
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gradient-morph LiCoO2 single crystals with stabilized energy density above 3400 W h L−1†
Zhi
Zhu‡
a,
Daiwei
Yu‡
 a,
Zhe
Shi
a,
Rui
Gao
a,
Xianghui
Xiao
b,
Iradwikanari
Waluyo
b,
Mingyuan
Ge
b,
Yanhao
Dong
a,
Weijiang
Xue
a,
Guiyin
Xu
a,
Wah-Keat
Lee
b,
Adrian
Hunt
b and
Ju
Li
*a
a,
Zhe
Shi
a,
Rui
Gao
a,
Xianghui
Xiao
b,
Iradwikanari
Waluyo
b,
Mingyuan
Ge
b,
Yanhao
Dong
a,
Weijiang
Xue
a,
Guiyin
Xu
a,
Wah-Keat
Lee
b,
Adrian
Hunt
b and
Ju
Li
*a
aDepartment of Nuclear Science and Engineering, Department of Materials Science and Engineering & Department of Electrical Engineering and Computer Science, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: liju@mit.edu
bNational Synchrotron Light Source II, Brookhaven National Laboratory, Upton, NY 11973, USA
First published on 20th May 2020
Abstract
The cycling stability of LiCoO2 under high voltages (>4.5 V) was plagued by hybrid anion- and cation-redox (HACR) induced oxygen escape and uncontrolled phase collapse. With DEMS and in situ XANES mapping at the NSLS-II, we demonstrate that oxygen escape triggers irreversible transformations into “bad” surface phases that rapidly propagate inward. Enabling HACR but stopping global oxygen migration is key to a stable high-energy cathode. Therefore, we developed ∼10 μm single crystals with LiCoO2 in the bulk smoothly transitioning to Co-free LiMn0.75Ni0.25O2 at the surface. By means of initial electrochemical formation, a semi-coherent LiMn1.5Ni0.5O4 spinel-like shell was established in operando with little oxygen loss to integrally wrap the LiCoO2 bulk. Then we obtained gradient-morph LiCoO2 single crystals to prevent the percolating migration of oxygen out of the particle and achieved enhanced HACR reversibility at high voltages. The gradient-morph HACR cathode undergoes substantially stabilized cycling when charged to above 4.6 V, and hence a stable cyclic volumetric energy density of >3400 W h L−1 has been achieved in a pouch full-cell coupled with a commercial graphite anode and lean electrolyte (2 g A h−1), exhibiting up to 2906 W h L−1 even after 300 cycles.
Though diverse transition-metal (TM) oxides have been investigated as cathode materials for Li-ion batteries (LIB), LiCoO2 (LCO) still takes the dominant position in the current LIB market for consumer electronics ever after Goodenough's1 and SONY's2 developments, due to the good rate performance, simple scalability and high compressed density of above 4.1 g cc−1. However, LCO is only working within a limited voltage range (<4.35 V) or capacity (<165 mA h g−1) at present in order to maintain the cycling stability,3 which is far from the theoretical capacity of 274 mA h g−1 when Li is fully extracted, so it still provides great potential to achieve much higher energy density.4 The cycling stability of LCO to high charging voltages (>4.5 V) has been investigated for decades.4–6 Some literature studies reported7–10 that bulk phase transformations (including O3 → H1-3 → O1) occurring at above 4.5 V caused rapid degradation of LCO, while others reported that the highly delithiated Li1−xCoO2 (x > 0.5) aggravated the interfacial side reactions with the electrolyte.11–13 However, we have argued that the exact sequence of events in bulk vs. surface degradation initiators need careful analysis.14
Because of the heavy Co3d–O2p hybridization (Fig. 1a, note that its energy resonance is much more than that of Ni3d–O2p or Mn3d–O2p, indicating stronger covalency), deep delithiation from LCO must extract electrons from both Co and O ions. Manthiram et al.15 reported that when L1−xCoO2 was delithiated beyond x = 0.5, further delithiation requires the oxidation of O2− (O2− → Oα−, α < 2), which has lately been verified by soft X-ray absorption spectroscopy (sXAS).16,17 That means hybrid anion- and cation-redox (HACR) reactions must be involved when charging LCO to above ∼150 mA h g−1. Although utilizing the last one-half of LCO's theoretical capacity (from 150 mA h g−1 to 274 mA h g−1) in the high-voltage range is extremely enticing, the involvement of oxygen anion-redox and the associated oxygen mobility (Oα− has a much lower migration energy barrier than O2−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18–20) and escape,20 causing lattice collapse and side reactions with the necessarily lean electrolyte in industrial batteries, is a killer problem.
18–20) and escape,20 causing lattice collapse and side reactions with the necessarily lean electrolyte in industrial batteries, is a killer problem.
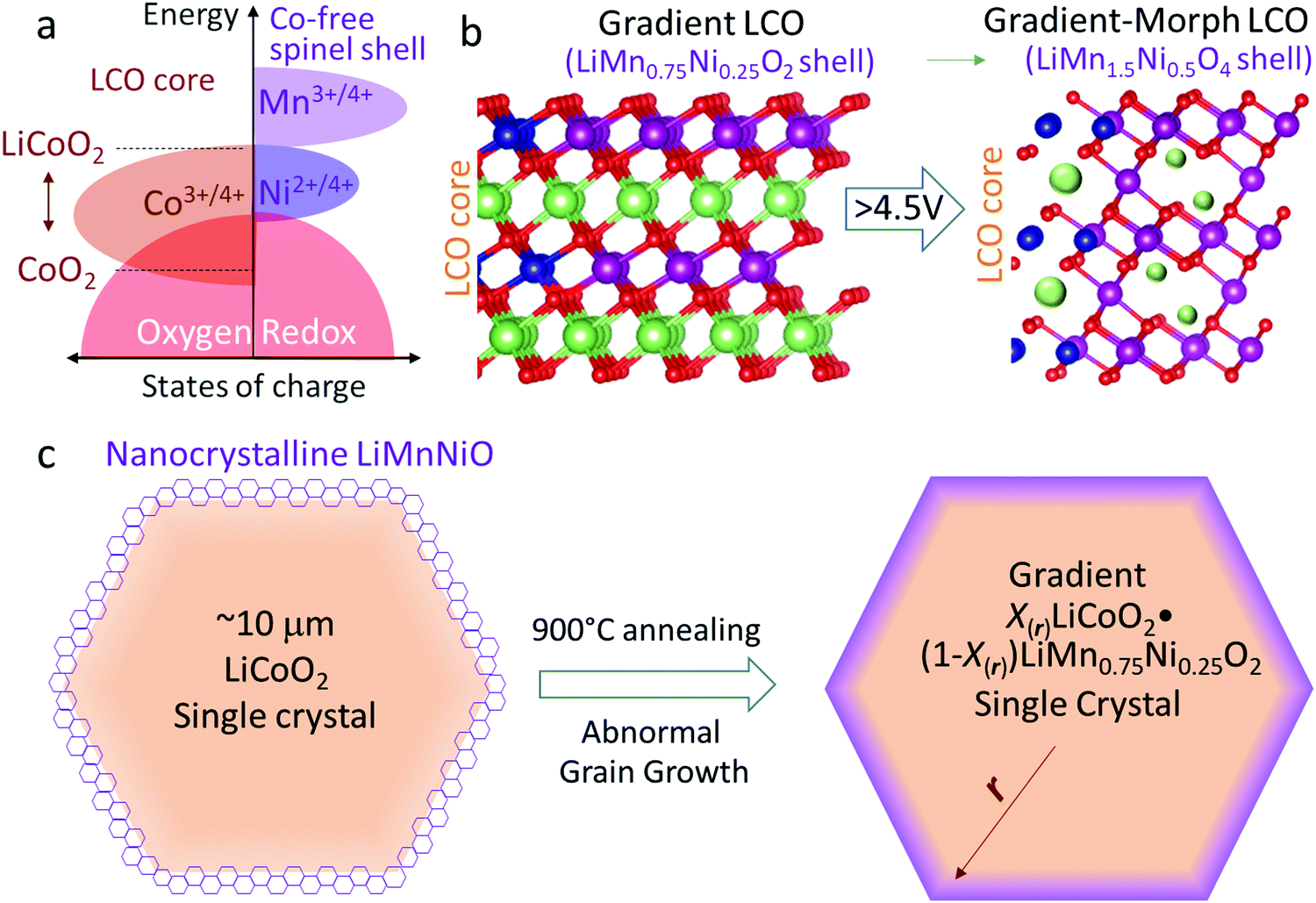 | ||
| Fig. 1 Electronic bands, lattice structures and preparation process of LiCoO2 and the gradient X(r)LiCoO2·(1 − X(r))LiMn0.75Ni0.25O2 single-crystal. (a) The band structures in LiCoO2 and Co-free LMO (M = Mn, Ni etc.). (b) The surface reconstruction of the gradient X(r)LiCoO2·(1 − X(r))LiMn0.75Ni0.25O2 crystal in the initial electrochemical formation. In this process, the oxygen sublattice maintains lattice-coherency during the layered LiMn0.75Ni0.25O2 → spinel LiMn1.5Ni0.5O4 transformation, with preserved Mn:Ni:O ratio and no oxygen loss. The blue balls represent Co ions, while the violet, green and red balls represent Mn/Ni, Li and O ions, respectively. (c) Schematic showing the creation of the gradient X(r)LiCoO2·(1 − X(r))LiMn0.75Ni0.25O2 single-crystal with an abnormal grain growth at high temperature. | ||
The significant HACR activities entail O2− oxidization, which would make oxygen ions more mobile18–20 and enable oxygen migration (OM). OM can be local or global: global OM (GOM) means the oxygen ions can move long-range in the particle all the way to the surface and be released from the particle surface, possibly reacting with the liquid electrolyte and causing oxygen depletion with a particularly pernicious irreversible phase transformation (IPT) CoO2 → Co3O4.15,21–23 On average, O2− ions only need to be minorly oxidized in Li1−xCoO2 when charged to 4.5 V (x = 0.7).15–17 However in charging the particle surface always delithiates more to drive Li ions diffusing from the bulk to the surface,22 so that GOM and IPT can be more easily switched on from the particle surface. In contrast to the well-known high-rate spinel LiMn2O424 or high-voltage spinel LiNi0.5Mn1.5O425 phases, even though the Co3O4 phase also has a spinel structure, it is a “bad spinel” which blocks Li ions, as both the tetrahedral and octahedral sites are taken by Co ions. The oxygen loss (CoO2 → Co3O4) with cobalt reduction (Co4+ → Co2.7+) and electrolyte oxidation is irreversible. Furthermore, this appears to be not a self-limiting (self-passivating) transformation, as oxygen loss produces rampant oxygen vacancies (VO) that “infect” the interior,18,26 producing flaws and microcracks3, further driving GOM and IPT propagating deep into the interior.21 In this work, we will demonstrate the oxygen release and visualize the IPT with DEMS and an in situ XANES mapping while fully charging LCO. Therefore, enabling HACR but stopping GOM is key to developing a reversible high-energy cathode. Surface passivation was tried by coating with metal oxides, fluorides, phosphates, etc.13,27,28 These foreign coatings improved the cycling stability, but whether they fully wet the surface and cut off GOM is questionable due to the structural incoherency.
Thus it is of key significance to establish a robust shell that can fully prevent GOM and IPT in the LCO particles to maintain facile Li+ and polaron transport in high-voltage cycling. The shell should meet the following requirements: (1) to completely shut down GOM, an integral lattice-coherent shell is required to fully enclose the LCO lattice (complete wetting) instead of a coating with island growth, otherwise the prolific grain boundaries and/or exposed surfaces will be preferential locations for GOM and stress-corrosion cracking (SCC) in the electrolyte. (2) The shell should remain highly conductive for both Li+ and electrons in cycling to promote fast electrochemical kinetics. (3) The shell should not participate in oxygen anion-redox and exclude oxygen vacancy in cycling to eliminate percolation paths for GOM. According to the band diagram in Fig. 1a, the shell should be Co-free (e.g. M can be Mn, Ni etc., which is not highly hybridized with O) to avoid the oxygen oxidation at high voltages. These requirements point to a high-voltage spinel LiMn(+4)1.5Ni(+2)0.5O4 phase25 as a likely candidate for the shell. The LiMn1.5Ni0.5O4 shell is well-known to be very stable up to 5 V,25,29–31 so one should expect a stable high-voltage cycling, when the spinel shell completely encloses and protects the LCO core.
However, so far researchers have never achieved a complete-wetting and lattice-coherent LiMn1.5Ni0.5O4 shell to fully wrap around the LCO lattice. With most of the coating methods, one tends to get islands-coating instead of an integral shell. The islands-coating cannot shut down GOM in cycling. So in this work, we developed a special way to exactly establish such an integral LiMn1.5Ni0.5O4 spinel shell to fully enclose LCO and shut down GOM in the high-voltage cycling.
In this work, we first developed lattice-coherent LiCoO2 and LiMn0.75Ni0.25O2 single crystals. Because LiMn0.75Ni0.25O2 is isostructural with LCO (both layered), it is easily able to fully and integrally enclose the LCO bulk by high-temperature annealing. As is known, such a Mn-rich LiMn0.75Ni0.25O2 layered phase is not electrochemically stable and thus rarely prepared for batteries, but here it is just coated as a precursor for the eventual integral LiMn1.5Ni0.5O4 shell. As shown in Fig. 1b and c, the pre-prepared particles are composed of X(r)LiCoO2·(1 − X(r))LiMn0.75Ni0.25O2. The core, with X(rcore) = 1, is pure LCO, and X(r) gradually decreases to 0 (X(rsurface) = 0) from the bulk to the surface. That is, the composition in the particle changes from LCO to LiMn0.75Ni0.25O2 from the core to the shell, but all M and O ions share a coherent layered crystal lattice (Fig. 1b), constituting a gradient LiMn0.75Ni0.25O2 → LiCoO2 single crystal (G-LCO). Then by means of the initial electrochemical cycling, the LiMn0.75Ni0.25O2 shell with epitaxial layered crystal structure can quickly transform into the desirable fully wetting LiMn1.5Ni0.5O4 shell with spinel crystal structure,32,33 as illustrated in Fig. 1b. The LiMn1.5Ni0.5O4 spinel-like shell that in operando transformed from the lattice-coherent LiMn0.75Ni0.25O2 by Jahn–Teller distortion in room-temperature cycling will still keep oxygen sublattice-coherency with the LCO bulk and fully enclose LCO. Crucially, although the final LiMn1.5Ni0.5O4 has a cubic crystal structure while the LiMn0.75Ni0.25O2 precursor has a layered structure, unlike LixCoO2 → Co3O4, this LiMn0.75Ni0.25O2 → LiMn1.5Ni0.5O4 transformation preserves the O:Mn:Ni ratios and requires neither oxygen loss nor VO injection into the particle that would enhance OM, as only the O : Li ratio changes from 2 to 4. This is the secret for the orderly and controlled LiMn0.75Ni0.25O2 → LiMn1.5Ni0.5O4 surface transformation (TM/O all in place and only displacive Jahn–Teller distortion is needed) upon formation by electrochemical delithiation, and the destination of this surface transformation, LiMn1.5Ni0.5O4, is unarguably the best high-voltage coating phase as it enables quick Li+ conduction but prevents GOM.
Though LiMn1.5Ni0.5O4 is cubic spinel while LCO has a layered structure, the spinel LiMn1.5Ni0.5O4 shell established in this way still shares the same oxygen-sublattice with the LCO bulk, so we would like to call the particle after the battery-formation process a gradient-morph LCO single crystal (GM-LCO, Fig. 1b). GM-LCO can achieve the following unique performances in prolonged high-voltage cycling: (1) with ∼10 μm single crystals, 4.1 g cc−1 compressed density and high electronic conductivity, the cathode can cycle to ∼190 mA h g−1, ∼230 mA h g−1 and ∼270 mA h g−1 when charged to 4.5 V, 4.6 V and 4.7 V vs. Li, respectively. While the spinel-like shell contributes little capacity, it is very stable and can always keep crystal dense in cycling.31–33 Though the O2− ions in the LCO bulk participate in HACR and intend to migrate when charged to above 4.5 V, they are completely enclosed and can hardly trespass the shell. GOM and IPT can thus be efficiently prevented, which “tames” the oxygen ions in the LCO core to be a highly-reversible anion-redox (“solid oxygen” HACR concept34,35 like the Li–sulfur chemistry36) in the high-voltage cycling. In this work, we will show that the cycling stability of G-LCO is highly respectable for consumer electronics applications even when cycled to 4.7 V. (2) Substantially different from the surface passivation, the LiMn1.5Ni0.5O4 shell conducts Li+ and polarons exceptionally well,24 preserving the facile kinetics of LCO in cycling, despite the large single crystals for high compressed density. (3) By keeping the Mn valence at +4 in cycling, the LiMn1.5Ni0.5O4 shell prevents both Co and Mn dissolutions, which can further stabilize the cycling of the practical full-cells versus graphite anodes. Furthermore, the new particles also have better compatibility with the commercial carbonate electrolyte at high voltages, so we will show that G-LCO has ultra-stable high-voltage cycling in a pouch full-cell with a commercial graphite anode and very lean electrolyte (2 g A h−1).
Preparation of X(r)LiCoO2·(1 − X(r))LiMn0.75Ni0.25O2 gradient single crystals
As the Mn valence in the LiMn0.75Ni0.25O2 precursor is below +4, such a layered crystal was rarely prepared in air as bulk material, as it is thermodynamically unstable. However, we will demonstrate that a thin coherent LiMn0.75Ni0.25O2 shell can be created on the surface of a huge LCO particle, which acts as a giant seed for the rapid grain coarsening that consumes the original Li/Mn/Ni oxide nanograins, with a composition-gradient region between the LCO core and the LiMn0.75Ni0.25O2 shell. Such epitaxial substrate has a stablizing effect on the surface layered phase, as the elimination of phase boundary energies and grain boundary energies compensates for the increased bulk energy, as long as the LiMn0.75Ni0.25O2-like shell is thinner than the coherency limit. In this work, we created a lattice-coherent LiMn0.75Ni0.25O2 shell on the pristine LCO (P-LCO) particle with a wet-coating process followed by high-temperature annealing (Method). The deposited Li/Mn/Ni has an opportunity to self-nucleate to other crystals (e.g. spinel) and grow into islands on the LCO particle surface. However, when we deposit only tens of nanometers thick layers of Li/Mn/Ni precursors on the surface of a ∼10 μm primary LCO particle, the huge LCO crystal will easily coarsen away the thin Li/Mn/Ni oxide nanocrystals in the calcination process due to grain coarsening, as shown in Fig. 1c. This means that the coated Li/Mn/Ni oxides can directly grow into the same layered lattice with the LCO substrate following the path of the abnormal grain growth (AGG).37,38 Because LiMn0.75Ni0.25O2 is isostructural with LCO, the two components can mutually diffuse and form a lattice-coherent solid solution single crystal when annealed at high temperature. Therefore, the new particle can remain single-crystalline, but one can expect pure LCO in the core and Mn/Ni/Co gradient concentration in the shell.We characterize the G-LCO particles before the electrochemical cycling, which was produced with 94 wt% LCO and 6 wt% LiMn0.75Ni0.25O2. The X-ray diffraction (XRD) pattern in Fig. 2a indicated that G-LCO particles were still composed of a single phase, as there was no other peak observed than the R3m phase peak. Fig. 2b and c show the scanning electron microscopy (SEM) images of P-LCO and G-LCO particles. It was clear that P-LCO particles had a well-crystallized morphology and smooth surface with a particle size of 10–15 μm in diameter (ESI,† Fig. S1), but the particles became round and were completely enclosed after the formation of the lattice-coherent shell. The STEM image of the particle cross-section (after FIB) in Fig. 2d indicated that there was no phase boundary in G-LCO particles. The STEM-HAADF images in Fig. 2e and f further proved the single-crystallinity of the particle, as the (001) lattice plane arranged well from the deep core to the outermost surface. EDS mapping under SEM in the ESI,† Fig. S2, verified the uniform and full conformal coverage of the Mn shell on the LCO particle, and that under STEM in Fig. 2d accurately indicated that the TM element in the core was only Co and the content gradually decreased in the ∼150 nm field near the surface, where Mn and Ni were concentrated. There was not a sharp edge between the Mn/Ni and Co distributions, indicating that the two compositions of LiMn0.75Ni0.25O2 and LCO formed a solid solution with a smooth gradient transition20,39 that promoted the lattice-coherency between the LiMn0.75Ni0.25O2 shell and LCO core. The Mn/Co/Ni distribution in G-LCO was quantified with EELS line-scan from the shell to core (Fig. 2g and h), and the result showed that Co was ∼33% (consistent with LCO) in the bulk but gradually decreased in the ∼150 nm surface region and to 0% (Co-free) in the ∼10 nm outermost surface, where Mn and Ni gradually concentrated from 0% to ∼25% and ∼8% (consistent with LiMn0.75Ni0.25O2) while O remained constant at ∼67% across the whole particle.
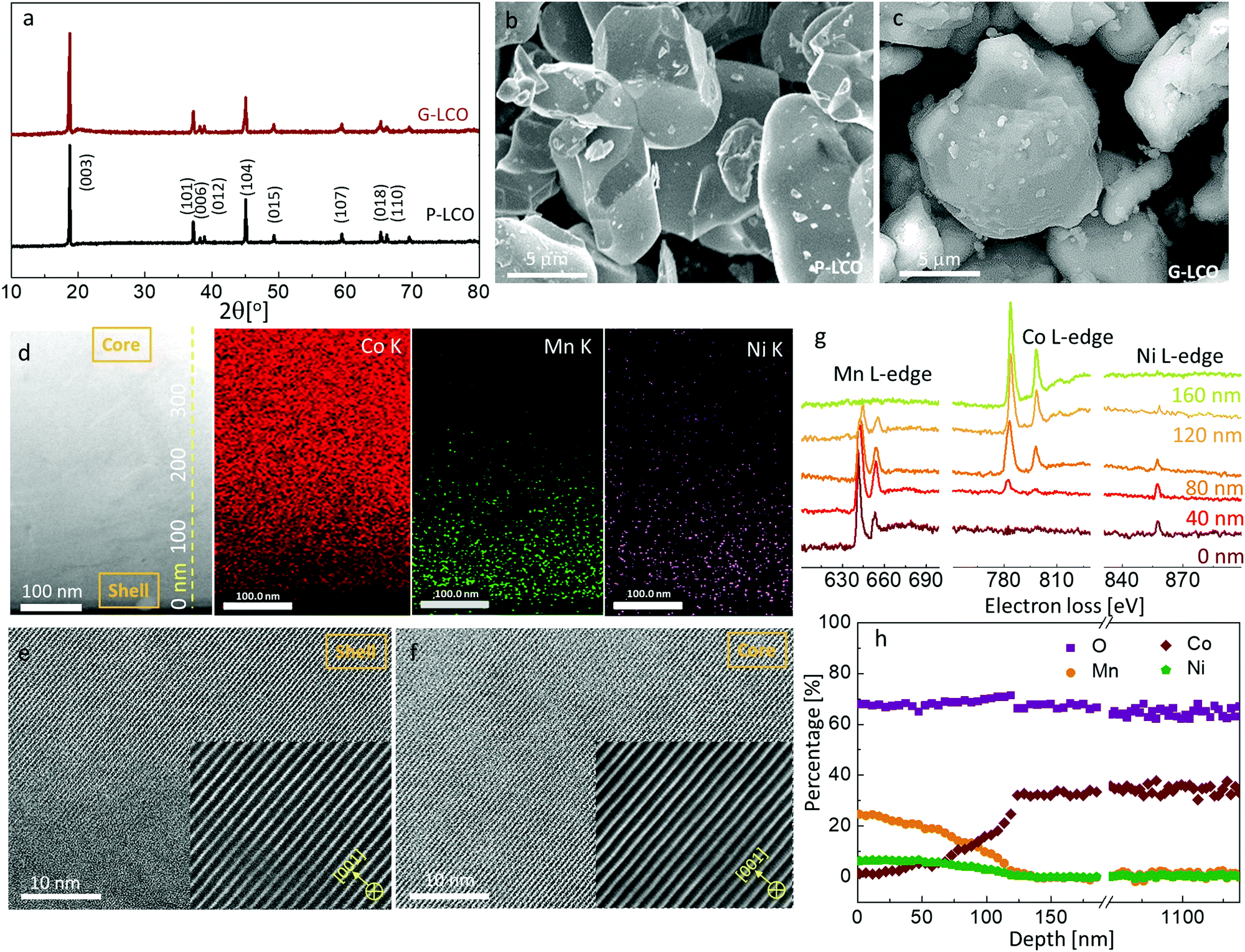 | ||
| Fig. 2 Characterization of the G-LCO particles. (a) The X-ray diffraction pattern of the P-LCO and G-LCO particles. (b and c) The SEM images of P-LCO (b) and G-LCO (c) particles. (d) The STEM images of the G-LCO particle after FIB preparation and the EDS mapping of Co, Mn and Ni elements. (e and f) STEM-HAADF images of the crystal lattices within the G-LCO particle from the LiMn0.75Ni0.25O2 shell (e) to the LCO bulk (f). (g) Selected EELS patterns to show the Mn L-edge, Co L-edge and Ni L-edge at different depths from the G-LCO particle surface. (h) The composition of elements at different depths in the G-LCO particle calculated from the EELS line-scan data. | ||
The above measurements indicated the successful preparation of the gradient X(r)LiCoO2·(1 −X(r))LiMn0.75Ni0.25O2 single crystals, with X(rcore) = 1 and X(rsurface) = 0. Additionally, according to the sXAS analysis in the ESI,† Fig. S3, the Mn valence can be quantified as +3.4 at the surface, quite close to the theoretical value in LiMn(+3.33)0.75Ni(+2)0.25O2, which further verified the composition of LiMn0.75Ni0.25O2 at the particle surface. Generally, the composition profile of G-LCO can be further optimized by adjusting the ratio of Li/Mn/Ni, M elements and annealing temperatures etc. to get better electrochemical performances.
Again we want to emphasize that the layered LiMn(+3.33)0.75Ni(+2)0.25O2 is not a typical compound that people can easily synthesize or use electrochemically in the bulk. But here by virtue of the large LCO single crystal as the seed and substrate, we stabilized this LiMn(+3.33)0.75Ni(+2)0.25O2 phase as a thin epitaxial surface layer, that serves as the precursor for the future LiMn0.75Ni0.25O2 → LiMn1.5Ni0.5O4 transformation by O and TM preserving displacive Jahn–Teller distortion. Because of the pre-positioning of Mn:Ni:O in the right ratio, and because the good spinel can still share the same oxygen sublattice with the LCO bulk, only minimum structural disruption is needed to achieve a conformal protection with the “good spinel”, in a highly organized and well-controlled surface transformation during the initial cycles.
Stabilized high cyclic energy density up to 3400 W h L−1 in pouch full-cells
We prepared the cathode electrodes with a standard industrial procedure, comprising 96 wt% active sample, 2 wt% carbon black and 2 wt% PVDF, with ∼17 mg cm−2 loading of active material on an Al foil. The P-LCO and G-LCO cathodes were firstly cycled in half-cells between 3.0–4.6 V. As shown in Fig. 3a, P-LCO and G-LCO had similar capacity in the 1st cycle: P-LCO was charged to 252.1 mA h g−1 and discharged to 224.5 mA h g−1, whereas G-LCO was charged to 251.2 mA h g−1 and discharged to 226.9 mA h g−1. However, Fig. 3a shows that the charge/discharge profile of P-LCO totally faded after 100 cycles, while that of G-LCO kept a similar shape to the initial cycle after 100 cycles. The cycling performance in Fig. 3b shows that the capacity of P-LCO decayed rapidly in the initial 30 cycles, and only 45% of the initial capacity and 34% of the initial energy density were retained after 40 cycles. However, G-LCO retained more than 80% of the initial capacity and energy density after 100 cycles. The cyclic voltammetry of P-LCO and G-LCO after different cycles can be found in the ESI,† Fig. S4.
 | ||
| Fig. 3 The electrochemical performances of P-LCO and G-LCO cathodes in coin-type half-cells and pouch full-cells. (a) The charge/discharge profiles of P-LCO and G-LCO cathodes within 3.0–4.6 V in coin-cell half-cells under a constant current of 50 mA g−1. (b) The cycling of capacity, energy density and coulombic inefficiency of P-LCO and G-LCO cathodes within 3.0–4.6 V in coin-cell half-cells. (c) The cycling of capacity of P-LCO and G-LCO cathodes at different rates within 3.0–4.6 V in coin-cell half-cells. (d) The cycling performance of the capacity and energy density of the P-LCO and G-LCO materials in pouch full-cells with commercial graphite anodes with a constant charge current of 100 mA g−1 when cycled to 4.55 V, and then 20 mA g−1 when cycled again to 4.55 V, constant discharge current of 100 mA g−1 when cycled to 3.0 V. (e) Comparison of the volumetric energy density between different LCO cathode–electrodes vs. commercial graphite anode. | ||
We use the coulombic inefficiency (CI ≡ 100% − CE, which is defined as the difference between coulombic efficiency and 100%) in half-cells to compare the capacity loss from the cathode in each cycle.40 As shown in Fig. 3b, the CI of G-LCO was only about half that of P-LCO in each cycle, indicating a ∼50% suppressed capacity loss in each discharge from the G-LCO cathode. The suppressed CI of G-LCO not only indicated thermodynamic stability, but could also indicate a maintained kinetics of G-LCO during each charging and discharging,41 while the higher CI of P-LCO reflected a serious kinetic retardation in each cycle when the growth of “bad” spinel Co3O4 in charging immediately added more impedance for the subsequent discharging. Therefore the difference in cyclic capacity between P-LCO and G-LCO is enlarged at higher rates. As shown in Fig. 3c, G-LCO had a similar capacity to P-LCO in the initial cycles at 20 mA g−1, but the capacity retention of P-LCO decreased considerably at high rates, while that of G-LCO decreased little. In particular, at 400 mA g−1 after 40 cycles, G-LCO can still discharge 175 mA h g−1, three-times higher than that of P-LCO (41 mA h g−1). When reducing the rate to 20 mA g−1, the capacity of G-LCO reverted to 210 mA h g−1 whereas that of P-LCO restored to 163 mA h g−1. That is, the kinetic capacity loss of G-LCO, when increasing the rate from 20 mA g−1 to 400 mA g−1, was only about 1/4 to that of P-LCO after 50 cycles. This result clearly indicated that the impedance of G-LCO was stabilized in the high-voltage cycling.
We then tested P-LCO and G-LCO cathodes in pouch full-cells with matched commercial graphite anodes (Method). As shown in Fig. 3d, though both P-LCO and G-LCO can discharge to about 205 mA h g−1 and 780 W h kg−1 in the initial cycles when cycled within 3.0–4.50 V, either the capacity or energy density of P-LCO suffered rapid decay in 60 cycles and totally died within 200 cycles. However, G-LCO had very little decay in the cycling, as the capacity remained stable above 191.2 mA h g−1 with an energy density of 736.3 W h kg−1 after 300 cycles. When cycled within 3.0–4.55 V, G-LCO discharged to 216 mA h g−1 and 844 W h kg−1 in the initial cycles, and retained 176.3 mA h g−1 with 664.7 W h kg−1 after 300 cycles. The previous highest energy density of LCO obtained in pouch full-cells vs. graphite anode was also around 800 W h kg−1, reported by Zhang et al,5 but the cyclic retention decayed to below 700 W h kg−1 in 70 cycles. The G-LCO in this work has achieved energy-density retention of 736.3 W h kg−1 after 300 cycles. Additionally, as we used only a very lean electrolyte (2 g A h−1) in the full-cell, the good cycling also proved that G-LCO had much better compatibility with the regular carbonate electrolyte at high voltages.42
The stabilized high energy density significantly improved the volumetric energy density of the cathode. Both the compressed densities of the P-LCO and G-LCO electrodes were measured as 4.1 ± 0.1 g cm−3 (ESI,† Table S1), the same as that provided by the current commercial manufacturers. Then the volumetric energy density of P-LCO and G-LCO cathodes can be compared in Fig. 3e, calculated from the cycling performance in the full-cell (Fig. 3d). Though the initial energy densities of the P-LCO and G-LCO cathodes were similar when charged to 4.50 V, both around 3100 W h L−1, P-LCO decayed to almost 0 after 300 cycles. However, the volumetric energy density of the G-LCO cathode can remain as high as 2906 W h L−1 after 300 cycles. When cycled to 4.55 V, G-LCO can even achieve 3404 W h L−1 and retain 2793 W h L−1 after 300 cycles. As far as we know, this is the first time that one achieved such a high cyclic volumetric energy density of cathode after 300 cycles, which is ∼30% higher than that of the commercial LCO on the market cycled within 4.35 V.
Prevention of GOM and Co3O4 in the full-delithiated G-LCO particles
The rapid degradation of P-LCO in the high-voltage cycling will be shown to be caused by GOM and the accumulated IPT. Firstly, we used differential electrochemical mass spectroscopy (DEMS) to monitor the O2 and CO2 release while charging the cathodes to 4.60 V vs. Li. As shown in Fig. 4a, O2 started to evolve from P-LCO at 4.35 V (∼175 mA h g−1), and the amount linearly increased afterward and especially sped up when holding the cathode at 4.60 V. The accompanying CO2 release also verified the rampant decomposition of the precious electrolyte when oxygen was released from the cathode. In contrast, GOM was substantially prevented in G-LCO particles at high voltages. As shown in Fig. 4b, little O2 was released from G-LCO even when holding the cathode at 4.60 V for more than 1 hour. The elimination of CO2 release also indicated a stabilized interface between the G-LCO particles and the carbonate electrolyte when OM was prevented, which will not only favor the stabilization of the particle surface, but also prevent the depletion of the precious electrolyte to stabilize the cycling, as according to the industrial practice, the total weight of the liquid electrolyte injected is only ∼40% of the cathode weight and must be able to wet the anode, cathode and separator even after thousands of cycles. | ||
| Fig. 4 Tracking of OM with DEMS and IPT with in situ 2D XANES mapping in the P-LCO and G-LCO particles while charging. (a and b) The DEMS in a charging process of the P-LCO cathode (a) and G-LCO cathode to 4.6 V (b). (c–f) The in situ 2D Co valence mapping (c–e) and XANES Co K edge (f) in P-LCO particles at different voltages in the charging process. (g–j) The in situ 2D Co valence mapping (g–i) and XANES Co K-edge (j) in G-LCO particles at different voltages in the charging process. The color scheme indicates the Co valence change. The in situ XANES Co K-edges at different voltages in (f) and (j) were picked from site S at the P-LCO particle surface in (c) and from site T at the G-LCO particle surface in (g). | ||
The occurrence of OM would immediately result in IPT in the P-LCO particle, producing the bad spinel Co3O4, where the average Co valence is below +3. Hereby, we utilized an in situ nano-XANES mapping with synchrotron-based full-field X-ray imaging at the NSLS-II of the Brookhaven National Laboratory, which can monitor the spatial distribution of Co valence when charging the cathode (Methods). Thanks to its high spatial resolution (∼21 nm) and high temporal resolution,43–45 we can track the detailed information of IPT from the operando spatial Co valence change during charging. Fig. 4c–e showed the in situ Co valence distribution in P-LCO at different states of charges (SOCs). Firstly, we can verify that the particle surface always delithiated more than the bulk while charging, as Fig. 4d shows that the Co valence is higher (in green) at the surface than in the bulk (in yellow). When only focusing on site S at the particle surface, we can note that Co was firstly oxidized when charging P-LCO from 3.0 V (Fig. 4c) to 4.2 V (Fig. 4d), indicating the routine delithiation of LiCoO2 → Li1−xCoO2. However, Co was reduced instead of being further oxidized when it was charged to 4.6 V (Fig. 4e and f). This can only be explained by Oα− (α < 2) reaching the surface, and subsequently reacting with the electrolyte or escaping as O2, leaving the electrons to Co. Since the XANES Co K edge at the outermost surface at 4.6 V was very close to the standard Co3O4 (ESI,† Fig. S5a), it can be inferred that Co3O4 was produced at 4.6 V at site S. This is the first time that, using an in situ characterization method, one confirmed that GOM and IPT (CoO2 → Co3O4) started from some specific patches at the LCO particle surface at high voltages. Meanwhile, we also obtained a gradient Co-valence distribution near site S at 4.6 V (Fig. 4e and ESI,† Fig. S5a), which showed that the Co ions in the core were able to be oxidized to around +4, but those near the surface were gradually reduced to +2.7. The gradient distribution of Co valence indicated the growth of Co3O4 inward in the bulk. When Co3O4 wrapped the P-LCO particles in the prolonged cycles, both the capacity and voltage were seen to be severely degraded, as shown in Fig. 3. Very differently, the stopped GOM prevented catastrophic uncontrolled changes to “bad” surface phases in the G-LCO particle. As shown in Fig. 4g–j, all Co ions in the particle were continuously oxidized from +3 to about +4 while charging G-LCO from 3 V to 4.6 V. There was no patch showing reduced Co at the particle surface (ESI† Fig. S5b) in the whole charging process, indicating the prevention of Co3O4 formation at high voltages. The prevented GOM and Co reduction in G-LCO while charging maintained high kinetics, thus promoting a well suppressed CI within 0.5% and enhanced capacity retention in the prolonged high-voltage cycling as shown in Fig. 3.
To fundamentally understand the prevention of GOM in G-LCO particles, it is essential to investigate the shell reconstruction in the high-voltage cycling. Because of Jahn–Teller distortion,23,24 the layered Mn-based LMO (like LiMnO2) would prefer transforming into a spinel-like phase when sufficiently delithiated by charging to above 4.0 V. In this work, the STEM-HAADF images and FFT patterns in Fig. 5a–f clearly indicated that the Co-free LiMn0.75Ni0.25O2 shell transformed into a LiMn1.5Ni0.5O446 spinel-like shell (G-LCO → GM-LCO) after the initial cycles, whereas the LCO core still kept a layered structure.47 Though this LiMn0.75Ni0.25O2 → LiMn1.5Ni0.5O4 transformation that occurred in the shell was also irreversible, the operando-created LiMn1.5Ni0.5O4 shell kept a semi-coherent lattice with the LCO core instead of producing phase boundaries or microcracks (Fig. 5d), as both the LCO core and LiMn1.5Ni0.5O4 shell kept a coherent oxygen sublattice (ESI,† Fig. S6, Table S2 and Discussion S1). Additionally, neither this phase transformation nor the subsequent high-voltage cycling involves oxygen loss (no O2− was oxidized or released) in the spinel-like shell, so neither VO nor flaws were pumped into the particle. The quantitative sXAS analysis in Fig. 5g, showing that the Mn valence at the discharge state at the G-LCO particle surface changed from +3.4 to +4 after a few initial cycles, was absolutely consistent with the LiMn(+3.33)0.75Ni(+2)0.25O2 → LiMn(+4)1.5Ni(+2)0.5O4 transformation, and verified the formation of a VO-free shell (if there was VO in the LiMn1.5Ni0.5O4 spinel, it must contain Mn3+ ions48,49). Therefore, the robust coherent VO-free LiMn1.5Ni0.5O4 shell can stop GOM in the LCO lattice, even though HACR and O2− oxidation still occur very actively inside the LCO core. Unlike the spinel Co3O4 in P-LCO, the well-known high-voltage spinel LiMn1.5Ni0.5O4 shell does not block Li+ diffusion or electron conductivity, but supplies a 3D 8a-16c-8a path to promote fast Li+ diffusion in cycling.24,25
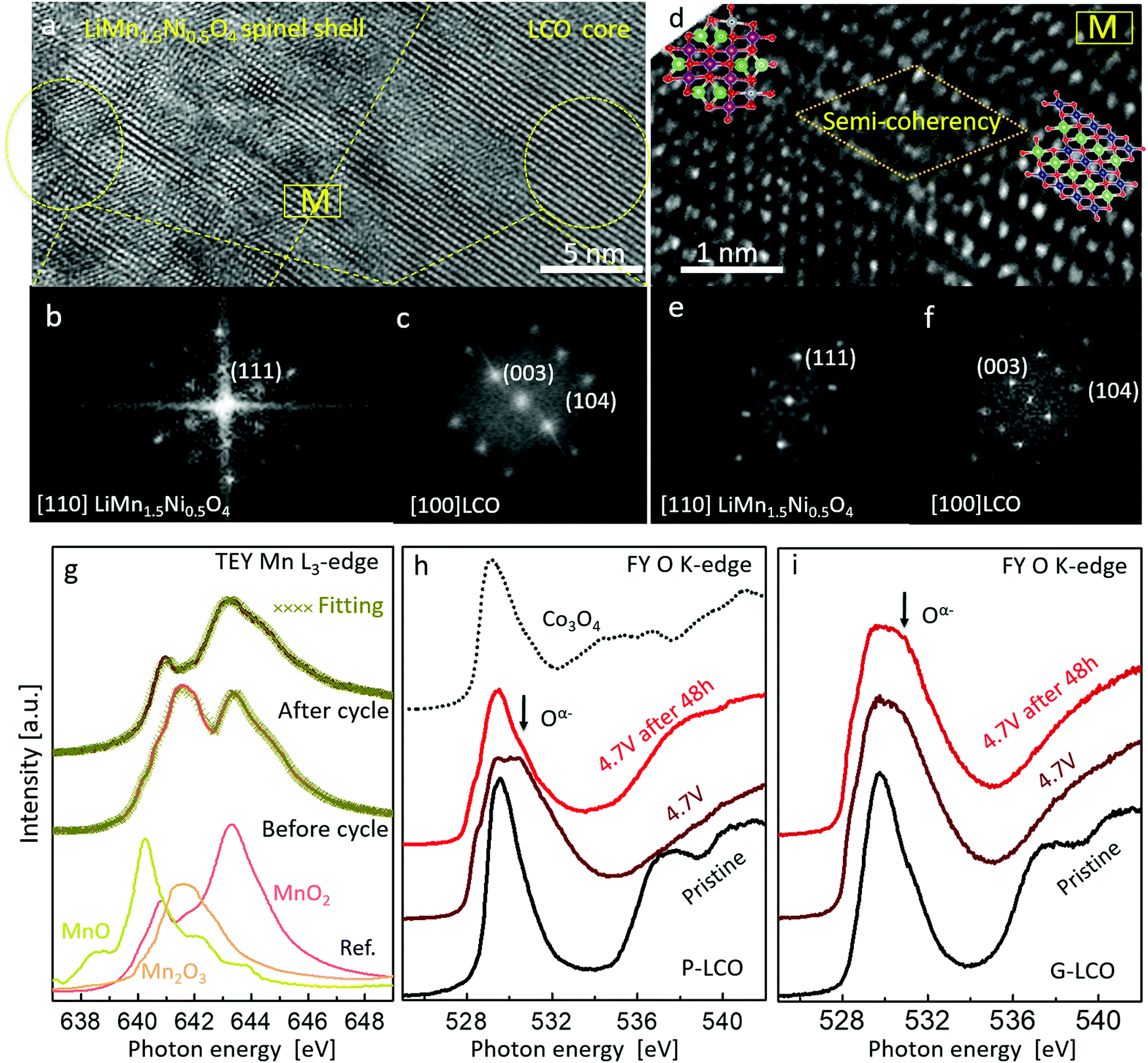 | ||
| Fig. 5 Surface reconstruction in the battery formation process and prevented oxygen migration in G-LCO particles. (a) STEM image of the G-LCO particle, showing the phase transformation into a spinel-like phase in the shell, while the core still keeping a layered structure in the G-LCO particle. (b and c) FFT patterns transformed from the spinel surface (b) and LCO bulk (c), as marked in (a) with yellow circles. (d) STEM-HAADF image of the intersection of the shell and the core, showing the semi-coherency between the operando-created spinel-like shell and the LCO core. Inserted are the simulated spinel LiMn1.5Ni0.5O4 structure (left) and LiCoO2 structure (right), where the green and red balls represent Li ions and oxygen ions, while the navy-blue, violet and grey balls represent Co, Mn and Ni ions, respectively. (e and f) FFT patterns transformed from the spinel surface (e) and LCO bulk (f), picked up from the left region and right region in (d). (g) sXAS TEY Mn L3-edge of G-LCO at the discharge state before cycling and after 2 cycles; the Mn valence was quantitatively analyzed by linear-fitting from the standard MnO, Mn2O3 and MnO2 references. (h and i) sXAS FY O K-edge spectra of P-LCO (h) and G-LCO (i) at different states of charge in the initial cycle. | ||
sXAS was performed to directly investigate HACR by tracking the oxidation states of O at high-voltages. Both P-LCO and G-LCO cathodes were charged to 4.7 V and then held for 48 h, and the FY sXAS O K-edges were recorded in this charge process. As shown in Fig. 5h and i, both the FY sXAS O K-edges of P-LCO and G-LCO were broadened toward higher energy when charged to 4.7 V, indicating that both O2− ions were oxidized to Oα− (α < 2)5,17 in the two materials. However, after holding at 4.7 V for 48 h, the peak intensity due to Oα− on the FY O K-edge of P-LCO decreased a lot, indicating that partial Oα− ions were reduced to O2− due to GOM (e.g. Oα− → O2− + O2). But the O K-edge of G-LCO remained nearly unchanged after 48 h, confirming that the oxidized Oα− was significantly stabilized in the G-LCO particle. The sXAS analysis strongly proved the prevention of GOM in the G-LCO particles at high voltages, which agreed well with the DEMS and in situ XANES mapping results in Fig. 4. The cycling stability of the fully-charged G-LCO to 4.7 V was also much improved compared to that of P-LCO (ESI,† Fig. S7).
When GOM was prevented in the G-LCO particles by the operando-created semi-coherent spinel-like nanoshell, the layered single crystal structure of the LCO core could be maintained in the high-voltage cycling, while IPT (Co3O4) continuously grew into the bulk of P-LCO. Fig. 6a shows the SEM image of P-LCO after 200 cycles between 3.0–4.60 V in half cells, which indicated that severely cracked fragments can be found everywhere at the particle surface. The collapsed surface phase (IPT) can grow thick enough towards the bulk and seriously block the charge transfer reaction. As shown in the HRTEM image of the cross-section of P-LCO after FIB preparation (Fig. 6b and c), the spinel Co3O4 almost spread across the whole particle from the surface (site A) to the center (site B). There were also lots of oxygen voids (in red circles in Fig. 6c) observed in the particle bulk because of the continuous oxygen migration and escape in the high-voltage cycling.18 The selected area electron diffraction (SAED) pattern in Fig. 6d clearly verified that lots of the layered structure had transformed into spinel Co3O4 in P-LCO after 200 cycles. However, very differently, from the SEM image in Fig. 6e, G-LCO still kept robust single-crystal morphology after 200 cycles between 3.0–4.6 V, without crack or fragment at the particle surface. The HRTEM image of G-LCO after FIB in Fig. 6f and g further indicated the well maintained layered structure throughout the whole LCO core after cycling, as the (001) lattice plane was well arranged from the deep core (site D) up to the shell (site C). The SAED pattern in Fig. 6h additionally proved that most of the layered structure was maintained in the LCO core.
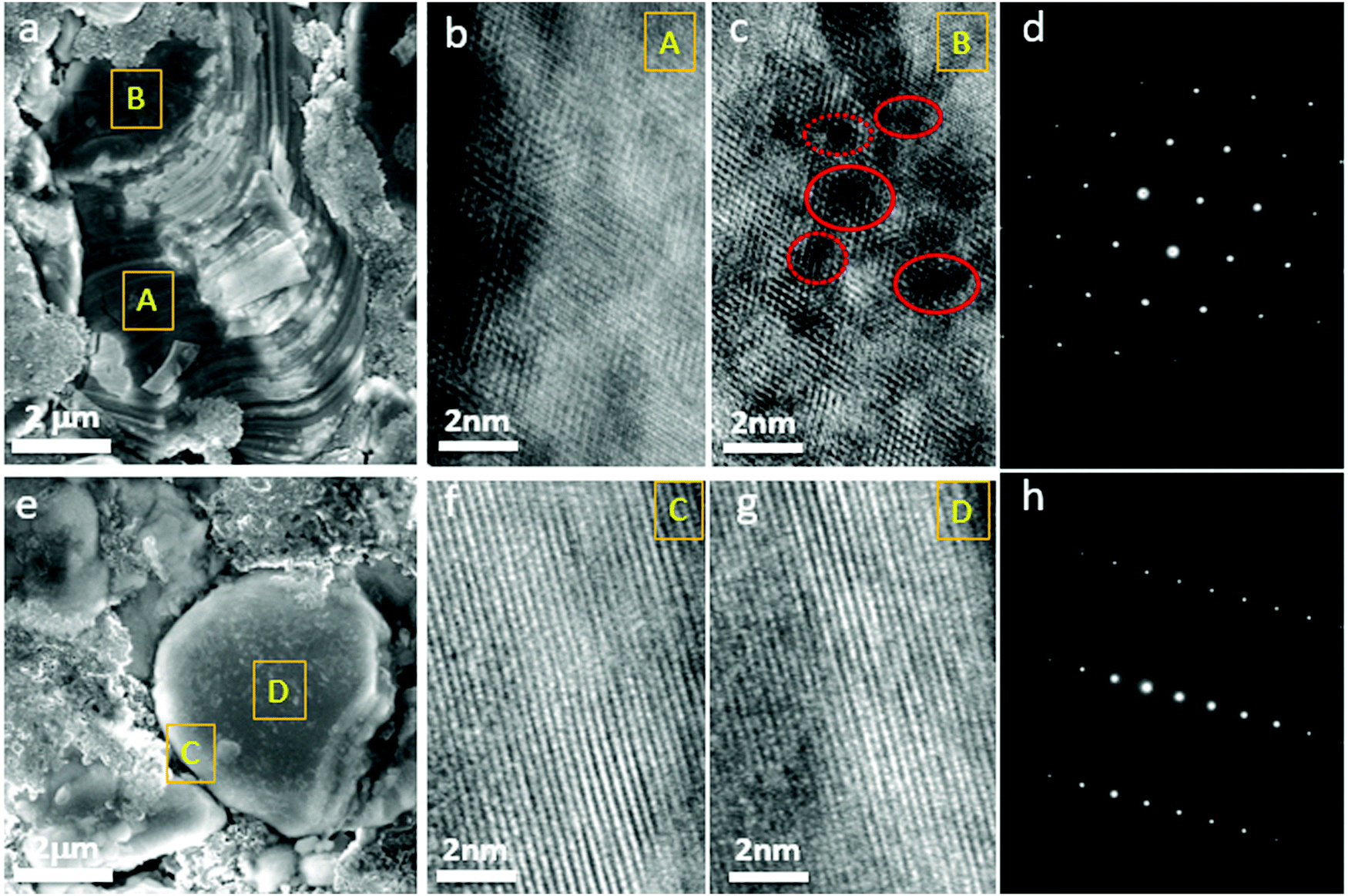 | ||
| Fig. 6 The phases of the cathode materials after long high-voltage cycles. (a) The SEM image of the P-LCO electrode after 200 cycles. (b and c) The HRTEM images of the P-LCO particles after FIB preparation, selected from site A near the particle surface and from site B in the particle bulk in (a). The red circle marks the oxygen voids in the particle after oxygen release. (d) SAED pattern of the cores in P-LCO particles. (e) The SEM image of the G-LCO electrode after 200 cycles. (f and g) The HRTEM images of the LCO core in G-LCO particles after FIB preparation, selected from site C near the shell and from site D in the particle bulk in (e). (h) SAED pattern of the cores in G-LCO particles. | ||
The stabilized phase in G-LCO would maintain good kinetics in the high-voltage cycling, which can be better understood by comparing the Li+ diffusivity (ĎLi) between P-LCO and G-LCO in the full-cells. The potentiostatic intermittent titration technique (PITT) was used to quantify ĎLi in the charging process. As shown in Fig. 7a, the ĎLi values of P-LCO and G-LCO were similar in the 3rd charging, both between 10−12–10−11 cm2 s−1; however ĎLi of P-LCO decreased to around 10−14 cm2 s−1 while that of G-LCO still remained around 10−12 cm2 s−1 after 200 cycles. Note that ĎLi here refers to the diffusion of Li+ and polarons in the full-cell, so ĎLi here can significantly indicate the practical electrochemical kinetics of the full-cell.
 | ||
| Fig. 7 Li+ diffusivity and EDX mapping of the graphite anodes after cycling. (a) The Li+ diffusivity of the full-cells assembled with P-LCO and G-LCO cathodes respectively and commercial graphite anode in the 3rd cycle and 200th cycle. (b and c) The EDX mapping of C (in yellow), F (in red) (b) and Co (c) at the graphite anode after 300 cycles in the full-cell with the P-LCO cathode. (d–f) The EDX mapping of C (in yellow), F (in red) (d), Co (e) and Mn (f) at the graphite anode after 300 cycles in the full-cell with the G-LCO cathode. | ||
Besides the cathode, the detailed condition of the graphite anode can also affect the kinetics when cycling the full-cell. The solid electrolyte interface (SEI) at the anode was analyzed by EDX mapping, as shown in Fig. 7b–f. It clearly indicated that while a similar amount of F was present at the graphite anode by comparing Fig. 7b and d, there was a massive amount of Co deposited on the surface of the graphite anode after cycling with P-LCO (Fig. 7c), which was greatly suppressed at the anode after cycling with G-LCO (Fig. 7e). The suppression of Co dissolution from the cathode and deposition at the anode significantly maintained the stability of the SEI and interfacial Li+ transfer.13,50 Additionally, as we expected, there was also very little Mn detected on the surface of the graphite anode after cycling with G-LCO (Fig. 7f), because the Mn in the shell always kept +4 in the high voltage cycling (Fig. 5g) in LiMn(+4)1.5Ni(+2)0.5O4, which have little dissolution in the carbonate electrolyte51,52 unlike the Mn3+ or Mn2+ cations.
Conclusions
We have investigated the global oxygen migration (GOM) and irreversible phase transformation (IPT) while charging LCO to high voltages by DEMS and in situ XANES mapping, and demonstrated that the reduction of transition metal cations started from some patches at the pristine LCO particle surface upon charging to above 4.3 V, which immediately triggered IPT into a bad spinel (CoO2 → Co3O4) to block the Li-ion transport, resulting in a rapid cycling fading. In order to prevent the harmful GOM and uncontrollable phase transformations at high voltages, we intentionally created a thin LiMn(+3.33)0.75Ni(+2)0.25O2 layer epitaxially wrapping the LCO single crystal seed by high-temperature processing and abnormal grain growth, which templates a future O-and-TM-preserving surface transformation into the good spinel LiMn1.5Ni0.5O4 that shares the same oxygen sublattice with LCO. We have proved that the oxidized oxygen ions were significantly stabilized in the LCO core when it was even fully-charged. The elimination of GOM stabilizes hybrid anion- and cation-redox (HACR) reactions in the high-voltage cycling, and preserves the good kinetics of LCO single crystals. The test in pouch full-cells with a commercial graphite anode further showed an extremely stable cycling performance when cycled to 4.55 V. Therefore, a cathode electrode with a super high volumetric energy density of 2906 W h L−1 has been achieved after 300 cycles in a full-cell.For generalization, this gradient-morph (GM-LMO) single-crystal structure can also be applied to other cathode materials as a general strategy to establish a desirable specific shell that cannot be directly created on a particle. We can firstly coat the particle with an intermediate layer that is isostructural with the bulk to realize the integral wrapping by a regular high-temperature reaction (prepare G-LMO first), and then it can transform in operando into the eventual desirable shell (e.g. spinel, G-LMO → GM-LMO) to fully enclose and protect the particle in prolonged cycling.
Methods
Preparation of the LiMn0.75Ni0.25O2 → LiCoO2 gradient morph single crystals
The pristine LiCoO2 single crystals were synthesized by a solid-reaction method. Firstly, Co3O4 (≥99%, Sigma-Aldrich) and Li2CO3 (ACS Reagent, ≥99%, Sigma-Aldrich) were sufficiently mixed with a mole ratio of 1:1.5 (with 5% excess of Li2CO3); then the mixture was heated at 1000–1100 °C for 10 hours with a heating and cooling rate of 5 °C min−1 to get the pristine LCO particles (P-LCO).
The pristine LCO particles were centrifuged with water to remove the small particles with size below micro-meters, and then ultrasonicated in an ethanol solution with LiCOOCH3 (Reagent Plus®, ≥99%, Sigma-Aldrich), Mn(COOCH3)2 (Reagent Plus®, ≥99%, Sigma-Aldrich) and Ni(COOCH3)2 (Reagent Plus®, ≥99%, Sigma-Aldrich) dissolved with a mole ratio of 1.05:0.75:0.25. After that, the mixture was dried in a 60–80 °C water bath with stirring. Finally, the obtained powder was heated at 900 °C for 8 hours to get the gradient-morph single-crystal product (G-LCO).
Electrochemical test
The cathode electrode was made of 96wt% active material, 2wt% carbon black, and 2wt% polyvinylidene fluoride (PVDF) binder, which was pasted on an Al current collector and compressed under 20 MPa. R2032 coin cells were fabricated with the above cathode, Li metal anode, a Celgard 2400 polymeric separator and a commercial electrolyte solution of 1.2 M LiPF6 dissolved in a mixture of EC and DEC with a volume ratio of 1:1, and 2wt% vinylene carbonate additive. Pouch full-cells were fabricated with the above cathodes, a commercial graphite anode (double-side coated), a separator of Celgard 2400 polymer and a commercial electrolyte solution of 1.2 M LiPF6 dissolved in a mixture of EC and DEC with a volume ratio of 1:1, and 2wt% vinylene carbonate additive. The loading density of LCO cathode was about ∼17 mg cm−2 with ∼3.5 mA h cm−2 (tested in half-cell under 0.1C); the commercial graphite anode had a loading density of ∼12 mg cm−2 with ∼3.8 mA g cm−2 (tested in half-cell under 0.1C). The pouch full cells were fabricated with double layers of electrodes (one graphite anode foil with both sides coated and two LCO cathode foils with a single side coated). The amount of electrolyte added was about 2 g A h−1 in the pouch cell. A LAND CT2001A 8-channel automatic battery test system (Wuhan Lanhe Electronics) was used for charging/discharging of the cells. An electrochemical workstation (Gamry Instr, Reference 3000) was used for the cyclic voltammetry (CV) test between 3.0 V and 5.0 V with 0.05 mV s−1 scanning rate, and for the potentiostatic intermittent titration technique (PITT) with constant potential for 200 seconds followed by 1800 seconds of relaxation with a voltage-step of 40 mV from 3.8 V to 4.6 V. The electrochemical tests were all carried out at room temperature. A self-made quantitative DEMS was used to detect and analyze the gas during the cell testing. Two glued polyether ether ketone (PEEK) capillary tubes were used as the inlet and outlet of gas. The cell was fabricated in a glove box where O2 <0.1ppm. Then, the output tube was connected to a Thermo Scientific mass spectrometer (MS). High-purity Ar gas was used as the carrier gas with a flow rate of 3 mL min−1 during the cycling process. In the constant current charging process, the current was 50mA g−1, and MS spectra were collected every 30 seconds.
Characterizations
X-ray diffraction was performed on a PANalytical X’Pert PRO X-ray diffractor using a Cu target under 45 kV and 40 mA. The data were collected with 2° min−1 and analyzed with the HighScore Plus software. Scanning electron microscopy was performed on a Zeiss Merlin high-resolution scanning electron microscope with X-ray spectroscopy (EDX). High-angle annular dark field (HAADF) images, energy dispersive X-ray spectroscopy (EDS) and electron energy loss spectroscopy (EELS) spectra were acquired from a JEOL ARM 200F scanning transmission electron microscope (STEM) operated at 200 keV, equipped with a cold field emission gun and integrated aberration (Cs) corrector. Dual EELS spectrometers were used to collect both the low-loss and high-loss spectra, and the low-loss spectra were utilized to correct the drift of the zero-loss peak (ZLP). Based on the full-width at half-maximum (FWHM) of the zero-loss peak (ZLP), the optimal energy-resolution of EELS was about 1 eV. The STEM sample was prepared using an FEI Helios NanoLab 600 DualBeam FIB/SEM equipped with a Ga ion source. A Pt layer was deposited on top to protect the cathode particle. High-resolution transmission electron microscopy (HRTEM) images and selected area electron diffraction (SAED) patterns were taken on a JEOL 2010F transmission electron microscope operated at 200 kV.In situ nano-XANES mapping
In situ nano-XANES mapping was performed at the FXI beamline (18-ID) at the National Synchrotron Light Source II (NSLS-II), Brookhaven National Laboratory. Pouch-cell configurations with P-LCO and G-LCO as cathodes were used for measurements. Each pouch cell was charged at a constant current of 8 mA and stopped at different voltages (4.2 V, 4.4 V and 4.6 V) while collecting XANES spectra. A volume in the middle of the cell was randomly chosen for imaging. XANES images were taken at different energies across the Co absorption edge (7.588–8.153 keV, 1 eV interval). The effective pixel size of each image was 21 nm. Standard samples (CoO and LiCoO2) were used to extract the reference absorption spectra for Co2+ and Co3+ oxidation states.Soft X-ray absorption spectroscopy (sXAS) measurements
Soft X-ray absorption spectroscopy (sXAS) measurements were carried out at the IOS beamline (23-ID-2) at the National Synchrotron Light Source II (NSLS-II), Brookhaven National Laboratory. Spectra were acquired in total electron yield (TEY) and partial fluorescence yield (PFY) modes. The estimated incident X-ray energy resolution was ∼0.05eV at the O K-edge. The monochromator absorption features and beam instabilities were normalized out by dividing the detected PFY and TEY signals by the drain current of a clean gold I0 mesh placed in the incident beam. TEY spectra were recorded from the drain current of the sample and PFY data were acquired using a Vortex EM silicon drift detector. The sXAS spectra for the O K-edge were recorded over a wide energy range from 520 to 565 eV covering energies well below and above sample absorptions. The normalization of the O K-edge was performed:53 (1) I0 normalization: the sample signal was divided by the incident intensity measured from the sample drain current from a freshly coated Au mesh inserted into the beam path before the X-rays could impinge on the sample. (2) The linear, sloping background was removed by fitting a line to the flat low energy region (520–524 eV) of the sXAS spectrum. (3) The spectrum was normalized by setting the flat low energy region to zero and the post-edge to unity (unit edge-jump). The photon energy selected for the post edge was 560 eV, beyond the region of any absorption (peaks).
List of abbreviations
| TM | Transition metal |
| LIB | Li ion batteries |
| LCO | LiCoO2 |
| HACR | Hybrid anion- and cation-redox |
| OM | Oxygen migration |
| GOM | Global oxygen migration |
| sXAS | Soft X-ray absorption spectroscopy |
| IPT | Irreversible phase transformation |
| SCC | Stress-corrosion cracking |
| G-LCO | Gradient LiMn0.75Ni0.25O2 → LiCoO2 single crystal |
| GM-LCO | Gradient-morph LCO single crystal |
| P-LCO | Pristine LCO |
| AGG | Abnormal grain growth |
| XRD | X-ray diffraction |
| SEM | Scanning electron microscopy |
| HRTEM | High resolution transmission electron microscopy |
| STEM-HAADF | Scanning transmission electron microscopy-high angle annular dark field |
| CI | Coulombic inefficiency |
| FIB | Focused ion beam |
| DEMS | Differential electrochemical mass spectroscopy |
| SOCs | States of charges |
| XANES | X-ray absorption near edge structure |
| SAED | Selected area electron diffraction |
| Ď Li | Li+ diffusivity |
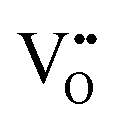
| Oxygen vacancy |
| FFT | Fast Fourier transform |
Author contributions
Z. Z. and J. L. conceived and designed the experiments. Z. Z. synthesized the materials, did the material characterization and performed the electrochemical measurements. D. Y. performed the aberration corrected STEM imaging, STEM-EDS mapping, EELS line scan, HRTEM imaging and in situ nano-XANES mapping. R. G. did the FIB sample preparation for STEM characterizations. Z. S. performed the lattice coherency calculation and analysis. I. W. measured the sXAS experiments. Z. Z., D. Y. and J. L. wrote the paper. All authors analyzed the data, discussed the results and commented on the manuscript.Data availability
The data that support the plots in this paper and other findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We acknowledge the support from the Watt Energy Technology Lab gift fund. This research used resources of the beamlines 18-ID (FXI) and 23-ID-2 (IOS) of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. This work was performed in part at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Coordinated Infrastructure Network (NNCI), which is supported by the National Science Foundation under NSF award No. 1541959.References
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Lixcoo2 (O Less-Than X, Less-Than-Or-Equal-To 1) - a New Cathode Material for Batteries Of High-Energy Density, Solid State Ionics, 1981, 3-4, 171–174, DOI:10.1016/0167-2738(81)90077-1.
- K. Ozawa, Lithium-Ion Rechargeable Batteries with Licoo2 And Carbon Electrodes - the Licoo2 C System, Solid State Ionics, 1994, 69, 212–221, DOI:10.1016/0167-2738(94)90411-1.
- S. Kalluri, et al., Surface Engineering Strategies of Layered LiCoO2 Cathode Material to Realize High-Energy and High-Voltage Li-Ion Cells, Adv. Energy Mater., 2017, 7, 1601507 CrossRef.
- Q. Liu, et al., Approaching the capacity limit of lithium cobalt oxide in lithium ion batteries via lanthanum and aluminium doping, Nat. Energy, 2018, 3, 936–943, DOI:10.1038/s41560-018-0180-6.
- J. N. Zhang, et al., Trace doping of multiple elements enables stable battery cycling of LiCoO2 at 4.6V, Nat. Energy, 2019, 4, 594–603, DOI:10.1038/s41560-019-0409-z.
- J. W. Qian, et al., Electrochemical surface passivation of LiCoO2 particles at ultrahigh voltage and its applications in lithium-based batteries, Nat. Commun., 2018, 9, 4918, DOI:10.1038/S41467-018-07296-6.
- A. Van der Ven, M. K. Aydinol, G. Ceder, G. Kresse and J. Hafner, First-principles investigation of phase stability in LixCoO2, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 2975–2987, DOI:10.1103/PhysRevB.58.2975.
- H. Xia, L. Lu, Y. S. Meng and G. Ceder, Phase transitions and high-voltage electrochemical behavior of LiCoO2 thin films grown by pulsed laser deposition, J. Electrochem. Soc., 2007, 154, A337–A342, DOI:10.1149/1.2509021.
- M. D. Radin, et al., Narrowing the Gap between Theoretical and Practical Capacities in Li-Ion Layered Oxide Cathode Materials, Adv. Energy Mater., 2017, 7, 1602888, DOI:10.1002/Aenm.201602888.
- T. Ohzuku and A. Ueda, Solid-State Redox Reactions Of Licoo2 (R(3)over-Bar-M) for 4 Volt Secondary Lithium Cells, J. Electrochem. Soc., 1994, 141, 2972–2977, DOI:10.1149/1.2059267.
- X. W. Yu and A. Manthiram, Electrode-electrolyte interfaces in lithium-based batteries, Energy Environ. Sci., 2018, 11, 527–543, 10.1039/c7ee02555f.
- D. Takamatsu, et al., First In Situ Observation of the LiCoO2 Electrode/Electrolyte Interface by Total-Reflection X-ray Absorption Spectroscopy, Angew. Chem., Int. Ed., 2012, 51, 11597–11601, DOI:10.1002/anie.201203910.
- A. Yano, M. Shikano, A. Ueda, H. Sakaebe and Z. Ogumi, LiCoO2 Degradation Behavior in the High-Voltage Phase Transition Region and Improved Reversibility with Surface Coating, J. Electrochem. Soc., 2017, 164, A6116–A6122, DOI:10.1149/2.0181701jes.
- M. Yoon, et al., Unveiling Nickel Chemistry in Stabilizing High-Voltage Cobalt-Rich Cathodes for Lithium-Ion Batteries, Adv. Funct. Mater., 2019, 1907903, DOI:10.1002/adfm.201907903.
- S. Venkatraman, Y. Shin and A. Manthiram, Phase relationships and structural and chemical stabilities of charged Li1−xCoO2-delta and Li1−xNi0.85Co0.15O2-delta cathodes, Electrochem. Solid-State Lett., 2003, 6, A9–A12, DOI:10.1149/1.1525430.
- W. S. Yoon, et al., Oxygen contribution on Li-ion intercalation-deinterealation in LiCoO2 investigated by OK-edge and Co L-edge X-ray absorption spectroscopy, J. Phys. Chem. B, 2002, 106, 2526–2532, DOI:10.1021/jp013735e.
- W. S. Yoon, et al., Oxygen contribution on Li-ion intercalation-deintercalation in LiAlyCo1−yO2 investigated by O K-edge and Co L-edge X-ray absorption spectroscopy, J. Electrochem. Soc., 2002, 149, A1305–A1309, DOI:10.1149/1.1503074.
- P. F. Yan, et al., Injection of oxygen vacancies in the bulk lattice of layered cathodes, Nat. Nanotechnol., 2019, 14, 602, DOI:10.1038/s41565-019-0428-8.
- E. Lee and K. A. Persson, Structural and Chemical Evolution of the Layered Li-Excess LixMnO3 as a Function of Li Content from First-Principles Calculations, Adv. Energy Mater., 2014, 4, 1400498, DOI:10.1002/aenm.201400498.
- Z. Zhu, et al., Gradient Li-rich oxide cathode particles immunized against oxygen release by a molten salt treatment, Nat. Energy, 2019, 4, 1049–1058, DOI:10.1038/s41560-019-0508-x.
- G. G. Amatucci, J. M. Tarascon and L. C. Klein, CoO2, the end member of the LixCoO2 solid solution, J. Electrochem. Soc., 1996, 143, 1114–1123, DOI:10.1149/1.1836594.
- J. Kikkawa, et al., Chemical States of Overcharged LiCoO2 Particle Surfaces and Interiors Observed Using Electron Energy-Loss Spectroscopy, J. Phys. Chem. C, 2015, 119, 15823–15830, DOI:10.1021/acs.jpcc.5b02303.
- T. Bartsch, et al., Gas Evolution in All-Solid-State Battery Cells, ACS Energy Lett., 2018, 3, 2539–2543, DOI:10.1021/acsenergylett.8b01457.
- M.-J. Lee, et al., Low-Temperature Carbon Coating of Nanosized Li1.015Al0.06Mn1.925O4 and High-Density Electrode for High-Power Li-Ion Batteries, Nano Lett., 2017, 17, 3744–3751, DOI:10.1021/acs.nanolett.7b01076.
- L. Suo, et al., Fluorine-donating electrolytes enable highly reversible 5-V-class Li metal batteries, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156, DOI:10.1073/pnas.1712895115.
- A. Singer, et al., Nucleation of dislocations and their dynamics in layered oxide cathode materials during battery charging, Nat. Energy, 2018, 3, 641–647, DOI:10.1038/s41560-018-0184-2.
- L. de Biasi, et al., Chemical, Structural, and Electronic Aspects of Formation and Degradation Behavior on Different Length Scales of Ni-Rich NCM and Li-Rich HE-NCM Cathode Materials in Li-Ion Batteries, Adv. Mater., 2019, 31, 1900985, DOI:10.1002/Adma.201900985.
- S. Kalluri, et al., Surface Engineering Strategies of Layered LiCoO2 Cathode Material to Realize High-Energy and High-Voltage Li-Ion Cells, Adv. Energy Mater., 2017, 7, 1601507 CrossRef.
- Z. Zhu, D. Zhang, H. Yan, W. Li and Q. Lu, Precise preparation of high performance spherical hierarchical LiNi0.5Mn1.5O4 for 5 V lithium ion secondary batteries, J. Mater. Chem. A, 2013, 1, 5492–5496, 10.1039/c3ta10980a.
- Z. Zhu, H. Yan, D. Zhang, W. Li and Q. Lu, Preparation of 4.7 V cathode material LiNi0.5Mn1.5O4 by an oxalic acid-pretreated solid-state method for lithium-ion secondary battery, J. Power Sources, 2013, 224, 13–19, DOI:10.1016/j.jpowsour.2012.09.043.
- Z. Zhu, Q. Lu, D. Zhang and H. Y. Yu, Preparation of spherical hierarchical LiNi0.5Mn1.5O4 with high electrochemical performances by a novel composite co-precipitation method for 5 V lithium ion secondary batteries, Electrochim. Acta, 2014, 115, 290–296, DOI:10.1016/j.electacta.2013.10.167.
- J. N. Reimers, E. W. Fuller, E. Rossen and J. R. Dahn, Synthesis And Electrochemical Studies Of Limno2 Prepared Of Low-Temperatures, J. Electrochem. Soc., 1993, 140, 3396–3401, DOI:10.1149/1.2221101.
- I. Koetschau, M. N. Richard, J. R. Dahn, J. B. Soupart and J. C. Rousche, Orthorhombic Limno2 as a High-Capacity Cathode for Li-Ion Cells, J. Electrochem. Soc., 1995, 142, 2906–2910, DOI:10.1149/1.2048663.
- Z. Zhu, et al., Anion-redox nanolithia cathodes for Li-ion batteries, Nat. Energy, 2016, 1, 16111, DOI:10.1038/Nenergy.2016.111.
- Y. Qiao, K. Jiang, H. Deng and H. Zhou, A high-energy-density and long-life lithium-ion battery via reversible oxide–peroxide conversion, Nat. Catal., 2019, 2, 1035–1044, DOI:10.1038/s41929-019-0362-z.
- W. Xue, et al., Intercalation-conversion hybrid cathodes enabling Li–S full-cell architectures with jointly superior gravimetric and volumetric energy densities, Nat. Energy, 2019, 4, 374–382, DOI:10.1038/s41560-019-0351-0.
- Y. H. Zhen and J. F. Li, Abnormal grain growth and new core-shell structure in (K,Na)NbO3-based lead-free piezoelectric ceramics, J. Am. Ceram. Soc., 2007, 90, 3496–3502, DOI:10.1111/j.1551-2916.2007.01977.x.
- B. K. Lee, S. Y. Chung and S. J. L. Kang, Grain boundary faceting and abnormal grain growth in BaTiO3, Acta Mater., 2000, 48, 1575–1580, DOI:10.1016/S1359-6454(99)00434-6.
- Y.-K. Sun, et al., Nanostructured high-energy cathode materials for advanced lithium batteries, Nat. Mater., 2012, 11, 942–947, DOI:10.1038/nmat3435.
- Y. Jin, et al., Self-healing SEI enables full-cell cycling of a silicon-majority anode with a coulombic efficiency exceeding 99.9%, Energy Environ. Sci., 2017, 10, 580–592, 10.1039/c6ee02685k.
- J. Kasnatscheew, et al., The truth about the 1st cycle coulombic efficiency of LiNi1/3Co1/3Mn1/3O2 (NCM) cathodes, Phys. Chem. Chem. Phys., 2016, 18, 3956–3965, 10.1039/c5cp07718d.
- P. Oh, et al., High-Performance Heterostructured Cathodes for Lithium-Ion Batteries with a Ni-Rich Layered Oxide Core and a Li-Rich Layered Oxide Shell, Adv. Sci., 2016, 3, 1600184, DOI:10.1002/Advs.201600184.
- M. Y. Ge, et al., One minute nano-tomography using hard X-ray full-field transmission microscope, Appl. Phys. Lett., 2018, 113, 083109, DOI:10.1063/1.5048378.
- D. S. Coburn, et al., Design, characterization, and performance of a hard x-ray transmission microscope at the National Synchrotron Light Source II 18-ID beamline, Rev. Sci. Instrum., 2019, 90, 053701 CrossRef PubMed.
- X. Xiao, et al., Full-field X-ray Imaging, a Workhorse Microscopy Beamline at NSLS II for Material Science Researches, Microsc. Microanal., 2019, 25, 386–387 CrossRef.
- J. M. Zheng, et al., Enhanced Li+ ion transport in LiNi0.5Mn1.5O4 through control of site disorder, Phys. Chem. Chem. Phys., 2012, 14, 13515–13521, 10.1039/c2cp43007j.
- Z. Li, et al., Structural study of epitaxial LiCoO2 films grown by pulsed laser deposition on single crystal SrTiO3 substrates, Thin Solid Films, 2016, 612, 472–482, DOI:10.1016/j.tsf.2016.05.017.
- J. Song, et al., Role of Oxygen Vacancies on the Performance of Li[Ni0.5−xMn1.5+x]O-4 (x = 0, 0.05, and 0.08) Spinel Cathodes for Lithium-Ion Batteries, Chem. Mater., 2012, 24, 3101–3109, DOI:10.1021/cm301825h.
- J. Xiao, et al., High-Performance LiNi0.5Mn1.5O4 Spinel Controlled by Mn3+ Concentration and Site Disorder, Adv. Mater., 2012, 24, 2109–2116, DOI:10.1002/adma.201104767.
- Y. S. Jung, et al., Enhanced Stability of LiCoO2 Cathodes in Lithium-Ion Batteries Using Surface Modification by Atomic Layer Deposition, J. Electrochem. Soc., 2010, 157, A75–A81, DOI:10.1149/1.3258274.
- C. Zhan, T. P. Wu, J. Lu and K. Amine, Dissolution, migration, and deposition of transition metal ions in Li-ion batteries exemplified by Mn-based cathodes - a critical review, Energy Environ. Sci., 2018, 11, 243–257, 10.1039/c7ee03122j.
- W. Choi and A. Manthiram, Comparison of metal ion dissolutions from lithium ion battery cathodes, J. Electrochem. Soc., 2006, 153, A1760–A1764, DOI:10.1149/1.2219710.
- K. Luo, et al., Charge-compensation in 3d-transition-metal-oxide intercalation cathodes through the generation of localized electron holes on oxygen, Nat. Chem., 2016, 8, 684–691, DOI:10.1038/nchem.2471.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee00231c |
| ‡ Z. Z. and D. Y. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |