
Rewiring photosynthesis: a photosystem I-hydrogenase chimera that makes H2in vivo†

Abstract
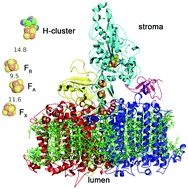
Harnessing the power of photosynthesis to catalyze novel light-driven redox chemistry requires a way to intercept electron flow directly from the photosynthetic electron transport chain (PETC). As a proof of concept, an in vivo fusion of photosystem I (PSI) and algal hydrogenase was created by insertion of the HydA sequence into the PsaC subunit. The PSI and hydrogenase portions are co-assembled and active in vivo, effectively creating a new photosystem. Cells expressing only the PSI-hydrogenase chimera make hydrogen at high rates in a light-dependent fashion for several days. In these engineered cells, photosynthetic electron flow is directed away from CO2 fixation and towards proton reduction, demonstrating the possibility of driving novel redox chemistries using electrons from water splitting and the photosynthetic electron transport chain.
- This article is part of the themed collection: Energy & Environmental Science Cover Art
 Please wait while we load your content...
Please wait while we load your content...

