
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical looping beyond combustion – a perspective
Xing
Zhu
ab,
Qasim
Imtiaz†
cd,
Felix
Donat†
 c,
Christoph R.
Müller
*c and
Fanxing
Li
*a
c,
Christoph R.
Müller
*c and
Fanxing
Li
*a
aDepartment of Chemical and Biomolecular Engineering, North Carolina State University, 911 Partners Way, Raleigh, NC 27695-7905, USA. E-mail: fli5@ncsu.edu
bState Key Laboratory of Complex Nonferrous Metal Resources Clean Utilization, Faculty of Metallurgical and Energy Engineering, Kunming University of Science and Technology, Kunming 650093, China
cDepartment of Mechanical and Process Engineering, Laboratory of Energy Science and Engineering, ETH Zurich, Leonhardstrasse 21, 8092, Zurich, Switzerland. E-mail: muelchri@ethz.ch
dDepartment of Chemistry and Chemical Engineering, Lahore University of Management Sciences, D.H.A. Lahore Cantt., 54792 Lahore, Pakistan
First published on 13th January 2020
Abstract
As a promising approach for carbon dioxide capture, chemical looping combustion has been extensively investigated for more than two decades. However, the chemical looping strategy can be and has been extended well beyond carbon capture. In fact, significant impacts on emission reduction, energy conservation, and value-creation can be anticipated from chemical looping beyond combustion (CLBC). This article aims to demonstrate the versatility and transformational benefits of CLBC. Specifically, we focus on the use of oxygen carriers or redox catalysts for chemical production – a $4 trillion industry that consumes 40.9 quadrillion BTU of energy. Compared to state-of-the-art chemical production technologies, we illustrate that chemical looping offers significant opportunities for process intensification and exergy loss minimization. In many cases, an order of magnitude reduction in energy consumption and CO2 emission can be realized without the needs for carbon dioxide capture. In addition to providing various CLBC examples, this article elaborates on generalized design principles for CLBC, potential benefits and pitfalls, as well as redox catalyst selection, design, optimization, and redox reaction mechanism.
Broader contextThe chemical industry consumes more than 40 quadrillion BTU of energy while emitting 2.5 Gigatons of carbon dioxide each year. Meanwhile, many established chemical processes are extensively optimized, providing limited space for further energy savings and emission reductions. Chemical looping offers exciting new opportunities to the aforementioned challenges through process intensification. Facilitated by a redox catalyst, a rationally designed chemical looping scheme can reduce the number of unit operations and significantly decrease the exergy loss for chemical production. In some cases, the chemical looping strategy can facilitate a chemical conversion that would be otherwise infeasible in a conventional reaction scheme. While a number of chemical looping based chemical production approaches have shown promising results, this concept can be further expanded to a significantly wider sets of products using a variety of feedstock and energy sources. Given the exciting opportunities offered by chemical looping beyond combustion (CLBC) and the emissions and energy demand in the current chemical production practice, the CLBC design principles and redox catalyst selection/optimization strategies covered in this article can provide useful information for researchers in the general areas of chemical looping, catalysis, particle technology, and process intensification. |
1. Introduction
Although the term “chemical looping” was first minted by Richter and Knoche in 1983 in the context of reducing exergy loss in fossil fuel combustion,1 the concept of chemical looping (CL), i.e. decomposition of a chemical reaction into multiple sub-reactions facilitated by solid reaction intermediates, was investigated long before that.2,3 Owing to the pressing demand for carbon emissions reduction,4,5 chemical looping combustion (CLC) has been studied extensively over the past three decades as a new technology for power generation with integrated CO2 capture,6–22 as evidenced by more than 2400 peer-reviewed publications to date (based on the chemical abstracts service). In comparison, research related to chemical looping strategies prior to 1983 was scattered and often dealt with concepts other than power generation and CO2 capture.2,23–25 A good example is the commercially implemented steam-iron process for hydrogen production, which was later replaced by methane reforming in the 1930s.23 However, besides the limitations from the lack of global policies on fighting global warming via carbon capture and storage (CCS)-related technologies, the lack of fundamental understanding and absence of effective strategies to design and optimize the reaction intermediates, also known as (oxygen) carriers, have largely hindered the research progress in the “pre-CLC” era. We note that the extensive investigation of the CLC technology26–37 has built up significant background knowledge and fundamental insights for potential breakthroughs in chemical looping applications well beyond combustion and power generation.24,25,38–51 It is, therefore, not too surprising to see that the extension of the chemical looping strategy has drawn significant attention over the past decade. In fact, journal articles covering such topics increased by nearly 10-fold over this period.Although there have been quite a few excellent reviews and books covering the topic of chemical looping, they tend to focus on CLC26–35 and, in some cases, chemical looping reforming (CLR) both with and without being integrated with CO2 or H2O splitting.38–53 A recent review by Zeng et al.21 provided comprehensive discussions of the various aspects of chemical looping including chemical looping for the production of chemicals. However, CLC and CLR related topics were still the primary focus, and many chemical production cases, as summarized in Table 1, have not been covered. Another review by Bayham et al.22 gave an overview of CLC for power production with CO2 capture and CLR for hydrogen or syngas production with carbon capture. It mainly focused on the potential process configurations (reaction systems and reactor design) and applications. As a highly versatile and effective process intensification strategy, the chemical looping concept can be applied to many applications beyond CO2 capture and reforming with significant merits in efficiency improvements, emission reduction, and potential cost savings. This article aims to offer a perspective on various potential applications of chemical looping beyond combustion (CLBC) and the generalized design principles for oxygen carrier or redox catalysts in chemical looping processes.
| Feedstock | Products | Product generation | Balance of the loop | Name of the processa | Sample oxygen/nitrogen carriers |
|---|---|---|---|---|---|
| a CLAS, chemical looping air separation; CL-OCM, chemical looping oxidative coupling of methane; CL-DHA, chemical looping dehydroaromatization; CL-ODH, chemical looping oxidative dehydrogenation; CL-WGS, chemical looping water–gas shift; CL-RWGS, chemical looping reverse water–gas shift. b Exemplary, promising CLBC schemes that has yet to be investigated. | |||||
| Air | Air separation | (R1) MeOx → MeOx−1 + 1/2O2 | (R2) MeOx−1 + 1/2O2 → MeOx | CLAS | CuO,49 perovskite54,55 |
| Methane | Synthesis gas | (R3) CH4 + MeOx → MeOx−1 + CO + 2H2 | (R4) MeOx−1 + 1/2O2 → MeOx | CLR | NiO,56,57 FeOx58–62 CeO2,63–65 perovskite,66–68 hexaaluminate69,70 |
| (R5) MeOx−1 + H2O → MeOx + H2 | |||||
| (R6) MeOx−1 + CO2 → MeOx + CO | |||||
| Ethylene | (R7) CH4 + MeOx → MeOx−1 + 1/2C2H4 + H2O | (R8) MeOx−1 + 1/2O2 → MeOx | CL-OCM | Alkali modified Mn, Fe oxides71,72 | |
| Benzene | (R9) 2/3CH4 + MeOx → MeOx−1 + 1/9C6H6 + H2O | (R10) MeOx−1 + 1/2O2 → MeOx | CL-DHA | Mo/Zn ZSM-5 + Fe oxide73,74 | |
| Methanol | (R11) CH4 + MeOx → MeOx−1 + CH3OH | (R12) MeOx−1 + H2O → MeOx + H2 | CL-Selective oxidation | Copper-exchanged zeolites75,76 | |
| Methanol | Formaldehyde | (R13) CH3OH + MeOx → MeOx−1 + CH2O + H2O | (R14) MeOx−1 + 1/2O2 → MeOx | CL-Selective oxidation | FeMoO4–Fe2O377 |
| C2+ hydrocarbon | Ethylene oxide | (R15) C2H4 + MeOx → MeOx−1 + C2H4O | (R19) MeOx−1 + 1/2O2 → MeOx or (R21) MeOx−1 + CO2 → MeOx + CO | CL-Epoxidation | Ag/SrFeO378 |
| Ethylene | (R16) C2H6 + MeOx → MeOx−1 + C2H4 + H2O | CL-ODH | Alkali modified Mn and Fe oxides79–83 | ||
| Propylene | (R17) C3H8 + MeOx → MeOx−1 + C3H6 + H2O | CL-ODH | Mn and La perovskite,84 and VOx85 | ||
| Propionaldehyde | (R18) 1/2C3H8 + MeOx → MeOx1 + 1/2C3H6O + 1/2H2O | CL-Selective oxidation | —b | ||
| Maleic anhydride | (R20) 1/7C4H10 + MeOx → MeOx−1 + 1/7C4H2O3 + 4/7H2O | CL-Selective oxidation | Vanadium phosphorous oxygen (VPO)86 | ||
| Butadiene | (R22) C4H8 + MeOx → MeOx−1 + C4H6 + H2O | CL-ODH | — | ||
| Ethylene | (R23) C6H14 + MeOx → MeOx−1 + 3C2H4 + H2O | CL-Oxidative cracking | Alkali modified Mn and Fe oxides87 | ||
| Aromatic | Styrene | (R24) C8H10 + MeOx → MeOx−1 + C8H8 + H2O | (R26) MeOx−1 + 1/2O2 → MeOx | CL-ODH | — |
| Methylstyrene | (R25) C9H12 + MeOx → MeOx−1 + C9H10 + H2O | — | |||
| Biomass | Synthesis gas | (R27) CxHyOz + MeOx → MeOx−1 + synthesis gas | (R28) MeOx−1 + 1/2O2 → MeOx | CLR | Fe oxide44 |
| H2O | Hydrogen | (R29) H2O + MeOx−1 → MeOx + H2 | (R30) MeOx → MeOx−1 + 1/2O2 | Thermochemical splitting | Perovskite,88 CeO2,64 Fe oxide89 |
| (R31) MeOx + CO → CO2 + MeOx−1 | CL-WGS | Fe oxide90–92 | |||
| CO2 | CO | (R32) CO2 + MeOx−1 → MeOx + CO | (R33) MeOx → MeOx−1 + 1/2O2 | Thermochemical splitting | Perovskite,67 CeO2,93 Fe oxide94 |
| (R34) MeOx + H2→ H2O + MeOx−1 | CL-RWGS | Perovskite,95 Fe oxide96,97 | |||
| N2/H2 | NH3 | (R35) 3/2H2 + MeNx → MeNx−1 + NH3 | (R36) 1/2N2 + MeNx−1 → MeNx | CL-ammonia synthesis | Transition metal nitride98 |
| (R37) H2O + MeNx → MeO + xNH3 + (1 − 3x/2)H2 | (R38) MeO + [2/(4m + n)]CmHn → Me + [n/(4m + n)]H2O + [2m/(4m + n)]CO2 | Metal nitride/metal oxide/metal99–101 | |||
| (R39) x/2N2 + Me → MeNx | |||||
| (R40) 3/2H2 + 1/βMeHαNβ → 1/βMeHα + NH3 | (R41) 1/βMeHα + 1/2N2 → 1/βMeHαNβ | Nitrogen-containing hydride/hydride98–105 | |||
2. The case for chemical looping beyond combustion
2.1. Chemical looping – the general principle
As illustrated in Table 1, chemical looping can take various forms for numerous applications. Generalized schematics and representative reactions of different chemical looping processes are summarized in Fig. 1 and Table 1, respectively. As can be seen, reactive separation facilitated by solid (oxygen, nitrogen, and hydrogen) carrier particles represents an important underlying feature of all chemical looping reactions and processes. In chemical looping processes, oxygen carrier, usually composed of metal oxides, acts as an oxygen reservoir for oxygen donation and regeneration.21 Nitrogen carrier composed of nitride or nitride–hydride mixed compounds enables the storage and release of N or/and H in the redox cycle for chemical looping ammonia synthesis.98 In a similar manner, hydride based hydrogen carrier allows the storage and release of hydrogen to participate in in chemical reactions or shift the reaction equilibrium.106 Although recent research on nitrogen,98–105,107,108 and hydrogen106 carriers offer significant new opportunities, the vast majority of the carriers investigated to date are oxygen carrying agents. In order to maintain coherency, we elected to focus exclusively on chemical looping approaches for chemical production using oxygen carriers, also known as redox catalysts. Table 1 summarizes both the key CLBC schemes reported to date and promising CLBC schemes for future investigations. To further illustrate the thermodynamic favorability of the various potential reaction pathways, Fig. 2 summarizes potential reaction pathways and Gibbs free energy changes (ΔG) among various carbon-containing feedstock and products. To maintain consistency, 1 bar oxygen partial pressure (PO2) was assumed in all cases. Fig. 2 is intended to be used as a screen tool to determine the thermodynamically favorable reactants, products, and CLBC schemes.49 It is also important to note that while one can anticipate similar trends in relative ease of product formation irrespective to the oxygen carrier used in CLBC, the equilibrium PO2 of oxygen carriers, which is dependent upon both the composition/phase of the redox pair and the reaction temperature, can span more than 20 orders of magnitudes. This large degree of freedom in PO2 and the ability of chemical looping to decouple a single overall reaction into multiple redox steps, provides unique flexibility in “manipulating” the extent and feasibility of certain chemical reactions, as will be illustrated in Section 2.2. Besides redox based CLBC schemes, this article also covers the use of CO2 sorbents (i.e. calcium looping) to enhance the oxygen carrier-based chemical looping approaches, as will be elaborated in Section 4.4.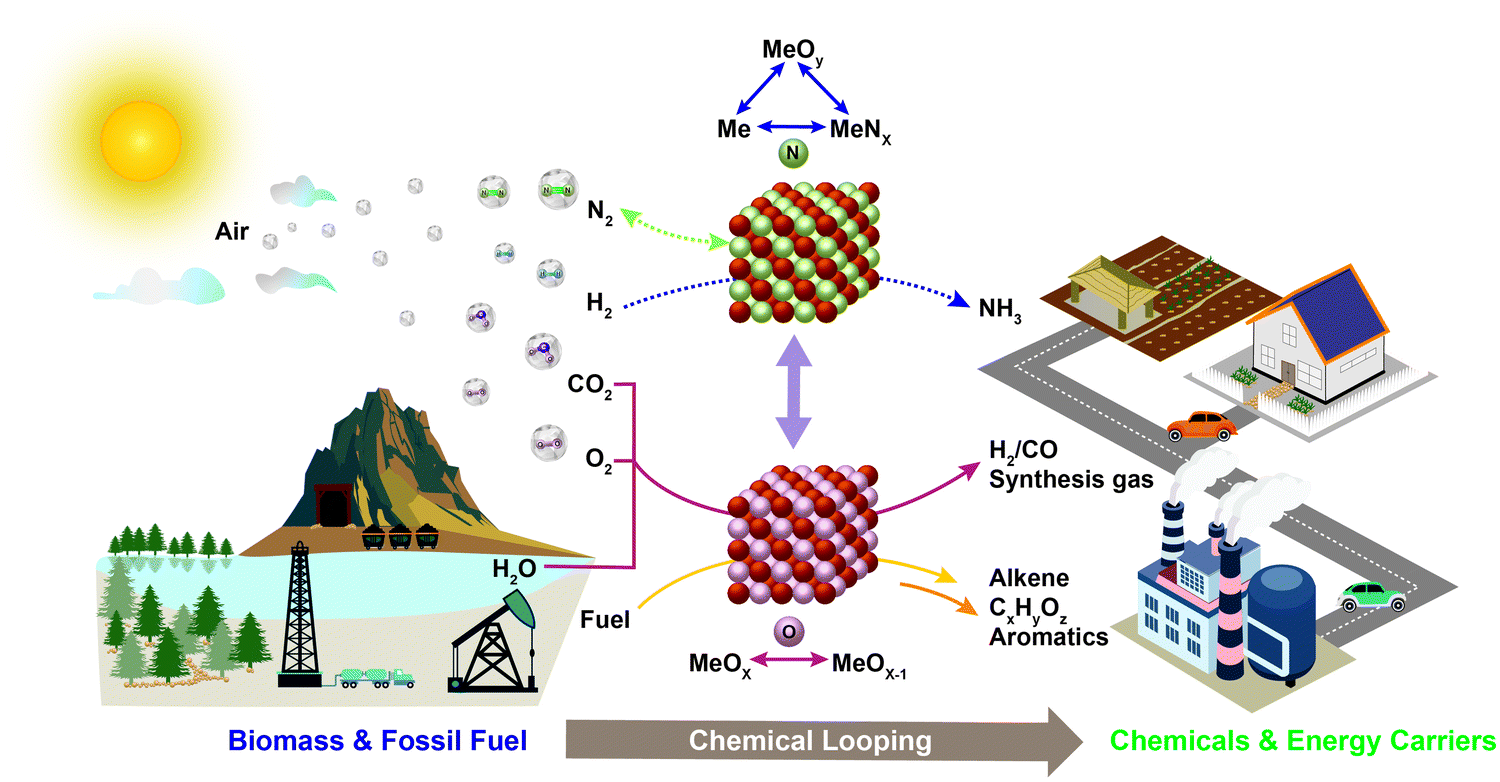 | ||
| Fig. 1 Schematic illustration of the chemical looping strategy and its potential applications. | ||
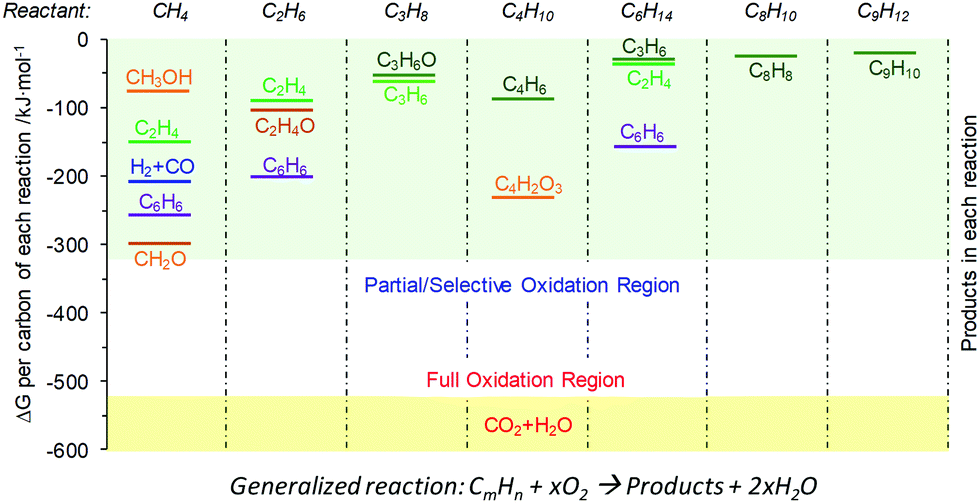 | ||
| Fig. 2 Normalized reaction Gibbs free energy change (ΔG) per mole of carbon at 700 °C for various potential chemical products from C1–C9 hydrocarbons. Bars with different colors represent the ΔG for the partial oxidation of hydrocarbons to the corresponding products. For consistency, 1 atm gaseous oxygen is assumed to be the oxidant. To calculate ΔG with a specific redox pair as the oxidant (e.g. Mn3O4/MnO), one can use the corresponding equilibrium oxygen partial pressure (PO2) of the redox pair at the temperature to account for the oxygen chemical potential change relative to 1 atm O2. | ||
A typical chemical looping process involving oxygen-carrying agents is composed of two or more reduction and oxidation steps that form a redox loop. In its simplest form, an oxygen carrier or redox catalyst first donates its lattice oxygen under a low oxygen partial pressure environment. The reduced oxygen carrier is subsequently exposed to an oxidant for the replenishment of its lattice oxygen, thereby completing a two-step redox loop. As illustrated in Table 1, the use of oxygen carrier offers significant flexibility and opportunity for process intensification by breaking an overall reaction into sub-reactions because: (i) besides acting as a reactant, the oxygen carrier also acts as a mass separation agent since the looping reactions prevent the mixing among the products from each sub-step; (ii) the carrier's oxygen donation properties can be tailored for improved thermal management of the overall process; and (iii) the reduction or oxidation steps can be further broken into multiple sub-steps.15,21 When designed properly, an oxygen carrier can be compatible with various oxidizing and reducing agents to facilitate the generation of multiple value-added products with minimal separation requirements.
From a thermodynamic standpoint, the oxygen donation ability of oxygen carriers/redox catalysts can be evaluated by the equilibrium oxygen partial pressure (PO2) of the reaction MeOx → MeOx−1 + 1/2O2.49 Redox pairs with high PO2s, which are suitable for chemical looping air separation (CLAS), chemical looping with oxygen uncoupling (CLOU), or CLC, can only be regenerated with air. As applications summarized in Table 1, CLBC can take advantage of redox pairs with a significantly wider range of PO2s. As such, valuable products can be produced in both the oxygen carrier reduction and re-oxidation steps. In the reduction or oxygen donation step, CLBC can valorize light alkanes, aromatics or biomass into chemicals via chemical looping partial oxidation (CLPOx). Although CO2 and H2O are the most stable forms of the oxidation products (other than carbonates), many chemical products, including hydrogen, synthesis gas, alkene, aromatic, alcohol, aldehyde, epoxide, or maleic anhydride, are thermodynamically feasible as illustrated in Fig. 2b. The key challenge for high yields towards these partial oxidation products resides in the design of selective redox catalysts to minimize COx and other byproduct formation, as will be elaborated in Section 3.2. In the regeneration step, hydrogen can be produced via water splitting or CO from CO2 splitting. Selective hydrogenation reactions can also be carried out during the re-oxidation of tailored redox catalysts. Although the chemical looping cycles can be more complex than conventional processes in the presence of heterogeneous catalysts, they do offer multiple potential advantages from both product selectivity and process efficiency viewpoints, as will be further illustrated in the following sections.
2.2. The case for chemical looping beyond combustion – a second law perspective
As discussed in Section 2.1, a common feature of chemical looping is the ability to facilitate chemical reactions with in situ product separation. With separation being the most energy-intensive step in the chemical industry, reactive separation enabled by chemical looping offers excellent opportunities for energy savings and emission reductions. The potential benefits of chemical looping, however, goes well-beyond the simplification of product separations. With strategically designed reaction scheme and intermediates, chemical looping can be intrinsically advantageous over conventional approaches from a second law of thermodynamics perspective. The potential second law (exergy) savings via chemical looping result from the following aspects:![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 stoichiometric ratio of 2:1, would consume at least 5.8 kJ of energy in air separation alone for each mole of methane converted. Methane POx, on the other hand, is thermodynamically highly favored with ΔG0 of −86.7 kJ mol−1 of methane converted. Chemical looping POx can take advantage of the large thermodynamic driving force for methane POx and integrate it for air separation. Using Mn2O3 as an example, the two chemical looping sub-reactions are each favored thermodynamically:
:O2 stoichiometric ratio of 2:1, would consume at least 5.8 kJ of energy in air separation alone for each mole of methane converted. Methane POx, on the other hand, is thermodynamically highly favored with ΔG0 of −86.7 kJ mol−1 of methane converted. Chemical looping POx can take advantage of the large thermodynamic driving force for methane POx and integrate it for air separation. Using Mn2O3 as an example, the two chemical looping sub-reactions are each favored thermodynamically:| Sub-reaction 1: CH4 + 3Mn2O3 → CO + 2H2 + 2Mn3O4 ΔG01 = −9.8 kJ mol−1 |
| Sub-reaction 2: 2Mn3O4 + 1/2O2 → 3Mn2O3 ΔG02 = −76.9 kJ mol−1 |
| Overall reaction: CH4 + 1/2O2 → CO + 2H2 ΔG0 = −86.7 kJ mol−1 |
b. Exergy savings via in situ conversion of byproducts. The production of value-added chemicals can be intensified by chemical looping, via selectively oxidizing the reaction byproducts. As exemplified in Table 1, this principle has been applied to olefin production via oxidative coupling of methane, oxidative dehydrogenation, and naphtha oxidative cracking. From a second law standpoint, chemical looping can lead to exergy savings in the following two ways. On one hand, the in situ oxidation of byproducts such as hydrogen can compensate for the thermal energy required by endothermic reactions, e.g. the non-oxidative coupling of methane or ethane cracking/dehydrogenation. Compared to conventional approaches where hydrocarbon fuels are combusted to neutralize the reaction endothermicity via indirect heat transfer through cracker furnaces, in situ combustion of hydrogen is intrinsically advantageous due to its lower exergy rate, i.e. the ratio between the exergy and enthalpy of a fuel compared to that of hydrocarbons. Fig. 3 illustrates the exergy loss for ideal steam cracking and chemical looping oxidative dehydrogenation (CL-ODH) steps,111 assuming no heat loss in neither case. When practical factors, such as the temperature difference for indirect heat transfer, are considered, a higher exergy loss can be anticipated for conventional cracking.
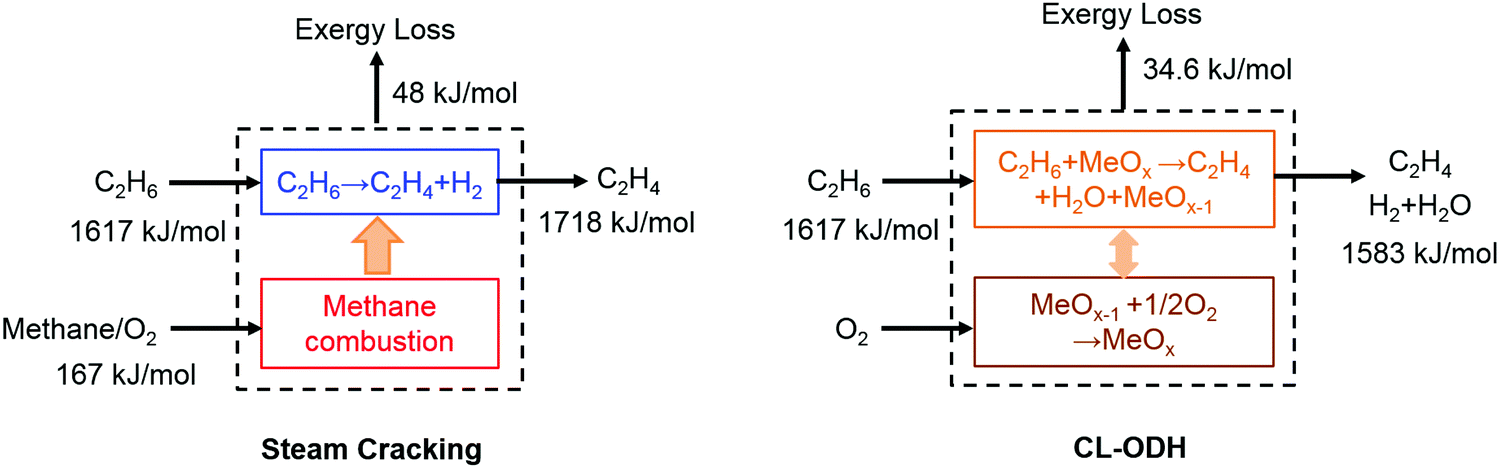 | ||
| Fig. 3 Availability analysis and exergy loss for idealized steam cracking and chemical looping-oxidative dehydrogenation (CL-ODH) schemes. | ||
A second aspect of exergy savings via byproduct oxidation is through enhanced product yields. For instance, steam cracking of both ethane and naphtha are equilibrium-limited.112In situ combustion of the hydrogen byproduct significantly increases the olefin yield from 50% to nearly 70% in a lab-scale reactor at 850 °C while decreasing the volumetric flow rate of non-condensable product gases by ∼40%.81 The corresponding energy savings in product compression and separation alone correspond to 554 kJth mol−1 of ethylene produced.113 Due to these reasons, process analyses indicate that the chemical looping approach can lead to more than 80% energy savings for ethane conversion and 50% energy savings for naphtha cracking.112,113
c. Ability to circumvent second law limitations by the redox loop. Another potential advantage of dissecting an overall reaction into sub-reactions resides in a higher degree of freedom in terms of thermodynamic characteristics for the individual sub-reactions. Moreover, the operating conditions for each sub-step, e.g. temperature and pressure, can be adjusted independently to maximize the product yields from an equilibrium standpoint. A good example of using the chemical looping strategy to circumvent second law limitations of an overall reaction is thermochemical water-splitting to produce hydrogen:
| H2O → H2 + 1/2O2 |
| Sub-reaction 1: H2O + Ce2O3 → H2 + 2CeO2 ΔH01 = −126.6 kJ mol−1 ΔS01 = −82.3 J K−1 ΔG01 = −104.1 kJ mol−1 |
| Sub-reaction 2: CeO2 → Ce2O3 + 1/2O2 ΔH02 = 368.2 kJ mol−1 ΔS02 = 125.9 J K−1 ΔG02 = 333.7 kJ mol−1 |
| Overall reaction: H2O → H2 + 1/2O2 ΔH0 = 241.5 kJ mol−1 ΔS0 = 43.5 J K−1 ΔG0 = 229.6 kJ mol−1 |
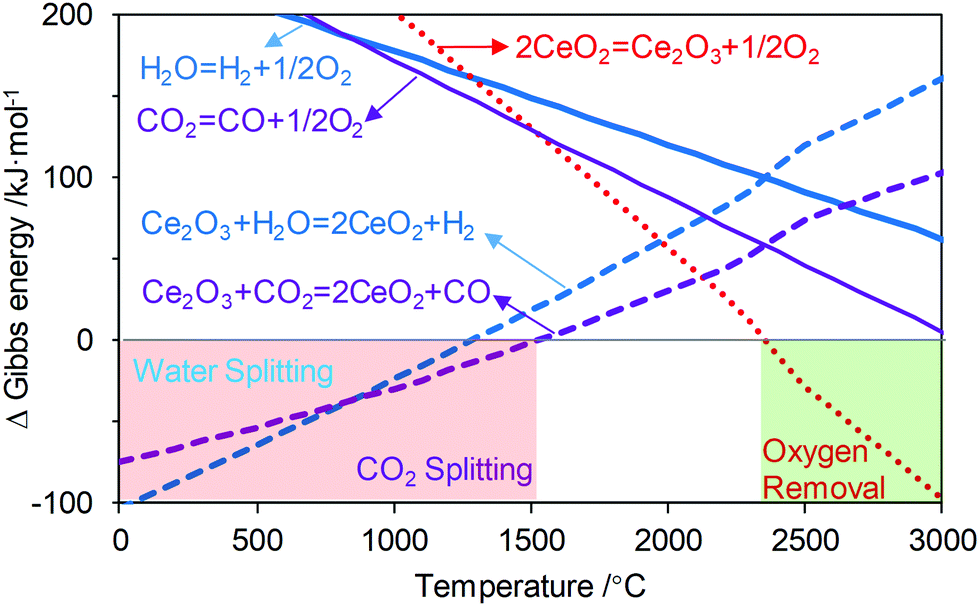 | ||
| Fig. 4 Ellingham diagram of thermal and chemical looping water and CO2 splitting. Light green and red regions represent thermodynamically spontaneous (K ≥ 1) reduction and oxidation reactions, respectively. In practice, a reaction would occur when K < 1. | ||
d. Tunable exo-/endothermicity of sub-reactions for improved energy integration. Chemical looping would not alter the enthalpy or entropy changes of the overall reaction under (near) isothermal conditions. However, the distributions of the reaction enthalpy/entropy can be modified by varying the type of the oxygen carrier/redox catalyst and the chemical looping sub-reactions. As such, the heat release/demand of individual chemical looping reactors can be adjusted. This additional degree of freedom can enable significantly improved energy integration and hence reduced exergy/energy losses.122 With an increased understanding of PO2, phase transition, and catalytic properties for the mixed oxide-based oxygen carriers, the tunabilities of oxygen carriers and hence the corresponding sub-reactions are becoming an increasingly attractive route for efficiency improvements. Fig. 5 summarizes the potential advantages of CLBC over conventional approaches.
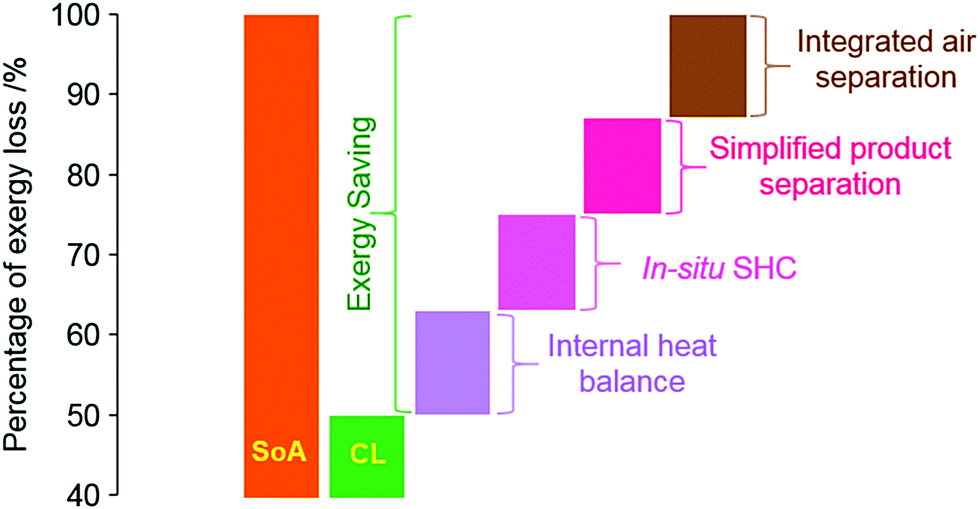 | ||
| Fig. 5 Schematic illustration of the exergy loss for conventional and chemical looping approaches. SoA: state-of-the-Art (SoA) approaches are typically based on thermal cracking or partial oxidation with oxygen co-feed. SHC refers to selective combustion of hydrogen byproduct, e.g. in CL-ODH, chemical looping oxidative coupling of methane (CL-OCM), and chemical looping dehydroaromatization (CL-DHA). | ||
2.3 Reasons for CLBC – the practical drivers
Besides the abovementioned thermodynamic “drivers”, a strong case can be made for CLBC from a practical standpoint. Commercial implementation of the CLC technology needs to address both technical challenges, in terms of scale-up, and economic challenges, resulting from the low margin of the utility industry and the relatively low economic incentive of the current carbon tax structure.22,36 A CLC power plant will be more complex and costlier than a standard pulverized coal combustion plant, due to large scale circulating fluidized bed operations, high solids circulation rates, and the need to continuously replenish degraded oxygen carrier particles.4 Meanwhile, low-cost and long-lifetime oxygen carrier material is required to make CLC cost effective.4 In addition, concerns over carbon sequestration, a critical downstream step for any carbon capture technology, also limits the wide spread utilization of CLC as a promising carbon capture process.123–127 CLBC, on the other hand, has the potential to address all the limitations identified above. In contrast to CLC whose economics is likely to be policy driven, CLBC for chemicals production is market driven because the products are of higher value than electric power. As illustrated in Fig. 6, the CLBC concept can be applied to produce chemical products that are significantly more valuable than electricity, thereby increasing the profit margin. In addition, CLBC based on partial oxidation is intrinsically more efficient from an oxygen carrier utilization standpoint. Complete combustion of carbonaceous fuels such as methane consumes a significant amount of lattice oxygen for power generation (four [O] are required for each carbon combusted in CH4). In comparison, the same amount of lattice oxygen is able to transform eight carbon atoms in ethane into more valuable ethylene products under a CL-ODH scheme (assuming 100% ethylene selectivity whereas ∼90% olefin selectivity has been reported experimentally).79,81,112,128 If oxygen carriers/redox catalysts with identical oxygen storage capacities are used, the CL-ODH plant would correspond to an eight-fold decrease in solids circulation rate compared to a CLC plant with identical feedstock processing capacity on a weight basis. In addition, at least an order of magnitude increase in reaction kinetics was observed for ethane CL-ODH when compared to methane CLC based on the experience at North Carolina State University.80,81 Considering that ethylene production could significantly increase the gross margin compared to power generation, the “breakeven” plant size for CL-ODH would likely to be one or more order of magnitude smaller than that for CLC from process economics, reaction stoichiometry, and kinetics standpoints. An even stronger case can be made for CLBC of heavier feedstock/products: applying the chemical looping strategy for butane to butadiene, if successful, would lead to a 16 times higher lattice oxygen utilization efficiency than for methane CLC. This simple case study clearly demonstrates the significant advantages of CLBC from process scale up and economic standpoints. Another interesting point to make is that many CLBC processes themselves do not offer the primary benefit of CLC, i.e. CO2 capture, since the target products are chemicals instead of heat (Table 1). However, the process intensification and second law advantages offered by CLBC can still lead to significant CO2 reductions by producing less CO2 per unit amount of product generated. A recent study published by Haribal et al.113 indicated that CL-ODH can reduce the CO2 emissions for ethylene production by 84% when compared to the commercial ethane cracking processes. Such integrated carbon savings without CO2 capture (except for the removal of a small amount of CO2 byproducts via conventional stripping) eliminates the needs for geological CO2 sequestration, a potential technical challenge for any CO2 capture process including CLC.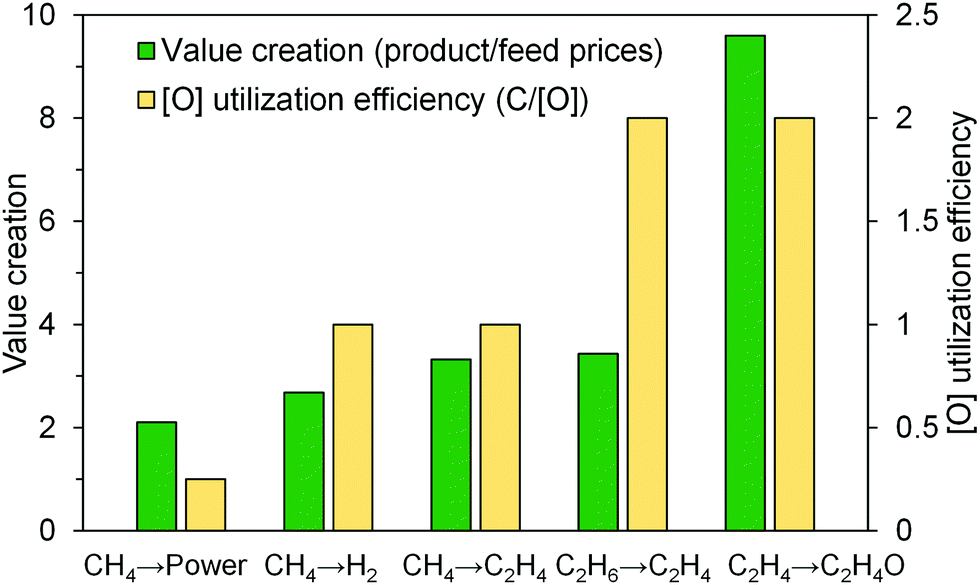 | ||
| Fig. 6 Valorization and efficiency of oxygen utilization in some typical chemical looping processes. | ||
3. General design strategies for CLBC
3.1 Design considerations for CLBC schemes and limitations
While chemical looping offers various potential advantages as stated in Section 2, it is subjected to a number of limitations, which occasionally are overlooked in CL process analyses. This is somewhat “understandable” considering the complexity of CL processes, which involve dynamic and intertwined gas–solid reactions, e.g. the extent of redox catalyst conversion in the reduction step(s) often affects the equilibrium conversion and practical performance of the oxidation step(s). As such, we think it is important to summarize the uniqueness of CLBC as well as its limitations and potential pitfalls in design and analysis of CLBC schemes. Compared to conventional catalytic oxidation or reduction processes, the chemical looping strategy is capable of:(i) Providing inherently separated product streams to eliminate or alleviate the needs for separation;
(ii) Enhancing process safety by avoiding the direct mixing between a gaseous oxidant (e.g. O2) and reductant (e.g. a hydrocarbon);
(iii) Redistributing the overall heat of reaction into two or more steps to simplify heat integration;
(iv) Breaking the “equilibrium barrier” of an overall reaction by carefully selecting redox catalysts, redox pairs91 and carrying out individual chemical looping steps at substantially different conditions and/or gas–solids contacting modes. For example, the chemical equilibrium limitations in the WGS reaction under a CO–H2O co-feed mode can be overcome using the chemical looping strategy over a non-stoichiometric mixed oxide, since such an oxide exhibits a large range of equilibrium PO2 as a function of oxygen non-stoichiometry.91 Metcalfe et al.91 investigated such a scheme using a perovskite (La0.6Sr0.4FeO3−δ) as the oxygen carrier. Using a packed bed reactor operated under a countercurrent contacting mode, a significantly higher overall CO to H2 conversion (compared to WGS equilibrium conversion) was demonstrated. The unique reactor design and oxygen carrier properties enabled the production of near pure H2 and CO2 as separate product streams.
(v) Potentially achieving higher product selectivity by using lattice oxygen as opposed to gaseous oxidants.
However, one must be aware of the following limitations for chemical looping when designing chemical looping schemes or performing process analyses:
(i) The sum of Gibbs free energies of the products from all the chemical looping sub-steps should be smaller than that of the feedstock;
(ii) For a two-step CL process with a single metal/metal oxide redox pair, products from an (oxide) reduction reactor 1 should not be able to oxidize products from an oxidation reactor 2. This principle would apply for both co-currently and counter-currently operated fluidized bed, fixed bed, and moving bed reactors. Using WSG/RWGS reactions129 as an example, one cannot produce a substantially pure stream of CO2 or H2O from reactor 1 while also producing a substantially pure stream of H2 or CO from reactor 2 if a single redox pair is used. We have to make a choice between a lower CO2/H2O yield in reactor 1 and a decreased CO/H2 yield from reactor 2. Note that this is different from the case illustrated by Metcalfe et al., in which a non-stoichiometric metal oxide was used as the carrier;91
(iii) A three-step CL process can allow products from reactor 1 to oxidize products from reactor 2 (since the two reactors and redox pairs do not form a complete loop). However, this is only feasible with an oxidant in step 3 that, when combined with the products from step 2, exhibits a higher equilibrium oxygen partial pressure (PO2) than do the products from step 1;
(iv) Kinetically, chemical looping reactions are generally much slower than conventional catalytic oxidation/reduction reactions. This is due to the fact that the rates of redox reactions are limited by the removal and replenishment of lattice oxygen from/to the bulk crystal structure of the redox catalyst. One exception is the CL-ODH operated at thermal cracking temperatures (>700 °C). In that case, the rate of hydrogen combustion by the redox catalysts can often match or exceed the hydrogen generation rate from the cracking reactions.80,81
We note that many mixed oxide-based redox catalysts, e.g. perovskites, can accommodate significant lattice defects prior to phase transition.129,130 As such, the redox thermodynamics of these redox catalysts are unique and cannot be categorized as redox pairs. That is, the equilibrium oxygen partial pressure of the oxide is a continuous function of the oxide's (oxygen) vacancy concentration without incurring a distinct phase change.131 The unique redox properties of these mixed oxides, coupled with counter-current and/or multiple step chemical looping reactor design, can lead to interesting process configurations that are not subjected to limitations (ii) and (iii) presented above. Fig. 7 illustrates a hypothetical case where a mixed oxide redox catalyst with varying equilibrium PO2s as a function of oxygen storage capacity can be used to break the RWGS equilibrium in a counter-currently operated two-step chemical looping scheme. A comparable effect can be achieved by using more than one redox pairs with different equilibrium PO2s. In reality, however, one must consider the complex relationships between the oxygen non-stoichiometry of the mixed oxide, which dictates the oxygen mass balance of the redox reactions, and the equilibrium PO2, which determines the redox thermodynamics, in addition to kinetic and transport effects.
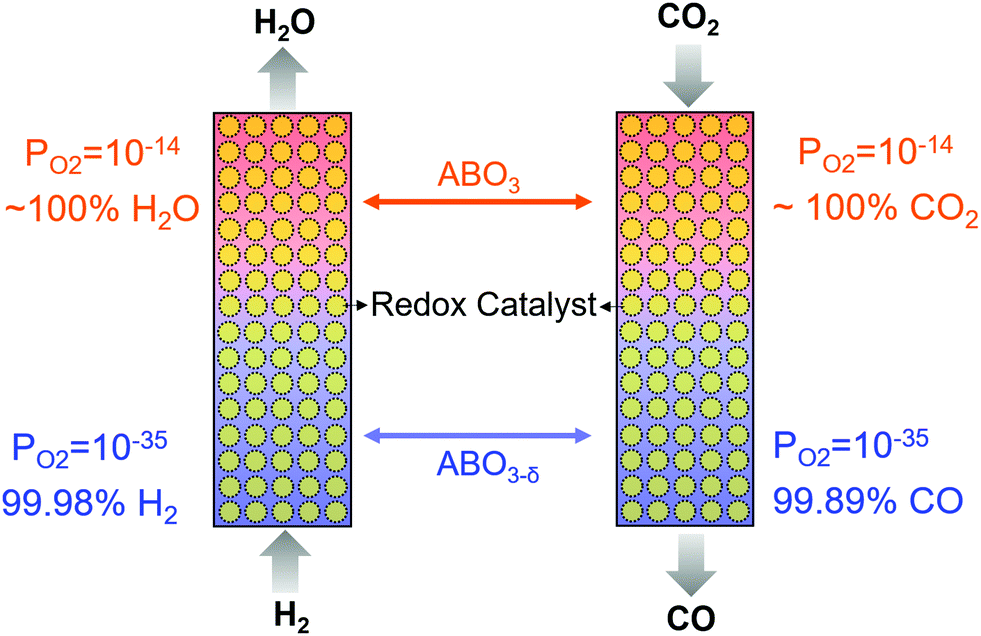 | ||
| Fig. 7 RWGS in a counter-currently operated two-step chemical looping scheme using ABO3 perovskite as a redox catalyst at 500 °C. | ||
3.2 Oxygen carrier design
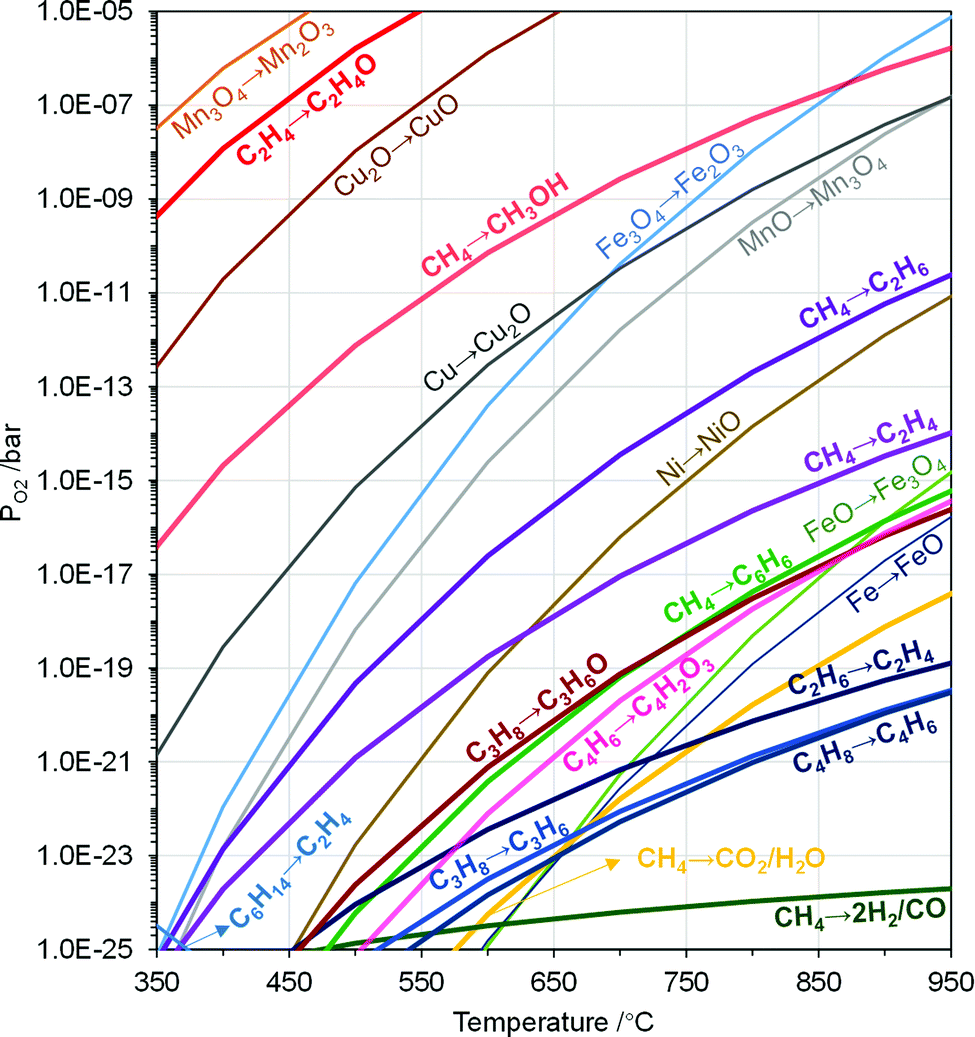 | ||
| Fig. 8 Modified Ellingham diagram of metal oxide redox and POx reactions. MeOx → MeOy and hydrocarbons POx reactions correspond to the PO2 when ΔG = 0. Reactions in pathways of MeOx → MeOy and hydrocarbons POx are given in Table 2. | ||
The thermodynamic criteria discussed above are intended for narrowing down the design space of the redox catalysts. Possessing suitable thermodynamic properties alone typically would not ensure high product selectivity. A rare exception is synthesis gas generation. As illustrated in Fig. 2b, synthesis gas is one of the thermodynamically favored oxidation product except for CO2 and water. As such, synthesis gas selectivity and yield can be optimized by tuning the PO2s of redox catalysts using Gibbs free energy minimization, as illustrated in a few recent publications.49,66,67,132 For other POx products with higher Gibbs free energies, however, such a thermodynamically based strategy would not work since the spontaneous decomposition or oxidation of oxygenates to synthesis gas and/or CO2/H2O is both kinetically and thermodynamically favored at elevated temperatures.
Aside from being a redox catalyst selection tool, equilibrium PO2s illustrated in the Ellingham diagram can be used to manage heat distributions among the redox steps involved in CLBC. Due to the endothermicity in the oxygen release step of redox catalysts, a large fraction of the fuel oxidation reactions with redox catalysts in CLBC are endothermic, even though oxidation of a fuel with oxygen would be exothermic. Oxides with higher PO2s tend to decrease the endothermicity in the fuel oxidation step while decreasing the exothermicity of the redox catalyst re-oxidation step. As such, rationally selected redox pairs can facilitate optimal distribution of the reaction heat in the redox steps to simplify heat integrations. In CLC, a bi-metallic Fe–Cu oxygen carrier composed of CuO and Fe2O3 was proposed to shift the endothermic reduction to exothermic or heat neutral.133,134 This PO2 tuning strategy has also been applied to CLBC, e.g. in ethane CL-ODH.82,83,128
Redox thermodynamic data for monometallic oxides are readily available, as illustrated in Fig. 8. However, the number of suitable redox pairs are limited when considering the aforementioned thermodynamic criteria. Mixed oxides such as hexaaluminates, spinels, or perovskites, offer significantly increased tunability in terms of redox properties. Moreover, equilibrium PO2s of many mixed oxides are often variable without incurring phase changes, as discussed in Section 3.1. As such, their multidimensional redox behavior can not be illustrated by a simple redox reaction or a curve in the Ellingham diagram. These unique properties make mixed oxides an intriguing option for CLBC, as illustrated by a number of recent publications.61,70,135 Among the various mixed oxides, perovskite-type oxides represent a large family of materials that is particularly promising for CLBC due to their highly tunable structural, compositional, and redox properties.136–138 A recent perspective provided a comprehensive discussion on perovskites as redox catalysts for CLBC.49
On the other hand, decreased COx formation can be achieved by facilitating more facile surface oxygen removal compared to the lattice oxygen flux since the formation of electrophilic oxygen species can be minimized. To ensure a relatively faster surface oxygen removal rate, one can either decelerate the O2− flux or enhance the surface activity of the redox catalyst, as illustrated in Fig. 9. Recent studies have confirmed the effectiveness of both approaches. Gao et al.79,128 investigated a LaxSr2–xFeO4−δ-based redox catalyst for CL-ODH of ethane to ethylene. While LaxSr2–xFeO4−δ promoted the complete combustion of ethane, alkali metal oxide promoters enriched on the surface of the mixed oxide and significantly inhibited the O2− flux from the bulk. As a result, 90% ethylene selectivity was achieved. Besides alkali metal oxide promoters, alkali salt modification also play a similar role to depress O2− flux of the redox catalyst for the selective combustion of hydrogen (SHC) in CL-ODH of ethane to ethylene.81–83 Enhancement of redox catalyst's surface activity was reported for methane partial oxidation for synthesis gas generation. Shafiefarhood et al.139 reported that impregnating Rh on a CaMnO3-based redox catalyst decreased the methane activation energy on the oxide surface by more than 95%, leading to a decrease in methane conversion temperature by 300 °C and a significantly increased synthesis gas selectivity. Those practices reveal that tuning of the oxygen transfer rate from bulk of a redox catalyst to the catalytically active species on the surface is of great importance for POx. In a redox-involved context, Machida at Kumamoto University found that surface CeO2 could act as a gateway for oxygen storage in and release from various oxides acting as an oxygen reservoir, such as Fe2O3 or La2O2SO4.141–143
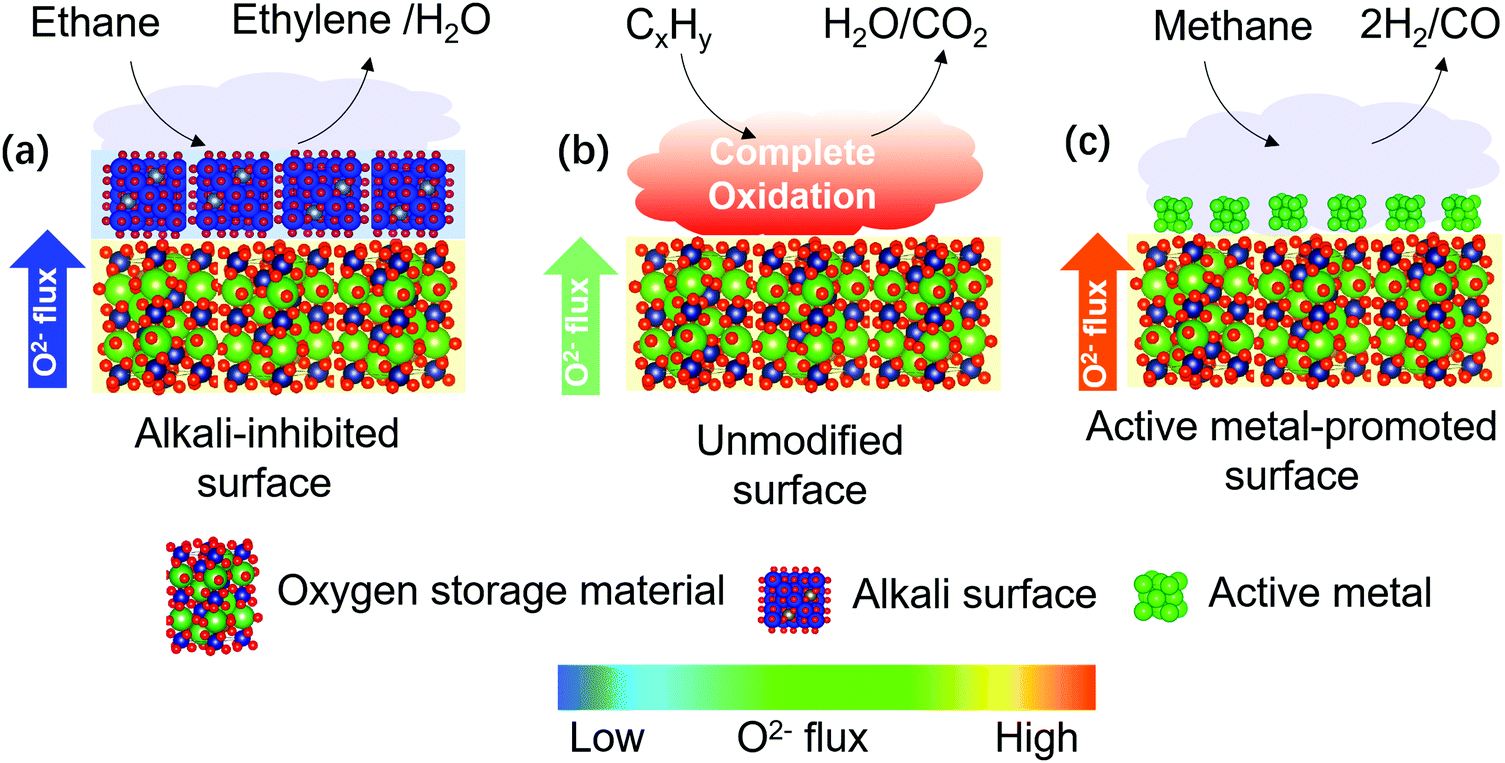 | ||
| Fig. 9 Schematic illustration of surface modifications of redox catalysts to improve product selectivity by changing the relative rates of surface oxygen removal and bulk O2− flux. The arrows in the three cases (a–c) correspond to the O2− fluxes and the colors correspond to the relative intensity of the O2− fluxes. | ||
Although the abovementioned strategy for catalyst selectivity optimization has been shown to be effective, we note that it still represents a rather simplified view of the redox catalyst. It would be desirable, from both scientific and engineering standpoints, to determine the active sites and elementary reaction pathways of the chemical looping reactions, but such tasks can be daunting due to the highly dynamic nature of chemical looping reactions. In fact, both the bulk and surface of the redox catalysts can dynamically rearrange throughout the redox cycle.144 The utilization of modern catalysis and surface science techniques in a few recent studies have already started to reveal fundamental insights on the mechanisms of the redox reactions.78,128,145–151 However, significant progress still needs to be made to gain additional fundamental insights and this should be a topic of focus for redox catalyst studies within the foreseeable future.
In principle, one can also combine heterogeneous catalysts or active sites with an oxygen storage material to achieve combined catalytic and chemical looping functions. For instance, as will be discussed in Section 4.2.2, Chan et al. demonstrated the CL epoxidation of ethylene over Ag/SrFeO3 redox catalyst where Ag acts as the epoxidation catalyst and SrFeO3 acts as the oxygen storage material for oxygen donation.78 Ethylene was converted to ethylene oxide (with CO2 and H2O by products) in the reduction step and the reduced redox catalyst was then replenished with air. Many other CLBC systems can potentially take advantage of such a strategy. Examples include alkane ODH, methane dehydroaromatization, methane oxidative coupling, among others. These topics are discussed in Sections 4.1 and 4.2.
We note that the above discussions are intended to cover the key strategies and the underlying principles for the design of CLBC redox catalysts and reaction schemes. From a practical standpoint, the redox catalyst's activity, available oxygen storage capacity, stability, mechanical strength,152,153 heat management,154 cost and environmental risk should all be taken into account to optimize the redox catalyst. The higher margin in the chemical industry over the utility industry can afford significantly higher redox catalyst cost in CLBC. While the raw material cost of some redox catalysts can be high (e.g. ceria-based materials), the additional value created for chemical production can potentially accommodate higher redox catalyst costs. Under CLBC schemes, redox catalysts functions similar to heterogeneous catalysts in chemical production processes.155 It is further noted that a number of CLBC redox catalysts have shown chemical productivities (in terms of weight hourly space velocity) comparable to commercial catalysts.111 The aforementioned factors should also be considered along with reactor design parameters, operating conditions as well as the reactor/process economics for CLBC process development.
4. CLPOx/CLBC process schemes
4.1 Chemical looping for C1 valorization
CH4 is the main constituent of natural gas and an important feedstock for the production of commodity chemicals and liquid fuels.156 It can be converted into methanol, NH3 and other higher hydrocarbons either directly via a one-step process or indirectly via a synthesis gas (a mixture of CO and H2) intermediate. At present, the only cost-effective route to convert CH4 into valuable chemicals is via synthesis gas.157 Synthesis gas can be obtained from CH4 using the following three reactions: steam reforming (R62), dry reforming (R63) and partial oxidation (R49).158 At the industrial scale, the catalytic steam methane reforming (SMR) is used predominantly to produce synthesis gas. Although SMR is highly optimized, it is a very energy-intensive process and releases large quantities of CO2 into the atmosphere.131 To reduce the energy demand of SMR, a part of CH4 is combusted internally with oxygen or air. The resulting so-called autothermal reforming process enhances CH4 conversion and reduces CO2 emissions, but at the expense of lowered synthesis gas yields.57 Recently, dry reforming of methane (DRM) has received a lot of attention due to its potential to convert two greenhouse gases (i.e. CH4 and CO2) into an equimolar mixture of CO and H2 desired for Fischer–Tropsch synthesis.159,160 Although this concept has many environmental and economic incentives, there is currently no commercial process for DRM. Unlike reforming reactions, methane POx is mildly exothermic allowing for auto-thermal operation at temperatures exceeding 1000 °C. Methane POx can be performed either at elevated temperatures (>1200 °C) without a catalyst or at lower temperatures (<900 °C) in the presence of a platinum group metal (PGM) based catalyst (e.g., Pt, Pd, Rh, Ru, etc.).161,162 Commercial methane POx processes incur a significant efficiency loss due to high operating temperatures. The catalytic route can be costly due to the high cost of the PGM and safety concerns. Both approaches also are challenged by the high cost and energy intensity of pure O2.In 2000, Lyon and Cole163 proposed a chemical looping approach to transfer oxygen from air to a fuel using solid oxygen carriers for a number of applications such as pollution control, production of inert gases, delivery of heat to endothermic reactions, etc. Since then various chemical looping cycles have been proposed to address the limitations of conventional CH4 conversion processes (see in Table 2). Sections 4.1.1 and 4.1.2 provide a brief summary of the chemical looping approaches related to indirect and direct CH4 conversion routes, respectively.
| Category | Pathway | Reaction |
|---|---|---|
| Metal oxide redox pairs | Mn3O4 → Mn2O3 | (R42) 4Mn3O4 + O2(g) → 6Mn2O3 |
| MnO → Mn3O4 | (R43) 6MnO + O2(g) → 2Mn3O4 | |
| FeO → Fe3O4 | (R44) 6FeO + O2(g) → 2Fe3O4 | |
| Cu2O → CuO | (R45) 2Cu2O + O2(g) → 4CuO | |
| Cu → CuO | (R46) 2Cu + O2(g) → 2CuO | |
| Ni → NiO | (R47) 2Ni + O2(g) → 2NiO | |
| Combustion | CH4 → Power | (R48) 1/2CH4(g) + O2(g) → 1/2CO2(g) + H2O(g) |
| Partial oxidation/selective oxidation | CH4 → Synthesis gas | (R49) 2CH4(g) + O2(g) → 2CO(g) + 4H2(g) |
| CH4 → CH3OH | (R50) 2CH4(g) + O2(g) → 2CH3OH(g) | |
| C2H4 → C2H4O | (R51) 2C2H4(g) + O2(g) → 2C2H4O(g) | |
| C3H8 → C3H6O | (R52) C3H8(g) + O2(g) → C3H6O(g) + H2O(g) | |
| C4H6 → C4H2O3 | (R53) 2/5C4H6(g) + O2(g) → 2/5C4H2O3(g) + 4/5H2O(g) | |
| Oxidative coupling | CH4 → C2H4 | (R54) 2CH4(g) + O2(g) → C2H4(g) + 2H2O(g) |
| CH4 → C2H6 | (R55) 4CH4(g) + O2(g) → 2C2H6(g) + 2H2O(g) | |
| Oxidative dehydroaromatization (DHA) | CH4 → C6H6 | (R56) 4/3CH4(g) + O2(g) → 2/9C6H6(g) + 2H2O(g) |
| Oxidative dehydrogenation or cracking | C2H6 → C2H4 | (R57) 2C2H6(g) + O2(g) → 2C2H4(g) + 2H2O(g) |
| C3H8 → C3H6 | (R58) 2C3H8(g) + O2(g) → 2C3H6(g) + 2H2O(g) | |
| C4H8 → C4H6 | (R59) 2C4H8(g) + O2(g) → 2C4H6(g) + 2H2O(g) | |
| C6H14 → C2H4 | (R60) 2C6H14(g) + O2(g) → 6C2H4(g) + 2H2O(g) | |
| C6H14 → C3H6 | (R61) 2C6H14(g) + O2(g) → 4C3H6(g) + 2H2O(g) | |
| Reforming | CH4 → Synthesis gas | (R62) CH4(g) + H2O(g) → 3H2(g) + CO(g) |
| (R63) CH4(g) + CO2(g) → 2H2(g) + 2CO(g) | ||
| Methane cracking | CH4 → H2 | (R64) CH4(g) → C(s) + 2H2(g) |
| Dehydroaromatization | CH4 → C6H6 | (R65) 6CH4(g) → C6H6(g) + 9H2(g) |
| Anaerobic conversion | CH4 → CH3OH | (R66) CH4(g) + H2O(g) → CH3OH(g) + H2(g) |
| Super-dry reforming | CH4/CO2 → CO/H2O | (R67) CH4(g) + 3CO2(g) → 4CO(g) + 2H2O(g) |
| Calcium/carbonate looping | CaO ↔ CaCO3 | (R68) CaO(s) + CO2(g) ↔ CaCO3(s) |
| (R69) CH4(g) + 2H2O (g)+ CaO → CaCO3(s) + 4H2(g) | ||
| (R70) 2CH4(s) + 4CuO(s) + 4NiO(s) + CaCO3(s) → CaO(s) + 4Cu(s) + 4Ni(s) + 3CO2(g) + 4H2O(g) | ||
| RWGS | CO2/H2→ CO/H2O | (R71) CO2(g) + H2(g) → CO(g) + H2O(g) |
| Methanol oxidation | CH3OH→ CH2O | (R72) 2CH3OH(g) + O2(g) → 2CH2O(g) + 2H2O(g) |
:H2 ratio of 1:2) and high-purity H2 are produced inherently, thus intensifying the production of high-purity H2 from CH4. It is noted here that CL-SMR would become the CL-WGS process if CO instead of CH4 were used to reduce the oxygen carrier.42,96,170,171 Both CL-SMR and CL-WGS are also referred to as CL-water-splitting processes.47,172 Recently the CL-DRM cycle, in which CO2 is used to regenerate the oxygen carrier (via (R6)), has been proposed to maximize the conversion of CO2, captured from various emission sources, to CO.96,162,167,173–175 The modified CL-methane POx coupled with H2O–CO2 splitting176 for the oxygen carrier regeneration allows the production of syngas with identical compositions in both the reduction and regeneration steps.121,177,178
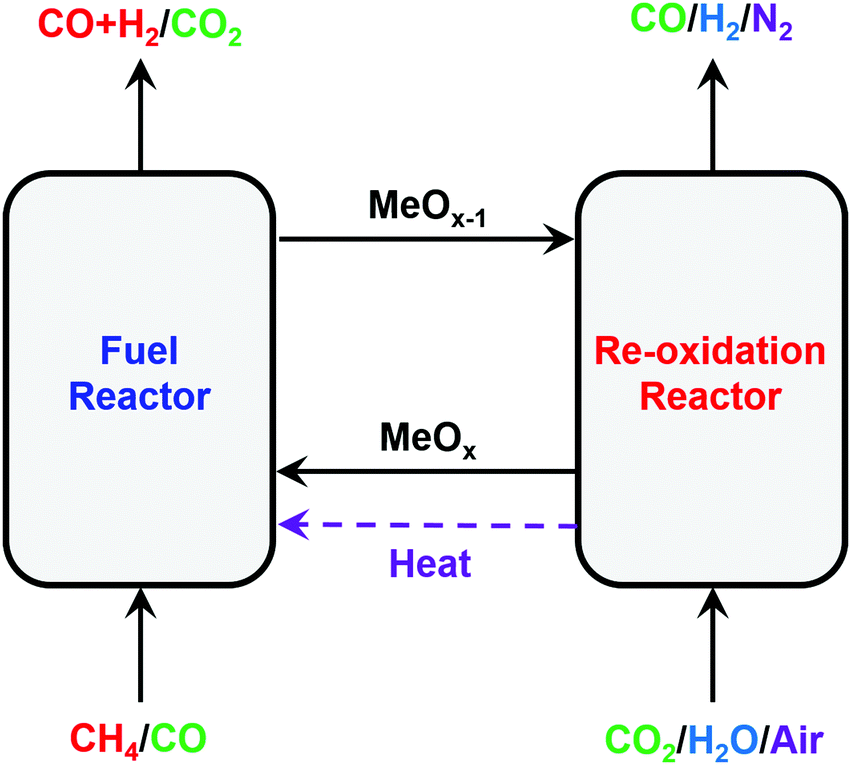 | ||
| Fig. 10 Schematic and reaction schemes of different CL cycles for conversion of CH4 to synthesis gas. | ||
The oxygen carriers used in the above-mentioned chemical looping processes are often called redox catalysts because they simultaneously act as an oxygen donor (i.e. as a reactant) and as a catalyst. NiO, Fe2O3, CeO2, spinel and certain perovskite-based oxides possess suitable thermodynamic properties that permit their use as redox catalysts for chemical looping conversion of CH4 to synthesis gas.41,179–184 Among various oxygen carriers, Ni-based oxygen carriers are particularly attractive for CLR owing to their high catalytic activity for CH4 reforming.56,185–191 NiO possesses a low selectivity towards synthesis gas, but as NiO reduces to Ni the selectivity towards synthesis gas increases.56 However, Ni is highly prone to deactivation via carbon deposition under low oxygen partial pressures187 and the formation of sulfides in the presence of feedstock containing sulfur species.192 Moreover, Ni is a known carcinogenic, thus possessing significant health and safety risks.193 Fe-Based redox catalysts are also very promising for CL-methane POx owing to their suitable thermodynamic properties for both CH4 reforming and H2O/CO2 splitting, low cost and minimal environmental impact.90,194 However, compared to Ni-based oxygen carriers, Fe-based redox catalysts possess a low reactivity with CH4 and a low selectivity towards synthesis gas.195 Because of their high activity and thermal stability, La-based perovskites with Fe, Mn, or Co as B-site cations have been widely investigated in recent years for CL-methane POx and CL-SMR.58–60,64,66,88,89,139,179,196–198 As mentioned above, the structural and compositional tailoring of the redox catalysts can open up extensive possibilities for improvements of CLR processes. Since the objective of this part of the perspective is not to present a comprehensive review of the redox catalysts used for CLR, we briefly summarize some of the strategies that have been demonstrated to enhance the reactivity and selectivity of redox catalysts in the following sections.
Stabilization by a mixed conductive support. The redox catalysts typically comprise of a primary metal oxide as an oxygen reservoir and an inert support to stabilize the metal oxide. The appropriate combinations of oxygen carriers and supports can potentially increase the selectivity of synthesis gas and reduce catalyst deactivation.199 For example, it has been demonstrated that the use of a mixed conductive support (such as La0.8Sr0.2FeO3−δ or Ca0.8Sr0.2Ti0.8Ni0.2O3) enables fast solid state O2− and electron/hole exchange92,131,200 between the metal oxide crystallites and the surface of oxygen carrier particle, thereby allowing effective oxygen removal and restoration during reactions (R3) and (R4).201,202 Importantly, supports that promote the outwards diffusion of active metal should be avoided because such supports can potentially deactivate the redox catalysts via surface enrichment and agglomeration of the active metal oxide crystallites.203
Core–shell architecture. The spatial distribution of support in an oxygen carrier particle also significantly affects its activity and selectivity.204 For example, Shafiefarhood et al.59 demonstrated that an Fe2O3@LaxSr1−xFeO3 (LSF) core–shell material was 10–200 times more active, selective and resistant to carbon formation than composite iron oxide-based materials comprising inert (Al2O3 and MgAl2O4), ionic-conductive (YSZ), and mixed-conductive (LSF) supports. Neal et al.58,60 and Shafiefarhood et al.205 investigated the mechanism for CH4 partial oxidation over an Fe2O3@LSF core–shell material and found four distinct regimes, viz. (i) full oxidation of CH4 to CO2, (ii) competing full oxidation to CO2 and partial oxidation to CO, (iii) partial oxidation of CH4 to CO, and (iv) CH4 decomposition. During deep oxidation in region (i) “loose” lattice oxygen from the iron oxide core transports and evolves into surface oxygen species. In region (ii), both selective and non-selective (i.e. loose) oxygen species compete for CH4 oxidation, with O2− flux and overall oxygen availability determining the product ratio. In region (iii), dissociatively adsorbed CHx species are partially oxidized by O2− species on the surface, resulting in an increase in metallic iron species along with a decrease in the concentration of oxygen surface species. Coke formation in region (iv) results from the depletion of O2− in the iron oxide core and CH4 decomposition over metallic iron precipitated from the LSF lattice. Transient pulse studies showed that oxygen anion conduction and/or its evolution to electrophilic surface oxygen species is the rate-limiting step in all the reduction regions of interest.205 Oxygen atoms maintain a modified Mars–van Krevelen (MvK) mechanism throughout the reaction, while the mechanism of CH4 oxidation changes from the Eley–Rideal in region (i) to a Langmuir–Hinshelwood-like mechanism in region (iii). These findings indicate that redox catalyst architectures that inhibit the formation of non-selective surface oxygen species while maintaining a steady supply of lattice oxygen to the surface can potentially lead to an improved performance for CL-methane POx.
Use of promotors. Ni- and Fe-based oxygen carriers have a high catalytic activity for the decomposition of CH4, via (R64). Promotion of redox catalysts with a transition metal that is inactive for the dissociation of CH4, e.g. Cu, Ag, or Au, can suppress the formation of carbon during the CH4 reforming step.206 In this regard, Imtiaz et al.207 demonstrated that doping of Fe2O3 with CuO significantly lowers its propensity for coke deposition in a CH4 atmosphere. The high resistance to carbon deposition of Cu modified oxygen carriers was found to be due to the (partial) coverage of surface Fe with Cu. Some transition metals (e.g. Ru, Rh, Ir, etc.) can expedite the dissociation of C–H bonds in CH4206 and can therefore enhance the reforming activity of redox catalysts. Shafiefarhood et al.139 reported a significant improvement in the performance of Rh-promoted CaMnO3 and La2Ce2O7 redox catalysts for CH4 reforming. It was found that a higher concentration of CHx species on the surface of Rh-promoted catalysts accelerates the extraction of facile oxygen resulting in an enhanced O2− conduction through the bulk of the redox catalyst. Therefore, a higher redox activity and synthesis gas selectivity was observed for Rh promoted redox catalysts at temperatures as low as 500 °C than for pristine catalysts. Similar findings were reported by Palchelva et al.208 for Rh promoted La0.8Sr0.2(Fe0.8Co0.2)1–xGaxO3, which showed a 40% enhancement in CO selectivity for methane conversion at 600 °C. Noble metals, modification with CeO2 nanoparticles can also improve the oxygen storage capacity and mobility in perovskite68 or iron oxides,63,64,141,209 which in turn can enhance the availability of lattice oxygen for the methane POx to synthesis gas.
For CL-based CH4 partial oxidation, gaseous O2 is substituted by lattice oxygen. This eliminates the expensive air separation, ensures safer operation and inhibits non-selective oxidation reactions in the gas phase when carried out at high temperatures.212,213 In addition, the use of lattice oxygen is also shown to potentially benefit the product selectivity.214,215 Challenges in the design of suitable redox catalysts relate to the following aspects of CL-based CH4 oxidation processes:213 (i) CL would likely to require higher operating temperature than co-feed which may negate the selectivity benefit of using lattice oxygen, and (ii) the surface and bulk of the CL redox catalyst tend to undergo notable changes during the redox reactions, which makes maintaining a high product selectivity challenging. Below we briefly discuss several CH4 conversion schemes that were shown to be feasible in chemical looping mode.
Oxidative coupling. The oxidative coupling of methane (OCM) is a method proposed in the early 1980s to convert CH4 directly into ethane and ethylene according to reactions (R55) and (R54) at temperatures >600 °C.216 The exothermic reactions can be carried out either in “normal” catalytic fashion where oxygen is co-fed with CH4, or in a cyclic redox (chemical looping) operation expressed by reactions (R7) and (R8).212,217,218 For either mode, an upper limit of 25–30% yield for C2+ hydrocarbon products has been supposed based on mechanistic considerations,215,217,219 which is near the yields required for economic competitiveness.220–222 Various engineering means have been suggested to improve yields, but no breakthroughs have been achieved to date.213,223,224 It is worth noting that early studies by Atlantic Richfield company, which operated a 12 year program on OCM focusing on the redox mode, reported an approximately 10% higher C2+ selectivity under the chemical looping mode compared to the conventional catalytic route.215
The OCM reaction occurs via a complex mechanism including coupled heterogeneous and homogeneous steps (depending on the catalyst) and is initiated by breaking one of the four equivalently strong C–H bonds of the methane molecule to form methyl radicals.224,225 The low yields for C2+ hydrocarbons obtained so far are largely due to secondary oxidation reactions of these reactive products with oxygen weakly bound to the surface of the catalyst and with oxygen in the gas phase, forming a significant amount of COx.71,222,226,227 More recently, the upper yield limit for C2+ products in the absence of gas phase reactions was estimated to be as high as 60% at elevated pressure, which could possibly be achieved via chemical looping.225,228 Experiments with doped-Mg6MnO8 and Mn/Na2WO4/SiO2, a catalyst previously shown to attain single pass C2+ product yields of ∼25% in co-feed mode,229,230 gave C2+ product yields of 23.2% and 25%, respectively under a chemical looping mode.231–234 In these studies, the selectivity toward the coupling reaction was high, but the H2 molecules formed through the dehydrogenation reaction of ethane competed with CH4 molecules for reacting with activated oxygen on the surface of the catalyst, resulting in a decreasing rate of CH4 activation and consequently lower C2+ yields than expected.222 Hence, improving the activity of the redox catalysts without negatively affecting their selectivity is one important task. In addition, the reaction conditions (e.g. temperature and pressure) probably need to be revised to surpass C2+ product yields of 30%.235 The importance of dopants in Mg6MnO8 was highlighted recently,234 and it was found that Li-doping-induced oxygen vacancies reduce the adsorption energy of methyl radicals and increase the C–H activation barrier, resulting in an increased selectivity towards C2+. For doped Na2WO4 on SiO2-supported MOx catalysts (where M = V, Cr, Mn, Fe, Co or Zn) it was found previously that their performance is correlated with their electrical conductivity.236
Non-oxidative dehydroaromatization. Non-oxidative dehydroaromatization (DHA) is a promising route to produce C6H6 directly from CH4. Conversion of CH4 to C6H6via reaction (R65) was first reported in 1993 using a ZSM-5 zeolite modified with Mo or Zn.237 Since then, a wide range of metals and zeolites have been investigated for DHA of CH4, but Mo-modified ZSM-5-based catalysts still remain the most effective.238,239
Despite being a potentially attractive pathway to selectively produce high-value aromatics, CH4 DHA has not yet been commercialized due to the following reasons: Firstly, CH4 DHA (R65) is endothermic and limited by the thermodynamic equilibrium. From Fig. 11, it can be seen that the amount of C6H6 produced is only significant at high temperatures (i.e. >600 °C). Secondly, the inevitable decomposition of CH4 (R64) under reaction conditions not only lowers the equilibrium yield of C6H6 substantially but also causes coking and catalyst deactivation.241 Consequently, C6H6 selectivity is typically between 60–80% at CH4 conversions of ∼10%, corresponding to net C6H6 yields of <10% under conditions of practical interest.239 Several strategies have been proposed to overcome these barriers, e.g. the use of membrane reactors to remove H2 produced during reaction242 or the periodic regeneration of the coked catalyst.73 Recently, a CL approach was suggested to potentially address all three limitations of conventional CH4 DHA process as following (Fig. 11):145 (i) the reactive separation of H2 from the products of DHA reaction to enhance the equilibrium CH4 conversion and C6H6 yield, (ii) heat generation by selective H2 combustion to decrease the overall heat demand, and (iii) in situ steam generation to reduce coking. However, care must be taken because excessive presence of steam can negatively affect catalyst selectivity. Moreover, re-oxidation requirements for CL may affect the active MoCx species on Mo/ZSM-5. Using Mo/HZSM-5 (as the DHA catalyst) and Ce0.9Gd0.1Oy (as the SHC redox catalyst) in a composite bed, an approximately three times higher aromatics yield was demonstrated compared to the conventional process employing only Mo/HZSM-5 without periodic regeneration with oxygen.145
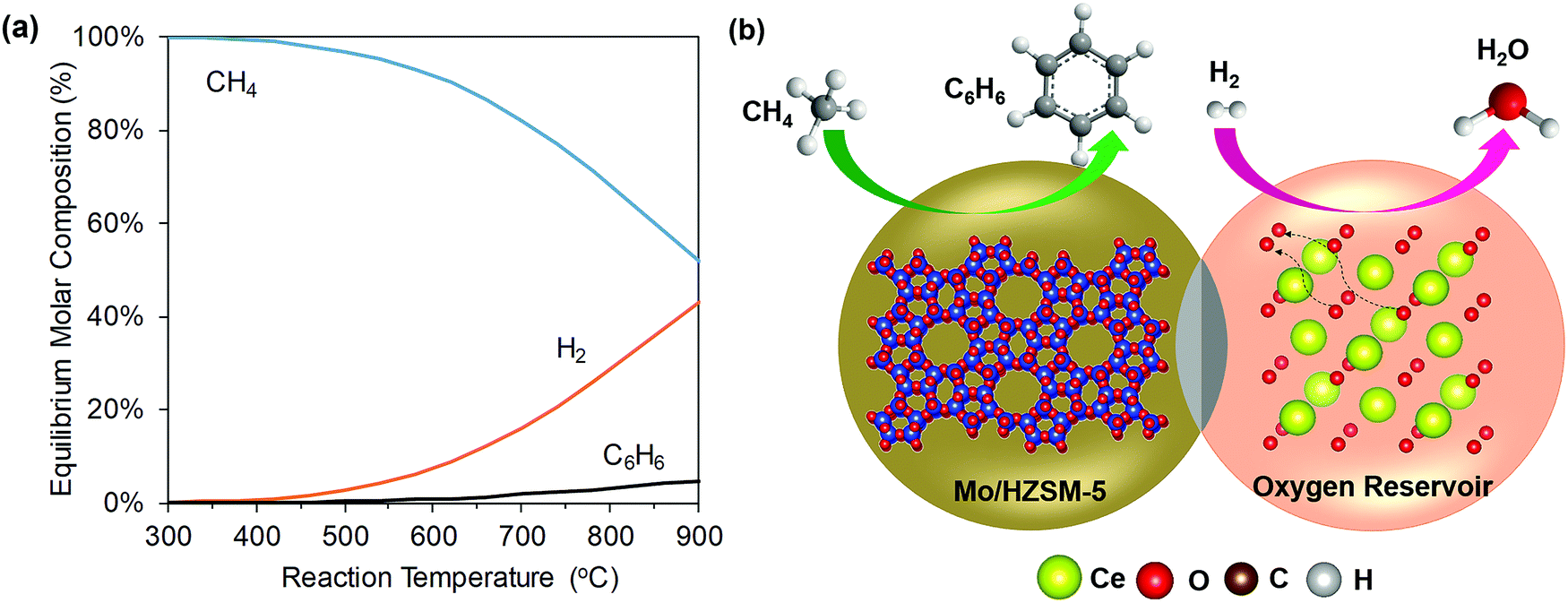 | ||
| Fig. 11 (a) Equilibrium product composition of CH4 DHA as a function of reaction temperature. (b) A potential configuration of methane CL-DHA process (the air regeneration step is not shown).240 | ||
Besides the two-step approach, Brady et al.240 proposed a four-step approach for CL-DHA. In the first step, DHA reaction is performed on a Mo/H-ZSM-5 catalyst at 700 °C to obtain a mixture of CH4, C6H6 and H2. Subsequently, H2 present in the DHA effluent is combusted selectively using Fe3O4. The H2O produced in step two is then removed using a high-temperature water adsorbent, e.g. Zeolite 5A, which can be regenerated using a temperature swing. Finally, the reduced oxygen carrier is re-oxidized back to Fe3O4 using steam. To increase the conversion of CH4 and the yield of C6H6, the effluent from step three is fed again to the DHA unit in the subsequent cycle. Brady et al.240 demonstrated each of these above steps. Based on extrapolation, the upper limit of aromatics yield for such a process was estimated to be ∼43%.240 However, an integrated CL-DHA process under such a multi-step scheme has yet to be demonstrated. Furthermore, the oxidation of Fe to Fe3O4 with steam is thermodynamically limited. It is also to be noted that the regeneration of Fe with water instead of air renders the overall process endothermic. The use of water for regeneration by Brady et al.240 may have been (partially) motivated by the lack of H2 combustion selectivity of Fe2O3. Although this CL-enhanced DHA scheme is quite innovative, it could be simplified while accounting for the aforementioned design considerations. For this, other SHC redox catalysts with higher H2 combustion selectivities should be considered in addition to iron oxides.83,145
Direct conversion of CH4 to CH3OH. Direct conversion of CH4 to CH3OH (and/or formaldehyde) via (R50) is thermodynamically feasible. However, despite over a century of research the development of such a process remains a challenge owing to (i) a large energy barrier associated with the activation of the C–H bonds of CH4, and (ii) a lower stability of CH3OH than CH4, resulting in further oxidation of CH3OH to CO2.243 Consequently, CH3OH is currently produced on a commercial scale from synthesis gas derived from coal or CH4.244
To date, no heterogeneous catalyst exists that can activate CH4 according to reaction (R50) while simultaneously inhibiting CH3OH oxidation at practical yields.245 Interestingly, in nature monooxygenase (MMO) enzymes convert CH4 selectively into CH3OH at ambient temperature.246 The active sites in these enzymes, i.e. binuclear iron and copper centers, can be mimicked using zeolite frameworks. However, so far with Fe and Cu-based zeolites a high selectivity for CH3OH is only achievable at very low CH4 conversions (<0.1%).247,248 The low CH3OH yields of the direct aerobic conversion of CH4 to CH3OH process is a thermodynamic feature that cannot be overcome by the choice of catalyst. Nonetheless, this thermodynamic constraint can be circumvented by using a CL approach and soft oxidants such as H2O or N2O.75,76,249–251 For example, using a Cu-exchanged mordenite zeolite, Sushkevich et al.252,253 reported an anaerobic CL process based on the reaction (R66) to avoid the over-oxidation of CH3OH. In the proposed three-step process, the Cu-exchanged mordenite zeolite was first activated at 400 °C in a flow of either dry O2 or He, followed by exposure to CH4 at 200 °C and 7 bar to form methoxy species. Subsequently, the methoxy species were desorbed in the form of CH3OH under a flow of a H2O/He mixture. Once the desorption step was complete, the second reaction cycle was started by heating the zeolite again to 400 °C under a flow of dry O2 or He. This process resulted in a normalized CH3OH production rate of ∼20 μmole CH3OH gcatalyst−1 h−1 (based on a cycle time of 6.7 h) with a selectivity of 97%, which is considerably higher than the CH3OH production rate of 1.81 μmole CH3OH gcatalyst−1 h−1 reported for a continuous process using O2 as the oxidant.254
4.2 Chemical looping for C2+ valorization
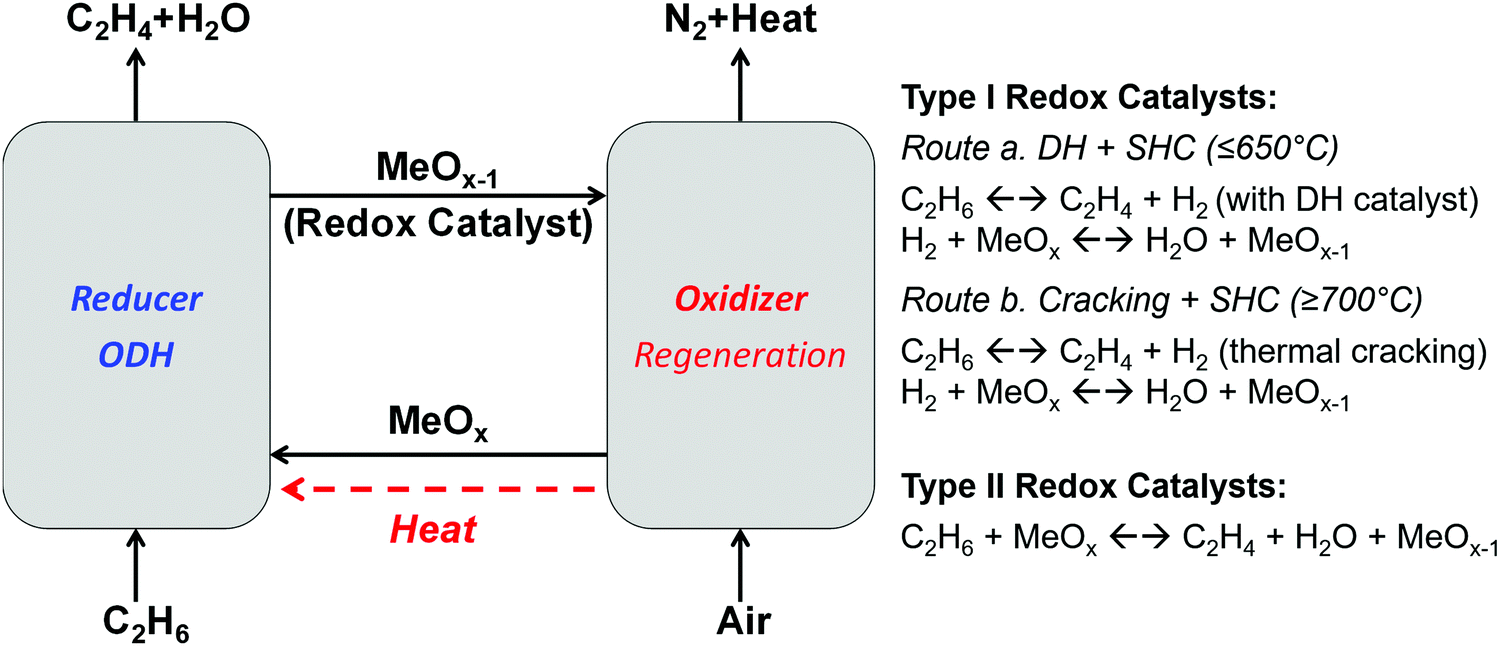 | ||
| Fig. 12 Schematic of CL-ODH, redox catalyst types, and reaction schemes. Ethane is used as an example for light alkanes for illustration purpose. | ||
The chemical looping operations illustrated in Fig. 12 can be realized by two types of redox catalysts with distinct functions.266 The Type I redox catalysts function primarily as SHC materials. Using Type I catalysts, ethylene production is realized by a combination of dehydrogenation (DH) or cracking reaction (for olefin and hydrogen production) and SHC reaction (for the in situ oxidation of the hydrogen byproduct, reaction (R16)). In comparison, Type II redox catalysts catalyze ethane ODH reactions while donating their active lattice oxygen.
Type I: SHC redox catalysts. While it could be argued that Type II redox catalysts are more desirable due to their multi-functionality, Type I redox catalysts could offer better flexibility since they can be tuned independently for various operating conditions and reactor configurations. For instance, SHC redox catalysts have been explored to enhance both DH and thermal cracking reactions with a temperature range varying from 450 to 900 °C.82,83
The use of SHC to enhance catalytic DH reactions (Route a in Fig. 12) was proposed in the late 1990s by Imai et al.267,268 in the context of propane dehydrogenation. While both co-feeding oxygen269 and redox mode operation using lattice oxygen269 were investigated, the latter was surmised to be superior based on both experimental data and reactor simulations.268,270,271 Resulting from their significant potential, various oxides were explored as SHC redox catalysts in the temperature range of 550–800 °C. Representative materials showing high (>90%) SHC selectivities, defined as the conversion of the hydrogen divided by the conversion of all combustible species, include Sb2O4, In2O3, WO3, Bi2O3, PbCrO4, doped ceria, ion-exchanged ZSM-5, etc.268,269,272–277 Bi, In, Pb, and Sb oxides often suffer from a poor redox stability owing to relatively low melting temperatures of their reduced states. Ceria doped with W, Bi, Cr or Pb, were found to be stable in the temperature range 500–600 °C with selectivities of up to 98% for hydrogen combustion in the presence of C2 and C3 hydrocarbons and oxygen storage capacities as high as 2 wt%.273,278–282 Mn- and La-containing perovskites also showed a high SHC selectivity and stability in the presence of C3 hydrocarbons at 550 °C.84 It is noted, however, that the SHC selectivity tends to decrease with increasing temperature due to the lower activation energy for hydrogen combustion compared to hydrocarbon oxidation. Therefore, these early SHC studies focusing mostly on operating temperatures <650 °C were exclusively geared towards dehydrogenation applications in combination with a DH catalyst such as supported PGM (e.g. Pt) and chromium oxides.259,278,283 Another challenge with the DH and SHC concept resides in the incompatibility of the catalysts and their operating conditions. While physical and chemical interactions between the catalysts themselves can be avoided by spatially separating them (e.g. in a packed bed with alternating DH catalyst and SHC redox catalyst layers), active sites and dominating reaction pathways on DH catalysts can be affected by the steam generated from the SHC material. For instance, steam can lead to undesirable reforming reactions on PGM-containing catalysts. For oxide-based DH catalysts, steam (or air during the regeneration step) can affect the oxidation state of the active metal, induce changes in the surface compositions, and hence negatively affecting the DH performance. Except for very few studies,269 the aforementioned studies used hydrogen and a hydrocarbon mixture (e.g. hydrogen, ethane, and ethylene) to evaluate the redox catalysts’ SHC performances. The incompatibility issues between DH and SHC have largely been unaddressed to date. Development of DH catalysts that are stable under redox conditions and in the presence of steam is therefore the key to apply the DH + SHC approach for CL-ODH.
SHC redox catalysts can also be integrated with thermal cracking reactions (Route b in Fig. 12). Unlike surface-catalyzed DH reactions, thermal cracking proceeds via gas-phase radical reactions and so steam generated by SHC would not negatively affect cracking reactions. Under this approach, the CL-ODH reactions proceed via parallel gas-phase and surface pathways, i.e. hydrogen generated from gas phase cracking reactions is selectively combusted on the surface of the redox catalysts.80,269 In some cases, the redox catalyst surface can also function as a radical initiator to enhance the gas phase reactions (i.e. surface-initiated homogeneous reactions).87 It is noted that the initiation of radicals from alkanes typically requires temperatures close to or higher than 700 °C. While such a temperature range is not uncommon for CLC, the inverse relationship between SHC selectivity and reaction temperature for most oxide redox catalysts dictates that many of the SHC catalysts reported for DH + SHC, which are geared towards operating at 650 °C or lower, would not be compatible with thermal cracking reactions. The high operating temperature can also be challenging for oxides or metals with low melting points or high vapor pressures. To address these challenges, Dudek et al.83 investigated a number of Mn-containing redox catalysts including CaMnO3, SrMnO3, and Mg6MnO8 for selective hydrogen combustion using hydrogen and ethylene mixtures at temperatures up to 850 °C. The effect of promoters was also studied by impregnating Na2WO4 on each of these oxides. In general, Na2WO4 was effective in inhibiting the formation of COx, but negatively affected the redox activity of the mixed oxide. The operating temperature windows, i.e. the optimal temperature ranges with a high activity and selectivity towards hydrogen combustion, for the six redox catalysts investigated ranged between 550 and 850 °C. The large and tunable operating temperature window opens up the potential opportunity for integration of a SHC redox catalyst with thermal cracking. The effectiveness of CL-ODH under thermal cracking conditions was confirmed with Na2WO4 promoted Mn–Mg and Mn–Si oxides.284 Resulting from their high activity and high temperature SHC selectivity, ethylene yields up to 68% were demonstrated with COx selectivities as low as 1.9%. Negligible coking was observed in these studies. This is not surprising considering the redox catalysts’ high activity for lattice oxygen donation, which can actively inhibit coke formation. Na2WO4 was important for obtaining a high olefin selectivity since it enriches on the oxide surface and suppresses the deep oxidation of hydrocarbons.285 In the case of the Mg6MnO8 system, a mixed oxide with a cation deficient rocksalt structure, near surface Mn4+ was suppressed by more than 85%.80,81 Further characterizations by low energy ion scattering (LEIS), in situ XRD, and differential scanning calorimetry indicated that the Na2WO4 is likely to form a molten layer on the oxide surface under the operating conditions and thus acts as a physical blocker to suppress surface catalyzed C–H bond activation.286 The corresponding process analysis further highlighted the advantages of CL-ODH of ethane over steam cracking.113 These include higher single-pass yields, a reduction in the total energy demand by >80% and a reduction in CO2 emissions by >80%. Importantly, CL-ODH is self-sufficient from a process heat requirement point of view, making it a highly efficient and low emission technology ideal for distributed systems.
Type II: ODH redox catalysts. Many heterogeneous catalysts used in conventional ethane ODH (where oxygen is co-fed with the ethane) are redox-active.287 In the presence of gaseous oxygen (or, as was shown recently, mild oxidants such as steam),288 the reaction proceeds through a MvK mechanism289 in which ethane is activated and dehydrogenated by surface metal-oxide species, thereby reducing the metal oxide and creating oxygen vacancies. The vacancies on the surface are subsequently replenished by gaseous oxygen, which is dissociated and incorporated at surface sites different from the ODH reaction sites.290–292 The overall reaction is therefore facilitated by the migration of oxygen anions through the lattice and/or on the surface. The most common redox-active heterogeneous catalysts for ODH of light alkanes are based on vanadium oxide293–295 with different supports, such as alumina,296–299 alumina modified with zirconia300,301 or silica.302–305 Nickel oxides306,307 and cobalt oxides308,309 have also been examined.
Since a key characteristic for the MvK mechanism is that the overall reaction can be carried out under a “swing mode”, i.e. separating the reduction and oxidation into two sequential steps identical to chemical looping operations, it is not surprising that many of the redox-active oxide catalysts can act as redox catalysts in CL-ODH schemes. For example, Hossain and de Lasa developed a number of cyclically stable VOx/γ-alumina-based catalysts for the CL-ODH of ethane in a continuous fluidized bed process consisting of an ODH reactor and a catalyst regenerator.298,299,310 Ethylene selectivities ranging from 80–90% at temperatures between 550–600 °C were reported. The corresponding ethane conversions, however, were relatively low (<10%).300,311,312 The same class of catalysts was also used for the CL-ODH of propane in the simulated fluidized bed process and propylene selectivities of >90% were obtained at up to 25% conversion of propane.313–315 The good performance of the most promising redox catalyst, VOx/CaO–γ-Al2O3, was attributed to an intermediate acidity of the catalyst and moderate active site–support interactions.85 Encouraging results for ethane CL-ODH were also obtained with Mo-based redox catalysts, which gave ethylene selectivties of 93–95% and conversion up to 66.5% at 600 °C.297,316 The oxygen storage capacity of these Mo-based redox catalysts was relatively low (<0.4 wt%), resulting in a decreasing conversion of ethane with time. Nonetheless, in a continuously operating riser reactor system, an ethane conversion of 12% at ethylene selectivities >90% was obtained with a mixed Mo–Te–V–Nb oxide supported on γ-Al2O3 when the contact time of ethane feed and redox catalysts was 1–2 s at 500–575 °C.317 The same authors also showed that the performance of Mo-based redox catalysts is strongly affected by the support material (Al2O3, Ga2O3 or Y2O3), leading to an increased ethane conversion and ethylene selectivity with a decrease in the content of MoO3; the best results were obtained with 5 wt% MoO3 on alumina, giving 90% selectivity for ethylene at an ethane conversion of 30%.318,319 Novotny et al.284 investigated the effect of impregnating MoO3 onto an α-Fe2O3-based redox catalyst, which improved the oxygen storage capacity from 0.28 wt% to 0.62 wt%. A MoO3 and Fe2(MoO4)3 enriched surface layer was formed upon calcination. At 600 °C, the ethylene selectivity was as high as 62% while CO2 formation was suppressed when compared to pure α-Fe2O3. Supported chromium oxide catalysts are widely used in catalytic dehydrogenation,320e.g. the commercial CATOFIN® process. Chromium oxides were also shown to perform reasonably well in CL-ODH of propane with potential use in a moving bed process.321–323 The oxygen storage capacity was determined to be crucial for the performance of the redox catalyst, and slightly higher oxygen capacities (up to ∼0.5 wt%) resulted in higher yields of propylene (∼46% at 630 °C).324 More recently, Chen et al. used to use Mo–V–O mixed oxides as the redox catalyst for CL-ODH of propane to propylene.325 Attractive propylene yield (89% propylene selectivity at 36% pro-pane conversion) over 100 redox cycles was obtained at 500 °C due to the tailored bulk lattice oxygen via atomic-scale doping of Mo in the redox catalyst. Although promising results were obtained from these redox catalysts, the toxicity of Cr and V and thermal stabilities of Mo and V oxides could affect their practical applicability especially in circulating bed systems.
Aside from the redox-active oxides commonly found in heterogeneous DH and ODH catalysts, alkali metal-promoted, Ruddlesden–Popper structured, LaxSr2−xFeO4−δ redox catalyst was recently shown to be active for CL-ODH of ethane.79,128 Up to 90% ethylene selectivity and 61% conversion were observed at 700 °C, with near 100% H2O selectivity. An important finding from this study was that electrophilic surface oxygen species (such as O− and O2−), which limit the selectivity of the ODH reaction and account for the deep oxidation of the alkanes and olefins, can be suppressed by enrichment of alkali metal oxide on the surface of the redox catalyst. A thin layer of Li2O on the surface likely served as a barrier to inhibit the outward diffusion of O2− and its evolution into electrophilic oxygen species on the surface.
A common challenge with these Type II ODH redox catalysts is the lack of oxygen storage capacity. Most of the redox catalysts reported to date exhibit <1 wt%, and in many cases <0.5 wt%, oxygen storage capacity. As noted above, the utilization of lattice oxygen in CL-ODH is significantly more efficient than that in CLC. For instance, combusting one mole of propane under CLC consumes ten moles of lattice oxygen whereas under CL-ODH it consumes only one. The order of magnitude increase in oxygen utilization efficiency make these low capacity redox catalysts potentially feasible for industrial applications, provided that high olefin selectivity, redox activity, and stability are demonstrated. Another challenge is the trade-off between conversion and selectivity especially in many of the V- and Mo-based redox catalysts, i.e. higher alkane conversion (e.g. by increasing the residence time) often leads to a significantly decreased olefin selectivity. This could result from the re-adsorption of the olefin products and/or the presence of distinct active sites for selective and non-selective reactions. In-depth understanding of the active sites and reaction pathways would be crucial for rational optimization of the redox catalysts. Although a large body of literature is available on conventional, catalytic ODH systems, the exact mechanisms for many redox-active ODH catalysts are still under debate.259,326 The dynamic nature of redox catalysts undergoing chemical looping reactions also makes mechanistic investigation on CL-ODH even more challenging than catalytic ODH. Therefore, further investigation on the mechanistic aspects of the redox catalysts is an important, yet challenging task. Although we did not separately discuss ethane and propane CL-ODH in this section, we note that propane (and C4+) is significantly less stable than ethane. The tendency for C–C bond cleavage makes it more difficult to develop highly selective redox catalysts for CL-ODH of propane and C4+ to olefins. Thus, a redox catalyst optimized for CL-ODH of ethane may not automatically be applicable for propane and C4+ conversion.
So far, only the epoxidation of ethylene has been realized industrially using molecular oxygen as the sole oxidant.328 The selectivity to ethylene oxide is typically in the range of 85–90% at ∼10% ethylene conversion with optimized silver-based catalysts.329–331 A chemical looping approach was recently explored to undertake the epoxidation reaction by substituting co-fed molecular oxygen with lattice oxygen (Fig. 13).78 Here, Ag (15 wt%) was impregnated onto the perovskite SrFeO3 that possesses an oxygen storage capacity of up to ∼2 wt%. The main rationale for selecting SrFeO3 was its excellent ability to spontaneously release O2 at relatively low temperatures.136 The authors proposed that oxygen was transported from subsurface SrFeO3 to the surface of the redox catalyst and thus formed oxygen adatoms on the silver surface. The vital role of atomic oxygen adsorbed on the silver surface for catalyzing the oxidation of ethylene has been discussed extensively in the literature,331,332 as well as the availability of subsurface oxygen to achieve a high ethylene oxide selectivity.333 Furthermore, it was found in previous studies that higher selectivities could be obtained with low surface area supports,334,335 a characteristic of most redox catalysts used in chemical looping applications. The selectivities and conversions reported by Chan et al.78 were, however, very low (the ethylene oxide selectivity was <20% and the ethylene conversion was <4% at 270 °C). Previous work showed that the dispersion of Ag, the morphology and temperature treatment of the support all affect, besides operational parameters, the performance of the catalyst.336–338 It is also possible that the exposed surface of SrFeO3 is largely responsible for non-selective oxidation reactions and the activity of the oxide substrate to catalyze non-selective oxidation reactions should not be overlooked. Another challenge observed by Chan et al. was the low rate of oxidation when the redox catalyst was regenerated in air, causing a gradual loss in oxygen capacity.78 A similar problem was reported for vanadium phosphorous oxide redox catalysts, as will be discussed in Section 4.2.3. The problem was overcome by modifying the SrFeO3 with 5 mol% CeO2, which also improved the cyclic stability and ultimately gave 60% selectivity towards ethylene oxide at 10% ethylene conversion.339 The authors proposed that CeO2 may have acted as oxygen gateway (see Section 3.2.2) and thus improved the re-oxidation of the redox catalyst. The challenge for further improvement will thus be to find oxygen storage materials with sufficient reduction/regeneration activities at temperatures <300 °C while suppressing over-oxidation of ethylene. Despite the relatively low reaction temperatures of 200–280 °C typically employed for the epoxidation of ethylene, Ag-based catalysts tend to deactivate owing to the sintering of the silver particles,340–342 and this may be even more problematic when the redox catalysts are regenerated in an exothermic oxidation reaction.
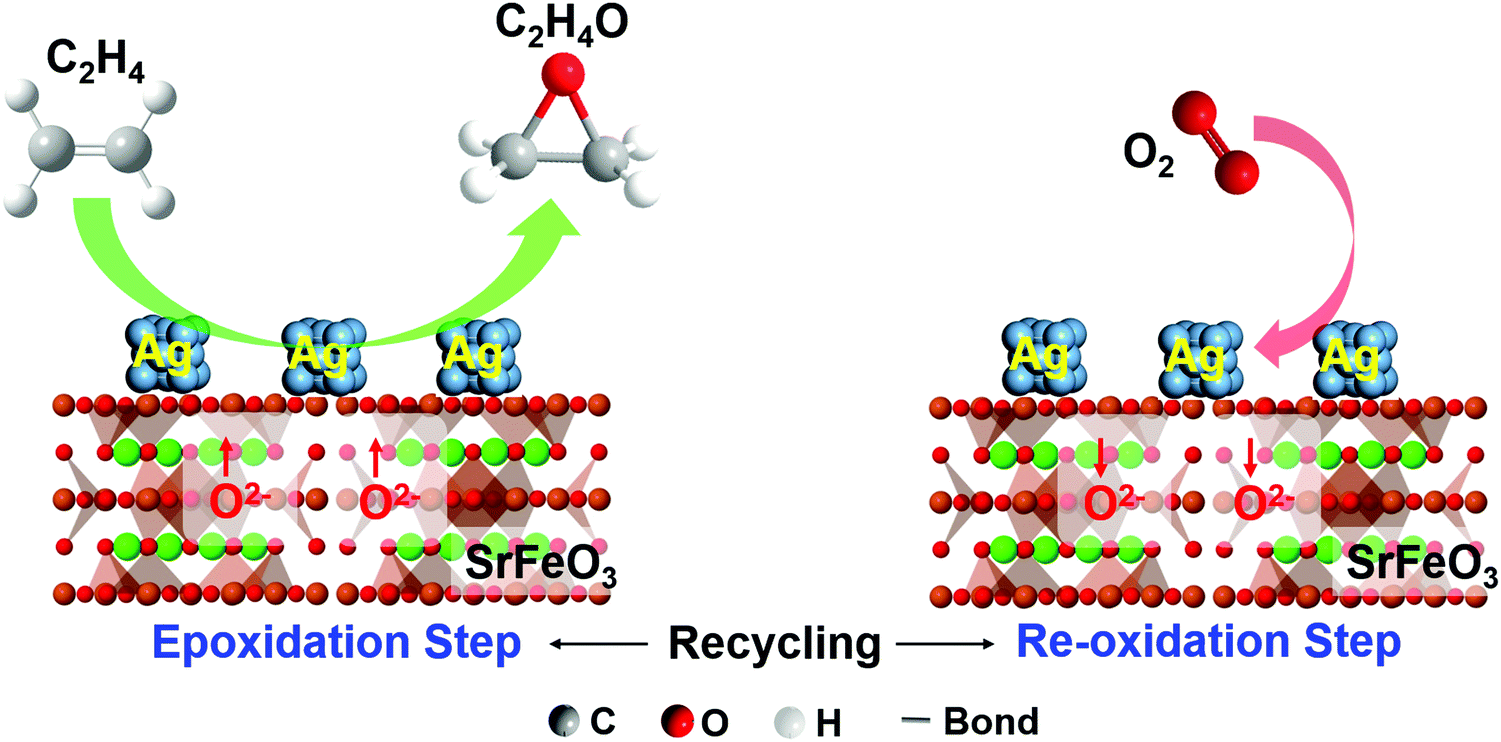 | ||
| Fig. 13 The reaction mechanism of chemical looping epoxidation using a silver-modified SrFeO3−δ redox catalyst. | ||
In principle, the chemical looping epoxidation approach can certainly also be applied to the selective oxidation of propylene, where it was shown recently that mixed metal oxides are catalytically active in a conventional oxygen co-fed mode. For a mixed MoO3/Bi2SiO5 catalysts supported on SiO2, propylene oxide selectivities of 55% were reported at a propylene conversion of 22% at 400 °C for a diluted feed with a molar ratio of O2/C3H6 of 4:1,343 whereas high formation rates of propylene oxide were observed for a RuO2/CuO catalyst supported on SiO2.344 Using a copper–manganese mixed metal oxide catalyst, Seubsai et al.345 found selectivities >30% towards propylene oxide at up to 1.5% propylene conversion at 300 °C. These catalyst systems are structurally and chemically similar to typical chemical looping redox catalysts, indicating the potential for future work in this field.
4.3 Chemical looping involving liquid and solid fuels
(i) The solid fuel can be gasified in situ in the presence of a redox catalyst using steam or CO2 as the fluidizing gas. This strategy not only eliminates the need for a separate gasification unit but also enhances the rate of gasification.375,376 However, owing to the inherently slow nature of the gasification reaction, it is difficult to achieve complete gasification of the solid fuel in situ.
(ii) Depending upon their thermodynamic characteristics, certain oxygen carriers can undergo spontaneous decomposition at high temperatures to release molecular oxygen.377,378 The use of such oxygen carriers allows the direct combustion of a solid fuel via a gas–solid reaction, i.e. without an intermediate gasification step. This process configuration, typically referred to as chemical looping with oxygen uncoupling (CLOU), significantly increases the rate of fuel oxidation, thereby allowing the complete combustion of the solid fuel. Furthermore, instead of fully oxidizing the solid fuel it is also possible to gasify it by controlling the partial pressure of oxygen released by the redox catalyst.379
(iii) Recently, it has been proposed to use molten oxygen carriers or reaction system for CL-methane POx, CLC, and CL separation to circumvent the challenges associated with the solid-based chemical looping systems, such as carbon deposition, thermal sintering and low reaction rates.380 Wang et al.381 proposed a molten slat alkali carbonate based CL-methane POx system for the coproduction of synthesis gas and metallic Zn. The molten reaction system enables to intensify heat and mass transfer during the cyclic redox, increasing the energy utilization efficiency. Sarafraz et al.382–384 investigated the thermodynamic feasibility of using liquid oxygen carriers for the partial oxidation of fuel to synthesis gas and identified Cu, Pb and Sb as the most promising candidates. Upham et al.385 demonstrated a molten bromide-oxide based chemical looping for the reactive separation of HBr in a halogen-based natural gas conversion. Nickel-alkali bromide and oxide were switched in molten reactor for the conversion of HBr to Br through the MeBr/MeO redox pair. Although the use of molten redox catalysts avoids the aforementioned issues and enhances the rate of reaction, it also introduces new challenges such as evaporation or solidification of liquid oxygen carriers, continuous circulation of liquid metals/oxides, large pressure drops across the reactors, etc.
(iv) For solid fuels with high volatile contents, e.g. biomass or low-rank coal, it is possible that a redox catalyst can be used to generate value-added products via pyrolysis-type of processes – an area that has not been investigated extensively by the chemical looping community.44,386 It is noted that pyrolysis products with less oxygenates are typically desired, which may conflict with typical chemical looping operations.
4.4 Synergy between oxide and calcium/carbonate looping
CaO-Based sorbents, often in the form of naturally-occurring lime, are used widely in applications such as flue gas desulfurization387–389 and sorption-enhanced steam methane reforming (SE-SMR) to produce H2.390–393 In SE-SMR, CO2 is removed continuously in the WGS reaction step, thus shifting the equilibrium to the product side according Le Chatelier's principle.394 The H2 yield can thereby be increased to ∼97 vol% at 550 °C.395 The WGS reaction is relevant also in gasification reactions, and generally in schemes where mixtures of CO, CO2, H2 and H2O co-exist at high temperature, and so sorbents for CO2 are conventionally used.396 Lately, CaO-based sorbents have also gained considerable attention for carbonate looping (CaL),397–405 a post-combustion CO2 capture technique.402,406 A key advantage of using CaO for the sorption of CO2 is its high theoretical uptake of 0.78 g CO2 per g CaO, according to reaction (R68) in Table 2. The carbonation reaction is exothermic whereas the reverse reaction, known as the calcination reaction, is endothermic, giving a stream of concentrated CO2.407 Carbonate and oxide (chemical) looping processes can be combined to improve efficiencies of both processes, as discussed below.Chemical looping with sorbent enhancement for heat-neutral sorbent regeneration. Besides permitting higher yields of H2 in the SE-SMR/gasification scheme, the exothermic carbonation reaction can provide a large amount of heat for the strongly endothermic methane reforming or the gasification reaction. Meanwhile, the regeneration of the sorbent via the highly endothermic calcination reaction is critical and requires high reaction temperatures (>750 °C). Performing the calcination reaction in the presence of an exothermic reaction would balance the heat requirements. Such exothermic reaction could be the oxidation of a reduced oxygen carrier, as suggested by Lyon and Cole,163,408e.g. the simultaneous oxidation of the Ni-based reforming catalyst. However, a relatively high ratio of Ni to CaCO3 (>0.75) would be required to balance the heats of reaction, resulting in an increase in the catalyst cost, and the CO2 produced is diluted with N2 when air is used for the re-oxidation of Ni. In addition, the oxidation of the reforming catalyst at high temperature is likely to promote its sintering and deactivation.
Chemical looping with sorbent enhancement for CO2 capture. The simultaneous oxidation and calcination reactions eliminate the benefit of CaO-based sorbents to produce a pure stream of CO2. In CaO-based CO2 capture architectures, the calcination reaction can be performed in a reactor operated in an oxy-fuel mode, where the heat is provided by combusting additional fuel (e.g. natural gas) in an atmosphere containing O2 diluted with CO2.409–411 Replacing molecular oxygen with an oxygen carrier that reduces in an exothermic reaction while fully combusting the fuel would enable both the balancing of the endothermicity of the calcination reaction and generating a pure stream of CO2.412 A few redox pairs, e.g. CuO/Cu or Mn2O3/Mn3O4, can be reduced exothermally with CH4 and other hydrocarbons.39 This feature can be applied to integrate the heat released from the CLC reduction step with the endothermic calcination step to produce concentrated CO2. A reaction scheme where CuO-based chemical looping combustion is combined with SE-SMR is shown in Fig. 14a and at least three reactors are required to obtain separate streams of H2 and CO2.413
 | ||
| Fig. 14 (a) Reaction scheme with three reactors where CuO-based chemical looping combustion is integrated into the SE-SMR process. (b) Reaction scheme of the super-dry reforming of methane with two reactors, in which the reforming catalyst (Ni) is spatially separated from the oxygen carrier (Fe3O4) and the CO2 sorbent. | ||
For carbonate looping as a CO2 capture technique, heat integration is vital to be economically competitive with other CO2 capture techniques. In analogy to the SE-SMR process, CuO-based oxygen carriers have mostly been investigated to render the heat exothermic during the exothermic calcination step.414–417
Unlike the CLC reduction step that is often endothermic, the regeneration step is always exothermic for monometallic oxides. Therefore, integration of the heat released in the CLC regeneration step with the sorbent calcination step can also result in concentrated CO2 if a heat transfer agent is used to avoid direct contact between the air (in the regeneration step and the oxidizer) and CO2 (from calcination in the calciner). Both ALSTOM and General Electric proposed and investigated such schemes (details on these two processes have been summarized by Fan122). In ALSTOM's Hybrid Combustion–Gasification Process for hydrogen generation, a CaSO4 oxygen carrier is employed to generate the heat for the calcination of the CaO sorbent. Instead of oxidizing the oxygen carrier in the vicinity of the CaCO3, bauxite is used to transfer the heat from the oxidation reactor to the calcination reactor. Similarly, General Electric's Fuel-Flexible Advanced Gasification–Combustion Process also adopted CLC reactions to provide the heat for calcination. In this case, an iron oxide-based oxygen carrier was used as the heat transfer agent.
Sorption-enhanced methane reforming coupled with chemical looping conversion schemes. The thermodynamic properties of some oxygen carriers permit the splitting of CO2 or H2O to produce CO or H2 upon oxidation (see Section 4.1.1). Since the rate of reduction of many oxygen carriers, e.g. Fe2O3, with primary fuels such as CH4 is low, it was proposed to convert CH4 first to a synthesis gas over a catalysts (e.g. Ni; in analogy to the dry reforming of methane (DRM)) and use the synthesis gas to reduce the oxygen carrier.94,418 Depending on the oxidant used (CO2 or H2O) for the subsequent regeneration of the oxygen carrier, CO or H2 can readily be produced.
This reaction concept can be extended by adding a CaO-based sorbent (Fig. 14b).419,420 Analogously to SE-SMR, the heat of sorption from the carbonation reaction (reaction (R68)) balances the endothermic reforming and reduction reactions. In the following oxidation step, CO2 is released from the sorbent and oxidizes the oxygen carrier while producing CO. The chemical looping concept, where in theory three moles of CO2 per mole CH4 are converted to four moles of CO, has been termed super-dry reforming of methane (R67). Importantly, in such a process the formation of an equilibrium mixture of CO2, CO, H2O and H2 is circumvented during the reforming step, thus avoiding a loss in CO yield through the RWGS reaction (R71). So far, the process has been demonstrated by mixing different materials, viz. CaO supported on Al2O3 as CO2 sorbent, Fe2O3 supported on MgAl2O4 as oxygen carrier, and NiO supported on MgAl2O4 as the CH4 reforming catalyst, rather than multifunctional redox catalysts. While the theoretical advantages over conventional dry-reforming of CH4 are apparent, there is still scope for improvement (besides improving the materials used). For example, it was proposed to run the process isothermally. Equilibrium thermodynamics dictate that the release of CO2 from the CO2 sorbent requires very low CO2 partial pressures and for that, an inert gas would likely be needed in the oxidation step (where Fe is oxidized by the CO2 released from the sorbent). This will inevitably dilute the product gas CO. The product gas CO is diluted further with CO2 because the conversion of CO2 to CO over Fe is thermodynamically limited and the ratio of CO/CO2 in the product gas will decrease further as the Fe-based oxygen carrier is oxidized via FeO to Fe3O4.
Three active components make the design of suitable, “tri-functional” materials rather challenging;433 current research thus focuses largely on the development and optimization of separate particle systems434–436 or bi-functional Ca–Cu422,437–440 or Ca–Ni441–443 composites and uses commercially available Ni-based catalyst particles.444,445 The CO2 capture capacity of the CaO-based sorbents is linked strongly with the available surface area, which is known to decay due to sintering when the sorbents undergo cycling of carbonation and calcination.446–448 Producing stable particles with a porous CaO structure in the vicinity of low surface area CuO in a Ca–Cu composite material requires appropriate support materials, e.g. MgO449 or Ca12Al14O33.450 Recent works405,414,451 at ETH Zürich have shown how such support materials function as structural stabilizers at the microscopic level and how subtle phase transitions affect the cyclic performance of the CaO-sorbents (Fig. 15). Another recent study reported an interesting phase transition based approach to effectively enhance sorbent stability for the hydrogen production from bio-glycerol.452
 | ||
| Fig. 15 Morphological changes during cyclic operation of a Ca–Mg mixed oxide-based sorbent. (a–c) SEM imges of FIB cross-sections. (d–f) HAADF-STEM images with EDX maps of Ca and Mg at different stages of CO2 capture/regeneration cycles. Scale bars: 1 μm for (a–c) and 700 nm for (d–f).451 | ||
Conventional design strategies aim to limit the mobility of active species and their growth by using inert support materials that act as physical barriers.199 New approaches have shown that the active species in a sorbent or redox catalyst may well be mobile, but their self-diffusion is limited if the simultaneous formation of a new phase with the active species is thermodynamically more preferred.453 For example, Dang et al.452 demonstrated that calcium cobalt, a layered bi-functional material acting as both sorbent and catalyst for the sorbent enhanced reforming of glycerol could effectively be stabilized because the components CaCO3 and CoO homogenized into calcium cobalt on an atomic level in every calcination stage of the cyclic process instead of remaining segregated and sintering. Well-ordered structures, such as layered oxides, spinels or perovskites are particularly suitable for such approaches.452,454
For reaction schemes such as the super-dry reforming of methane, catalyst and oxygen storage material are usually separated spatially.455 Work by Hosseini et al. demonstrated that both the reforming of methane through the catalyst and the reduction of the oxygen storage material can be realized by the same redox catalyst particle provided it is designed carefully in a core–shell structure.456
5. Summary and perspective
The chemical and petrochemical industry generates an annual revenue in excess of $4 trillion while consuming 40.9 quadrillion BTU of energy and emitting 2.5 Gigatons of CO2 (2014 numbers provided by IEA).457 The significant energy intensity and CO2 emissions associated with this important industrial sector offer excellent opportunities for process intensification, energy conservation, emission reduction, as well as profitability enhancement. We believe that CLBC represents a promising new frontier in chemical reaction engineering, catalysis, and particle technology to address the aforementioned challenges and opportunities. As discussed in Section 2, the unique advantages of CLBC largely reside in its ability to significantly improve a chemical (and energy) conversion process from a thermodynamic second law standpoint: a strategically designed CLBC scheme, when coupled with tailored oxygen carrier or redox catalyst, can significantly reduce process exergy loss and/or facilitate chemical transitions that would otherwise be impossible under a conventional scheme. From an economic standpoint, reactive separation enabled by oxygen carriers in CLBC offers excellent potential to simplify conventional processes and to reduce the number of unit operations. With the continued technical progress in CLC, a promising carbon capture technology that addresses the carbon intensive, $1.4 trillion power generation sector, we argue that chemical looping beyond combustion offers equally and perhaps even more exciting opportunities due to their potential for more efficient lattice oxygen utilization, higher product value, and relative ease for scale-up compared to full scale utility plants.Although CLBC has attracted increasing attentions over the past 5–10 years, we note this emerging research area is far from being adequately explored particularly considering its significant complexity and excellent potential. From scientific and engineering standpoints, CLBC represents an “ultimate” multidisciplinary and multiscale challenge that requires expertise in various areas including, but not limited to, chemical looping, heterogeneous catalysis, material science, particle technology, reaction and reactor engineering, and process and system engineering. This is due to the uniquely intertwined nature of CLBC. For instance, redox catalyst performance cannot be treated in an isolated manner since it is significantly affected by reactor design and gas–solids contacting patterns as well as the multi-step redox scheme configuration. Meanwhile, the redox catalyst can undergo significant changes in its surface and bulk properties within a CLBC cycle that exceeds complexities typically observed in heterogeneous catalysts. Such complexities, while daunting, are closely associated with the unique advantages CLBC has to offer. They can and should be addressed through continued research via interdisciplinary efforts. From a practical standpoint, redox catalysts with high stability and satisfactory long-term performance are critical for CLBC. Moreover, reactor designs that facilitate suitable redox catalyst particle circulation, gas–solids contacting patterns, residence time distributions, and heat integration between multiple reactors are important to enable these promising CLBC technologies.
This article aims to cover all the CLBC schemes to the best of our knowledge. Although a number of promising approaches such as oxidative dehydrogenation of light alkanes and alkene epoxidation are under investigation, significantly greater opportunities still exist both within and beyond these CLBC topics. From a reaction chemistry standpoint, the chemical looping approach is analogous to the MvK mechanism commonly encountered in catalytic oxidation reactions. As illustrated in Fig. 16, CLBC carries out catalytic oxidation cycles via a step-wise MvK with macroscopic spatial or temporal separation of the lattice oxygen removal and replenishment events. This is in contrast with conventional MvK, where the catalytic cycle is completed on the surface of an oxide catalyst under steady-state operations. The step-wise operations in CLBC offers both significant advantages (reactive separation, process intensification, and energy/cost reduction) and potential challenges (surface and/or phase changes of the redox catalytic under redox cycles). Considering their similarities, it would not be unreasonable to anticipate catalytic reactions operated under MvK mechanism to be potentially suitable candidates for CLBC. Table 3 summarizes a number of such candidate reactions. Compared to the limited CLBC reactions investigated to date, a significantly number of potential reactions are yet to be explored. In addition, the regeneration step in CLBC, which was demonstrated to be effective for H2 and CO production via CO2/H2O splitting, could be investigated for hydrogenation or carbonylation reactions. Carriers other than oxygen, e.g. nitrogen carriers, also offer exciting potential for chemical syntheses. Aside from these, significant progress still needs to be made towards in-depth understanding of redox reaction mechanisms, catalyst design and optimizations, reactor and process design and operations, as well as process scale up for the existing CLBC approaches. To summarize, we believe that CLBC represents a largely uncharted and extraordinarily exciting area of research and potential applications.
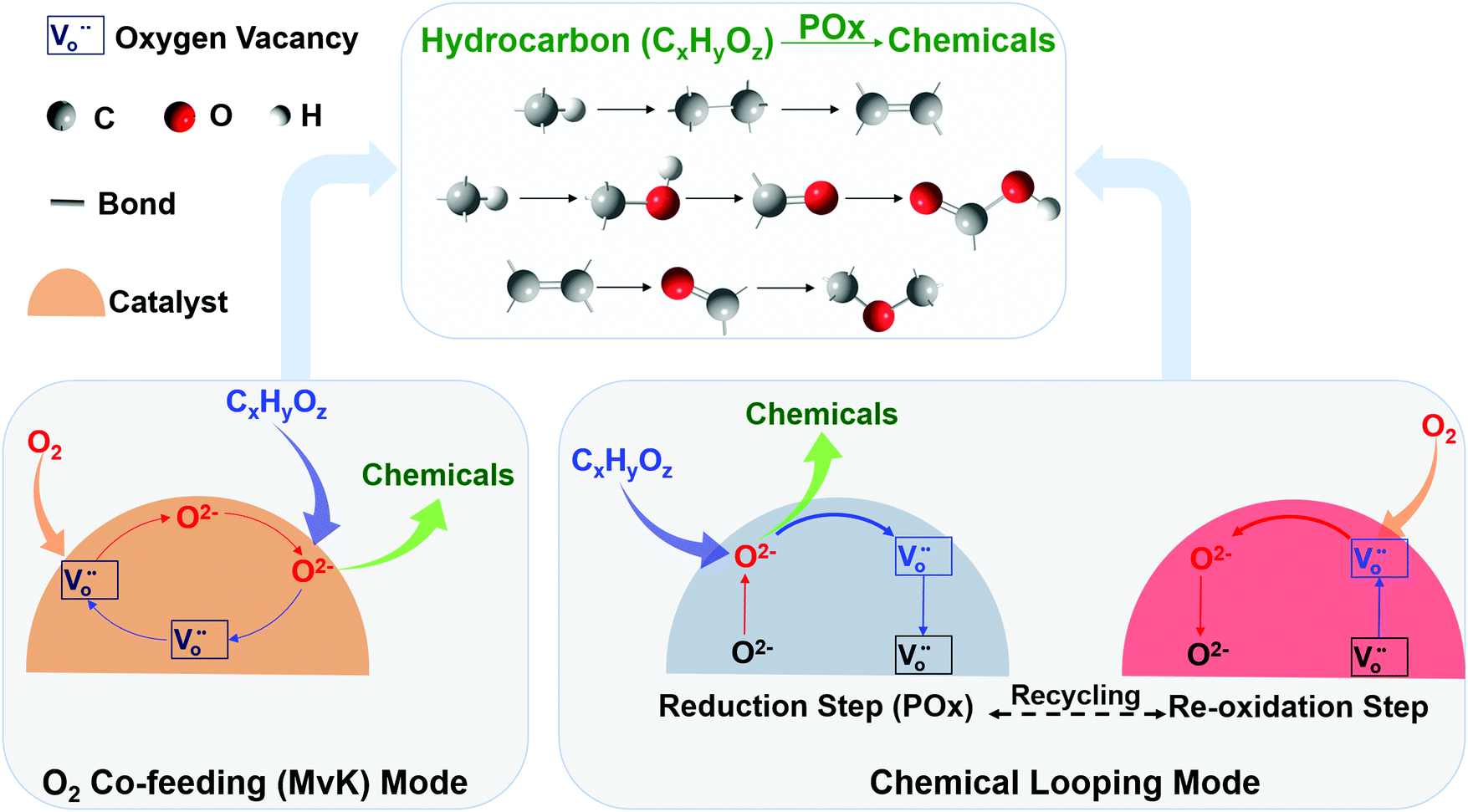 | ||
| Fig. 16 Comparison of O2 co-feeding (MvK) and chemical looping modes for partial oxidations and their applications on conversions of hydrocarbon to chemicals (transformation between alkane, alkene, alcohol, aldehyde, ketone, acid, and aromatics). | ||
| Feedstock | Products | Reaction | Heterogeneous catalysts |
|---|---|---|---|
| Methane | Formaldehyde | (R73) CH4 + 2CO2 → CH2O + 2CO + H2O | V2O5/SiO2458 |
| Methanol | Formaldehyde | (R74) CH3OH + 1/2O2 → CH2O + H2O | Fe2(MoO4)3459,460 |
| Methyl formate | (R75) 2CH3OH + O2 → HCOOCH3 + 2H2O | V2O5/TiO2,461 SnO2–MoO3462 | |
| Ethanol | Acetic acid | (R76) C2H5OH + O2 → CH3COOH + H2O | V2O5/TiO2463 |
| Acetaldehyde | (R77) C2H5OH + 1/2O2 → CH3CHO + H2O | ||
| 1,2-Ethanediol | Ethanedial | (R78) C2H6O2 + O2 → C2H2O2 + 2H2O | P-Doped Fe–Mo mixed oxide464 |
| Propane | Acrolein | (R79) C3H8 + 3/2O2 → C3H4O + 2H2O | NiMoO4/MoO3/Te2MoO7/P2O5465 |
| Acrylic acid | (R80) C3H8 + 2O2 → C3H4O2 + 2H2O | NiMoO4/MoO3/Te2MoO7/P2O5465 | |
| Propene | Acrolein | (R81) C3H6 + O2 → C3H4O + H2O | Fe–Sb mixed oxide,466 V2O5/Nb2O5467 |
| Acrylonitrile | (R82) C3H6 + NH3 + 3/2O2 → C3H3N + 3H2O | Fe–Sb mixed oxide466 | |
| 1,5-Hexadiene | (R83) 2C3H6 + 1/2O2 → C6H10 + H2O | Bi2M0.1V0.9O5.35 (M = Cu or Co)468 | |
| Acrylic acid | (R84) C3H6 + 3/2O2 → C3H4O2 + H2O | As2O3–Nb2O5–MoO3469 | |
| Butane | 1-Butene | (R85) C4H10 + 1/2O2 → C4H8 + H2O | VOx/Al2O3,470 VOx/Mg–Al spinel,470 VOx/Mg–Al hydrotalcite,470 VMgO,470 Mg3(VO4)2–MgO,471 Mg2V2O7,471 (VO)2P2O7471 |
| 1,3-Butadiene | (R86) C4H10 + O2 → C4H6 + 2H2O | ||
| Acetic acid | (R87) C4H10 + 5/2O2 → 2CH3COOH + H2O | V2O5/TiO2472 | |
| 1-Butene | 1,3-Butadiene | (R88) C4H8 + 1/2O2 → C4H6 + H2O | VOx/Al2O3,470 VOx/Mg–Al spinel,470 VOx/Mg–Al hydrotalcite,470 VMgO,470 USb3O10,473 USbO5,473 MO3/SiO2 (M = Cr, Mo or W)474 |
| Cyclopentene | Glutaraldehyde | (R89) C5H8 + O2 → C5H6O2 | WO3/SiO2475 |
| Ethyl lactate | Ethyl pyruvate | (R90) C5H10O3 + 1/2O2 → C5H8O3 + H2O | Mesoporous VOx/TiO2476 |
| Cyclohexane | Cyclohexanone | (R91) C6H12 + 1/2O2 → C6H10O + H2O | Mn0.5Ce0.5Ox477 |
| Cyclohexanol | (R92) C6H12 + 1/2O2 → C6H12O | ||
| Glucose | Lactic acid | (R93) C6H12O6 + O2 → C3H6O3 + 3CO2 + 3H2O | LaCoO3478 |
| 5-Hydroxymethylfurfural | 2,5-Furandicarboxylic acid | (R94) C6H6O3 + 3/2O2 → C6H4O5 + H2O | Co–Mn mixed oxide479 |
| Benzene | Maleic acid | (R95) C6H6 + 9/2O2 → C4H4O4 + 2CO2 + 2H2O | V2O5–MoO3480 |
| Toluene | Benzaldehyde | (R96) C7H8 + O2 → C7H6O + H2O | V2O5/TiO2,481 V2O5–MoO3/TiO2481 |
| Benzoic acid | (R97) C7H8 + 3/2O2 → C7H6O2 + H2O | V2O5/TiO2,481 V2O5–MoO3/TiO2481 | |
| Benzyl alcohol | Benzaldehyde | (R98) C7H8O + 1/2O2 → C7H6O + H2O | Au/CeO2482 |
| p-Cresol | p-Hydroxybenzyl alcohol | (R99) C7H8O + 1/2O2 → C7H8O2 | MWCNT–Mn3O4 composite483 |
| Ethylbenzene | Styrene | (R100) C8H10 + 1/2O2 → C8H8 + H2O | MnxTi1−xO2 (x = 0–0.15)484 |
| o-Xylene | Phthalic anhydride | (R101) C8H10 + 3O2 → C8H4O3 + 3H2O | V2O5485 |
| Naphthalene | Phthalic anhydride | (R102) C10H8 + 9/2O2 → C8H4O3 + 2CO2 + 2H2O | V2O5-Based catalysts486 |
| Amines | Imines | (R103) R/R′-CH2-NH2 + O2 → R-CH = NH-CH2-R/R′ + H2O (R = aryl, R' = alkyl) | Meso-Cs/MnOx487 |
| Methyl bromide | Bromine | (R104) 2CH3Br + 7/2O2 → 2CO2 + 3H2O + Br2 | Amorphous MnOx488 |
| 1,2-Dichlorobenzene | Chlorine | (R105) C6H4Cl2 + 7O2 → 6CO2 + 2H2O + Cl2 | Ru/Co3O4489 |
| Formaldehyde | Carbon dioxide | (R106) CH2O + O2 → CO2 + H2O | Au/α-MnO2490 |
| Benzene | Carbon dioxide | (R107) C6H6 + 15/2O2 → 6CO2 + 3H2O | MCo2O4 (M = Cu, Ni or Mn)491 |
| Toluene | Carbon dioxide | (R108) C7H8 + 9O2 → 7CO2 + 4H2O | LaMnO3,492 La0.9Sr0.1MnO3,492 La0.9Sr0.1Mn0.9Fe0.1O3,492 Ag, Pt or Pd doped CoAl layered double hydroxide493 |
| Ammonia | Nitrogen | (R109) 2NH3 + 3/2O2 → N2 + 3H2O | Fe2O3,494 Cr2O3,494 CeO2494 |
| Nitric oxide | (R110) NH3 + 5/4O2 → NO + 3/2H2O | Fe2O3,494 Cr2O3,494 CeO2494 | |
| Hydrogen sulfide | Sulfur | (R111) H2S + 1/2O2 → 1/nSn + H2O | La1−xCexFeO3 (x = 0, 0.2, 0.4, 0.6 and 0.8),495 Cu–V and Cu–V–Mo mixed oxides496 |
Conflicts of interest
Authors have no conflict of interest to declare.Acknowledgements
This work was supported by the U.S. National Science Foundation (Award No. CBET-1604605 and CBET CBET-1510900), US Department of Energy (Award No. FE0031521 and RAPID Subaward DE-EE0007888-05-6) and the Kenan Institute for Engineering, Technology and Science at NC State University. The Scholarship provided by the State Key Laboratory of Complex Nonferrous Metal Resources Clean Utilization at Kunming University of Science and Technology is gratefully acknowledged. F. D and C. R. M. acknowledge the Swiss Office of Energy and the European Commission for financial support (BFE SI/501590-01 and ACT Grant Agreement No. 691712 under the Horizon 2020 programme).References
- H. J. Richter and K. F. Knoche, Efficiency and Costing, American Chemical Society, 1983, ch. 3, vol. 235, pp. 71–85 Search PubMed.
- A. Messerschmitt, Process of Producing Hydrogen, US Pat., US971206A, 1910 Search PubMed.
- H. Lane, Process for the production of hydrogen, US Pat., US1078686A, 1913 Search PubMed.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- E. J. B. Anthony, Coal in the 21st Century: Energy Needs, Chemicals and Environmental Controls, The Royal Society of Chemistry, 2018, pp. 198–215 Search PubMed.
- M. Ishida, D. Zheng and T. Akehata, Energy, 1987, 12, 147–154 CrossRef CAS.
- M. Ishida and H. Jin, Energy, 1994, 19, 415–422 CrossRef CAS.
- M. Ishida and H. Jin, Ind. Eng. Chem. Res., 1996, 35, 2469–2472 CrossRef CAS.
- H. Jin and M. Ishida, Int. J. Hydrogen Energy, 2001, 26, 889–894 CrossRef CAS.
- P. Gupta, L. G. Velazquez-Vargas, F. Li and L.-S. Fan, Proc. – Annu. Int. Pittsburgh Coal Conf., 2006, 23rd, 37.33/31-37.33/38.
- F. He, H. Wang and Y. Dai, J. Nat. Gas Chem., 2007, 16, 155–161 CrossRef CAS.
- F. Li and L.-S. Fan, Energy Environ. Sci., 2008, 1, 248–267 RSC.
- L.-S. Fan and F. Li, Ind. Eng. Chem. Res., 2010, 49, 10200–10211 CrossRef CAS.
- L.-S. Fan, L. Zeng and S. Luo, AIChE J., 2015, 61, 2–22 CrossRef CAS.
- L.-S. Fan, Cambridge Series in Chemical Engineering, Cambridge University Press, 2017 Search PubMed.
- C. Chung, L. Qin, V. Shah and L.-S. Fan, Energy Environ. Sci., 2017, 10, 2318–2323 RSC.
- S. Bhavsar, M. Najera, A. More and G. Veser, Reactor and Process Design in Sustainable Energy Technology, ed. F. Shi, Elsevier, Amsterdam, 2014, pp. 233–280 Search PubMed.
- L. C. Buelens, H. Poelman, G. B. Marin and V. V. Galvita, Ind. Eng. Chem. Res., 2019, 58, 16235–16257 CrossRef CAS.
- T. Mattisson, M. Keller, C. Linderholm, P. Moldenhauer, M. Rydén, H. Leion and A. Lyngfelt, Fuel Process. Technol., 2018, 172, 1–12 CrossRef CAS.
- G. Veser and C. R. Müller, Energy Technol., 2016, 4, 1127–1129 CrossRef.
- L. Zeng, Z. Cheng, J. A. Fan, L.-S. Fan and J. Gong, Nat. Rev. Chem., 2018, 2, 349–364 CrossRef CAS.
- S. C. Bayham, A. Tong, M. Kathe and L.-S. Fan, Wiley Interdiscip. Rev.: Energy Environ., 2016, 5, 216–241 CAS.
- Development of the steam-iron process for hydrogen production, 1979.
- S. Bhavsar, M. Najera, R. Solunke and G. Veser, Catal. Today, 2014, 228, 96–105 CrossRef CAS.
- Q. Wang, J. Guo and P. Chen, J. Energy Chem., 2019, 36, 25–36 CrossRef.
- M. M. Hossain and H. I. de Lasa, Chem. Eng. Sci., 2008, 63, 4433–4451 CrossRef CAS.
- H. Jin, H. Hong and T. Han, Chin. Sci. Bull., 2009, 54, 906–919 CAS.
- L.-S. Fan, L. Zeng, W. Wang and S. Luo, Energy Environ. Sci., 2012, 5, 7254–7280 RSC.
- Q. Imtiaz, D. Hosseini and C. R. Mueller, Energy Technol., 2013, 1, 633–647 CrossRef CAS.
- A. Lyngfelt, Appl. Energy, 2014, 113, 1869–1873 CrossRef CAS.
- P. Wang, N. Means, D. Shekhawat, D. Berry and M. Massoudi, Energies, 2015, 8, 10605–10635 CrossRef CAS.
- M. Matzen, J. Pinkerton, X. Wang and Y. Demirel, Int. J. Greenhouse Gas Control, 2017, 65, 1–14 CrossRef CAS.
- J. Li, H. Zhang, Z. Gao, J. Fu, W. Ao and J. Dai, Energy Fuels, 2017, 31, 3475–3524 CrossRef CAS.
- T. Gauthier, M. Yazdanpanah, A. Forret, B. Amblard, A. Lambert and S. Bertholin, Powder Technol., 2017, 316, 3–17 CrossRef CAS.
- T. Song and L. Shen, Int. J. Greenhouse Gas Control, 2018, 76, 92–110 CrossRef CAS.
- A. Lyngfelt, A. Brink, Ø. Langørgen, T. Mattisson, M. Rydén and C. Linderholm, Int. J. Greenhouse Gas Control, 2019, 88, 38–56 CrossRef CAS.
- Z. Haibo, T. Xin, M. Jinchen, S. Mingze, W. Baowen and M. Daofeng, Int. J. Greenhouse Gas Control, 2020, 93, 102898 CrossRef.
- A. Thursfield, A. Murugan, R. Franca and I. S. Metcalfe, Energy Environ. Sci., 2012, 5, 7421–7459 RSC.
- J. Adanez, A. Abad, F. Garcia-Labiano, P. Gayan and L. F. de Diego, Prog. Energy Combust. Sci., 2012, 38, 215–282 CrossRef CAS.
- B. Moghtaderi, Energy Fuels, 2012, 26, 15–40 CrossRef CAS.
- M. Tang, L. Xu and M. Fan, Appl. Energy, 2015, 151, 143–156 CrossRef CAS.
- G. Voitic and V. Hacker, RSC Adv., 2016, 6, 98267–98296 RSC.
- L. Protasova and F. Snijkers, Fuel, 2016, 181, 75–93 CrossRef CAS.
- X. Zhao, H. Zhou, V. S. Sikarwar, M. Zhao, A.-H. A. Park, P. S. Fennell, L. Shen and L.-S. Fan, Energy Environ. Sci., 2017, 10, 1885–1910 RSC.
- P. T. Krenzke, J. R. Fosheim and J. H. Davidson, Sol. Energy, 2017, 156, 48–72 CrossRef CAS.
- J. Hu, V. V. Galvita, H. Poelman and G. B. Marin, Materials, 2018, 11, 1187 CrossRef PubMed.
- M. Luo, Y. Yi, S. Wang, Z. Wang, M. Du, J. Pan and Q. Wang, Renewable Sustainable Energy Rev., 2018, 81, 3186–3214 CrossRef CAS.
- Z. Cheng, L. Qin, J. A. Fan and L.-S. Fan, Engineering, 2018, 4, 343–351 CrossRef CAS.
- X. Zhu, K. Li, L. Neal and F. Li, ACS Catal., 2018, 8, 8213–8236 CrossRef CAS.
- A. Mishra and F. Li, Curr. Opin. Chem. Eng., 2018, 20, 143–150 CrossRef.
- S. Laassiri, Alternative Catalytic Materials: Carbides, Nitrides, Phosphides and Amorphous Boron Alloys, The Royal Society of Chemistry, 2018, pp. 120–132 Search PubMed.
- J. A. Medrano, A. Helmi, V. Spallina, M. van Sint Annaland and F. Gallucci, Membrane Engineering for the Treatment of Gases: Volume 2: Gas-separation Issues Combined with Membrane Reactors (2), The Royal Society of Chemistry, 2018, vol. 2, pp. 30–57 Search PubMed.
- D. S. Baser, S. G. Nadgouda, A. S. Joshi and L. S. Fan, Ind. Eng. Chem. Res., 2019, 58, 16407–16416 CrossRef CAS.
- J. Vieten, B. Bulfin, F. Call, M. Lange, M. Schmucker, A. Francke, M. Roeb and C. Sattler, J. Mater. Chem. A, 2016, 4, 13652–13659 RSC.
- J. Dou, E. Krzystowczyk, A. Mishra, X. Liu and F. Li, ACS Sustainable Chem. Eng., 2018, 6, 15528–15540 CrossRef CAS.
- F. X. Chiron, G. S. Patience and S. Rifflart, Chem. Eng. Sci., 2011, 66, 6324–6330 CrossRef CAS.
- L. Silvester, A. Antzara, G. Boskovic, E. Heracleous, A. A. Lemonidou and D. B. Bukur, Int. J. Hydrogen Energy, 2015, 40, 7490–7501 CrossRef CAS.
- L. M. Neal, A. Shafiefarhood and F. Li, ACS Catal., 2014, 4, 3560–3569 CrossRef CAS.
- A. Shafiefarhood, N. Galinsky, Y. Huang, Y. Chen and F. Li, ChemCatChem, 2014, 6, 790–799 CrossRef CAS.
- L. Neal, A. Shafiefarhood and F. Li, Appl. Energy, 2015, 157, 391–398 CrossRef CAS.
- C. Huang, J. Wu, Y.-T. Chen, M. Tian, A. I. Rykov, B. Hou, J. Lin, C.-R. Chang, X. Pan, J. Wang, A. Wang and X. Wang, Commun. Chem., 2018, 1, 55 CrossRef.
- C. Lu, K. Li, H. Wang, X. Zhu, Y. Wei, M. Zheng and C. Zeng, Appl. Energy, 2018, 211, 1–14 CrossRef CAS.
- K. Li, H. Wang, Y. Wei and D. Yan, Appl. Catal., B, 2010, 97, 361–372 CrossRef CAS.
- X. Zhu, Y. Wei, H. Wang and K. Li, Int. J. Hydrogen Energy, 2013, 38, 4492–4501 CrossRef CAS.
- J. R. Fosheim, B. J. Hathaway and J. H. Davidson, Energy, 2019, 169, 597–612 CrossRef CAS.
- V. P. Haribal, F. He, A. Mishra and F. Li, ChemSusChem, 2017, 10, 3402–3408 CrossRef CAS PubMed.
- J. Zhang, V. Haribal and F. Li, Sci. Adv., 2017, 3, e1701184 CrossRef PubMed.
- Y. Zheng, K. Li, H. Wang, D. Tian, Y. Wang, X. Zhu, Y. Wei, M. Zheng and Y. Luo, Appl. Catal., B, 2017, 202, 51–63 CrossRef CAS.
- Q. Shen, F. Huang, M. Tian, Y. Zhu, L. Li, J. Wang and X. Wang, ACS Catal., 2019, 9, 722–731 CrossRef CAS.
- Y. Zhu, W. Liu, X. Sun, X. Ma, Y. Kang, X. Wang and J. Wang, AIChE J., 2018, 64, 550–563 CrossRef CAS.
- S. Parishan, E. Nowicka, V. Fleischer, C. Schulz, M. G. Colmenares, F. Rosowski and R. Schomäcker, Catal. Lett., 2018, 148, 1–17 CrossRef.
- W. Riedel, L. Thum, J. Möser, V. Fleischer, U. Simon, K. Siemensmeyer, A. Schnegg, R. Schomäcker, T. Risse and K.-P. Dinse, J. Phys. Chem. C, 2018, 122, 22605–22614 CrossRef CAS.
- N. Kosinov, F. J. Coumans, E. Uslamin, F. Kapteijn and E. J. Hensen, Angew. Chem. Int. Ed., 2016, 55, 15086–15090 CrossRef CAS PubMed.
- C. Brady, B. Murphy and B. Xu, ACS Catal., 2017, 7, 3924–3928 CrossRef CAS.
- T. Sheppard, C. D. Hamill, A. Goguet, D. W. Rooney and J. M. Thompson, Chem. Commun., 2014, 50, 11053–11055 RSC.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS PubMed.
- R. Blume, M. Hävecker, S. Zafeiratos, D. Techner, A. Knop-Gericke, R. Schlögl, L. Gregoratti, A. Barinov and M. Kiskinova, in Nanostructured Catalysts: Selective Oxidations, ed. C. Hess and R. Schlögl, The Royal Society of Chemistry, 2011, pp. 248–265 Search PubMed.
- M. S. C. Chan, E. Marek, S. A. Scott and J. S. Dennis, J. Catal., 2018, 359, 1–7 CrossRef CAS.
- Y. Gao, L. M. Neal and F. Li, ACS Catal., 2016, 6, 7293–7302 CrossRef CAS.
- L. M. Neal, S. Yusuf, J. A. Sofranko and F. Li, Energy Technol., 2016, 4, 1200–1208 CrossRef CAS.
- S. Yusuf, L. M. Neal and F. Li, ACS Catal., 2017, 7, 5163–5173 CrossRef CAS.
- S. Yusuf, L. Neal, V. Haribal, M. Baldwin, H. H. Lamb and F. Li, Appl. Catal., B, 2018, 232, 77–85 CrossRef CAS.
- R. B. Dudek, Y. Gao, J. Zhang and F. Li, AIChE J., 2018, 64, 3141–3150 CrossRef CAS.
- J. Beckers, R. Drost, Z. I. Van, P. F. Collignon and G. Rothenberg, ChemPhysChem, 2010, 9, 1062–1068 CrossRef PubMed.
- A. A. Ayandiran, I. A. Bakare, H. Binous, S. Al-Ghamdi, S. A. Razzak and M. M. Hossain, Catal. Sci. Technol., 2016, 6, 5154–5167 RSC.
- D. K. Hood and O. M. Musa, Handbook of Maleic Anhydride Based Materials, 2016, pp. 3–55 Search PubMed.
- R. B. Dudek, X. Tian, M. Blivin, L. M. Neal, H. Zhao and F. Li, Appl. Catal., B, 2019, 246, 30–40 CrossRef CAS.
- F. He and F. Li, Energy Environ. Sci., 2015, 8, 535–539 RSC.
- F. He, J. Trainham, G. Parsons, J. S. Newman and F. Li, Energy Environ. Sci., 2014, 7, 2033–2042 RSC.
- A. Murugan, A. Thursfield and I. Metcalfe, Energy Environ. Sci., 2011, 4, 4639–4649 RSC.
- I. S. Metcalfe, B. Ray, C. Dejoie, W. Hu, C. de Leeuwe, C. Dueso, F. R. García-García, C.-M. Mak, E. I. Papaioannou, C. R. Thompson and J. S. O. Evans, Nat. Chem., 2019, 11, 638–643 CrossRef CAS PubMed.
- C. Dueso, C. Thompson and I. Metcalfe, Appl. Energy, 2015, 157, 382–390 CrossRef CAS.
- D. Arifin and A. W. Weimer, Sol. Energy, 2018, 160, 178–185 CrossRef CAS.
- V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Appl. Catal., B, 2015, 164, 184–191 CrossRef CAS.
- B. J. Hare, D. Maiti, S. Ramani, A. E. Ramos, V. R. Bhethanabotla and J. N. Kuhn, Catal. Today, 2019, 323, 225–232 CrossRef CAS.
- M. Wenzel, L. Rihko-Struckmann and K. Sundmacher, Chem. Eng. J., 2018, 336, 278–296 CrossRef CAS.
- N. V. R. A. Dharanipragada, L. C. Buelens, H. Poelman, E. De Grave, V. V. Galvita and G. B. Marin, J. Mater. Chem. A, 2015, 3, 16251–16262 RSC.
- G. Wenbo, G. Jianping, W. Peikun, W. Qianru, C. Fei, P. Qijun, Z. Weijin, L. Lin and C. Ping, Nat. Energy, 2018, 3, 1067–1075 CrossRef.
- R. Michalsky, B. J. Parman, V. Amanor-Boadu and P. H. Pfromm, Energy, 2012, 42, 251–260 CrossRef CAS.
- Y. Wu, G. Jiang, H. Zhang, Z. Sun, Y. Gao, X. Chen, H. Liu, H. Tian, Q. Lai, M. Fan and D. Liu, Chem. Commun., 2017, 53, 10664–10667 RSC.
- Y. Gao, Y. Wu, Q. Zhang, X. Chen, G. Jiang and D. Liu, Int. J. Hydrogen Energy, 2018, 43, 16589–16597 CrossRef CAS.
- R. Michalsky, A. M. Avram, B. A. Peterson, P. H. Pfromm and A. A. Peterson, Chem. Sci., 2015, 6, 3965–3974 RSC.
- I. AlShibane, A. Daisley, J. S. J. Hargreaves, A. L. Hector, S. Laassiri, J. L. Rico and R. I. Smith, ACS Sustainable Chem. Eng., 2017, 5, 9214–9222 CrossRef CAS.
- S. Laassiri, C. D. Zeinalipour-Yazdi, C. R. A. Catlow and J. S. J. Hargreaves, Appl. Catal., B, 2018, 223, 60–66 CrossRef CAS.
- J. S. J. Hargreaves and D. McKay, J. Mol. Catal. A: Chem., 2009, 305, 125–129 CrossRef CAS.
- N. A. A. Rusman and M. Dahari, Int. J. Hydrogen Energy, 2016, 41, 12108–12126 CrossRef CAS.
- R. Michalsky, P. H. Pfromm and A. Steinfeld, Interface Focus, 2015, 5, 20140084 CrossRef PubMed.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- A. Darde, R. Prabhakar, J.-P. Tranier and N. Perrin, Energy Procedia, 2009, 1, 527–534 CrossRef CAS.
- R. L. Cornelissen and G. G. Hirs, Energy Convers. Manage., 1998, 39, 1821–1826 CrossRef CAS.
- L. M. Neal, V. P. Haribal and F. Li, iScience, 2019, 19, 894–904 CrossRef CAS PubMed.
- V. P. Haribal, Y. Chen, L. Neal and F. Li, Engineering, 2018, 4, 714–721 CrossRef CAS.
- V. P. Haribal, L. M. Neal and F. Li, Energy, 2017, 119, 1024–1035 CrossRef CAS.
- Z. Zhao, M. Uddi, N. Tsvetkov, B. Yildiz and A. F. Ghoniem, J. Phys. Chem. C, 2016, 120, 16271–16289 CrossRef CAS.
- N. Gokon, T. Suda and T. Kodama, Energy, 2015, 90, 1280–1289 CrossRef CAS.
- C. L. Muhich, B. D. Ehrhart, V. A. Witte, S. L. Miller, E. N. Coker, C. B. Musgrave and A. W. Weimer, Energy Environ. Sci., 2015, 8, 3687–3699 RSC.
- Z. Zhao, M. Uddi, N. Tsvetkov, B. Yildiz and A. F. Ghoniem, Phys. Chem. Chem. Phys., 2017, 19, 25774–25785 RSC.
- J. R. Scheffe and A. Steinfeld, Mater. Today, 2014, 17, 341–348 CrossRef CAS.
- C. L. Muhich, B. D. Ehrhart, I. Al-Shankitit, B. J. Ward, C. B. Musgrave and A. W. Weimer, Wiley Interdiscip. Rev.: Energy Environ., 2016, 5, 261–287 CAS.
- C. Agrafiotis, M. Roeb and C. Sattler, Renewable Sustainable Energy Rev., 2015, 42, 254–285 CrossRef CAS.
- C. Ruan, Z.-Q. Huang, J. Lin, L. Li, X. Liu, M. Tian, C. Huang, C.-R. Chang, J. Li and X. Wang, Energy Environ. Sci., 2019, 12, 767–779 RSC.
- L.-S. Fan, Chemical Looping Systems for Fossil Energy Conversions, Wiley, 2010 Search PubMed.
- Z. Sarshar, F. Kleitz and S. Kaliaguine, Energy Environ. Sci., 2011, 4, 4258–4269 RSC.
- F. Li, Z. Sun, S. Luo and L.-S. Fan, Energy Environ. Sci., 2011, 4, 876–880 RSC.
- F. Li, S. Luo, Z. Sun, X. Bao and L.-S. Fan, Energy Environ. Sci., 2011, 4, 3661–3667 RSC.
- S. Wang, G. Wang, F. Jiang, M. Luo and H. Li, Energy Environ. Sci., 2010, 3, 1353–1360 RSC.
- Q. Song, W. Liu, C. D. Bohn, R. N. Harper, E. Sivaniah, S. A. Scott and J. S. Dennis, Energy Environ. Sci., 2013, 6, 288–298 RSC.
- Y. Gao, F. Haeri, F. He and F. Li, ACS Catal., 2018, 8, 1757–1766 CrossRef CAS.
- D. Maiti, B. J. Hare, Y. A. Daza, A. E. Ramos, J. N. Kuhn and V. R. Bhethanabotla, Energy Environ. Sci., 2018, 11, 648–659 RSC.
- A. Mishra, T. Li, F. Li and E. E. Santiso, Chem. Mater., 2019, 31, 689–698 CrossRef CAS.
- A. Thursfield, A. Murugan, R. V. Franca and I. S. Metcalfe, Energy Environ. Sci., 2012, 5, 7421–7459 RSC.
- F. Donat, Y. Xu and C. R. Müller, Energy Technol., 2020 DOI:10.1002/ente.201900655.
- W. Benincosa, R. Siriwardane, H. Tian and J. Riley, Appl. Energy, 2017, 203, 522–534 CrossRef CAS.
- F. He, N. Galinsky and F. Li, Int. J. Hydrogen Energy, 2013, 38, 7839–7854 CrossRef CAS.
- I. Teknetzi, P. Nessi, V. Zaspalis and L. Nalbandian, Int. J. Hydrogen Energy, 2017, 42, 26231–26242 CrossRef CAS.
- C. Y. Lau, M. T. Dunstan, W. Hu, C. P. Grey and S. A. Scott, Energy Environ. Sci., 2017, 10, 818–831 RSC.
- A. H. Bork, E. Povoden-Karadeniz and J. L. M. Rupp, Adv. Energy Mater., 2017, 7, 1601086 CrossRef.
- M. Kubicek, A. H. Bork and J. L. M. Rupp, J. Mater. Chem. A, 2017, 5, 11983–12000 RSC.
- A. Shafiefarhood, J. Zhang, L. M. Neal and F. Li, J. Mater. Chem. A, 2017, 5, 11930–11939 RSC.
- X. P. Dai, R. J. Li, C. C. Yu and Z. P. Hao, J. Phys. Chem. B, 2006, 110, 22525–22531 CrossRef CAS PubMed.
- M. Machida, T. Kawada, H. Fujii and S. Hinokuma, J. Phys. Chem. C, 2015, 119, 24932–24941 CrossRef CAS.
- D. Zhang, T. Kawada, F. Yoshioka and M. Machida, ACS Omega, 2016, 1, 789–798 CrossRef CAS PubMed.
- M. Machida, M. Ueno, T. Omura, S. Kurusu, S. Hinokuma, T. Nanba, O. Shinozaki and H. Furutani, Ind. Eng. Chem. Res., 2017, 56, 3184–3193 CrossRef CAS.
- Z. Zhao, M. Uddi and A. F. Ghoniem, Combust. Flame, 2019, 207, 71–88 CrossRef CAS.
- Y. Zhang and H. Jiang, Chem. Commun., 2018, 54, 10343–10346 RSC.
- N. S. Yüzbasi, A. M. Kierzkowska, Q. Imtiaz, P. M. Abdala, A. Kurlov, J. L. M. Rupp and C. R. Müller, J. Phys. Chem. C, 2016, 120, 18977–18985 CrossRef.
- Q. Imtiaz, P. M. Abdala, A. M. Kierzkowska, W. van Beek, S. Schweiger, J. L. M. Rupp and C. R. Müller, Phys. Chem. Chem. Phys., 2016, 18, 12278–12288 RSC.
- Q. Imtiaz, A. Kurlov, J. L. M. Rupp and C. R. Müller, ChemSusChem, 2015, 8, 2055–2065 CrossRef CAS PubMed.
- J. W. Bennett, X. Huang, Y. Fang, D. M. Cwiertny, V. H. Grassian and S. E. Mason, J. Phys. Chem. C, 2019, 123, 6450–6463 CrossRef CAS.
- N. S. Yüzbasi, P. M. Abdala, Q. Imtiaz, S. M. Kim, A. M. Kierzkowska, A. Armutlulu, W. van Beek and C. R. Müller, Phys. Chem. Chem. Phys., 2018, 20, 12736–12745 RSC.
- K. J. Warren and J. R. Scheffe, J. Phys. Chem. C, 2019, 123, 13208–13218 CrossRef CAS.
- T. Hatanaka and Y. Yukihiro, Powder Technol., 2019, 356, 974–979 CrossRef CAS.
- Y. De Vos, M. Jacobs, P. Van Der Voort, I. Van Driessche, F. Snijkers and A. Verberckmoes, Chem. Eng. J., 2017, 309, 824–839 CrossRef CAS.
- K. Li, X. Cheng, N. Li, X. Zhu, Y. Wei, K. Zhai and H. Wang, J. Mater. Chem. A, 2017, 5, 24232–24246 RSC.
- H. Bahzad, N. Shah, N. M. Dowell, M. Boot-Handford, S. M. Soltani, M. Ho and P. S. Fennell, Int. J. Hydrogen Energy, 2019, 44, 21251–21263 CrossRef CAS.
- K. Aasberg-Petersen, I. Dybkjær, C. V. Ovesen, N. C. Schjødt, J. Sehested and S. G. Thomsen, J. Nat. Gas Sci. Eng., 2011, 3, 423–459 CrossRef CAS.
- A. I. O. Suarez, Á. Szécsényi, E. J. M. Hensen, J. Ruiz-Martínez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef.
- S. Arora and R. Prasad, RSC Adv., 2016, 6, 108668–108688 RSC.
- B. Abdullah, N. A. Abd Ghani and D.-V. N. Vo, J. Cleaner Prod., 2017, 162, 170–185 CrossRef CAS.
- S. M. Kim, P. M. Abdala, T. Margossian, D. Hosseini, L. Foppa, A. Armutlulu, W. van Beek, A. Comas-Vives, C. Copéret and C. Müller, J. Am. Chem. Soc., 2017, 139, 1937–1949 CrossRef CAS PubMed.
- B. C. Enger, R. Lødeng and A. Holmen, Appl. Catal., A, 2008, 346, 1–27 CrossRef CAS.
- V. P. Haribal, X. Wang, R. Dudek, C. Paulus, B. Turk, R. Gupta and F. Li, Adv. Energy Mater., 2019, 1901963 CrossRef CAS.
- R. K. Lyon and J. A. Cole, Combust. Flame, 2000, 121, 249–261 CrossRef CAS.
- S. Bhavsar and G. Veser, RSC Adv., 2014, 4, 47254–47267 RSC.
- D. Li, R. Xu, Z. Gu, X. Zhu, S. Qing and K. Li, Energy Technol., 2019 DOI:10.1002/ente.201900925.
- H. C. Mantripragada and G. Veser, Energy Technol., 2020 DOI:10.1002/ente.201900580.
- D. R. Barcellos, M. D. Sanders, J. Tong, A. H. McDaniel and R. P. O’Hayre, Energy Environ. Sci., 2018, 11, 3256–3265 RSC.
- D. Hosseini, F. Donat, S. M. Kim, L. Bernard, A. M. Kierzkowska and C. R. Müller, ACS Appl. Energy Mater., 2018, 1, 1294–1303 CrossRef CAS.
- X. Zhu, K. Li, Y. Wei, H. Wang and L. Sun, Energy Fuels, 2014, 28, 754–760 CrossRef CAS.
- B. J. Hare, D. Maiti, Y. A. Daza, V. R. Bhethanabotla and J. N. Kuhn, ACS Catal., 2018, 8, 3021–3029 CrossRef CAS.
- M. Wenzel, N. V. R. Aditya Dharanipragada, V. V. Galvita, H. Poelman, G. B. Marin, L. Rihko-Struckmann and K. Sundmacher, J. CO2 Util., 2017, 17, 60–68 CrossRef CAS.
- S. Bock, R. Zacharias and V. Hacker, RSC Adv., 2019, 9, 23686–23695 RSC.
- S. M. Kim, P. M. Abdala, M. Broda, D. Hosseini, C. Copéret and C. Müller, ACS Catal., 2018, 8, 2815–2823 CrossRef CAS.
- J. Hu, V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Appl. Catal., B, 2019, 247, 86–99 CrossRef CAS.
- D. Kang, M. Lee, H. S. Lim and J. W. Lee, Fuel, 2018, 215, 787–798 CrossRef CAS.
- A. Farooqui, A. Bose, D. Ferrero, J. Llorca and M. Santarelli, J. CO2 Util., 2018, 27, 500–517 CrossRef CAS.
- Y. Chen, X. Zhu, K. Li, Y. Wei, Y. Zheng and H. Wang, ACS Sustainable Chem. Eng., 2019, 7, 15452–15462 CrossRef CAS.
- A. Farooqui, F. Di Tomaso, A. Bose, D. Ferrero, J. Llorca and M. Santarelli, Energy Convers. Manage., 2019, 186, 200–219 CrossRef CAS.
- A. Mishra, N. Galinsky, F. He, E. E. Santiso and F. Li, Catal. Sci. Technol., 2016, 6, 4535–4544 RSC.
- O. Vozniuk, N. Tanchoux, J. M. M. Millet, S. Albonetti, F. Di Renzo and F. Cavani, Spinel Mixed Oxides for Chemical-Loop Reforming: From Solid State to Potential Application, 2019 Search PubMed.
- T. Li, R. S. Jayathilake, D. D. Taylor and E. E. Rodriguez, Chem. Commun., 2019, 55, 4929–4932 RSC.
- D. Zeng, Y. Qiu, S. Peng, C. Chen, J. Zeng, S. Zhang and R. Xiao, J. Mater. Chem. A, 2018, 6, 11306–11316 RSC.
- M. Guo, Z. Cheng, Y. Liu, L. Qin, J. Goetze, J. A. Fan and L.-S. Fan, Catal. Today, 2019 DOI:10.1016/j.cattod.2019.06.016.
- Y. Kang, M. Tian, C. Huang, J. Lin, B. Hou, X. Pan, L. Li, A. I. Rykov, J. Wang and X. Wang, ACS Catal., 2019, 9, 8373–8382 CrossRef CAS.
- A. Antzara, E. Heracleous, L. Silvester, D. B. Bukur and A. A. Lemonidou, Catal. Today, 2016, 272, 32–41 CrossRef CAS.
- M. Johansson, T. Mattisson, A. Lyngfelt and A. Abad, Fuel, 2008, 87, 988–1001 CrossRef CAS.
- A. More, C. J. Hansen and G. Veser, Catal. Today, 2017, 298, 21–32 CrossRef CAS.
- A. More, S. Bhavsar and G. Veser, Energy Technol., 2016, 4, 1147–1157 CrossRef CAS.
- S.-K. Otto, K. Kousi, D. Neagu, L. Bekris, J. Janek and I. Metcalfe, ACS Appl. Energy Mater., 2019, 2, 7288–7298 CrossRef CAS.
- J. Huang, W. Liu, Y. Yang and B. Liu, ACS Catal., 2018, 8, 1748–1756 CrossRef CAS.
- D. Hosseini, F. Donat, P. M. Abdala, S. M. Kim, A. M. Kierzkowska and C. R. Müller, ACS Appl. Mater. Interfaces, 2019, 11, 18276–18284 CrossRef CAS PubMed.
- H. Tian, T. Simonyi, J. Poston and R. Siriwardane, Ind. Eng. Chem. Res., 2009, 48, 8418–8430 CrossRef CAS.
- A. R. Oller, M. Costa and G. Oberdöster, Toxicol. Appl. Pharmacol., 1997, 143, 152–166 CrossRef CAS PubMed.
- R. D. Solunke and G. T. Veser, Ind. Eng. Chem. Res., 2010, 49, 11037–11044 CrossRef CAS.
- S. K. Wilke and D. C. Dunand, Acta Mater., 2019, 162, 90–102 CrossRef CAS.
- K. Zhao, F. He, Z. Huang, G. Wei, A. Zheng, H. Li and Z. Zhao, Appl. Energy, 2016, 168, 193–203 CrossRef CAS.
- F. He, X. Li, K. Zhao, Z. Huang, G. Wei and H. Li, Fuel, 2013, 108, 465–473 CrossRef CAS.
- M. S. S. Khine, L. Chen, S. Zhang, J. Lin and S. P. Jiang, Int. J. Hydrogen Energy, 2013, 38, 13300–13308 CrossRef CAS.
- Y. Saito, F. Kosaka, N. Kikuchi, H. Hatano and J. Otomo, Ind. Eng. Chem. Res., 2018, 57, 5529–5538 CrossRef CAS.
- X.-Y. Wu and A. F. Ghoniem, Prog. Energy Combust. Sci., 2019, 74, 1–30 CrossRef.
- N. L. Galinsky, Y. Huang, A. Shafiefarhood and F. Li, ACS Sustainable Chem. Eng., 2013, 1, 364–373 CrossRef CAS.
- W. C. Cho, J. K. Lee, G. D. Nam, C. H. Kim, H.-S. Cho and J. H. Joo, Appl. Energy, 2019, 239, 644–657 CrossRef CAS.
- N. L. Galinsky, A. Shafiefarhood, Y. G. Chen, L. Neal and F. X. Li, Appl. Catal., B, 2015, 164, 371–379 CrossRef CAS.
- S. Ma, S. Chen, M. Zhu, Z. Zhao, J. Hu, M. Wu, S. Toan and W. Xiang, Int. J. Hydrogen Energy, 2019, 44, 6491–6504 CrossRef CAS.
- A. Shafiefarhood, J. C. Hamill, L. M. Neal and F. Li, Phys. Chem. Chem. Phys., 2015, 17, 31297–31307 RSC.
- M. S. Liao and Q. E. Zhang, J. Mol. Catal. A: Chem., 1998, 136, 185–194 CrossRef CAS.
- Q. Imtiaz, N. S. Yuzbasi, P. M. Abdala, A. M. Kierzkowska, W. van Beek, M. Broda and C. R. Muller, J. Mater. Chem. A, 2016, 4, 113–123 RSC.
- R. Palcheva, U. Olsbye, M. Palcut, P. Rauwel, G. Tyuliev, N. Velinov and H. H. Fjellvåg, Appl. Surf. Sci., 2015, 357, 45–54 CrossRef CAS.
- H. Li, K. Li, H. Wang, X. Zhu, Y. Wei, D. Yan, X. Cheng and K. Zhai, Appl. Surf. Sci., 2016, 390, 513–525 CrossRef CAS.
- J. L. G. Fierro, Catal. Lett., 1993, 22, 67–91 CrossRef CAS.
- S. Arndt, G. Laugel, S. Levchenko, R. Horn, M. Baerns, M. Scheffler, R. Schlogl and R. Schomacker, Catal. Rev., 2011, 53, 424–514 CrossRef CAS.
- Y. Amenomiya, V. I. Birss, M. Goledzinowski, J. Galuszka and A. R. Sanger, Catal. Rev., 1990, 32, 163–227 CrossRef CAS.
- E. V. Kondratenko and B. Manfred, Oxidative Coupling of Methane, Handbook of Heterogeneous Catalysis, 2008, DOI:10.1002/9783527610044.hetcat0152.
- K. Tabata, Y. Teng, T. Takemoto, E. Suzuki, M. A. Bañares, M. A. Peña and J. L. G. Fierro, Catal. Rev., 2002, 44, 1–58 CrossRef CAS.
- A. M. Gaffney, C. A. Jones, J. J. Leonard and J. A. Sofranko, J. Catal., 1988, 114, 422–432 CrossRef CAS.
- G. E. Keller and M. M. Bhasin, Chem. Informationsdienst, 1982, 73, 9–19 CrossRef CAS.
- J. A. Labinger, Catal. Lett., 1988, 1, 371–375 CrossRef CAS.
- J. S. Lee and S. T. Oyama, Catal. Rev., 1988, 30, 249–280 CrossRef CAS.
- Y. S. Su, J. Y. Ying and W. H. Green Jr, J. Catal., 2003, 218, 321–333 CrossRef CAS.
- A. E. Bishop, K. Sekiya, M. J. Salahuddin, J. H. Steel, M. Hedges, T. Domoto, R. Fischer-Colbrie, H. Winkler, M. A. Ghatei and S. R. Bloom, ChemCatChem, 2011, 3, 1935–1947 CrossRef.
- E. V. Kondratenko and M. Baerns, Catalysis of Oxidative Methane Conversions, 2011, ch. 3, pp. 35–55 Search PubMed.
- S. Parishan, P. Littlewood, A. Arinchtein, V. Fleischer and R. Schomäcker, Catal. Today, 2018, 311, 40–47 CrossRef CAS.
- A. Cruellas, T. Melchiori, F. Gallucci and M. V. S. Annaland, Catal. Rev.: Sci. Eng., 2017, 59, 234–294 CrossRef CAS.
- E. V. Kondratenko and M. Baerns, Focus on Catalysts, 2012, 8 Search PubMed.
- B. Beck, V. Fleischer, S. Arndt, M. G. Hevia, A. Urakawa, P. Hugo and R. Schomäcker, Catal. Today, 2014, 228, 212–218 CrossRef CAS.
- I. Kim, G. Lee, H. Bin Na, J. M. Ha and J. C. Jung, Mol. Catal., 2017, 435, 13–23 CrossRef CAS.
- V. Fleischer, R. Steuer, S. Parishan and R. Schomacker, J. Catal., 2016, 341, 91–103 CrossRef CAS.
- V. Fleischer, P. Littlewood, S. Parishan and R. Schomäcker, Chem. Eng. J., 2016, 306, 646–654 CrossRef CAS.
- Z. C. Jiang, H. Gong and S. B. Li, Stud. Surf. Sci. Catal., 1997, 112, 481–490 CrossRef CAS.
- S. Pak, P. Qiu and J. H. Lunsford, J. Catal., 1998, 179, 222–230 CrossRef CAS.
- V. Fleischer, P. Littlewood, S. Parishan and R. Schomacker, Chem. Eng. J., 2016, 306, 646–654 CrossRef CAS.
- E. Y. Chung, W. K. Wang, S. G. Nadgouda, D. S. Baser, J. A. Sofranko and L. S. Fan, Ind. Eng. Chem. Res., 2016, 55, 12750–12764 CrossRef CAS.
- V. Fleischer, U. Simon, S. Parishan, M. G. Colmenares, O. Gorke, A. Gurlo, W. Riedel, L. Thum, J. Schmidt, T. Risse, K. P. Dinse and R. Schomacker, J. Catal., 2018, 360, 102–117 CrossRef CAS.
- Z. Cheng, D. S. Baser, S. G. Nadgouda, L. Qin, J. A. Fan and L.-S. Fan, ACS Energy Lett., 2018, 3, 1730–1736 CrossRef CAS.
- S. Parishan, P. Littlewood, A. Arinchtein, V. Fleischer and R. Schomacker, Catal. Today, 2018, 311, 40–47 CrossRef CAS.
- A. Malekzadeh, A. Khodadadi, M. Abedini, M. Amini, A. Bahramian and A. K. Dalai, Catal. Commun., 2001, 2, 241–247 CrossRef CAS.
- L. Wang, L. Tao, M. Xie, G. Xu, J. Huang and Y. Xu, Catal. Lett., 1993, 21, 35–41 CrossRef CAS.
- Z. R. Ismagilov, E. V. Matus and L. T. Tsikoza, Energy Environ. Sci., 2008, 1, 526–541 RSC.
- J. J. Spivey and H. Graham, Chem. Soc. Rev., 2014, 43, 792–803 RSC.
- C. Brady, B. M. Murphy and B. Xu, ACS Catal., 2017, 7, 3924–3928 CrossRef CAS.
- J. Gao, Y. Zheng, J.-M. Jehng, Y. Tang, I. E. Wachs and S. G. Podkolzin, Science, 2015, 348, 686–690 CrossRef CAS PubMed.
- R. W. Borry, E. C. Lu, Y. H. Kim and E. Iglesia, Natural Gas Conversion V, 1998, vol. 119, pp. 403–410 Search PubMed.
- A. A. Latimer, A. Kakekhani, A. R. Kulkarni and J. K. Nørskov, ACS Catal., 2018, 8, 6894–6907 CrossRef CAS.
- T. Lunkenbein, F. Girgsdies, T. Kandemir, N. Thomas, M. Behrens, R. Schlögl and E. Frei, Angew. Chem., Int. Ed., 2016, 128, 12900–12904 CrossRef.
- M. Ravi and M. Ranocchiari, Angew. Chem. Int. Ed., 2017, 56, 16464 CrossRef CAS PubMed.
- A. C. Rosenzweig, C. A. Frederick, S. J. Lippard and P. Nordlund, Nature, 1993, 366, 537–543 CrossRef CAS PubMed.
- K. T. Dinh, M. M. Sullivan, P. Serna, R. J. Meyer, M. Dinca and Y. Roman-Leshkov, ACS Catal., 2018, 8, 8306–8313 CrossRef CAS.
- A. R. Kulkarni, Z. J. Zhao, S. Siahrostami, J. K. Norskov and F. Studt, Catal. Sci. Technol., 2018, 8, 114–123 RSC.
- B. Ipek and R. F. Lobo, Chem. Commun., 2016, 52, 13401–13404 RSC.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- J.-P. Lange, V. L. Sushkevich, A. J. Knorpp and J. A. van Bokhoven, Ind. Eng. Chem. Res., 2019, 58, 8674–8680 CAS.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 358, eaan5970 CrossRef PubMed.
- L. Artiglia, V. L. Sushkevich, D. Palagin, A. J. Knorpp, K. Roy and J. A. van Bokhoven, ACS Catal., 2019, 9, 6728–6737 CrossRef CAS.
- K. Narsimhan, K. Iyoki, K. Dinh and Y. Roman-Leshkov, ACS Cent. Sci., 2016, 2, 424–429 CrossRef CAS PubMed.
- P. Atkins, T. Overton and J. Rourke, Shriver and Atkins' Inorganic Chemistry, Oxford University Press, 2009 Search PubMed.
- L. Neal, V. Haribal, J. McCaig, H. H. Lamb and F. Li, J. Adv. Manuf. Proc., 2019, 1, e10015 CrossRef.
- F. Cavani and F. Trifiro, Catal. Today, 1995, 24, 307–313 CrossRef CAS.
- M. A. Banares, Catal. Today, 1999, 51, 319–348 CrossRef CAS.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- O. O. James, S. Mandal, N. Alele, B. Chowdhury and S. Maity, Fuel Process. Technol., 2016, 149, 239–255 CrossRef CAS.
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS.
- C. A. Gartner, A. C. van Veen and J. A. Lercher, ChemCatChem, 2013, 5, 3196–3217 CrossRef.
- S. Agouram, A. Dejoz, F. Ivars, I. Vázquez, J. M. López Nieto and B. Solsona, Fuel Process. Technol., 2014, 119, 105–113 CrossRef CAS.
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS.
- US Pat., 2017/0313637 A1, 2015.
- Y. Gao, L. Neal, D. Ding, W. Wu, C. Baroi, A. M. Gaffney and F. Li, ACS Catal., 2019, 8592–8621 CrossRef CAS.
- J. G. Tsikoyiannis, D. L. Stern and R. K. Grasselli, J. Catal., 1999, 184, 77–86 CrossRef CAS.
- R. K. Grasselli, D. L. Stern and J. G. Tsikoyiannis, in Studies in Surface Science and Catalysis, ed. A. Corma, F. V. Melo, S. Mendioroz and J. L. G. Fierro, Elsevier, 2000, vol. 130, pp. 773–778 Search PubMed.
- R. K. Grasselli, D. L. Stern and J. G. Tsikoyiannis, Calcif. Tissue Int., 1999, 189, 1–7 CAS.
- D. Creaser, B. Andersson, R. R. Hudgins and P. L. Silveston, J. Catal., 1999, 182, 264–269 CrossRef CAS.
- E. A. D. Graaf, G. Zwanenburg, G. Rothenberg and A. Bliek, Org. Process Res. Dev., 2005, 9, 397–403 CrossRef.
- L. M. van der Zande, E. A. de Graaf and G. Rothenberg, Adv. Synth. Catal., 2002, 344, 884–889 CrossRef CAS.
- E. A. de Graaf, A. Andreini, E. J. M. Hensen and A. Bliek, Appl. Catal., A, 2004, 262, 201–206 CrossRef CAS.
- J. Beckers and G. Rothenberg, Green Chem., 2009, 11, 1550 RSC.
- C.-H. Lin, K.-C. Lee and B.-Z. Wan, Appl. Catal., A, 1997, 164, 59–67 CrossRef CAS.
- L. Late, W. Thelin and E. A. Blekkan, Appl. Catal., A, 2004, 262, 63–68 CrossRef CAS.
- M. S. C. Chan, H. G. Baldovi and J. S. Dennis, Catal. Sci. Technol., 2018, 8, 887–897 RSC.
- J. Beckers, A. F. Lee and G. Rothenberg, Adv. Synth. Catal., 2009, 351, 1557–1566 CrossRef CAS.
- J. Beckers and G. Rothenberg, Dalton Trans., 2009, 5673–5682 RSC.
- J. H. Blank, J. Beckers, P. F. Collignon and G. Rothenberg, ChemPhysChem, 2007, 8, 2490–2497 CrossRef CAS PubMed.
- G. Rothenberg, E. A. de Graaf and A. Bliek, Angew. Chem., Int. Ed., 2003, 42, 3366–3368 CrossRef CAS PubMed.
- J. H. Blank, J. Beckers, P. F. Collignon, F. Clerc and G. Rothenberg, Chemistry, 2007, 13, 5121–5128 CrossRef CAS PubMed.
- E. A. D. Graaf, G. Rothenberg, P. J. Kooyman, A. Andreini and A. Bliek, Appl. Catal., A, 2005, 278, 187–194 CrossRef.
- P. Novotny, S. Yusuf, F. X. Li and H. H. Lamb, Catal. Today, 2018, 317, 50–55 CrossRef CAS.
- S. Yusuf, V. Haribal, D. Jackson, L. Neal and F. Li, Appl. Catal., B, 2019, 257, 117885 CrossRef CAS.
- S. Yusuf, L. Neal, Z. Bao, Z. Wu and F. Li, ACS Catal., 2019, 9, 3174–3186 CrossRef CAS.
- C. Zhao and J. A. Lercher, ChemCatChem, 2012, 4, 64–68 CrossRef CAS.
- H. Saito, H. Seki, Y. Hosono, T. Higo, J. G. Seo, S. Maeda, K. Hashimoto, S. Ogo and Y. Sekine, J. Phys. Chem. C, 2019, 123, 26272–26281 CrossRef CAS.
- P. Mars and D. W. V. Krevelen, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef CAS.
- R. Grabowski, Catal. Rev., 2006, 48, 199–268 CrossRef CAS.
- R. Schlöl, Angew. Chem. Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- D. Arne, S. C. Reinhard and A. T. Bell, Phys. Chem. Chem. Phys., 2009, 11, 6119–6124 RSC.
- N. Ballarini, A. Battisti, F. Cavani, A. Cericola, C. Cortelli, M. Ferrari, F. Trifirò and P. Arpentinier, Appl. Catal., A, 2006, 307, 148–155 CrossRef CAS.
- D. Creaser, B. Andersson, R. R. Hudgins and P. L. Silveston, Chem. Eng. Sci., 1999, 54, 4437–4448 CrossRef CAS.
- O. Rubio, J. Herguido and M. Menéndez, Chem. Eng. Sci., 2003, 58, 4619–4627 CrossRef CAS.
- I. A. Bakare, S. A. Mohamed, S. Al-Ghamdi, S. A. Razzak, M. M. Hossain and H. I. de Lasa, Chem. Eng. J., 2015, 278, 207–216 CrossRef CAS.
- S. N. Khadzhiev, N. Y. Usachev, I. M. Gerzeliev, V. P. Kalinin, V. V. Kharlamov, E. P. Belanova, A. V. Kazakov, S. A. Kanaev and T. S. Starostina, Pet. Chem., 2015, 55, 640–644 CrossRef CAS.
- S. Al-Ghamdi, M. Volpe, M. M. Hossain and H. de Lasa, Appl. Catal., A, 2013, 450, 120–130 CrossRef CAS.
- A. Qiao, V. N. Kalevaru, J. Radnik, A. Düvel, P. Heitjans, A. S. H. Kumar, P. S. S. Prasad, N. Lingaiah and A. Martin, Ind. Eng. Chem. Res., 2014, 53, 18711–18721 CrossRef CAS.
- A. H. Elbadawi, M. S. Ba-Shammakh, S. Al-Ghamdi, S. A. Razzak, M. M. Hossain and H. I. de Lasa, Chem. Eng. Sci., 2016, 145, 59–70 CrossRef CAS.
- A. H. Elbadawi, M. S. Ba-Shammakh, S. Al-Ghamdi, S. A. Razzak and M. M. Hossain, Chem. Eng. J., 2016, 284, 448–457 CrossRef CAS.
- N. Ballarini, F. Cavani, M. Ferrari, R. Catani and U. Cornaro, J. Catal., 2003, 213, 95–102 CrossRef CAS.
- N. Ballarini, F. Cavani, A. Cericola, C. Cortelli, M. Ferrari, F. Trifirò, R. Catani and U. Cornaro, Stud. Surf. Sci. Catal., 2004, 147, 649–654 CrossRef CAS.
- N. Ballarini, F. Cavani, A. Cericola, C. Cortelli, M. Ferrari, F. Trifirò, G. Capannelli, A. Comite, R. Catani and U. Cornaro, Catal. Today, 2004, 91, 99–104 CrossRef.
- K. Fukudome, N. O. Ikenaga, T. Miyake and T. Suzuki, Catal. Sci. Technol., 2011, 1, 987–998 RSC.
- D. Delgado, B. Solsona, A. Ykrelef, A. Rodríguez-Gómez, A. Caballero, E. Rodríguez-Aguado, E. Rodríguez-Castellón and J. M. López Nieto, J. Phys. Chem. C, 2017, 121, 25132–25142 CrossRef CAS.
- Z. Zhang, J. Ding, R. Chai, G. Zhao, Y. Liu and Y. Lu, Appl. Catal., A, 2018, 550, 151–159 CrossRef CAS.
- R. Koirala, R. Buechel, S. E. Pratsinis and A. Baiker, Appl. Catal., A, 2016, 527, 96–108 CrossRef CAS.
- Z. Li, A. W. Peters, A. E. Platero-Prats, J. Liu, C. W. Kung, H. Noh, M. R. DeStefano, N. M. Schweitzer, K. W. Chapman, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2017, 139, 15251–15258 CrossRef CAS.
- S. A. Al-Ghamdi, M. M. Hossain and H. I. de Lasa, Ind. Eng. Chem. Res., 2013, 52, 5235–5244 CrossRef CAS.
- M. Y. Khan, S. Al-Ghamdi, S. A. Razzak, M. M. Hossain and H. de Lasa, Mol. Catal., 2017, 443, 78–91 CrossRef CAS.
- A. H. Elbadawi, M. S. Ba-Shammakh, S. Al-Ghamdi, S. A. Razzak, M. M. Hossain and H. I. de Lasa, Chem. Eng. Res. Des., 2017, 117, 733–745 CrossRef CAS.
- J. Moreira, S. Al-Ghamdi and H. D. Lasa, Ind. Eng. Chem. Res., 2014, 53, 15317–15332 CrossRef.
- A. H. I. d. L. Samira Rostom, Ind. Eng. Chem. Res., 2017, 56, 13109–13124 CrossRef.
- M. M. Hossain, Ind. Eng. Chem. Res., 2017, 56, 4309–4318 CrossRef CAS.
- S. N. Khadzhiev, N. Y. Usachev, I. M. Gerzeliev, E. P. Belanova, V. P. Kalinin, V. V. Kharlamov, A. V. Kazakov, S. A. Kanaev, T. S. Starostina and A. Y. Popov, Pet. Chem., 2015, 55, 651–654 CrossRef CAS.
- I. M. Gerzeliev, A. Y. Popov and V. A. Ostroumova, Pet. Chem., 2016, 56, 724–729 CrossRef CAS.
- N. Y. Usachev, I. M. Gerzeliev, V. V. Kharlamov, V. P. Kalinin, E. P. Belanova, S. A. Kanaev, A. V. Kazakov and T. S. Starostina, Pet. Chem., 2016, 56, 841–845 CrossRef CAS.
- N. Y. Usachev, I. M. Gerzeliev, E. P. Belanova, A. V. Kazakov, V. P. Kalinin, V. V. Kharlamov, S. A. Kanaev and T. S. Starostina, Pet. Chem., 2016, 56, 846–851 CrossRef CAS.
- B. M. Weckhuysen and R. A. Schoonheydt, Catal. Today, 1999, 51, 223–232 CrossRef CAS.
- K. H. Kang, T. H. Kim, W. C. Choi, Y. K. Park, U. G. Hong, D. S. Park, C. J. Kim and I. K. Song, Catal. Commun., 2015, 72, 68–72 CrossRef CAS.
- T. H. Kim, M. Y. Gim, J. H. Song, W. C. Choi, Y.-K. Park, U. G. Hong, D. S. Park and I. K. Song, Catal. Commun., 2017, 97, 37–41 CrossRef CAS.
- S. Sim, S. Gong, J. Bae, Y.-K. Park, J. Kim, W. C. Choi, U. G. Hong, D. S. Park, I. K. Song, H. Seo, N. Y. Kang and S. Park, Mol. Catal., 2017, 436, 164–173 CrossRef CAS.
- T. H. Kim, K. H. Kang, M. Baek, J. H. Song, U. G. Hong, D. S. Park, W. C. Choi, Y. K. Park and I. K. Song, Mol. Catal., 2017, 433, 1–7 CrossRef CAS.
- S. Chen, L. Zeng, R. Mu, C. Xiong, Z.-J. Zhao, C. Zhao, C. Pei, L. Peng, J. Luo, L.-S. Fan and J. Gong, J. Am. Chem. Soc., 2019, 141, 18653–18657 CrossRef CAS PubMed.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, ACS Catal., 2014, 4, 3357–3380 CrossRef CAS.
- C. H. Bartholomew and R. J. Farrauto, Catalytic Oxidations of Inorganic and Organic Compounds, John Wiley & Sons, Inc., 2010 Search PubMed.
- J. R. Monnier, Appl. Catal., A, 2001, 221, 73–91 CrossRef CAS.
- S. J. Khatib and S. T. Oyama, Catal. Rev., 2015, 57, 306–344 CrossRef CAS.
- A. Chongterdtoonskul, J. W. Schwank and S. Chavadej, J. Mol. Catal. A: Chem., 2013, 372, 175–182 CrossRef CAS.
- M. O. Ozbek and R. A. van Santen, Catal. Lett., 2013, 143, 131–141 CrossRef.
- R. M. Lambert, F. J. Williams, R. L. Cropley and A. Palermo, J. Mol. Catal. A: Chem., 2005, 228, 27–33 CrossRef CAS.
- C. J. Bertole and C. A. Mims, J. Catal., 1999, 184, 224–235 CrossRef CAS.
- A. Chongterdtoonskul, J. W. Schwank and S. Chavadej, J. Mol. Catal. A: Chem., 2012, 358, 58–66 CrossRef CAS.
- P. T. Connor, S. Kovenklioglu and D. C. Shelly, Appl. Catal., 1991, 71, 247–263 CrossRef CAS.
- S. S. Hassani, M. R. Ghasemi, M. Rashidzadeh and Z. Sobat, Cryst. Res. Technol., 2010, 44, 948–952 CrossRef.
- A. Chongterdtoonskul, J. W. Schwank and S. Chavadej, Catal. Lett., 2012, 142, 991–1002 CrossRef CAS.
- L. Zhu, W. Zhang, J. Q. Zhu and D. J. Cheng, Appl. Catal., A, 2017, 538, 27–36 CrossRef CAS.
- E. J. Marek, S. Gabra, J. S. Dennis and S. A. Scott, Appl. Catal., B, 2020, 262, 118216 CrossRef CAS.
- G. L. Montrasi, G. R. Tauszik, M. Solari and G. Leofanti, Appl. Catal., 1983, 5, 359–369 CrossRef CAS.
- G. Boskovic, N. Dropka, D. Wolf, A. Bruckner and M. Baerns, J. Catal., 2004, 226, 334–342 CrossRef CAS.
- G. Boskovic, D. Wolf, A. Brückner and M. Baerns, J. Catal., 2004, 224, 187–196 CrossRef CAS.
- Y. Pang, X. Chen, C. Xu, Y. Lei and K. Wei, ChemCatChem, 2014, 6, 876–884 CrossRef CAS.
- T. Chukeaw, A. Seubsai, P. Phon-in, K. Charoen, T. Witoon, W. Donphai, P. Parpainainar, M. Chareonpanich, D. Noon, B. Zohour and S. Senkan, RSC Adv., 2016, 6, 56116–56126 RSC.
- A. Seubsai, M. Kahn, B. Zohour, D. Noon, M. Charoenpanich and S. Senkan, Ind. Eng. Chem. Res., 2015, 54, 2638–2645 CrossRef CAS.
- M. P. Dudukovic, Science, 2009, 325, 698–701 CrossRef CAS PubMed.
- G. S. Patience and R. E. Bockrath, Appl. Catal., A, 2010, 376, 4–12 CrossRef CAS.
- R. Contractor, R. J. Dry, C. White, Q. M. Mao, S. Konstantinidis and O. E. Potter, Powder Technol., 2000, 111, 132–144 CrossRef CAS.
- R. M. Contractor, Chem. Eng. Sci., 1999, 54, 5627–5632 CrossRef CAS.
- H. G. Lintz and A. Reitzmann, Catal. Rev., 2007, 49, 1–32 CrossRef CAS.
- R. R. Hudgins, P. L. Silveston, C.-Y. Li and A. A. Adesina, in Periodic Operation of Chemical Reactors, ed. P. L. Silveston and R. R. Hudgins, Butterworth-Heinemann, Oxford, 2013, pp. 79–122 Search PubMed.
- J. G. Yates and P. Lettieri, Catalytic Processes, 2016, vol. 26, pp. 23–65 Search PubMed.
- A. Godefroy, G. S. Patience, R. Cenni and J.-L. Dubois, Chem. Eng. Sci., 2010, 65, 261–266 CrossRef CAS.
- G. S. Patience and R. E. Bockrath, Appl. Catal., A, 2010, 376, 4–12 CrossRef CAS.
- M. G. Rosmaninho, F. C. C. Moura, L. R. Souza, R. K. Nogueira, G. M. Gomes, J. S. Nascimento, M. C. Pereira, J. D. Fabris, J. D. Ardisson, M. S. Nazzarro, K. Sapag, M. H. Araujo and R. M. Lago, Appl. Catal., B, 2012, 115, 45–52 CrossRef.
- E. Hormilleja, P. Duran, J. Plou, J. Herguido and J. A. Pena, Int. J. Hydrogen Energy, 2014, 39, 5267–5273 CrossRef CAS.
- R. Campo, P. Duran, J. Plou, J. Herguido and J. A. Pena, J. Power Sources, 2013, 242, 520–526 CrossRef CAS.
- C. Trevisanut, M. Mari, J. M. M. Millet and F. Cavani, Int. J. Hydrogen Energy, 2015, 40, 5264–5271 CrossRef CAS.
- J. V. Ochoa, C. Trevisanut, J. M. M. Millet, G. Busca and F. Cavani, J. Phys. Chem. C, 2013, 117, 23908–23918 CrossRef CAS.
- C. Trevisanut, F. Bosselet, F. Cavania and J. M. M. Millet, Catal. Sci. Technol., 2015, 5, 1280–1289 RSC.
- O. Vozniuk, S. Agnoli, L. Artiglia, A. Vassoi, N. Tanchoux, F. Di Renzo, G. Granozzi and F. Cavani, Green Chem., 2016, 18, 1038–1050 RSC.
- N. V. R. A. Dharanipragada, V. V. Galvita, H. Poelman, L. C. Buelens, C. Detavernier and G. B. Marin, Appl. Catal., B, 2018, 222, 59–72 CrossRef CAS.
- C. Wang, B. L. Dou, B. Jiang, Y. C. Song, B. G. Du, C. Zhang, K. Q. Wang, H. S. Chen and Y. J. Xu, Int. J. Hydrogen Energy, 2015, 40, 7037–7044 CrossRef CAS.
- P. Pimenidou, G. Rickett, V. Dupont and M. V. Twigg, Bioresource Technol., 2010, 101, 6389–6397 CrossRef CAS PubMed.
- A. Hoteit, A. Forret, W. Pelletant, J. Roesler and T. Gauthier, Oil Gas Sci. Technol., 2011, 66, 193–199 CrossRef CAS.
- P. Moldenhauer, M. Ryden, T. Mattisson and A. Lyngfelt, Int. J. Greenhouse Gas Control, 2012, 9, 1–9 CrossRef CAS.
- T. Mendiara, J. M. Johansen, R. Utrilla, P. Geraldo, A. D. Jensen and P. Glarborg, Fuel, 2011, 90, 1049–1060 CrossRef CAS.
- M. Keller, H. Leion, T. Mattisson and H. Thunman, Energy Fuels, 2014, 28, 3833–3840 CrossRef CAS.
- M. Keller, D. P. Anderson, H. Leion and T. Mattisson, Appl. Catal., A, 2018, 550, 105–112 CrossRef CAS.
- Z. Q. Wang, M. Zhu, T. He, J. Z. Zhang, J. L. Wu, H. J. Tian and J. H. Wu, Fuel, 2018, 222, 375–384 CrossRef CAS.
- Y. Sun, E. Jiang, X. Xu, J. Wang and Z. Li, ACS Sustainable Chem. Eng., 2018, 6, 14660–14668 CrossRef CAS.
- Q. Hu, Y. Shen, J. W. Chew, T. Ge and C.-H. Wang, Chem. Eng. J., 2020, 379, 122346 CrossRef CAS.
- T. Shen, H. Ge and L. Shen, Int. J. Greenhouse Gas Control, 2018, 75, 63–73 CrossRef CAS.
- Z. Sun, X. Wu, C. K. Russell, M. D. Dyar, E. C. Sklute, S. Toan, M. Fan, L. Duan and W. Xiang, J. Mater. Chem. A, 2019, 7, 1216–1226 RSC.
- J. S. Dennis and S. A. Scott, Fuel, 2010, 89, 1623–1640 CrossRef CAS.
- M. Keller, H. Leion, T. Mattisson and A. Lyngfelt, Combust. Flame, 2011, 158, 393–400 CrossRef CAS.
- Q. Imtiaz, D. Hosseini and C. R. Müller, Energy Technol., 2013, 1, 633–647 CrossRef CAS.
- G. Luo, X. Wang, M. R. Zachariah and R. Mishra, J. Phys. Chem. C, 2019, 123, 17644–17649 CrossRef CAS.
- P. Wang, N. Means, B. H. Howard, D. Shekhawat and D. Berry, Fuel, 2018, 217, 642–649 CrossRef CAS.
- M. Jafarian, M. Arjomandi and G. J. Nathan, Chem. Eng. Res. Des., 2017, 120, 69–81 CrossRef CAS.
- X. Ao, H. Wang and Y. Wei, Energy Convers. Manage., 2008, 49, 2063–2068 CrossRef CAS.
- M. M. Sarafraz, M. Jafarian, M. Arjomandi and G. J. Nathan, Int. J. Hydrogen Energy, 2017, 42, 16396–16407 CrossRef CAS.
- M. M. Sarafraz, M. Jafarian, M. Arjomandi and G. J. Nathan, Appl. Energy, 2017, 195, 702–712 CrossRef CAS.
- M. M. Sarafraz, M. Jafarian, M. Arjomandi and G. J. Nathan, Int. J. Hydrogen Energy, 2018, 43, 4195–4210 CrossRef CAS.
- D. C. Upham, Z. R. Snodgrass, M. T. Zavareh, T. B. McConnaughy, M. J. Gordon, H. Metiu and E. W. McFarland, Chem. Eng. Sci., 2017, 160, 245–253 CrossRef CAS.
- F. Li, L. Zeng and L.-S. Fan, Fuel, 2010, 89, 3773–3784 CrossRef CAS.
- J. S. D. a. A. N. Hayhurst, Chem. Eng. Sci., 1990, 45, 1175–1187 CrossRef.
- J. A. H. Oates, Gaseous Effluents, Wiley-VCH Verlag GmbH, 2007 Search PubMed.
- E. S. Rubin, S. Yeh, D. A. Hounshell and M. R. Taylor, Institute of Transportation Studies Working Paper, 2007, vol. 2, pp. 52–69.
- J. R. Hufton, S. G. Mayorga and S. Sircar, AIChE J., 1999, 45, 248–256 CrossRef CAS.
- D. P. Harrison, Ind. Eng. Chem. Res., 2008, 47, 6486–6501 CrossRef CAS.
- B. Marcin, M. Vasilije, I. Qasim, A. M. Kierzkowska, E. J. Anthony and C. R. Müller, Environ. Sci. Technol., 2013, 47, 6007–6014 CrossRef PubMed.
- D. P. Hanak, S. Michalski and V. Manovic, Energy Convers. Manage., 2018, 177, 428–452 CrossRef CAS.
- C. Han and D. P. Harrison, Chem. Eng. Sci., 1994, 49, 5875–5883 CrossRef CAS.
- B. Balasubramanian, A. Lopez Ortiz, S. Kaytakoglu and D. P. Harrison, Chem. Eng. Sci., 1999, 54, 3543–3552 CrossRef CAS.
- S. M. Kim, A. Armutlulu, A. Kierzkoska, D. Hosseini, F. Donat and C. R. Müller, Sustainable Energy Fuels, 2019 10.1039/C9SE00619B.
- A. Wang, N. Deshpande and L. S. Fan, Energy Fuels, 2015, 29, 321–330 CrossRef CAS.
- D. P. Connell, D. A. Lewandowski, S. Ramkumar, N. Phalak, R. M. Statnick and L.-S. Fan, Fuel, 2013, 105, 383–396 CrossRef CAS.
- N. Phalak, W. Wang and L.-S. Fan, Chem. Eng. Technol., 2013, 36, 1451–1459 CrossRef CAS.
- W. Peng, Z. Xu and H. Zhao, Fuel, 2016, 170, 226–234 CrossRef CAS.
- D. P. Hanak, E. J. Anthony and V. Manovic, Energy Environ. Sci., 2015, 8, 2199–2249 RSC.
- B. Duhoux, P. Mehrani, D. Y. Lu, R. T. Symonds, E. J. Anthony and A. Macchi, Energy Technol., 2016, 4, 1158–1170 CrossRef CAS.
- L. Zhou, L. Duan and E. J. Anthony, Appl. Energy, 2019, 235, 480–486 CrossRef CAS.
- M. Samari, F. Ridha, V. Manovic, A. Macchi and E. J. Anthony, Mitigation and Adaptation Strategies for Global Change, 2019 Search PubMed.
- F. Donat and C. R. Müller, Chem. Eng. J., 2018, 336, 544–549 CrossRef CAS.
- B. Arias, G. S. Grasa, M. Alonso and J. C. Abanades, Energy Environ. Sci., 2012, 5, 7353–7359 RSC.
- C. C. Dean, J. Blamey, N. H. Florin, M. J. Al-Jeboori and P. S. Fennell, Chem. Eng, Res. Des., 2011, 89, 836–855 CrossRef CAS.
- R. V. Kumar, R. K. Lyon and J. A. Cole, in Advances in Hydrogen Energy, ed. C. E. Grégoire Padró and F. Lau, Springer US, Boston, MA, 2002, pp. 31–45 Search PubMed.
- J. Kremer, A. Galloy, J. Ströhle and B. Epple, Chem. Eng. Technol., 2013, 36, 1518–1524 CrossRef CAS.
- B. Arias, M. E. Diego, J. C. Abanades, M. Lorenzo, L. Diaz, D. Martínez, J. Alvarez and A. Sánchez-Biezma, Int. J. Greenhouse Gas Control, 2013, 18, 237–245 CrossRef CAS.
- M.-H. Chang, W.-C. Chen, C.-M. Huang, W.-H. Liu, Y.-C. Chou, W.-C. Chang, W. Chen, J.-Y. Cheng, K.-E. Huang and H.-W. Hsu, Energy Procedia, 2014, 63, 2100–2108 CrossRef CAS.
- J. R. Fernández, J. M. Alarcón and J. C. Abanades, Catal. Today, 2019, 333, 176–181 CrossRef.
- I. Martínez, J. R. Fernández, M. Martini, F. Gallucci, M. van Sint Annaland, M. C. Romano and J. C. Abanades, Int. J. Greenhouse Gas Control, 2019, 85, 71–85 CrossRef.
- J. Chen, L. Duan, F. Donat, C. R. Müller, E. J. Anthony and M. Fan, Chem. Eng. J., 2018, 351, 1038–1046 CrossRef CAS.
- J. R. Fernández and J. C. Abanades, Curr. Opin. Chem. Eng., 2017, 17, 1–8 CrossRef.
- J. M. Alarcón and J. R. Fernández, Chem. Eng. Sci., 2015, 137, 254–267 CrossRef.
- F. N. Ridha, D. Lu, A. Macchi and R. W. Hughes, Fuel, 2015, 153, 202–209 CrossRef CAS.
- V. V. Galvita, H. Poelman and G. B. Marin, Top. Catal., 2011, 54, 907–913 CrossRef CAS.
- V. V. Galvita, H. Poelman and G. B. Marin, J. Power Sources, 2015, 286, 362–370 CrossRef CAS.
- L. C. Buelens, V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Science, 2016, 354, 449–452 CrossRef CAS PubMed.
- J. Chen, L. Duan, T. Shi, R. Bian, Y. Lu, F. Donat and E. J. Anthony, J. Mater. Chem. A, 2019, 7, 21096–21105 RSC.
- M. Vasilije and E. J. Anthony, Environ. Sci. Technol., 2011, 45, 10750–10756 CrossRef PubMed.
- K. Wang, P. T. Clough, P. Zhao and E. J. Anthony, J. Mater. Chem. A, 2019, 7, 9173–9182 RSC.
- F. N. Ridha, V. Manovic, A. Macchi, M. A. Anthony and E. J. Anthony, Fuel Process. Technol., 2013, 116, 284–291 CrossRef CAS.
- K. Wang, F. Gu, P. T. Clough, P. Zhao and E. J. Anthony, Energy Fuels, 2019, 33, 7550–7560 CrossRef CAS.
- X. Ma, Y. Li, L. Duan, E. Anthony and H. Liu, Appl. Energy, 2018, 225, 402–412 CrossRef CAS.
- L. Díez-Martín, J. M. López, J. R. Fernández, I. Martínez, G. Grasa and R. Murillo, Chem. Eng. J., 2018, 350, 1010–1021 CrossRef.
- J. C. Abanades, R. Murillo, J. R. Fernandez, G. Grasa and I. Martínez, Environ. Sci. Technol., 2010, 44, 6901–6904 CrossRef CAS PubMed.
- J. R. Fernández and J. C. Abanades, Chem. Eng. Sci., 2017, 166, 144–160 CrossRef.
- I. Martínez, M. C. Romano, J. R. Fernández, P. Chiesa, R. Murillo and J. C. Abanades, Appl. Energy, 2014, 114, 192–208 CrossRef.
- J. R. Fernández, J. C. Abanades, R. Murillo and G. Grasa, Int. J. Greenhouse Gas Control, 2012, 6, 126–141 CrossRef.
- M. Martini, A. van den Berg, F. Gallucci and M. van Sint Annaland, Chem. Eng. J., 2016, 303, 73–88 CrossRef CAS.
- L. Tan, C. Qin, Z. Zhang, J. Ran and V. Manovic, Energy Technol., 2018 DOI:10.1002/ente.201700894.
- C. Qin, B. Feng, J. Yin, J. Ran, Z. Li and V. Manovic, Chem. Eng. J., 2015, 262, 665–675 CrossRef CAS.
- L. Diez-Martin, G. S. Grasa, R. Murillo, A. Scullard and G. Williams, Ind. Eng. Chem. Res., 2018, 57, 2890–2904 CrossRef CAS.
- A. L. García-Lario, M. Aznar, I. Martinez, G. S. Grasa and R. Murillo, Int. J. Hydrogen Energy, 2015, 40, 219–232 CrossRef.
- V. Manovic and E. J. Anthony, Energy Fuels, 2011, 25, 4846–4853 CrossRef CAS.
- V. Manovic, Y. Wu, I. He and E. J. Anthony, Ind. Eng. Chem. Res., 2011, 50, 12384–12391 CrossRef CAS.
- A. M. Kierzkowska and C. R. Müller, Energy Environ. Sci., 2012, 5, 6061–6065 RSC.
- A. M. Kierzkowska and C. R. Müller, ChemPlusChem, 2013, 78, 92–100 CrossRef CAS.
- C. S. Martavaltzi and A. A. Lemonidou, Chem. Eng. Sci., 2010, 65, 4134–4140 CrossRef CAS.
- M. R. Cesário, B. S. Barros, C. Courson, D. M. A. Melo and A. Kiennemann, Fuel Process. Technol., 2015, 131, 247–253 CrossRef.
- A. L. García-Lario, G. S. Grasa and R. Murillo, Chem. Eng. J., 2015, 264, 697–705 CrossRef.
- M. V. Navarro, J. M. López, T. García, G. Grasa and R. Murillo, J. Power Sources, 2017, 363, 117–125 CrossRef CAS.
- G. Grasa, M. V. Navarro, J. M. López, L. Díez-Martín, J. R. Fernández and R. Murillo, Chem. Eng. J., 2017, 324, 266–278 CrossRef CAS.
- P. S. Fennell, R. Pacciani, J. S. Dennis, J. F. Davidson and A. N. Hayhurst, Energy Fuels, 2007, 21, 2072–2081 CrossRef CAS.
- J. Wang and E. J. Anthony, Ind. Eng. Chem. Res., 2005, 44, 627–629 CrossRef CAS.
- J. C. Abanades and D. Alvarez, Energy Fuels, 2003, 17, 308–315 CrossRef CAS.
- C. Qin, J. Yin, W. Liu, H. An and B. Feng, Ind. Eng. Chem. Res., 2012, 51, 12274–12281 CAS.
- S. S. Kazi, A. Aranda, L. D. Felice, J. Meyer, R. Murillo and G. Grasa, Energy Procedia, 2017, 114, 211–219 CrossRef CAS.
- M. A. Naeem, A. Armutlulu, Q. Imtiaz, F. Donat, R. Schäublin, A. Kierzkowska and C. R. Müller, Nat. Commun., 2018, 9, 2408 CrossRef PubMed.
- C. Dang, Y. Li, S. M. Yusuf, Y. Cao, H. Wang, H. Yu, F. Peng and F. Li, Energy Environ. Sci., 2018, 11, 660–668 RSC.
- O. Kwon, S. Sengodan, K. Kim, G. Kim, H. Y. Jeong, J. Shin, Y.-W. Ju, J. W. Han and G. Kim, Nat. Commun., 2017, 8, 15967 CrossRef CAS PubMed.
- J. Y. Do, N. Son, N.-K. Park, B. S. Kwak, J.-I. Baek, H.-J. Ryu and M. Kang, Appl. Energy, 2018, 219, 138–150 CrossRef CAS.
- J. Hu, V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Appl. Catal., B, 2018, 231, 123–136 CrossRef CAS.
- D. Hosseini, P. M. Abdala, F. Donat, S. M. Kim and C. R. Müller, Appl. Catal., B, 2019, 258, 117946 CrossRef CAS.
- https://www.iea.org/etp/tracking2017/chemicalsandpetrochemicals/ .
- T. Shimamura, K. Okumura, K. Nakagawa, T. Ando, N. O. Ikenga and T. Suzuki, J. Mol. Catal. A: Chem., 2004, 211, 97–102 CrossRef CAS.
- A. P. V. Soares, M. F. Portela and A. Kiennemann, Catal. Rev., 2005, 47, 125–174 CrossRef CAS.
- C. Brookes, P. P. Wells, N. Dimitratos, W. Jones, E. K. Gibson, D. J. Morgan, G. Cibin, C. Nicklin, D. Mora-Fonz, D. O. Scanlon, C. R. A. Catlow and M. Bowker, J. Phys. Chem. C, 2014, 118, 26155–26161 CrossRef CAS.
- P. Forzatti, E. Tronconi, G. Busca and P. Tittarelli, Catal. Today, 1987, 1, 209–218 CrossRef CAS.
- M. Ai, J. Catal., 1982, 77, 279–288 CrossRef CAS.
- V. V. Kaichev, Y. A. Chesalov, A. A. Saraev, A. Y. Klyushin, A. Knop-Gericke, T. V. Andrushkevich and V. I. Bukhtiyarov, J. Catal., 2016, 338, 82–93 CrossRef CAS.
- V. R. Chumbhale and P. A. Awasarkar, Appl. Catal., A, 2001, 205, 109–115 CrossRef CAS.
- A. Kaddouri, C. Mazzocchia and E. Tempesti, Appl. Catal., A, 1999, 180, 271–275 CrossRef CAS.
- M. D. Allen, S. Poulston, E. G. Bithell, M. J. Goringe and M. Bowker, J. Catal., 1996, 163, 204–214 CrossRef CAS.
- C. Zhao and I. E. Wachs, J. Phys. Chem. C, 2008, 112, 11363–11372 CrossRef CAS.
- A. Chetouani, B. Taouk and E. Bordes-Richard, Catal. Today, 2004, 91–92, 73–77 CrossRef CAS.
- W. E. Campbell, E. L. McDaniel, W. H. Reece, J. E. Williams and H. S. Young, Product R&D, 1970, 9, 325–334 Search PubMed.
- J. M. López Nieto, P. Concepción, A. Dejoz, H. Knözinger, F. Melo and M. I. Vázquez, J. Catal., 2000, 189, 147–157 CrossRef.
- P. M. Michalakos, M. C. Kung, I. Jahan and H. Kung, J. Catal., 1993, 140, 226–242 CrossRef CAS.
- J. Schulze and M. Homann, C4-Hydrocarbons and Derivatives: Resources, Production, Marketing, Springer Berlin Heidelberg, Berlin, Heidelberg, 1989, pp. 5–8 Search PubMed.
- J. M. Herrmann, J. Disdier, F. G. Freire and M. F. Portela, J. Chem. Soc., Faraday Trans., 1995, 91, 2343–2348 RSC.
- N. C. Ramani, D. L. Sullivan, J. G. Ekerdt, J.-M. Jehng and I. E. Wachs, J. Catal., 1998, 176, 143–154 CrossRef CAS.
- X. Xia, R. Jin, Y. He, J.-F. Deng and H. Li, Appl. Surf. Sci., 2000, 165, 255–259 CrossRef CAS.
- W. Zhang, G. Innocenti, P. Oulego, V. Gitis, H. Wu, B. Ensing, F. Cavani, G. Rothenberg and N. R. Shiju, ACS Catal., 2018, 8, 2365–2374 CrossRef CAS PubMed.
- P. F. Zhang, H. F. Lu, Y. Zhou, L. Zhang, Z. L. Wu, S. Z. Yang, H. L. Shi, Q. L. Zhu, Y. F. Chen and S. Dai, Nat. Commun., 2015, 6, 10 Search PubMed.
- X. K. Yang, L. S. Yang, W. Fan and H. F. Lin, Catal. Today, 2016, 269, 56–64 CrossRef CAS.
- K. T. V. Rao, J. L. Rogers, S. Souzanchi, L. Dessbesell, M. B. Ray and C. Xu, ChemSusChem, 2018, 11, 3323–3334 CrossRef CAS PubMed.
- A. Bielański and M. Najbar, Appl. Catal., A, 1997, 157, 223–261 CrossRef.
- H. K. Matralis, C. Papadopoulou, C. Kordulis, A. Aguilar Elguezabal and V. Cortes Corberan, Appl. Catal., A, 1995, 126, 365–380 CrossRef CAS.
- T. Li, F. Liu, Y. Tang, L. Li, S. Miao, Y. Su, J. Zhang, J. Huang, H. Sun, M. Haruta, A. Wang, B. Qiao, J. Li and T. Zhang, Angew. Chem. Int. Ed., 2018, 57, 7795–7799 CrossRef CAS PubMed.
- M. Biswal, V. V. Dhas, V. R. Mate, A. Banerjee, P. Pachfule, K. L. Agrawal, S. B. Ogale and C. V. Rode, J. Phys. Chem. C, 2011, 115, 15440–15448 CrossRef CAS.
- S. S. Negi, A. T. Venugopalan, T. Raja, A. P. Singh and C. S. Gopinath, RSC Adv., 2014, 4, 57087–57097 RSC.
- Y. I. Pyatnitskii and G. I. Golodets, React. Kinet. Catal. Lett., 1976, 5, 345–351 CrossRef CAS.
- A. F. D'Alessandro and A. Farkas, J. Colloid Sci., 1956, 11, 653–670 CrossRef.
- S. Biswas, B. Dutta, K. Mullick, C. H. Kuo, A. S. Poyraz and S. L. Suib, ACS Catal., 2015, 5, 4394–4403 CrossRef CAS.
- J.-C. Lin, J. Chen, S. L. Suib, M. B. Cutlip and J. D. Freihaut, J. Catal., 1996, 161, 659–666 CrossRef CAS.
- Y. Lao, N. Zhu, X. Jiang, J. Zhao, Q. Dai and X. Wang, Catal. Sci. Technol., 2018, 8, 4797–4811 RSC.
- J. Chen, D. Yan, Z. Xu, X. Chen, X. Chen, W. Xu, H. Jia and J. Chen, Environ. Sci. Technol., 2018, 52, 4728–4737 CrossRef CAS PubMed.
- X. Wang, W. Zhao, X. Wu, T. Zhang, Y. Liu, K. Zhang, Y. Xiao and L. Jiang, Appl. Surf. Sci., 2017, 426, 1198–1205 CrossRef CAS.
- X. Weng, W. L. Wang, Q. Meng and Z. Wu, Catal. Sci. Technol., 2018, 8, 4364–4372 RSC.
- S. Zhao, K. Li, S. Jiang and J. Li, Appl. Catal., B, 2016, 181, 236–248 CrossRef CAS.
- J. Perez-Ramirez and E. V. Kondratenko, J. Catal., 2007, 250, 240–246 CrossRef CAS.
- F. Zhang, X. Zhang, G. Jiang, N. Li, Z. Hao and S. Qu, Chem. Eng. J., 2018, 348, 831–839 CrossRef CAS.
- S. Yasyerli, G. Dogu, I. Ar and T. Dogu, Chem. Eng. Sci., 2004, 59, 4001–4009 CrossRef CAS.
Footnote |
| † QI and FD contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2020 |