (Mn6+/MnO42−), as it has been previously shown in a drinking water treatment context that manganate is thermodynamically unstable in circumneutral conditions, leading via disproportionation to MnO2 and permanganate.44 Though ICP-OES analysis demonstrated that MnO2 was predominantly generated, it was evident that disproportionation of manganate to the permanganate ion was also occurring. As demonstrated previously with respect to the lag period prior to rapid decrease in soluble manganese, the mathematical model (presented in section 3.1.3) is able to show that the lag period prior to rapid permanganate generation may also be decreased by altering several reactor design variables, such as the inter-electrode gap distance and the electrode dimensions, assuming a common mechanism is shared between the two processes.
Although only a small concentration of permanganate was quantified (∼1 μM) in the bulk water solution, additional permanganate may have been generated and rapidly reduced at the electrode surface when interacting with, and oxidising Mn2+, as it is known to do efficiently.11,13,16 Moreover, some evidence exists which would suggest that the permanganate concentration observed may be adequate in a water treatment process to serve as a useful pre-oxidant or residual disinfectant. Previous researchers have shown that significant degradation (>50%) of bisphenol A could be achieved by permanganate concentrations as low as 5 μM.45 Other phenol-type contaminants have been found to be significantly reduced when treated with a potassium permanganate concentration of 10 μM.24 Turbidity has also been shown to be effectively reduced by 34% using 1.7 μM permanganate as a pre-oxidant.46
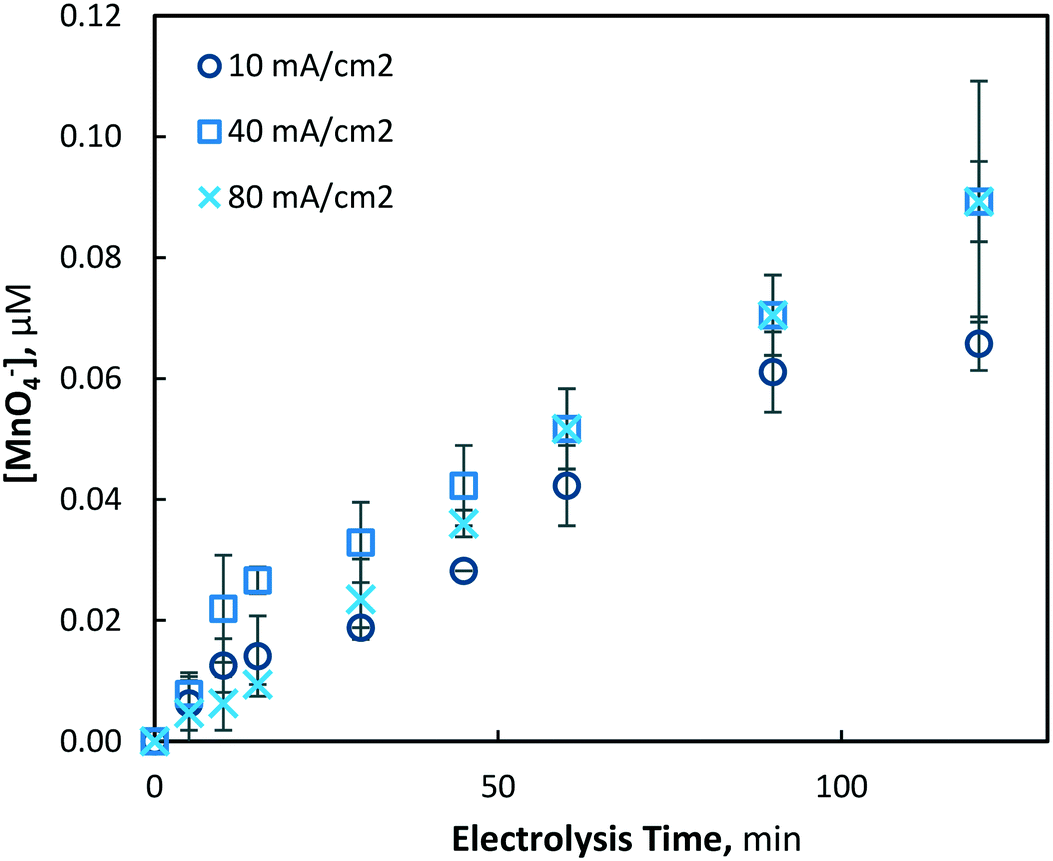
When the initial soluble manganese concentration was decreased to 39 μM (2.2 mg L−1), significantly decreased permanganate generation was observed (Fig. 9). Electrolysis under 40 and 80 mA cm−2 conditions generated very similar permanganate concentrations indicating a diffusion limited process, while 10 mA cm−2 operations produced slightly less permanganate. Although synthesised at reliably quantifiable concentrations, a permanganate dose of ∼0.1 μM may be too low to provide any significant benefit as an oxidant chemical.
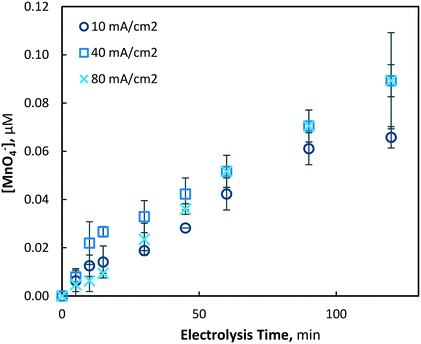 |
| | Fig. 9 Permanganate production under 10, 40 and 80 mA cm−2 electrolysis conditions over 120 min (pH = 7, [Mn2+]0 = 39 μM, T = 21.0 ± 0.8 °C). | |
4. Conclusion
In summary, a novel electrochemical process for the simultaneous removal of dissolved manganese and generation of useful permanganate oxidant for drinking water treatment, in neutral pH conditions, has been presented for the first time. While electrochemical techniques like electrocoagulation have been shown previously to remove manganese from groundwater, this study presents an effective electrochemical pre-oxidation technique suitable for small, remote and decentralised systems, as it completely eliminates the chemical supply chain associated with conventional pre-oxidation processes.
At initial dissolved manganese concentrations of 175 and 39 μM, reductions in total manganese were observed to be as high as approximately 50% in both cases. While dissolved manganese reduced with electrolysis time, an increased reduction was observed as the operating current density increased. For current densities of 10, 40 and 80 mA cm−2, the total manganese reduced by 7.2, 28.6 and 49.3% for an initial Mn2+ concentration of 175 μM, and 9.1, 38.7 and 57.4% for an initial Mn2+ concentration of 39 μM, respectively. At both initial Mn2+ concentrations, soluble manganese was observed to be removed at a similar rate. Furthermore, greater manganese removal at increased current densities is attributed to the increased hydroxyl radical formation, as indicated indirectly by the pseudo-first-order decay constants for methanol (˙OH radical scavenger): k′ = 2.77 × 10−2, 7.31 × 10−2 and 1.72 × 10−1 s−1, at 10, 40 and 80 mA cm−2 electrolysis, respectively.
A mathematical model was developed to describe the manganese removal, under mass transport limitations, and was used to compare the experimental results with those predicted by theory. The model assumed manganese to be in either a soluble (Mn2+) or insoluble (Mn3–5+) form and was developed through a mole balance of both the electrochemical reactor and the anolyte vessel (for the batch recycle system). Experimental and model results correlated well, with a coefficient of determination of R2 = 0.97 and 0.98 for the 80 and 40 mA cm−2 operations, respectively. Additionally, the model can provide the basis for increasing manganese removal with respect to reactor design considerations, such as electrode dimensions and inter-electrode gap.
Finally, the generation of permanganate from dissolved manganese (Mn2+) was explored at two Mn concentrations. Through cyclic voltammetric analysis, a mechanism via both direct and advanced oxidation was established. At the higher initial Mn2+ concentration (175 μM), greater permanganate formation was observed, particularly during 40 and 80 mA cm−2 electrolysis, whereby 0.921 and 0.930 μM MnO4− were generated after 120 and 90 min of electrolysis, respectively. Although relatively low MnO4− concentrations were generated under the test conditions, the work presented provides a completely novel synthesis pathway for permanganate generation. Compared to the conventional method of producing potassium permanganate via an energy intensive, two-step process, this work has demonstrated a potentially important in situ synthesis process, which exploits the presence of dissolved, low valence, ambient manganese in raw waters.
Conflicts of interest
There are no potential conflicts of interest to declare.
Acknowledgements
The authors wish to acknowledge the support of Imperial College London with a President's Scholarship for S. T. McBeath, as well as the support of the Natural Sciences and Engineering Research Council of Canada (NSERC), [PGSD3-516562-2018]. The authors would also like to thank Adrian Serrano Mora (University of British Columbia) for help with the cyclic voltammetry analysis.
References
- S. T. McBeath, M. Mohseni and D. P. Wilkinson, Pilot-scale iron electrocoagulation for natural organic matter removal, Environ. Technol., 2018, 1–9, DOI:10.1080/09593330.2018.1505965.
- S. E. Amrose, S. R. S. Bandaru, C. Delaire, C. M. van Genuchten, A. Dutta and A. DebSarkar,
et al. Electro-chemical arsenic remediation: Field trials in West Bengal, Sci. Total Environ., 2014, 488–489(1), 539–546, DOI:10.1016/j.scitotenv.2013.11.074.
- S. T. McBeath, D. P. Wilkinson and N. J. D. Graham, Application of Boron-Doped Diamond Electrodes for the Anodic Oxidation of Pesticide Micropollutants in a Water Treatment Process: A Critical Review, Environ. Sci.: Water Res. Technol., 2019, 5, 2090–2107 RSC.
- M. Panizza and G. Cerisola, Application of diamond electrodes to electrochemical processes, Electrochim. Acta, 2005, 51(2), 191–199 CrossRef CAS.
- M. H. P. Santana, L. A. D. Faria and J. F. C. Boodts, Electrochemical characterisation and oxygen evolution at a heavily boron doped diamond electrode, Electrochim. Acta, 2005, 50(10), 2017–2027 CrossRef CAS.
- A. M. Polcaro, A. Vacca, M. Mascia and F. Ferrara, Product and by-product formation in electrolysis of dilute chloride solutions, J. Appl. Electrochem., 2008, 38(7), 979–984 CrossRef CAS.
- P.-A. Michaud, M. Panizza, L. Ouattara and T. Diaco, Electrochemical oxidation of water on synthetic boron-doped diamond thin film anodes, J. Appl. Electrochem., 2003, 33, 151–154, DOI:10.1023/A:1024084924058.
- J. Jeong, J. Y. Kim and J. Yoon, The role of reactive oxygen species in the electrochemical inactivation of microorganisms, Environ. Sci. Technol., 2006, 40(19), 6117–6122 CrossRef CAS PubMed.
- X. Zhu, M. Tong, S. Shi, H. Zhao and J. Ni, Essential explanation of the strong mineralization performance of boron-doped diamond electrodes, Environ. Sci. Technol., 2008, 42(13), 4914–4920 CrossRef CAS PubMed.
-
WHO, Guidelines for Drinking-water Quality, World Health Organization, Geneva, 4th edn, 2017 Search PubMed.
- W. Yu, L. Campos, T. Shi, G. Li and N. Graham, Enhanced removal of manganese in organic-rich surface water by combined sodium hypochlorite and potassium permanganate during drinking water treatment, RSC Adv., 2015, 5(35), 27970–27977 RSC.
- B. O. J. Hao, A. P. Davis, A. Member and P. H. Chang, Kinetics of manganese(ii) oxidation with chlorine, J. Environ. Eng., 1991, 117(3), 359–374 CrossRef.
- J. E. Van Benschoten and W. Lin, Kinetic Modeling of Manganese(II) Oxidation by Chlorine Dioxide and Potassium Permanganate, Environ. Sci. Technol., 1992, 26(7), 1327–1333 CrossRef CAS.
- S. Y. Jasim and M. Mohseni, Ozone Application for Arsenic and Manganese Treatment at the City of White Rock, BC, Canada, Ozone: Sci. Eng., 2019, 41(4), 322–331, DOI:10.1080/01919512.2019.1608811.
- R. El Araby, S. Hawash and G. El Diwani, Treatment of iron and manganese in simulated groundwater via ozone technology, Desalination, 2009, 249(3), 1345–1349, DOI:10.1016/j.desal.2009.05.006.
- K. H. Carlson and W. R. Knocke, Modeling manganese oxidation with KMnO4 for drinking water treatment, J. Environ. Eng., 1999, 125(10), 892–896 CrossRef CAS.
- J. E. Goodwill, X. Mai, Y. Jiang, D. A. Reckhow and J. E. Tobiason, Oxidation of manganese(II) with ferrate: Stoichiometry, kinetics, products and impact of organic carbon, Chemosphere, 2016, 159, 457–464, DOI:10.1016/j.chemosphere.2016.06.014.
- V. W. Hoyland, W. R. Knocke, J. O. Falkinham, A. Pruden and G. Singh, Effect of drinking water treatment process parameters on biological removal of manganese from surface water, Water Res., 2014, 66(Ii), 31–39, DOI:10.1016/j.watres.2014.08.006.
- S. Omranpour Shahreza, N. Mokhtarian and S. Behnam, Optimization of Mn removal from aqueous solutions
through electrocoagulation, Environ. Technol., 2018, 1–11, DOI:10.1080/09593330.2018.1514071.
- L. Xu, G. Cao, X. Xu, S. Liu, Z. Duan and C. He,
et al. Simultaneous removal of cadmium, zinc and manganese using electrocoagulation: Influence of operating parameters and electrolyte nature, J. Environ. Manage., 2017, 204, 394–403, DOI:10.1016/j.jenvman.2017.09.020.
- P. Ganesan, J. Lakshmi, G. Sozhan and S. Vasudevan, Removal of manganese from water by electrocoagulation: Adsorption, kinetics and thermodynamic studies, Can. J. Chem. Eng., 2013, 91(3), 448–458 CrossRef CAS.
- N. Singh and D. G. Lee, Permanganate: A green and versatile industrial oxidant, Org. Process Res. Dev., 2001, 5(6), 599–603 CrossRef CAS.
- A. I. Omoike and D. Harmon, Slow-releasing permanganate ions from permanganate core-manganese oxide shell particles for the oxidative degradation of an algae odorant in water, Chemosphere, 2019, 223, 391–398, DOI:10.1016/j.chemosphere.2019.02.036.
- J. Jiang, Y. Gao, S. Y. Pang, Q. Wang, X. Huangfu and Y. Liu,
et al. Oxidation of bromophenols and formation of brominated polymeric products of concern during water treatment with potassium permanganate, Environ. Sci. Technol., 2014, 48(18), 10850–10858 CrossRef CAS PubMed.
- L. Hu, A. M. Stemig, K. H. Wammer and T. J. Strathmann, Oxidation of antibiotics during water treatment with potassium permanganate: Reaction pathways and deactivation, Environ. Sci. Technol., 2011, 45(8), 3635–3642 CrossRef CAS PubMed.
- C. Liu, Z. Qiang, C. Adams, F. Tian and T. Zhang, Kinetics and mechanism for degradation of dichlorvos by permanganate in drinking water treatment, Water Res., 2009, 43(14), 3435–3442, DOI:10.1016/j.watres.2009.05.001.
- L. Hu, H. M. Martin and T. J. Strathmann, Oxidation kinetics of antibiotics during water treatment with potassium permanganate, Environ. Sci. Technol., 2010, 44(16), 6416–6422 CrossRef CAS PubMed.
- E. Rodríguez, M. E. Majado, J. Meriluoto and J. L. Acero, Oxidation of microcystins by permanganate: Reaction kinetics and implications for water treatment, Water Res., 2007, 41(1), 102–110 CrossRef PubMed.
- X. Qi and F. Xie, Promotion effects of potassium permanganate on removal of Pb(II), Ni(II) and Cd(II) from hydrous manganese dioxide, Chem. Eng. J., 2018, 351, 22–30 CrossRef CAS.
- J. L. Cleasby, Iron and Manganese Removal - A Case Study, J. - Am. Water Works Assoc., 1975, 67(3), 147–149 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals in Aqueous Solution, J. Phys. Chem. Ref. Data, 1988, 17(2), 513–886 CrossRef CAS.
- Y. Lee, J. Yoon and U. Von Gunten, Spectrophotometric determination of ferrate(Fe(VI)) in water by ABTS, Water Res., 2005, 39(10), 1946–1953 CrossRef CAS PubMed.
- M. A. Cataldo-Hernández, A. May, A. Bonakdarpour, M. Mohseni and D. P. Wilkinson, Analytical quantification of electrochemical ferrates for drinking water treatments, Can. J. Chem., 2017, 95, 105–112 CrossRef.
- S. T. McBeath, D. P. Wilkinson and N. J. D. Graham, Analytical quantification of aqueous permanganate: Direct and indirect spectrophotometric determination for water treatment processes, Chemosphere, 2020, 251, 126626, DOI:10.1016/j.chemosphere.2020.126626.
- S. T. McBeath, A. Nouri-Khorasani, M. Mohseni and D. P. Wilkinson, In-situ determination of current density distribution and fluid modeling of an electrocoagulation process and its effects on natural organic matter removal for drinking water treatment, Water Res., 2020, 171, 115404 CrossRef CAS PubMed.
- P. C. Ricci, A. Anedda, C. M. Carbonaro, F. Clemente and R. Corpino, Electrochemically induced surface modifications in boron-doped diamond films: A Raman spectroscopy study, Thin Solid Films, 2005, 482(1–2), 311–317 CrossRef CAS.
- A. Kraft, Doped diamond: A compact review on a new, versatile electrode
material, Int. J. Electrochem. Sci., 2007, 2, 355–385 CAS.
- S. T. McBeath, D. P. Wilkinson and N. J. D. Graham, Exploiting water contaminants: In-situ electrochemical generation of ferrates using ambient raw water iron(Fe2+), J. Environ. Chem. Eng., 2020, 8, 103834 CrossRef CAS.
- A. Kapałka, B. Lanova, H. Baltruschat, G. Fóti and C. Comninellis, A DEMS study of methanol and formic acid oxidation on boron-doped diamond electrode, J. Electrochem. Soc., 2009, 156(11), 149–153 CrossRef.
- I. Kisacik, A. Stefanova, S. Ernst and H. Baltruschat, Oxidation of carbon monoxide, hydrogen peroxide and water at a boron doped diamond electrode: The competition for hydroxyl radicals, Phys. Chem. Chem. Phys., 2013, 15(13), 4616–4624 RSC.
- Y. Y. Zhan, Y. Zhang, Q. M. Li and X. Z. Du, A novel visible spectrophotometric method for the determination of methanol using sodium nitroprusside as spectroscopic probe, J. Chin. Chem. Soc., 2010, 57(2), 230–235 CrossRef CAS.
- M. E. H. Bergmann, J. Rollin and T. Iourtchouk, The occurrence of perchlorate during drinking water electrolysis using BDD anodes, Electrochim. Acta, 2009, 54(7), 2102–2107 CrossRef CAS.
- A. M. Polcaro, A. Vacca, M. Mascia, S. Palmas and R. J. Rodiguez, Electrochemical treatment of waters with BDD anodes: Kinetics of the reactions involving chlorides, J. Appl. Electrochem., 2009, 39(11), 2083–2092 CrossRef CAS.
- F. W. Zhao, X. Li and N. Graham, The application of potassium manganate in water treatment, Water Sci. Technol.: Water Supply, 2011, 11(5), 612–620 CAS.
- J. Zhang, B. Sun and X. Guan, Oxidative removal of bisphenol A by permanganate: Kinetics, pathways and influences of co-existing chemicals, Sep. Purif. Technol., 2013, 107, 48–53, DOI:10.1016/j.seppur.2013.01.023.
- R. Liu, L. Sun, R. Ju, H. Liu, J. Gu and G. Li, Treatment of low-turbidity source water by permanganate pre-oxidation: In situ formed hydrous manganese dioxide as filter aid, Sep. Purif. Technol., 2013, 117, 69–74, DOI:10.1016/j.seppur.2013.04.007.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ew00261e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
David P.
Wilkinson
b and
Nigel J. D.
Graham
a
*a,
David P.
Wilkinson
b and
Nigel J. D.
Graham
a