
High-purity pyrrole-type FeN4 sites as a superior oxygen reduction electrocatalyst†
Nan
Zhang‡
a,
Tianpei
Zhou‡
b,
Minglong
Chen‡
c,
Hu
Feng
b,
Ruilin
Yuan
b,
Cheng’an
Zhong
b,
Wensheng
Yan
a,
Yangchao
Tian
a,
Xiaojun
Wu
 c,
Wangsheng
Chu
a,
Changzheng
Wu
*b and
Yi
Xie
b
c,
Wangsheng
Chu
a,
Changzheng
Wu
*b and
Yi
Xie
b
aNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, Anhui 230029, P. R. China
bHefei National Laboratory for Physical Science at the Microscale, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), and CAS Key Laboratory of Mechanical Behavior and Design of Materials, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: czwu@ustc.edu.cn
cCAS Key Laboratory of Materials for Energy Conversion and Department of Material Science and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
First published on 23rd November 2019
Abstract
Atomically dispersed iron–nitrogen (FeN4) catalysts have emerged as the most promising alternative to costly Pt-based counterparts in proton exchange membrane fuel cells (PEMFCs), but often they suffer from high overpotential and poor stability due to the diverse iron–nitrogen coordination structure. Herein, we demonstrate high-purity pyrrole-type FeN4 sites for the first time, as a superior ORR electrocatalyst for PEMFCs. The high-purity pyrrole-type FeN4 catalyst exhibited extremely outstanding ORR activity with an ultra-high active area current density of 6.89 mA m−2 in acid medium, which exceeds that of most reported metal–nitrogen coordination catalysts. Experimental and theoretical analyses reveal that high-purity pyrrole-type coordination significantly modifies the atomic and electronic structures of FeN4 sites, bringing with it high intrinsic catalytic activity, preferable O2 adsorption energy and full four-electron reaction selectivity for ORR catalysis. Therefore, PEMFCs built with this high-purity FeN4 catalyst achieve a high open-circuit voltage (1.01 V) and a large peak power density (over 700 mW cm−2). High-purity iron–nitrogen coordination would give new insights into highly efficient electrocatalysts for PEMFCs.
Broader contextProton exchange membrane fuel cells (PEMFCs) have been targeted as promising clean power candidates with the merits of large power density, environmental compatibility and high efficiency. As the key reaction catalysts of PEMFCs, commercial Pt-based catalysts account for more than 26% of the total cell cost and the scarcity of Pt largely impedes their widespread commercialization. Recently, iron–nitrogen (FeN4) coordination sites have emerged as the most promising non-noble catalysts for PEMFCs, but often they suffer from high overpotential and instability. At the heart of the performance-determining parameters, a high-purity FeN4 structure has always been a researcher's dream since then. However, current methodologies for iron–nitrogen coordination sites usually require high-temperature pyrolysis, which inevitably leads to diverse structures of iron–nitrogen coordination. In this work, we report a high-purity pyrrole-type FeN4 structure as a superior cathode material for PEMFCs and unveil its structure–activity relationship at the atomic scale. Benefiting from the regulated atomic and electronic structure, the high-purity pyrrole-type FeN4 catalyst exhibited an extremely outstanding catalytic activity in a PEMFC system and exceeds most reported metal–nitrogen coordination catalysts. This work will open up a new avenue to design advanced electrocatalysts for PEMFCs. |
Proton exchange membrane fuel cells (PEMFCs) are considered to be a clean power candidate due to their high energy conversion efficiency, large power density and environmentally friendly features.1–3 As a key reaction of PEMFCs, the oxygen reduction reaction (ORR) at the cathode electrode suffers greatly from sluggish reaction kinetics and multiple proton-coupled electron transfer processes, which largely impedes the widespread commercialization of PEMFCs.4–6 Recently, iron–nitrogen coordination sites (FeN4) have emerged as the most promising non-noble catalysts for PEMFCs because of their encouraging ORR activity in acidic media.7–9 However, the FeN4 catalysts tend to be protonated or attacked by free radicals at the active sites, leading to high overpotential and insufficient stability of ORR catalysis.10,11 Therefore, development of highly active FeN4 sites with low overpotential and superior stability for PEMFCs is actively being pursued.
Over the past few years, much effort has been devoted to optimizing the FeN4 configurations at the atomic scale, such as increasing the density of iron–nitrogen sites,12,13 fabricating dual metal sites,14,15 anion doping16,17 and coordination number regulations.18,19 Despite the excellent results, most research studies indicate that the properties are derived from the synergy of multiple coordination forms.6,20 It is still a great challenge to figure out which coordination form is decisive for the performance.21,22 In this regard, developing high-purity FeN4 sites with defined structure is extremely important for understanding the structure–activity relationship at the atomic scale. In effect, theoretical calculations already gave us an inspiration that pyrrolic N can not only regulate the O2 adsorption energy of bonded Fe atoms but also activate the neighbouring C atoms as active sites for the ORR.23,24 However, the current synthesis of pyrrole nitrogen coordinated FeN4 structures remains a challenge, which seriously hampers the commercial application of FeN4 catalysts. Therefore, realizing high-purity pyrrole-type FeN4 structures has become a vital issue for designing advanced electrocatalysts for PEMFCs.
Herein, we highlight a high-purity pyrrole-type FeN4 structure for the first time, as the cathode material for PEMFCs. By removing additional carbon atoms with the assistance of ammonia gas, high-purity pyrrole-type FeN4 sites could be successfully achieved via chemical configuration transformation from pyridine nitrogen to pyrrole nitrogen. Benefiting from the regulated atomic and electronic structures, high-purity pyrrole-type FeN4 sites exhibit high intrinsic catalytic activity, preferable O2 adsorption energy and full four-electron reaction selectivity. As expected, the exclusive high-purity FeN4 structure as a reaction site boosts the ORR catalytic reaction, achieving a high open-circuit voltage and a large peak power density in PEMFCs. High-purity FeN4 sites will open up a new avenue for the design of highly-efficient electrocatalyts for PEMFC systems.
In this work, high-purity pyrrole-type FeN4 sites (denoted as HP-FeN4) were derived from the pyrolysis of a polyaniline precursor. By removing additional carbon atoms with the assistance of ammonia, the chemical configuration of pyridine nitrogen was transformed to pyrrole nitrogen and a pyrrole-type FeN4 structure was obtained (Fig. 1a). High-resolution transmission electron microscopy (Fig. 1b) shows highly disordered carbon structures with randomly oriented graphitic domains of HP-FeN4 and no obvious nanoparticles were found. Meanwhile, only two broad carbon diffraction peaks of (002) and (100) at 26° and 44° were observed in the X-ray diffraction (XRD) pattern of HP-FeN4 (Fig. S3, ESI†),25 which was similar to the NC material without Fe doping. These results suggest that no Fe-based compound existed in the HP-FeN4 catalyst. The Fe species were highly dispersed as tiny clusters or single sites on the graphene framework. The uniform dispersion of Fe atoms was confirmed by aberration-corrected high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) as shown in Fig. 1c. Bright spots corresponding to the atomically dispersed Fe sites were homogeneously distributed across the carbon framework. The associated energy-dispersive spectroscopy (EDS) mapping shows the uniform distribution of Fe, N and C atoms (Fig. 1d). For comparison, the characterization of traditional FeN4 catalysts synthesized under an Ar atmosphere, which are denoted as FeN4, is presented in Fig. S1–S5 of the ESI.† The traditional FeN4 catalyst exhibited similar features to HP-FeN4 in both carbon structure and element distribution. Therefore, this ammonia-assisted strategy keeps the morphology and elemental dispersion of the traditional FeN4 material and Fe atoms are uniformly dispersed as single sites or clusters in the HP-FeN4 material.
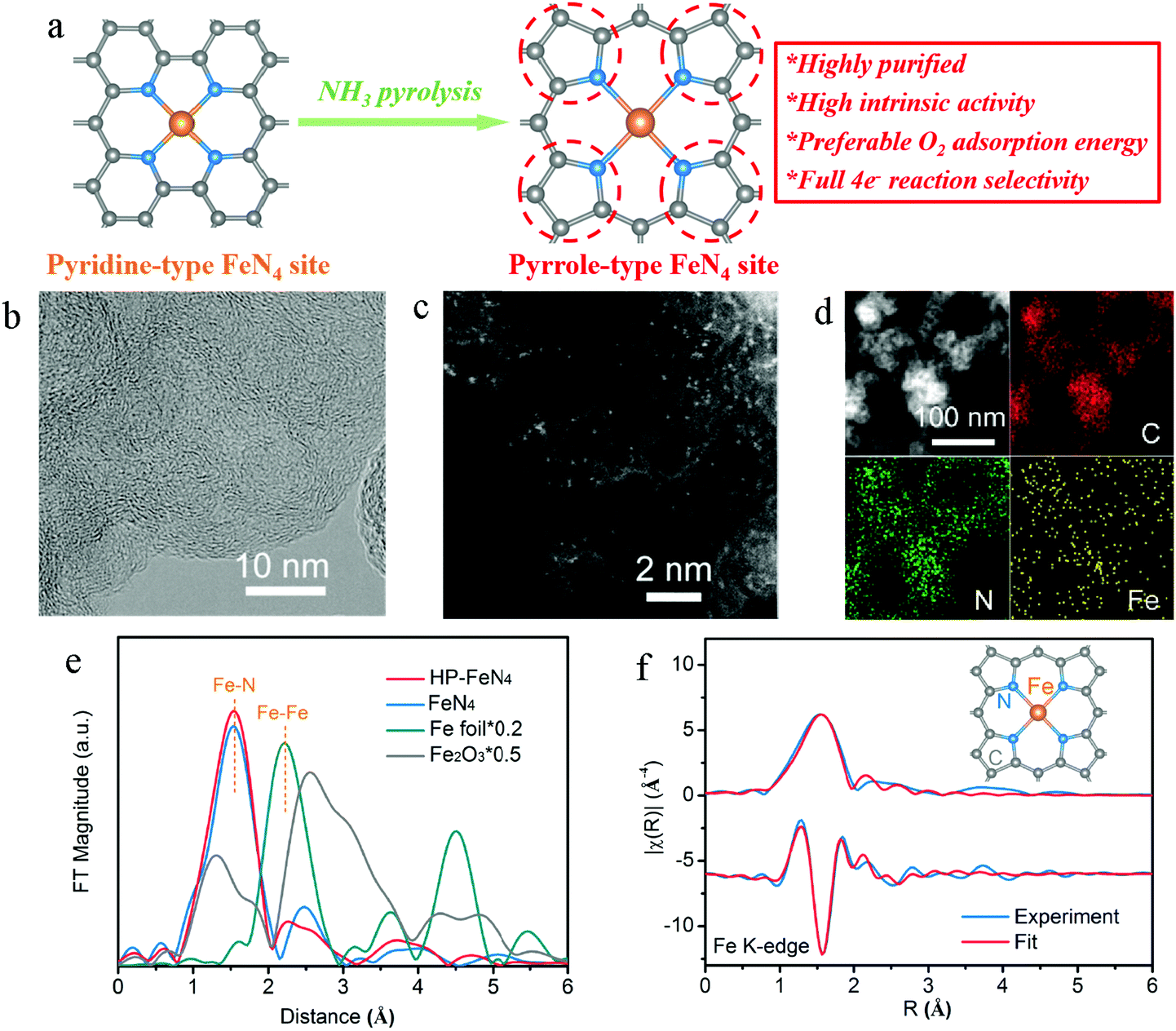 | ||
| Fig. 1 High-purity FeN4 sites. (a) Preparation process of high-purity pyrrole-type FeN4 structure. The balls in grey, blue and orange represent C, N and Fe atoms, respectively. (b) HRTEM and (c) HAADF-STEM image of HP-FeN4 material. (d) EDS elemental mapping profiles of HP-FeN4. (e) Fourier transformations of the EXAFS spectra for Fe foil, Fe2O3, HP-FeN4 and FeN4. (f) First-shell fitting of Fourier transformations of EXAFS spectra for HP-FeN4. Top and bottom spectra are magnitude and imaginary part, respectively. Inset: The structure of the iron site in the HP-FeN4 material. | ||
Synchrotron X-ray absorption spectroscopy (XAS) measurements were carried out to determine the electronic structure and local geometry around implanted Fe atoms. Fig. S6 (ESI†) shows Fe K-edge X-ray absorption near edge structure (XANES) spectra of HP-FeN4 catalyst and standard compounds. HP-FeN4 shows Fe K-edge absorption energy between Fe foil and Fe2O3, which demonstrates that the Fe atoms exhibited a positive valence state below +3. Besides, both HP-FeN4 and FeN4 show clear pre-edge peaks at 7113.5 eV, which are similar to the characteristics of Hemin, suggesting the typical FeN4 configurations.26Fig. 1e displays the Fourier transform of extended X-ray absorption fine structure (FT-EXAFS) spectra. The Fe–Fe interactions of Fe foil were not observed in the FT-EXAFS of HP-FeN4, which confirms the HAADF-STEM results and verifies the existence of atomically dispersed Fe sites on the graphene framework. The main peaks at approximately 1.55 Å can be ascribed to the single scattering of photoelectrons excited by an X-ray from Fe atoms to the nearest neighbored N atoms i.e., the Fe–N shell. Fig. 1f shows the best fit of the FT-EXAFS of HP-FeN4 compared with the experimental one. The fitting result shows that Fe was bonded by four nitrogen atoms with Fe–N bonds, suggesting the Fe–N4 coordination structure of this catalyst. Moreover, traditional FeN4 exhibits a similar local geometric structure as that of HP-FeN4. And the fitting parameters of both the samples are summarized in Fig. S7 and Table S1 (ESI†). In this regard, the ammonia-assisted strategy has little effect on the bond length and coordination number of iron–nitrogen sites. And Fe atoms of the HP-FeN4 material are basically in a coordination form with the Fe–N4 structure.
In order to eliminate the influence of carbon structure on material properties, Brunauer–Emmett–Teller (BET) characterization was performed to analyze the surface area and pore size distribution of the as-prepared catalysts. As shown in Fig. S10 (ESI†), HP-FeN4 and FeN4 exhibit BET specific surface areas of 609.9 and 527.7 m2 g−1, respectively. N2 sorption isotherms of both the HP-FeN4 and FeN4 catalysts possess a pronounced hysteresis loop, and can be identified as type-IV isotherms, suggesting the existence of a mesoporous structure in the carbon framework.27 Moreover, Barrett–Joyner–Halenda (BJH) pore size distribution curves derived from the adsorption branch confirm that these two catalysts were rich in mesopores with a diameter of 3–7 nm (Fig. S11, ESI†). Raman spectroscopy was also used to characterize the carbon structure of the two catalysts. As shown in Fig. S12 (ESI†), both HP-FeN4 and FeN4 show broad D and G peaks, revealing the disordered carbon structures. The ID/IG ratios of HP-FeN4 and FeN4 are 1.025 and 1.027, suggesting the similar structural defects of the two catalysts. Specifically, HP-FeN4 exhibits similar C 1s spectra compared to FeN4, which further reveals that HP-FeN4 and FeN4 exhibit similar surface oxidation degree (Fig. S13, ESI†). The graphitization degree and hierarchical pore structure were insensitive to the ammonia-assisted strategy. And the adjacent structure of FeN4 sites would play a key role in affecting the ORR performance. Moreover, a large number of mesopores in HP-FeN4 will promote the electrolyte wetting of the physical surface to increase the portion of electrochemically available active sites.28,29 Thus, the kinetic accessibility to active sites could be largely improved for the ORR process, which increased the utilization of active sites.
Iron–nitrogen sites of HP-FeN4 were confirmed to be high-purity as pyrrole-type coordination by virtue of Soft X-ray absorption spectrocopy (sXAS) based on the synchrotron radation facility, which is sensitive to the local chemical configuration around the probed element and can be employed to clarify at the electronic level the chemical physics of the coordinated low-z atoms, such as N in the FeN4 site. As shown in Fig. 2a, C K-edge XANES spectra present two typical spectroscopic features for HP-FeN4 and FeN4 materials: the 1s core electron of carbon into the π*C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (283.8 eV) and σ*C–C (290.5 eV).30,31 The HP-FeN4 material shows similar features to highly graphitized carbon. This result demonstrates that the introduction of Fe and N atoms maintains the electronic structure and high electron conductivity of carbon supports.32 Moreover, the N K-edge spectra of HP-FeN4 materials are dominated by two typical spectroscopic features: 1s → π* transition at the region of 393.0–401.0 eV and 1s → σ* transition at 401.0–410.0 eV, respectively.33 In the region of 1s → π* transition, two obvious resonances labeled as peak a and peak b can be observed. The resonance of peak a can be ascribed to the π* transition in the C–N–C portion of pyridinic N sites.34 And peak b is the π* transition of pyrrolic/graphitic type N-groups, consistent with previous reports.35 Notably, peak b split into double peaks for HP-FeN4 while FeN4 kept unchanged, indicating that the pyrrolic N was bonded to Fe atoms under NH3 pyrolysis. The formation of Fe–pyrrolic bonds will induce a charge transfer between the metal atom and the N-decorated graphene, thereby promoting the electron transfer capacity between active FeN4 sites and the carbon supports during the ORR process.36
C (283.8 eV) and σ*C–C (290.5 eV).30,31 The HP-FeN4 material shows similar features to highly graphitized carbon. This result demonstrates that the introduction of Fe and N atoms maintains the electronic structure and high electron conductivity of carbon supports.32 Moreover, the N K-edge spectra of HP-FeN4 materials are dominated by two typical spectroscopic features: 1s → π* transition at the region of 393.0–401.0 eV and 1s → σ* transition at 401.0–410.0 eV, respectively.33 In the region of 1s → π* transition, two obvious resonances labeled as peak a and peak b can be observed. The resonance of peak a can be ascribed to the π* transition in the C–N–C portion of pyridinic N sites.34 And peak b is the π* transition of pyrrolic/graphitic type N-groups, consistent with previous reports.35 Notably, peak b split into double peaks for HP-FeN4 while FeN4 kept unchanged, indicating that the pyrrolic N was bonded to Fe atoms under NH3 pyrolysis. The formation of Fe–pyrrolic bonds will induce a charge transfer between the metal atom and the N-decorated graphene, thereby promoting the electron transfer capacity between active FeN4 sites and the carbon supports during the ORR process.36
 | ||
| Fig. 2 Pyrrole-type FeN4 coordination. (a) sXAS spectra of the C K-edge and N K-edge for HP-FeN4 and FeN4. Deconvoluted features of (b) peak a and (c) peak b of the N K-edge spectra. (d) High-resolution N 1s XPS spectra of HP-FeN4 and FeN4. (e) Comparison of the pyrrolic N content (from XPS spectra) of the HP-FeN4 precursor pyrolysed for different time under NH3 atmosphere. | ||
To further confirm the coordination N structure of FeN4 sites, the region of 1s → π* transition for N K-edge sXAS was fitted and is shown in Fig. 2b and c. As shown in Fig. 2b, the high-resolution spectrum of peak a for the FeN4 material was deconvoluted into two peaks which can be ascribed to the pyridinic N (395.7 eV) and Fe–pyridinic N sites (396.4 eV). In particular, HP-FeN4 exhibits only one peak at 395.7 eV, which demonstrates that there is no Fe–pyridinic N coordination in the HP-FeN4 catalyst. In contrast, peak b of HP-FeN4 can be divided into three characteristic peaks at 397.5, 398.0 and 398.7 eV, which are assigned to pyrrolic N, Fe–pyrrolic N and graphitic N sites, respectively (Fig. 2c). Notably, only one peak of graphitic N for the FeN4 catalyst was observed, suggesting that NH3 pyrolysis results in the change of neighbouring N structure around the FeN4 sites and promotes the formation of Fe–pyrrolic N coordination structure. HP-FeN4 manifests an obvious Fe–pyrrolic N characteristic peak, and no Fe–pyridinic N site was detected in the sXAS spectra. The above results confirm that HP-FeN4 possesses a high-purity FeN4 structure and almost all of the FeN4 sites are pyrrole-type.
The coordination N structure of the HP-FeN4 catalyst can also be confirmed by the Fourier transform infrared (FT-IR) spectra and X-ray photoelectron spectroscopic (XPS) analysis. In Fig. S14 (ESI†), the FT-IR spectra of FeN4 and HP-FeN4 show three characteristic peaks at 1404, 1637 and 3459 cm−1, respectively. The wider band at ∼3459 cm−1 corresponds to C–OH and N–H stretching vibrations, whereas the peaks at around 1637 cm−1 and 1404 cm−1 could be assigned to the typical CN heterocycle stretching modes.37,38 Moreover, both HP-FeN4 and FeN4 show a peak at low wavenumbers of 833 cm−1, which can be related to the Fe–N bonds. Specifically, the peak at 873 cm−1 belongs to the typical Fe–N stretch (pyrrole), which is consistent with FePc.39 Therefore, the pyrrole-type FeN4 structure was successfully achieved via the ammonia assisted pyrolysis strategy.
Fig. S15 (ESI†) shows the high resolution spectra of N 1s for different catalysts. The N 1s spectra of NC can be deconvoluted into three peaks, which correspond to pyridinic N (398.5 eV), pyrrolic N (399.7 eV) and graphitic N (401.1 eV), respectively.40,41 Specifically, the pyridinic N of the FeN4 catalyst shifted to a higher binding energy compared with NC materials (Fig. S15(a), ESI†), which can be ascribed to the interaction between Fe atoms and pyridinic N species, suggesting the formation of Fe–pyridinic N bonds.42 In contrast, the binding energy of pyrrolic N increased from 399.7 eV to 399.9 eV for the HP-FeN4 catalyst (Fig. S15(b), ESI†), which demonstrates that Fe–pyrrolic N moieties have been formed during the NH3 pyrolysis. The different N species content can be obtained by calculating the area of the N species peak in XPS spectra. As shown in Fig. 2d and Table S2 (ESI†), HP-FeN4 exhibits significantly increased pyrrolic N content (22.1 at%) and decreased pyridinic N content (41.7 at%) compared with FeN4 synthesized under Ar (5.9 at% and 54.2 at%, respectively). This result reveals that the FeN4 structure was successfully converted from pyridine-type to pyrrole-type via the ammonia-assisted strategy and maintains its high purity.
The content of pyrrole-type FeN4 sites can also be tuned by controlling the annealing atmosphere and chemical reaction time. Fig. S15(c) (ESI†) displays the N 1s XPS spectra of the HP-FeN4 precursor annealed under NH3 for different lengths of time. When the NH3 annealing time was 0 min, few pyrrolic N species were observed and almost all Fe atoms were coordinated by pyridinic N. More pyrrole-type FeN4 sites were formed by increasing the NH3 annealing time. A high-purity pyrrole-type FeN4 structure was obtained with an annealing time of 60 min. As shown in Fig. 2e, it was obviously observed that the content of pyrrolic N increased with the pyrolysis time increasing, which further demonstrates that NH3 atmosphere plays an important role in forming the pyrrole-type FeN4 structure. Based on the above results, high-purity pyrrole-type FeN4 sites were successfully achieved by controlling the ammonia-assisted reaction conditions and it exhibits perfect atom utilization and efficient mass transfer capability, which makes it a promising electrocatalyst for the ORR process.
To theoretically evaluate the ORR activity and selectivity on the pyrrole-type FeN4 structure, density functional theory (DFT) calculations were carried out to investigate the free energetics of the ORR reaction mechanism. As shown in Fig. 3a and b, pyrrole-type FeN4 shows stronger electron depletion (blue area) around the Fe atom than pyridine-type FeN4, but weaker electron depletion is observed around the neighboring pyrrolic N. This result reveals that iron has a distinctly different charge distribution on both structures, and the valence state of iron in the pyrrole-type structure is more positive than that in the pyridine-type structure. In the free energy diagram of the ORR (Fig. 3c), lower Gibbs free energy differences between O2 and OOH* of pyrrole-type FeN4 suggest its preferable oxygen adsorption for the ORR.43 Moreover, pyrrole-type FeN4 exhibits lower thermodynamic overpotential (0.35 eV) from the initial state (O2) to the final state (H2O) than that of pyridine-type (0.67 eV) in a 4e− pathway, giving theoretical evidence for a highly efficient ORR catalytic activity. In particular, the thermodynamic onset potential of pyrrole-type FeN4 (0.88 V) is also higher than that of pyridine-type FeN4, which further confirms that pyrrole-type coordination is more conducive to the ORR reaction (Fig. S24, ESI†). At a potential of 1.23 V, the reduction of OOH* to H2O2 is endothermic by 1.77 eV on the pyrrole-type FeN4 structure, higher than that of pyridine-type (1.57 eV) (Fig. 3c), revealing that the 2e− reduction pathway is largely suppressed and pyrrole-type FeN4 would have high selectivity for the 4e− reduction process. Fig. 3d shows the atomic configuration details of the ORR reaction, which involves four electron transfer steps. Initially, an O2 molecule adsorbs on the top of Fe atom and then transfers into OOH*. Next, H2O is desorbed and O* is formed on the central Fe atom. Finally, OH* is formed and further converts to a H2O molecule. Overall, the DFT results reveal that pyrrole-type FeN4 could lower the limiting potential and is more conducive to 4e− reaction, which would be an ideal active site for ORR catalysis.
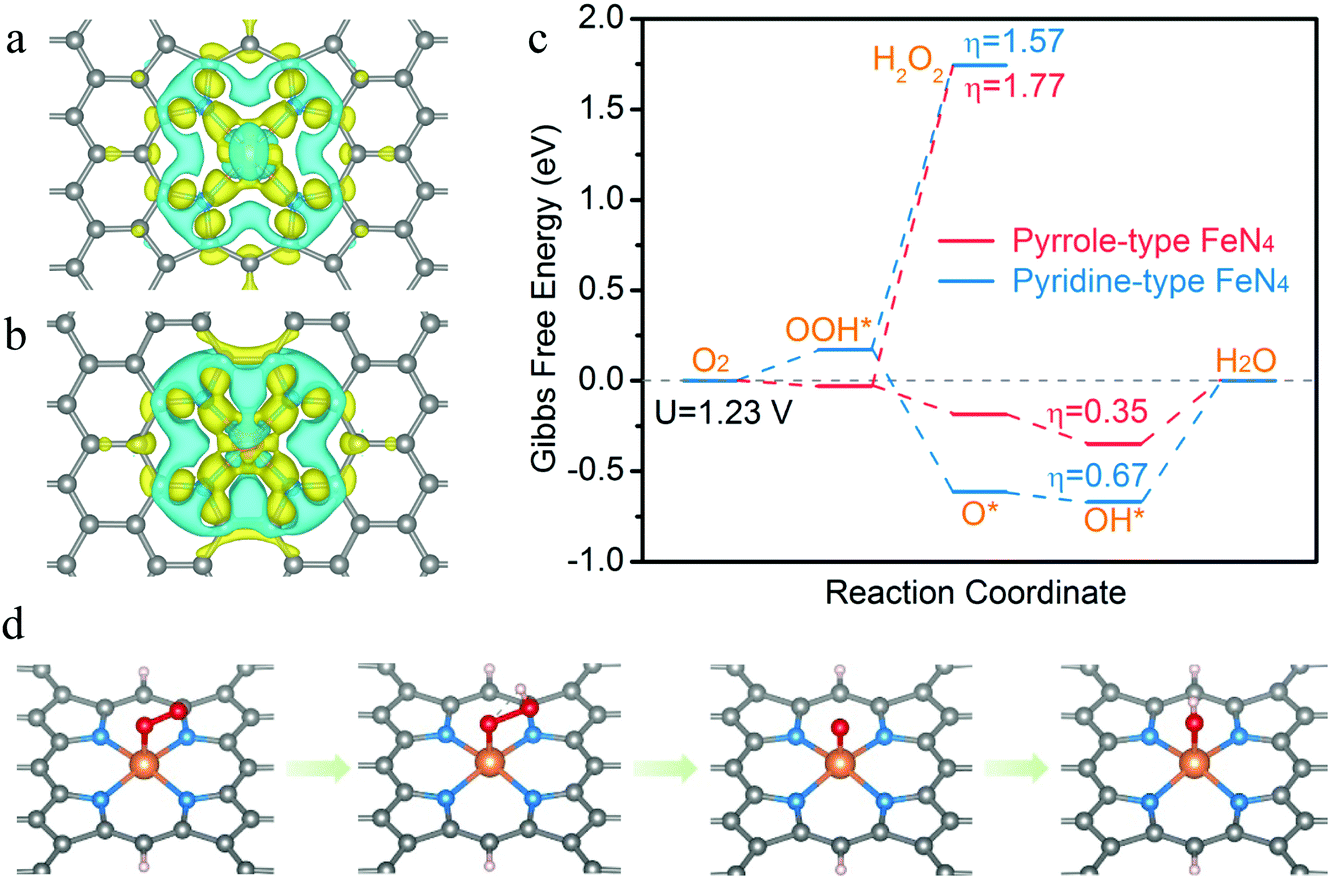 | ||
| Fig. 3 Theoretical calculations. Calculated charge density difference of (a) pyrrole-type FeN4 and (b) pyridine-type FeN4. Yellow and blue regions represent electron accumulation and electron depletion, respectively. Iso-surface 0.005 e− Å−3. (c) Free energy diagram of the oxygen reduction reaction on pyrrole-type FeN4 and pyridine-type FeN4. (d) Adsorption configurations of the intermediates during the ORR process on pyrrole-type FeN4 (the balls in grey, blue, orange, red and pink represent C, N, Fe, O and H atoms, respectively). | ||
To investigate the catalytic activity of high-purity pyrrole-type FeN4 sites, electrochemical performance of ORR is evaluated by employing a rotating disk electrode (RDE) method. As shown in Fig. 4a, HP-FeN4 exhibits higher onset potential (0.95 V vs. RHE) and more positive half-wave potential (0.80 V vs. RHE) than that of FeN4 (0.86 V and 0.71 V vs. RHE, respectively), which is comparable to the commercial Pt/C catalyst. Compared to both FeN4 (80 mV dec−1) and Pt/C (88 mV dec−1), a small Tafel slope of 72 mV dec−1 can also be observed for HP-FeN4, indicating its fast kinetics in the oxygen reduction process (Fig. 4b). The current density relative to the BET specific surface area at 0.8 V vs. RHE has also been compared. In Fig. 4c, HP-FeN4 exhibits the largest current density of 6.89 mA m−2, which is 6.9 and 54.3 times that of traditional FeN4 and NC, and it surpasses most reported metal–nitrogen coordination catalysts (Table S3, ESI†). The electrochemical activity has been greatly improved for HP-FeN4 while the specific surface area has not changed significantly, which demonstrates that high-purity pyrrole-type iron–nitrogen sites are the key factor for performance improvement. Moreover, the ORR activity of HP-FeN4 annealing under NH3 for different times was also evaluated. As shown in Fig. 4d, HP-FeN4 catalysts exhibit more positive half-wave potential and smaller Tafel slope with increasing NH3 annealing time. This result gives direct evidence for the high intrinsic catalytic activity of pyrrole-type FeN4 sites.
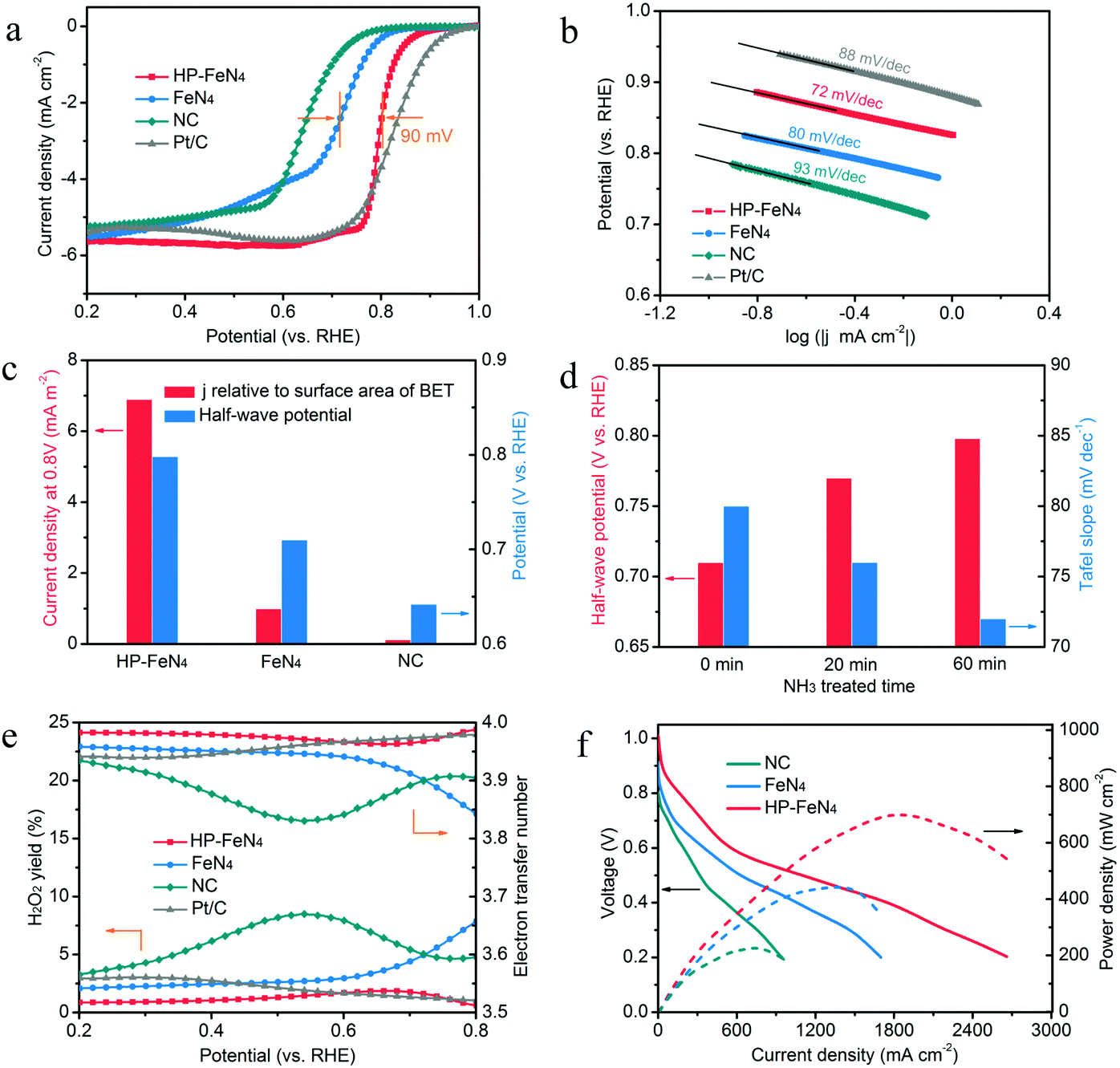 | ||
| Fig. 4 Electrocatalytic performance. (a) ORR polarization curves and (b) corresponding Tafel plots of HP-FeN4, FeN4 and NC catalysts in O2-saturated 0.5 M H2SO4 and 20% Pt/C in 0.1 M HClO4 under a rotating rate of 1600 rpm. (c) Comparison of current density relative to BET surface area at 0.8 V and half-wave potential for HP-FeN4, FeN4 and NC catalysts. (d) ORR catalytic activity of HP-FeN4 precursor pyrolysed for different times under NH3 atmosphere. (e) Peroxide yield and electron transfer number of HP-FeN4, FeN4, NC and Pt/C during the ORR process. (f) Polarization and power density curves of HP-FeN4, FeN4 and NC based membrane electrode assemblies in PEMFCs. Cell temperature: 80 °C; RH: 100%, H2/O2: 200 kPa. | ||
To verify the ORR catalytic pathways of these electrocatalysts, rotating ring-disk electrode (RRDE) measurements were performed to monitor the formation of peroxide species (H2O2) during the ORR process. As shown in Fig. 4e, the measured H2O2 yields of HP-FeN4, FeN4 and NC were below 10% over the potential range of 0.2–0.8 V, suggesting an electron transfer number over 3.8. Specifically, HP-FeN4 exhibited a Pt-like property with H2O2 yields below 2%, much lower than the FeN4 catalyst, which demonstrated higher product selectivity of pyrrole-type FeN4 sites. This result reveals that a pyrrole-type FeN4 structure could restrain the generation of H2O2, thereby reducing Fenton reactions during oxygen reduction and enhancing the stability of FeN4 sites. Electrochemical impedance spectroscopy (EIS) was also employed to characterize the interface reactions and electrode kinetics in the ORR. As shown in Fig. S21 (ESI†), HP-FeN4 exhibits a smaller charge transfer resistance than that of FeN4 and NC, suggesting that the high-purity pyrrole-type FeN4 structure possessed a lower charge transfer resistance and allowed much faster shuttling of electrons during the ORR process. The stability test of HP-FeN4 was also performed. As shown in Fig. S17 (ESI†), only 26 mV loss of half-wave potential after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential cycles in O2-saturated 0.5 M H2SO4 was observed, suggesting its excellent stability. Therefore, purity and coordination N structure essentially determine the activity and stability of the FeN4 site. Pyrrolic N coordination and high-purity FeN4 sites achieve significantly enhanced catalytic activity and stability and would favor improved performance of PEMFCs.
000 potential cycles in O2-saturated 0.5 M H2SO4 was observed, suggesting its excellent stability. Therefore, purity and coordination N structure essentially determine the activity and stability of the FeN4 site. Pyrrolic N coordination and high-purity FeN4 sites achieve significantly enhanced catalytic activity and stability and would favor improved performance of PEMFCs.
To corroborate the ORR activity revealed by the half-cell characterization, HP-FeN4, FeN4 and NC were assembled into the membrane electrode assembly (MEA) for evaluating the actual PEMFC performance. Fig. S29 (ESI†) shows the working principle scheme of the PEMFC and digital photographs of a single cell assembling process. As shown in Fig. S19 (ESI†), the open circuit voltage (OCV) of HP-FeN4 was as high as 1.01 V, better than 0.92 V of FeN4 and 0.84 V of NC, suggesting the good intrinsic catalytic performance of high-purity pyrrole-type FeN4 sites. Fig. 4f presents polarization and power density curves for the three catalysts. The HP-FeN4 catalyst was capable of generating current densities of 0.55 and 2.66 A cm−2 at 0.6 and 0.2 V with H2/O2 total pressure of 200 kPa, which outperformed the FeN4 and NC catalyst. The corresponding peak power density of HP-FeN4 was up to 700 mW cm−2, suggesting its excellent application prospects. Based on the above results, the greatly improved PEMFC performance of the HP-FeN4 catalyst can be ascribed to the following features: (1) high-purity FeN4 sites enable significantly enhanced intrinsic activity for the ORR. (2) The pyrrole-type FeN4 configuration possesses preferred O2 adsorption energy and ultra-high four-electron reaction selectivity. (3) The presence of a large number of mesopores greatly increased the utilization of active sites. Due to the combination of enhanced catalytic activity and high exposure of active sites, the ORR processes of HP-FeN4 are largely accelerated, and a high power density is achieved when integrated into a PEMFC system.
In conclusion, we have successfully developed a high-purity pyrrole-type FeN4 structure as a high-efficiency PEMFC electrocatalyst. The as-prepared pyrrole-type FeN4 sites exhibit significantly enhanced intrinsic activity, preferred O2 adsorption energy and complete four-electron reaction selectivity, leading to an excellent ORR activity and stability. As expected, the high-purity FeN4 catalyst exhibits an ultra-high active area current density of 6.89 mA m−2 in acid medium, which exceeds most reported metal–nitrogen coordination catalysts. Moreover, the high-purity FeN4 site based PEMFCs show excellent performance with a large power density and a high open circuit voltage, suggesting a wider perspective for practical applications. A high-purity iron–nitrogen configuration will open up new avenues to design advanced non-noble metal catalysts for reversible energy conversion systems.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the National Basic Research Program of China (2015CB932302), Natural Science Foundation of China (No. 91745113, 11621063), National Program for Support of Top-notch Young Professionals, and the Fundamental Research Funds for the Central Universities (No. WK 2060190084). We also appreciate the support from the Major/Innovative Program of Development Foundation of Hefei Center for Physical Science and Technology. This work was partially carried out at the USTC Center for Micro and Nanoscale Research and Fabrication. Minglong Chen and Xiaojun Wu thank the Supercomputing Center of USTC for the computational resources and the support of the Natural Science Foundation of China (No. 21573204, 21421063) and the Ministry of Science and Technology of the People's Republic of China (2018YFA0208603).Notes and references
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed.
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D.-J. Liu, Science, 2018, 362, 1276 CrossRef CAS PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552 CrossRef CAS PubMed.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230 CrossRef CAS PubMed.
- X. Wang, Z. Li, Y. Qu, T. Yuan, W. Wang, Y. Wu and Y. Li, Chem, 2019, 5, 1–26 Search PubMed.
- U. Martinez, S. Komini Babu, E. F. Holby, H. T. Chung, X. Yin and P. Zelenay, Adv. Mater., 2019, 1806545 CrossRef PubMed.
- Y. Shao, J.-P. Dodelet, G. Wu and P. Zelenay, Adv. Mater., 2019, 1807615 CrossRef PubMed.
- G. Wu, Front. Energy, 2017, 11, 286–298 CrossRef.
- J. Herranz, F. Jaouen, M. Lefèvre, U. I. Kramm, E. Proietti, J.-P. Dodelet, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, P. Bertrand, T. M. Arruda and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 16087–16097 CrossRef CAS PubMed.
- Y. He, S. Hwang, D. A. Cullen, M. A. Uddin, L. Langhorst, B. Li, S. Karakalos, A. J. Kropf, E. C. Wegener, J. Sokolowski, M. Chen, D. Myers, D. Su, K. L. More, G. Wang, S. Litster and G. Wu, Energy Environ. Sci., 2019, 12, 250–260 RSC.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- V. Yarlagadda, M. K. Carpenter, T. E. Moylan, R. S. Kukreja, R. Koestner, W. Gu, L. Thompson and A. Kongkanand, ACS Energy Lett., 2018, 3, 618–621 CrossRef CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- L. Lin, Z. K. Yang, Y.-F. Jiang and A.-W. Xu, ACS Catal., 2016, 6, 4449–4454 CrossRef CAS.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef PubMed.
- H. Shen, E. Gracia-Espino, J. Ma, K. Zang, J. Luo, L. Wang, S. Gao, X. Mamat, G. Hu, T. Wagberg and S. Guo, Angew. Chem., Int. Ed., 2017, 56, 13800–13804 CrossRef CAS PubMed.
- H. Shen, E. Gracia-Espino, J. Ma, H. Tang, X. Mamat, T. Wagberg, G. Hu and S. Guo, Nano Energy, 2017, 35, 9–16 CrossRef CAS.
- M. Xiao, H. Zhang, Y. Chen, J. Zhu, L. Gao, Z. Jin, J. Ge, Z. Jiang, S. Chen, C. Liu and W. Xing, Nano Energy, 2018, 46, 396–403 CrossRef CAS.
- Q. Jia, E. Liu, L. Jiao, S. Pann and S. Mukerjee, Adv. Mater., 2018, 1805157 Search PubMed.
- Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby, P. Zelenay and S. Mukerjee, ACS Nano, 2015, 9, 12496–12505 CrossRef CAS PubMed.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937 CrossRef CAS PubMed.
- L. Yang, D. Cheng, H. Xu, X. Zeng, X. Wan, J. Shui, Z. Xiang and D. Cao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6626 CrossRef CAS PubMed.
- K. Liu, G. Wu and G. Wang, J. Phys. Chem. C, 2017, 121, 11319–11324 CrossRef CAS.
- H. Yu, L. Shang, T. Bian, R. Shi, G. I. N. Waterhouse, Y. Zhao, C. Zhou, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2016, 28, 5080–5086 CrossRef CAS PubMed.
- Q. Liu, X. Liu, L. Zheng and J. Shui, Angew. Chem., Int. Ed., 2018, 57, 1204–1208 CrossRef CAS PubMed.
- M. Graglia, J. Pampel, T. Hantke, T.-P. Fellinger and D. Esposito, ACS Nano, 2016, 10, 4364–4371 CrossRef CAS PubMed.
- S. H. Lee, J. Kim, D. Y. Chung, J. M. Yoo, H. S. Lee, M. J. Kim, B. S. Mun, S. G. Kwon, Y.-E. Sung and T. Hyeon, J. Am. Chem. Soc., 2019, 141, 2035–2045 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479 CrossRef CAS PubMed.
- I. Y. Kim, S. Kim, X. Jin, S. Premkumar, G. Chandra, N.-S. Lee, G. P. Mane, S.-J. Hwang, S. Umapathy and A. Vinu, Angew. Chem., 2018, 130, 17381–17386 CrossRef.
- P. Chen, N. Zhang, S. Wang, T. Zhou, Y. Tong, C. Ao, W. Yan, L. Zhang, W. Chu, C. Wu and Y. Xie, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6635 CrossRef CAS PubMed.
- D. Deng, X. Chen, L. Yu, X. Wu, Q. Liu, Y. Liu, H. Yang, H. Tian, Y. Hu, P. Du, R. Si, J. Wang, X. Cui, H. Li, J. Xiao, T. Xu, J. Deng, F. Yang, P. N. Duchesne, P. Zhang, J. Zhou, L. Sun, J. Li, X. Pan and X. Bao, Sci. Adv., 2015, 1, e1500462 CrossRef PubMed.
- Y. Liang, H. Wang, J. Zhou, Y. Li, J. Wang, T. Regier and H. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef PubMed.
- N. Meng, J. Ren, Y. Liu, Y. Huang, T. Petit and B. Zhang, Energy Environ. Sci., 2018, 11, 566–571 RSC.
- Y. Tong, P. Chen, T. Zhou, K. Xu, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 7121–7125 CrossRef CAS PubMed.
- F. Arcudi, L. Đorđević and M. Prato, Angew. Chem., 2016, 128, 2147–2152 CrossRef.
- F. Wang, P. Chen, Y. Feng, Z. Xie, Y. Liu, Y. Su, Q. Zhang, Y. Wang, K. Yao, W. Lv and G. Liu, Appl. Catal., B, 2017, 207, 103–113 CrossRef CAS.
- Z. Zhang, M. Dou, J. Jing and W. Feng, Nano Energy, 2017, 34, 338–343 CrossRef CAS.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444 CrossRef CAS PubMed.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS PubMed.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, SEM, XPS, TEM and additional electrochemical data. See DOI: 10.1039/c9ee03027a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |