
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiorbital bond formation for stable oxygen-redox reaction in battery electrodes†
Takaaki
Sudayama
a,
Kazuki
Uehara
b,
Takahiro
Mukai
c,
Daisuke
Asakura
 a,
Xiang-Mei
Shi
b,
Akihisa
Tsuchimoto
b,
Benoit
Mortemard de Boisse
bd,
Tatau
Shimada
b,
Eriko
Watanabe
b,
Yoshihisa
Harada
e,
Masanobu
Nakayama
cdf,
Masashi
Okubo
bd and
Atsuo
Yamada
*bd
a,
Xiang-Mei
Shi
b,
Akihisa
Tsuchimoto
b,
Benoit
Mortemard de Boisse
bd,
Tatau
Shimada
b,
Eriko
Watanabe
b,
Yoshihisa
Harada
e,
Masanobu
Nakayama
cdf,
Masashi
Okubo
bd and
Atsuo
Yamada
*bd
aResearch Institute for Energy Conservation, National Institute of Advanced Industrial Science and Technology, Tsukuba, Ibaraki 305-8568, Japan
bDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: yamada@chemsys.t.u-tokyo.ac.jp
cFrontier Research Institute for Materials Science (FRIMS), Nagoya Institute of Technology, Showa-ku, Nagoya, Aichi 466-8555, Japan
dElements Strategy Initiative for Catalysts & Batteries (ESICB), Kyoto University, Nishikyo-ku, Kyoto 615-8245, Japan
eInstitute for Solid State Physics, The University of Tokyo, Kashiwa, Chiba 277-8581, Japan
fMi2i GREEN, National Institute of Materials Science (NIMS), Tsukuba, Ibaraki 305-0047, Japan
First published on 12th March 2020
Abstract
High-energy-density batteries have been a long-standing target toward sustainability, but the energy density of state-of-the-art lithium-ion batteries is limited in part by the small capacity of the positive electrode materials. Although employing the additional oxygen-redox reaction of Li-excess transition-metal oxides is an attractive approach to increase the capacity, an atomic-level understanding of the reaction mechanism has not been established so far. Here, using bulk-sensitive resonant inelastic X-ray scattering spectroscopy combined with ab initio computations, we demonstrate the presence of a localized oxygen 2p orbital weakly hybridized with transition metal t2g orbitals that was theoretically predicted to play a key role in oxygen-redox reactions. After oxygen oxidation, the hole in the oxygen 2p orbital is stabilized by the generation of either a (σ + π) multiorbital bond through strong π back-donation or peroxide O22− through oxygen dimerization. The multiorbital bond formation with σ-accepting and π-donating transition metals can thus lead to reversible oxygen-redox reaction.
Broader contextIncreasing the energy density of lithium-ion batteries is an urgent task toward achieving a sustainable society. However, the limited capacities of cathodes render lithium-ion batteries unsuitable for electric transport or grid-scale applications. As an effective way to increase the capacity of cathodes, additional oxygen-redox reactions have attracted much attention. Although the presence of “orphaned” oxygen 2p orbitals that are responsible for additional oxygen-redox reactions was theoretically proposed in 2016, no experimental verification has been reported to date. In this work, we experimentally verify the presence of a localized oxygen 2p orbital weakly hybridized with transition metal t2g orbitals in oxygen-redox battery cathodes using resonant inelastic X-ray scattering. We also clarify the stabilization mechanism of the oxidized phase through multiorbital bond formation of oxidized oxygen 2p orbitals. |
Introduction
The development of advanced electrochemical energy storage devices has been a topic of intensive research because it offers prospects of sustainability, for instance, reduction of fossil fuel reliance by realizing an electric vehicle range of 500 km per charge or integrating renewable energy sources to an electrical grid.1–3 Market-leading lithium-ion batteries (LIBs) efficiently store energy by lithium-ion (de)intercalation associated with redox reactions.4,5 However, their energy density has approached the theoretical limit, in part owing to the small capacity of the positive electrode materials, which severely hinders the wide deployment of LIBs.6 Therefore, it is important to find an alternative battery chemistry that can exceed the existing positive-electrode capacity limits.The use of redox reactions of oxygen in addition to conventional transition-metal redox reactions is an attractive way to increase the capacity of transition-metal oxides.7 In general, most oxygen 2p orbitals in conventional electrode materials LiMO2 (M: transition metal) form σ-type bonds with axial M 3d orbitals (eg orbitals in an Oh symmetry). As the atomic energy level of oxygen 2p orbitals is usually lower than that of M eg orbitals, the σ-type bonding orbitals are predominantly from oxygen 2p orbitals.8,9 Therefore, it is difficult to oxidize oxygen 2p orbitals that have a σ-type bonding character.10 However, research groups led by Bruce and Ceder postulated that oxygen in Li-excess transition-metal oxides Li1+xM1−xO2 (Fig. 1a) intrinsically have localized (“orphaned”) 2p orbital along the Li–O–Li axis without σ-type bonding characteristics, which can contribute to an additional oxygen-redox capacity.10,11 Indeed, large capacities exceeding the capacity limit of M redox reactions have been reported for many Li-excess transition-metal oxides including Li2MnO3–LiMO2 solid solutions,12,13 layered honeycomb Li2IrO3,14 random rock salt Li1.2Ti0.4Mn0.4O2,15 three-dimensional β-Li2IrO3,16 and anti-fluorite Li5FeO4.17 In addition, the electronic states of various oxygen-redox electrode materials have been intensively investigated using analytical methods such as X-ray photoelectron spectroscopy, X-ray absorption spectroscopy, X-ray emission spectroscopy (or resonant inelastic X-ray scattering (RIXS)), or density functional theory (DFT) calculations.18–22
 | ||
| Fig. 1 Resonant inelastic X-ray scattering (RIXS) spectra for delocalized and localized oxygen 2p orbitals. (a) Crystal structure of Li-excess layered transition-metal oxides Li[LixM1−x]O2 (M: transition metal). (b) π-type localized occupied/unoccupied states consisting of oxygen 2p and M t2g orbitals (b1/b1* bands in a C2v-symmetry OM2Li4 cluster) give a Raman-like RIXS peak with constant energy loss regardless of the excitation energy. (c) σ-type delocalized occupied/unoccupied states consisting of oxygen 2p and M eg orbitals (3a1/3a1* and 3b2/3b2* bands in a C2v-symmetry OM2Li4 cluster) give a fluorescence-like RIXS peak with variable energy loss depending on the excitation energy. | ||
However, despite the theoretical adequacy of the proposed “orphaned” oxygen 2p orbital hypothesis, the presence of the localized oxygen 2p orbitals in Li-excess transition-metal oxides has not been verified experimentally. Furthermore, although localized, the oxygen 2p orbital along the Li–O–Li axis still exhibits π-type interactions with the M t2g orbital, which should play an essential role in oxygen-redox reactions.23 It is important to emphasize that the rigid-band model, which has been frequently used for consideration of oxygen-redox electrodes,15,24 cannot qualitatively discuss M–O interaction (e.g., covalency), because it only considers a bandwidth and the band-center energy of each band. Therefore, for a thorough understanding of the reversible oxygen-redox reactions occurring in Li-excess transition-metal oxides, it is necessary to clarify the nature of the M–O bonds during the oxygen-redox reactions.25
In this work, we used oxygen K-edge resonant inelastic X-ray scattering (RIXS) spectroscopy combined with ab initio computations to demonstrate a criterion for stable oxygen-redox reactions.
Results and discussion
RIXS to detect localized orbitals
Oxygen K-edge RIXS spectroscopy was used to examine the existence of π-type localized oxygen 2p orbitals in Li2MnO3, a standard Li-excess layered transition-metal oxide. Li2MnO3 was synthesized by a conventional solid-state method, and the powder X-ray diffraction pattern and Rietveld refinement confirmed its successful synthesis (Fig. S1 and Table S1, ESI†). Li2MnO3 that contains only Mn4+ enables obtaining a simple RIXS spectrum, allowing for direct comparison with DFT calculations. Oxygen K-edge RIXS spectroscopy records the inelastic scattering during core excitation–relaxation (1s → 2p → 1s), in which the energy loss corresponds to valence excitation between occupied and unoccupied 2p bands.26 RIXS spectroscopy has a probing depth of approximately 100 nm (bulk-sensitive). Importantly, when occupied/unoccupied oxygen 2p bands show a localized character with weak orbital hybridization, the inelastic scattering becomes Raman-like and leads to constant energy loss regardless of the incident photon energy (Fig. 1b and Fig. S2a, ESI†).27 Conversely, delocalized occupied/unoccupied oxygen 2p bands with a large orbital hybridization give fluorescence-like inelastic scattering with variable energy loss depending on the incident photon energy (Fig. 1c and Fig. S2b, ESI†).27 Therefore, oxygen K-edge RIXS spectroscopy allows us to diagnose localized or delocalized oxygen 2p bands.Before obtaining the RIXS spectra, we conducted ab initio calculations using the HSE06 hybrid functional28 for Li2MnO3. Fig. 2a shows that a localized oxygen 2p orbital exists along the Li–O–Li axis with an orbital energy of approximately −0.9 to 0.0 eV against the Fermi energy. The RIXS spectra and the corresponding second differential RIXS map (Fig. 2b and c) exhibit two broad fluorescence-like peaks (the energy loss ranging from 3.5 to 8.0 eV), which could be related to the valence excitation between the σ-type wide oxygen 2p bands strongly hybridized with axial Mn eg orbitals (Fig. 1c). Based on the C2v symmetry of an OMn2Li4 cluster, these bands are labelled as 3a1/3b2 bands.8,23 More importantly, the RIXS spectra show an intense Raman-like peak with constant energy loss of approximately 2.5 eV. This Raman-like peak clearly indicates the existence of the π-type narrow oxygen 2p bands weakly hybridized with Mn t2g orbitals (b1/b1* bands based on the C2v-symmetry labelling, Fig. 1b). It is important to note that the Raman-like feature is not the simple projection of the d–d excitation of Mn L-edge, because O K-edge XAS and RIXS always involve charge-transfer processes through O–Mn interactions (Fig. S3, ESI†). Therefore, whether Raman- or fluorescence-like RIXS is observed depends primarily on the strength of the O–Mn interactions.
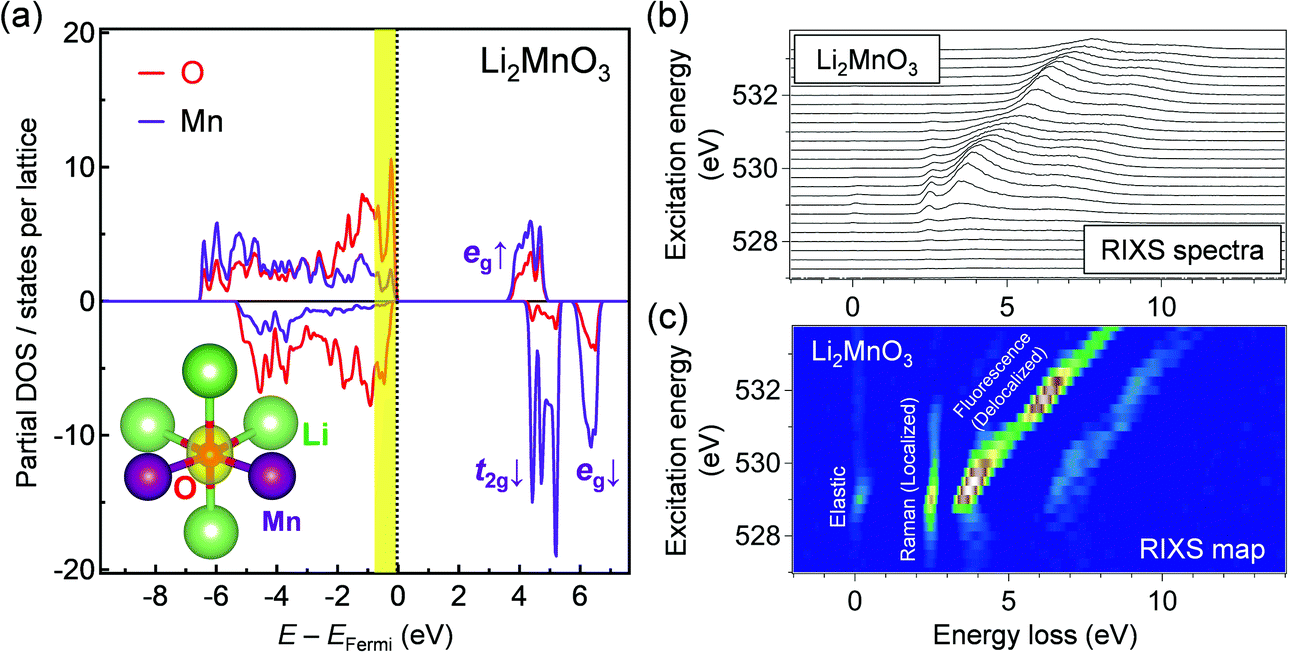 | ||
| Fig. 2 Direct observation of a localized oxygen 2p orbital hybridized with Mn t2g orbitals in Li2MnO3. (a) Calculated partial density of states for oxygen and manganese in Li2MnO3. The inset shows the spatial electron density at the energy range from −0.9 to 0 eV versus the Fermi energy. (b) Oxygen K-edge RIXS spectra, and (c) second differential RIXS map for Li2MnO3 with excitation photon energy from 527 to 533.75 eV. The Raman-like RIXS peak with constant energy loss of approximately 2.5 eV indicates the existence of a localized oxygen 2p orbital that is weakly hybridized with Mn t2g orbitals. | ||
In contrast, for a non-Li-excess layered oxide LiNi1/3Co1/3Mn1/3O2, the RIXS spectra show only fluorescence-like peaks with variable energy loss depending on the incident photon energy (Fig. S4, ESI†) because most oxygen 2p orbitals form σ-type wide bands with axial M 3d orbitals. Therefore, this is the first experimental verification of the localized oxygen 2p orbital weakly hybridized with M t2g orbitals in Li-excess transition-metal oxides, and oxygen K-edge RIXS spectroscopy can, thus, be considered as an effective diagnostic tool to detect electrochemically active oxygen states.
Oxidation of localized oxygen 2p orbital
Having experimentally confirmed the existence of the π-type narrow oxygen 2p bands, the contribution of the localized states to the oxygen-redox reactions was examined using ex situ oxygen K-edge RIXS spectroscopy. As a target Li-excess layered transition metal oxide, Li1.2Ni0.13Co0.13Mn0.54O2 was used as an oxygen-redox electrode material. Li1.2Ni0.13Co0.13Mn0.54O2 was synthesized by a conventional solid-state method, and the powder X-ray diffraction pattern and Rietveld refinement confirmed its successful synthesis (Fig. S5 and Table S2, ESI†). Before acquiring the RIXS spectra, ab initio calculations using the HSE06 hybrid functional were carried out for Li1.2−xNi0.13Co0.13Mn0.54O2 (Fig. 3a). The structural configuration of Li1.2Ni0.13Co0.13Mn0.54O2 was determined by a genetic algorithm (GA) approach that optimizes Li/Ni/Co/Mn arrangement (Fig. S6a and b, ESI†). For the delithiated states, Li and the vacancy configuration in Li1.2−xNi0.13Co0.13Mn0.54O2 was also determined by the GA approach (Fig. S6c, ESI†). The optimized structures for x = 0 and 1.2 are shown in the inset of Fig. 3b. The analysis of partial density of states (pDOS) for pristine Li1.2Ni0.13Co0.13Mn0.54O2 shows that Co and Ni 3d bands dominate the top of the valence band (−1 eV < E − EF < 0 eV), accompanied by a localized oxide band due to Li–O–Li at a slightly lower energy region (−2 eV < E − EF < 1 eV) (Fig. S7, ESI†). The averaged Bader charges of Ni and Co decrease only during the early stage of Li+ extraction (x < 0.4), whereas that of oxygen decreases continuously during the entire delithiation process, suggesting the occurrence of additional oxygen-redox reactions for x > 0.4 (Fig. 3a). This trend is also confirmed by the spin integration (SI) analysis, where the change of spin density of O (O oxidation) occurs at the delithiation process of x > 0.4 (Fig. S8, ESI†).29 Ni2+ and Co3+ ions are oxidized to Ni4+ and Co4+ respectively in the early stage of delithiation (x < 0.4), followed by oxidation of oxide ions (x > 0.4). The oxidation state of Mn4+ ions remains unchanged in the entire compositional range. Indeed, using the predicted oxygen-redox reactions, Li1.2Ni0.13Co0.13Mn0.54O2 delivers a large reversible capacity of approximately 250 mA h g−1, which exceeds the theoretical capacity (123 mA h g−1) of the Ni4+/Ni2+ and Co4+/Co3+ redox reactions (Fig. 3b).11,30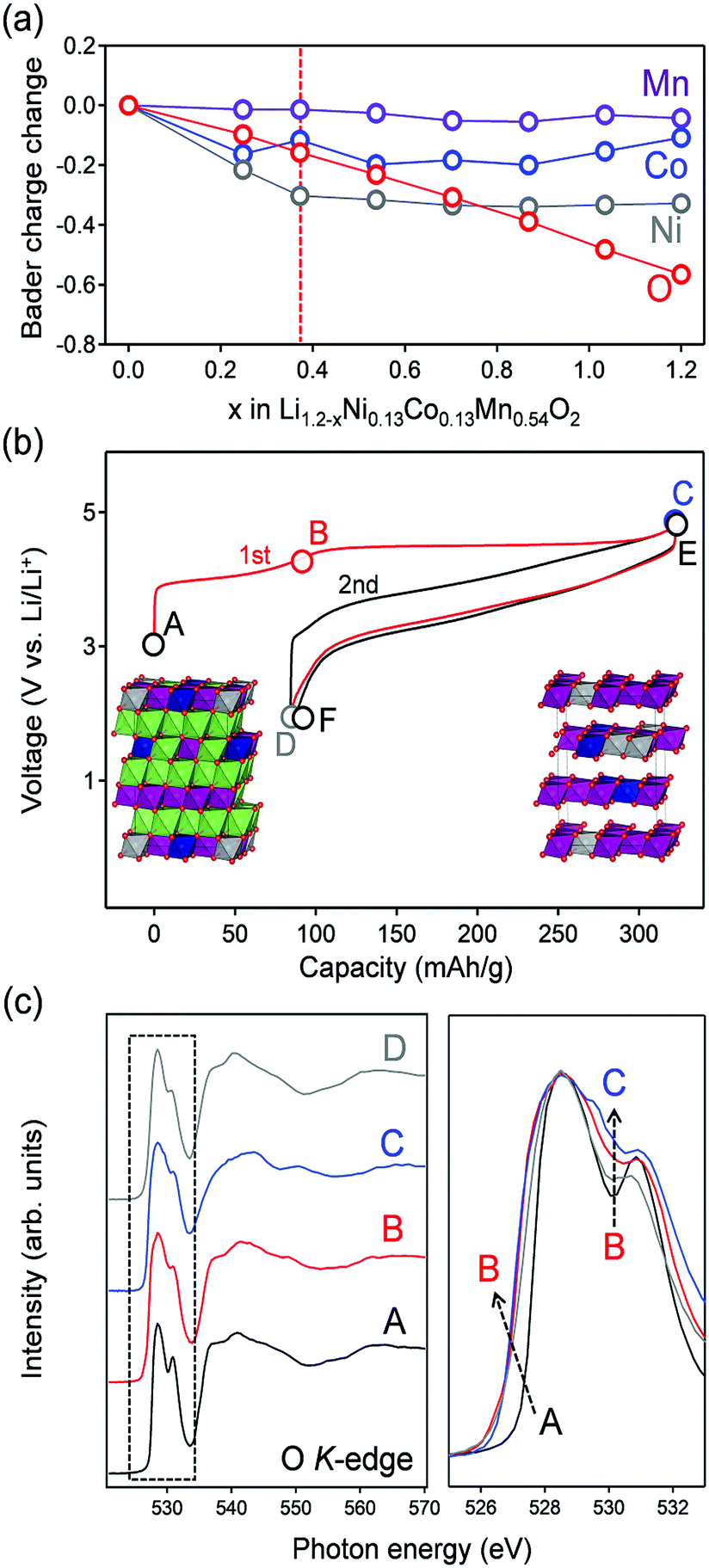 | ||
| Fig. 3 Oxygen-redox reaction in Li1.2−xNi0.13Co0.13Mn0.54O2. (a) The average Bader charge changes of Mn, Co, Ni, and O in Li1.2−xNi0.13Co0.13Mn0.54O2. (b) Charge–discharge curves for Li1.2Ni0.13Co0.13Mn0.54O2. The inset shows optimized Li/Ni/Co/Mn arrangements for Li1.20Ni0.13Co0.13Mn0.54O2, and Li0Ni0.13Co0.13Mn0.54O2. Green, grey, deep blue, and purple-colored octahedra correspond to LiO6, NiO6, CoO6, and MnO6 octahedra, respectively. (c) Oxygen K-edge X-ray absorption spectra in a bulk sensitive partial fluorescence yield mode for Li1.2−xNi0.13Co0.13Mn0.54O2 during the first cycle. The pre-edge region is enlarged for comparison. | ||
The oxygen K-edge X-ray absorption spectrum (XAS) obtained in the bulk-sensitive partial fluorescence yield mode for the pristine Li1.2Ni0.13Co0.13Mn0.54O2 (Fig. 3c) shows two absorption peaks (528.5 and 531 eV) for the 1s core-electron excitation to unoccupied σ-type bands (oxygen 2p-Mn e↓g/e↑g) and an unoccupied π-type band (oxygen 2p-Mn t↓2g). Upon charging to 4.4 V vs. Li/Li+ (A → B in Fig. 3b), a new XAS shoulder emerged at 527 eV owing to hole generation in σ-type (oxygen 2p-Ni eg) and π-type (oxygen 2p-Co t2g) bands. After further charging to 4.8 V vs. Li/Li+ (B → C in Fig. 3b), a new absorption peak emerged at a relatively high absorption-energy region of approximately 530.5 eV. Similar spectral changes were also observed during the second cycle (Fig. S9, ESI†). The XAS signal at 530.5 eV can be ascribed to a 1s core electron excitation to an unoccupied π-type band (oxygen 2p-Mn t↑2g). Emergence of the absorption peak at a relatively high energy region is reasonable because oxygen oxidation increases the effective nuclear charge, which lowers the initial energy level of an oxygen 1s core electron and hence raises the excitation energy.23 Similar changes in the XAS signals by oxidation (the emergence of new absorption peaks at a high energy region) are often observed in transition-metal K-edge absorption spectra.11,15,31 Another explanation for this new absorption is a 1s core electron excitation to an unoccupied σ* molecular orbital of peroxide O22− resulting from the dimerization of oxidized oxide ions, which will be discussed later. In either case, the emergence of the XAS signal at 530.5 eV is an experimental indicator for the occurrence of oxygen oxidation.
The RIXS spectra of pristine Li1.2Ni0.13Co0.13Mn0.54O2 (Fig. 4a) exhibit a Raman-like peak with constant energy loss corresponding to a π-type narrow oxygen 2p band (π → π*), in addition to the fluorescence-like peaks (σ → σ*). The ab initio calculations for pristine Li1.2Ni0.13Co0.13Mn0.54O2 supports the existence of localized oxygen 2p orbitals along the Li–O–Li axes with an orbital energy near the Fermi level (Fig. S7, ESI†). The π → π* RIXS peak intensified after charging to 4.4 V vs. Li/Li+ (transition-metal oxidation, Fig. 4b), which indicates accumulation of the localized oxygen 2p states to the Fermi level. The initial Li+ extraction associated with Co and Ni oxidation gives an oxygen coordination environment with a □–O–Li+ or □–O–□ axis (□: Li+ vacancy), which electrostatically raises the energy level of the localized oxygen 2p orbitals. Indeed, the fact that the energy loss of the Raman-like RIXS peak decreased from 2.5 to 2.0 eV supports a rise in the energy level of the localized oxygen 2p orbitals. We presume that accumulation of the localized oxygen 2p states to the Fermi level should be an essential preceding process that triggers oxygen-redox reactions. For example, it is well known that the electrochemical activity of Li2MnO3 is poor32 even though oxygen has a Li+–O–Li+ axis and hence a localized oxygen 2p orbital (Fig. 2a).10 Most likely, as Li2MnO3 cannot exhibit initial Li+ extraction owing to the electrochemically inactive Mn4+, it is difficult to generate the □–O–Li+ and □–O–□ axes, and the energy level of the localized oxygen 2p orbital remains too low to undergo oxidation within the liquid electrolyte stability window. Indeed, Li2MnO3 delivers large reversible capacities when cycled with a solid electrolyte that possesses a large electrochemical stability window.33 Conversely, when redox-active transition metals are present, such as in Li2MnO3–LiMO2 solid solution, □–O–Li+ and □–O–□ axes are easily formed by the initial Li+ extraction to raise the energy level of the localized oxygen 2p orbital, unlocking the redox activity of oxygen. Once the oxygen-redox reaction is triggered, the □–O–Li+ and □–O–□ axes are continuously generated to achieve a large oxygen-redox capacity.
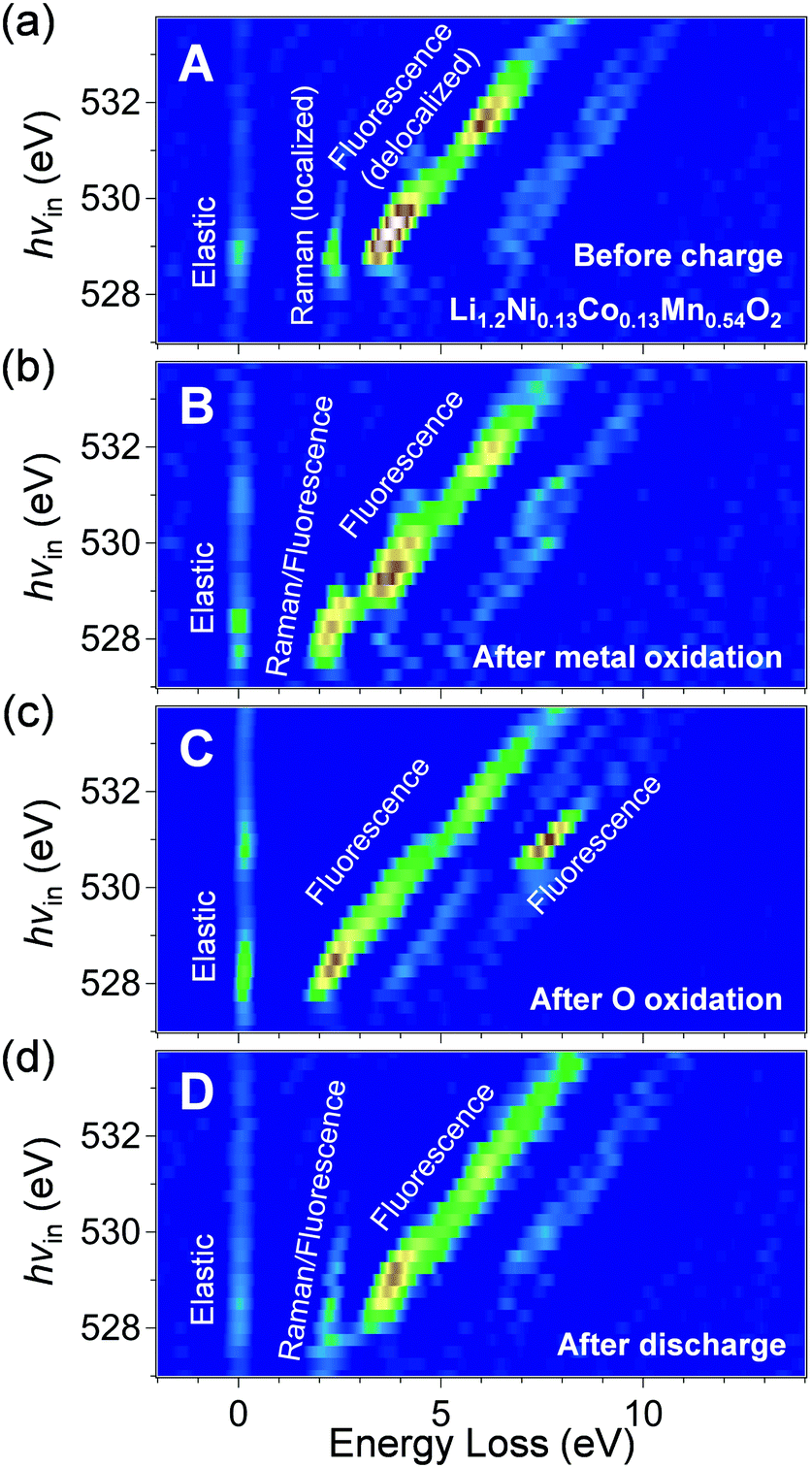 | ||
| Fig. 4 Monitoring oxygen 2p orbitals during oxygen-redox reaction of Li1.2Ni0.13Co0.13Mn0.54O2. Oxygen K-edge second differential RIXS map for the samples (a) before charge (sample A in Fig. 3b), (b) after transition-metal oxidation (sample B in Fig. 3b), (c) after oxygen oxidation (sample C in Fig. 3b), and (d) after discharge (sample D in Fig. 3b), with excitation photon energy from 527 to 533.75 eV. The corresponding RIXS spectra are shown in Fig. S10 (ESI†). | ||
After charging to 4.8 V vs. Li/Li+ (oxygen oxidation, Fig. 4c), the Raman-like π → π* RIXS peaks disappear, and all the RIXS peaks become fluorescence-like. Importantly, a new intense fluorescence-like peak emerges at an energy loss of approximately 7.5 eV with an incident photon energy of 530.5 eV. Assuming an incident photon of 530.5 eV excites a 1s core electron to an unoccupied π-type band (oxygen 2p-Mn t↑2g), resonant inelastic scattering with the 530.5 eV incident photon includes valence excitation between the π-type bands (π → π*). Therefore, a large energy loss of approximately 7.5 eV for this RIXS peak should correspond to π-type splitting. This large π-type splitting should be induced by hole generation in the antibonding π-type band which makes the Mn–O bonds more bonding (more delocalization/larger orbital hybridization). Therefore, the RIXS peak is observed as fluorescence-like. The enhanced π-type interaction stabilizes the oxidized oxygen, leading to reversible oxygen-redox reactions.
It is also important to note that the Raman-like RIXS peak of the localized oxygen 2p orbitals was not fully recovered after full discharge (Fig. 4d). The emission at the energy loss region of approximately 2 eV becomes weakly fluorescence-like after the 1st cycle. Presumably, structural degradation such as cation migration during the 1st charge modulates the coordination environment of oxide ions, making the RIXS peak weakly fluorescence-like. The irreversible structural change during the 1st charge could explain the experimental observation that the potential profile of the 2nd charge differs from that of the 1st charge (Fig. 3b). Nevertheless, in the 2nd cycle, the spectral shape change is reversible (Fig. S11, ESI†), suggesting stable occurrence of the oxygen-redox reactions.
Possible O–O bond formation in oxygen redox electrodes
Gent and Hong et al. also observed similar intense RIXS peaks for Li1.17−xNi0.21Co0.08Mn0.54O2 and Li2Ir0.75Sn0.25O3 after oxygen oxidation,34,35 and transition-metal migration and subsequent O–O bond formation were proposed to explain the emergence of the new XAS and RIXS peaks after oxygen oxidation. To examine this hypothesis, we acquired O K-edge XAS and RIXS spectra for WO2(O2)(H2O), which contains peroxide O22− coordinated to W6+.36 Based on the DFT calculations (Fig. 5a and b), WO2(O2)(H2O) possesses (i) a strong π-type LUMO band consisting of O 2p and large W 5d (t2g) (3.5 < E − EF < 4.3 eV) and (ii) an unoccupied σ* band of peroxide (6.1 < E − EF < 7.1 eV). The XAS spectrum shows a broad absorption arising from core-electron excitations to these unoccupied π-type and σ-type bands of W–O and peroxide (Fig. 5c). Importantly, two intense fluorescence-like emissions were observed in the RIXS map (Fig. 5d), which can be ascribed to valence excitations between the π-type bands of W–O and between the σ-type bands of O–O, respectively. The fluorescence-like feature of the emission from the π-type bands of W–O is explained by electron delocalization through strong π-type W t2g–O 2p interactions. The fluorescence-like emission between the σ-type bands of O–O takes place approximately with an energy loss of 6 eV at hvin = 530 eV, which differs slightly from that observed for fully charged Li1.2Ni0.13Co0.13Mn0.54O2 (the energy loss of 7.5 eV at hvin = 530.5 eV). Furthermore, the transition-metal migration and peroxide formation are one of possible explanations for the RIXS spectral changes for lithium-rich layered oxides, but cannot explain the almost identical RIXS spectral changes during the oxygen-redox reactions of many other sodium-based layered oxides (e.g., Na2/3[Mg0.28Mn0.72]O2)37 and Na2Mn3O7,38 where the transition-metal migration is known to be negligible due to large Na+ slabs. In fact, neither a short O–O distance below 2.2 Å nor significant MO6 distortion was found in the relaxed structures of delithiated phases (Fig. S12, ESI†). Although the possibility of the O–O bond formation cannot be excluded, another possible but less argued mechanism of hole generation in the antibonding π-type band will be discussed below. | ||
| Fig. 5 Electronic structure and XAS/RIXS spectra for WO2(O2)(H2O). (a) Partial density of states (pDOS) for W and O, and crystal orbital overlap population (COOP) diagrams for W–O and O–O bonds. (b) Spatial hole densities with the selected orbital energies (i) from 3.5 to 4.3 eV, and (ii) from 6.1 to 7.1 eV against the Fermi energy (dotted areas in pDOS). (c) O K-edge X-ray absorption (XAS) spectrum. (d) O K-edge differential RIXS map. While the RIXS peak for O22− can be referred to explain a mechanism of O redox cathodes, a care should be taken that the similar signal is also induced by M–O multi-orbital bond formation (see text). | ||
Multiorbital bond formation in oxygen redox electrodes
Crystal orbital overlap population (COOP) diagrams for selected Mn–O bonds from ab initio calculations for Li1.2Ni0.13Co0.13Mn0.54O2 (Fig. 6) quantify the reversible transformation of an oxygen 2p orbital characteristics during the oxygen-redox reaction. The COOP is an overlap-population-weighted density of states that serves as a bonding descriptor.39 The sign of COOP indicates bonding (positive), non-bonding (zero), or anti-bonding (negative) contributions between two selected atoms. Before charging, the spatial distribution of the electron at the energy range of −2.0 to −1.0 eV against the Fermi energy (Fig. 6a) takes up a π-type orbital shape consisting of oxygen 2p and Mn t2g orbitals (b1* in C2v). However, the oxygen 2p orbitals have no significant COOP with Mn t2g orbitals (black arrow, Fig. 6a), indicating a localized nature of the oxygen 2p orbitals. After oxygen oxidation, the spatial distribution of a hole at the energy range from +1.0 to +2.0 eV versus the Fermi energy also has a π-type orbital shape (Fig. 6b). On increasing the number of holes in the antibonding π-type orbital, Mn–O bonds show more bonding characteristics with a considerable COOP between the oxygen 2p and Mn t2↑g orbitals (Fig. S13, ESI†). The COOP diagram also indicates that the bonding states reside at the energy range of −7.0 to −6.0 eV against the Fermi energy (red arrows, Fig. 6b), which may correspond to the occupied π-type bonding b1 orbital. The calculated b1/b1* splitting (ca. 7.0 eV) agrees well with the energy loss of the intense fluorescence-like peak observed in the RIXS spectra (Fig. 4c). | ||
| Fig. 6 Enhancement of π-type interaction after oxygen oxidation. Partial density of states (pDOS) for oxygen and manganese, and crystal orbital overlap population (COOP) diagrams for Mn–O bonds in Li1.2−xNi0.13Co0.13Mn0.54O2 (a) before charge (x = 0) and (b) after oxygen oxidation (x = 1.2). The dotted areas denote states mainly consisting of oxygen 2p orbitals responsible for oxygen-redox reactions. The spatial electron or hole density with the selected orbital energy (dotted areas in pDOS) is depicted. | ||
To summarize the reaction mechanism, the Li+–O–Li+ coordination in the pristine state forms localized oxygen 2p orbital weakly hybridized with the Mn t2g orbital. When the initial Li+ extraction occurs in association with M redox reactions, the □–O–Li+ and □–O–□ coordination axes are generated, which raises the energy level of the localized oxygen 2p states to the Fermi level. After this accumulation, exclusive oxygen oxidation occurs; a hole is generated in the antibonding π-type orbital and orbital hybridization is enhanced to delocalize the hole within a Mn–O–Mn bond. Alternatively, or in parallel, unstable oxidized oxide ions dimerize to form peroxide O22−. The resulting either b1/b1* splitting of the Mn–O–Mn bond or σ/σ* splitting of the O–O bond stabilizes the oxidized oxygen, enabling a reversible oxygen-redox reaction.
From a conventional viewpoint of an electronic structure in transition-metal oxides, an M–O bond is formed mainly by σ donation from oxygen (σ bond), as the 2p orbitals of O2− are filled completely (Fig. 7a).9 However, hole generation on the outer sphere 2p orbital of oxygen is intrinsically unstable, and π back-donation from M occurs to stabilize the oxygen hole, giving rise to (σ + π) multiorbital bonds (Fig. 7b).8 Therefore, the reversible oxygen-redox reaction requires M to possess both σ-accepting and π-donating abilities for oxidized oxygen. For example, Ti4+ with a vacant t2g configuration has no π-donating ability whereas Sn4+ with a filled eg configuration has no σ-accepting ability. Therefore, Li2TiO3 and Li2SnO3 are not oxygen-redox active,40,41 and replacing transition metals with Ti4+ or Sn4+ could promote stabilizing oxidized oxygen, for instance, by peroxide formation.35 Conversely, Mn4+, Co4+, and Ni4+ with (partially) filled t2g and vacant eg configurations satisfy the requirement for (σ + π) multiorbital bond formation, leading to the high oxygen-redox activity of the Li2MnO3–LiMO2 solid solution such as Li1.2Ni0.13Co0.13Mn0.54O2. Large overlap between O 2p and M 4d/5d orbitals should give more stable (σ + π) multiorbital bonds. Indeed, a short Ru(V)–O distance of 1.85 Å (relative to the conventional Ru(V)–O distance of 2.0 Å) was observed for Li2Ru0.75Sn0.25O3 after oxygen oxidation.42 Owing to the enhanced multiorbital bonding, Li-excess 4d/5d transition-metal oxides exhibit highly reversible oxygen-redox reactions without oxygen gas release or surface side reactions.
 | ||
| Fig. 7 Multiorbital bond formation upon oxygen oxidation. (a) σ bond in conventional metal oxides. (b) (σ + π) multiorbital bond in oxygen-oxidized states. | ||
Conclusions
In this work, using oxygen K-edge RIXS spectroscopy, we investigated oxygen-redox battery electrodes, demonstrating the importance of the combined control of the (σ + π) interaction between transition metals and oxygen. In addition to the previously reported criteria,10,11,15 we have discussed a novel and crucial criterion related to the electronic configuration: transition metal ions with simultaneous σ-accepting and π-donating abilities can realize stable oxygen redox reaction toward higher-capacity, higher-energy-density cathode materials. Typical transition metals that satisfy these requirements are Mn4+ (t2↑3gt↓02ge↑0geg↓0; π-donating t↑2g and σ-accepting e↑g to O 2p) and Ru5+ (t↑32gt↓02ge↑0ge↓0g; significantly π-donating t↑2g and σ-accepting e↑g to O 2p). The exploration of Li-excess oxides including transition metals that satisfy these criteria could help surpass the existing LIB performance limits.Methods
Synthesis
Li2MnO3, LiNi1/3Co1/3Mn1/3O2, and Li1.2Ni0.13Co0.13Mn0.54O2 were prepared using stoichiometric amounts of Li2CO3 (Wako, 99%), NiO (Wako, 99%), Co3O4 (Kanto chemicals, 99%) and MnO2 (Wako, 99.5%). The precursors were ball-milled at 400 rpm for 6 h (cycles of 5 min ball milling followed by 5 min of resting) using a FRITSCH P-7. After recovery, the powders were pelletized and fired at 900 °C (heating rate of 5 °C min−1) for 12 h under an O2 atmosphere (ASAHI RIKA, ARF-MC). WO2(O2)(H2O) was synthesized by a procedure reported in literature.30 The resulting powder compounds were characterized by X-ray diffraction recorded using a Rigaku RINT TTR-III (Cu Kα radiation), and Rietveld refinements were performed using Jana 2006 software.43 The crystal structures were drawn using VESTA.44Electrochemistry
Electrochemical measurements were carried out in Li/LiPF6 in PC (1 mol L−1, Kishida Chemical)/positive electrode batteries assembled in 2032-type coin cells. The positive electrodes were prepared by mixing 80 wt% active material with 10 wt% acetylene black (Denka) and 10 wt% polyvinylidene difluoride (Sigma-Aldrich) in N-methyl pyrrolidone (Kanto Chemical) to form a slurry, which was then coated onto Al foil (mass loading of ca. 1 mg cm−2). The obtained films were dried at 70 °C for several hours before punching the electrodes (diameter: 16 mm), which were then further dried under vacuum at 120 °C overnight before placing them into a glovebox. The positive and negative electrodes (Li foils) were separated by a glass fiber separator soaked with the electrolyte. All electrochemical measurements were controlled and recorded using a TOSCAT-3100 battery tester.Sample preparation for X-ray absorption/emission spectroscopy
Samples for the X-ray absorption and emission spectroscopy experiments were prepared in 2032-type coin cells. The batteries were galvanostatically charged/discharged until a given voltage, and then the voltage was maintained until the residual current dropped to 0 mA. The batteries were recovered and disassembled in an Ar-filled glovebox and the electrodes were washed three to five times with anhydrous dimethyl carbonate and dried before being stored in air-tight containers. The samples were then transported to the synchrotron facility without exposure to air.X-ray absorption/emission spectroscopy
The XAS and RIXS experiments were performed at BL07LSU, SPring-8.45 For the oxygen K-edge XAS, the bulk-sensitive partial-fluorescence-yield (PFY) detection mode was employed. Flat-field type grazing incidence spectrometers were used for the oxygen K-edge RIXS. All the measurements were performed at room temperature. The samples were taken out from the cell in the glove box filled with Ar gas, and then they were transferred into a vacuum chamber without exposure to air using a transfer vessel.Ab initio calculations
Ab initio calculations were performed using the Vienna ab initio simulation package (VASP)46 using the projector-augmented-wave (PAW) method.47,48 For Li2MnO3, the range-separated hybrid functional introduced by Heyd, Scuseria, and Ernzerhof (HSE) was used throughout the calculations.28 The exact exchange fraction in the Hartree–Fock/density functional theory (DFT) hybrid functional and range-separation parameter are set at 0.25 and 0.2, respectively (HSE06). The DFT part of HSE06 employed a modified version of the Perdew–Burke–Ernzerhof functional (PBEsol)49 according to literature. The kinetic energy cutoff was set to 520 eV and the k-point was sampled on a (5 × 3 × 5) grid. The lattice parameters and atomic positions were relaxed until the forces on each atomic site were less than 0.01 eV Å−1. To model Li1.2Ni0.13Co0.13Mn0.54O2, Li29/24Ni3/24Co3/24Mn13/24O2 was considered and the structural configurations were determined by the genetic algorithm (GA) approach that optimizes the Li/Ni/Co/Mn arrangement. The on-site Coulomb correction (GGA+U) was included for localized electronic states, and the U value was chosen to be 6.0, 3.4 and 3.9 eV for the 3d states of Ni, Co and Mn, respectively, according to literature.50 During the GA procedure, we obtained the total energies using modified Perdew–Burke–Ernzerhof generalized gradient approximation (PBEsol–GGA)49 by reducing the energy cutoff and the number of k points in the reciprocal cell to 360 eV and unity (Γ point sampling), respectively. After the GA cycles, the obtained five lowest energy configurations were recalculated with a sufficient energy cutoff (500 eV) and k point mesh (2 × 2 × 2 k-point meshes) using the HSE06 hybrid functional. U was not employed in the HSE calculations. The lowest energy configuration among the five configurations was chosen for Li29/24Ni3/24Co3/24Mn13/24O2. Similarly, the Li and vacancy configurations for delithiated Li29/24(1−x)Ni3/24Co3/24Mn13/24O2 were also determined by optimizing the Li/vacancy arrangement using the GA approach, and recalculated using the HSE06 functional for selected configurations. The calculated formation energies correspond to zero-Kelvin conditions. The GA approach was detailed in the ESI.† For WO2(O2)(H2O), HSE06 hybrid functional was employed with the Grimme dispersion correction (DFT-D3)51 for hydrogen bonds. The energy cutoff was set to 520 eV with the k-point of 4 × 6 × 4. The COOP was calculated by LOBSTER software.52Author contribution
M. O. and A. Y. conceived and directed the project. K. U. and B. M. B. synthesized and characterized Li2MnO3, Li1.2Ni0.13Co0.13Mn0.54O2 and Na2Ru0.5Sn0.5O3. A. T. synthesized WO2(O2)(H2O). T. S., K. U., A. T., D. A., T. S., Y. H. and M. O. measured and analysed the XAS, XES, and RIXS spectra. T. M., X. M. S., T. S., E. W., and M. N. conducted the ab initio calculation. All authors wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan; Grant-in-Aid for Specially Promoted Research No. 15H05701 and the “Elemental Strategy Initiative for Catalysis and Batteries” (ESICB). M. O. was financially supported by JSPS KAKENHI Grant Number 19H05816, 18K19124, and the Iketani Science and Technology Foundation. The RIXS spectroscopy was performed by the joint research in Synchrotron Radiation Research Organization and the Institute for Solid State Physics, the University of Tokyo (Proposal No. 2019B7456, 2019A7452, 2018B7590, 2018A7560, 2017B7540, 2017A7525, 2016B7516 and 2015B7500). We also thank the Information Technology Center of Nagoya University for providing computing resources (CX400 and FX400). M. N. was supported by “Materials Research by Information Integration” Initiative (MI2I) project of the Support Program for Starting Up Innovation Hub from Japan Science and Technology Agency (JST), and JSPS KAKENHI Grant Number 19H05815. We are grateful to J. Miyawaki at the University of Tokyo for his support to the RIXS measurements.References
- R. V. Noorden, Nature, 2014, 507, 26–28 CrossRef PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- M. S. Whittingham, Chem. Rev., 2014, 114, 11414–11443 CrossRef CAS PubMed.
- R. A. Huggins, Advanced Batteries, Springer, New York, 2009 Search PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J. M. Tarascon, Nat. Mater., 2016, 15, 121–126 CrossRef CAS PubMed.
- I. B. Bersuker, Electronic Structure and Properties of Transition Metal Compounds, John Wily & Sons, New York, 1995 Search PubMed.
- P. A. Cox, Transition Metal Oxides, Oxford Press, New York, 1992 Search PubMed.
- D.-H. Seo, J. Lee, A. Urban, R. Malik, S. Y. Kang and G. Ceder, Nat. Chem., 2016, 8, 692–697 CrossRef CAS PubMed.
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y.-S. Liu, K. Edström., J.-H. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Nat. Chem., 2016, 8, 684–691 CrossRef CAS PubMed.
- Z. Lu, D. D. MacNeil and J. Dahn, ECS Solid State Lett., 2001, 4, A191–A194 CrossRef CAS.
- Z. Lu and J. Dahn, J. Electrochem. Soc., 2002, 149, A815–A822 CrossRef CAS.
- E. McCalla, A. M. Abakumov, M. Saubanère, D. Foix, E. J. Berg, G. Rousse, M.-L. Doublet, D. Gonbeau, P. Novák, G. Van Tendeloo, R. Dominko and J.-M. Tarascon, Science, 2015, 350, 1516–1521 CrossRef CAS PubMed.
- N. Yabuuchi, M. Nakayama, M. Takeuchi, S. Komaba, Y. Hashimoto, T. Mukai, H. Shiiba, K. Sato, Y. Kobayashi, A. Nakao, M. Yonemura, K. Yamanaka, K. Mitsuhara and T. Ohta, Nat. Commun., 2016, 7, 13814 CrossRef CAS PubMed.
- P. E. Pearce, A. J. Perez, G. Rousse, M. Saubanère, D. Batuk, D. Foix, E. McCalla, A. M. Abakumov, G. Van Tendeloo, M. L. Doublet and J. M. Tarascon, Nat. Mater., 2017, 16, 580–586 CrossRef CAS PubMed.
- C. Zhan, Z. P. Yao, J. Lu, L. Ma, V. A. Maroni, L. Li, E. Lee, E. E. Alp, T. P. Wu, J. G. Wen, Y. Ren, C. Johnoson, M. M. Thackeray, M. K. Y. Chan, C. Wolverton and K. Amine, Nat. Energy, 2017, 2, 963–971 CrossRef CAS.
- G. Assat, D. Foix, C. Delacourt, A. Ladecola, R. Dedryvere and J. M. Tarascon, Nat. Commun., 2017, 8, 2219 CrossRef PubMed.
- D. Eum, B. Kim, S. J. Kim, H. Park, J. Wu, S. P. Cho, G. Yoon, M. H. Lee, S. K. Jung, W. Yang, W. M. Seong, K. Ku, O. Tamwattana, S. K. Park, I. Hwang and K. Kang, Nat. Mater., 2020 DOI:10.1038/s41563-019-0572-4.
- R. A. House, U. Maitra, M. A. Perez-Osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Dude, A. Nag, A. Walters, K. J. Zhou, M. R. Roberts and P. G. Bruce, Nature, 2020, 577, 502–508 CrossRef CAS PubMed.
- M. B. Yahia, J. Vergnet, M. Saubanere and M. L. Doublet, Nat. Mater., 2019, 18, 496–502 CrossRef PubMed.
- J. Xu, M. Sun, R. Qiao, S. E. Renfrew, L. Ma, T. Wu, S. Hwang, D. Nordlund, D. Su, K. Amine, J. Lu, B. D. McCloskey, W. Yang and W. Tong, Nat. Commun., 2018, 9, 947 CrossRef PubMed.
- M. Okubo and A. Yamada, ACS Appl. Mater. Interfaces, 2017, 9, 36463–36472 CrossRef CAS PubMed.
- Y. Xie, M. Saubanere and M. L. Doublet, Energy Environ. Sci., 2017, 10, 266–274 RSC.
- G. Assat and J. M. Tarascon, Nat. Energy, 2018, 3, 373–386 CrossRef CAS.
- F. de Groot and A. Kotani, Core level spectroscopy of solids, CRC Press, Boca Raton, 2008 Search PubMed.
- V. Bisogni, S. Catalano, R. J. Green, M. Gilbert, R. Scherwitzl, Y. B. Huang, V. N. Strocov, P. Zubko, S. Balandeh, J. M. Triscone, G. Sawatzky and T. Schmitt, Nat. Commun., 2016, 7, 13017 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Reed and G. Ceder, Electrochem. Solid-State Lett., 2002, 5, A145–A148 CrossRef CAS.
- H. Koga, L. Croguennec, M. Menetrier, K. Douhil, S. Belin, L. Bourgeois, E. Suard, F. Weill and C. Delmas, J. Electrochem. Soc., 2013, 160, A786–A792 CrossRef CAS.
- B. Mortemard de Boisse, G. D. Liu, J. T. Ma, S. Nishimura, S. C. Chung, H. Kiuchi, Y. Harada, J. Kikkawa, Y. Kobayashi, M. Okubo and A. Yamada, Nat. Commun., 2016, 7, 11397 CrossRef CAS PubMed.
- A. D. Robertson and P. G. Bruce, Chem. Commun., 2002, 2790–2791 RSC.
- K. Hikima, K. Suzuki, S. Taminato, M. Hirayama, S. Yasuno and R. Kanno, Chem. Lett., 2019, 48, 192–195 CrossRef CAS.
- W. E. Gent, K. Lim, Y. F. Liang, Q. H. Li, T. Barnes, S.-J. Ahn, K. H. Stone, M. Mclntire, J. Y. Hong, J. H. Song, Y. Y. Li, A. Mehta, S. Ermon, T. Tyliszczak, D. Kilcoyne, D. Vine, J.-H. Park, S.-K. Doo, M. F. Toney, W. L. Yang, D. Prendergast and W. C. Chueh, Nat. Commun., 2017, 8, 2091 CrossRef PubMed.
- J. Hong, W. E. Gent, P. Xiao, K. Lim, D.-H. Seo, J. Wu, P. M. Csernica, C. J. Takacs, D. Nordlund, C. J. Sun, K. H. Stone, D. Passarello, W. L. Yang, D. Prendergast, G. Ceder, M. F. Toney and W. C. Chueh, Nat. Mater., 2019, 8, 256–265 CrossRef PubMed.
- B. Pecquenard, S. Castro-Garcia, J. Livage, P. Y. Zavalij, M. S. Whittingham and R. Thouvenot, Chem. Mater., 1998, 10, 1882–1888 CrossRef CAS.
- U. Maitra, R. A. House, J. Somerville, N. Tapia-Ruiz, J. G. Lozano, N. Guerrini, R. Hao, K. Luo, L. Y. Jin, M. A. Perez-Osorio, F. Massel, D. M. Pickup, S. Ramos, X. Y. Lu, D. E. McNally, M. R. Roberts and P. G. Bruce, Nat. Chem., 2018, 10, 288–295 CrossRef CAS PubMed.
- B. Mortemard de Boisse, S. Nishimura, E. Watanabe, L. Lander, A. Tsuchimoto, J. Kikkawa, E. Kobayashi, D. Asakura, M. Okubo and A. Yamada, Adv. Energy Mater., 2018, 8, 1800409 CrossRef.
- T. Hughbanks and R. Hoffmann, J. Am. Chem. Soc., 1983, 105, 3528–3537 CrossRef CAS.
- H. Shigemura, M. Tabuchi, H. Sakaebe, H. Kobayashi and H. Kageyama, J. Electrochem. Soc., 2003, 150, A638–A644 CrossRef CAS.
- M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M. L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. Ben Hassine, L. Dupont and J. M. Tarascon, Nat. Mater., 2013, 12, 827–835 CrossRef CAS PubMed.
- G. Assat, A. Iadecola, C. Delacourt, R. Dedryvere and J. M. Tarascon, Chem. Mater., 2017, 29, 9714–9724 CrossRef CAS.
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr., 2014, 229, 345–352 CAS.
- F. Monma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef.
- Y. Harada, M. Kobayashi, H. Niwa, Y. Senba, H. Ohashi, T. Tokushima, Y. Horikawa, S. Shin and M. Oshima, Rev. Sci. Instrum., 2012, 83, 013116 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Hautier, S. P. Ong, A. Jain, C. J. Moore and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155208 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee04197d |
| This journal is © The Royal Society of Chemistry 2020 |