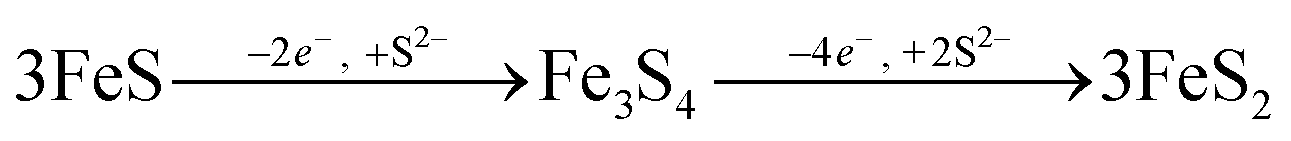Organic matter influences transformation products of ferrihydrite exposed to sulfide†
Received
17th April 2020
, Accepted 18th September 2020
First published on 9th October 2020
Abstract
In redox-dynamic environments, sorption to poorly-crystalline, nanometer-sized Fe(III)-(oxyhydr)oxides like ferrihydrite influences the biogeochemical cycling of nutrients and trace elements. Under sulfate-reducing conditions, the reductive dissolution of ferrihydrite leads to the release of associated constituents, which may be re-immobilized via sorption to secondary Fe minerals. To date, studies following the kinetics and transformation pathways of Fe(III)-(oxyhydr)oxides upon exposure to dissolved sulfide (S(−II)) have largely focused on pure Fe minerals. However, in nature, Fe(III)-(oxyhydr)oxides are often found in association with organic matter (OM). Because ferrihydrite–OM associations exhibit characteristics and biogeochemical reactivity differing from those of pure ferrihydrite, in this study, we compared sulfidization kinetics and transformation pathways of a pure ferrihydrite to those of ferrihydrite coprecipitated with contrasting organic ligands; polygalacturonic acid, galacturonic acid, and citric acid (C/Fe molar ratio ∼0.55). Incorporating aqueous- and solid-phase S and Fe speciation analyses (via wet chemistry techniques and S and Fe X-ray absorption spectroscopy) in addition to X-ray diffraction and electron microscopy, we studied both rapid (<7 days) and long-term (12 months) mineral transformations as well as the impact of varying S(−II)/Fe molar ratios at neutral pH. Our results showed that at low S(−II)/Fe molar ratios (=0.1), poorly-crystalline Fe sulfide minerals (e.g. mackinawite) did not form in any (co)precipitate. In contrast, at higher S(−II)/Fe molar ratios (=0.5), mackinawite rapidly precipitated, with higher contributions detected in the coprecipitates than in the pure ferrihydrite. Aging of the samples led to further mineral transformations, including divergent pyrite and greigite precipitation, and an overall increase in the crystallinity of secondary mineral phases. Still, the fraction of residual ferrihydrite at 12 months was higher in the OM-containing coprecipitates, with the most ferrihydrite preservation observed in coprecipitates comprising carboxyl-poor ligands (galacturonic acid and citric acid). This suggests that the presence of OM inhibited S(−II)-induced ferrihydrite mineral transformations and that the composition of the associated OM influenced mineral transformation pathways. Collectively, these results suggest that further studies regarding sulfidization pathways should include OM in order to better represent environmental conditions.
Environmental significance
Ferrihydrite is a poorly-crystalline, nanometer-sized Fe(III)-oxyhydroxide that is found in abundance in natural soils and sediments, often in association with organic matter. With a high specific surface area, ferrihydrite is an important sorbent for nutrients and trace elements. Under reducing conditions, sulfidization of ferrihydrite can lead to the release of sorbed constituents. Following the transformation of ferrihydrite during sulfidization is therefore critical to understanding nutrient and trace element cycling in redox-dynamic environments. Our results show that mineral transformation pathways and kinetics in pure ferrihydrite are vastly differ from those seen in ferrihydrite–organic matter coprecipitates and are likely an effect of both changes in available surface-sites as well the chemical composition of the coprecipitated ligand.
|
Introduction
Iron is the fourth most abundant element in the Earth's crust1 and iron redox chemistry influences, directly or indirectly, the biogeochemical cycles of most elements and nutrients on Earth.2 In soils and sediments, iron is commonly found as Fe(III)-(oxyhydr)oxides. With high specific surface areas and points of zero charge (PZC) near pH 7–8, poorly crystalline Fe(III)-(oxyhydr)oxides like ferrihydrite are often linked to high adsorption of nutrients (C, N, P, S) and trace elements (e.g. As, Zn) under oxic conditions. However, under sulfate-reducing conditions, microbially derived sulfide (S(−II)) may cause the rapid reductive dissolution of ferrihydrite and the release of associated nutrients and trace elements.3–5 Released nutrients and trace elements may re-adsorb onto or become incorporated into secondary iron mineral phases.3,6–8 However, the mechanism and extent of re-immobilization is controlled by the availability of surface adsorption sites on the secondary iron mineral phases as well as the respective trace element and/or nutrient's adsorption affinity, both of which differ in the presence of varying Fe(III)-(oxyhydr)oxides.3,6,8 Thus, under prolonged anoxic conditions, the mobility of nutrients and trace elements is controlled by both the kinetics and transformation pathways of poorly crystalline Fe(III)-(oxyhydr)oxides reacted with S(−II).
Sulfidization of pure iron mineral phases like ferrihydrite (Fe(OH)3), lepidocrocite (γ-FeOOH), and goethite (α-FeOOH) has been extensively studied9–19 and is understood to proceed via rapid electron transfer between surface-complexed sulfide and the Fe(III) mineral phase, resulting in elemental S (S(0)) and surface-associated Fe(II):9
| | | 2Fe(OH)3 + H2S + 4H+ ↔ 2Fe2+ + S(0) + 6H2O | (1) |
Under anoxic conditions, thiosulfate (S
2O
32−) may form through the partial oxidation of sulfide by Fe(
III)-(oxyhydr)oxides (
eqn (2)),
4,18,20 while secondary reactions involving S(0) may lead to intermediate sulfur species, like polysulfides (S
n2−), or sulfate (SO
42−):
14,16,21| | | 8Fe(OH)3 + 2HS− + 16H+ = 8Fe2+ + S2O32− + 21H2O | (2) |
| | | (n − 1)S(0) + HS− ↔ Sn2− + H+ | (3) |
| | | 2Fe2+ + S(0)n + 4H2O ↔ 2FeOOH + Sn2− + 6H+ | (4) |
| | | S(0) + 6Fe3+ + 4H2O → SO42− + 6Fe2+ + 8H+ | (5) |
In addition, aqueous Fe(
II) may react with excess sulfide, precipitating nano-crystalline mackinawite (FeS,
eqn (6))
9 which, in the presence of excess sulfide (
eqn (7))
22 or intermediate sulfur species
23–26 (
e.g. polysulfides,
eqn (8)), may eventually convert to pyrite (FeS
2):
| | | FeS + Sn2− → FeS2 + Sn−12− | (8) |
For pure iron mineral phases, the kinetics and transformation pathways during sulfidization are influenced by mineral structure and the S(−
II)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
:
surface site ratio.
9,11,13,15–17 However, pure iron mineral phases rarely form in natural environments. Rather, the ubiquitous presence of natural organic matter (OM) in soils and sediments promotes the formation of mineral–organic associations.
27–31 Because adsorbed OM may reduce the number of available surface sites and coprecipitation with OM changes the resulting mineral's particle size, morphology, PZC, and aggregation tendency,
32–38 it stands to reason that sulfidization of OM-associated Fe(
III)-(oxyhydr)oxides may proceed differently compared to OM-free Fe(
III)-(oxyhydr)oxides. Moreover, recent evidence suggests that the chemical composition of the associated OM (
e.g. molecular weight (MW)
39 and carboxyl content
38) impacts transformation products during interactions between Fe(
II) and ferrihydrite. Because sulfidization reactions similarly result in (solid-associated) Fe(
II) (
eqn (1) (
ref. 9)), it seems likely that the kinetics and pathways of sulfide-induced transformations of ferrihydrite may also vary according to the chemical composition of the associated organic ligand.
To date, however, the impact of OM on the sulfidization kinetics and mineral transformation pathways of Fe(III)-(oxyhydr)oxides has received limited attention.40–42 Henneberry et al.40 reacted Fe–OM flocs comprising natural OM collected from agricultural drainage waters with S(−II) (0.05 or 0.6 mM, molar ratios S(−II)/Fe = 0.014–0.46, C/Fe = 1.9–3.5). Results from X-ray diffraction (XRD) analyses did not indicate any change in floc crystallinity over 14 days.40 Because an OM-free Fe-floc reacted under the same conditions transformed to goethite within 5 days, the authors suggested that the presence of OM hindered the reductive recrystallization of floc-Fe.40 In contrast, previous work8 showed that sulfidization of ferrihydrite and nano-lepidocrocite in naturally occurring Fe-rich, organic flocs (5 mM S(−II), molar ratios S(−II)/Fe = 0.75–1.62, C/Fe = 2.2–7.5) resulted in the rapid (<7 days) (neo)formation of mackinawite, lepidocrocite, and goethite. For the Fe-rich, organic flocs, increasing the S(−II)/Fe molar ratios to ≥1 resulted in complete transformation of floc-ferrihydrite.8 Because dissolved sulfide concentrations reported for porewaters of OM-rich wetland and estuarine sediments vary widely (e.g. 0.023–5.0 mM),43–49 the role of OM in hindering or promoting the recrystallization or transformation of poorly-crystalline iron mineral phases in the presence of sulfide needs to be addressed.
Therefore, in this study, we monitored iron mineral transformations and sulfur speciation during exposure of ferrihydrite–OM coprecipitates to sulfide at neutral pH, utilizing well-characterized coprecipitates38 comprising contrasting organic ligands chosen to represent a range of MWs and carboxyl-group contents which affect the charge characteristics of the resulting coprecipitate and are expected to result in varying transformation products.38 Specifically, polygalacturonic acid (PGA, (C6H8O6)n, MW = 25–50 kDa, pKa = 3.48 (25 °C)), which consists of linear chains of partially methylated (1–4) linked α-D-galacturonic acid monomers, was chosen as a proxy for acidic carbohydrates found in naturally-occurring iron–organic precipitates and comprises an abundance of carboxyl groups.29,50 Galacturonic acid (GA, C6H9O7, MW = 194 Da, pKa = 3.48 (25 °C)) is the monosaccharide equivalent of PGA, and represents a low MW organic acid with one carboxyl group, and citric acid (CA, C6H5O7, MW = 192 Da, pKa1,2,3 = 3.13, 4.76, 6.4 (25 °C)), an α-hydroxy acid, contains exactly three carboxyl groups. Reaction kinetics and transformation products of the coprecipitates were compared to those of a pure ferrihydrite. Solid-phase Fe and S speciation, derived from XRD patterns and X-ray absorption spectroscopy (XAS), was complemented by electron microscopy (EM), while wet-chemical techniques determined the concentration and speciation of Fe and S in solution. Collectively, our results suggest that the impact of OM on sulfidization kinetics and secondary mineral formation is varied and dependent upon S(−II)/Fe molar ratios as well as chemical structures of the coprecipitated organic ligands, thus highlighting the need to re-assess Fe mineral sulfidization kinetics in natural environments where OM is omnipresent.
Materials and methods
(Co)precipitate synthesis and characterization
Synthesis of ferrihydrite–OM coprecipitates followed methods previously published,8,38,50 and is therefore presented in summary in the ESI.† Likewise, detailed characterization of the (co)precipitates included in this study can be found in ref. 38 as well as in the ESI.† This information includes total element and impurity contents, specific surface area (SSA, Tables S1–S3†), and confirmation of coprecipitate mineralogy via XRD (Fig. S2†). Briefly, the synthetic ferrihydrite coprecipitates containing polygalacturonic acid (PGA), citric acid (CA), and galacturonic acid (GA), hereafter referred to as Fh-PGA, Fh-CA, and Fh-GA (respectively), all have similar C/Fe molar ratios (0.55 ± 0.04, Table S2†). This molar ratio was chosen because at C/Fe molar ratio >0.7, Fe(III)–organic complexes may form to extents above spectroscopic detection limits (>5 mole%) during ferrihydrite coprecipitation with citrate.35 All of the coprecipitates are comparable to the OM-free Fh in terms of structural disorder and aggregate morphology (Fig. S3†).38 All organic ligands in the coprecipitates comprise hydroxyl and carboxyl functional groups, the latter of which participates in ligand exchange with hydroxyl groups of the precipitating ferrihydrite.51 For GA, the cyclic hemiacetal form is more thermodynamically stable than the open-chain form,52 and thus only trace fractions of unprotected carbonyl groups may potentially react with S(−II). However, significant formation of organic S species via reactions between S(−II) and mono- or polysaccharides are not expected at the temperatures or timescales of the current study.53–55
Sulfidation experiment setup
All solutions used in this experiment were prepared from doubly deionized (DDI) water (Milli-Q®, Millipore, 18.2 MΩ cm). Experiments were completed in triplicates in an anoxic glovebox (N2 atmosphere, <1 ppm (v/v) O2), where all glassware and N2-purged solutions were equilibrated for >2 days to remove trace O2. In order to assess both rapid and long-term sulfidization of ferrihydrite–OM coprecipitates, two experimental set-ups were employed in this study. Firstly, dried (co)precipitate material amounting to 10 mmol Fe(III)/L was weighed into 100 mL glass serum bottles wrapped in Al foil, resuspended in 90 mL of anoxic 50 mM 3-(N-morpholino)propanesulfonic acid (MOPS) buffer adjusted to pH 7, and equilibrated overnight. The experiment was started when aliquots of an anoxic sulfide stock solution (10 or 50 mM Na2S·9H2O) were added to the reaction bottles to obtain sulfide concentrations of 1 or 5 mM (Table S3†). Prior to addition, the concentration of the sulfide stock solutions was determined with iodometric titration. Following initial pH adjustments in the first 5 minutes (<1 mL of 1 M HCl, Ultrapur, Merck), serum bottles were subsequently crimp-sealed and set on an orbital shaker (250 rpm) at room temperature (24 ± 1 °C) for 1 week, during which time aliquots (5 mL) were removed for sampling at 6, 24, 48, and 168 hours. During sampling, the headspace was exchanged with the glovebox atmosphere. Sulfide is not expected to be detectable after 30 minutes during reactions with ferrihydrite at circumneutral pH,15 thus the additional headspace introduced during regular sampling is not expected to affect sulfide partitioning. Although sulfide oxidation is rapid, interactions may continue between the (secondary) iron minerals and sulfide oxidation reaction products (eqn (2)–(8)). In order to assess the extent of continued mineral transformations during aging, a second series of Al-wrapped reaction bottles (total volume = 20 mL) containing 15 mL of the (co)precipitate-sulfide slurry were prepared in the same way as described above. Following initial pH adjustments (<100 μL 1 M HCl), these serum bottles were also crimp-sealed and set on the orbital shaker. The aged slurries were not sampled until 12 months after the reaction start. Sulfide-free controls were included for each treatment to quantify element releases originating from solid-phase dissolution.
Aqueous sulfur and Fe concentrations and speciation
Dissolved sulfide, including H2S, HS−, and S2−, was measured in 0.22 μm filtrates with the methylene blue method56 (λ = 665 nm, Cary 60 UV-vis, Agilent). Aqueous sulfate and thiosulfate concentrations were determined in 0.22 μm filtrates by HPLC-ICP-MS/MS (Agilent Technologies 1290 quaternary pump coupled to an ICP-QQQ 8800) using an anion-exchange column (Hamilton PRP-X100, 125 × 4.0 mm, 10 μm with the appropriate guard column) and isocratic elution with a carbonate buffer (100 mM, pH 9.5). The ICP-QQQ instrument was tuned daily before analysis with a multi-element solution and operated in tandem mode with a mixture of O2 and H2 as reaction gas. A solution containing Sc and Y (1 ppm and 50 ppb, respectively) was continuously supplied post-column through a T-piece to monitor its sensitivity during analysis. Elemental measurements were all made in mass shift mode (m/z 32 to 48, 45 to 61 and 89 to 105 for S, Sc and Y, respectively). Detailed instrumental parameters and examples of chromatograms are provided in ESI.† Data evaluation was performed with the Masshunter software (Agilent). Detection limits were 0.1 and 0.5 μmol L−1 for thiosulfate and sulfates, respectively.
Elemental S (S(0)) was determined for both unfiltered samples and 0.22 μm filtrates, however, concentrations in the 0.22 μm filtrates were negligible, in agreement with previous iron mineral sulfidization studies.8,11 For these analyses, a modified version of the method published by Kamyshny et al.21 was used, whereby sulfide was precipitated by adding 25 μL of 200 g L−1 Zn acetate (Zn(CH3COO)2·2H2O, Merck) to 725 μL of an (un)filtered sample. Zinc acetate also reacts with the sulfide group of polysulfides, leading to the conversion of zerovalent S in polysulfides to S(0) and consequently the codetermination of polysulfide-bound zerovalent S. The fixed samples were stored frozen until the extraction of S(0) was performed by addition of 600 μL of chloroform (CHCl3, Fluka). After shaking for 2 h, chloroform extracts were analyzed by HPLC-UV-vis (Agilent 1100) using a C18 column (Thermo Scientific, Hypersil GOLD, 50 × 2.1 mm, 1.9 μm). The eluent was 90% methanol (H3COH, VWR) at a flow rate of 0.5 mL min−1. The injection volume was 1 μL, and the detection wavelength was 265 nm. Calibration standards were prepared by dissolving elemental S powder (>99.998%, Sigma-Aldrich) in toluene (C6H5CH3, VWR). Total dissolved Fe (Feaq) and the speciation of Feaq (Fe(II) and Fe(totals)) at 1 week and 12 months were determined on 0.22 μm filtrates via ICP-MS and UV-vis (1,10-phenanthroline method;57λ = 510 nm), respectively.
S(−II)-reacted solid-phase sampling and characterization
Material for solid-phase analyses was collected on 0.45 μm cellulose-acetate filters and thoroughly rinsed with anoxic DDI water. The filter residues were covered and dried in the glovebox atmosphere. Triplicate samples were combined, homogenized with a mortar and pestle, and stored in darkness until further analyses. Total C and S contents were determined with an elemental analyzer (CHNS-932, LECO, n = 2). Total Fe contents were determined with atomic absorption spectrometry (AAS, Varian AA240FS). For these measurements, ∼2 mg of (co)precipitate material was dissolved in 1 mL of 30% HCl and diluted with 1% HCl to 30 mL. Qualitative mineral phase analyses were performed by X-ray diffraction (XRD, D8 Advance, Bruker). For these analyses, ∼2 mg dried sample material was re-suspended in ethanol (∼30 μL, Merck) and pipetted onto a polished silicon wafer (Sil'tronix Silicon Technologies, France) under glovebox atmosphere. After drying, the wafers were inserted into anoxic sample holders, removed from the glovebox, and measured in Bragg–Brentano geometry using Cu Kα radiation (λ = 1.5418 Å, 40 kV and 40 mA) and a high-resolution energy dispersive 1-D detector (LYNXEYE). Diffractograms were recorded from 10 to 70° 2θ with a step size of 0.02° 2θ and 4 s acquisition time per step. Sulfide-reacted samples were additionally investigated with electron microscopy (EM). For these analyses, ∼2 mg of solid-phase material was resuspended in 10 μL of DDI water and drop-deposited onto a 200 mesh Cu grid coated with a holey C-coated support film (SPI supplies) under anoxic conditions. Loaded grids were transported to the microscope anoxically and imaged within 3 hours. Microscopy images were obtained with a dedicated scanning transmission electron microscope (STEM, 2700Cs, Hitachi) operated at an acceleration voltage of 200 kV. A secondary electron (SE) and high angular annular dark field (HAADF) detector were used for image acquisition. Elemental analyses were conducted with an energy dispersive X-ray (EDX) analysis system (EDAX, USA) coupled to the microscope. The spectra were recorded and processed using Digital Micrograph (v.1.85, Gatan Inc., USA).
Synchrotron measurements
Speciation of solid-phase Fe and S in the coprecipitates before and after reaction with S(−II) were analyzed by bulk Fe and S K-edge (7112 and 2472 eV, respectively) X-ray absorption spectroscopy (XAS) at the XAFS beamline of ELETTRA (Fe, Trieste, Italy) and LUCIA beamline of SOLEIL (Fe and S, Saint-Aubin, France). For Fe measurements, (un)reacted (co)precipitate material was pressed into 1.0 or 1.3 cm pellets and sealed with Kapton® tape. Sulfide-reacted samples were prepared and transferred to the beamline under anoxic conditions. At ELETTRA, Fe X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra were recorded in transmission mode at ∼80 K using a N2(1) cryostat and a Si(111) monochromator calibrated to the first-derivative maximum of the K-edge absorption spectrum of a metallic Fe foil (7112 eV). The foil was continuously monitored to account for small energy shifts (<1 eV) during the sample measurements. Higher harmonics in the beam were eliminated by detuning the monochromator by 30% of its maximal intensity. Three scans per sample were collected and averaged. Additional Fe samples, measured at SOLEIL, were recorded in transmission mode at ∼70 K using a He(1) cryostat. The Si(111) monochromator was calibrated to the first-derivative maximum of the K-edge absorption spectrum of a metallic Fe foil (7112 eV). Higher harmonics in the beam were eliminated by mirrors. All spectra were energy calibrated, pre-edge subtracted, and post-edge normalized in Athena.58 For Fe XANES analysis, the edge-energy, E0, was defined as zero-crossing in the second XANES derivatives. Linear combination fit (LCF) analyses of k3-weighted Fe K-edge EXAFS spectra were performed over a k-range of 2–12 Å−1 with the E0 of all spectra and reference compounds set to 7128 eV. No constraints were imposed during LCF analyses, and initial fit fractions were recalculated to a compound sum of 100% and adjusted to represent the total fraction of Fe in the solid-phase. Iron reference compounds for LCF analysis were selected after principal component analysis and target-transform testing (PCA-TT, Tables S4 and S5 and Fig. S4†).
For S XANES measurements, samples were diluted with BN to obtain ∼500 mg kg−1 S and pressed into 1.0 cm pellets. XANES spectra were recorded in fluorescence mode at ∼70 K using a mono-element X Flash 6160 detector and a He(l) cryostat. The Si(111) monochromator was calibrated relative to the absorption maximum of S(0) (2472.7 eV).59 Higher harmonics in the beam were eliminated by mirrors. Two to 4 scans per sample were collected and averaged. Data were processed and normalized in Athena as described in ref. 8 and deconvolution of S XANES was performed using WinXAS 3.09,60 following a modified version of the fitting approaches detailed in Manceau and Nagy59 and Shakeri Yetka et al.61 Additional details are presented in the ESI.†
Results and discussion
Trends in aqueous S and Fe
Sulfide is expected to be rapidly consumed during reactions with ferrihydrite,15 and was not detected in solution at any sampling point during the experiment (detection limit = 2.5 μM, data not shown). The addition of sulfide resulted in various color changes of the experimental solutions in all treatment bottles. With the addition of 5 mM S(−II), solutions turned black nearly immediately. In contrast, with the 1 mM S(−II) addition, all treatment bottles required slightly longer (up to 3 minutes) before a color change was apparent, after which solution colors ranged from black (Fh and Fh-PGA) to dark brown (Fh-CA and Fh-GA). Black coloration is typically attributed to the formation of FeS during sulfidization reactions.8,13,14 However, an excess of Fe(II) may also induce secondary Fe(II)-catalyzed mineral transformations or recrystallization in ferrihydrite at circumneutral pH.37,50,62–65 Depending on the Fe(II)/Fe(III) molar ratio, such reactions may result in magnetite (Fe3O4) accumulation,63,66 which may also account for the change in solution color to black/brown. After 3 days, the black coloration of the Fh sample reacted with 1 mM S(−II) disappeared, while all other samples retained a dark brown/black coloration. A similar change in suspension color was reported during sulfidization of lepidocrocite and goethite after 2 weeks (pH = 7, S(−II)/Fe molar ratios of 3.69 and 3.28, respectively), which was suggested to reflect small particle size (<100 nm) of the secondary minerals formed.15 Samples aged for 12 months retained similar coloration as was visible at 1 week.
During the first week of reaction, dissolved sulfate concentrations were similar among the treatments (5.1–10.4 and 22.9–30.0 μM for 1 and 5 mM S(−II)spikes, respectively, Fig. S6A and B and Table S7†). Similarly, concentrations of dissolved thiosulfate were stable during the first week (29.7–51.3 and 146.6–174.5 μM for 1 and 5 mM S(−II)spikes, respectively, Fig. S6C and D and Table S7†). After 12 month aging, increases in sulfate and thiosulfate were recorded (+0 to +63%, ![[x with combining overline]](https://www.rsc.org/images/entities/i_char_0078_0305.gif) = 28% and +10 to +59%, = 34%, respectively). While increases in sulfate may be ascribed to secondary reactions between S(0) and Fe3+ (eqn (5)),16 in our anoxic system, thiosulfate increases may suggest the presence of very low concentration of HS− (eqn (2)).4,18,20 At all timepoints, ratios of dissolved thiosulfate to sulfate were similar amongst all (co)precipitates (6.1 ± 0.7, ± σ, Table S7†). Total sulfate and thiosulfate concentrations were ∼3.6 times higher with the 5 mM S(−II) addition compared to the 1 mM S(−II) addition. In general, the similarities between dissolved sulfate and thiosulfate concentrations amongst all (co)precipitates and negligible S(0) determined in 0.22 μm filtrates suggest that dissolved (partially-)oxidized S species are not key factors controlling secondary mineral formation. Solid-phase-associated S(0) is, however, expected to be the most dominant oxidation product formed during dissolution of Fe(III) to Fe(II) via interactions with S(−II).9,10 Trends in S(0) determined via wet-chemistry in unfiltered samples are shown in Fig. S7.† In general, extracted S(0) concentrations were similar during the first week of the experiment. After 12 months, S(0) concentrations in the 1 mM S(−II)spike treatments were lower, while higher S(0) contributions were recorded for all OM-containing coprecipitates exposed to 5 mM S(−II). Because orthorhombic sulfur was not detected in any sample via XRD (Fig. 1), this may suggest that the majority of S(0) determined via wet chemistry resulted from polysulfide-bound zerovalent S formed viaeqn (3) and (4).
= 28% and +10 to +59%, = 34%, respectively). While increases in sulfate may be ascribed to secondary reactions between S(0) and Fe3+ (eqn (5)),16 in our anoxic system, thiosulfate increases may suggest the presence of very low concentration of HS− (eqn (2)).4,18,20 At all timepoints, ratios of dissolved thiosulfate to sulfate were similar amongst all (co)precipitates (6.1 ± 0.7, ± σ, Table S7†). Total sulfate and thiosulfate concentrations were ∼3.6 times higher with the 5 mM S(−II) addition compared to the 1 mM S(−II) addition. In general, the similarities between dissolved sulfate and thiosulfate concentrations amongst all (co)precipitates and negligible S(0) determined in 0.22 μm filtrates suggest that dissolved (partially-)oxidized S species are not key factors controlling secondary mineral formation. Solid-phase-associated S(0) is, however, expected to be the most dominant oxidation product formed during dissolution of Fe(III) to Fe(II) via interactions with S(−II).9,10 Trends in S(0) determined via wet-chemistry in unfiltered samples are shown in Fig. S7.† In general, extracted S(0) concentrations were similar during the first week of the experiment. After 12 months, S(0) concentrations in the 1 mM S(−II)spike treatments were lower, while higher S(0) contributions were recorded for all OM-containing coprecipitates exposed to 5 mM S(−II). Because orthorhombic sulfur was not detected in any sample via XRD (Fig. 1), this may suggest that the majority of S(0) determined via wet chemistry resulted from polysulfide-bound zerovalent S formed viaeqn (3) and (4).
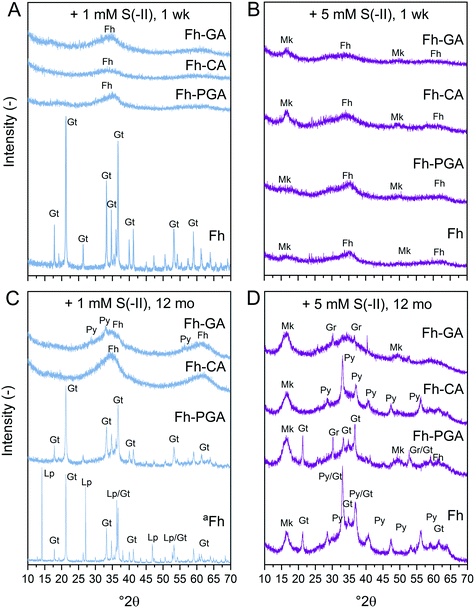 |
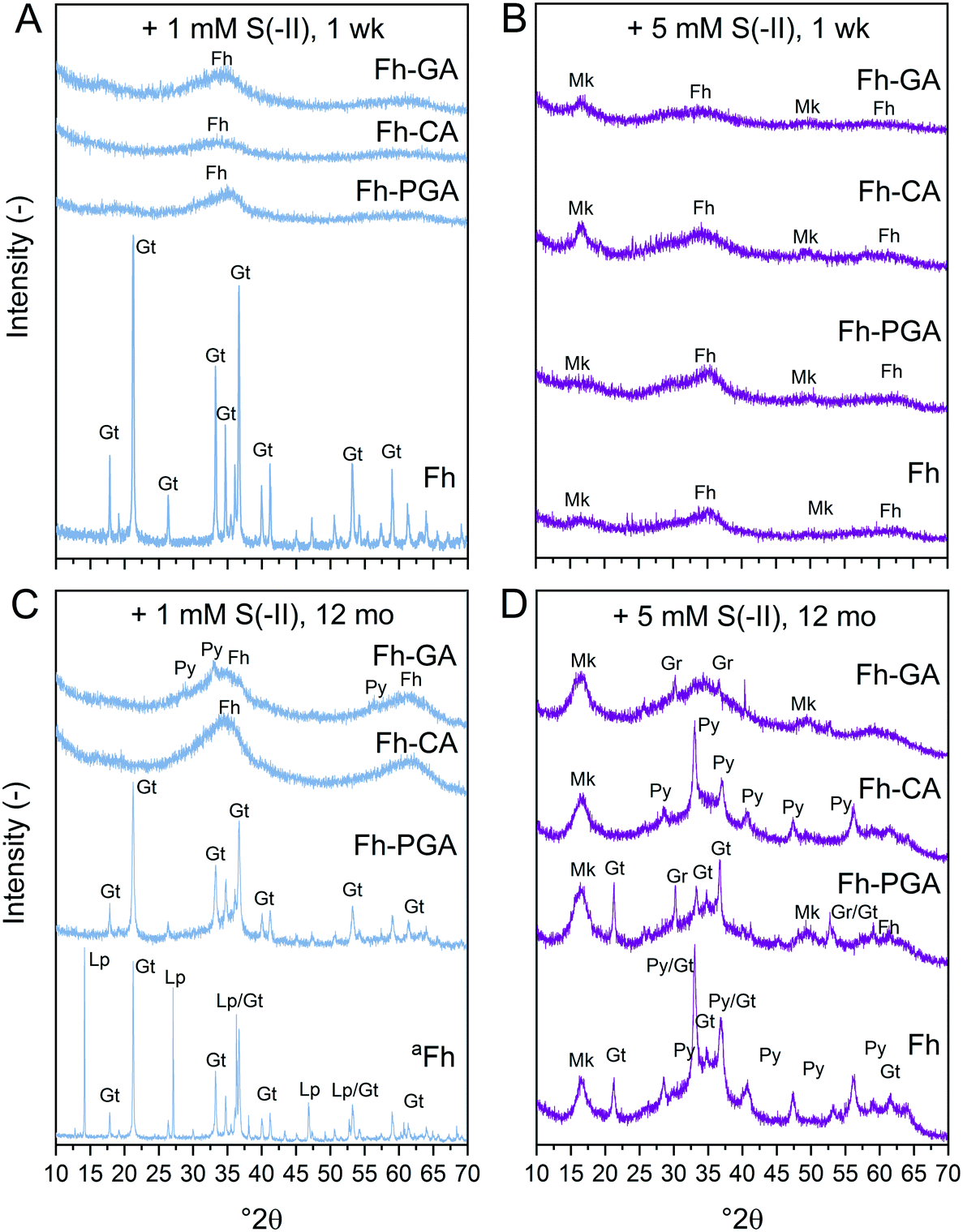
| | Fig. 1 X-ray diffraction patterns of (co)precipitates reacted for 1 week (A and B) or 12 months (C and D) with (A and C) 1 and (B and D) 5 mM S(−II). aSpectrum of Fh reacted with 1 mM S(−II) for 12 months is scaled down 2 times. Abbreviations: Fh = ferrihydrite, Gr = greigite, Gt = goethite, Lp = lepidocrocite, Mk = mackinawite, Py = pyrite. | |
Trends in total dissolved Fe (<0.22 μm, Feaq), which may include aqueous FeS clusters,42 are shown in Fig. S6E and F.† Overall, Feaq concentrations were low, accounting for <5 and ≤9% of total Fe present in the system after 1 week and 12 months, respectively. It is likely that significant Fe(II) fractions may be found as solid-phase bound, nonsulfide-associated Fe(II).14,15 However, the HCl extractions commonly used to quantify this fraction tend to overestimate its contribution when ferrihydrite or lepidocrocite are involved, as Fe(II) from solid-phase FeSs and Fe(II) associated with polysulfides (FeSn,s) may be simultaneously liberated.11,15 Therefore, in this study, we discuss only Feaq as determined in 0.22 μm filtrates. As Fig. S6† shows, for both 1 and 5 mM S(−II) additions, Feaq concentrations in all treatments increased within the first 6 hours, consistent with the rapid dissolution of Fe(III)-minerals.15 In contrast to Kumar et al.,16 who reported that Feaq measured after 1 day reaction time increased with increasing S(−II) spikes up to a S/Fe molar ratio of unity, no consistent trends in Feaq could be deciphered between 1 and 5 mM S(−II) treatments at any timepoint in our study (Fig. S6E and F†). However, Feaq concentrations were consistently higher in Fh-CA and Fh-GA compared to Fh and Fh-PGA; possibly reflecting the impact of slight differences in particle size and crystallinity38 on coprecipitated-ferrihydrite reactivity. Analyses of dissolved Fe species at 1 week and 12 months (data not shown) revealed that Fe(II) accounted for 98 ± 5% of Feaq, suggesting that the release of organically-complexed Fe(III) was negligible. Dissolved Fe concentrations in the sulfide-free controls was very low (<detection limit, i.e. 0.02 mM), indicating that microbial activity during the experiment period was negligible, and likely did not affect iron mineral transformations.
Rapid mineral transformations: solid-phase Fe and S speciation at 1 week
X-ray diffraction patterns of (co)precipitates reacted with 1 or 5 mM S(−II) for 1 week revealed that crystalline goethite only formed from pure Fh reacted with 1 mM S(−II) (Fig. 1A). Coprecipitates reacted under the same conditions showed XRD features indicating only ferrihydrite (2.54 and 1.49 Å, ca. 35 and 62° 2θ, respectively) (Fig. 1A and B). With the 5 mM S(−II) spike, all samples showed additional broad features consistent with poorly-crystalline mackinawite at 5.00, 2.97, and 1.81 Å (ca. 17, 30, and 50° 2θ). Variations in secondary mineral phases crystallinity at 1 week are a direct result of the experimental S(−II)/Fe molar ratios applied in this study. At higher S(−II)/Fe molar ratios, residual S(−II) reacts with Fe(II) to form mackinawite (eqn (6)).9,15 At low S(−II)/Fe molar ratios, Fe(II) forms in excess to S(−II),15 resulting in low FeS precipitation and dissolved Fe(II) in solution.16 It is likely that the excess Fe(II) then interacts with the mineral surface via iron atom exchange, thereby catalyzing the transformation of pure ferrihydrite to lepidocrocite and goethite;62,64,66 a process which can be severely inhibited in the presence of organic matter.37–39,50,63,65
Because XRD targets primarily the crystalline mineral fraction, samples were additionally investigated by Fe K-edge XAS and LCF including a suite of reference spectra deemed suitable through PCA-TT (see ESI†). For the 1 mM S(−II)spike samples, the pre-edge features, energy position, and intensities of the white-lines of Fe K-edge XANES spectra match those of the ferrihydrite reference, suggesting that no change in solid-phase Fe oxidation state occurred for (co)precipitates reacted with 1 mM S(−II) within 1 week (Fig. S8†). In contrast, for all 5 mM S(−II) reacted samples, the appearance of a slight shoulder near 7120 eV as well as shifts in the pre-edge feature towards lower eV (Fig. S8†) indicated the presence of solid-phase Fe(II). Still, LCFs, illustrated in Fig. 2A and S9,† indicated that all samples were dominated by combinations of Fe(III) minerals (Fig. 2B and Table 1). In agreement with XRD patterns, for the pure Fh, exposure to 1 mM S(−II) over 1 week resulted in near complete transformation to goethite. The remaining coprecipitates comprised primarily ferrihydrite. Noteworthy is the extent of ferrihydrite preservation in the Fh-CA and Fh-GA samples compared to Fh-PGA (96 vs. 75%). Previously, we showed that Fe(II)-catalyzed mineral transformations in carboxyl-rich coprecipitates (e.g. Fh-PGA) were significantly less inhibited compared to their carboxyl-poor counterparts (e.g. Fh-CA and Fh-GA).38 Because sulfidization reactions involving iron minerals also produce Fe(II) (eqn (1)),9 these results may suggest that, following the initial oxidation of S(−II), further mineral transformations are strongly influenced by dissolved Fe(II) (cp. Fig. S6†) interactions with the mineral surface. Mackinawite contributed to all 5 mM S(−II)spike samples (15–30%), in addition to lesser fractions of goethite, lepidocrocite, and magnetite. For the latter three minerals, the lack of corresponding XRD peaks (Fig. 1A and B) indicates that these phases may be poorly-crystalline. This is in agreement with reports of X-ray amorphous, nanometer-sized clusters of secondary mineral phases forming during reactions between dissolved Fe(II) and ferrihydrite in the presence of OM at circumneutral pH.37–39,50,63
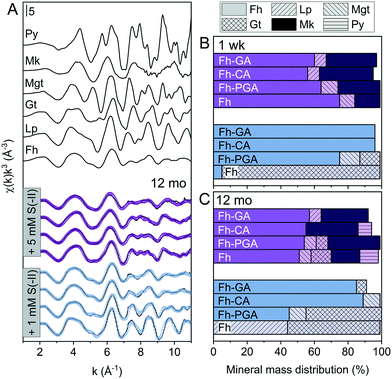 |
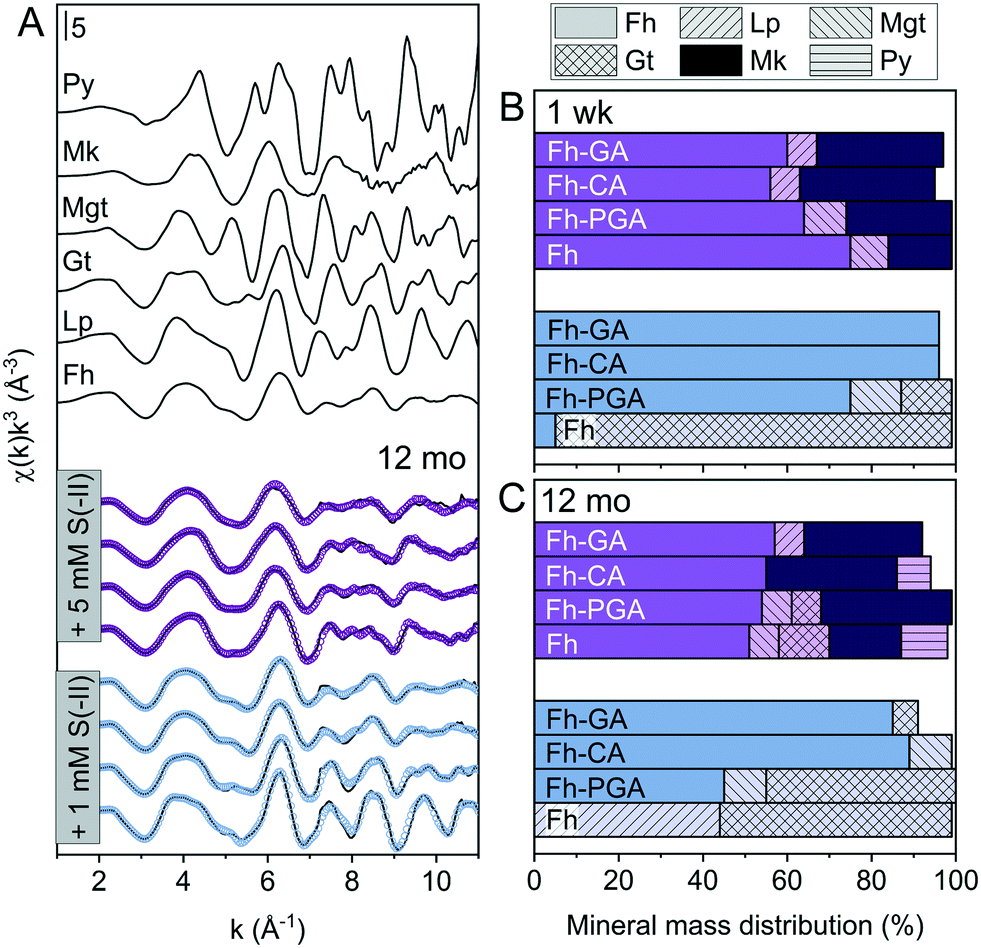
| | Fig. 2 (A) Iron K-edge EXAFS spectra of references and LCFs of 12 month aged S(−II)-reacted (co)precipitates. Experimental data and model fits are shown as solid and dotted lines, respectively. (B and C) Results from linear combination fit analyses of k3-weighted EXAFS spectra of both 1 week (B) and 12 month (C) samples reacted with 1 or 5 mM S(−II), shown in blue and purple, respectively. Values are adjusted to represent total solid-phase Fe measured at 1 week and 12 months, respectively (compare to [Feaq] in Fig. S6†). Additional spectra and corresponding fits for the 1 week samples are found in the ESI,† Fig. S8. Fit parameters are detailed in Table 1. Abbreviations: Fh = ferrihydrite, Gt = goethite, Lp = lepidocrocite, Mgt = magnetite, Mk = mackinawite, Py = pyrite. | |
Table 1 Linear combination fit results for Fe K-edge EXAFS spectra prior to and after reaction with 1 or 5 mM S(−II) for 1 week or 12 months. Values are adjusted to represent the fraction of total solid-phase Fe at each time point (compare. to [Feaq] in Fig. S6†)
| Sample |
Fh |
Mk |
Lp |
Mgt |
Gt |
Py |
NSSRa |
Red.bχ2 |
| (%) |
(−) |
|
NSSR: normalized sum of squared residuals (100 × ∑i(datai − fiti)2/∑idata2).
Fit accuracy; reduced χ2 = (Nidp/Npts)∑i((datai − fiti)/εi)2(Nidp − Nvar)−1. Nidp, Npts and Nvar are, respectively, the number of independent points in the model fit (16), the total number of data points (201), and the number of fit variables (1–4). εi is the uncertainty of the ith data point.83
Sample spectra were previously published.38
Evidence of pyrite in XRD patterns. Abbreviations: Fh = ferrihydrite, Gt = goethite, Lp = lepidocrocite, Mgt = magnetite, Mk = mackinawite, Py = pyrite.
|
| Initial |
Fhc |
100 |
|
|
|
|
|
1.6 |
0.080 |
| Fh-PGAc |
100 |
|
|
|
|
|
1.1 |
0.002 |
| Fh-CAc |
100 |
|
|
|
|
|
1.3 |
0.003 |
| Fh-GAc |
100 |
|
|
|
|
|
1.7 |
0.004 |
| +1 mM S(−II) 1 week |
Fh |
5 |
|
|
|
94 |
|
0.3 |
0.007 |
| Fh-PGA |
75 |
|
|
12 |
12 |
|
0.2 |
0.004 |
| Fh-CA |
96 |
|
|
|
|
|
0.8 |
0.002 |
| Fh-GA |
96 |
|
|
|
|
|
0.8 |
0.002 |
| +5 mM S(−II) 1 week |
Fh |
75 |
15 |
|
9 |
|
|
1.9 |
0.003 |
| Fh-PGA |
64 |
25 |
|
10 |
|
|
2.6 |
0.005 |
| Fh-CA |
56 |
32 |
7 |
|
|
|
2.1 |
0.003 |
| Fh-GA |
60 |
30 |
7 |
|
|
|
1.7 |
0.003 |
| +1 mM S(−II) 12 mo |
Fh |
|
|
44 |
|
55 |
|
1.7 |
0.006 |
| Fh-PGA |
45 |
|
|
10 |
45 |
|
1.8 |
0.004 |
| Fh-CA |
89 |
|
|
10 |
|
|
1.2 |
0.002 |
| Fh-GA |
85 |
|
|
|
6 |
?d |
1.2 |
0.002 |
| +5 mM S(−II) 12 mo |
Fh |
51 |
17 |
|
7 |
12 |
11 |
1.3 |
0.003 |
| Fh-PGA |
54 |
31 |
|
7 |
7 |
|
1.6 |
0.003 |
| Fh-CA |
55 |
31 |
|
|
|
8 |
1.8 |
0.003 |
| Fh-GA |
57 |
28 |
7 |
|
|
|
3.9 |
0.006 |
The importance of non-sulfur-associated Fe(II) during sulfidization reactions was also noted by Peiffer et al.,15 who demonstrated that conditions which favored its formation in excess hindered FeS precipitation. Similarly, Kumar et al.16 suggested that a minimum S(−II)/Fe molar ratio of 0.5 was required for mackinawite accumulation. In agreement with this, in our experiments, mackinawite was only detected in the 5 mM S(−II) treatments, accounting for 15% of total Fe in the system in the pure Fh sample, and 25, 32, and 30% in Fh-PGA, Fh-CA, and Fh-GA, respectively (Table 1). Considering that a S(−II)/Fe molar ratio of 1.5 required to stoichiometrically reduce all ferrihydrite-Fe(III) to FeS (eqn (1) and (6)),9 FeS precipitation in the coprecipitates approached the theoretical stoichiometric maximum (∼33%) as calculated for the high S(−II) treatments (S(−II)/Fe = 0.5, Table S3†). Because sulfidization rates in pure iron oxides depend on the mineral structure and the S(−II):surface site ratio,9,11,13,15–17 higher rates of FeS precipitation in the coprecipitates may suggest that the presence of OM increased the S(−II):surface site ratio,15 possibly through OM-induced aggregation,67,68 or by blocking adsorption sites69 and/or micropores.36 Complete blockage of mineral surface sites by adsorbed or coprecipitated OM is not expected at the C/Fe molar ratio of the coprecipitates in this study.69 Still, considering that mackinawite formed via sulfidization of Fe(III)-(oxyhydr)oxides is expected to be nano-crystalline,8–10,25 in total, the nano-crystalline fractions (e.g. mackinawite + ferrihydrite) determined at 1 week were similar for both OM-containing and OM-free samples (91 ± 4%, Table 1). It should be noted, that despite the rapid formation of pyrite reported during similar Fe(III)-(oxyhydr)oxide sulfidization studies,11,14,15 pyrite was not detected in any 1 week sample.
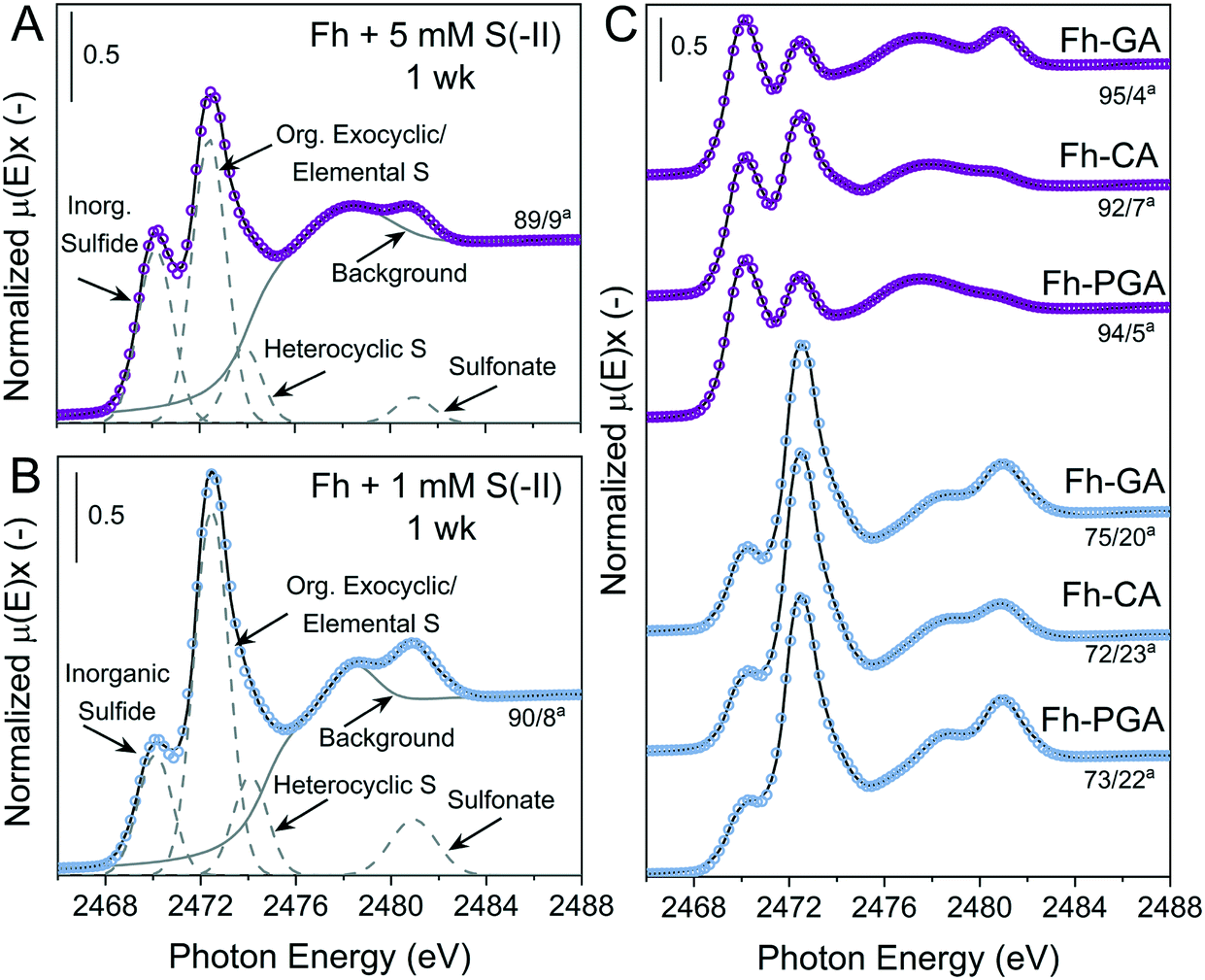
Examples of S K-edge XANES spectra and deconvolutions from 1 week are shown in Fig. 3, and the results from the deconvolution of all spectra are summarized in Tables S8 and S9.† For all samples, solid-phase S speciation was dominated by ‘reduced’ S species; inorganic sulfide, organic exocyclic and elemental S, and heterocyclic S (∼2470–2475 eV). It should be noted that the small difference between white-line energies of organic exocyclic and elemental S (<1 eV) renders the differentiation between these species impossible. However, formation of organic S species is not expected with the ligands, temperature, or timescales used in this study.53–55 Furthermore, all ‘reduced’ S species show strong post-edge adsorption features in the range 2475–2482 eV,70 thus the fitting of Gaussian curves in this energy range can lead to misinterpretation of ‘intermediate oxidized’ S species in samples dominated by ‘reduced’ S.59 Therefore, ‘intermediate oxidized’ S species (sulfoxide and sulfone) were not included in the deconvolutions. In some samples, a feature distinct from the ‘reduced’ S species post-edge adsorption was fit with a Gaussian at ca. 2481 eV and determined to be the ‘oxidized’ S species sulfonate.
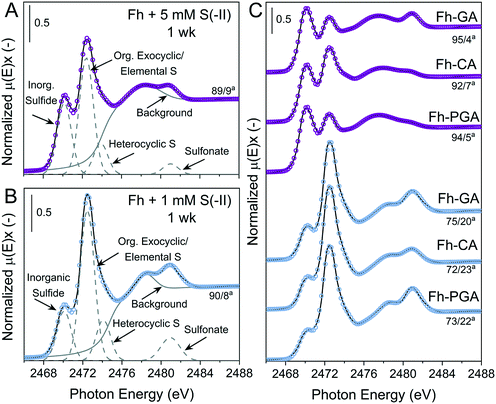 |
| | Fig. 3 Deconvolution of normalized bulk S K-edge XANES spectra are exemplarily illustrated for of Fh reacted with (A) 5 mM S(−II) or (B) 1 mM S(−II) for 1 week. (C) Normalized S K-edge XANES spectra and deconvolution fits for the coprecipitates (Fh-PGA, Fh-CA, and Fh-GA) reacted with 1 or 5 mM S(−II) for 1 week, shown in blue and purple, respectively. The spectra were decomposed into 4 Gaussians and 1 arctangent functions. The broad peak feature at ∼2475–2479 eV in is a postedge absorption feature of the reduced S species59,70 and thus was included in the background. aContributions from [inorganic sulfide]/[org. exocyclic/elemental S]. Fit parameters are detailed in Table S8.† | |
In general, solid-phase S species detected at 1 week were most similar amongst samples exposed to the same S(−II) addition (Fig. 3). Compared to samples reacted with 1 mM S(−II), relative contributions from inorganic sulfide were higher in 5 mM S(−II)-reacted samples ( = 77 vs. = 93%), with the highest fractions of inorganic sulfide determined in the 5 mM S(−II)-reacted OM-containing coprecipitates (89 (Fh) vs. = 94% (coprecipitates)). This is in agreement with the detectible formation of FeS in only 5 mM S(−II) reacted samples and higher rates of mackinawite accumulation in the coprecipitates (Fig. 1B and 2 and Table 1). Sulfur XANES spectra of 1 mM S(−II)-reacted samples showed that these samples comprised comparatively more organic exocyclic/elemental S ( = +12%), while all samples contained small fractions of sulfonate (<1.4%).
Impact of OM on ageing of sulfidized ferrihydrite
Sulfide oxidation by Fe(III)-(oxyhydr)oxides is rapid (timescale of hours),15 and the vast majority of sulfidization studies have focused on the subsequent Fe(III)-(oxyhydr)oxide transformations that occur within 2 weeks.8–19,40 However, under extended anoxic conditions, (secondary) iron minerals may continue to interact with sulfide oxidation reaction products (e.g. thiosulfate and (surface-associated) S(0), polysulfides, and Fe(II), eqn (2)–(8)), resulting in continued mineral transformation over longer time scales. For example, in a study lasting 157 days, Wan et al. reported pyrite formation during sulfidization of goethite (S(−II):Fe(III) molar ratio = 4.4) only after 120 days.11 Similar to this, X-ray diffraction patterns and electron microscopy images of the 1 and 5 mM S(−II)-reacted samples at 12 months show that ageing resulted in further mineral transformations and an increase in crystallinity of secondary mineral phases in all (co)precipitates (Fig. 1C and D, 4 and 5). An exception is noted for Fh-CA reacted with 1 mM S(−II), where only ferrihydrite was detected in XRD patterns at 12 months (Fig. 1C). For all other samples, of note is the formation of new iron sulfide minerals (greigite; Fe3S4, and pyrite) of varying morphologies (Fig. 4 and 5 and Table 2) and an increase in crystallinity of mackinawite in the aged 5 mM S(−II) treatments; as is visible in the (001) reflection (ca. 17° 2θ).71 Quantitative analyses of solid-phase Fe mineral contributions at 12 months (LCF, Table 1) revealed that, for all samples, the fraction of mackinawite established at 1 week (15–32% of total Fe) remained constant for the experiment duration (17–31% of total Fe at 12 months), during which time mackinawite crystal growth may have proceeded via iron isotope exchange72 facilitated by the low Fe(II) concentrations present in solution (Fig. S6E and F†). In contrast, the residual ferrihydrite fraction decreased in all samples during ageing (up to 30% of total Fe), mostly in favor of new goethite formation (+6 to +33% of total Fe) but also pyrite (+8 to +11% of total Fe). Traces of pyrite were also detected in the XRD pattern of Fh-GA reacted with 1 mM S(−II) for 12 months (Fig. 1C), however its contribution was below the detection limits of XAS (>5 mole%). Although greigite was identified in Fh-PGA and Fh-GA reacted with 5 mM S(−II) using XRD, a greigite reference spectra was not available for LCF analysis. However, initial fit fractions (97 ± 8%) suggest that the greigite contribution was small (<10 mole%).
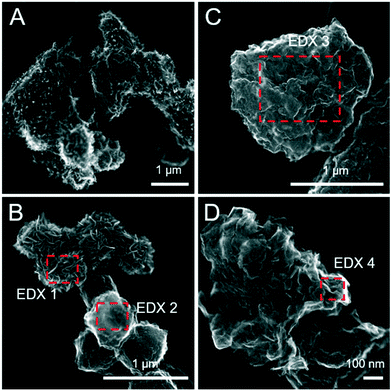 |
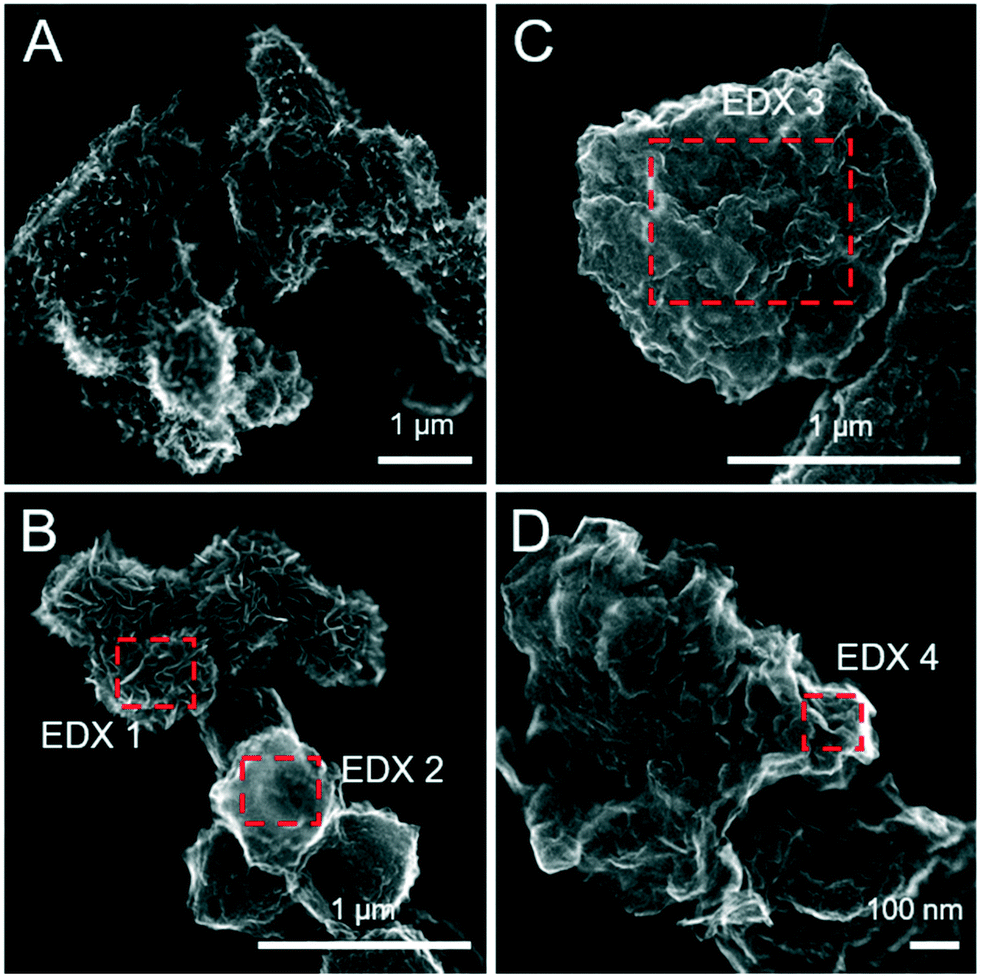
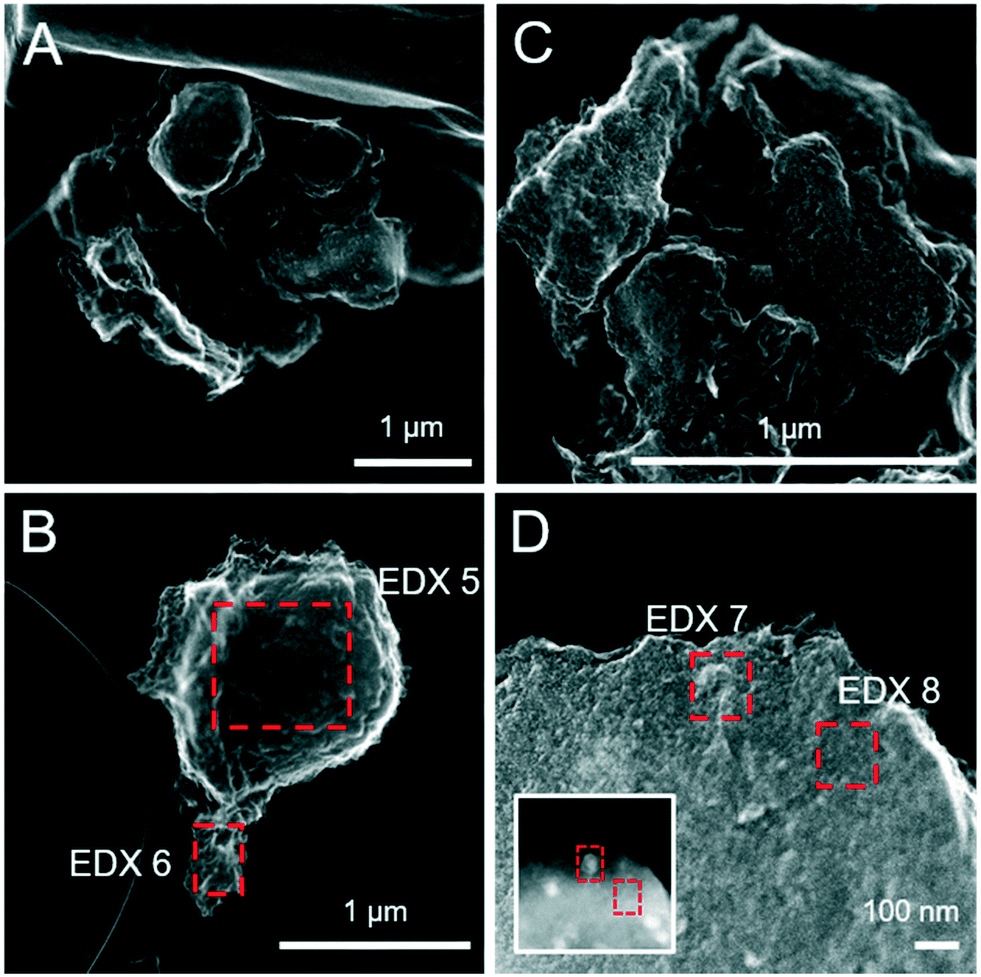
| | Fig. 4 Secondary electron (SE) images of the pure ferrihydrite (A and B) and Fh-PGA (C and D) reacted with 5 mM S(−II) for 12 months. Intensity ratios of S Kα:Fe Kα (to a first approximation reflecting elemental ratios) derived from the EDX spectra are detailed in Table 2. | |
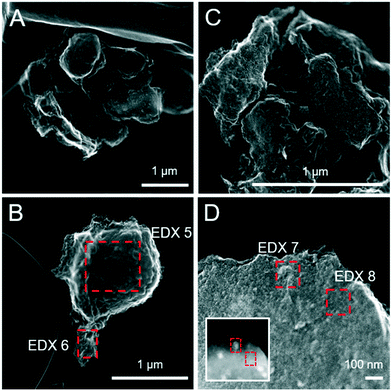 |
| | Fig. 5 Secondary electron (SE) images of the pure Fh-CA (A and B) and Fh-GA (C and D) reacted with 5 mM S(−II) for 12 months. The inset in panel D was imaged with HAADF detector and highlights differences in the average atomic weight of mineral phases not visible on the mineral surface. Intensity ratios of S Kα:Fe Kα (to a first approximation reflecting elemental ratios) derived from the EDX spectra are detailed in Table 2. | |
Table 2 Fe:S ratios approximated based on EDX spectra
| Fig. |
Sample |
EDX number |
Crystal habit |
Fe:S molar ratio |
|
Theoretical Fe:S molar ratios of pure mineral phases: mackinawite (FeS) = 1, pyrite (FeS2) = 0.5, greigite (Fe3S4) = 0.75.
|
|
4B
|
Fh + 5 mM S(−II), 12 mo |
1 |
Rosette flakes |
0.6 |
| 2 |
Protoframboid |
0.4 |
|
4C
|
Fh-PGA + 5 mM S(−II), 12 mo |
3 |
Morphologically indistinct |
1.2 |
|
4D
|
4 |
Morphologically indistinct |
0.5 |
|
5B
|
Fh-CA + 5 mM S(−II), 12 mo |
5 |
Protoframboid |
0.3 |
| 6 |
Morphologically indistinct |
0.8 |
|
5D (+ inset) |
Fh-GA + 5 mM S(−II), 12 mo |
7 |
Cubic |
1.6 |
| 8 |
Morphologically indistinct |
4.8 |
The formation of greigite and its relevance for pyrite formation is debated. Greigite may form through the oxidation of mackinawite,73 or as an intermediate species during pyrite formation via the mackinawite pathway, either through reactions with excess S(−II) (eqn (9)) or from an iron loss pathway (eqn (10)):74,75
| | 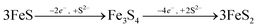 | (9) |
| | 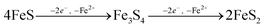 | (10) |
Because our experiment was conducted under anoxic conditions and no subsequent additions of S(−
II) were included, it is most likely that greigite formed
viaeqn (10), whereby the oxidation of Fe in FeS may have been facilitated by intermediate S species (S(0) or polysulfides
23) formed during S(−
II) interactions with ferrihydrite (Fig. S7
†). However, greigite is no longer considered a necessary precursor to pyrite formation. Indeed, examples of spatial de-coupling between pyrite and greigite in natural sediments suggests that greigite and pyrite may represent separate end members to the Fe–S mineralization pathway.
76,77 For example, Wan
et al.11 recently proposed a novel pathway for pyrite formation in which surface-bound, non-sulfur-associated Fe(
II) (>Fe(
II)OH
2+), formed through surface complexation reactions (
eqn (11)–(13) (
ref. 17)), reacts with sulfide radicals to form surface-bound Fe(
II)S
2− species (
eqn (14) (
ref. 11)), prompting pyrite nucleation in conditions below supersaturation for pyrite formation by creating a new equilibrium with the aqueous phase (
eqn (15) (
ref. 11)):
| | | >Fe(III)OH + HS− ↔ H2O + >Fe(III)S− | (11) |
| | | >Fe(III)S− ↔ >Fe(II)S˙ | (12) |
| | | >Fe(II)S˙ + H2O ↔ >Fe(II)OH2+ + S˙− | (13) |
| | | >Fe(II)OH2+ + 2S˙− → H2O + >Fe(II)S2− | (14) |
| | | >Fe(II)S2− + H+ → FeS2,aq + new surface site | (15) |
In our experiments, the formation of greigite and pyrite did not occur in the same sample; greigite formed from Fh-PGA and Fh-GA (+5 mM S(−
II)), while pyrite formed from Fh and Fh-CA (+5 mM S(−
II)) and Fh-GA (+1 mM S(−
II),
Fig. 1C and D), suggesting that different Fe–S mineralization pathways occurred in the various treatments. In controlled laboratory experiments, Rickard
et al.78 demonstrated that the presence of aldehydic carbonyls acted as a ‘mineral switch’; altering the end products (greigite
vs. pyrite) which formed during reactions between mackinawite and sulfide at higher temperatures (40–100 °C). This was explained through aldehyde carbonyl moiety interactions with FeS mineral surface sites, which suppressed the dissolution of amorphous FeS and initiated Fe(
II) oxidation, consequently promoting greigite formation.
78 In the absence of aldehydic carbonyls or in the presence of formic acid, ethanoic acid or acetone, pyrite formed.
78 Although the organic ligands used in the current study do not contain aldehydic carbonyl groups (Fig. S1
†), it is reasonable to consider that they could also act as ‘mineral switches’. For example, in a previous study involving the same coprecipitates (Fh-PGA, Fh-CA and Fh-GA), we showed that the presence of free carboxyl groups in coprecipitates was important to the formation of secondary crystalline mineral phases during exposure to aqueous Fe(
II); no mineral transformations were recorded in the absence of free carboxyl groups (in Fh-GA).
38
The presence of OM also increased the fraction of residual ferrihydrite at 12 months for both the low and high S(−II) treatments, with ferrihydrite accounting for = 64% of total Fe in the coprecipitates at 12 months, compared to = 26% in the OM-free ferrihydrite. Moreover, the fraction of crystalline minerals (lepidocrocite + magnetite + goethite + pyrite) at 12 months was higher in the absence of OM ( = 65% vs. = 16%). Collectively, these findings support Henneberry et al.40 and suggest that, similar to Fe(II)-catalyzed reactions,37–39,50,63 OM inhibits ferrihydrite mineral transformations and the formation of secondary crystalline phases in the presence of reduced S species.
However, the role that specific reduced S species play during ferrihydrite ageing is not clear. As Fig. S10† shows, solid-phase S speciation determined at 12 months shows significantly more variation amongst the samples than was visible at 1 week (cp. to Fig. 3). Although contributions from organic exocyclic/elemental S remain higher with the 1 mM S(−II) addition compared to the 5 mM S(−II) addition ( = 15% vs. = 9%), slight shifts (⋍0.4 eV) in the position of the Gaussian in pyrite-containing samples (Fig. 1C and D) are noted, suggesting contributions from S−1-species, which cannot be well discerned in mixtures with inorganic sulfide and elemental S.79 All samples contained similarly small fractions of sulfonate at 12 months (<1%), indicating negligible re-oxidation of ‘reduced’ or ‘intermediate’ S species during ageing.
Environmental implications
The results presented in this study suggest that our extensive understanding of sulfidization and pyritization kinetics involving pure iron mineral phases9–19 may be incomplete considering the omnipresence of OM in natural systems. The impact of OM on sulfidization and pyritization pathways is varied and dependent upon both S(−II)/Fe molar ratios as well as chemical structures of the coprecipitating organic ligands. It should also be noted, that the C/Fe molar ratio of the coprecipitates included in this study (∼0.55) is relatively low compared to naturally-occurring iron–organic precipitates.29 In the presence of Fe(II), increasing C/Fe molar ratios result in decreasing ferrihydrite mineral transformations.37,63 Therefore, it is possible that, at higher C/Fe molar ratios, the impact of OM on sulfidization and pyritization kinetics and pathways may similarly be increasingly inhibited. Moreover, in the current study, simple organic ligands were chosen which are not expected to interact directly or significantly with S(−II). However, natural OM is heterogeneous and capable of both oxidizing and incorporating S(−II) in anoxic environments at rates comparable to those of S(−II) reactions with Fe(III)-(oxyhydr)oxides.80–82 Because Fe((III)-(oxyhydr)oxide)–organic associations are widespread and important to the biogeochemical cycling of nutrients and trace elements, further studies are needed to probe the mechanisms through and assess the extent to which OM influences interactions between S(−II) and Fe mineral phases.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We are grateful to K. Barmettler and P. Kälin (ETH Zurich) for assisting with laboratory analyses and R. Ossola (ETH Zurich) for helpful discussions. We acknowledge ELETTRA (Proposal no. 20180024) and SOLEIL (Proposal no. 20190784) for the provision of synchrotron radiation facilities and thank G. Aquilanti (ELETTRA, XAFS beamline) and D. Vantelon (SOLEIL, LUCIA beamline) for their support during the synchrotron measurements. We acknowledge the Scientific Center for Optical and Electron Microscopy (ScopeM) of the ETH Zurich for providing access to their microscopes. This work was funded by ETH Zürich and received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant agreement No. 788009-IRMIDYN-ERC-2017-ADG).
References
- K. H. Wedepohl, The composition of the continental crust, Geochim. Cosmochim. Acta, 1995, 59, 1217–1232 CrossRef CAS.
- T. Borch, R. Kretzschmar, A. Kappler, P. Van Cappellen, M. Ginder-Vogel, A. Voegelin and K. Campbell, Biogeochemical redox processes and their impact on contaminant dynamics, Environ. Sci. Technol., 2010, 44, 15–23 CrossRef CAS.
- B. D. Kocar, T. Borch and S. Fendorf, Arsenic repartitioning during biogenic sulfidization and transformation of ferrihydrite, Geochim. Cosmochim. Acta, 2010, 74, 980–994 CrossRef CAS.
- S. L. Saalfield and B. C. Bostick, Changes in iron, sulfur, and arsenic speciation associated with bacterial sulfate reduction in ferrihydrite-rich systems, Environ. Sci. Technol., 2009, 43, 8787–8793 CrossRef CAS.
- E. D. Burton, S. G. Johnston and R. T. Bush, Microbial sulfidogenesis in ferrihydrite-rich environments: Effects on iron mineralogy and arsenic mobility, Geochim. Cosmochim. Acta, 2011, 75, 3072–3087 CrossRef CAS.
- M. L. Farquhar, J. M. Charnock, F. R. Livens and D. J. Vaughan, Mechanisms of arsenic uptake from aqueous solution by interaction with goethite, lepidocrocite, mackinawite, and pyrite: An X-ray absorption spectroscopy study, Environ. Sci. Technol., 2002, 36, 1757–1762 CrossRef CAS.
- Y. Wang, G. Morin, G. Ona-Nguema, F. Juillot, F. Guyot, G. Calas and G. E. Brown, Evidence for different surface speciation of arsenite and arsenate on green rust: An EXAFS and XANES study, Environ. Sci. Technol., 2010, 44, 109–115 CrossRef CAS.
- L. K. ThomasArrigo, C. Mikutta, R. Lohmayer, B. Planer-Friedrich and R. Kretzschmar, Sulfidization of organic freshwater flocs from a minerotrophic peatland: Speciation changes of iron, sulfur, and arsenic, Environ. Sci. Technol., 2016, 50, 3607–3616 CrossRef CAS.
- S. W. Poulton, M. D. Krom and R. Raiswell, A revised scheme for the reactivity of iron (oxyhydr)oxide minerals towards dissolved sulfide, Geochim. Cosmochim. Acta, 2004, 68, 3703–3715 CrossRef CAS.
- S. W. Poulton, Sulfide oxidation and iron dissolution kinetics during the reaction of dissolved sulfide with ferrihydrite, Chem. Geol., 2003, 202, 79–94 CrossRef CAS.
- M. Wan, C. Schroder and S. Peiffer, Fe(III): S(-II) concentration ratio controls the pathway and the kinetics of pyrite formation during sulfidation of ferric hydroxides, Geochim. Cosmochim. Acta, 2017, 217, 334–348 CrossRef CAS.
- S. Peiffer, M. D. Afonso, B. Wehrli and R. Gachter, Kinetics and mechanism of the reaction of H2S with lepidocrocite, Environ. Sci. Technol., 1992, 26, 2408–2413 CrossRef CAS.
- M. Wan, A. Shchukarev, R. Lohmayer, B. Planer-Friedrich and S. Peiffer, Occurrence of surface polysulfides during the interaction between ferric (hydr)oxides and aqueous sulfide, Environ. Sci. Technol., 2014, 48, 5076–5084 CrossRef CAS.
- K. Hellige, K. Pollok, P. Larese-Casanova, T. Behrends and S. Peiffer, Pathways of ferrous iron mineral formation upon sulfidation of lepidocrocite surfaces, Geochim. Cosmochim. Acta, 2012, 81, 69–81 CrossRef CAS.
- S. Peiffer, T. Behrends, K. Hellige, P. Larese-Casanova, M. Wan and K. Pollok, Pyrite formation and mineral transformation pathways upon sulfidation of ferric hydroxides depend on mineral type and sulfide concentration, Chem. Geol., 2015, 400, 44–55 CrossRef CAS.
- N. Kumar, J. L. Pacheco, V. Noel, G. Dublet and G. E. Brown, Sulfidation mechanisms of Fe(III)-(oxyhydr)oxide nanoparticles: a spectroscopic study, Environ. Sci.: Nano, 2018, 5, 1012–1026 RSC.
- M. D. Afonso and W. Stumm, Reductive dissolution of iron(III) (hydr)oxides by hydrogen-sulfide, Langmuir, 1992, 8, 1671–1675 CrossRef.
- A. J. Pyzik and S. E. Sommer, Sedimentary iron monosulfides: Kinetics and mechanism of formation, Geochim. Cosmochim. Acta, 1981, 45, 687–698 CrossRef CAS.
- D. T. Rickard, Kinetics and mechanism of sulfidation of goethite, Am. J. Sci., 1974, 274, 941–952 CrossRef CAS.
- R. Lohmayer, A. Kappler, T. Lösekann-Behrens and B. Planer-Friedrich, Sulfur species as redox partners and electron shuttles for ferrihydrite reduction by Sulfurospirillum deleyianum, Appl. Environ. Microbiol., 2014, 80, 3141–3149 CrossRef.
- A. Kamyshny, C. G. Borkenstein and T. G. Ferdelman, Protocol for quantitative detection of elemental sulfur and polysulfide zero-valent sulfur distribution in natural aquatic samples, Geostand. Geoanal. Res., 2009, 33, 415–435 CrossRef CAS.
- D. Rickard and G. W. Luther, Kinetics of pyrite formation by the H2S oxidation of iron(II) monosulfide in aqueous solutions between 25 and 125°C: The mechanism, Geochim. Cosmochim. Acta, 1997, 61(1), 135–147 CrossRef CAS.
- R. T. Wilkin and H. L. Barnes, Pyrite formation by reactions of iron monosulfides with dissolved inorganic and organic sulfur species, Geochim. Cosmochim. Acta, 1996, 60, 4167–4179 CrossRef CAS.
- M. A. A. Schoonen and H. L. Barnes, Reactions forming pyrite and marcasite from solution: II: Via FeS precursors below 100°C, Geochim. Cosmochim. Acta, 1991, 55, 1505–1514 CrossRef CAS.
- D. Rickard and G. W. Luther, Chemistry of iron sulfides, Chem. Rev., 2007, 107, 514–562 CrossRef CAS.
- G. W. Luther, Pyrite synthesis via polysulfide compounds, Geochim. Cosmochim. Acta, 1991, 55, 2839–2849 CrossRef CAS.
- K. Eusterhues, C. Rumpel and I. Kögel-Knabner, Organo-mineral associations in sandy acid forest soils: importance of specific surface area, iron oxides and micropores, Eur. J. Soil Sci., 2005, 56, 753–763 CAS.
- I. Köegel-Knabner, G. Guggenberger, M. Kleber, E. Kandeler, K. Kalbitz, S. Scheu, K. Eusterhues and P. Leinweber, Organo-mineral associations in temperate soils: Integrating biology, mineralogy, and organic matter chemistry, J. Plant Nutr. Soil Sci., 2008, 171, 61–82 CrossRef.
- L. K. ThomasArrigo, C. Mikutta, J. Byrne, K. Barmettler, A. Kappler and R. Kretzschmar, Iron and arsenic speciation and distribution in organic flocs from streambeds of an arsenic-enriched peatland, Environ. Sci. Technol., 2014, 48, 13218–13228 CrossRef CAS.
- K. Lalonde, A. Mucci, A. Ouellet and Y. Gélinas, Preservation of organic matter in sediments promoted by iron, Nature, 2012, 483, 198–200 CrossRef CAS.
- U. Schwertmann and E. Murad, The nature of an iron oxide-organic iron association in a peaty environment, Clay Miner., 1988, 23, 291–299 CrossRef CAS.
- K. Eusterhues, F. E. Wagner, W. Häusler, M. Hanzlik, H. Knicker, K. U. Totsche, I. Kögel-Knabner and U. Schwertmann, Characterization of ferrihydrite-soil organic matter coprecipitates by X-ray diffraction and Mössbauer spectroscopy, Environ. Sci. Technol., 2008, 42, 7891–7897 CrossRef CAS.
- U. Schwertmann, F. Wagner and H. Knicker, Ferrihydrite-humic associations: Magnetic hyperfine interactions, Soil Sci. Soc. Am. J., 2005, 69, 1009–1015 CrossRef CAS.
- C. Mikutta, X-ray absorption spectroscopy study on the effect of hydroxybenzoic acids on the formation and structure of ferrihydrite, Geochim. Cosmochim. Acta, 2011, 75, 5122–5139 CrossRef CAS.
- C. Mikutta, J. Frommer, A. Voegelin, R. Kaegi and R. Kretzschmar, Effect of citrate on the local Fe coordination in ferrihydrite, arsenate binding, and ternary arsenate complex formation, Geochim. Cosmochim. Acta, 2010, 74, 5574–5592 CrossRef CAS.
- C. Mikutta, R. Mikutta, S. Bonneville, F. Wagner, A. Voegelin, I. Christl and R. Kretzschmar, Synthetic coprecipitates of exopolysaccharides and ferrihydrite. Part 1: Characterization, Geochim. Cosmochim. Acta, 2008, 72, 1111–1127 CrossRef CAS.
- C. Chen, R. K. Kukkadapu and D. L. Sparks, Influence of coprecipitated organic matter on Fe2+(aq)-catalyzed transformation of ferrihydrite: Implications for carbon dynamics, Environ. Sci. Technol., 2015, 49, 10927–10936 CrossRef CAS.
- L. K. ThomasArrigo, R. Kaegi and R. Kretzschmar, Ferrihydrite growth and transformation in the presence of ferrous Fe and model organic ligands, Environ. Sci. Technol., 2019, 53, 13636–13647 CrossRef CAS.
- Z. Zhou, D. E. Latta, N. Noor, A. Thompson, T. Borch and M. M. Scherer, Fe(II)-catalyzed transformation of organic matter-ferrihydrite coprecipitates: A closer look using Fe isotopes, Environ. Sci. Technol., 2018, 52, 11142–11150 CrossRef CAS.
- Y. K. Henneberry, T. E. C. Kraus, P. S. Nico and W. R. Horwath, Structural stability of coprecipitated natural organic matter and ferric iron under reducing conditions, Org. Geochem., 2012, 48, 81–89 CrossRef CAS.
- J. W. Morse and Q. W. Wang, Pyrite formation under conditions approximating those in anoxic sediments: II. Influence of precursor iron minerals and organic matter, Mar. Chem., 1997, 57, 187–193 CrossRef CAS.
- V. Noël, N. Kumar, K. Boye, L. Barragan, J. S. Lezama-Pacheco, R. Chu, N. Tolic, G. E. Brown and J. Bargar, FeS colloids-formation and mobilization pathways in natural waters, Environ. Sci.: Nano, 2020, 7, 2102–2116 RSC.
- S. H. Bottrell, D. Hatfield, R. Bartlett, M. J. Spence, K. D. Bartle and R. J. G. Mortimer, Concentrations, sulfur isotopic compositions and origin of organosulfur compounds in pore waters of a highly polluted raised peatland, Org. Geochem., 2010, 41, 55–62 CrossRef CAS.
- J. Boulegue, C. J. Lord and T. M. Church, Sulfur speciation and associated trace-metals (Fe, Cu) in the pore waters of Great Marsh, Deleware, Geochim. Cosmochim. Acta, 1982, 46, 453–464 CrossRef CAS.
- G. W. Luther, T. M. Church, J. R. Scudlark and M. Cosman, Inorganic and organic sulfur cycling in salt-marsh pore waters, Science, 1986, 232, 746–749 CrossRef CAS.
- C. M. Koretsky, J. R. Haas, N. T. Ndenga and D. Miller, Seasonal variations in vertical redox stratification and potential influence on trace metal speciation in minerotrophic peat sediments, Water, Air, Soil Pollut., 2006, 173, 373–403 CrossRef CAS.
- C. M. Koretsky, M. Haveman, L. Beuving, A. Cuellar, T. Shattuck and M. Wagner, Spatial variation of redox and trace metal geochemistry in a minerotrophic fen, Biogeochemistry, 2007, 86, 33–62 CrossRef CAS.
- G. C. Wallace, M. Sander, Y. P. Chin and W. A. Arnold, Quantifying the electron donating capacities of sulfide and dissolved organic matter in sediment pore waters of wetlands, Environ. Sci.: Processes Impacts, 2017, 19(5), 758–767 RSC.
- E. D. Burton, R. T. Bush and L. A. Sullivan, Fractionation and extractability of sulfur, iron and trace elements in sulfidic sediments, Chemosphere, 2006, 64(8), 1421–1428 CrossRef CAS.
- L. K. ThomasArrigo, C. Mikutta, J. Byrne, A. Kappler and R. Kretzschmar, Iron(II)-catalyzed iron atom exchange and mineralogical changes in iron-rich organic freshwater flocs: An iron isotope tracer study, Environ. Sci. Technol., 2017, 51, 6897–6907 CrossRef CAS.
- B. H. Gu, J. Schmitt, Z. H. Chen, L. Y. Liang and J. F. McCarthy, Adsorption and desorption of natural organic matter on iron oxide: Mechanisms and models, Environ. Sci. Technol., 1994, 28, 38–46 CrossRef CAS.
-
W. H. Brown, B. L. Iverson, E. Anslyn and C. Foote, Organic Chemistry, Brookes Cole Cengage Learning, United States, 2012 Search PubMed.
- J. S. S. Damste, M. D. Kok, J. Koster and S. Schouten, Sulfurized carbohydrates: an important sedimentary sink for organic carbon?, Earth Planet. Sci. Lett., 1998, 164, 7–13 CrossRef.
- I. Ciglenecki, B. Cosovic, V. Vojvodic, M. Plavsic, K. Furic, A. Minacci and F. Baldi, The role of reduced sulfur species in the coalescence of polysaccharides in the Adriatic Sea, Mar. Chem., 2000, 71, 233–249 CrossRef CAS.
- B. E. van Dongen, S. Schouten, M. Baas, J. A. J. Geenevasen and J. S. S. Damste, An experimental study of the low-temperature sulfurization of carbohydrates, Org. Geochem., 2003, 34, 1129–1144 CrossRef CAS.
- J. D. Cline, Spectrophotometric determination of hydrogen sulfide in natural waters, Limnol. Oceanogr., 1969, 14, 454–458 CrossRef CAS.
- W. B. Fortune and M. G. Mellon, Determination of iron with o-phenanthroline - A spectrophotometric study, Ind. Eng. Chem., Anal. Ed., 1938, 10, 60–64 CrossRef CAS.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- A. Manceau and K. L. Nagy, Quantitative analysis of sulfur functional groups in natural organic matter by XANES spectroscopy, Geochim. Cosmochim. Acta, 2012, 99, 206–223 CrossRef CAS.
- T. Ressler, WinXAS: A program for X-ray absorption spectroscopy data analysis under MS-Windows, J. Synchrotron Radiat., 1998, 5, 118–122 CrossRef CAS.
- S. Shakeri Yekta, J. Gustavsson, B. H. Svensson and U. Skyllberg, Sulfur K-edge XANES and acid volatile sulfide analyses of changes in chemical speciation of S and Fe during sequential extraction of trace metals in anoxic sludge from biogas reactors, Talanta, 2012, 89, 470–477 CrossRef.
- D. Boland, R. Collins, C. Miller, C. Glover and T. D. Waite, Effect of solution and solid-phase conditions on the Fe(II)-accelerated transformation of ferrihydrite to lepidocrocite and goethite, Environ. Sci. Technol., 2014, 48, 5477–5485 CrossRef CAS.
- L. K. ThomasArrigo, J. Byrne, A. Kappler and R. Kretzschmar, Impact of organic matter on iron(II)-catalyzed mineral transformation in ferrihydrite-OM coprecipitates, Environ. Sci. Technol., 2018, 52, 12316–12326 CrossRef CAS.
- H. D. Pedersen, D. Postma, R. Jakobsen and O. Larsen, Fast transformation of iron oxyhydroxides by the catalytic action of aqueous Fe(II), Geochim. Cosmochim. Acta, 2005, 69, 3967–3977 CrossRef CAS.
- A. M. Jones, R. N. Collins, J. Rose and T. D. Waite, The effect of silica and natural organic matter on the Fe(II)-catalysed transformation and reactivity of Fe(III) minerals, Geochim. Cosmochim. Acta, 2009, 73, 4409–4422 CrossRef CAS.
- C. M. Hansel, S. G. Benner and S. Fendorf, Competing Fe(II)-induced mineralization pathways of ferrihydrite, Environ. Sci. Technol., 2005, 39, 7147–7153 CrossRef CAS.
- L. Gentile, T. Wang, A. Tunlid, U. Olsson and P. Persson, Ferrihydrite nanoparticle aggregation induced by dissolved organic matter, J. Phys. Chem. A, 2018, 122, 7730–7738 CrossRef CAS.
- H. Guénet, M. Davranche, D. Vantelon, J. Gigault, S. Prévost, O. Taché, S. Jaksch, M. Pédrot, V. Dorcet, A. Boutier and J. Jestin, Characterization of iron-organic matter nano-aggregate networks through a combination of SAXS/SANS and XAS analyses: impact on As binding, Environ. Sci.: Nano, 2017, 4, 938–954 RSC.
- C. M. Chen, J. J. Dynes, J. Wang and D. L. Sparks, Properties of Fe-organic matter associations via coprecipitation versus adsorption, Environ. Sci. Technol., 2014, 48, 13751–13759 CrossRef CAS.
- B. Morgan, E. D. Burton and A. W. Rate, Iron monosulfide enrichment and the presence of organosulfur in eutrophic estuarine sediments, Chem. Geol., 2012, 296, 119–130 CrossRef.
- J. A. Bourdoiseau, M. Jeannin, R. Sabot, C. Rémazeilles and P. Refait, Characterisation of mackinawite by Raman spectroscopy: Effects of crystallisation, drying and oxidation, Corros. Sci., 2008, 50(11), 3247–3255 CrossRef CAS.
- R. Guilbaud, I. B. Butler, R. M. Ellam and D. Rickard, Fe isotope exchange between Fe(II)aq and nanoparticulate mackinawite (FeSm) during nanoparticle growth, Earth Planet. Sci. Lett., 2010, 300(1–2), 174–183 CrossRef CAS.
- S. Boursiquot, M. Mullet, M. Abdelmoula, J. M. Génin and J. J. Ehrhardt, The dry oxidation of tetragonal FeS1-x mackinawite, Phys. Chem. Miner., 2001, 28(9), 600–611 CrossRef CAS.
- S. Hunger and L. G. Benning, Greigite: a true intermediate on the polysulfide pathway to pyrite, Geochem. Trans., 2007, 8, 1 CrossRef.
- R. T. Wilkin and H. L. Barnes, Formation processes of framboidal pyrite, Geochim. Cosmochim. Acta, 1997, 61, 323–339 CrossRef CAS.
- E. D. Burton, R. T. Bush, S. G. Johnston, L. A. Sullivan and A. F. Keene, Sulfur biogeochemical cycling and novel Fe-S mineralization pathways in a tidally re-flooded wetland, Geochim. Cosmochim. Acta, 2011, 75(12), 3434–3451 CrossRef CAS.
- A. F. Keene, S. G. Johnston, R. T. Bush, L. A. Sullivan, E. D. Burton, A. E. McElnea, C. R. Ahern and B. Powell, Effects of hyper-enriched reactive Fe on sulfidisation in a tidally inundated acid sulfate soil wetland, Biogeochemistry, 2011, 103(1–3), 263–279 CrossRef CAS.
- D. Rickard, I. B. Butler and A. Oldroyd, A novel iron sulphide mineral switch and its implications for Earth and planetary science, Earth Planet. Sci. Lett., 2001, 189(1–2), 85–91 CrossRef CAS.
- J. Prietzel, A. Botzaki, N. Tyufekchieva, M. Brettholle, J. Thieme and W. Klysubun, Sulfur speciation in soil by S K-edge XANES spectroscopy: Comparison of spectral deconvolution and linear combination fitting, Environ. Sci. Technol., 2011, 45, 2878–2886 CrossRef CAS.
- T. Heitmann and C. Blodau, Oxidation and incorporation of hydrogen sulfide by dissolved organic matter, Chem. Geol., 2006, 235, 12–20 CrossRef CAS.
- Z.-G. Yu, S. Peiffer, J. Göttlicher and K.-H. Knorr, Electron transfer budgets and kinetics of abiotic oxidation and incorporation of aqueous sulfide by dissolved organic matter, Environ. Sci. Technol., 2015, 49, 5441–5449 CrossRef CAS.
- M. Hoffmann, C. Mikutta and R. Kretzschmar, Bisulfide reaction with natural organic matter enhances arsenite sorption: Insights from X-ray absorption spectroscopy, Environ. Sci. Technol., 2012, 46, 11788–11797 CrossRef CAS.
-
S. D. Kelly, D. Hesterberg and B. Ravel, Analysis of soils and minerals using X-ray absorption spectroscopy, in Methods of Soil Analysis, Part 5, Minerological Methods, ed. A. L. Ulery and L. R. Drees, Soil Science Society of America, Madison, 2008, pp. 378–463 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details to experimental materials and setup, aqueous S and Fe concentrations, supplementary XAS analyses. See DOI: 10.1039/d0en00398k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Sylvain
Bouchet
*a,
Sylvain
Bouchet