
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactions of pyrrole, imidazole, and pyrazole with ozone: kinetics and mechanisms†
Agnes
Tekle-Röttering‡
a,
Sungeun
Lim‡
 bc,
Erika
Reisz
d,
Holger V.
Lutze
efgj,
Mohammad Sajjad
Abdighahroudi
e,
Sarah
Willach
e,
Winfried
Schmidt
af,
Peter R.
Tentscher
h,
Daniel
Rentsch
i,
Christa S.
McArdell
b,
Torsten C.
Schmidt
*efg and
Urs
von Gunten
*bc
bc,
Erika
Reisz
d,
Holger V.
Lutze
efgj,
Mohammad Sajjad
Abdighahroudi
e,
Sarah
Willach
e,
Winfried
Schmidt
af,
Peter R.
Tentscher
h,
Daniel
Rentsch
i,
Christa S.
McArdell
b,
Torsten C.
Schmidt
*efg and
Urs
von Gunten
*bc
aWestphalian University of Applied Sciences, Department of Environmental Engineering, Neidenburgerstraße 10, 45897 Gelsenkirchen, Germany
bEawag, Swiss Federal Institute of Aquatic Science and Technology, Überlandstrasse 133, 8600 Dübendorf, Switzerland. E-mail: urs.vongunten@eawag.ch
cSchool of Architecture, Civil and Environmental Engineering (ENAC), École Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland
dUniversity “Politehnica” of Timişoara, Faculty of Industrial Chemistry and Environmental Engineering, Bulevardul Vasile Pârvan Nr. 6, 300233 Timişoara, Romania
eUniversity of Duisburg-Essen, Faculty of Chemistry, Instrumental Analytical Chemistry, Universitätsstraße 5, 45141 Essen, Germany. E-mail: torsten.schmidt@uni-due.de
fCentre for Water and Environmental Research (ZWU), University of Duisburg-Essen, Universitätsstraße 2, 45141 Essen, Germany
gIWW Water Centre, Moritzstraße 26, 45476 Mülheim an der Ruhr, Germany
hDepartment of Chemistry and Bioscience, Aalborg University, 9220 Aalborg East, Denmark
iEMPA, Swiss Federal Laboratories for Materials Science and Technology, 8600 Dübendorf, Switzerland
jTechnical University Darmstadt, Franziska-Braun-Straße 3, 64287 Darmstadt, Germany
First published on 31st January 2020
Abstract
Five-membered nitrogen-containing heterocyclic compounds (azoles) belong to potential moieties in complex structures where transformations during ozonation can occur. This study focused on the azole–ozone chemistry of pyrrole, imidazole, and pyrazole as model compounds. Reaction kinetics and ozonation products were determined by kinetic and analytical methods including NMR, LC-HRMS/MS, HPLC-UV, and IC-MS. Analyses of reactive oxygen species (1O2, ˙OH, H2O2), quantum chemical computations (Gibbs energies), and kinetic simulations were used to further support the proposed reaction mechanisms. The species-specific second-order rate constants for the reactions of ozone with pyrrole and imidazole were (1.4 ± 1.1) × 106 M−1 s−1 and (2.3 ± 0.1) × 105 M−1 s−1, respectively. Pyrazole reacted more slowly with ozone at pH 7 (kapp = (5.6 ± 0.9) × 101 M−1 s−1). Maleimide was an identified product of pyrrole with a 34% yield. Together with other products, formate, formamide, and glyoxal, C and N mass balances of ∼50% were achieved. Imidazole reacted with ozone to cyanate, formamide, and formate (∼100% yields per transformed imidazole, respectively) with a closed mass balance. For pyrazole, a high ozone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pyrazole molar stoichiometry of 4.6 was found, suggesting that the transformation products contributed to the over-stoichiometric consumption of ozone (e.g., hydroxypyrazoles). Glyoxal and formate were the only identified transformation products (C mass balance of 65%). Overall, the identified major products are suspected to hydrolyze and/or be biodegraded and thereby abated by a biological post-treatment typically following ozonation. However, as substructures of more complex compounds (e.g., micropollutants), they might be more persistent during biological post-treatment.
:pyrazole molar stoichiometry of 4.6 was found, suggesting that the transformation products contributed to the over-stoichiometric consumption of ozone (e.g., hydroxypyrazoles). Glyoxal and formate were the only identified transformation products (C mass balance of 65%). Overall, the identified major products are suspected to hydrolyze and/or be biodegraded and thereby abated by a biological post-treatment typically following ozonation. However, as substructures of more complex compounds (e.g., micropollutants), they might be more persistent during biological post-treatment.
Water impactAzoles are commonly found structures in biomolecules and micropollutants. However, their fate during ozonation is not well understood. This study provides information on the kinetics and mechanisms of ozone-reactions with pyrrole, imidazole, and pyrazole as model compounds. The findings of this study lead to a better understanding of the fate of micropollutants containing azole moieties during water treatment with ozone. |
Introduction
Micropollutants are ubiquitously present in the aquatic environment.1,2 Although micropollutants are present in surface waters only in the μg L−1 to ng L−1 range, they may be harmful to the aquatic ecosystem and/or pose a potential risk to human health through contamination of drinking water.3 Ozonation has been proven to successfully abate micropollutants in drinking waters and wastewater effluents.4–7 During ozonation, micropollutants are not mineralized but converted to transformation products. Often, biologically active micropollutants lose their effects upon transformation during ozonation,8–11 however in some cases, transformation products with higher toxicity than their parent compounds were formed.12–16 To predict the transformation products and assess their toxicities upon ozonation, a detailed knowledge of ozone reaction mechanisms is needed. To this end, several compilations of known reaction pathways are available,17,18 which recently resulted in an ozone pathway prediction tool.19 Many well-known ozone-reactive groups, such as phenols, olefins and nitrogen-containing moieties are commonly found as structural units of micropollutants. Several studies were carried out to understand ozone–nitrogen interactions for primary, secondary and tertiary amines and anilines.18,20,21 Information is also available for some heterocyclic amines such as N-heterocyclic aromatics, piperidine, piperazine, and morpholine.22–24 Nevertheless, there is still lack of information on the reactivity and/or transformation reactions of five-membered nitrogen-containing heterocyclic compounds (or azoles) such as pyrrole, imidazole, and pyrazole (Fig. 1), which are commonly present in natural and synthetic organic compounds. Essential amino acids, histidine and tryptophan, contain an imidazole or a fused pyrrole moiety, respectively.25 The nucleobases, adenine and guanine, contain a fused imidazole moiety, as part of the DNA.25 Naturally occurring pigments, chlorophyll a (green pigments in plant) and heme (red pigments in blood) contain four pyrrole units which jointly form a larger ring system that complexes metal ions in the center.25 Azoles are also of great significance as synthetic compounds. They have been widely used as core units of pharmaceuticals, pesticides, dyes, plastics, etc.26–28 | ||
| Fig. 1 Azole model compounds investigated in this study: chemical structures of pyrrole, imidazole, and pyrazole. | ||
Because of their widespread occurrences and numerous applications in medical and material sciences, azoles are likely to be present in municipal and industrial wastewaters. Among more than 500 environmentally relevant micropollutants for the aquatic environment,7 approximately 50% are heterocyclic compounds and about 20% of the heterocycles contain pyrrole, imidazole, or pyrazole moieties, based on our assessment. Ionic liquids, considered as promising replacements for traditional organic solvents, contain cationic head groups such as imidazolium. They have received an increasing attention as emerging contaminants in the aquatic environment because of their chemical and thermal stability and high solubility in water.29,30 Pyrazole has recently been detected in the rivers Meuse and Rhine with exceptionally high concentrations (∼100 μg L−1).31 The source was identified to be industrial processes related to acrylonitrile synthesis which produces pyrazole as a by-product.
Recently, haloazoles, azoles substituted with one or more halogen atoms, have been identified as a new class of disinfection by-products.30 Tribromopyrroles were detected during chlorine disinfection of drinking water and wastewater containing high bromide levels.32,33 2,3,5-Tribromopyrrole was found to be a strong cytotoxin and genotoxin in mammalian cells.32 Tetrapyrroles, metabolites of the above-mentioned natural pigments, were suggested to be precursors.33 Tri- and di-bromo-1-methylimidazoles were detected in swimming pool and spa waters treated by bromine.34 Histidine introduced by urine, sweat, and personal care products containing an imidazole moiety was proposed as a possible precursor.
Despite the importance of understanding the fate of azoles in water and wastewater treatment, only limited information is available regarding the kinetics and mechanisms of their reactions with oxidants and in particular with ozone. Early investigations on the oxidation of azoles, which date back to the 1960s, were mostly carried out in organic solvents.35–38 More recently, six-membered heterocyclic amines, such as pyridine, pyridazine, pyrimidine and pyrazine were thoroughly investigated in terms of their reactions with ozone in aqueous solution.23 They were found to react slowly with ozone with second-order rate constants <102 M−1 s−1.23 On the contrary, imidazole and histidine showed very high reactivity towards ozone with second-order rate constants >105 M−1 s−1.39 This implies that different reaction mechanisms are involved in the reactions of ozone with five-membered heterocyclic amines in comparison to the six-membered ones.
In this study the kinetics and mechanisms of the reactions of five-membered heterocycles, i.e., pyrrole, imidazole, and pyrazole with ozone were investigated (Fig. 1). The second-order rate constants of the reactions of azoles with ozone were determined by competition kinetics with 3-buten-2-ol as a competitor or by direct monitoring of the ozone decrease. Transformation products formed during the azole–ozone reactions were identified by nuclear magnetic resonance (NMR), liquid chromatography coupled with high resolution tandem mass spectrometry (LC-HRMS/MS), ion chromatography coupled with conductivity detection (IC-CD) or mass spectrometry (IC-MS), and high performance liquid chromatography coupled with UV detection (HPLC-UV). Additionally, reactive oxygen species including singlet oxygen, hydroxyl radical, and hydrogen peroxide were quantified. Finally, kinetic simulations and quantum chemical computations were conducted to obtain additional mechanistic insights. The combination of the results of all these methods and approaches were used to derive reaction mechanisms of the azole–ozone reactions.
Materials and methods
Chemical reagents
A list of chemicals used in this study is shown in Table S1 in the ESI.†Ozone reaction kinetics
All the applied kinetic methods are described in Text S1 (ESI†) and examples of competition kinetic and pseudo-first-order kinetic plots are shown in Fig. S1 (ESI†). With the determined kapp, kinetic simulations were performed by the Kintecus software40 based on the models described in Text S2 (ESI†). Apparent second-order rate constants (kapp) for the reactions of pyrrole and imidazole with ozone in the pH range 0.5 to 12 were determined by a competition kinetic method with 3-buten-2-ol as a competitor.41kapp for the reaction of pyrazole with ozone at pH 7 was determined by following the pyrazole decrease by HPLC-UV under pseudo-first-order conditions of excess ozone. Additionally, kapp for the reactions of maleimide and 4-hydroxypyrazole (an identified and a suspected transformation product, respectively) with ozone at pH 7 were determined by following the ozone decrease by measurement with indigo-trisulfonate42 under pseudo-first-order conditions of excess substrate. All kinetic experiments were carried out in presence of dimethylsulfoxide (DMSO) or t-butanol, as ˙OH scavengers.Ozonation experiments for target compounds abatement and determination of transformation products
The required initial concentrations of azoles and ozone varied depending on subsequent analyses for product identification. Accordingly, various ozonation conditions were applied in this study and are described in detail in Text S3 (ESI†). For most analyses including LC-HRMS/MS and IC-CD, ozonation experiments were conducted by preparing approximately 100 μM of azole and 50 mM of t-butanol (˙OH scavenger) in 10 mM phosphate buffer solution at pH 7 with a total volume of 10 mL. Ozone stock solutions were prepared by sparging ice-cooled water with ozone-containing oxygen.42 The concentrations of ozone (typically 1.4–1.6 mM) were determined spectrophotometrically with ε260nm = 3200 M−1 cm−1.18 Aliquots of the ozone stock solution were added to the azole solutions under rapid stirring to initiate ozone reactions with ozone doses ranging from 0 to 200 μM. Ozonated solutions were left at room temperature for 4–6 hours to ensure complete consumption of ozone and stored at 4 °C prior to further analyses carried out within a week. For HPLC-UV quantifying formamide, higher concentrations of azoles and ozone were applied (Text S3(b), ESI†). For NMR identifying products from the pyrrole–ozone solution, ozone-containing oxygen-gas was directly sparged to the pyrrole solution (Text S3(d), ESI†). Some experiments were conducted in ultrapurified water without buffering agents (Text S3(c) and (d), ESI†), which resulted in significant differences in pH before and after ozonation (differed by maximum 2 pH units). Under such conditions, ozone reactivity of azoles and their transformation products might change over the course of the reaction, especially for dissociating compounds, and thus the evolution of products might be different from the results obtained under buffered conditions.Analytical methods
N-(3-Oxo-1-propen-1-yl)formamide (TP1), 5-hydroxy-1,5-dihydro-2H-pyrrol-2-one (TP2), and maleimide were identified by NMR. Pyrrole, imidazole, pyrazole, 1-benzylimidazole, 1-benzylpyrazole, maleimide, urea, and N-benzylformamide were analyzed by LC-HRMS/MS. Cyanate was analyzed by IC-CD as well as IC-MS. Formate was analyzed by IC-CD. Formamide was analyzed by HPLC-UV as well as GC-MS. Total N-nitrosamines were analyzed by a UV-photolysis–chemiluminescence system.43 Glyoxal was analyzed by a derivatization method using o-phenylendiamine and the derivatized product, quinoxaline, was determined by HPLC-UV.44 Singlet oxygen (1O2) was quantified by detecting the phosphorescence emitted by 1O2 at 1270 nm by a near-infrared photomultiplier tube.20 Hydroxyl radicals (˙OH) were quantified by using the ˙OH quencher t-butanol. Formaldehyde, a product of the reaction of t-butanol with ˙OH with a reported yield of ∼50%,45 was quantified by the Hantzsch method.46 Formaldehyde formed during the reaction of pyrazole with ozone was taken into account for calculating its ˙OH yield. For pyrrole and imidazole, formaldehyde formation from the direct ozone reactions was minimal and thus disregarded. Hydrogen peroxide (H2O2) was quantified spectrophotometrically with Allen's reagent (molybdate-activated iodide).47,48 More details on the methods are provided in Texts S4 and S5, Fig. S3 to S10, Tables S5 to S7 (ESI†). The limit of quantifications (LOQ) and precisions of the methods are summarized in Table S6 (ESI†).Quantum chemical computations
All computations were performed with Gaussian09 vD.01.49 The thermodynamic feasibility of the proposed reactions was tested with free energy calculations, along with the connectivity of intermediates on the potential energy surface.50 The LC-ωPBE51 functional was chosen for the description of the initial ozone reactivity52 and for its generally sound description of the thermochemistry.51 The SMD implicit solvation model was used.53 Free energy differences are reported with respect to the isolated molecules, ozone and an azole. Details are given in Text S6 (ESI†).Results and discussion
1. Ozone reaction kinetics
Apparent second-order rate constants (kapp) for the reactions of ozone with pyrrole, imidazole, and pyrazole show that pyrrole and imidazole react fast with ozone with kapp > 103 M−1 s−1 for pH 0.5–12 and pyrazole reacts moderately fast with kapp = (5.6 ± 0.9) × 101 M−1 s−1 at pH 7 (Table 1, Table S2, ESI†). kapp of pyrrole was hardly affected by pH (Fig. S2, ESI†), with averaged kapp of (1.4 ± 1.1) × 106 M−1 s−1. kapp of imidazole changed over two orders of magnitude in the pH range 2–12. This is due to the acid–base speciation of imidazole with a pKa of 7.0, which lies in the investigated pH range where pyrrole is present as a neutral species54 (note that generally deprotonated species react faster with ozone than protonated ones).18 The determined kapp values of imidazole were fitted by the expression kapp = kNH+(1 − α) + kNα and α = 1/(1 + 10pKa-pH) (Fig. S2, ESI†). kNH+ and kN are the species-specific second-order rate constants for the reactions of ozone with the protonated and the neutral imidazole, respectively, and α is the fraction of the neutral imidazole. Based on this approach, the species-specific second-order rate constant for the reaction of ozone with the neutral imidazole was determined to be (2.3 ± 0.1) × 105 M−1 s−1 (Table 1), identical to the previously reported value of 2.4 × 105 M−1 s−1.39 The species-specific second-order rate constant for the protonated imidazole is (1.5 ± 0.1) × 103 M−1 s−1 (Table 1). The kapp values determined in absence of an ˙OH scavenger were similar to those determined in presence of an ˙OH scavenger (Table S2, ESI†). Maleimide, an identified product of the pyrrole–ozone reaction, reacts fast with ozone with kapp = (4.2 ± 0.2) × 103 M−1 s−1 at pH 7. The moderate to high ozone reactivity of azoles is especially remarkable when compared to six-membered heterocyclic amines such as pyridine, pyrazine, and pyridazine with second-order rate constants for the reactions with ozone of <2 M−1 s−1.23 The high ozone reactivity of pyrrole probably relates to the effect of the nitrogen atom in the π-aromatic system, which increases the electron density of the ring.25 Consequently, ozone reacts faster with pyrrole than with typical, unsubstituted aromatic rings, e.g., benzene, of which kO3 = 2 M−1 s−1.55 In contrast, the nitrogen in pyridine withdraws the electron density from the ring,25 rendering the nitrogen more susceptible to the ozone attack, rather than the ring. This agrees with the high yield of pyridine N-oxide determined after ozonation.23 Interestingly, these reactions show much lower kinetics than reactions of tertiary amines with ozone (typical kO3 ≥ 106 M−1 s−1),20,56 which also form N-oxides as major products.| Compound | pKaa | pHb | k, M−1 s−1 | k app,pH7, M−1 s−1 | Ref. |
|---|---|---|---|---|---|
| a pKa values were based on the protonated form for pyrrole, imidazole, and pyrazole (i.e., NH2+ → NH)54 and based on the neutral form for maleimide (NH → N−).84 b Experimental pH range. c Obtained by averaging kapp for the pH range 0.5–12 (Table S2). d Obtained by fitting kapp for the pH range 2–12 with the expression kapp = kNH+(1 − α) + kNα and α = 1/(1 + 10pKa-pH) (dotted line in Fig. S2, ESI†). e See Text S1 (ESI†) for the estimation. | |||||
| Pyrrole | −3.8 | 0.5–12 | (1.4 ± 1.1) × 106c | (8.6 ± 0.7) × 105 | This study |
| Imidazole | 7.0 | 2–12 | (2.3 ± 0.1) × 105 (as kN)d | (1.7 ± 0.2) × 105 | This study |
| (1.5 ± 0.1) × 103 (as kNH+)d | This study | ||||
| 2.4 × 105 (as kN) | 39 | ||||
| 2.1 × 102 (as kNH+) | 39 | ||||
| Pyrazole | 2.5 | 7 | ND | (5.6 ± 0.9) × 101 | This study |
| Maleimide | 9.5 | 7 | ND | (4.2 ± 0.2) × 103 | This study |
| 4-Hydroxypyrazole | Not available | 7 | ND | >9 × 104e | This study |
2. Reactive oxygen species
Reactive oxygen species (ROS) such as singlet oxygen (1O2), hydroxyl radical (˙OH), and hydrogen peroxide (H2O2) can be used to elucidate reaction mechanisms.18 In this study, they were quantified for the reactions of azoles with ozone with molar excess of azole relative to ozone ([azole]0/[O3] > 1) and the corresponding yields are summarized in Table 2. The ROS yields per consumed ozone (Δ[ROS]/Δ[O3]) varied significantly for pyrrole, imidazole, and pyrazole. All measured data for 1O2, ˙OH, and H2O2 are summarized in Table S7, Fig. S9 and S10 (ESI†), respectively. Pyrrole produced mostly 1O2 (11%) and H2O2 (7%) and only minor quantities of ˙OH (2%), whereas pyrazole produced mostly ˙OH (8%) and only traces of 1O2 (2%) and H2O2 (2%). Imidazole showed almost no formation of any type of ROS (all ≤1%). The sum of all ROS yields per consumed ozone was only up to 20% for azoles, which is much smaller than 60–76%, the sum of the 1O2 and ˙OH yields per consumed ozone determined for aliphatic amines.20 The low ROS yields for the azole–ozone reactions are most likely due to reaction pathways, which do not form any ROS (see below). The ROS yields per consumed ozone were also multiplied by the ozone:azole molar stoichiometry to calculate the ROS yields per consumed azole (Δ[ROS]/Δ[azole]) (Table 2). These yields will be compared with the yields of the formed transformation products below.
| Azole | Product (X) | X typea | Ozone:azole molar stoichiometryb |
Δ[X]/Δ[O3]c | Δ[X]/Δ[azole]d | Δ[X]/Δ[N]e | Δ[X]/Δ[C]e |
|---|---|---|---|---|---|---|---|
|
a ROS, TP_N, and TP_C represent reactive oxygen species, nitrogenous transformation products, and carbonaceous transformation products, respectively.
b Ozone:azole molar stoichiometry values were obtained by inverting the slopes from the linear regression of the measured concentrations and taking the absolute values (Fig. S11, ESI†).
c For ˙OH, H2O2, maleimide, formate, glyoxal, cyanate, and N-benzylformamide, the yields per consumed ozone were determined by slopes from the linear regression of the measured concentrations at molar ratios of [O3]/[azole]0 ≤ 1. For 1O2, the yields were averaged for 6 replicates at a fixed molar ratio of [O3]/[azole]0 = 0.03–0.04. For formamide, the yields were averaged for duplicate experiments at fixed molar ratios of [O3]/[azole]0 = 2.0 for the pyrrole–ozone reaction and at [O3]/[azole] = 0.4 and 0.8 for the imidazole–ozone reaction.
d Yields per consumed azoles were calculated by multiplying yields per ozone (Δ[X]/Δ[O3]) by the ozone:azole molar stoichiometry.
e Yields per nitrogen (or carbon) were calculated by multiplying the number of nitrogen (or carbon) of the product and dividing the number of nitrogen (or carbon) of the azole, with the yields per abated azole.
f Total represents nitrogen or carbon mass balance.
|
|||||||
| Pyrrole | ˙OH | ROS | 1.2 | 2% | 2% | — | — |
| H2O2 | ROS | 1.2 | 7% | 8% | — | — | |
| 1O2 | ROS | 1.2 | 11% | 13% | — | — | |
| Maleimide | TP_N | 1.2 | 29% | 34% | 34% | 34% | |
| Formamide | TP_N | 1.2 | 12% | 14% | 14% | 3.5% | |
| Formate | TP_C | 1.2 | 46% | 54% | 0% | 14% | |
| Glyoxal | TP_C | 1.2 | 4% | 5% | 0% | 2.5% | |
| Total | 48% | 54% | |||||
| Imidazole | ˙OH | ROS | 1.0 | 1% | 1% | — | — |
| H2O2 | ROS | 1.0 | 0.2% | 0.1% | — | — | |
| 1O2 | ROS | 1.0 | 0.1% | 0.1% | — | — | |
| Formamide | TP_N | 1.0 | 105% | 104% | 52% | 35% | |
| Cyanate | TP_N | 1.0 | 114% | 113% | 57% | 38% | |
| Formate | TP_C | 1.0 | 98% | 97% | 0% | 32% | |
| Glyoxal | TP_C | 1.0 | <LD | <LD | <LD | <LD | |
| Total | 109% | 105% | |||||
| 1-Benzylimidazole | N-Benzylformamide | TP_N | 1.0 | 93% | 91% | 46% | 30% |
| Pyrazole | ˙OH | ROS | 4.6 | 8% | 35% | — | — |
| H2O2 | ROS | 4.6 | 2% | 7% | — | — | |
| 1O2 | ROS | 4.6 | 2% | 8% | — | — | |
| Formamide | TP_N | 4.6 | <LD | <LD | <LD | <LD | |
| Nitrosamines | TP_N | 4.6 | <LD | <LD | <LD | <LD | |
| Formate | TP_C | 4.6 | 28% | 126% | 0% | 42% | |
| Glyoxal | TP_C | 4.6 | 7% | 34% | 0% | 23% | |
| Total | 0% | 65% | |||||
| 1-Benzylpyrazole | N-Benzylformamide | TP_N | 6.5 | 1% | 9% | 4.5% | 3% |
3. Transformation products and reaction pathways
Reaction pathways of the azole–ozone reactions were investigated by identifying transformation products and taking into consideration the formation of ROS. Quantum chemical computations for Gibbs energies of reaction pathways were additionally performed to elucidate the plausibility of the proposed mechanisms.Quantified products. To identify transformation products from the pyrrole–ozone reaction, pyrrole was oxidized by ozone in presence of t-butanol as a ˙OH scavenger mostly at pH 7 (10 mM phosphate buffer) except for the samples prepared for NMR analysis and for glyoxal determination (no buffer, Text S3, ESI†). Fig. 2 shows the abatement of pyrrole as a function of the molar ozone
:pyrrole ratio (initial concentrations) ([O3]/[pyrrole]0). The ozonated pyrrole samples were analyzed by various methods including NMR, LC-HRMS/MS, HPLC-UV, and IC-CD. Based on the combination of these analyses, the formation of six transformation products were confirmed: N-(3-oxo-1-propen-1-yl)formamide (TP1, hereafter), 5-hydroxy-1,5-dihydro-2H-pyrrol-2-one (TP2, hereafter), maleimide, formamide, formate, and glyoxal (for chemical structures see Fig. 3). Maleimide, formamide, formate, and glyoxal for which reference standards are available, were quantified at varying ozone doses. The formation trends are shown in Fig. 2 and the product yields are summarized in Table 2. Product concentrations in Fig. 2 are expressed as pyrrole-normalized relative product concentrations, because the initial pyrrole concentrations were different for the various analytical methods. The stoichiometry for the reaction of ozone with pyrrole was calculated by the slope from a linear regression of the pyrrole concentrations measured with substoichiometric doses of ozone ([O3]/[pyrrole]0 < 1). The fitting results for pyrrole as well as other azoles are shown in Fig. S11 (ESI†). Pyrrole decreased with an ozone:pyrrole molar stoichiometry of 1.2, close to 1. Maleimide was gradually formed with a yield of 34% per consumed pyrrole up to about one molar equivalent of ozone ([O3]/[pyrrole]0 = 1) and then decreased at higher molar ratios of [O3]/[pyrrole]0. The concentrations of pyrrole and maleimide were predicted by a kinetic simulation by the Kintecus software,40 with measured apparent second-order rate constants for the reactions with ozone at pH 7 (8.6 × 105 M−1 s−1 and 4.2 × 103 M−1 s−1 for pyrrole and maleimide, respectively, Table 1). The model includes the stoichiometric coefficient for maleimide formation of 0.34, based on the measured maleimide yield (Table S3, ESI†). The predicted concentrations are shown as lines in Fig. 2 and agree well with the measured data. Formamide was formed with a yield of 14% per consumed pyrrole at a ratio of [O3]/[pyrrole]0 = 2. It was not possible to quantify formamide at lower molar ratios of [O3]/[pyrrole]0 of 1.1 and 0.8, because of an interference of neighboring peaks in the HPLC-UV method. Formate shows a gradual formation over the entire range of molar ratios of [O3]/[pyrrole]0 with a yield of 54% per consumed pyrrole. Glyoxal was quantified as a minor product with a yield of 5% per consumed pyrrole. The product yields per consumed pyrrole can be further assessed in terms of nitrogen or carbon mass balances. For example, the yield of formamide was 14% per consumed pyrrole, which corresponds to 3.5% per carbon, as formamide contains only 1/4 of carbons in comparison to pyrrole. The yields of other products were converted likewise, and the sum of the yields per nitrogen and per carbon for all products were 48% and 54%, respectively (Table 2). This means about half of nitrogen and carbon mass balances were completed based on the quantified products. The other missing half of the mass balances can be partially explained by TP1 and TP2 for which the formation was only qualitatively confirmed (see below). The formation of maleimide and TP2 involve various intermediates (Fig. 4) which may also contribute to the mass balances. Product yields determined under the different pH conditions (pH 2 and 11) are summarized in Fig. S12 and Table S8 (ESI†). Maleimide was formed with similar yields at pH 2 and 7. Formate was formed with a higher yield at pH 11. This may indicate that the reaction proceeds differently at higher pH, but further investigations are needed to confirm this.
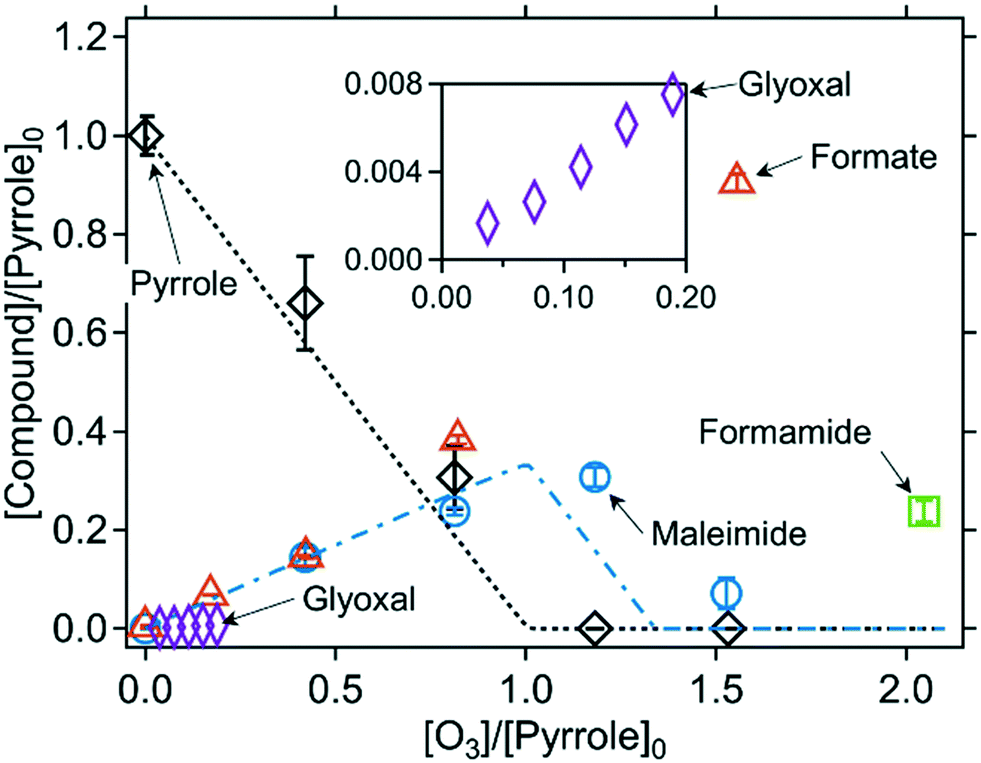 | ||
| Fig. 2 Relative abatement of pyrrole and the formation (and consumption) of transformation products as a function of the molar ratio of [O3]/[pyrrole]0. The inset has the same axis labels as the main figure. The symbols indicate the measured concentrations (single measurement for glyoxal and duplicate/triplicate for the other compounds) and the lines indicate simulated concentrations based on the kinetic models described in Table S3 (ESI†). All measurements were carried out in presence of t-butanol (0.05–1 M). 10 mM phosphate buffer was used for all measurements but glyoxal. The initial pyrrole concentrations were around 110 μM for determining pyrrole, maleimide, and formate, 0.8–2.0 mM for determining formamide, and 1 mM for determining glyoxal (Text S3, ESI†). | ||
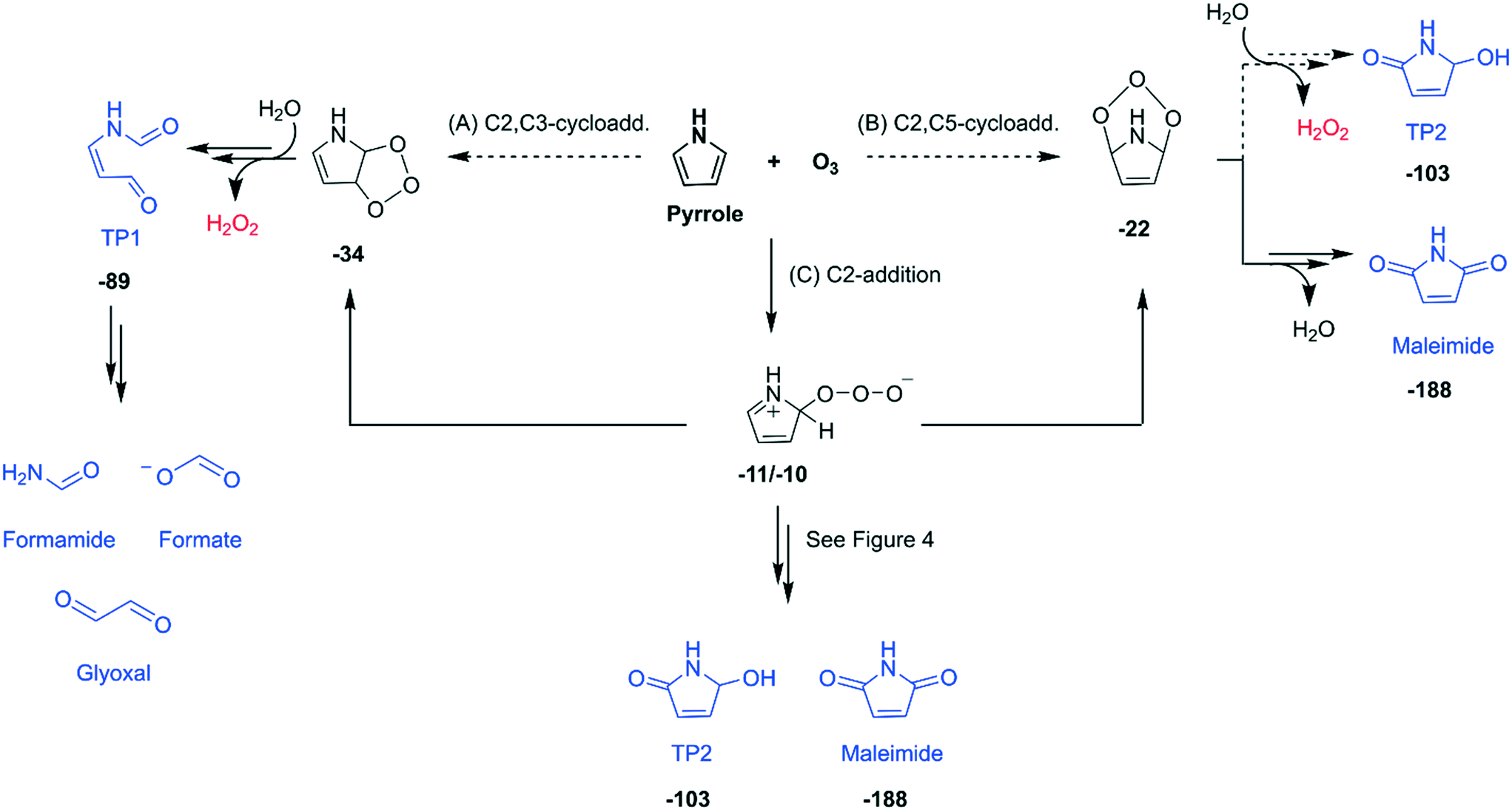 | ||
| Fig. 3 Initial reaction pathways (A–C) proposed for the reaction of pyrrole with ozone based on the identified transformation products (highlighted in blue) and reactive oxygen species (in red). Bold numbers below the products and the intermediates are calculated Gibbs energies with respect to the initial reactants (pyrrole and ozone) in kcal mol−1. Double arrows represent pathways involving more than one reaction step. Dashed arrows indicate that the transition structures have not been found or investigated with quantum chemical computations. The full descriptions of the initial pathways including computed transition structures are shown in Fig. S27–S29 (ESI†). | ||
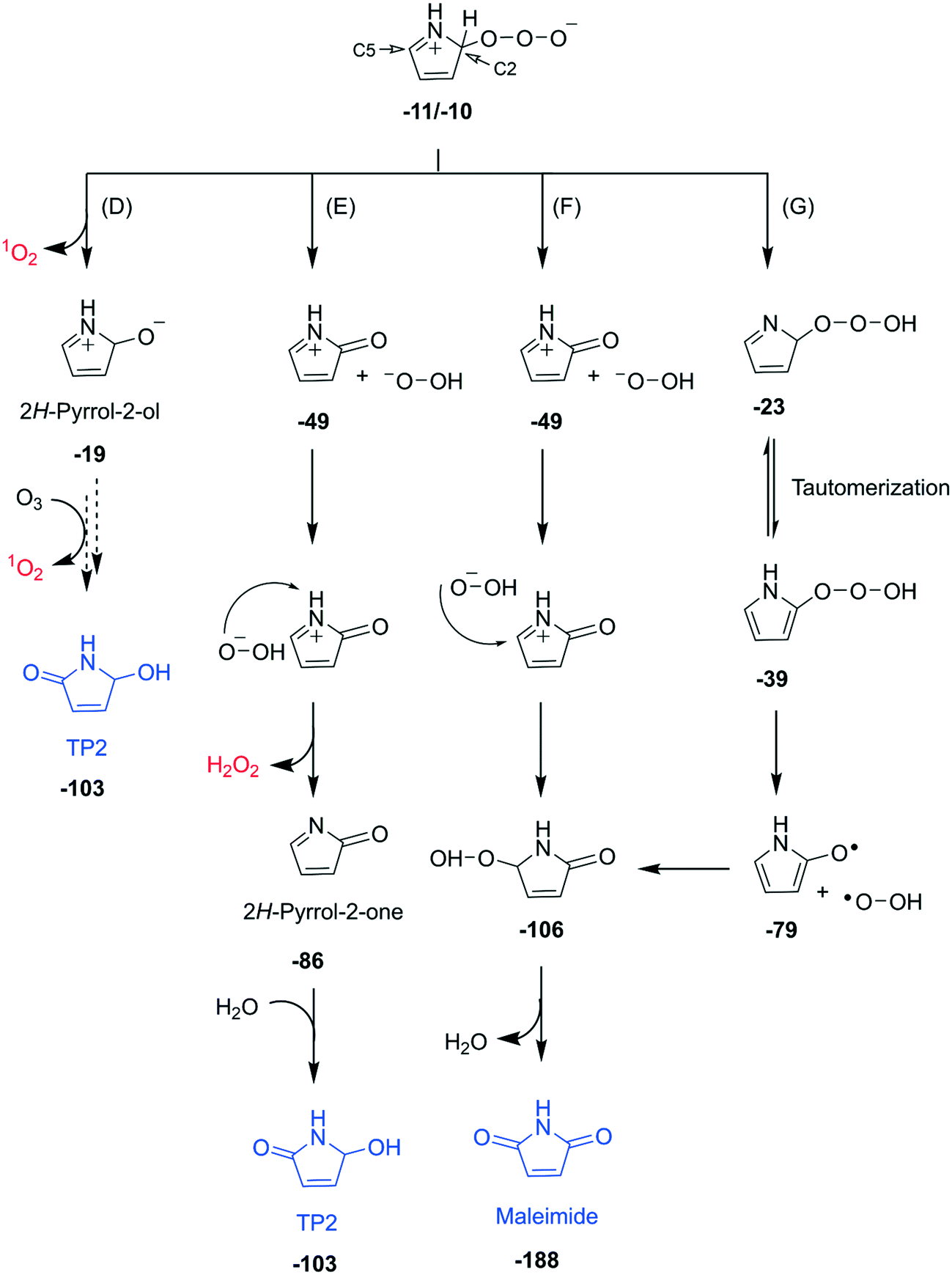 | ||
| Fig. 4 Detailed reaction mechanisms of the subsequent transformation of the zwitterion formed via C2-addition (pathway C in Fig. 3) of the pyrrole–ozone reaction (pathways D–G). Bold numbers below the products and the intermediates are calculated Gibbs energies with respect to the initial reactants (pyrrole and ozone) in kcal mol−1. Dashed arrows indicate that the corresponding reactions have not been investigated by quantum chemical computations. The full descriptions of the pathways including computed transition structures are shown in Fig. S30–S32 (ESI†). | ||
Identified but not quantified products. The formation of TP1, TP2, and maleimide were qualitatively confirmed by NMR (Text S7, ESI†). Assignments of 1H and 13C NMR data of TP1, TP2, and maleimide are summarized in Table S9 (ESI†) and the recorded NMR spectra are shown in Fig. S14 to S17 (ESI†). Because of the lack of reference standards, quantification of TP1 and TP2 was not possible. Moreover, the isolated solid from the ozonated pyrrole sample was not fully soluble in acetone-d6 (NMR solvent), hindering an estimation of the relative quantities of TP1 and TP2 based on the comparison of 1H signal intensities (Text S8, ESI†). The NMR sample containing TP1 and TP2 was also analyzed by LC-HRMS/MS to additionally support their presence and identification. Both TPs have an exact mass of m/z = 100.0393 as [M + H]+ (molecular formula of C4H5NO2) that showed up as two peaks at retention times of 4.2 min and 6.3 min (C4H5NO2_4.2 and C4H5NO2_6.3, Fig. S18b, ESI†) with distinct MS2 fragmentation patterns (Fig. S19, ESI†). C4H5NO2_4.2 produced mostly a fragment ion of m/z = 83.0125, corresponding to C4H2O2 (loss of NH3), whereas C4H5NO2_6.3 produced mostly m/z = 72.0443, corresponding to C3H5NO (loss of CO). TP1 with a carbonyl group at the end of the open-chain structure appears to be able to more readily release CO and form C3H5NO during fragmentation (Fig. S20b, ESI†) than TP2 with a cyclic structure (Fig. S21b, ESI†). In contrast, TP2 might more easily produce C4H2O2 than TP1, because TP1 requires an intramolecular rearrangement to release NH3 (Fig. S21a, ESI† for TP2 and Fig. S20a, ESI† for TP1). Examples of possible fragmentation pathways predicted by the mass spectra interpretation software (Mass Frontier™, Thermo Scientific) are illustrated in Fig. S20 and S21 (ESI†). Based on the structural characteristics and the distinct MS2 fragmentation patterns, C4H5NO2_4.2 and C4H5NO2_6.3 are likely to correspond to TP2 and TP1, respectively.
The C4H5NO2_4.2 and C4H5NO2_6.3 peaks were not only observed in the sample prepared for NMR analysis but also in the samples with varying molar ratios of [O3]/[pyrrole]0 during the pyrrole abatement and the maleimide formation (Fig. 2). The integrated peak areas as a function of the molar ratios of [O3]/[pyrrole]0 are plotted in Fig. S22 (ESI†). Both peaks were gradually formed reaching a maximum formation at about one molar equivalent of ozone ([O3]/[pyrrole]0 = 1) and decrease at higher ozone doses, similarly to maleimide (Fig. 2). Considering the similar formation trends, TP1, TP2, and maleimide seem to be formed in parallel via different reaction pathways.
Unidentified products. A non-target screening with LC-HRMS/MS of the ozonated pyrrole samples revealed two additional peaks at retention times of 5.4 min and 7.0 min (C4H5NO_5.4 and C4H5NO_7.0, Fig. S23a, ESI†), which showed significant intensities for the ozonated pyrrole samples in comparison to the ozonated blank (no pyrrole) samples. Both peaks correspond to the exact mass of C4H5NO with a mass deviation of 3 ppm. 1,5-Dihydro-pyrrole-one (see the structure in Fig. S23b, ESI†) was suspected, based on previous studies reporting it as an oxidation product of pyrrole.35,57 However, the reference standard, which is commercially available, resulted in too many peaks, probably due to the presence of impurities or isomers, which made it ambiguous to match the retention times (Fig. S23b, ESI†). The fragmentation patterns look similar for the ozonated pyrrole sample and the standard with the same fragment ion of m/z = 56.0500 (Fig. S24, ESI†).
The C4H5NO_5.4 and C4H5NO_7.0 peaks are plotted against the molar ratio of [O3]/[pyrrole]0 (Fig. S25, ESI†). They were formed at a molar ratio of [O3]/[pyrrole]0 of 0.5, followed by a gradual decrease, concurrent with the increase of C4H5NO2 (Fig. S26, ESI†). This implies C4H5NO was formed at an earlier stage of the pyrrole–ozone reaction and could be a precursor of C4H5NO2.
Initial reaction pathways. Based on the product identification, possible formation pathways for the transformation products formed during the reactions of pyrrole with ozone are proposed (Fig. 3). Ozone initially attacks at a double bond of pyrrole via a Criegee-type mechanism (pathways A and B) or addition on C2-position (pathway C). Such concerted cycloadditions have been studied in depth for reactions of simple unsaturated hydrocarbons (e.g., ethene and ethyne) with ozone in aqueous phase by a recent quantum chemical study.52 The pyrrole–nitrogen is excluded as a possible attack site, because its lone electron pair is delocalized in the π-system of the ring. A recent study demonstrated that the addition of atomic oxygen on the pyrrole–N is very unlikely compared to the addition on C, based on quantum chemical computations of the energies of the corresponding products (+39 kcal mol−1 for N-addition and −19 kcal mol−1 for C2-addition).58 Pathways A and B are cycloadditions of ozone to different positions of the ring. The ozone addition to the C2,C3-positions may lead to the ring-opened product, TP1, which could further degrade into formamide, formate, and glyoxal. In contrast, the addition to the C2,C5-positions could result in TP2 or maleimide. Besides cycloadditions, ozone can also attack at the C2 position to form a zwitterionic adduct (pathway C). The zwitterion can be rearranged to the C2,C3- or C2,C5-cycloadducts or follow other routes such as via cleavage of an O–O bond. The latter is discussed below in detail.
Addition of ozone to the C2,C5-positions of the five-membered ring was proposed for the reaction of furan with ozone.59 Additionally, earlier studies which investigated the oxidation of substituted pyrroles with singlet oxygen suggested both C2,C3- and C2,C5-cycloadditions.60–62 These studies found the product with two oxygen atoms added to the C2,C5-positions (equivalent to TP2) and the ring-opened product (equivalent to TP1). Compounds containing a maleimide, formamide, or an epoxide moiety were also identified or hypothesized as minor products.61,62 The minor product containing a formamide moiety was presumably formed by subsequent degradation of the ring-opened product,61 which could also explain the formation of formamide as well as formate and glyoxal observed in our study.
Quantum chemical computations. Additional evidence supporting the proposed pathways was obtained by quantum chemical computations of the energies of the products and their transition structures. All initial pathways (A–C) are exergonic reactions with −10 to −34 kcal mol−1 for the calculated energies of the products with respect to the initial reactants, pyrrole and ozone (Fig. 3 and more details in Fig. S27–S29, ESI†). However, our calculations found transition structures only for pathway C, but not for pathways A nor B (presented as dashed arrows in Fig. 3). Even though this implies pathway C as the only plausible reaction, we cannot exclude the possibility of pathways A and B, because ozone is typically considered as a difficult molecule to be treated by quantum chemical computations.50,63,64 If the cycloaddition pathways A and B occur, pathway A seems more likely than pathway B, because the ring strain of the C2,C5-cycloadduct is probably more pronounced than that of the C2,C3-cycloadduct (Text S10, ESI†). For pathway C and the rearrangement to the cycloadducts, transition structures were found between the zwitterion and the C2,C3-cycloadduct and between the zwitterion and C2,C5-cycloadduct (Fig. S27, ESI†). Once the cycloadducts are formed, further reactions to the identified products, TP1 or maleimide, are exergonic with defined transition structures (Fig. S28 and S29, ESI†). The reaction of the C2,C5-cycloadduct to form TP2 could not be assessed, because of lack of details about the corresponding reaction mechanism. The full assessment of the proposed initial reaction pathways on the basis of the computation results are summarized in Texts S10–S12 (ESI†).
Subsequent reactions from pathway C. The zwitterion, the initial product of pathway C, can undergo diverse subsequent reactions, with a cleavage of an O–O bond and production of 1O2, hydrogen peroxide anion, or hydroperoxyl radical as transient oxygen species (Fig. 4). The cleavage of the O–O bond to form 1O2 would produce 2H-pyrrole-2-ol, an isomer of 1,5-dihydro-pyrrole-one with the molecular formula of C4H5NO, which could further react with ozone to produce TP2 and another 1O2 (pathway D). The formation of 1,5-dihydro-pyrrole-one was confirmed by other studies during the oxidation of pyrrole with hydrogen peroxide.35,57 Our analyses suggest the formation of 1,5-dihydro-pyrrole-one by the detection of its exact mass by LC-HRMS, but could not clearly identify it (Fig. S23, ESI†). Pathways E and F involve a cleavage of the O–O bond associated with H-transfer from the C2 position of the ring to form a hydrogen peroxide anion and a cationic form of 2H-pyrrole-2-one. The ensuing ions can be recombined by two ways. First, the hydrogen peroxide anion can abstract H+ from N–H of the cation to form H2O2 and 2H-pyrrole-2-one (pathway E). The latter can react with water to form TP2. Second, the hydrogen peroxide anion can also attack the C5 position of the ring to eventually form maleimide (pathway F). Such recombinations seem to occur as cage reactions based on the minimal free energy barrier (Fig. S31, ESI†). The cleavage of the O–O bond can also occur homolytically by forming the hydroperoxyl radical (pathway G). The radical further reacts with the ring at the C5 position to form maleimide. The maleimide formation pathways forming the hydrogen peroxide anion or the hydroperoxyl radical followed by its subsequent attack on the C5 position of the ring (pathways F and G) do not produce any reactive oxygen species. These pathways can explain the low total ROS yields of 23% measured per consumed pyrrole as the sum of ˙OH, 1O2, and H2O2 (Table 2). All pathways depicted in Fig. 4 are thermodynamically feasible based on quantum chemical computations and the corresponding Gibbs energies are shown in bold numbers underneath the chemical structures (kcal mol−1). Detailed discussion and results including transition structures for each pathway are presented in Texts S13–S15 (ESI†).
Quantified products. The abatement of imidazole and the formation of the ozone-induced transformation products are shown in Fig. 5. Imidazole reacts with ozone with an ozone
:imidazole molar stoichiometry of 1 (Fig. S11, ESI†). Cyanate, formamide, and formate were identified as major products with high yields of (113 ± 5)%, (104 ± 5)%, and (97 ± 4)%, respectively, per consumed imidazole (Table 2). Urea also seems to be formed as a minor products with a yield of 8% (Text S16 and Fig. S33, ESI†). However, its formation and yield were determined by a semi-quantitative approach and therefore require further confirmation (Text S16†). The total yields of cyanate, formamide, and formate were (109 ± 4)% per nitrogen and (105 ± 3)% per carbon, resulting in almost perfect nitrogen and carbon mass balances. The three products were formed concurrently, as all of them reached the maximum formation at one molar equivalent of ozone ([O3]/[imidazole]0 = 1). In addition, a kinetic simulation (lines in Fig. 5) could well describe the evolution of imidazole and the products as a function of the ozone doses, based on the apparent second-order rate constants for the reactions of ozone with imidazole (k = 1.7 × 105 M−1 s−1 (Table 1)), cyanate (k ≤ 10−2 M−1 s−1),65 formamide (k ≤ 10−2 M−1 s−1, estimated), and formate (k = 46 M−1 s−1)66 at neutral pH (Table S4, ESI†). The second-order rate constant for the reaction of formamide with ozone was estimated to be small, considering its similar evolution as for cyanate. Under the condition where imidazole was completely consumed ([O3]/[imidazole]0 > 1), formate shows a gradual decrease because its ozone reactivity is much higher than for cyanate and formamide. At a high ozone dose ([O3]/[imidazole]0 ∼ 2), the measured concentration of formate was higher than the predicted concentration, due to currently unknown reasons. The yield of formate decreased significantly at pH 2 (Fig. S12, ESI†), which suggests that the proposed mechanism may not be applicable to acidic conditions. However, more information (e.g., cyanate and formamide yields at low pH) is needed to confirm these findings.
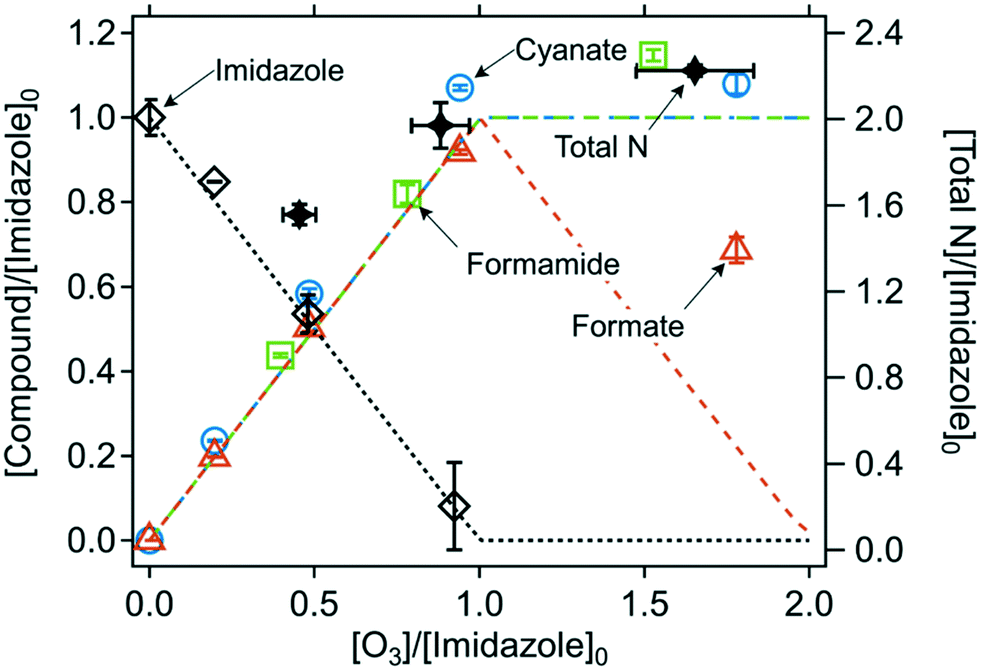 | ||
| Fig. 5 Relative abatement of imidazole and the formation of transformation products as a function of the molar ratio of [O3]/[imidazole]0. The symbols indicate the measured concentrations (duplicate or triplicate) and the lines indicate simulated concentrations based on the kinetic models described in Table S4 (ESI†). Total N indicates the sum of the concentrations of the nitrogen in the residual imidazole and all nitrogen-containing transformation products. All measurements were carried out in presence of t-butanol at pH 7 (10 mM phosphate buffer). The initial imidazole concentration was around 100 μM for all experiments except for formamide determination where it was 1–4 mM (Text S3, ESI†). | ||
The formation of formamide as a major product was also observed for another imidazole-containing compound (1-benyzlimidazole) in this study. Similar to imidazole, 1-benzylimidazole was fully consumed at a 1:1 molar ratio of ozone to 1-benzylimidazole and transformed into N-benzylformamide with a 91% yield (Fig. S35, ESI†). This additionally supports the efficient conversion of imidazole to formamide upon ozonation and implies that a similar reaction can also occur in more complex compounds.
Reaction mechanisms. Three key findings can be summarized based on the product identification of the imidazole–ozone reaction: (1) upon ozonation, imidazole was degraded into three compounds, namely cyanate, formamide, and formate; (2) the reaction was complete with one molar equivalent of ozone; (3) the reaction produced almost no reactive oxygen species. Based on these findings, a mechanism for the imidazole–ozone reaction is proposed in Fig. 6. Ozone initially attacks at the C–C double bond of imidazole to form an ozonide intermediate, similarly to pathway A in the pyrrole–ozone reaction. However, different from the pyrrole case, the zwitterion derived from the ozonide intermediate is rearranged into a 3H-1,2,4-dioxazole ring to form a dioxazole intermediate, as hypothesized in Fig. 6. The dioxazole intermediate then further decomposes to formamide and formyl isocyanate with the aid of a base and a proton. The resulting formyl isocyanate further hydrolyzes into formate and cyanate. Based on this mechanism, all oxygen atoms in ozone are transferred to the products, with no reactive oxygen species being formed.
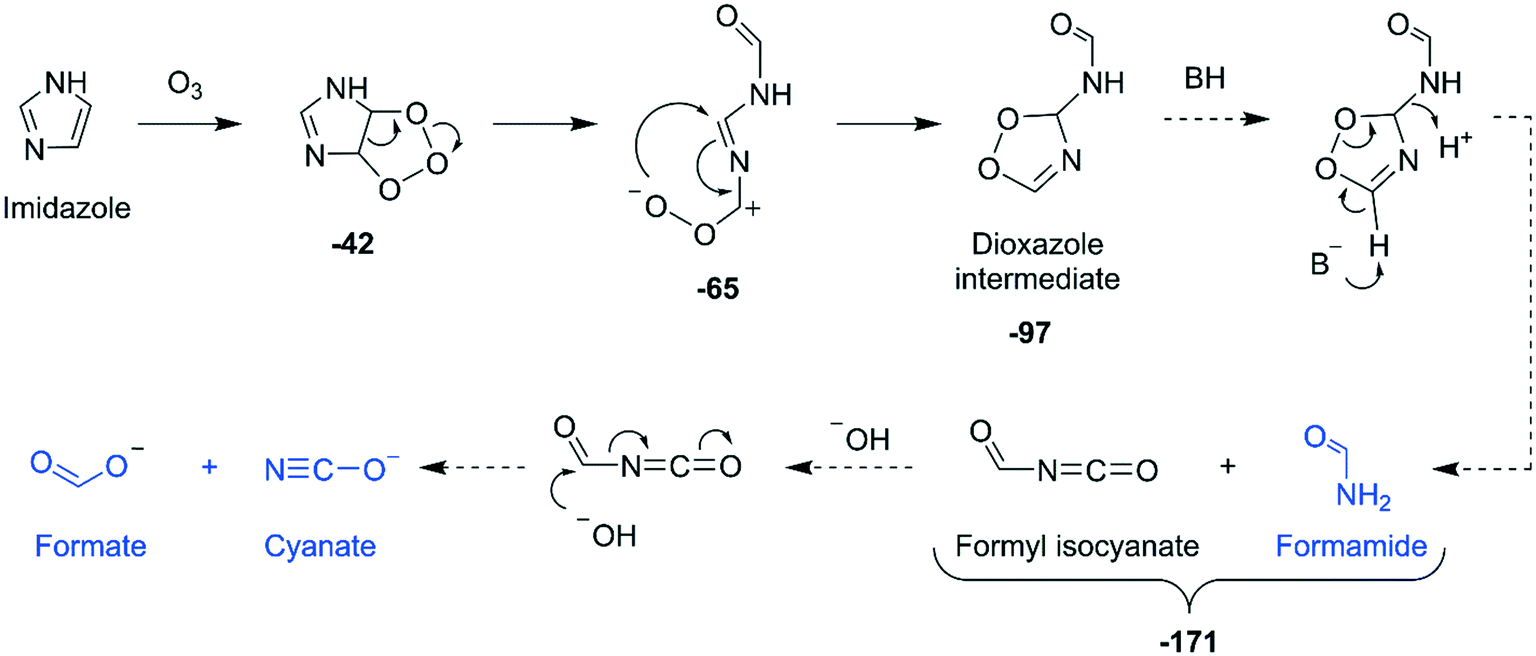 | ||
| Fig. 6 Proposed mechanisms for the reaction of imidazole with ozone based on the identified transformation products (highlighted in blue). Bold numbers below the products and the intermediates are calculated Gibbs energies with respect to the initial reactants (imidazole and ozone) in kcal mol−1. Dashed arrows indicate that the corresponding reactions have not been investigated by quantum chemical computations. The full description of the pathway including computed transition structures is shown in Fig. S36 (ESI†). | ||
Quantum chemical computations. The proposed reaction mechanisms were partially investigated by quantum chemical computations. The cycloaddition on the C–C double bond, the ring cleavage, and the rearrangement of the zwitterion to the hypothesized dioxazole intermediate are exergonic reactions with −42, −65, and −97 kcal mol−1, respectively, for the calculated energies with respect to the initial reactants, imidazole and ozone (Fig. 6). The corresponding transition structures to the investigated reaction steps were found by the computations and are presented in Fig. S36 (ESI†). The subsequent reactions from the dioxazole intermediate were not investigated in detail, but expected to be exergonic (Text S17, ESI†).
Information on oxidation products of imidazole is not easily available. A few studies investigated the oxidation of histidine, an amino acid containing an imidazole moiety, but no mechanistic consensus has been obtained.67–70 The carbon between the two nitrogens in the imidazole ring was suggested as a main attack site of the oxidation of histidine in metal-catalyzed systems69,70 and by ozone.67 This led to the formation of 2-oxohistidine as a final product. However, a product with three oxygens added to histidine was also reported during the oxidation of histidine with ozone,68 which could be formed by an ozone attack on the C–C double bond of the imidazole ring. However, the previous ozonation experiments67,68 were performed without ˙OH scavenger, and thus it is unclear whether the products were formed by the reaction with ozone or with ˙OH. Our findings strongly suggest the ozone attack at the double bond of the ring rather than at the carbon between the two nitrogens, according to the concomitant formation of the three major products by one molar equivalent of ozone, as discussed above.
Quantified products. The abatement of pyrazole as a function of the molar ratio of [O3]/[pyrazole]0 is shown in Fig. 7. Pyrazole required about 5 molar equivalents of ozone for its full abatement (Fig. S11, ESI†), which is much higher than for the pyrrole- and imidazole–ozone reactions. The ˙OH yield was also particularly high (35% per consumed pyrazole, Table 2) in comparison to the other azole–ozone reactions. The ˙OH yield was even higher at pH 11 (87%, Fig. S12 and Table S8, ESI†). The ozone reactivity of hydroxide ion (k = 70 M−1 s−1)71 is similar to that of pyrazole. Therefore, at high pH where hydroxide ions are predominantly present, the reaction of hydroxide ion with ozone can outcompete the pyrazole–ozone reaction and result in forming excess ˙OH. Formate and glyoxal were the only quantifiable transformation products, with yields of 126% and 34% per consumed pyrazole, respectively. The total yield of formate and glyoxal achieved 65% of the carbon mass balance. Formate yields remained similarly high for pH 2, 7, and 11 (Fig. S12, ESI†). No nitrogen-containing products were identified. Formamide was not detected (LOQ = 1.6 μM) even for high molar [O3]/[pyrazole]0 ratio of 5, for which pyrazole should be completely abated according to the ozone
:pyrazole molar stoichiometry. N-Nitroso compounds were suspected because of the N–N bond present in pyrazole, but they were not detected for any molar [O3]/[pyrazole]0 ratios by the analysis of the total concentrations of N-nitroso compounds (LOQ = 0.7 μM).43 A non-target screening using LC-HRMS/MS resulted in three exact masses of interest, showing increases in the peak intensities with increasing ozone doses (Fig. S37 and S38, ESI†). However, molecular formulas assigned for these masses are not reasonable (e.g., containing five nitrogens for m/z = 201.1009, Table S13, ESI†). Moreover, MS2 spectra were not measurable, probably due to the low intensity, which hindered obtaining further structural evidence. It seems that the products formed during the pyrazole–ozone reactions were not retained well on the applied LC column, poorly ionized by ESI-MS, or had masses under the scan range of the MS detector (m/z = 50 as the lower limit), since pyrazole itself has only a mass of 68.
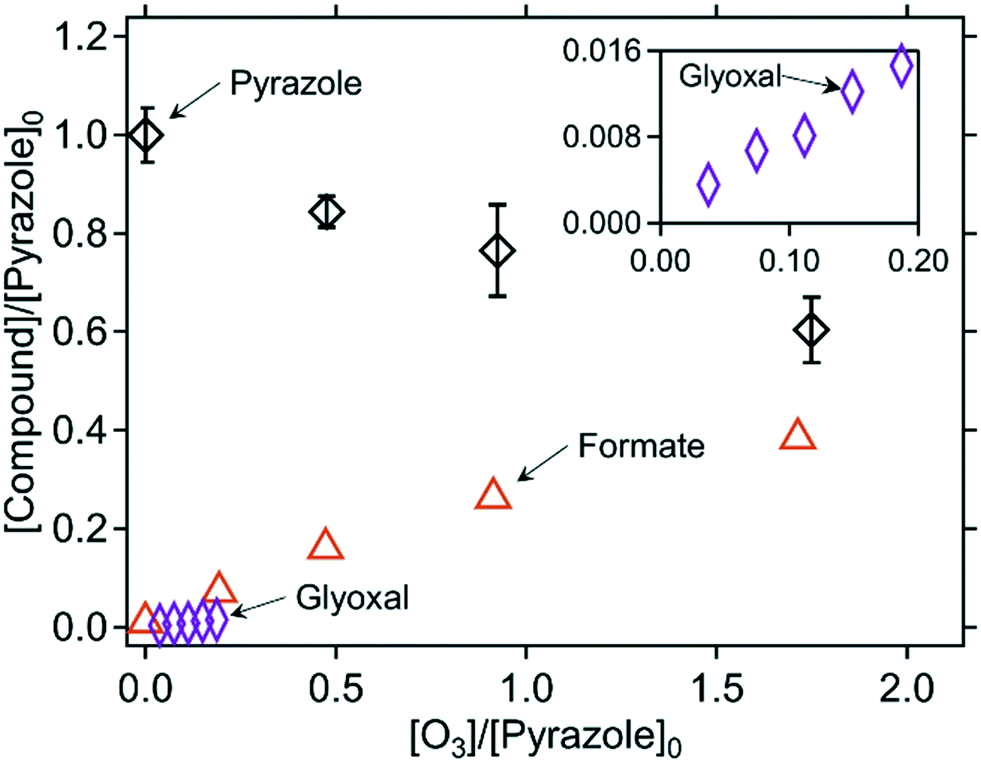 | ||
| Fig. 7 Relative abatement of pyrazole and the formation of formate as a function of the molar ratio of [O3]/[pyrazole]0. The inset has the same axis labels as the main figure. The symbols indicate the measured concentrations (triplicate for pyrazole and single measurements for formate and glyoxal). All measurements were carried out in presence of t-butanol. 10 mM phosphate buffer was used for all measurements but glyoxal. The initial pyrazole concentrations were around 100 μM for determining pyrazole and formate, and 1 mM for determining glyoxal (Text S3, ESI†). | ||
Benzylpyrazole-ozone reaction. To facilitate the product identification by LC-HRMS/MS, 1-benzylpyrazole, a substituted pyrazole with a benzyl group, was chosen as an alternative model compound. A similarly inefficient abatement was observed as for the pyrazole–ozone reaction, with an ozone
:1-benzylpyrazole molar stoichiometry of 6.5 (Table 2 and Fig. S39, ESI†). N-Benzylformamide was detected as a minor product with a yield of 9% per consumed 1-benzylpyrazole. N-Nitroso compounds were not detected at molar [O3]/[1-benzylpyrazole]0 ratios of 2 and 4. A non-target screening found three masses with m/z = 191.0813, 163.0864, and 151.0864 (as [M + H]+), corresponding to the molecular formulas C10H10N2O2, C9H10N2O, and C8H10N2O, respectively, with a mass deviation of less than 2 ppm (note that the molecular formula of 1-benzylpyrazole is C10H10N2). All masses are characterized by a similar formation trend, increasing in peak intensities for increasing molar ratios of [O3]/[1-benzylpyrazole]0, reaching a maximum at 1 < [O3]/[1-benzylpyrazole]0 < 2, and then slightly decreasing at [O3]/[1-benzylpyrazole]0 > 2.5 (Fig. S40, ESI†). The evolution of these compounds is quite similar, implying they were formed in parallel and not sequentially.
To elucidate their structures, MS2 spectra of m/z = 191.0813, 163.0864, and 151.0864 were measured (Fig. S41–S43, ESI†) and compared with simulated fragment ions of candidate structures by an in silico fragmenter, MetFrag.72 The candidate structures for each mass were found by searching the exact mass (e.g., m/z = 191.0813 as [M + H]+) in a compound database, PubChem,73 and were further processed by MetFrag72 to generate a list of theoretical fragments based on a bond dissociation approach. More details of procedures and results are described in Text S18, ESI.† The MetFrag analyses proposed two types of transformation products: a ring product with the addition of two oxygens in ketone and enol forms, corresponding to m/z = 191.0813 (shown as candidate 191a, Fig. 8 and S41D, ESI†) and a ring-opened product containing a hydrazide moiety, corresponding to m/z = 151.0864 (shown as candidate 151c, Fig. 8 and S43D, ESI†). For m/z = 163.0864, no valid hit was found.
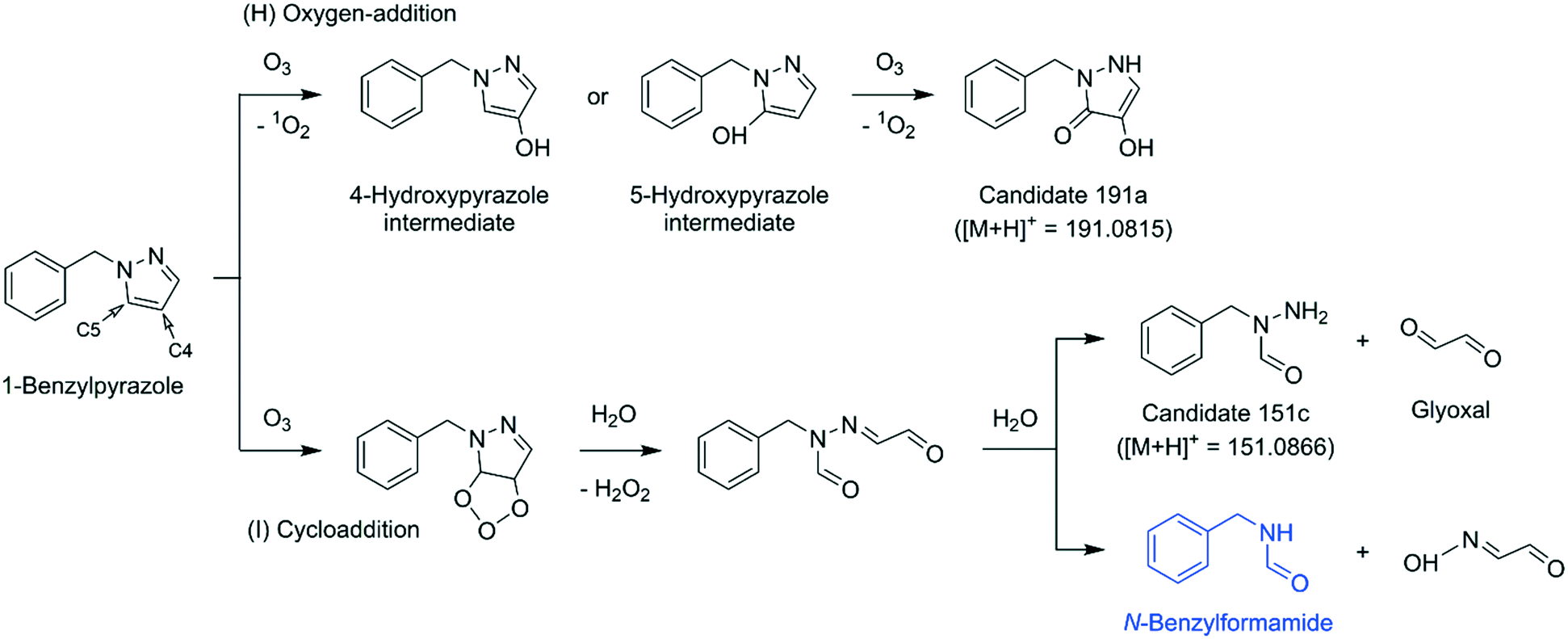 | ||
| Fig. 8 Proposed mechanisms for the reaction of 1-benzylpyrazole with ozone based on the identified (highlighted in blue) and the proposed products. Glyoxal was confirmed by a reference standard for the reaction of pyrazole with ozone. Candidates 191a and 151c were proposed based on the exact masses detected by LC-HRMS/MS analyses. Theoretical exact masses are shown in parentheses. | ||
Reaction mechanism. Based on the proposed transformation products, a potential mechanism of the reactions of 1-benzylpyrazole with ozone is proposed in Fig. 8. The first pathway (H) explains the formation of the ring product (candidate 191a) via a series of oxygen-addition mechanisms. The second pathway (I) describes the formation of the ring-opened product (candidate 151c) or N-benzylformamide via a Criegee-type mechanism followed by hydrolysis of the resulting hydrazone. Both pathways are initiated by an ozone attack on the pyrazole moiety rather than on the benzene moiety. This can be substantiated because of the higher ozone reactivity of pyrazole, higher than of benzene (kO3 = 56 M−1 s−1 for pyrazole (Table 1) and 2 M−1 s−1 for benzene55).
Candidate 191a contains two oxygen atoms added to the C4 and C5 positions of the pyrazole ring. Therefore, it is speculated that a reaction intermediate containing a hydroxypyrazole moiety preceded candidate 191a (e.g., 4-hydroxypyrazole or 5-hydroxypyrazole intermediates, Fig. 8). The ozone reactivity of 4-hydroxypyrazole was determined as a representative case for hydroxypyrazoles. Its apparent second-order ozone rate constant at pH 7 was estimated to be higher than 9 × 104 M−1 s−1 (Table 1 and Text S1, ESI†), much higher than for benzene. Thus, the hydroxypyrazole moiety is significantly more reactive to ozone than the benzene moiety, wherefore, it can further react with ozone to form candidate 191a as a secondary ozonation product. The exact mass of the 4-hydroxypyrazole intermediate was not detected by LC-HRMS/MS for the ozonated benzylpyrazole samples. If the 4-hydroxypyrazole intermediate was formed, it would have been quickly oxidized to the subsequent product and escaped the detection. The fate of the resulting candidate 191a, whether it would be hydrolyzed or further oxidized, is unknown. Candidate 151c formed by the cycloaddition pathway (I) accompanies glyoxal as the other product fragmented from the pyrazole ring (Fig. 8). Glyoxal was not measured for the benzylpyrazole–ozone reaction, but measured for the pyrazole–ozone reaction (34% glyoxal per consumed pyrazole, Table 2). If a similarly high yield of glyoxal is assumed for the benzylpyrazole–ozone reaction, the reaction seems to proceed to a large extent through the cycloaddition pathway.
By extrapolating the findings of the benzylpyrazole–ozone reaction to the pyrazole–ozone reaction, pyrazole would be transformed into 4,5-dihydroxypyrazole and formylhydrazine, equivalent to the candidates 191a and 151c, respectively. However, the corresponding exact masses of the products (101.0346 and 61.0396 as [M + H]+) were not detected by LC-HRMS/MS for any ozonated pyrazole samples. Nevertheless, their formation cannot be entirely ruled out. In addition to the proposed oxygen-addition and cycloaddition pathways, electron transfer pathways seem to be involved in the pyrazole–ozone reaction, based on the particularly high ˙OH yield (35% per consumed pyrazole, Table 2). The primary ozone attack on pyrazole or secondary ozone reactions of transient reaction intermediates may undergo electron transfer pathways and form excessive ˙OH. However, the product information is not sufficient to propose a plausible reaction mechanism.
The high molar ozone:azole stoichiometry of the pyrazole and 1-benzylpyrazole is probably attributed to the formation of reaction intermediates at least as reactive with ozone as pyrazole, consuming ozone over-stoichiometrically. Potential ozone consuming products could be hydroxypyrazoles which have been suggested on the basis of the LC-HRMS/MS results. However, their formation upon ozonation still needs to be confirmed by reference standards. Moreover, the formation of hydroxypyrazole alone cannot fully explain the high stoichiometry and more unknown intermediates must have been involved.
4. Implications
Pyrrole, imidazole, and pyrazole are frequently present as substructures of biomolecules or micropollutants. Upon ozonation, effective abatement of pyrrole- and imidazole-moieties is expected according to the high ozone reactivity determined in this study (k > 103 M−1 s−1), which warrants the formation of the identified products. Pyrrole and imidazole derivatives would be transformed by ozone into the products containing a maleimide, formamide, or cyanate moiety with a moderate to high yield, according to our findings (34% maleimide from the pyrrole–ozone reaction, ∼100% formamide and ∼100% cyanate from the imidazole–ozone reaction). However, as substructures of more complex molecules, they may behave differently during ozonation. Kinetics and products can be altered by substitutions by other functional groups. Examples are sartan drugs containing imidazole. Depending on the substitution on imidazole, the ozone reactivities of the sartans differ up to four orders of magnitude (e.g., k = 23 M−1 s−1 for irbesartan and k = 2.1 × 105 M−1 s−1 for losartan).7 Despite the possible low ozone reactivity due to the substitution effect, micropollutants containing pyrrole or imidazole were typically well abated during ozonation at a full scale wastewater treatment plant (Table S14, ESI†).7 In this case, this is mostly due to reactions with ˙OH formed during ozonation and a different set of products is expected in this case.As single compounds, the ozone products maleimide, cyanate, and formamide do not appear to be persistent in the aquatic environment, but are rather hydrolyzed74,75 or biodegraded.76 Since ozonation is typically followed by a biological post-treatment,5 they might be readily abated during the post-treatment. The C–C double bond in maleimide with two neighboring carbonyl groups is highly susceptible to nucleophilic additions. Therefore, maleimide reacts fast and selectively with thiol groups.77 Such selectivity may induce toxic effects by converting cysteine residues of proteins, in analogy to α,β-unsaturated enedials and oxoenals produced by oxidation of phenols.78 Formamide is polar and biodegradable and is less prone to be bioaccumulated in aquatic organisms.76 However, some studies reported toxic effects possibly associated with a formamide moiety in more complex compounds. An increased toxicity observed in phototransformation studies of herbicides was assigned to N-formylated products.79,80 Similarly, a recent study on phototransformation of ionic liquids containing an imidazole moiety reported an increased toxicity possibly induced by N-formyl transformation products.81 Compared to pyrrole and imidazole, pyrazole reacts slower with ozone (kapp = 56 M−1 s−1 at pH 7) and exhibits a much higher ozone:azole molar stoichiometry. Compounds with low ozone reactivity can be still abated during an ozonation process by ˙OH. Examples are irbesartan and acesulfame which are not highly reactive towards ozone (kO3 = 23 M−1 s−1 for irbesartan7 and 88 M−1 s−1 for acesulfame82), but still significantly or reasonably well abated under a conventional ozonation condition (75% for irbesartan and ∼50% for acesulfame).7 The ˙OH reactivity of pyrazole lies between irbesartan and acesulfame (k˙OH = ∼1010 M−1 s−1 for irbesartan,7 4.6 × 109 M−1 s−1 for acesulfame,82 and ∼8 × 109 M−1 s−1 for pyrazole83). Therefore, at least a moderate abatement of pyrazole is expected during ozonation process. Ozonation products of pyrazole are still largely unknown. Transformation products, at least as reactive towards ozone as pyrazole (e.g., hydroxypyrazoles), are postulated, but none of them could be confirmed by reference standards, which requires further investigations.
Conclusion
All azole–ozone reactions investigated in this study were initiated by an ozone attack on the C–C double bond in the ring. From the initial attack, the reactions went through distinct pathways clearly differentiated among pyrrole, imidazole, and pyrazole. Pyrrole reacted with ozone via multiple mechanisms (Criegee and oxygen-addition) to form ring products (5-hydroxy-1,5-dihydro-2H-pyrrol-2-one and maleimide) as well as a ring-opened product (N-(3-oxo-1-propen-1-yl)formamide). The major quantified products from the pyrrole–ozone reaction were maleimide, formamide, and formate with yields of 34%, 14%, and 54%, respectively, per consumed pyrrole. The total yields of all identified products accounted for 48% and 54% of the nitrogen and carbon mass balances, respectively.Imidazole reacted with ozone predominantly via a Criegee mechanism with ring cleavage to form three fragments (formamide, cyanate, and formate). The fragmentation was initiated by only a single attack of ozone, and all oxygen atoms ended up in the products, producing almost no reactive oxygen species. The sum of the yields of the three products was ∼100% per nitrogen and per carbon, completely closing the mass balance of imidazole.
Pyrazole is the most persistent compound against ozonation among the studied azoles, showing an ozone reactivity at least three orders magnitude lower than those of pyrrole and imidazole. Also, pyrazole requires about five molar equivalents of ozone for a complete abatement. The exceptionally high stoichiometry suggests the formation of primary products, such as hydroxypyrazoles, reacting fast with ozone. Hydroxypyrazoles and hydrazides were postulated, possibly formed via an oxygen-addition and a cycloaddition pathway. Glyoxal and formate were identified as major products, completing 65% of the carbon mass balance. No nitrogenous products were identified from the pyrazole–ozone reaction.
All confirmed nitrogenous transformation products (maleimide, formamide, and cyanate) are expected to be readily hydrolyzed and/or biodegraded. Therefore, they are supposed to be well removed by a biological post-treatment typically following ozonation. They do not appear to pose a direct threat to the aquatic environment as single substances, but as subunits of more complex compounds (e.g., micropollutants), they might be persistent during the post-treatment, with unknown (eco)toxicological consequences.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The NMR hardware was partially granted by the Swiss National Science Foundation (SNSF, grant no. 206021_150638/1). We thank Elisabeth Salhi for her support with the ozonation experiments and IC-CD measurements, Rebekka Gulde, Jennifer Schollée, and Marc Suter for sharing their expertise on LC-HRMS/MS data analyses, Cornel Niederhauser and Samuel Derrer for the help on the work-up for NMR samples. We would also like to thank Laura Wiegand and Klaus Kerpen for their support in IC-measurements as well as HPLC-measurements and Natalia Tabler for her help by translation of Russian literature.References
- A. Pal, K. Y.-H. Gin, A. Y.-C. Lin and M. Reinhard, Impacts of emerging organic contaminants on freshwater resources: Review of recent occurrences, sources, fate and effects, Sci. Total Environ., 2010, 408(24), 6062–6069 CrossRef PubMed.
- R. P. Schwarzenbach, B. I. Escher, K. Fenner, T. B. Hofstetter, C. A. Johnson and U. von Gunten, et al., The Challenge of Micropollutants in Aquatic Systems, Science, 2006, 313(5790), 1072–1077 CrossRef PubMed.
- Human Pharmaceuticals, Hormones and Fragrances - The Challenge of Micropollutants in Urban Water Management, ed. A. Joss and T Ternes, IWA Publishing, 2006 [cited 2018 Jul 25], Available from: https://iwaponline.com/ebooks/book/19/ Search PubMed.
- T. A. Ternes, J. Stüber, N. Herrmann, D. McDowell, A. Ried and M. Kampmann, et al., Ozonation: a tool for removal of pharmaceuticals, contrast media and musk fragrances from wastewater?, Water Res., 2003, 37(8), 1976–1982 CrossRef PubMed.
- U. von Gunten, Oxidation Processes in Water Treatment: Are We on Track?, Environ. Sci. Technol., 2018, 52(9), 5062–5075 CrossRef PubMed.
- R. I. L. Eggen, J. Hollender, A. Joss, M. Schärer and C. Stamm, Reducing the Discharge of Micropollutants in the Aquatic Environment: The Benefits of Upgrading Wastewater Treatment Plants, Environ. Sci. Technol., 2014, 48(14), 7683–7689 CrossRef PubMed.
- M. Bourgin, B. Beck, M. Boehler, E. Borowska, J. Fleiner and E. Salhi, et al., Evaluation of a full-scale wastewater treatment plant upgraded with ozonation and biological post-treatments: Abatement of micropollutants, formation of transformation products and oxidation by-products, Water Res., 2018, 129, 486–498 CrossRef.
- M. C. Dodd, H.-P. E. Kohler and U. von Gunten, Oxidation of Antibacterial Compounds by Ozone and Hydroxyl Radical: Elimination of Biological Activity during Aqueous Ozonation Processes, Environ. Sci. Technol., 2009, 43(7), 2498–2504 CrossRef.
- M. M. Huber, T. A. Ternes and U. von Gunten, Removal of Estrogenic Activity and Formation of Oxidation Products during Ozonation of 17α-Ethinylestradiol, Environ. Sci. Technol., 2004, 38(19), 5177–5186 CrossRef PubMed.
- F. Lange, S. Cornelissen, D. Kubac, M. M. Sein, J. von Sonntag and C. B. Hannich, et al., Degradation of macrolide antibiotics by ozone: A mechanistic case study with clarithromycin, Chemosphere, 2006, 65(1), 17–23 CrossRef PubMed.
- M. C. Dodd, D. Rentsch, H. P. Singer, H.-P. E. Kohler and U. von Gunten, Transformation of β-Lactam Antibacterial Agents during Aqueous Ozonation: Reaction Pathways and Quantitative Bioassay of Biologically-Active Oxidation Products, Environ. Sci. Technol., 2010, 44(15), 5940–5948 CrossRef.
- A. Magdeburg, D. Stalter, M. Schlüsener, T. Ternes and J. Oehlmann, Evaluating the efficiency of advanced wastewater treatment: Target analysis of organic contaminants and (geno-)toxicity assessment tell a different story, Water Res., 2014, 50, 35–47 CrossRef.
- B. I. Escher, N. Bramaz and C. Ort, JEM Spotlight: Monitoring the treatment efficiency of a full scale ozonation on a sewage treatment plant with a mode-of-action based test battery, J. Environ. Monit., 2009, 11(10), 1836 RSC.
- D. Stalter, A. Magdeburg and J. Oehlmann, Comparative toxicity assessment of ozone and activated carbon treated sewage effluents using an in vivo test battery, Water Res., 2010, 44(8), 2610–2620 CrossRef CAS.
- U. von Gunten, E. Salhi, C. K. Schmidt and W. A. Arnold, Kinetics and Mechanisms of N-Nitrosodimethylamine Formation upon Ozonation of N, N-Dimethylsulfamide-Containing Waters: Bromide Catalysis, Environ. Sci. Technol., 2010, 44(15), 5762–5768 CrossRef CAS.
- C. K. Schmidt and H.-J. Brauch, N,N -Dimethylsulfamide as Precursor for N -Nitrosodimethylamine (NDMA) Formation upon Ozonation and its Fate During Drinking Water Treatment, Environ. Sci. Technol., 2008, 42(17), 6340–6346 CrossRef CAS.
- Y. Lee and U. von Gunten, Advances in predicting organic contaminant abatement during ozonation of municipal wastewater effluent: reaction kinetics, transformation products, and changes of biological effects, Environ. Sci.: Water Res. Technol., 2016, 2(3), 421–442 RSC.
- C. von Sonntag and U. von GuntenChemistry of Ozone in Water and Wastewater Treatment, IWA Publishing, 2012 [cited 2018 Mar 29], p. 320, Available from: https://www.iwapublishing.com/books/9781843393139/chemistry-ozone-water-and-wastewater-treatment Search PubMed.
- M. Lee, L. C. Blum, E. Schmid, K. Fenner and U. von Gunten, A computer-based prediction platform for the reaction of ozone with organic compounds in aqueous solution: kinetics and mechanisms, Environ. Sci.: Processes Impacts, 2017, 19(3), 465–476 RSC.
- S. Lim, C. S. McArdell and U. von Gunten, Reactions of aliphatic amines with ozone: Kinetics and mechanisms, Water Res., 2019, 157, 514–528 CrossRef CAS.
- A. Tekle-Röttering, C. von Sonntag, E. Reisz, C. Eyser vom, H. V. Lutze and J. Türk, et al., Ozonation of anilines: Kinetics, stoichiometry, product identification and elucidation of pathways, Water Res., 2016, 98, 147–159 CrossRef.
- E. Borowska, M. Bourgin, J. Hollender, C. Kienle, C. S. McArdell and U. von Gunten, Oxidation of cetirizine, fexofenadine and hydrochlorothiazide during ozonation: Kinetics and formation of transformation products, Water Res., 2016, 94, 350–362 CrossRef CAS.
- A. Tekle-Röttering, E. Reisz, K. S. Jewell, H. V. Lutze, T. A. Ternes and W. Schmidt, et al., Ozonation of pyridine and other N-heterocyclic aromatic compounds: Kinetics, stoichiometry, identification of products and elucidation of pathways, Water Res., 2016, 102, 582–593 CrossRef.
- A. Tekle-Röttering, K. S. Jewell, E. Reisz, H. V. Lutze, T. A. Ternes and W. Schmidt, et al., Ozonation of piperidine, piperazine and morpholine: Kinetics, stoichiometry, product formation and mechanistic considerations, Water Res., 2016, 88, 960–971 CrossRef.
- J. A. Joule and K. Mills, Heterocyclic Chemistry At A Glance, Wiley, 2nd edn, 2012 [cited 2018 Jun 1]. Available from: https://www.wiley.com/en-us/Heterocyclic+Chemistry+At+A+Glance%2C+2nd+Edition-p-9780470971222 Search PubMed.
- A. Ansari, A. Ali, M. Asif and S. Shamsuzzaman, Review: biologically active pyrazole derivatives, New J. Chem., 2017, 41(1), 16–41 RSC.
- V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, Pyrrole: a resourceful small molecule in key medicinal hetero-aromatics, RSC Adv., 2015, 5(20), 15233–15266 RSC.
- B. Narasimhan, D. Sharma and P. Kumar, Biological importance of imidazole nucleus in the new millennium, Med. Chem. Res., 2011, 20(8), 1119–1140 CrossRef CAS.
- M. Amde, J.-F. Liu and L. Pang, Environmental Application, Fate, Effects, and Concerns of Ionic Liquids: A Review, Environ. Sci. Technol., 2015, 49(21), 12611–12627 CrossRef CAS.
- S. D. Richardson and T. A. Ternes, Water Analysis: Emerging Contaminants and Current Issues, Anal. Chem., 2018, 90(1), 398–428 CrossRef CAS.
- K. Baken, A. Kolkman, P. van Diepenbeek, H. Ketelaars and A. van Wezel, Signalering van ‘overige antropogene stoffen’, en dan? De pyrazool-casus, H2O/Waternetwerk, 2016, [cited 2019 Jul 22]. Available from: https://www.h2owaternetwerk.nl/vakartikelen/signalering-van-overige-antropogene-stoffen-en-dan-de-pyrazool-casus Search PubMed.
- S. D. Richardson, A. D. Thruston, C. Rav-Acha, L. Groisman, I. Popilevsky and O. Juraev, et al., Tribromopyrrole, Brominated Acids, and Other Disinfection Byproducts Produced by Disinfection of Drinking Water Rich in Bromide, Environ. Sci. Technol., 2003, 37(17), 3782–3793 CrossRef CAS.
- M. Yang and X. Zhang, Halopyrroles: A New Group of Highly Toxic Disinfection Byproducts Formed in Chlorinated Saline Wastewater, Environ. Sci. Technol., 2014, 48(20), 11846–11852 CrossRef CAS.
- E. J. Daiber, D. M. DeMarini, S. A. Ravuri, H. K. Liberatore, A. A. Cuthbertson and A. Thompson-Klemish, et al., Progressive Increase in Disinfection Byproducts and Mutagenicity from Source to Tap to Swimming Pool and Spa Water: Impact of Human Inputs, Environ. Sci. Technol., 2016, 50(13), 6652–6662 CrossRef CAS.
- V. Bocchi, L. Chierici, G. P. Gardini and R. Mondelli, On pyrrole oxidation with hydrogen peroxide, Tetrahedron, 1970, 26(17), 4073–4082 CrossRef CAS.
- J. P. Wibaut and J. W. P. Boon, Über die Einwirkung von Ozon auf Pyrazol sowie einige Homologe und Derivate, Helv. Chim. Acta, 1961, 44(4), 1171–1190 CrossRef CAS.
- J. P. Wibaut, Ozonolysis of Pyrroles, Furans, and γ-Pyrones in Connection with Reactivity of Bonds in Ring System, in Ozone Chemistry and Technology, American Chemical Society, 1155 Sixteenth St., N.W. Washington 6, D.C., 1959, pp. 153–161 [cited 2019 May 7]. Available from: http://pubs.acs.org/doi/abs/10.1021/ba-1959-0021.ch024 Search PubMed.
- H. H. Wasserman, J. U. Yoo and R. W. DeSimone, Singlet Oxygen Reactions from the Adducts of Ozone with Heterocyclic Substrates, J. Am. Chem. Soc., 1995, 117(38), 9772–9773 CrossRef CAS.
- W. A. Pryor, D. H. Giamalva and D. F. Church, Kinetics of ozonation. 2. Amino acids and model compounds in water and comparisons to rates in nonpolar solvents, J. Am. Chem. Soc., 1984, 106(23), 7094–7100 CrossRef CAS.
- J. C. Ianni, Kintecus, 2017, Windows Version 6.01, www.kintecus.com.
- F. Muñoz and C. von Sonntag, Determination of fast ozone reactions in aqueous solution by competition kinetics, J. Chem. Soc., Perkin Trans. 2, 2000,(4), 661–664 RSC.
- H. Bader and J. Hoigné, Determination of ozone in water by the indigo method, Water Res., 1981, 15(4), 449–456 CrossRef CAS.
- F. Breider and U. von Gunten, Quantification of Total N -Nitrosamine Concentrations in Aqueous Samples via UV-Photolysis and Chemiluminescence Detection of Nitric Oxide, Anal. Chem., 2017, 89(3), 1574–1582 CrossRef CAS.
- S. Ohmori, M. Mori, M. Kawase and S. Tsuboi, Determination of methylglyoxal as 2-methylquinoxaline by high-performance liquid chromatography and its application to biological samples, J. Chromatogr. B: Biomed. Sci. Appl., 1987, 414, 149–155 CrossRef CAS.
- R. Flyunt, A. Leitzke, G. Mark, E. Mvula, E. Reisz and R. Schick, et al., Determination of ˙OH, O2˙−, and Hydroperoxide Yields in Ozone Reactions in Aqueous Solution, J. Phys. Chem. B, 2003, 107(30), 7242–7253 CrossRef CAS.
- T. Nash, The colorimetric estimation of formaldehyde by means of the Hantzsch reaction, Biochem. J., 1953, 55(3), 416 CrossRef CAS.
- A. O. Allen, C. J. Hochanadel, J. A. Ghormley and T. W. Davis, Decomposition of Water and Aqueous Solutions Under Mixed Fast Neutron and Gamma Radiation, J. Phys. Chem., 1952, 56, 575–586 CrossRef.
- K. Kitsuka, A. M. Mohammad, M. I. Awad, K. Kaneda, M. Ikematsu and M. Iseki, et al., Simultaneous Spectrophotometric Determination of Ozone and Hydrogen Peroxide, Chem. Lett., 2007, 36(11), 1396–1397 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- P. R. Tentscher, M. Lee and U. von Gunten, Micropollutant Oxidation Studied by Quantum Chemical Computations: Methodology and Applications to Thermodynamics, Kinetics, and Reaction Mechanisms, Acc. Chem. Res., 2019, 52(3), 605–614 CrossRef CAS.
- O. A. Vydrov and G. E. Scuseria, Assessment of a long-range corrected hybrid functional, J. Chem. Phys., 2006, 125(23), 234109 CrossRef.
- D. Trogolo, J. S. Arey and P. R. Tentscher, Gas-Phase Ozone Reactions with a Structurally Diverse Set of Molecules: Barrier Heights and Reaction Energies Evaluated by Coupled Cluster and Density Functional Theory Calculations, J. Phys. Chem. A, 2019, 123(2), 517–536 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS.
- Dissociation constants of organic acids and bases, in CRC Handbook of Chemistry and Physics, ed. J. R. Rumble, CRC Press/Taylor & Francis, Boca Raton, FL, 100th edn, 2019 Search PubMed.
- J. Hoigné and H. Bader, Rate constants of reactions of ozone with organic and inorganic compounds in water—I: non-dissociating organic compounds, Water Res., 1983, 17(2), 173–183 CrossRef.
- F. Muñoz and C. von Sonntag, The reactions of ozone with tertiary amines including the complexing agents nitrilotriacetic acid (NTA) and ethylenediaminetetraacetic acid (EDTA) in aqueous solution, J. Chem. Soc., Perkin Trans. 2, 2000,(10), 2029–2033 RSC.
- J. H. Atkinson, R. S. Atkinson and A. W. Johnson, The structure and reactions of some pyrrolin-2-ones, J. Chem. Soc., 1964, 5999–6009 RSC.
- Y. Zhang, R. Song, Y. Sun, J. Sun, Y. Tang and R. Wang, Mechanistic and kinetic study on the reaction of Pyrrole (C4H5N) with O(3P), Chem. Phys., 2018, 513, 50–57 CrossRef CAS.
- P. S. Bailey and H. O. Colomb, 1,4-addition of ozone to furans and pyrroles, J. Am. Chem. Soc., 1957, 79(15), 4238–4238 CrossRef CAS.
- D. L. Boger and C. M. Baldino, Singlet oxygen mediated oxidative decarboxylation of pyrrole-2-carboxylic acids, J. Org. Chem., 1991, 56(24), 6942–6944 CrossRef CAS.
- D. A. Lightner and C.-S. Pak, Dye-sensitized photooxygenation of tert-butylpyrroles, J. Org. Chem., 1975, 40(19), 2724–2728 CrossRef CAS.
- H. H. Wasserman, R. Frechette, V. M. Rotello and G. Schulte, Singlet oxygen reactions of 2-carbalkoxy-3-methoxypyrroles, Tetrahedron Lett., 1991, 32(51), 7571–7574 CrossRef CAS.
- S. E. Wheeler, D. H. Ess and K. N. Houk, Thinking Out of the Black Box: Accurate Barrier Heights of 1,3-Dipolar Cycloadditions of Ozone with Acetylene and Ethylene, J. Phys. Chem. A, 2008, 112(8), 1798–1807 CrossRef CAS.
- Y. Zhao, O. Tishchenko, J. R. Gour, W. Li, J. J. Lutz and P. Piecuch, et al., Thermochemical Kinetics for Multireference Systems: Addition Reactions of Ozone, J. Phys. Chem. A, 2009, 113(19), 5786–5799 CrossRef CAS.
- J. Hoigné, H. Bader, W. R. Haag and J. Staehelin, Rate constants of reactions of ozone with organic and inorganic compounds in water—III. Inorganic compounds and radicals, Water Res., 1985, 19(8), 993–1004 CrossRef.
- E. Reisz, A. Fischbacher, S. Naumov, C. von Sonntag and T. C. Schmidt, Hydride Transfer: A Dominating Reaction of Ozone with Tertiary Butanol and Formate Ion in Aqueous Solution, Ozone: Sci. Eng., 2014, 36(6), 532–539 CrossRef CAS.
- B. S. Berlett, R. L. Levine and E. R. Stadtman, Comparison of the Effects of Ozone on the Modification of Amino Acid Residues in Glutamine Synthetase and Bovine Serum Albumin, J. Biol. Chem., 1996, 271(8), 4177–4182 CrossRef CAS.
- T. Kotiaho, M. N. Eberlin, P. Vainiotalo and R. Kostiainen, Electrospray mass and tandem mass spectrometry identification of ozone oxidation products of amino acids and small peptides, J. Am. Soc. Mass Spectrom., 2000, 11(6), 526–535 CrossRef CAS.
- C. Schöneich, Mechanisms of metal-catalyzed oxidation of histidine to 2-oxo-histidine in peptides and proteins, J. Pharm. Biomed. Anal., 2000, 21(6), 1093–1097 CrossRef.
- K. Uchida, Histidine and lysine as targets of oxidative modification, Amino Acids, 2003, 25(3–4), 249–257 CrossRef CAS.
- G. Merényi, J. Lind, S. Naumov and C. von Sonntag, The Reaction of Ozone with the Hydroxide Ion: Mechanistic Considerations Based on Thermokinetic and Quantum Chemical Calculations and the Role of HO4− in Superoxide Dismutation, Chem. – Eur. J., 2010, 16(4), 1372–1377 CrossRef PubMed.
- C. Ruttkies, E. L. Schymanski, S. Wolf, J. Hollender and S. Neumann, MetFrag relaunched: incorporating strategies beyond in silico fragmentation, Aust. J. Chem., 2016, 8(1), 3 Search PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He and S. He, et al., PubChem 2019 update: improved access to chemical data, Nucleic Acids Res., 2019, 47(Database issue), D1102–D1109 CrossRef.
- National Library of Medicine (US), HSDB: Potassium Cyanate, 2004 [cited 2019 Jul 16]. Available from: http://toxnet.nlm.nih.gov/cgi-bin/sis/search2/r?dbs+hsdb:@term+@DOCNO+1757 Search PubMed.
- S. Matsui, Aida H. Hydrolysis of some N-alkylmaleimides, J. Chem. Soc., Perkin Trans. 2, 1978,(12), 1277 RSC.
- National Library of Medicine (US), HSDB: Formamide, 2006 [cited 2019 Jul 16]. Available from: http://toxnet.nlm.nih.gov/cgi-bin/sis/search2/r?dbs+hsdb:@term+@DOCNO+88 Search PubMed.
- T. Miyadera and E. M. Kosower, Receptor site labeling through functional groups. 2. Reactivity of maleimide groups, J. Med. Chem., 1972, 15(5), 534–537 CrossRef CAS.
- C. Prasse, B. Ford, D. K. Nomura and D. L. Sedlak, Unexpected transformation of dissolved phenols to toxic dicarbonyls by hydroxyl radicals and UV light, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(10), 2311–2316 CrossRef CAS.
- F. Galichet, G. Mailhot, F. Bonnemoy, J. Bohatier and M. Bolte, Iron(III) photo-induced degradation of isoproturon: correlation between degradation and toxicity, Pest Manage. Sci., 2002, 58(7), 707–712 CrossRef CAS.
- C. Tixier, L. Meunier, F. Bonnemoy and P. Boule, Phototransformation of three herbicides: chlorbufam, isoproturon, and chlorotoluron. Influence of irradiation on toxicity, Int. J. Photoenergy, 2000, 2(1), 1–8 CrossRef CAS.
- P. Calza, D. Vione, D. Fabbri, R. Aigotti and C. Medana, Imidazolium-Based Ionic Liquids in Water: Assessment of Photocatalytic and Photochemical Transformation, Environ. Sci. Technol., 2015, 49(18), 10951–10958 CrossRef CAS.
- H.-P. Kaiser, O. Köster, M. Gresch, P. M. J. Périsset, P. Jäggi and E. Salhi, et al., Process Control For Ozonation Systems: A Novel Real-Time Approach, Ozone: Sci. Eng., 2013, 35(3), 168–185 CrossRef CAS.
- A. I. Cederbaum and L. Berl, Pyrazole and 4-methylpyrazole inhibit oxidation of ethanol and dimethyl sulfoxide by hydroxyl radicals generated from ascorbate, xanthine oxidase, and rat liver microsomes, Arch. Biochem. Biophys., 1982, 216(2), 530–543 CrossRef CAS.
- K. R. Darnall, L. B. Townsend and R. K. Robins, The Structure of Showdomycin, a Novel Carbon-Linked Nucleoside Antibiotic Related to Uridine, Proc. Natl. Acad. Sci. U. S. A., 1967, 57(3), 548–553 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ew01078e |
| ‡ Co-primary authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2020 |