
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultra-rapid uptake and the highly stable storage of methane as combustible ice†
Gaurav
Bhattacharjee‡
 ,
Marcus N.
Goh‡
,
Sonia E. K.
Arumuganainar
,
Ye
Zhang
and
Praveen
Linga
*
,
Marcus N.
Goh‡
,
Sonia E. K.
Arumuganainar
,
Ye
Zhang
and
Praveen
Linga
*
Department of Chemical and Biomolecular Engineering, National University of Singapore, Singapore 117 585, Singapore. E-mail: praveen.linga@nus.edu.sg
First published on 27th October 2020
Abstract
The continuously increasing trend of natural gas (NG) consumption due to its clean nature and abundant availability indicates an inevitable transition to an NG-dominated economy. Solidified natural gas (SNG) storage via combustible ice or clathrate hydrates presents an economically sound prospect, promising high volume density and long-term storage. Herein, we establish 1,3-dioxolane (DIOX) as a highly efficient dual-action (thermodynamic and kinetic promoter) additive for the formation of clathrate (methane sII) hydrate. By synergistically combining a small concentration (300 ppm) of the kinetic promoter L-tryptophan with DIOX, we further demonstrated the ultra-rapid formation of hydrates with a methane uptake of 83.81 (±0.77) volume of gas/volume of hydrate (v/v) within 15 min. To the best of our knowledge, this is the fastest reaction time reported to date for sII hydrates related to SNG technology and represents a 147% increase in the hydrate formation rate compared to the standard water–DIOX system. Mixed methane–DIOX hydrates in pelletized form also exhibited incredible stability when stored at atmospheric pressure and moderate temperature of 268.15 K, thereby showcasing the potential to be industrially applicable for the development of a large-scale NG storage system.
Broader contextThe emergence of natural gas as a key player in the current energy landscape presents a rare opportunity for the development of new and robust gas storage technologies. Gas hydrates or combustible ice-based solidified natural gas (SNG) technology can realize the compact and safe long-term storage of natural gas using eco-friendly water as the major raw material (>94%). However, its practical application has been limited by problems in forming natural gas hydrates at a rapid rate, and subsequently ensuring their prolonged stability. Herein, we report 1,3-dioxolane (DIOX) as a dual-action chemical promoter for the formation of methane sII hydrate, offering excellent thermodynamic and kinetic enhancement ability. A small amount (300 ppm) of kinetic promoter, L-tryptophan, was added to the scheme to further help in achieving ultra-rapid hydrate formation rate. A mixed methane–DIOX hydrate pellet stored at atmospheric pressure in a conventional freezer for 8 days remained highly stable, thereby demonstrating the industrial applicability, scalability and ease of operation of this approach. |
Introduction
As economic progress and population growth drive the global energy demand, fossil fuels continue to remain strategically important and natural gas (NG) will play a vital role in society.1 The exploration of abundant NG reserves available in unconventional form (shale, hydrates and coal-bed) together with NG being the cleanest burning fossil fuel compared to gasoline and coal, makes this resource, which is primarily methane,2 economically competitive and environment-friendly. Accordingly, 2018 witnessed a 4.6% increase in NG consumption with a projected average annual increase of 0.9% over the next decade.1 An increase in the demand and reliance on NG imports require the development of safe and reliable large-scale gas storage technology by NG importing countries to cater for energy security, resilience and redundancy. Although liquefied natural gas (LNG) is the best option to transport NG where pipelines are not possible, it is not considered suitable for long-term storage due to the extremely low temperature (111.2 K) required and associated boil-off. Compressed natural gas (CNG) is not suitable for large-scale storage systems due to its explosive nature and high-pressure requirement. On the other hand, adsorbed natural gas (ANG) has attracted increasing scientific attention in recent years with advances in the synthesis of polymeric materials such as MOFs and graphene, and even the emergence of new porous organic framework materials.3–7Nature has stored methane gas in the form of natural gas hydrates for millions of years, although in a slow manner, resulting in the accumulation of a huge energy resource.8–10 Gas hydrates are crystalline inclusion compounds, where under suitable conditions, cages made of water molecules may host guest gas molecules.11 Gas hydrates made of methane or natural gas are also known as combustible ice. With the appropriate tuning of the formation conditions and the identification of suitable promoters, the formation of gas hydrates can be accelerated. Thus, NG stored in the form of hydrate as solidified natural gas (SNG) has emerged as an option for its large volume and long-term storage.12–14 Tetrahydrofuran (THF) has proven to be a stable thermodynamic promoter for H2 storage via the formation of clathrate hydrates.15,16 Recently, THF has also been demonstrated as a dual-action (thermodynamic and kinetic promotion) promoter for methane storage.17 sII methane hydrate is formed rapidly in presence of THF over a wide temperature range17 and has been shown to be stable near the ambient pressure of 0.15 MPa and at 271.5 K.18
However, despite the use of THF in many industrial chemical processes,19,20 its role in the formation of hydrates has often been questioned considering its carcinogenicity,21 high volatility,22,23 and corrosive nature,24 which impede its utilization in the development of large-scale technology. Thus, it is necessary to identify cleaner alternatives to THF without compromising the vital dual functionality. Accordingly, 1,3-dioxolane (DIOX) is a heterocyclic compound closely related to THF, where the carbon atom at the 3-position of THF is replaced with an oxygen atom.25 It has similar water solubility as THF, but is less volatile and toxic (Table S1 in the ESI†). DIOX on its own can stabilize the sII hydrate.26 However, in the literature, there is only one study on the phase equilibrium of the methane–DIOX system, where 5.0 mol% of DIOX was found to be optimal as a thermodynamic promoter among the investigated concentrations in the range of 0.99 mol% to 20.02 mol%.27
Herein, we report the rapid methane uptake for the formation of mixed methane–DIOX hydrates via a detailed kinetic study, and elucidate the mechanism for this rapid enhancement by combining crystal morphology and in situ Raman Sepctroscopy observations. Further, we report the ultra-rapid formation of mixed methane–DIOX hydrates under mild operating conditions with exceptionally high gas uptake, which was achieved together with the use of L-tryptophan. The hydrate formed was also analyzed using the powder X-ray diffraction (p-XRD) technique for structure identification. Finally, this study demonstrated the production of an sII methane–DIOX hydrate pellet together with the monitoring of its stability over an eight-day period for the first time.
Results and discussion
Phase equilibrium measurement for methane–DIOX/water system
Fig. 1 presents the thermodynamic phase equilibrium data for the methane–DIOX/water system with DIOX used in a stoichiometric concentration (5.56 mol%). An isothermal pressure search method was employed at three fixed temperatures of 283.15 K, 288.15 K and 293.15 K. These temperatures were chosen in accordance with the experimental temperature employed for the kinetic measurements in this study. The data indicates that DIOX is an excellent thermodynamic promoter for the formation of methane hydrates. Specifically, the equilibrium pressure for the formation of pure methane hydrate at 283.15 K is 7.2 MPa,28 whereas that for the methane–DIOX system is less than 1.0 MPa. This shift demonstrates the potential of DIOX since its thermodynamic promotion ability directly correlates to hydrate formation under milder operating conditions compared to that for pure methane (sI) hydrates. The average equilibrium pressures for the methane–DIOX/water system based on two individual measurements each at the three temperatures studied are presented in Table S2 in the ESI.†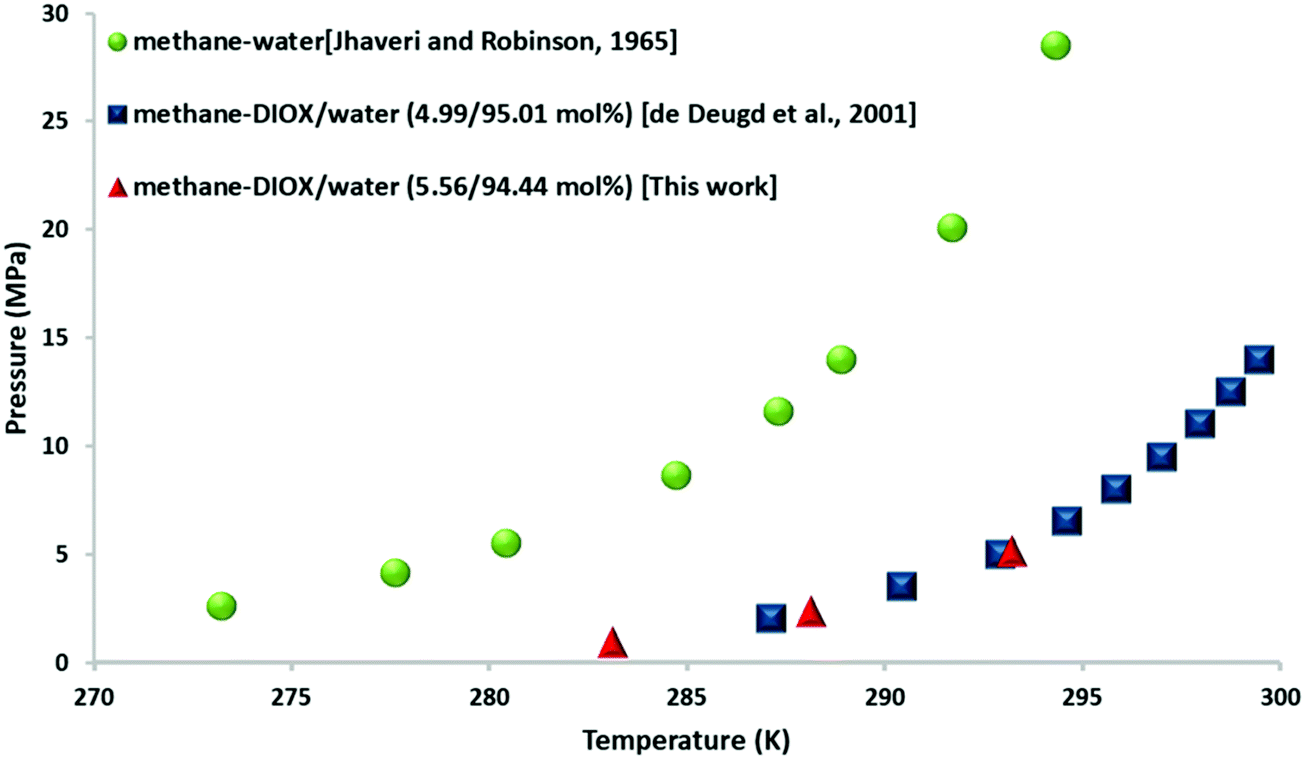 | ||
| Fig. 1 Three-phase (H–Lw–V) equilibrium points for methane–water29 and methane–DIOX/water systems27 together with the experimental data obtained in the present study for the methane–DIOX/water system containing a stoichiometric concentration of DIOX (5.56 mol%). | ||
Mixed methane–DIOX hydrate growth under moderate conditions
The hydrate growth (gas uptake) curve for the methane–DIOX/water system operated under quiescent (unstirred) conditions is presented in Fig. 2a. The experimental conditions used were 283.15 K and 7.2 MPa (initial pressure). The experimental procedure and the calculation to estimate the methane uptake for hydrate formation is explained in the Methods section. Hydrate formation was based on the hybrid combinatorial reactor (HCR) approach, which is described in detail in the literature.30 In the HCR approach, the system is stirred (400 rpm in the present case) for a very short time until nucleation occurs, which in the current study occurred within 2 min for all the experiments performed. The HCR approach offers the advantage of controlling the stochasticity of hydrate nucleation, while eliminating the need for mixing during the hydrate growth phase.30Fig. 2a shows fast hydrate growth and high methane uptake in a simple unstirred configuration, which was maintained during the hydrate growth phase. Within 45 min, the formation process was completed, with methane uptake of 87.03 (±0.23) volume of gas/volume of hydrate (v/v) achieved for quiescent growth. Hydrate growth reached 90% completion (t90 refers to the time required for 90% completion of the process) in 32.45 (±3.70) min. The shaded regions in this figure represent the remarkable reproducibility of the experimental data (standard error of 3 experimental runs). The kinetic performance data for individual experiment runs presented in Fig. 2a is included in Table S3 in the ESI.†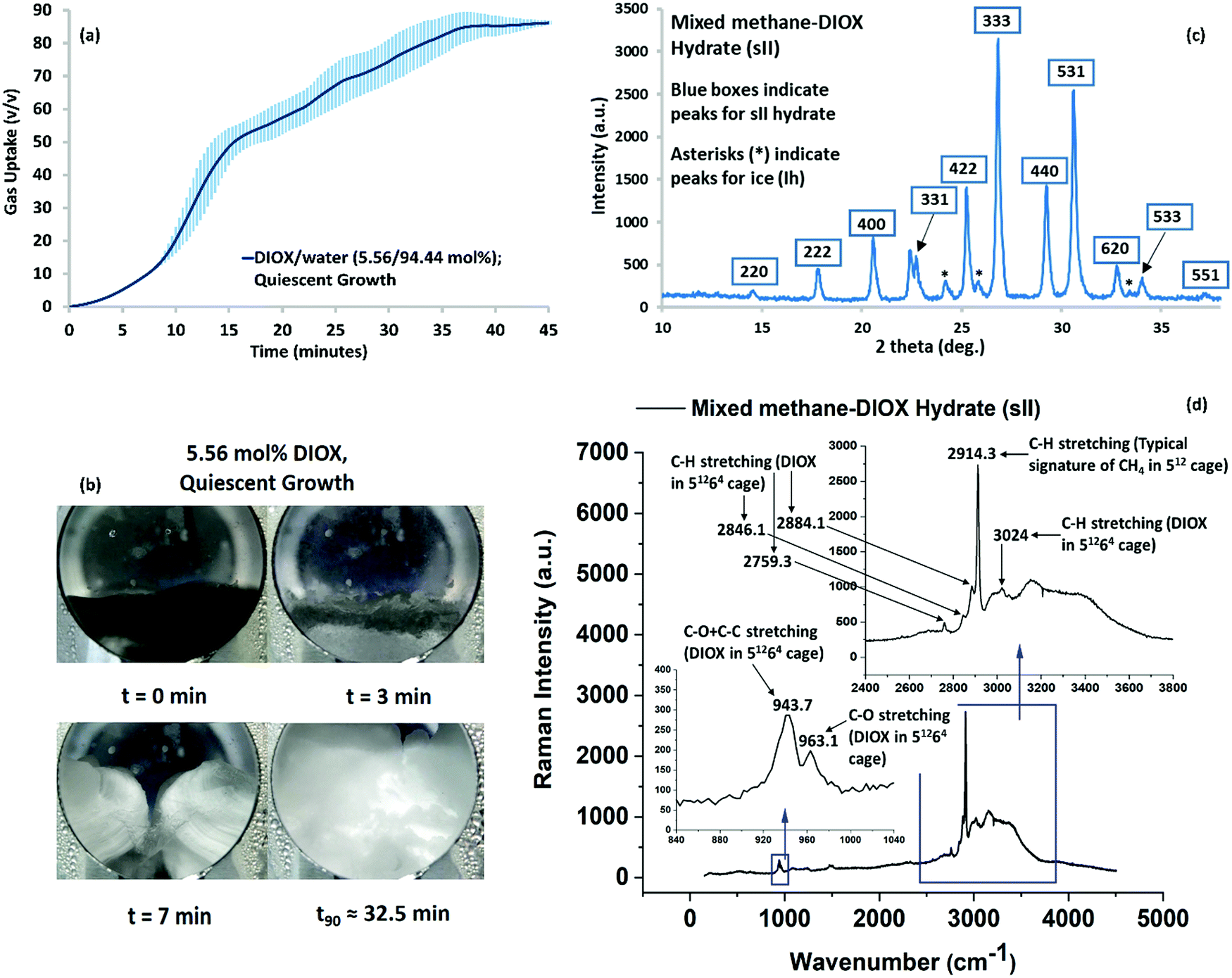 | ||
| Fig. 2 (a) Mixed methane–DIOX hydrate growth under quiescent conditions (in blue) at 283.15 K and an initial pressure of 7.2 MPa, where the continuous line represents the average data of three experiments and the vertical shaded regions represent the standard deviation of three experimental data sets. (b) Morphology images of a typical experiment of mixed methane–DIOX hydrate formation under quiescent hydrate growth conditions. (c) p-XRD pattern of methane–DIOX mixed hydrate sample synthesized using 5.56 mol% DIOX aqueous solution at 283.15 K and an initial pressure of 7.2 MPa, where the peaks corresponding to the sII hydrate are inside blue boxes, while the asterisks indicate the presence of ice (Ih). (d) Complete Raman spectrum obtained for hydrates formed from methane–DIOX/water system at 283.15 K and an initial pressure of 7.2 MPa, where the DIOX concentration used was 5.56 mol%. | ||
The visual images captured during a typical hydrate formation experiment for the methane–DIOX/water system is presented in Fig. 2b, which provide insight into the physical hydrate growth patterns. Additionally, a video of the visual observation of hydrate formation is presented in the ESI† (Video SV1). For the methane–DIOX/water system under quiescent growth conditions, hydrate masses propagated from the three-phase (solid–liquid–gas) interface points on both sides of the reactor (as observed through the viewing window, see visual image at t = 7 min) inwards towards the center, where they eventually meet, and thereon grew as bulk hydrates. Interestingly, at about 7 min in Fig. 2b, a significant amount of hydrates can be observed in the reactor, but the methane uptake is only about 9.12 v/v, [11.1% (±0.76%)] of the total methane uptake. Subsequently, 88.9% (±0.76%) of the methane uptake occurs after 7 min of the formation experiment. These calculations are based on the assumption that the methane uptake at the 35 minute mark is the total methane uptake since beyond this point (Fig. 2b), the hydrate growth observed is minimal. It should be noted that this particular pattern of methane uptake and hydrate growth was consistent for all the experiments, as seen by the very small standard error in Fig. 2a.
For comparison, we also conducted experiments using the traditional stirred-tank operation mode for the growth phase, and the results are presented as Fig. S2 in the ESI.† As observed in Fig. S2 (ESI†), the inclusion of stirring does not provide significant advantages to the hydrate growth characteristics, with the hydrate growth rate being slightly better for the stirred system compared to quiescent growth. Specifically, t90 for the stirred growth condition is 22.89 (±0.69) min, whereas this is achieved in 32.45 (±3.70) min for the quiescent (unstirred) growth condition. The kinetic performance data for individual experiment runs conducted in the stirred-tank operation mode is presented in Table S3 in the ESI.† A video of the visual observation of hydrate formation in the stirred-tank operation is also presented in the ESI† (Video SV2). As can be seen in Video SV2 (ESI†), within 2 min the reactor was full of hydrates, but the average methane uptake for this system at this point was only 12.56 v/v, [15.2% (±0.94%)] of the total methane uptake.
For the experimental conditions employed for hydrate formation (283.15 K and 7.2 MPa), from a thermodynamic viewpoint, it would be practically impossible to form pure methane (sI) hydrates (refer Fig. 1). Thus, only mixed methane–DIOX (sII) hydrate formation could occur. To confirm this independently, the produced hydrate was analyzed using both the powder X-ray diffraction (p-XRD) and in situ Raman spectroscopy techniques, and the results are presented in the following section.
Characterization of mixed methane–DIOX hydrates
The rapid and repeatable kinetic data obtained demonstrates the fantastic potential of DIOX to be established as a remarkably efficient dual-action promoter for SNG technology. Thus, it is important to understand micro-scale data such as the structure of the hydrates formed in the presence of DIOX. Accordingly, to confirm the structure of the hydrates formed using the methane–DIOX/water system, they were characterized using the powder X-ray diffraction (p-XRD) technique. The XRD pattern obtained is shown in Fig. 2c, which matches perfectly with the standard patterns of sII hydrates in the literature,15,31 thus presenting strong evidence that only sII hydrates were formed in the present study. For the p-XRD analysis, the hydrate sample was recovered under liquid nitrogen temperature, which caused the unreacted water present in the sample to appear as ice peaks in the XRD pattern.We independently analysed the hydrates formed from the methane–DIOX/water system under the current experimental conditions (283.15 K and 7.2 MPa) via in situ Raman spectroscopy. The obtained spectrum is presented in Fig. 2d, which confirms the formation of only sII hydrates using the investigated methane–DIOX/water system under the aforementioned experimental conditions. The presence of sII hydrates was determined by the signature C–H stretching at 2914.3 cm−1 of methane gas in the small (512) cages of sII hydrates. The absence of any other specific methane signatures such as the C–H stretching at 2904.85 (±0.33) cm−1, which is a characteristic of methane occupancy in the large (51262) cages of sI hydrate,32 confirms the presence of only sII hydrates. This in situ Raman spectroscopy study provided the first complete Raman spectrum and associated analysis for mixed methane–DIOX hydrates. The detailed analysis of the observed Raman spectra is presented in the ESI† (refer to pages S5–S9).
Comparison with methane–THF mixed hydrate system
Fig. 3a compares the gas uptake profiles (quiescent hydrate growth) obtained under the same initial driving force for the methane–DIOX/water and methane–THF/water systems. Keeping the temperature fixed, the initial experimental pressure was varied for the two systems according to their respective phase equilibrium measurements (Fig. 3b), thus ensuring the initial driving force remained uniform. The gas uptake profiles for the DIOX/water and THF/water systems are presented in Fig. 3a, which are generally similar, with the final gas uptake for the DIOX/water system about 13.69% higher after 45 min. The kinetic performance parameters for the mixed methane–THF hydrate formation experiments are presented in Table S4 (ESI†).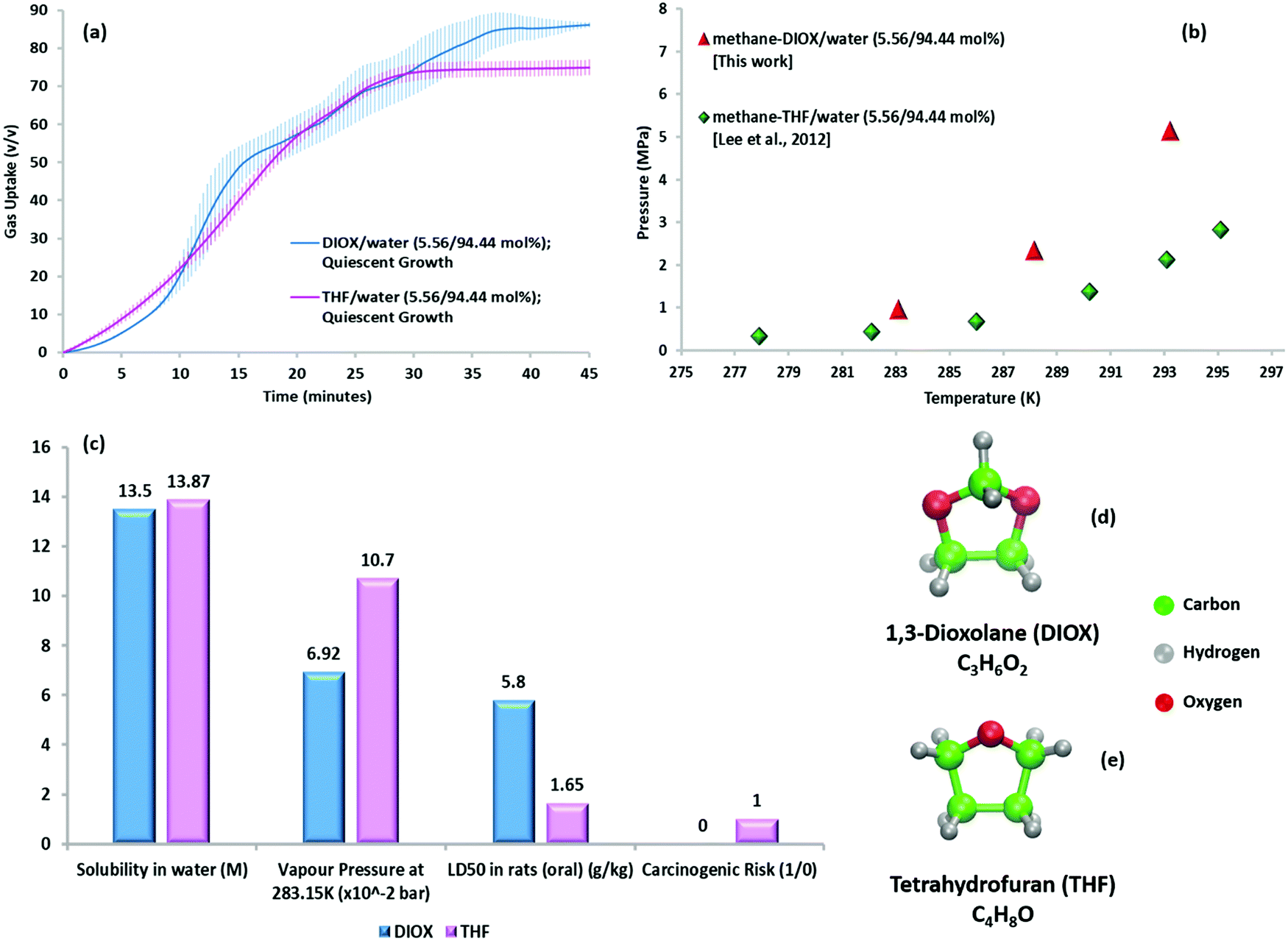 | ||
| Fig. 3 (a) Hydrate growth under quiescent conditions for methane–THF hydrates (in pink) and methane–DIOX hydrates (in blue) at 283.15 K and an initial driving force of ∼6.2 MPa, where the continuous lines represent the average data of three experiments and the vertical shaded regions represent the standard deviation of three experimental data sets. (b) Three-phase (H–Lw–V) equilibrium points for methane–DIOX/water (5.56/94.44 mol%) and methane–THF/water (5.56/94.44 mol%) systems.33 (c) Comparison of the relevant physical and safety parameters of DIOX34–36 and THF21,23 for their use as dual-action promoters for hydrate formation. (d) Molecular structure and formula of DIOX and (e) molecular structure and formula of THF. | ||
The kinetic gas uptake data in Fig. 3a evidently indicates that the presence of DIOX results in rapid methane hydrate growth similar to that observed with THF. However, this observation can only feasibly be put into practice if DIOX exhibits other clear advantages compared to THF, for example, with regards to the operational and safety hazards of THF outlined in the Introduction. Accordingly, Fig. 3c compares various relevant physical and safety parameters of DIOX and THF for their application in this technology. The data presents similar water solubility for both compounds, which is expected owing to their similar molecular structures (Fig. 3d and e for DIOX and THF, respectively), but more importantly, the significantly lower volatility and toxicity of DIOX compared to THF. Moreover, DIOX is classified as non-carcinogenic36 compared to THF, which is a confirmed animal carcinogen with unknown relevance to humans21 (refer to Table S1, ESI† for details of the properties presented in Fig. 3c). Lower toxicity and non-carcinogenicity of DIOX imply obvious safety benefits, and furthermore, its lower volatility indicates both safety and possible recyclability advantages due to lower promoter loss between continuous cycles. Therefore, considering all the available information, the DIOX/water system appears to be an attractive alternative to the significantly more toxic THF for utilization in SNG technology for gas storage application.
Two-step hydrate growth mechanism for mixed methane–DIOX hydrate system
We observed a distinctive pattern of hydrate growth behavior for the methane–DIOX/water system. When hydrates nucleate, all three types of molecules present in the system (water, methane and DIOX) participate in the ensuing hydrate formation phenomenon. However interestingly, once hydrate nucleation occurred, sluggish gas uptake kinetics was observed for an initial period of the hydrate growth phase (up to 7 min in Fig. 2a), accompanied with a simultaneous visual marker of a substantial amount of hydrate mass growth (see Fig. 2b and Video SV1, ESI†). This indicates that during the initial growth period under consideration, the primary guest molecule entering the hydrate structure was DIOX (occupying the large cages of sII hydrate), together with a small (relatively lesser) amount of methane going into the small cages of the formed sII hydrate. This preferential DIOX enclathration stage, which occurs at the beginning of the hydrate growth process, was termed by us as Step 1 of the growth mechanism for the mixed methane–DIOX hydrate system.Fig. S3 (ESI†) presents images of the methane–DIOX hydrate system undergoing growth, in both the quiescent and stirred regimes, together with quantified methane uptake volumes and percentages for the related time periods. As seen in Fig. S3a (ESI†) representing quiescent growth, at about 7 min, although there was visibly lots of hydrates in the reactor, the average methane uptake at this stage is only about 9.12 v/v, [11.1% (±0.76%)] of the total methane uptake. Step 1 of growth (as described in the preceding paragraph) was also observed for the stirred system (see Fig. S3b, ESI†), where at 2 min of hydrate growth (the time around when the stirring of the contents ceased, refer to Video SV2, ESI†), the reactor already appeared to contain a considerable amount of hydrate mass, while the average methane uptake was only 12.56 v/v, [15.2% (±0.94%)] of the total methane uptake. If we assume that the enclathration (or cage loading) of DIOX and methane occurs at the same rate in the large (for DIOX) and small (for methane) cages, i.e. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (DIOX:methane), the methane uptake presented in Step 1 in Fig. S3 (ESI†) (refer to the bar charts) corresponds to a conversion of ∼2.62 mL (for quiescent growth) and 3.53 mL (for stirred growth) of the 32.4 mL solution present in the reactor. However, contradictorily, we observed considerable amount of hydrate mass in the reactor for both operating modes (quiescent and stirred).
:2 (DIOX:methane), the methane uptake presented in Step 1 in Fig. S3 (ESI†) (refer to the bar charts) corresponds to a conversion of ∼2.62 mL (for quiescent growth) and 3.53 mL (for stirred growth) of the 32.4 mL solution present in the reactor. However, contradictorily, we observed considerable amount of hydrate mass in the reactor for both operating modes (quiescent and stirred).
In Step 2 of the hydrate growth process, accelerated enclathration of methane molecules in the hydrate structure (small cages of sII hydrate) occurs. The visual images together with the methane uptake presented in Fig. S3 (ESI†) indicate the occupation of methane molecules in the small cages of the formed sII hydrates during Step 2 of hydrate growth. Quantifiably, on average, 88.9% (±0.76%) and 84.8% (±0.94%) of the total methane uptake in the hydrate phase for quiescent growth and stirred growth, respectively, occurred during Step 2. The stirred system ensures better gas–liquid contact due to rigorous mixing right after nucleation, and thus it is plausible to expect that the transition from Step 1 to Step 2 would be much quicker for mixed methane–DIOX hydrate growth under the fully stirred condition compared to that under the quiescent condition.
In situ Raman spectroscopy experiments were independently carried out on the mixed methane–DIOX hydrate system for an independent perspective on the proposed two-step hydrate growth mechanism. A detailed discussion on the same is presented in the ESI† (pages S5–S9). The time-dependent Raman spectra obtained at various intervals for the first 30 min of mixed methane–DIOX hydrate growth at 7.2 MPa pressure and 283.2 K are presented in Fig. S5 (ESI†). As mentioned previously, when the hydrates nucleate, we expect both the DIOX and CH4 molecules to start moving into the hydrate structure and stabilize the hydrate cages, i.e. both DIOX and CH4 trigger hydrate nucleation and initial growth. Actually, this is something we also observed in our prior work on a similar mixed methane–THF (sII) hydrate system.18,37 As seen in Fig. S5b (ESI†), at the 1 min mark post-nucleation, a small signal appeared at 2914.3 cm−1, indicating methane occupancy in the small cages of the sII hydrates, thus confirming the formation of hydrates in the system. The signatures for DIOX enclathration in the large cages were also observed in the Raman spectra, as shown in Fig. S5a (ESI†), and are discussed in detail in the ESI.† It can be seen in Fig. S5b (ESI†) that the peak intensity for methane in the 512 cages increases significantly throughout hydrate growth from 1 min after nucleation, as reflected by the Raman spectra obtained at selected periods during the hydrate growth process. This suggests significant enclathration of methane molecules into the solid hydrate phase as hydrate growth proceeds right up to the 30 min upper limit mark, as shown in Fig. S5 (ESI†), and this molecular level observation is consistent with the results of the methane uptake experiments performed at the macroscopic scale for similar periods. On the other hand, the various peak intensities for DIOX in the 51264 cages did not exhibit a significant increase during this period, indicating that a significant percentage of the DIOX enclathration in the hydrates may have already occurred during the short initial hydrate growth period right after nucleation. The independent conclusions drawn from the in situ Raman spectroscopy study are consistent with the observed two-step growth mechanism for mixed methane–DIOX hydrate formation.
Thus, based on the methane uptake, visual observations, and in situ Raman spectra analysis for methane loading in the hydrate structure, a schematic to describe the two-step hydrate growth for the mixed methane–DIOX hydrate system is presented in Fig. 4. The visual observation of the reactor contents, methane uptake and Raman spectra for the DIOX and methane signals in sII hydrate structure for 2 min and 30 min post-hydrate nucleation (stirred growth condition) are also presented in Fig. 4 to support the proposed mechanism of hydrate growth. The two-step hydrate growth mechanism comprises a preferential DIOX over methane enclathration step (Step 1) and a subsequent rapid and sustained methane enclathration step (Step 2). The methane uptake across both periods represented independently is consistent with the corresponding methane peak intensity changes. Similarly, the lack of significant change in the DIOX peaks in the respective Raman spectra across the represented periods also corroborates the large volumes of the hydrates visually observed immediately after nucleation.
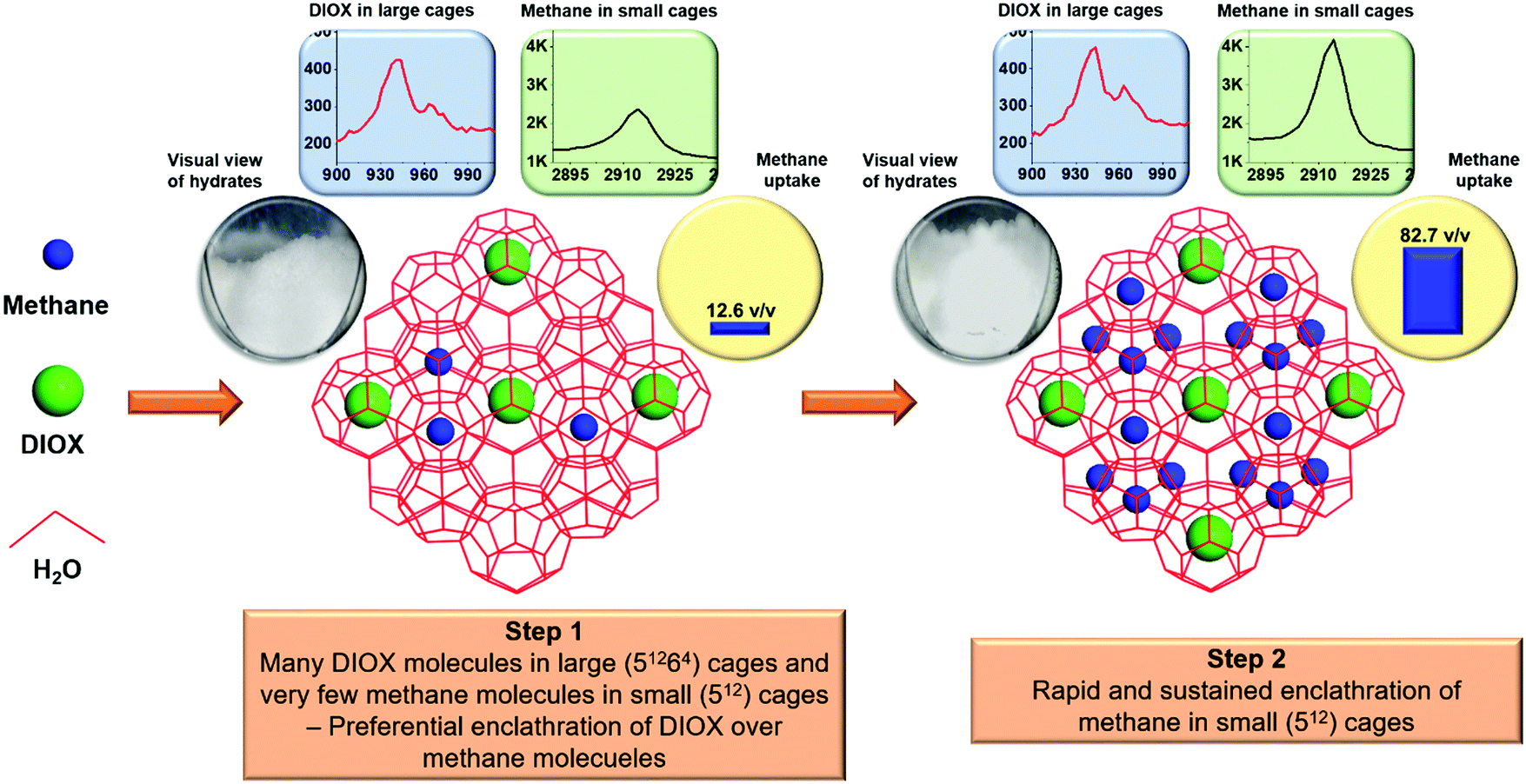 | ||
| Fig. 4 Two-step hydrate growth mechanism for mixed methane–DIOX sII hydrate formation comprising a preferential DIOX over methane enclathration step (Step 1) and a subsequent rapid and sustained methane enclathration step (Step 2). | ||
Ultra-rapid mixed methane–DIOX hydrate growth
We further examined the DIOX/water system to form sII hydrates at ultra-rapid rates. To obtain ultra-rapid rates of hydrate formation, we explored the use of small concentrations of bio-friendly amino acids as kinetic promoters. Recently, amino acids have exhibited potential application to kinetically promote gas hydrate growth formation owing to the peculiar morphology pattern for crystal formation.38 Thus, we studied the possibility of enhancing the kinetics of quiescent hydrate growth for the methane–DIOX system by introducing the hydrophobic amino acid and benign kinetic promoter L-tryptophan into the mixture.Fig. 5a represents the gas uptake (quiescent hydrate growth) obtained for a methane–DIOX/water system containing 300 ppm L-tryptophan and its comparison with the gas uptake (quiescent hydrate growth) for the standard system in the present study, i.e. “the methane–DIOX/water system without any additional kinetic promoter”. As observed, the presence of L-tryptophan greatly boosted the hydrate formation kinetics by inducing ultra-rapid hydrate growth even under quiescent operation. Fig. 5b presents a comparison of the t90 and methane uptake rate between the standard case and the solution with 300 ppm L-tryptophan. In the presence of L-tryptophan, mixed methane–DIOX hydrate formation reached 90% completion (t90) in 12.11 (±0.79) min after nucleation (also see Table S5 in the ESI†), which is faster by a factor of 2.7 times compared to the standard system without any L-tryptophan. The normalized gas uptake rate comparison for the t90 periods between the two systems, also presented in Fig. 5b, indicates a 147% increase in the hydrate formation rate for the DIOX/water/L-tryptophan mixture compared to the DIOX/water standard system.
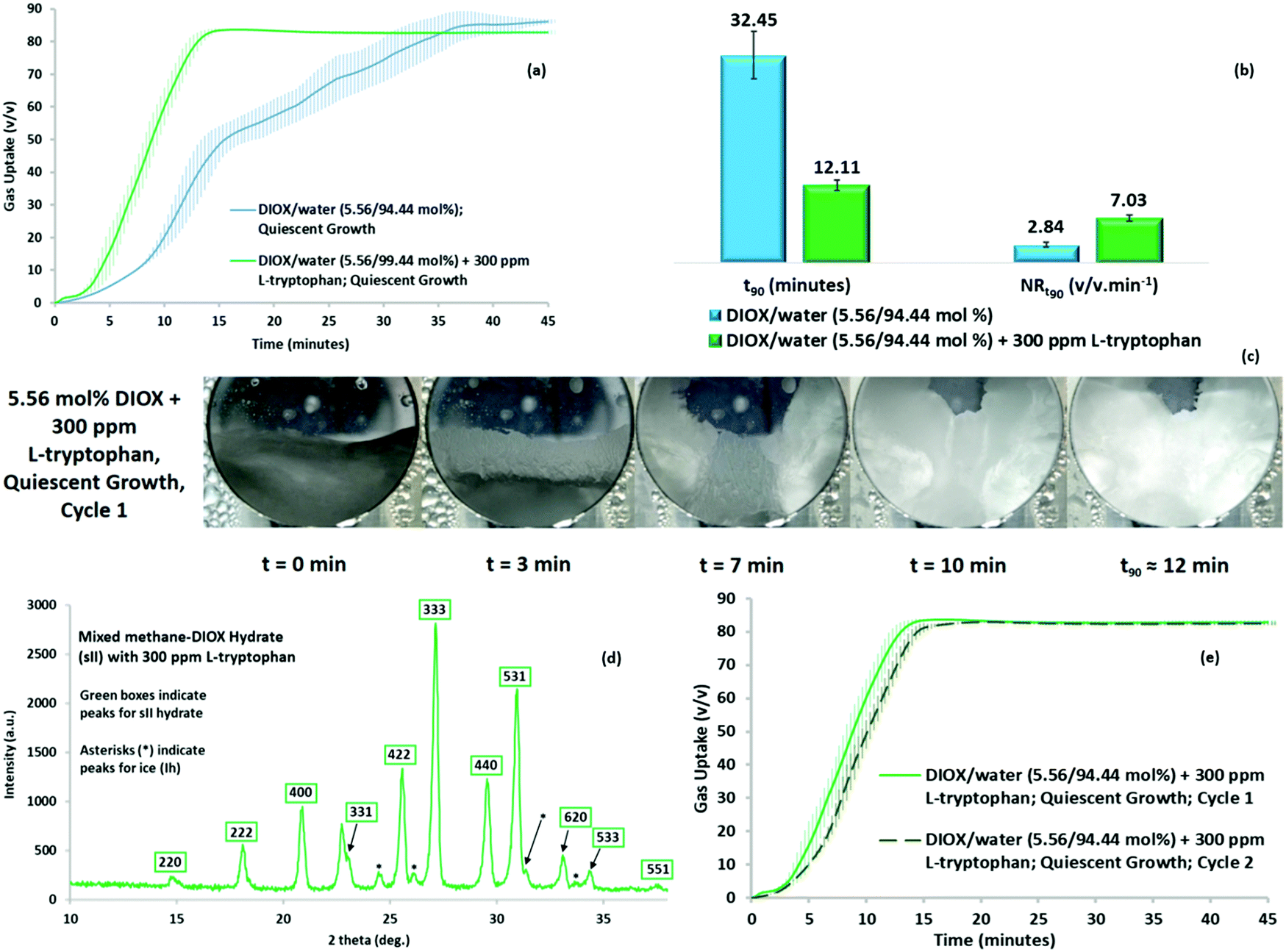 | ||
| Fig. 5 (a) Mixed methane–DIOX hydrate growth under quiescent condition without L-tryptophan (in blue) and with 300 ppm of L-tryptophan (in green) at 283.15 K and initial pressure of 7.2 MPa, where the continuous lines represent the average of three experiments and the vertical shaded regions represent the standard deviation of three experimental data sets. (b) Comparison of the t90 and normalized gas uptake rate for the t90 periods (average and standard deviation of three experiments) for the methane–DIOX/water standard system and methane/DIOX/water system in the presence of 300 ppm L-tryptophan. (c) Visual observations for mixed methane–DIOX hydrate formation in the presence of 300 ppm L-tryptophan under quiescent hydrate growth condition. (d) p-XRD pattern of methane–DIOX mixed hydrate sample synthesized using 5.56 mol% DIOX aqueous solution at 283.15 K and an initial pressure of 7.2 MPa in the presence of 300 ppm of L-tryptophan, where the peaks corresponding to the sII hydrate are inside green boxes, while the asterisks indicate the presence of ice (Ih). (e) Mixed methane–DIOX hydrate growth under quiescent condition for cycle 1-fresh (in solid light green) and cycle 2-repeat (in dashed dark green) runs using DIOX/water/L-tryptophan (300 ppm) solution at 283.15 K and an initial pressure of 7.2 MPa, where the continuous lines (solid or dashed) represent the average of three experiments and the vertical shaded regions represent the standard deviation of three experimental data sets. | ||
Upon the introduction of L-tryptophan, the morphology observed during quiescent growth (Fig. 5c) indicates a porous and flexible hydrate structure; this is the distinctive crystal morphology characteristic to which we attribute the excellent kinetic promotion observed for mixed methane–DIOX hydrate formation when L-tryptophan was added to the system. The images of the system at 7 and 10 min post-nucleation show a hydrate structure that appears strongly consolidated near the three-phase interface points (reactor walls) and loosely stacked towards the centre of the reactor. This is indicative of a mechanism known as “capillary suction”, which is unique to porous hydrates, wherein underlying water or solution from the aqueous phase is drawn through channels present in the hydrate microstructure towards the top of the hydrate layer, thus facilitating further hydrate growth. The propagation of this growth in the present case clearly occurred from the three-phase (solid–liquid–gas) interface points, i.e. reactor walls, towards the centre of the reactor. Two videos are presented in the ESI† (Videos SV3 and SV4) to illustrate the ultra-rapid hydrate growth in the presence of L-tryptophan under the quiescent condition. As can be seen in the videos, the hydrates were formed extremely fast and exhibited a porous and flexible nature. The aforementioned capillary suction mechanism manifested as the underlying water or solution migrating to the top of the hydrate layer, facilitating further gas–water contact and ensuring fast, sustained hydrate formation, can actually be observed in Videos SV3 and SV4 (ESI†). Although the underlying unreacted water or solution is transported through the channels present in the hydrate microstructure towards the top of the hydrate layer, the overlying gas may use the same channels to move into the already formed hydrate structure. This would further enhance the rate of gas uptake, and consequently, enhance the overall kinetics of hydrate formation. Thus, the kinetic promotion activity of L-tryptophan is mainly based on its ability to enable the easy migration of both gas and water/solution through the system via the formation of a distinctive porous and flexible hydrate crystal structure. It should be noted that the porous hydrate crystal structure observed for the system containing the amino acid L-tryptophan in the present study is a classical signature of amino acid kinetic promotion.38
Although the presence of 300 ppm L-tryptophan resulted in ultra-rapid mixed methane–DIOX hydrate formation, the final gas uptake achieved exhibited a minute drop of about 3.7% (see Fig. 5a). The slight dip observed can be attributed to the mass transfer resistance to methane induced by the ultra-rapid rate of hydrate formation in the presence of L-tryptophan, which is also corroborated by the huge mass of hydrates observed in the morphology videos (Videos SV3 and SV4, ESI†). Subsequently, p-XRD characterization was performed for the hydrates formed using the methane–DIOX/water/L-tryptophan system, with the typical sII hydrate pattern observed again. This confirms the fact that even in the presence of the kinetic promoter L-tryptophan, only sII mixed methane–DIOX hydrates were formed under the experimental conditions (283.15 K temperature and initial pressure of 7.2 MPa). The representative XRD pattern is shown as Fig. 5d. Further augmenting the findings from the p-XRD characterization are the results from the in situ Raman spectroscopy experiment carried out for hydrate formation using the methane–DIOX/water/L-tryptophan system, keeping the experimental temperature and pressure conditions constant. The details of this experiment together with the time-dependent in situ Raman spectra obtained at regular intervals during the hydrate growth process are included in the ESI,† Fig. S7, and its associated discussion. According to Fig. S7 (ESI†), it can be further confirmed that the presence of L-tryptophan in the system did not change the hydrate structure formed (mixed methane–DIOX (sII) hydrates in the present case), but rather only provides pure kinetic enhancement to the process.
The recyclability of the DIOX/water/L-tryptophan solution for mixed methane hydrate formation is presented in Fig. 5e. Under quiescent operation, ultra-rapid hydrate growth and a predictable pattern were observed for both the fresh (cycle 1) and repeat (cycle 2) solution states. The t90 for the repeat runs of the DIOX/water/L-tryptophan system is 13.44 (±0.42) min post-nucleation, with a gas uptake at t90 of 75.11 (±0.48) v/v. In comparison, for the fresh runs, the t90 and gas uptake at t90 are 12.11 (±0.79) min post-nucleation and 75.43 (±0.69) v/v, respectively. The final gas uptake achieved for cycles 1 and 2 using the DIOX/water/L-tryptophan solution is also similar, i.e. 83.81 (±0.77) v/v for the fresh runs and 83.46 (±0.53) v/v for the repeat runs. The closeness in the kinetic data obtained for the fresh and repeat cycles using the DIOX/water/L-tryptophan system firmly establishes its exceptional recyclability potential. This should a vital contributor towards the successful scale-up of the technology under consideration currently since recycling the solution implies considerable economic conservation. The individual kinetic performance parameters for the repeat runs of the DIOX/water/L-tryptophan system are provided in Table S6 in the ESI.† With regards to the hydrate growth pattern of the guest molecules, the methane–DIOX system with L-tryptophan also appears to follow the two-step mechanism presented for the methane–DIOX system. This can be clearly seen from the initial slow methane uptake rates in Fig. 5e (<5 min), while Videos SV3 and SV4 (ESI†) reveal the presence of a considerable amount of hydrates at 5 min post-nucleation, following which an ultra-rapid and sustained methane uptake can be observed. The majority of the methane uptake (>90%) into the hydrate phase evidently occurs after 5 min of hydrate growth. Moreover, the evolution trends in the time-dependent in situ Raman spectra for the methane–DIOX/water/L-tryptophan system presented in Fig. S7 (ESI†) show strong similarity to that observed for the in situ Raman spectroscopy runs conducted for the methane–DIOX/water systems (Fig. S5 and S6, ESI†). It can be observed in Fig. S7 (ESI†) that while the peak intensity for methane trapped in the small cages of the sII hydrates increases significantly right from the start of hydrate growth up to the 15 min upper limit hydrate growth mark shown in this figure, the various peak intensities for the DIOX trapped in the large cages of the sII hydrates do not exhibit a significant increase over the same period. This indicates that the majority of the DIOX enclathration into the hydrate phase occurred during an initial short hydrate growth period immediately following hydrate nucleation, whereas methane enclathration into the hydrate phase occurred throughout the hydrate growth process. Combining the gas uptake trends obtained with the evolution of the time-dependent in situ Raman spectra observed, it can be concluded for hydrate formation from the methane–DIOX/water/L-tryptophan system that, similar to the methane–DIOX/water system, hydrate formation follows the distinct two-step hydrate growth mechanism. This includes, (a) a preferential DIOX over methane enclathration step (Step 1) immediately after hydrate nucleation, where the majority of the large cages of the formed sII hydrates get filled with DIOX, and only a small amount of methane molecules get enclathrated into the hydrate phase during this period, occupying a few of the small cages of the hydrates, and (b) a rapid and sustained methane enclathration step (Step 2), in which the majority of methane uptake in the small cages of the formed sII hydrates occurs.
We also tested the recyclability of the DIOX/water/L-tryptophan system for multiple hydrate formation cycles, where two sets of experiments were conducted, demonstrating 10 (1C1 to 1C10) and 7 (2C1 to 2C7) hydrate formation cycles. The results revealed that the gas uptake at the end of 45 min of hydrate growth remained practically the same for these systems, even after undergoing multiple cycles of hydrate formation. For cycles 1C1 to 1C10, the average gas uptake achieved at the end of 45 min of hydrate growth was 84.06 (±0.95) v/v, whereas that for cycles 2C1 to 2C7 was 84.02 (±0.65) v/v. These findings demonstrate the strong recyclability characteristics of the DIOX/water/L-tryptophan system. The individual kinetic performance parameters of the two sets of multiple cycle experiments (1C1 to 1C10 and 2C1 to 2C7) performed for the DIOX/water/L-tryptophan system are presented in the ESI,† Table S7.
Two supplemental studies were conducted to culminate the investigation on mixed methane–DIOX hydrate formation in the presence of L-tryptophan. In the first study, the effect of L-tryptophan concentration on the mixed methane–DIOX hydrate formation kinetics was considered, and it was found that 300 ppm is the optimum L-tryptophan concentration for the current experimental investigation. The results and relevant discussion are presented in the ESI† (Fig. S8 and Table S8). The second supplemental study involved the comparison of the kinetic performance parameters of mixed methane–DIOX hydrate formation and mixed methane–THF hydrate formation, each in the presence of 300 ppm L-tryptophan. The overall conclusion from this study is that considering that the initial driving force for hydrate formation is kept constant, the methane–DIOX/water/L-tryptophan system kinetically outperforms the methane–THF/water/L-tryptophan system, with a significant advantage obtained in the t90 period for the system containing DIOX and L-tryptophan. The results and relevant discussion are presented in the ESI† (Fig. S9 and Table S9).
Highly stable storage characteristics
The stability of the hydrates formed for SNG technology is an extremely important characteristic that needs to be established. SNG formed via sII hydrates can be readily stored at atmospheric pressure and at conventional freezer temperatures of about 271.15 to 268.15 K.18 The thermodynamic stability of pure sI hydrates at atmospheric pressure is 193 K.12 Thus, sII mixed methane hydrates formed with DIOX or THF do not have to rely on the self-preservation effect to remain stable, which occurs at 253.15 K at atmospheric pressure for sI hydrates.12We investigated the stability of mixed methane–DIOX hydrate pellets stored at atmospheric pressure and a moderate storage temperature of 268.15 K. Cylindrical mixed methane–DIOX hydrate pellets in the presence of L-tryptophan were synthesized using a unique, custom-designed bench-scale SNG technology prototype available in our lab. The details of the apparatus used, and the procedure followed for the synthesis of these pellets are provided in the ESI.† Specifically, a pellet having a length of 5.4 cm (Fig. 6a), diameter of 5.0 cm (Fig. 6b), and weight of 91.57 g (Fig. 6c) was produced and stored in a separate storage vessel, maintained at atmospheric pressure and storage temperature of 268.15 K, and monitored to demonstrate the stability of the mixed methane–DIOX hydrates. We observed a volumetric gas uptake of 82.47 v/v for the pellet based on the gas uptake analysis at the end of hydrate formation.
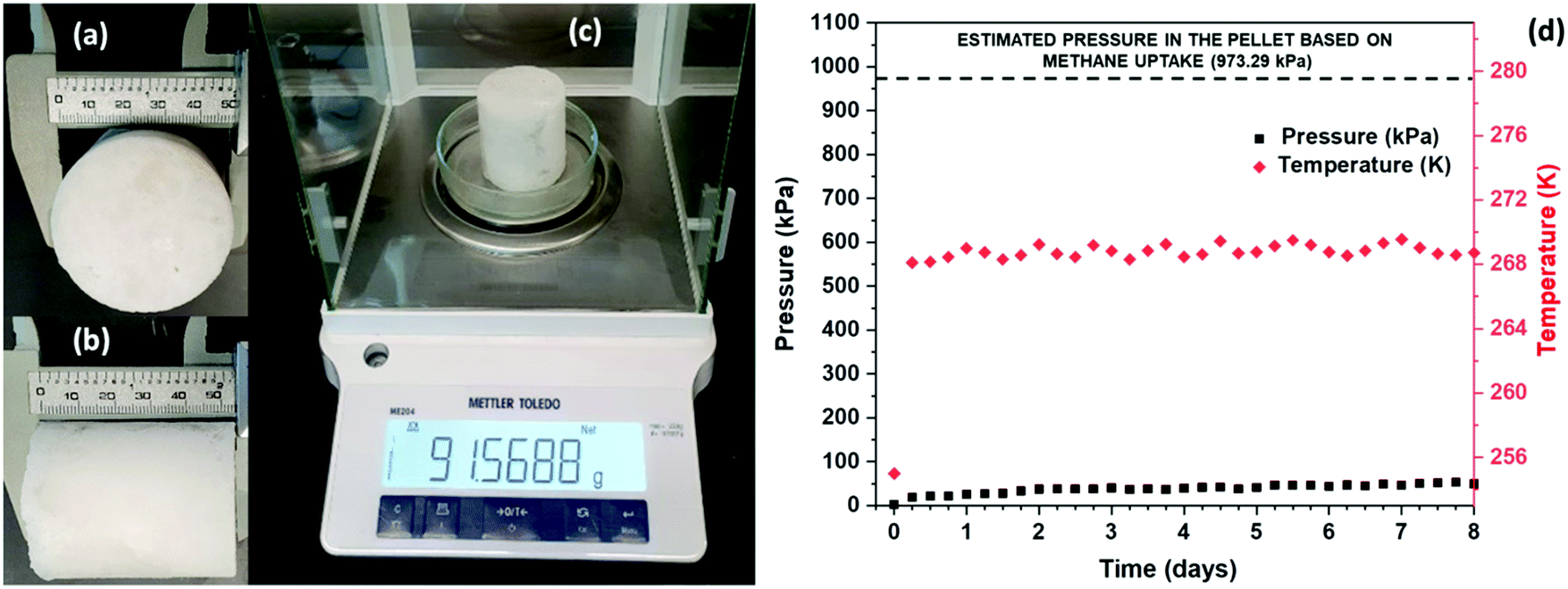 | ||
| Fig. 6 (a) Diameter, (b) length and (c) weight of the synthesized mixed methane–DIOX hydrate pellet. (d) Demonstration of the stable storage of the mixed methane–DIOX hydrate pellet at atmospheric pressure and moderate temperature of 268.15 K. | ||
If a hydrate pellet was unstable under the aforementioned storage pressure and temperature conditions, it would dissociate, thus releasing the trapped gas into the storage vessel, which would consequently lead to an increase in the pressure inside the vessel. The amount of gas in the stored hydrate pellet was 82.47 v/v, which would translate to a pressure increase in the storage vessel of 973.29 kPa on complete dissociation. Conversely, no or a gradual pressure increase in the storage vessel would indicate good stability of the hydrate under the experimental storage conditions. Fig. 6d shows the stability of the mixed methane–DIOX hydrate pellet monitored over a period of eight days. At the first data point (beginning of the storage period), the temperature of the storage vessel was recorded as 255 K, while the pressure inside the storage vessel was 3.25 kPa (gauge pressure). The initial low temperature in the vessel is due to the fact that the storage vessel was pre-cooled at 253.15 K before transferring the produced hydrate pellet into it and subjecting the vessel containing the pellet to the pre-determined storage conditions (vessel closed at atmospheric pressure and kept inside a freezer maintained at the storage temperature). Quickly the temperature inside the storage vessel reached the desired experimental storage temperature of 268.15 K, and we also observed a slight increase in the pressure inside the storage vessel at this point (19.22 kPa; second data point in Fig. 6d depicting the hydrate storage period), which can be predominantly attributed to residual gas expansion due to the temperature increase rather than hydrate dissociation. From here on, both the temperature and pressure inside the storage vessel remained largely constant until the end of the 8 day hydrate storage period, thus signifying that the hydrate pellet was highly stable for the tested period. For the last five days of storage, the pressure in the storage vessel was extremely stable at 44.87 (±5.09) kPa. This demonstrates the exceptional stability of the mixed methane–DIOX hydrate pellet. It should be noted that the dotted pressure line at the top of Fig. 6d represents the expected pressure if all the gas stored in the pellet dissociates and evolves into the storage vessel.
Thus, for the first time, we demonstrate the highly stable storage of mixed methane–DIOX hydrate pellets at atmospheric pressure. Through the identification of DIOX as a dual-action promoter and synergistically combining it with a small amount of L-tryptophan, we addressed both the bottlenecks pertaining to SNG technology by (a) ensuring ultra-rapid hydrate formation under moderate pressure and temperature conditions and (b) ensuring highly stable storage of formed hydrates under moderate pressure and temperature conditions.
Comparison with competing technologies
With regards to the conventional natural gas storage methods, CNG (compressed natural gas) is a commercially employed technology. However, its high pressure requirement and safety aspects make it economically unfeasible for large-scale storage. On the other hand, ANG (adsorbed natural gas)-based technologies have been making constant progress, albeit on the material scale mostly for on-board storage application. Although inexpensive adsorbent materials such as activated carbon are available, the performance of activated carbon is not as good as promising materials such as metal–organic frameworks (MOFs) and other polymeric materials. In the context of on-board application, at the material scale, a recent breakthrough work by Rozyyev et al.5 reported 0.625 g g−1 gas uptake for their best performing material COP-150. In the same study, Rozyyev et al.5 reported the larger scale testing of methane storage using a 142 mL stainless steel pressure vessel (similar volume to that used in the present study, refer to Methods) maintained at 293.15 K and filled with 30 g (88.24 mL) of COP-150 material. The authors report a gravimetric gas uptake of 0.137 g g−1 at the working pressure of 6.5 MPa (methane), which is the closest operating point to 7.2 MPa, the operating pressure used in the present study. Using the correlations provided by the authors, the volumetric gas uptake capacity of COP-150 was estimated to be around 64.20 v/v. The authors also reported that COP-150 outperformed other popular porous materials such as HKUST-1 and polystyrene in their scale-up study. In our work, a volumetric gas storage capacity of 83.81 (±0.77) v/v was achieved for the methane–DIOX/water/L-tryptophan system on a similar scale. Table S10 in the ESI† presents the average volumetric and gravimetric (g of gas/g of hydrate) gas uptake (methane storage capacities) obtained after 45 min of hydrate growth for the various systems studied presently for mixed methane (sII) hydrate formation. It should be noted that because gas hydrates are made up mainly of water or in the present study, water and the dual-function promoter DIOX or THF, the gravimetric methane storage capacity achieved for gas hydrate formation is generally low. The theoretical maximum gravimetric gas storage capacity achievable for mixed methane (sII) hydrate formation assuming full occupancy of the large cages by the dual-function promoter, in other words, assuming a dual-function promoter is used at a stoichiometric concentration, is 0.078 g g−1. For a large-scale gas storage and transportation system at an industrial scale, volumetric storage is an important aspect, for example LNG and crude oil storage tanks.Clathrate hydrates can be synthesized as three structures, namely sI, sII and sH. Methane by itself forms sI hydrates, while in order to form sII and sH hydrates, methane requires a thermodynamic promoter (or co-guest). In our work, we illustrated DIOX as a co-guest for mixed methane (sII) hydrate formation. According to the knowledge that the only hydrate structure to exclusively house methane is sI, it becomes obvious that the storage capacity for sI hydrates is the highest, followed by sII and sH hydrates, respectively. A detailed comparison of the storage capacity (volumetric) and storage temperature at 1 atmosphere (atm) pressure for the three hydrate structures with methane is presented in Table S11 in the ESI.† As seen in this table, the key advantage of methane storage as sII hydrates is the requirement of much milder conditions for these hydrates to remain stable and non-reliance on the self-preservation effect, as demonstrated in our mixed methane-DIOX hydrate stability test. Moreover, it is also possible to tune the methane uptake in the large cages of sII hydrates through innovation in experiment process design and optimization of the promoter concentration,16 which can lead to an increase in the gas storage capacity offered by this particular hydrate structure.
Similar to CNG, adsorption-based large-scale gas storage ANG systems will need to be kept at high pressures. This is the great advantage of SNG, where SNG formed with sII hydrates is extremely stable at atmospheric pressure and moderate temperature. For the first time, we demonstrated the exceptionally stable storage of mixed methane–DIOX SNG pellets at atmospheric pressure in the present work. From an economic viewpoint, the cost of a storage vessel made of stainless steel for methane storage as ANG or CNG (pressure rating of 7.5 MPa) works out to be in excess of 5.5 times the cost of a vessel for SNG storage as sII mixed methane hydrate (pressure rating of 1.0 MPa) having identical volume, dimensions, orientation and materials.39 These details are included in the ESI† (Table S12). The practical use of large-scale high-pressure storage tanks is not advisable due to the explosive nature of these systems. The scale-out (such as CNG for laboratory use) approach will result in an increase in the cost for both CNG and ANG for large-scale operation. On the other hand, there are industry standards for large-scale storage tanks that can be readily adopted for SNG, for example, liquefied natural gas (stored at 0.2–0.5 MPa and 111.2 K) storage tanks are designed for a pressure of 1.0 MPa. Another significant advantage of SNG technology compared to ANG is that the former mainly uses water as the solvent (>94 mol%) with the addition of two small quantities of thermodynamic and kinetic promoters. Even if industrial grade water is produced from seawater by desalination, its cost is less than USD$1.13 per tonne (or per m3),40 making the economics of SNG technology highly feasible considering the raw materials.
Thus, in addition to the ultra-rapid formation of SNG via sII methane–DIOX hydrates, the low cost of its storage tank, its high degree of stability under mild storage conditions and safety firmly cement SNG technology as an efficient option for large-scale methane storage.
Conclusion
We presented DIOX as a clean and competent dual-action chemical promoter for SNG technology via combustible ice or clathrate hydrates, providing both thermodynamic and kinetic enhancement to the process. Rapid mixed methane–DIOX hydrate formation was achieved under the experimental conditions of 7.2 MPa and 283.15 K. We highlighted the existence of a synergistic effect between DIOX and methane and proposed a two-step hydrate growth mechanism for the methane–DIOX hydrate system, wherein the introduction of hydrophobic methane into the system catalyzes the initial formation of sII hydrate with initial preferential incorporation of DIOX, followed by rapid incorporation of methane into the molecular water (host) framework. Further, by synergistically combining a small concentration (300 ppm) of kinetic promoter, L-tryptophan, we established ultra-rapid rate of hydrate formation using the methane–DIOX/water system, with complete hydrate formation being achieved within 15 min following hydrate nucleation, together with a high methane uptake of 83.81 (±0.77) volume of gas/volume of hydrate (v/v). We demonstrated the recyclability of this system in 10 cycles, showcasing its consistent kinetic performance and reproducibility. Finally, we demonstrated the extraordinary stability of a mixed methane–DIOX hydrate pellet synthesized using a bench-scale SNG technology prototype and stored at atmospheric pressure and in a conventional freezer at 268.15 K. Our current findings on mixed methane–DIOX sII hydrate formation address and overcome the two major bottlenecks of SNG technology and present exciting prospects for its application in industry.Experimental
Materials
Methane gas (99.995%) was purchased from Air Liquide Singapore Pte Ltd. Anhydrous 1,3-dioxolane (DIOX) (99.75% containing 75 ppm of BHT (butylated hydroxytoluene)) and reagent grade L-tryptophan (98%) were purchased from Sigma Aldrich, and tetrahydrofuran (THF, 99.7%) was purchased from VWR Chemicals. Ultrapure deionized water (Merck Millipore) was used for all experiments.Methods
Phase equilibrium experiments
An isothermal pressure search method was used for the phase equilibrium experiments conducted in the present study. Once the internal temperature of the reactor reached the desired set point, the reactor was pressurized with pure methane gas to approximately 2.5 MPa above the expected equilibrium point and the system was left untouched for 15 min to allow the reactor temperature to stabilize. At the end of the 15 min stabilization period, stirring at 400 rpm was initiated to induce hydrate formation. Once nucleation was observed through the viewing window, the stirring action was stopped, and excess methane gas was vented from the reactor. Dissociation of the hydrates as a result of depressurization was allowed to occur until no visible crystals remained. Close observation of the morphological changes of the contents inside the reactor through the viewing window during the hydrate dissociation phase allowed a rough estimation of the equilibrium hydrate pressure at the predetermined experimental temperature. Once complete dissociation of the hydrates was accomplished, the reactor was re-pressurized with pure methane gas up to approximately 2–2.5 MPa above the expected equilibrium point and stirring was initiated to induce hydrate formation again. When nucleation was observed, stirring was stopped and the reactor was depressurised to the estimated equilibrium pressure. Thereafter, the system was left to reach a state of equilibrium such that an infinitesimally small amount of hydrate crystals remained stable for a sufficiently long period; typically, any period upwards of 5 h. If hydrate crystals were not observed to persist for a sufficiently long period, i.e. complete hydrate dissociation was observed to have taken place within 5 h, the reactor was pressurized once again to reform the hydrates, and subsequently depressurized, this time to a pressure higher than the earlier attempts. If hydrate crystals were observed to persist for a sufficiently long period, the reactor temperature and pressure were noted and the reactor was depressurized slightly, by 20–50 kPa, and allowed to equilibrate for at least another 5 h. If hydrates continued to persist, the above step was repeated. Otherwise, the last recorded reactor temperature and pressure where infinitesimally small amounts of hydrates were observed to persist for a sufficiently long period were noted to be the equilibrium temperature and pressure, respectively. Since the temperature of the system was regulated using an external chiller, the final recorded reactor temperature, i.e. the final equilibrium temperature, was always very close to the predetermined experimental temperature. However, there was always some deviation in the final temperature recorded owing to experimental error and the inherently endothermic nature of hydrate dissociation. Considering the fact that DIOX was investigated as an alternative to THF as a dual-action (thermodynamic and kinetic) promoter for SNG hydrate formation, the thermodynamic phase equilibrium data for the methane–DIOX/water (5.56/94.44 mol%) system was estimated only at three fixed temperatures of 283.15 K, 288.15 K and 293.15 K (as already mentioned).Hydrate formation under quiescent (unstirred) growth condition
After the internal temperature of the reactor reached the desired set value, the reactor was pressurized with pure methane gas to a predetermined experimental pressure over a period of 3–5 min to ensure that the reactor temperature remained stable. The reactor was then sealed tightly and no further gas injection to the reactor was allowed to take place during the experiment, thus essentially making the operation a batch experiment. For both the DIOX- and THF-based experiments, the predetermined set temperature was 283.15 K, whereas the predetermined initial experimental pressures were 7.2 MPa and 6.7 MPa, respectively, thus ensuring an even initial driving force of ∼6.2 MPa throughout the study. Subsequent to gas injection, the system was left undisturbed for 15 min to allow the temperature inside the system to stabilize. At the end of the 15 min stabilization period, stirring at 400 rpm was initiated to induce hydrate formation. At the first instance of observable nucleation (simultaneous indicators including visual observation using the viewing window and characteristic exothermic temperature spike), stirring was stopped and the stirring time was recorded. In the present study, the stirring time taken to induce nucleation was defined as the induction time. Thereafter, the entire hydrate growth phase proceeded in an unstirred (quiescent) manner. This is known as the hybrid combinatorial reactor (HCR) approach for hydrate formation, which offers the benefit of eliminating the stochasticity of hydrate nucleation, while maintaining the superior kinetics of hydrate formation at minimal energy expenditure owing to the significantly reduced stirring time. The HCR approach was discussed in great detail in our previously reported studies.30,41 For the experiments involving THF/water and THF/water/L-tryptophan solutions, the 15 min equilibration time was not part of the experimental procedure since prior knowledge of this system indicated that hydrates would nucleate during the equilibration period.17 Hence, utmost care was taken to ensure that the temperature inside the system remained stable and as close as possible to the desired experimental temperature during gas pressurization. Once the desired experimental pressure was reached, stirring at 400 rpm was immediately initiated to induce hydrate formation and then stopped at the first instance of observable nucleation. Subsequently, the entire hydrate growth occurred in the unstirred (quiescent) mode, thus cumulatively following the HCR approach for hydrate formation.Pressure and temperature data were recorded over the course of hydrate formation using a data acquisition system. The pressure drop inside the system was used to calculate the amount of gas consumed due to hydrate formation. As hydrate formation proceeded, the pressure inside the system dropped as more and more gas was incorporated into the solid hydrate phase. Since the experiments were carried out in batch mode, i.e. the system pressure was not replenished at any point during the experiments, hydrate formation ceased when there was not sufficient driving force in the system to sustain the process. Once hydrate formation was completed, the excess gas in the system was vented over a period of 3–5 min and an external chiller was used to regulate the reactor temperature back up to 298.15 K (ambient temperature) to facilitate hydrate dissociation. The reactor pressure and temperature were likewise recorded during hydrate dissociation, which occurred over a period of 1.5–2 h. This concluded the first cycle of formation and dissociation, and for any solution used, was referred to as the fresh run. When a solution previously used for a hydrate formation-dissociation cycle was used in a subsequent cycle, the second run for the same solution was referred to as the repeat run.
Hydrate formation under stirred growth condition
The only difference between the stirred and quiescent hydrate growth conditions employed in the present study was that in the case of stirred hydrate growth, stirring at 400 rpm was allowed to continue throughout the duration of the experiment compared to being turned off at the onset of hydrate nucleation in the case of the HCR approach for quiescent hydrate growth. On completion of hydrate formation, stirring was switched off, the excess gas present in the reactor was vented and hydrate dissociation was allowed to occur in the same manner, as explained in the previous section.Stability test of mixed methane–DIOX hydrates
Herein, the mixed methane–DIOX hydrate formation experiments were conducted in the SNG technology prototype to obtain a compact hydrate pellet, which was then stored to demonstrate the stability of the mixed methane–DIOX (sII) hydrates. Accordingly, 100 mL of a 5.56 mol% DIOX aqueous solution together with 1000 ppm L-tryptophan was prepared and introduced into the hydrate formation zone of the SNG prototype using the solution entry port. This corresponded to target masses of 81.38 g and 19.74 g for water and DIOX, respectively, while L-tryptophan was weighed relative to the total weight of liquid used. We observed at the 32.4 mL solution scale that the kinetic promotion provided by the presence of L-tryptophan when used in a concentration of up to 1000 ppm did not make a significant difference to the overall gas uptake. The hydrate formation experiments were conducted at a temperature of 283.15 K and initial pressure of 7.2 MPa, i.e. the hydrate formation zone was operated under these temperature and pressure conditions. The experimental temperature and pressure were so chosen as it would be virtually impossible to form pure methane (sI) hydrates under these conditions (refer to Fig. 1). Prior to the pelletization process, the hydrate pelletization zone was cooled to the storage temperature of 268.15 K. Once hydrate formation was completed and the hydrate pelletization zone had cooled to the desired hydrate storage temperature, the excess gas inside the hydrate formation zone was vented and the ball valve separating the two zones was opened. The piston present then extruded the formed hydrate particles into the hydrate pelletization zone followed by compacting them into a solid hydrate pellet, as described in the previous paragraph. The hydrate pelletization zone was then opened and the compacted hydrate pellet was pushed and collected into a pre-cooled storage vessel. The storage vessel having a total volume of 1029.58 mL was then transferred to a conventional freezer maintained at a storage temperature of 268.15 K, where the storage vessel containing the compacted hydrate pellet was left untouched so as to study the stability of the mixed methane–DIOX hydrate pellet formed. The storage vessel was equipped with a pressure transducer, WIKA A-10, and thermocouple, Omega T type, connected to a data logger, which were used to record the pressure and temperature inside the storage vessel at 20 s intervals throughout the hydrate pellet storage period, respectively. The pressure increase inside the storage vessel was representative of hydrate pellet dissociation, which involves the release of free gas into the storage vessel. For the current work, we reported the stability data for the mixed methane–DIOX hydrate pellet for a storage period of 8 days.
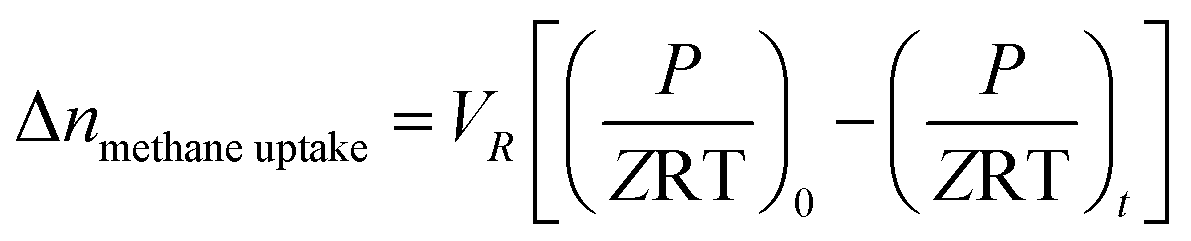 | (1) |
uptake is the number of moles of methane participating in hydrate formation at any given time “t” during the experiment and nwater is the total number of moles of water used for hydrate formation.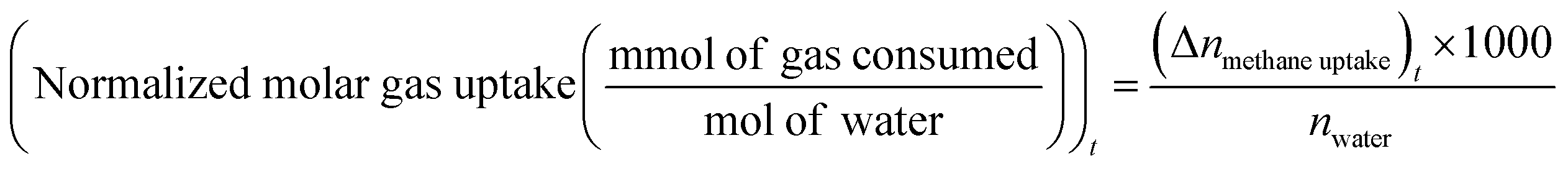 | (2) |
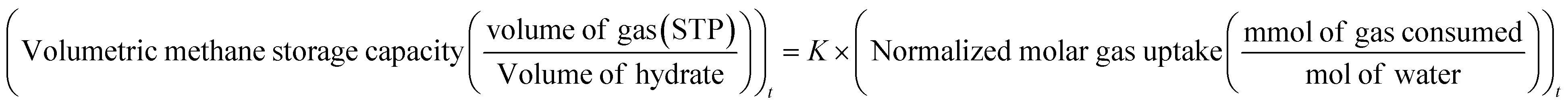 | (3) |
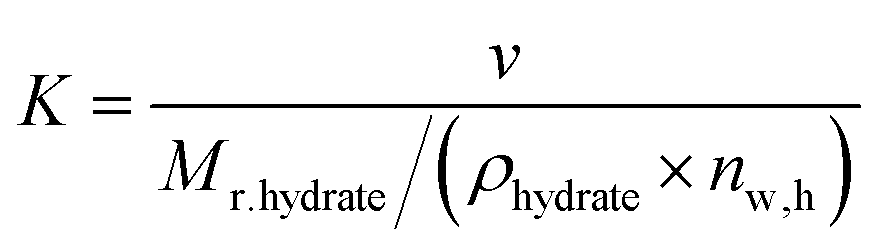 | (4) |
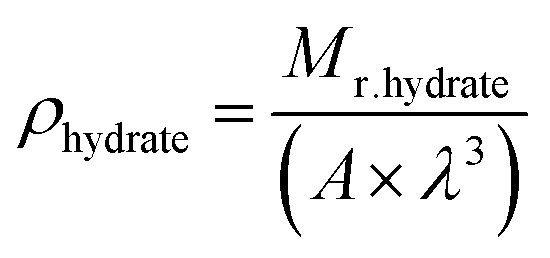 | (5) |
| Mr.hydrate = (136 × 18) + (8 × 74.08) + (16 × 16), |
| ∴ρhydrate = 1.057 (g cm−3) |
| Normalized gas uptake rate, NRt = Rt × K (v v−1 h−1) | (6) |
Procedure for powder X-ray diffraction (p-XRD) analysis
Powder X-ray diffraction (p-XRD) characterization was performed to obtain information about the crystal structure of the resulting mixed methane–DIOX hydrate. The p-XRD measurements were carried out using a BRUKER D8 Advance diffractometer (40 kV, 30 mA) capable of analysing the hydrate samples at atmospheric pressure and low temperature conditions.18 Mixed methane–DIOX hydrates were synthesised using a high-pressure reactor, as discussed earlier. After hydrate formation (as indicated by the pressure no longer decreasing), the excess gas in the reactor was quickly vented, the reactor was opened, and the formed hydrates were instantaneously quenched using liquid nitrogen. This allowed easy recovery of the hydrates, while ensuring that they remained stable at atmospheric pressure conditions. The hydrate samples were then ground to a uniform powder using a mortar and pestle in a liquid nitrogen environment and quickly transferred to the p-XRD unit for subsequent analysis. Each p-XRD pattern was collected with a total 2θ scan time of 1.5 min in the 2θ range of 10–40° at a step size of 0.02° and rate of 0.055° s−1 using CuKα radiation (λ = 1.542 Å).44,45 The obtained p-XRD patterns were then compared with the standard hydrate patterns available in the literature to realize the type of hydrate structure formed using the methane–water/DIOX system.Apparatus used and procedure for in situ Raman analysis
The details and representative schematic of the equipment used for the in situ Raman spectroscopy measurements can be found in the literature.18,46 Briefly, the apparatus consisted of a ∼226 mL high-pressure jacketed crystallizer connected to an external refrigerator, which maintained the correct experimental temperature throughout the trial. Highly sensitive pressure transducer (PT) and thermocouple mounts were employed to monitor the pressure and temperature inside the crystallizer at all times, respectively. The PT and thermocouple were connected to a data acquisition (DAQ) system, which recorded the requisite data at 20 s intervals. The high pressure crystallizer was also coupled with a Raman probe, which was responsible for providing the in situ Raman spectrum. A dispersive laser Raman spectrometer was used for real-time Raman characterization and analysis of the mixed methane–DIOX hydrate formation. Specific details of the spectrometer used can also be found in the literature.18,46The in situ Raman spectroscopy experiments were conducted in fully stirred operation (600 rpm) at an experimental temperature of 283.15 K and initial pressure of 7.2 MPa (similar to the experimental conditions used for the gas uptake experiments). Also, the same as the gas uptake experiments, a solution volume of 32.4 mL comprising target masses of 26.37 g and 6.39 g for water and DIOX, respectively, was loaded into the crystallizer for in situ Raman spectroscopy measurements. For mixed methane–DIOX hydrate formation in the presence of L-tryptophan, the solution additionally contained 300 ppm L-tryptophan as a kinetic promoter, which was calculated relative to the total weight of the liquid used to make up the solution. Once the solution loading was complete, the crystallizer was tightly closed and allowed to reach the desired experimental temperature (making use of the external refrigerator) prior to the injection of methane gas. The location of the Raman probe was pre-fixed before the solution was introduced into the crystallizer and was chosen to ensure that the probe was as close to the solution interface as possible, while also being fully submerged in the solution. The eventual selected location of the Raman probe in the present study was consistent with that used in a previous study published by our group, and thus a representative schematic illustration of it is available in the literature.46 A 3 cm stirrer bar controlled using a magnetic stirring plate positioned underneath the crystallizer was used to provide agitation to the system. Care was taken to ensure that the stirrer bar did not interfere with the Raman probe present inside the system. Once the desired experimental temperature was reached, the crystallizer was flushed with methane gas through rapid pressurization and depressurization cycles to remove any air present inside the system, following which methane gas injection was carried out slowly until the desired experimental pressure was reached, making sure that the temperature of the system remained close to the desired experimental temperature. When methane gas pressurization was completed, the system was isolated, i.e. the gas inlet valve was closed (refer schematic available in the literature)46 and data acquisition was started. Additionally, at this point, both stirring of the system (600 rpm) and Raman signal acquisition were also simultaneously initiated. The real-time Raman spectrometer was set to record Raman spectra at 20 s intervals throughout the hydrate formation process. Hydrate nucleation was determined using three simultaneous markers, i.e. the familiar characteristic pressure drop and temperature spike of hydrate nucleation, and the appearance of characteristic Raman spectral signatures for one or more hydrate guests (methane or DIOX) incorporated into the hydrate structure.
Author contributions
PL supervised the project. PL and GB conceptualized the work and designed the experiments. MNG, GB and SEKA carried out the kinetic experiments. MNG and GB analysed the data. GB and MNG performed the p-XRD characterization. GB and YZ performed the in situ Raman study and analysis. GB and MNG performed the stability study. GB and MNG wrote the original manuscript draft. PL edited the manuscript and contributed to the final version. All authors read the final manuscript and consented to the submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded in part under the Energy Innovation Research Programme (EIRP, Award No. NRF2015EWTEIRP02-002) and MoE AcRF Tier 1 (R-279-000-542-114). EIRP is administrated by the Energy Market Authority (EMA) of Singapore and funded by the National Research Foundation (NRF), Singapore. The authors would like to acknowledge Professor Rajnish Kumar (IIT Madras) for the valuable suggestions related to in situ Raman data analysis.References
- U.S. Energy Information Administration, International Energy Outlook 2019, 2019.
- K. Huang, J. B. Miller, G. W. Huber, J. A. Dumesic and C. T. Maravelias, Joule, 2018, 2, 349–365 CrossRef CAS.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi, J. R. Long and A. I. L. A. P. S. Argonne National Lab, Nature, 2015, 527, 357–361 CrossRef CAS.
- C. M. Simon, J. Kim, D. A. Gomez-Gualdron, J. S. Camp, Y. G. Chung, R. L. Martin, R. Mercado, M. W. Deem, D. Gunter, M. Haranczyk, D. S. Sholl, R. Q. Snurr, B. Smit, B. C. A. Lawrence Berkeley National Lab and M. M. N. N. M. G. C. Univ. of Minnesota, Energy Environ. Sci., 2015, 8, 1190–1199 RSC.
- V. Rozyyev, D. Thirion, R. Ullah, J. Lee, M. Jung, H. Oh, M. Atilhan and C. T. Yavuz, Nat. Energy, 2019, 4, 604–611 CrossRef CAS.
- T. Ben, C. Pei, D. Zhang, J. Xu, F. Deng, X. Jing and S. Qiu, Energy Environ. Sci., 2011, 4, 3991–3999 RSC.
- S. Bracco, D. Piga, I. Bassanetti, J. Perego, A. Comotti and P. Sozzani, J. Mater. Chem. A, 2017, 5, 10328–10337 RSC.
- R. Boswell and T. S. Collett, Energy Environ. Sci., 2011, 4, 1206–1215 RSC.
- Z. R. Chong, S. H. B. Yang, P. Babu, P. Linga and X. S. Li, Appl. Energy, 2016, 162, 1633–1652 CrossRef.
- P. L. Stanwix, N. M. Rathnayake, F. P. P. de Obanos, M. L. Johns, Z. M. Aman and E. F. May, Energy Environ. Sci., 2018, 11, 1828–1840 RSC.
- E. D. Sloan, Nature, 2003, 426, 353–363 CrossRef CAS.
- H. P. Veluswamy, A. Kumar, Y. Seo, J. D. Lee and P. Linga, Appl. Energy, 2018, 216, 262–285 CrossRef CAS.
- W. Wang, C. L. Bray, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 11608–11609 CrossRef CAS.
- F. Ning, Y. Yu, S. Kjelstrup, T. J. H. Vlugt and K. Glavatskiy, Energy Environ. Sci., 2012, 5, 6779–6795 RSC.
- L. J. Florusse, C. J. Peters, J. Schoonman, K. C. Hester, C. A. Koh, S. F. Dec, K. N. Marsh and E. D. Sloan, Science, 2004, 306, 469–471 CrossRef CAS.
- H. Lee, J.-W. Lee, D. Y. Kim, J. Park, Y.-T. Seo, H. Zeng, I. L. Moudrakovskr, C. I. Ratcliffe and J. A. Ripmeester, Nature, 2005, 434, 743–746 CrossRef CAS.
- H. P. Veluswamy, S. Kumar, R. Kumar, P. Rangsunvigit and P. Linga, Fuel, 2016, 182, 907–919 CrossRef CAS.
- A. Kumar, H. P. Veluswamy, P. Linga and R. Kumar, Fuel, 2019, 236, 1505–1511 CrossRef CAS.
- D.-L. Zhong, Z. Li, Y.-Y. Lu, J.-L. Wang and J. Yan, Appl. Energy, 2015, 158, 133–141 CrossRef CAS.
- O. A. Abdelrahman, D. S. Park, K. P. Vinter, C. S. Spanjers, L. Ren, H. J. Cho, D. G. Vlachos, W. Fan, M. Tsapatsis, P. J. Dauenhauer and I. Energy Frontier Research Centers. Catalysis Center for Energy, ACS Sustainable Chem. Eng., 2017, 5, 3732–3736 CrossRef CAS.
- National Center for Biotechnology Information, PubChem Database. Tetrahydrofuran, CID = 8028, https://pubchem.ncbi.nlm.nih.gov/compound/8028, (accessed March 12, 2020).
- National Institute of Standards and Technology, Tetrahydrofuran, https://webbook.nist.gov/cgi/cbook.cgi?ID=C109999&Mask=4, (accessed March 12, 2020).
- BASF, Tetrahydrofuran (THF) storage and handling, https://wayback.archive-it.org/all/20080309162147/http://www.basf.com/diols/pdfs/thf_brochure.pdf, (accessed June 15, 2020).
- F. W. Mackison, R. S. Stricoff and L. J. Partridge, Occupational health guidelines for chemical hazards, 1981 Search PubMed.
- J. P. Torré, D. Haillot, S. Rigal, R. de Souza Lima, C. Dicharry and J.-P. Bedecarrats, Chem. Eng. Sci., 2015, 126, 688–697 CrossRef.
- A. Venkateswaran, J. Easterfield and D. Davidson, Can. J. Chem., 1967, 45, 884–886 CrossRef CAS.
- R. M. de Deugd, M. D. Jager and J. de Swaan Arons, AIChE J., 2001, 47, 693–704 CrossRef CAS.
- T. Maekawa, Fluid Phase Equilib., 2008, 267, 1–5 CrossRef CAS.
- J. Jhaveri and D. B. Robinson, Can. J. Chem. Eng., 1965, 43, 75–78 CrossRef CAS.
- H. P. Veluswamy, A. Kumar, R. Kumar and P. Linga, Appl. Energy, 2017, 188, 190–199 CrossRef CAS.
- W. L. Mao, H.-K. Mao, A. F. Goncharov, V. V. Struzhkin, Q. Guo, J. Hu, J. Shu, R. J. Hemley, M. Somayazulu and Y. Zhao, Science, 2002, 297, 2247–2249 CrossRef CAS.
- A. K. Sum, R. C. Burruss and E. D. Sloan, J. Phys. Chem. B, 1997, 101, 7371–7377 CrossRef CAS.
- Y.-J. Lee, T. Kawamura, Y. Yamamoto and J.-H. Yoon, J. Chem. Eng. Data, 2012, 57, 3543–3548 CrossRef CAS.
- National Institute of Standards and Technology, 1,3-Dioxolane, https://webbook.nist.gov/cgi/cbook.cgi?ID=C646060&Mask=4, (accessed March 12, 2020).
- National Center for Biotechnology Information, PubChem Database. 1,3-Dioxolane., https://pubchem.ncbi.nlm.nih.gov/compound/12586, (accessed March 12, 2020).
- European Chemicals Agency, 1,3-Dioxolane, https://echa.europa.eu/registration-dossier/-/registered-dossier/15807/7/8, (accessed March 12, 2020).
- A. Kumar, N. Daraboina, R. Kumar and P. Linga, J. Phys. Chem. C, 2016, 120, 29062–29068 CrossRef CAS.
- H. P. Veluswamy, Q. W. Hong and P. Linga, Cryst. Growth Des., 2016, 16, 5932–5945 CrossRef CAS.
- R. Turton, R. C. Bailie, W. B. Whiting and J. A. Shaeiwitz, Analysis, Synthesis, and Design of Chemical Processes, Prentice Hall, Upper Saddle River, N.J., 3rd edn, 2009 Search PubMed.
- PUB, Water Price, https://www.pub.gov.sg/watersupply/waterprice, (accessed June 15, 2020).
- G. Bhattacharjee, H. Prakash Veluswamy, R. Kumar and P. Linga, Appl. Energy, 2020, 269, 115142 CrossRef CAS.
- P. Linga, H. P. Veluswamy, A. Kumar and M. Khurana, Singapore Pat., WO 2020/117129 A1, 2019.
- E. D. Sloan and C. A. Koh, Clathrate Hydrates of Natural Gases, CRC Press, Boca Raton, FL, 3rd edn, 2008 Search PubMed.
- S. Takeya and J. A. Ripmeester, Angew. Chem., Int. Ed., 2008, 47, 1276–1279 CrossRef CAS.
- S. Takeya and J. A. Ripmeester, ChemPhysChem, 2010, 11, 70–73 CrossRef CAS.
- A. Kumar, R. Kumar and P. Linga, iScience, 2019, 14, 136–146 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee02315a |
| ‡ Equal contribution from authors. |
| This journal is © The Royal Society of Chemistry 2020 |