
Fabrication of practical catalytic electrodes using insulating and eco-friendly substrates for overall water splitting†
Weiju
Hao‡
ab,
Renbing
Wu‡
 a,
Hao
Huang
c,
Xin
Ou
*c,
Lincai
Wang
d,
Dalin
Sun
a,
Xiaohua
Ma
a and
Yanhui
Guo
*a
a,
Hao
Huang
c,
Xin
Ou
*c,
Lincai
Wang
d,
Dalin
Sun
a,
Xiaohua
Ma
a and
Yanhui
Guo
*a
aDepartment of Material Science, Fudan University, Shanghai, 200433, China. E-mail: gyh@fudan.edu.cn
bCollege of Science, University of Shanghai for Science and Technology, Shanghai 200093, P. R. China
cState Key Laboratory of Functional Materials for Informatics, Shanghai Institute of Microsystem and Information Technology, CAS, 20050, Shanghai, China. E-mail: ouxin@mail.sim.ac.cn
dResearch Center of Resource Recycling Science and Engineering, Shanghai Polytechnic University, Shanghai, 201209, China
First published on 21st November 2019
Abstract
The development of efficient and cost-effective catalytic electrodes is of great importance to electrolysis. Herein, a strategy of fabricating practical catalytic electrodes by depositing conductive catalysts on inexpensive and easily accessible insulating substrates of paper, textiles and sponge has been realized and well developed. These electrodes are found to be highly active toward overall water splitting. As a distinctive example, the Ni–P–B/paper electrode affords 50 mA cm−2 at overpotentials of only 76 mV for the hydrogen evolution reaction and 263 mV for the oxygen evolution reaction, and can survive at large current density of 1000 mA cm−2 for over 240 h without apparent performance degradation in 1.0 M KOH. A two-electrode cell constructed by this paper electrode, which is only 1/5 the weight of a traditional metal electrode, delivered 50 mA cm−2 water-splitting current at a cell voltage of only 1.661 V, rivalling the integrated state-of-the-art Pt–C/Ni and IrO2/Ni electrode. Moreover, a functional Ni–P–B/paper ring electrode with in situ separation function has been constructed, enabling simultaneous generation, separation and collection of hydrogen and oxygen. This discovery may enable a large extension toward practical catalytic electrodes that are also active, cheap, light, flexible, earth-abundant and recyclable.
Broader contextThe development of efficient and cost-effective catalytic electrodes is of great importance to electrolysis. Currently, electrolysis of water that can produce hydrogen using a cheap source of electrical energy is driving extensive research efforts to address increasing global energy challenges and related environmental issues. Normally, a catalytic electrode is fabricated by deposition of active electrocatalysts on conductive substrates of metal, carbon cloth and semiconductor. Despite the advanced progress in the development of electrocatalysts, an extension of the substrate to inexpensive and eco-friendly materials will be of great practical interest and environmental benefit. A family of catalytic electrodes fabricated on insulating substrates of paper, cloth and sponge which bring dramatic advantages of high performance, low cost, light weight, eco-friendliness, flexibility, and simple and scalable fabrication, were developed. One of the electrolyzers assembled by the bifunctional Ni–P–B/paper electrode, which is only 1/5 the weight of the electrode with a common Ni foil substrate, delivered a current density of 50 mA cm−2 at a cell voltage of only 1.661 V for overall water splitting, rivalling most of the electrodes constructed by traditional conductive substrates. Meanwhile, the novel electrode can also survive at large current density of 1000 mA cm−2 for over 240 h without apparent performance degradation in 1.0 M KOH. |
Electrolysis is an important technique with a large variety of applications. Currently, electrolysis of water that can produce hydrogen using a cheap source of electrical energy is driving extensive research efforts to address increasing global energy challenges and related environmental issues.1–3 As one of the key challenges for scalable and sustainable water electrolysis, the development of an efficient, durable, and economic catalytic electrode has become a highly active and exciting area.4,5 Normally, a catalytic electrode is fabricated by deposition of active electrocatalysts on a conductive substrate.6–8 Based on many excellent efforts, various highly active and low-cost electrocatalysts for water electrolysis have been developed, however, the substrates remain quite limited, mainly metal such as nickel (Ni),9 titanium (Ti),10 iron (Fe)11 in the form of foil12 or foam,13–15 or carbon cloth (CC).16,17 Despite the advantages of satisfactory electric conductivity and mechanical properties, these substrates also have some of the intrinsic disadvantages of high cost, poor flexibility, heaviness and environmental concerns.18,19 Therefore, an extension of the substrate to inexpensive, abundant, flexible and eco-friend substrates will be of great practical interest and environmental benefit. Among the range of substrates, the materials of paper, textiles, and sponge are potential easily accessible candidates,20,21 except that these materials are normally insulating, and cannot provide the conductive pathways between the electrical circuit and the electrocatalyst. Thus, they have rarely been explored for electrocatalysis since this fundamental requirement cannot be achieved.22,23 It has been reported that insulating materials of paper and cotton fabric can be transformed to a conducting current collector first and then functionalized as a substrate of an efficient catalytic electrode.3,24 Recently, a number of conductive transition metal based electrocatalysts (e.g., metal alloys, borides, phosphides, nitrides, and carbides),25 which can serve as the conductive electrode layer themselves, have been developed, allowing a more straightforward construction of novel catalytic electrodes using an insulating substrate.26 Among the various conductive earth-abundant electrocatalysts recently discovered, the transition metal based complexes containing B and/or P have emerged as an interesting family of low-cost materials with high catalytic activity toward overall water splitting.27–30 It is therefore possible that efficient and cost-effective catalytic electrodes can be constructed by depositing these conductive electrocatalysts on an insulating substrate.31
To realize workable electrodes using an insulating substrate, the deposition of a continuous and uniform conductive electrocatalyst layer on the substrate is of fundamental importance.32 Several techniques to make the insulating substrate conductive, such as ink-jet, screen, vacuum filtration, sputtering techniques and the electroless plating method, etc. may be applicable.26,33 However, the involved conductive materials are normally not efficient electrocatalysts. Moreover, some of these techniques require complicated instruments or incorporation of additives which will negatively affect the catalytic performance of the catalytic layer.34 Therefore, the development of a versatile technique to deposit electrocatalysts on insulating substrates will be meaningful.3,35 Herein, we show a facile, effective, and straightforward technique to deposit various conductive electrocatalysts on a wide range of insulating substrates, and the outstanding performance of these novel catalytic electrodes for overall water splitting. Electrochemical measurements confirm that these novel bifunctional electrodes have satisfactory conductivity, remarkable catalytic activity and long-term stability at a current density up to 2000 mA cm−2 during both the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), rivalling the traditional electrode using a conductive substrate. Meanwhile, a multifunctional electrode constructed by a flexible catalytic paper electrode has achieved in situ separation function, enabling simultaneous generation, separation and collection of H2 and O2 gas. This strategy of fabricating catalytic electrodes on cheap, easily accessible, flexible and eco-friendly substrates may promote the development of cost-effective and practical electrodes toward water electrolysis as well as other electrochemical fields.
To prepare the electrode, the insulating substrate is activated by alternately dipping the substrates into NiSO4 solution and NaBH4 solution (Fig. 1a). After this, nickel boride nanoparticles can be deposited on the substrate.36 Then, the catalytic electrode is prepared by depositing an even layer of electrocatalyst on the pre-treated substrate using the electroless plating (EP) technique (Fig. 1a). In an optimized synthetic process, a small piece of pre-treated filter paper (1 × 0.5 cm) is immersed in an aqueous EP bath containing NiSO4, NaH2PO2, Et2NHBH3, and Na2SO4 for 60 min at 10 °C to fabricate a Ni–P–B/paper electrode.
 | ||
| Fig. 1 (a) Schematic synthetic process for the Ni–P–B/paper electrode. SEM images of (b) bare paper, (c) activated paper, and (d) Ni–P–B/paper electrode; (e) cross section SEM image of the Ni–P–B/paper electrode. (f) HRTEM image of the Ni–P–B catalyst (inset, SAED pattern). (g) Photographs of the Ni–P–B/paper electrode (diameter = 12 cm). (h) Photograph of the conductive Ni–P–B/paper electrode with a sign of ‘FDU’ (Fudan University). | ||
To clearly observe the evolution of the catalyst layer during the deposition process, deposition of the catalyst on a paper substrate as well as a single fiber extracted from the substrate were studied. The field-emission scanning electron microscope (FESEM) images of the paper substrate indicate a network of interwined fibers with relatively clean and smooth surface (Fig. 1b), and the original diameter of the single fiber is 17.4 μm (Fig. S1, ESI†). After activation, many nanoparticles with an average particle size of ∼20 nm (0.1 mg cm−2) are uniformly deposited on the paper as well as the single fiber as shown in Fig. 1c and Fig. S2, ESI.† It has been proved that no catalyst can be deposited on the paper during the EP process without this pre-treatment. While with the assistance of the nano-activator, a continuous deposit of catalyst (3.15 mg cm−2) is formed after dipping the activated substrate in the EP bath for 30 min (Fig. S2, ESI†). The diameter of the single fiber grows to 20.4 μm, which means that a 1.5 μm thick catalyst layer is deposited on the fiber (Fig. S1b, ESI†). An extension of the plating time to 60 min leads to the development of an electrode with the best performance (Fig. S2f and S3, ESI†), which is denoted as Ni–P–B/paper electrode. It is confirmed that the fibers on the top of the paper substrate as well as the single fiber are uniformly coated by a layer of electrocatalyst (Fig. 1d and Fig. S1b, ESI†). The amount of the catalyst loaded on the paper substrate is determined to be 6.15 mg cm−2, and the thickness of the catalyst layer on the single fiber is measured to be around 2.6 μm (Fig. S1b, ESI†). Meanwhile, the TEM image (Fig. S4, ESI†) of a fiber drawn from the as-prepared Ni–P–B/paper electrode, and the cross section SEM images of the electrode (Fig. 1e and Fig. S5, ESI†) suggest uniform coating of the Ni–P–B catalyst on its surface, and a close contact between the catalyst and the fiber. It is also observed that the surface of the fiber inside the as-prepared paper electrode is covered by many small nano particles (Fig. 1e). Moreover, the space between the fibers is found to be filled with the Ni–P–B catalyst, which joins these fibers up (Fig. S5, ESI†). Therefore, a catalyst conductive network may be constructed in the Ni–P–B/paper electrode. To demonstrate the scalability and adaptability of this technique, a large Ni–P–B/paper electrode of 12 cm in diameter (Fig. 1g) and paper electrode with different configurations (Fig. 1h and Fig. S6, ESI†) have been prepared. It is noted that the paper electrode is bendable and even foldable, which enables much safe and easy operation in practical applications.
X-ray diffraction (XRD) studies (Fig. S7a, ESI†) show the disappearance of the diffraction peaks for the substrate and an amorphous phase of the as-prepared electrode, which may be the result of full coverage of the amorphous catalyst on the surface of the paper substrate. Energy dispersive spectroscopy (EDS) elemental mapping images (Fig. S7b, ESI†) demonstrate homogeneous element distributions of Ni, P and B elements on the electrode. Furthermore, the atomic ratio of Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P:B is determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analysis to be 16.0:1.0:2.1 (Table S1, ESI†). A disordered lattice and amorphous phase of the Ni–P–B catalyst is further confirmed by its high-resolution transmission electron microscopy (HRTEM) image and corresponding selected-area electron diffraction (SAED) pattern as shown in Fig. 1f.
:P:B is determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analysis to be 16.0:1.0:2.1 (Table S1, ESI†). A disordered lattice and amorphous phase of the Ni–P–B catalyst is further confirmed by its high-resolution transmission electron microscopy (HRTEM) image and corresponding selected-area electron diffraction (SAED) pattern as shown in Fig. 1f.
Investigation on chemical states of each element of the catalyst is carried out using high-resolution XPS spectra (Fig. S8, ESI†). Two peaks in the Ni 2p level with binding energy (BE) at 852.7 and 869.4 eV indicate the presence of elemental nickel (Ni0). The separated peaks for the P 2P level at 128.7 (P 2p3/2) and 129.5 eV (P 2p1/2) are attributed to the metallic phosphorous, and the BE of B1s at 187.1 eV is assigned to elemental boron. By comparing the binding energy of Ni with those of Ni–P and Ni–B species, the peak of the Ni 2p3/2 state at 852.7 eV for the Ni–P–B catalyst stays right between the corresponding BE in Ni–P (853.1 eV) and Ni–B (852.1 eV).37 Meanwhile, the BE of the P 2p3/2 peak at 128.7 eV (Fig. S8b, ESI†) for the Ni–P–B catalyst is 0.5 eV lower than that of the Ni–P catalyst (129.2 eV),38 and the BE of the B 1s peak is 0.1 eV higher than that of the Ni–B catalyst (187.7 eV).36 These results clearly suggest the formation of Ni–P–B catalyst rather than a physical mixture of Ni–P and Ni–B during the EP process. Moreover, oxidization of Ni as well as P and B on the surface of the catalyst is also evidenced by the presence of the Ni 2p 3/2 peak at 856.0 eV with a satellite peak at 860.8 eV, P 2p peak at 133.1 eV and B 1s peak at 190.1 eV, respectively, which seems inevitable for this kind of catalyst.6,39,40
The conductivity of the Ni–P–B/paper electrode is demonstrated by series connecting these electrodes with a light emitting diode (LED) in a circuit (Fig. 1h and Fig. S6b, ESI†). The illuminated LED clearly suggests that the paper electrode meets the basic requirement of conductivity, which may be attributed to the conductive network in the Ni–P–B/paper electrode. Quantitative studies show that the sheet resistance of the Ni–P–B/paper electrode is only 0.42 Ω sq−1 (Table S2, ESI†), which is comparable to the efficient Ni–P/paper current collector (4.2 Ω sq−1)3 and CNT coated cotton (1–4 Ω sq−1),41,42 indicating easy electron transport within the conductive network of the paper electrode. It should be noted that further prolonging the Ni–P–B plating time can result in more catalyst deposition of 10.9 mg cm−2 on the paper substrate and a thicker catalyst layer (3.2 μm) on the single fiber at 90 min (Fig. S1a and b, ESI†). Although the sheet resistance of the electrode further reduces a little (Table S2, ESI†), the internal spaces of the catalyst layer are almost blocked to form a much compact catalyst layer on the paper substrate (Fig. S2e, ESI†). This unfavourable structure evolution would consequently lead to the decrease of the active sites on the electrode (Fig. S1c, S3c and d, ESI†), which is probably responsible for the HER and OER activity decay as shown in Fig. S2f, ESI.†
Electrochemical performance of the Ni–P–B/paper electrode toward electrolysis of water is evaluated using a three-electrode system in 1 M KOH solution. It is observed that negligible HER activity is achieved by bare paper or with Pt–C catalyst on it as shown in Fig. 2a, probably due to the poor inherent HER activity and electric conductivity of paper. Thus, paper is normally an unsuitable substrate for catalytic electrodes. Encouragingly, the as-prepared Ni–P–B/paper electrode shows remarkable HER activity, and its onset overpotential (vs. RHE) is as low as 76 and 276 mV at the current density of 50 mA cm−2 and 500 mA cm−2, respectively, superior to that of Ni–P–B/Ni foil, and Pt–C/Ni foil. Moreover, the Tafel slope of the electrode (39.2 mV dec−1) is also comparable to that of the Ni–P–B/Ni foil (46.5 mV dec−1), and Pt–C/Ni foil electrodes (93.2 mV dec−1) (Fig. S9a, ESI†). The turnover frequency (TOF) of the Ni–P–B catalyst is calculated to be 3.8 s−1 at an overpotential of 100 mV in 1.0 M KOH, assuming that all of the active materials in the catalysts are catalytically active for the HER. The TOF value is superior to that of Pt–C/Ni foil (1.24 s−1) electrodes, suggesting the excellent inherent HER activity of the Ni–P–B catalyst. It is also observed that the HER performance of the as-prepared Ni–P–B/paper electrode has surpassed those of many recently reported non-noble electrodes with a medium catalytic loading amount (Table S3, ESI†). The electrochemically active surface area (EASA) for the Ni–P–B catalyst is estimated from the electrochemical double-layer capacitance (Cdl) by measuring the cyclic voltammograms in a non-faradaic region of 0.02–0.12 V vs. RHE (Fig. S10, ESI†). The Cdl of the Ni–P–B/paper electrode (280 mF cm−2) indicates a quite large EASA, which is a distinctive feature for this electrode. Moreover, the charge transfer resistance (Rct) of the Ni–P–B/paper sample is significantly reduced to only 4 Ω for the optimized Ni–P–B/paper electrode after 60 min EP (Fig. S11, ESI†), suggesting that charge transfer can be largely facilitated in this electrode. Accordingly, the remarkable performance of the Ni–P–B/paper electrode toward the HER may be the result of an integration of highly active electrocatalyst, large EASA, and small reaction resistance (Fig. S12, ESI†).
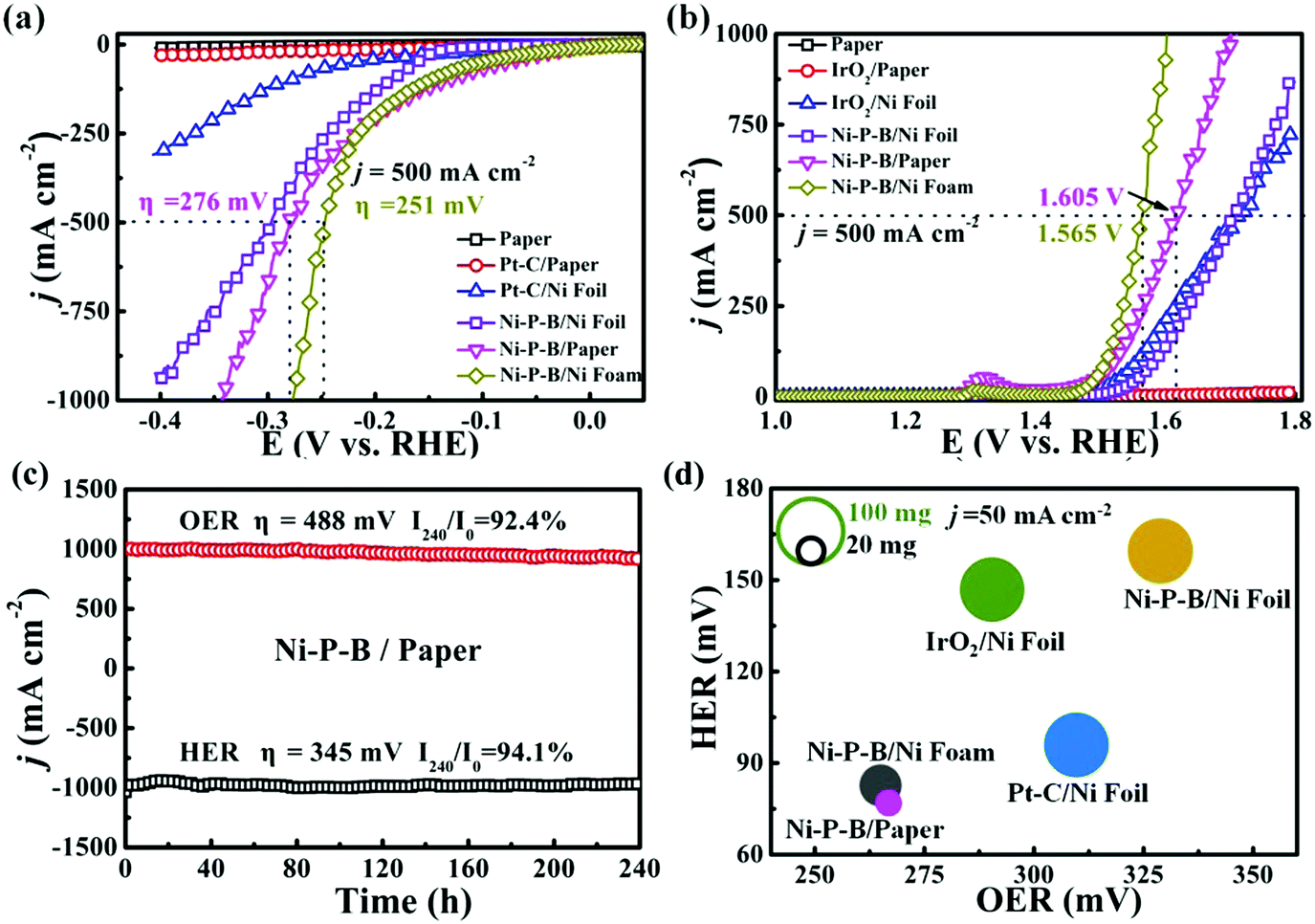 | ||
| Fig. 2 (a) iR-corrected polarization curves of bare paper, Ni–P–B/paper, Ni–P–B/Ni foil, Ni–P–B/Ni foam and Pt–C/paper electrodes at a scan rate of 2 mV s−1 for the HER. (b) iR-corrected polarization curves of bare paper, Ni–P–B/paper, Ni–P–B/Ni foil, Ni–P–B/Ni foam and commercial IrO2/paper electrodes at a scan rate of 2 mV s−1 for OER. (c) Chronopotentiometric measurements of long-term stability of Ni–P–B/paper at the current density of 1000 mA cm−2 for 240 h. (d) The HER and OER overpotential at 50 mA cm−2 and the weight of Ni–P–B/paper, Ni–P–B/Ni foil, Pt–C/Ni foil, IrO2/Ni foil and Ni–P–B/Ni foam electrodes. | ||
Concerning the OER performance, the Ni–P–B/paper electrode is continuously cycled between 1.1 V and 1.4 V vs. RHE at 2 mV s−1 until reproducible voltammograms are obtained before the test. It is found (Fig. 2b) that the electrode requires overpotentials of only 263 mV at 50 mA cm−2 and 375 mV at 500 mA cm−2 in 1 M KOH solution, superior to that of paper, IrO2/paper, Ni–P–B/Ni foil, and even IrO2/Ni foil electrodes. The Tafel slope of the Ni–P–B/paper electrode for the OER is measured to be 70.6 mV dec−1, which is lower than that of IrO2/Ni foil (82.7 mV dec−1), and Ni–P–B/Ni foil (74.3 mV dec−1) as shown in Fig. S9b, ESI.† The outstanding performances of Ni–P–B/paper on the OER ranks it among one of the best non-noble electrodes compared to most of the previous electrodes shown in Table S4, ESI.† The excellent inherent OER activity of the Ni–P–B catalyst is evidenced by its large TOF value of 3.45 s−1 at an overpotential of 300 mV compared to that of IrO2/Ni (2.37 s−1). Meanwhile, the Cdl of the Ni–P–B/paper electrode for the OER (89.1 mF cm−2) indicates a large EASA and thus benefits its OER activity (Fig. S13, ESI†). Furthermore, a very small Rct for the Ni–P–B/paper electrode is determined (5 Ω) (Fig. S14, ESI†) compared with many other electrodes (Table S5, ESI†). Thus, the Ni–P–B electrode can also achieve an integration of high activity, large EASA, and small reaction resistance toward the OER. To shed light on the significance of the composition and structure of the Ni–P–B/paper electrode, two extra Ni–B/paper and Ni–P/paper electrodes are also prepared by a similar method with about 6.0 mg cm−2 corresponding catalyst deposited on the paper substrate.
It is also observed that both the Ni–B/paper electrode (η50 = −117 mV for HER and η50 = 271 mV for OER) and Ni–P/paper electrode (η50 = −147 mV for HER and η50 = 282 mV for OER) exhibit excellent HER and OER performances as shown in Fig. S15, ESI.† However, these two electrodes are less efficient than the Ni–P–B/paper electrode toward both the HER and OER, suggesting the efficiency of simultaneous deposition of P and B toward the promoted performance. Apart from the favourable composition of the catalyst, the unique porous structure of the Ni–P–B/paper electrode may provide several advantages, including a large electrochemically active surface area and a large number of vent channels for the gas produced within the electrode. The Brunauer–Emmett–Teller (BET) surface area of the paper substrate is measured to be 0.77 m2 g−1, while the surface area of Ni foil is only 0.002 m2 g−1. Therefore, the porosity in the insulating substrate enables a larger surface area compared with a Ni foil substrate, which benefits the distribution of the catalyst and consequently leads to large surface area of the resulting Ni–P–B/paper electrode (8.2 m2 g−1 as shown in Fig. S16, ESI†), so that more active sites can be exposed to the electrolyte. According to further study on the Cdl of Ni–P–B/Ni foil electrode, it is evidenced that the EASA of Ni–P–B/paper electrode is much bigger than that of Ni–P–B/Ni foil with a similar catalyst and deposition amount (Cdl = 101.5 mF cm−2 for HER and Cdl = 36.7 mF cm−2 for OER, Fig. S10, S13, ESI†). Since the paper substrate is neither electrically conductive nor catalytically active, the large EASA feature of the Ni–P–B/paper electrode may be responsible for its superior performance over the Ni–P–B/Ni foil electrode. Furthermore, it is found that the paper electrode may have two beneficial factors toward a large number of active sites evidenced by the comparison on the structures of the Ni–P–B/paper and Ni–P–B/Ni foil electrodes (Fig. S17, ESI†). Firstly, the paper substrate has a macroporous structure which enables a larger accessible surface area than that of Ni foil; secondly, the catalyst layer on the paper substrate has rough surface morphology due to the formation of the nano electrocatalyst, rather than a compact catalyst layer formed on the Ni foil substrate. The efficiency of using the network substrate has already been proved on metal substrates,43,44 and is also evidenced by the Ni–P–B/Ni foam electrode constructed by a simple EP method. It is found that the bifunctional Ni–P–B/Ni foam electrode with the deposition amount of 6.1 mg cm−2 achieved the optimized performance, requiring overpotentials of only 251 mV and 335 mV to deliver a current density of 500 mA cm−2 for the HER and OER (Fig. 2 and Fig. S18, ESI†). Meanwhile, this electrode also has a low Tafel slope and satisfactory durability (Fig. S9 and S19, ESI†). Further studies reveal that large EASA (Cdl = 315.5 mF cm−2 for HER and Cdl = 95.6 mF cm−2 for OER, Fig. S10, S13, ESI†) and small Rct (Fig. S18, ESI†) have been achieved by the Ni–P–B/Ni foam electrode. A detailed comparison between the Ni–P–B/Ni foam electrode and Ni–P–B/paper electrode has been provided in Table S5, ESI.† Owing to the conductive nature and favourable porous 3D structure of the Ni foam substrate, the resulting electrode has achieved smaller reaction resistance and larger EASA as compared to a Ni–P–B/paper electrode. Consequently, the Ni–P–B/Ni foam electrode shows slightly better performance compared to the Ni–P–B/paper electrode at large current density.
Given the high activity of the Ni–P–B/paper electrode for both the HER and OER, we further studied the durability of the electrode which will be of great concern toward its practical applications. Encouragingly, satisfactory durability of the Ni–P–B/paper electrode for both the HER and OER in 1 M KOH solution is confirmed by 5000-cycles of continuous chronoamperometry scans in 1.0 M KOH (Fig. S20–S21, ESI†), and long-term stability tests at a large current density of 1000 mA cm−2 for 240 h (Fig. 2c) and 2000 mA cm−2 for 24 h (Fig. S22, ESI†). It is evidenced that the polarization curve after 5000 cycles shows negligible difference compared to the initial one after the OER and HER tests. The slight degradation of the current density at around 1000 mA cm−2 for 240 h at a set overpotential for both the HER and OER (HER: η1000 = 345 mV, I240/I0 = 94.1% and OER: η1000 = 488 mV, I240/I0 = 92.4%) suggests reliable high performance of the paper electrode for a long lifespan. The stability of the paper electrode can also be evidenced by the fact that negligible change in its morphology and structure is present during the 24 h HER and OER test at a current density of 1000 mA cm−2 (Fig. S23–S25, ESI†). Firstly, no obvious change in the cross section structure of the electrode is found after the test as shown in Fig. S23, ESI.† Secondly, there is still a layer of nano particles on the fibers of the paper electrode (Fig. S24 and S25, ESI†). Further investigation of degradation with the use of the electrode is necessary as its degradation mode may be different to the electrode with a conductive scaffold, to enable deeper understanding of the degradation mechanism and to promote further studies to achieve more practical electrodes. It is also noticed that the paper electrode has a unique advantage of being light weight compared with traditional electrodes with a metal substrate. It is shown that the Ni–P–B/paper electrode (21.5 mg) is able to achieve remarkable HER and OER performance with only ca. 1/5 the weight of the Ni–P–B/Ni foil (108.5 mg), Pt–C/Ni foil and IrO2/Ni foil electrodes, and ca. 1/2 the weight of the Ni–P–B/Ni foam (44.2 mg, Table S5 and S6, ESI†) with the same size of 1 × 1 cm2 (Fig. 2d).
In addition to the well-retained morphology of the Ni–P–B/paper electrode, the amorphous phase of the electrode does not change after both the HER and OER according to the XRD measurements (Fig. S26, ESI†). However, it is found that the chemical states of the elements on the surface of the as-prepared Ni–P–B/paper electrode changed apparently after both the HER and OER according to the XPS measurements (Fig. 3 and Fig. S27, ESI†). For the post-HER samples (Fig. 3a1–a4), the elemental nickel (Ni0) peak, and peaks for boron in both the oxidized and reduced states weakened gradually with prolonging the working time and almost disappeared after 24 h operation.
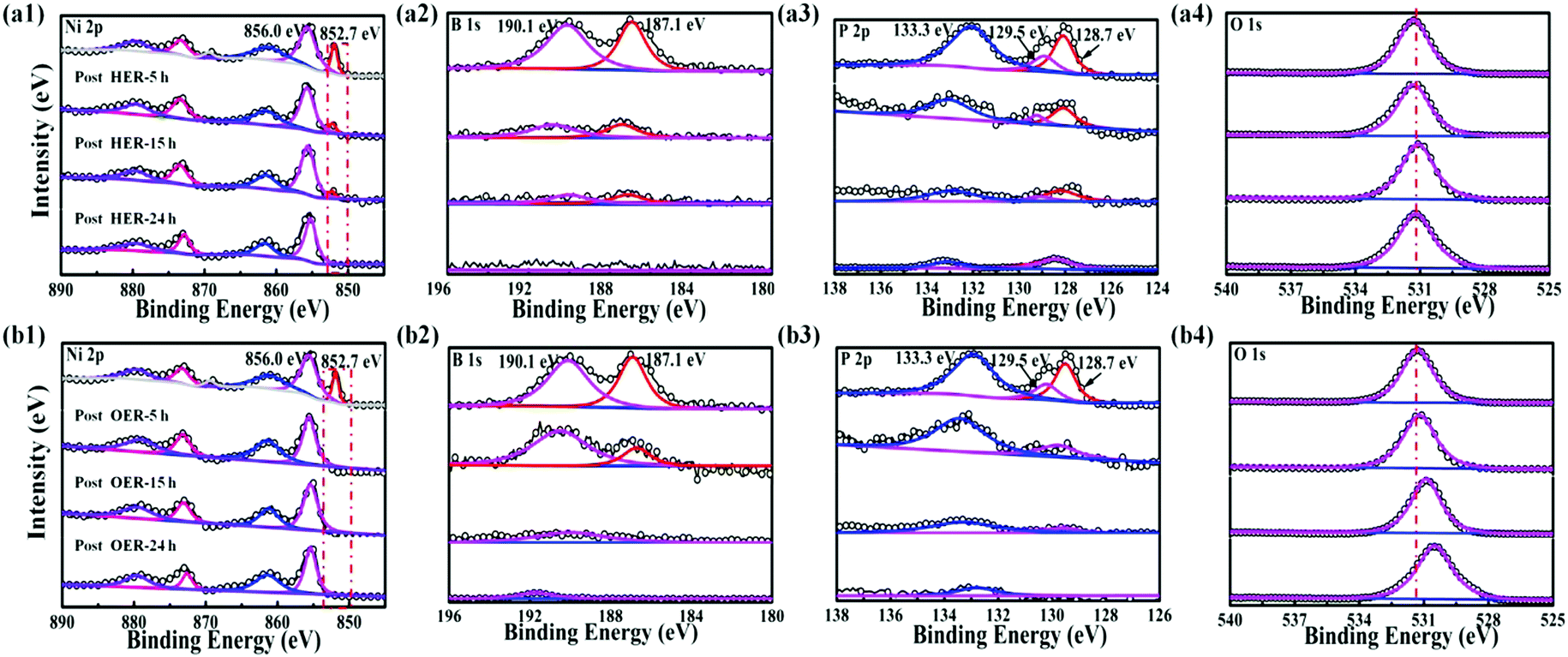 | ||
| Fig. 3 High-resolution XPS spectra of Ni 2p3/2, B1s, P 2p and O 1s for the Ni–P–B/paper electrode before and after HER (a1–a4) and OER (b1–b4) stability tests at 1000 mA cm−2 for 5 h, 15 h and 24 h. | ||
XPS results further reveal that nickel on the surface of the catalyst is mainly present in Ni(II) state, and peaks for phosphorous weaken after the HER test for 24 h, while B is almost leached out. Thus, it is supposed that the Ni–P–B catalyst will be covered by a layer of metal oxide after a long-term HER test. Concerning the post-OER electrode, the initial elemental Ni, P and B on the surface of the as-prepared electrode can no longer be observed during OER operation at 5, 15 and 24 h according to the XPS results (Fig. 3b1–b4). All the three elements are in their oxidized states after the OER. This may be the result of anodic oxidation on the surface of the catalyst. The O 1s spectra of the electrodes show a transition of surface dominated adsorbed oxygen species with the main O 1s peaks from 531.7 eV (Ni(OH)2) to 530.1 eV (NiOOH) (Fig. 3b4), which suggests that the surface of the post-OER electrode is mainly covered with a nickel oxyhydroxide (NiOOH) layer during the OER.45 The ICP-AES results (Table S1, ESI†) of post-HER (Ni:P:B = 18.1:1.0:2.0) and post-OER (Ni:P:B = 17.5:1.0:2.1) indicate only a little decrease of both P and B contents after the test as compared to the initial sample (Ni:P:B = 16:1.0:2.1), suggesting that the transformation would proceed only on the superficial area of the pre-catalyst. These phenomena agree well with many other electrodes.46,47
The above results strongly indicate that the as-prepared Ni–P–B phase act as a pre-catalyst that will transform to the catalytically active structures under electrochemical conditions. Meanwhile, P and B play crucial roles that can either facilitate the favourable transformation or promote the intrinsic activity of the catalyst. Under cathodic working conditions, elemental Ni is liable to be oxidized in the form of Ni–B48 and Ni–P,3 and thus can be slowly oxidized by the oxygen released from the anode to produce probably a layer of metal hydroxide on the Ni–P–B phase during the transformation.3 In the meantime, B and P on the surface were slowly leached out.49,50 The resulting layer could protect inner Ni–P–B from further oxidation. Meanwhile, the oxidation layer on the metal may also facilitate a “chimney effect” on the interface between the metal and oxidation layer for the HER, which could promote the catalytic activity of the composites.44 The Ni–P–B phase under the oxidation layer which is responsible for the conductivity of the electrode may provide real active sites of elemental P,49,51,52 Ni, and B on the interface of the composite for the HER as indicated in the previous reports.53–55 Moreover, the charge transfer within Ni–P–B would promote the intrinsic HER activity of the active sites. As compared to binding energies of pure B (187.1 eV, B 1s), Ni (852.6 eV, Ni 2p3/2) and P (130.1 eV, P 2p3/2), the elemental B and Ni in the as-prepared catalyst are positively shifted by 0.7 eV and 0.1 eV, while the elemental P in the catalyst is negatively shifted by 1.4 eV. These results suggest electron transfer from B and Ni to the vacant p-orbital of P making formers electron deficient while later enriched with electrons. Moreover, elemental P in Ni–P–B is more electron enriched than that of the Ni–P catalyst as indicated by the negative shift of 0.5 eV in their corresponding P elemental peaks,38 which is probably due to the existence of elemental B. It has been proved that the electronically enriched P atom sites are highly active for a catalytic reaction.56 As the elemental P is retained in the post-HER Ni–P–B catalyst (Fig. 3a3), the more electron enriched P in Ni–P–B/paper may be responsible for its better HER activity over the Ni–P/paper electrode (Fig. S15a, ESI†).
Concerning the OER, NiOOH species are developed on the surface of Ni–P–B during the OER test as evidenced by our results (Fig. 3). The surface NiOOH layer can play a dual role of acting as catalytic sites toward the OER57 and as a barrier coating to prevent complete oxidation of the conductive Ni–P–B layer on paper. It is noteworthy that the corresponding transformation can proceed easily under electrochemical conditions due to the presence of evident Ni2+ → Ni3+ redox peaks once the potential reaches around 1.3 V (RHE) as shown in Fig. 2b. During the transformation, B would promote the oxidation of nickel as suggested in previous reports, facilitating the formation of an active intermediate for the OER.48 Meanwhile, it is observed that the B and P elements at the surface are slowly leached out during the transformation. The Ni–P–B phase under the oxidation layer will mainly serve as the current collector of the electrode. Encouraged by the outstanding performances of the Ni–P–B/paper electrode for both the HER and OER, an electrolyzer in a two-electrode setup using the Ni–P–B/paper as both cathode and anode (Ni–P–B/paper‖Ni–P–B/paper) is made. Meanwhile, Paper‖Paper, Pt–C/paper‖IrO2/paper and Ni–P–B/Ni‖Ni–P–B/Ni alkaline water electrolyzers are also made for comparison. Owing to the poor conductivity of paper, neither paper nor the Pt–C/paper system works, while Ni–P–B/Ni foil, Ni–P–B/Ni foam and Ni–P–B/paper turn out to be excellent workable electrodes (Fig. 4a). Impressively, the novel Ni–P–B/paper system is much more efficient than the Ni–P–B/Ni foil system and comparable to the Ni–P–B/Ni foam system, delivering a current density of 50 mA cm−2 at a cell voltage of only 1.661 V, and can be driven by a 1.5 V commercial battery to produce evolution of both O2 and H2 (Fig. S28, ESI†).
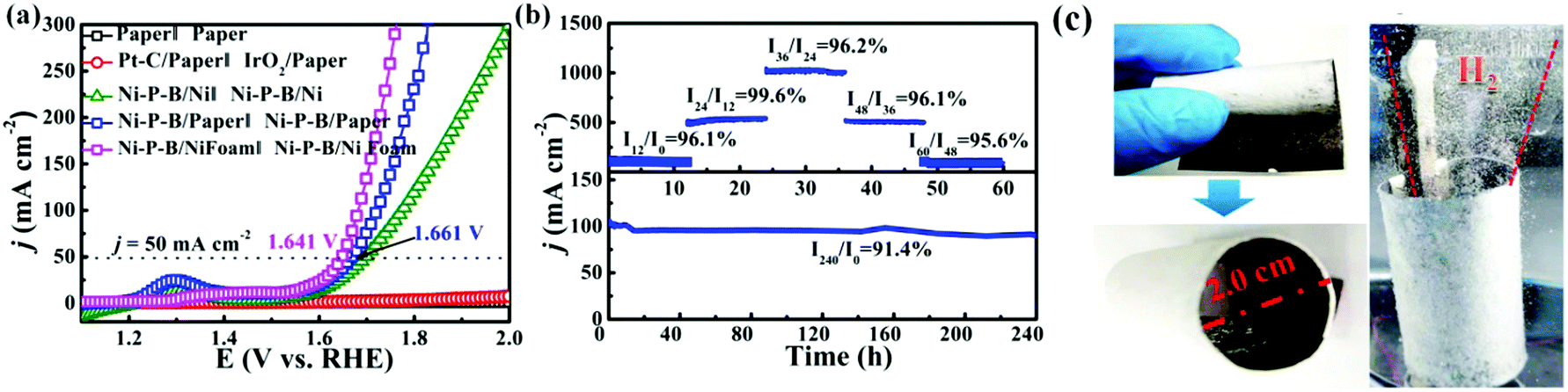 | ||
| Fig. 4 (a) Polarization curves of overall water splitting using the different electrodes as both cathode and anode. (b) Multistep chronopotentiometric test at 200, 500, 1000, 500 and 200 mA cm−2 for 12 h each, and long-term stability test carried out at 100 mA cm−2 for 240 h. (c) Fabrication and working photograph of the Ni–P–B/paper ring electrode. | ||
A multistep chronopotentiometric test for the Ni–P–B/paper electrolyzer from 200 to 1000 mA cm−2 is carried out as shown in Fig. 4b, and the retention of current density at each step is higher than 95%, implying the high stability of the Ni–P–B/paper electrode within a wide range of current densities, which will facilitate its practical application driven by an unstable power source. Moreover, the Ni–P–B/paper‖Ni–P–B/paper system also exhibits remarkably high stability with a current retention of 97% at 10 mA cm−2 after 24 h, and 91.4% at 100 mA cm−2 after 240 h electrolysis operation in 1.0 M KOH (Fig. 4b and Fig. S29, ESI†). The faradaic efficiency (FE) of the system is investigated and the cathode/anode shows stable hydrogen/oxygen evolution rates that match well with the theoretical values, implying a nearly 100% faradaic efficiency of the Ni–P–B/paper electrode (Fig. S30, ESI†). It is noteworthy that the Ni–P–B/paper electrode is comparable to most of the reported bifunctional electrodes with traditional substrates according to a detailed comparison of their overall water splitting performance in Table S7, ESI.† Moreover, a Ni–P–B/paper ring electrode (diameter = 2 cm) with in situ separation function has been constructed by deposition of the Ni–P–B catalyst on only one side of the filter paper substrate (Fig. 4c). The unique structure allows ions and water molecules but little gas product to pass through the electrode. It can be seen from the photo that the working ring electrode with a Ni–P–B catalyst on its inner surface enabled simultaneous generation, and collection of the gas product within the ring. Meanwhile, this structure may also protect the catalyst layer from the out-side environment. Currently, a small amount of gas is still observed on the outer side of the ring electrode after long time operation, but it is believed that the gas penetration can be minimized by further coating a gas-blocking layer on the outer side of the paper. Finally, the environment issues of the paper electrode can also be minimized as the metal materials on the electrode can be easily removed and recycled through the process shown in Fig. S31, ESI.†
To demonstrate the versatility of this strategy, several other electrodes of Co–B/paper, Ni–Co–B/paper, Ni–P–B/cloth, Co–B/cloth, Ni–Co–B/cloth, Ni–P–B/PU (PU: polyurethane sponge), Co–B/PU and Ni–Co–B/PU are prepared using a similar strategy. The digital photographs and FESEM images of the as-prepared electrodes indicate uniform coating of the boride-based catalysts on the surface of the insulating substrates, regardless of the composition of the catalyst and the material and geometry of the substrates (Fig. 5a–c). Their ICP-AES results are listed in Table S1, ESI,† which suggest the deposition of the corresponding catalysts on these insulating substrates. To give further perspective of their performances, the electrocatalytic activities of the boride-based electrodes are tested as shown in Fig. S32–S34, ESI.† According to these results, most of the as-prepared boride-based electrodes are highly active toward both the HER and OER, and can deliver 500 mA cm−2 current at relatively low overpotentials (Fig. S35, ESI†).
 | ||
| Fig. 5 Digital photographs and FESEM images (inset) of (a) Ni–P–B/paper, (b) Co–B/cloth and (c) Ni–Co–B/PU electrodes. (d) Comparison of the cell voltage of the as-prepared electrodes and other reported electrodes to drive overall water splitting in an alkaline electrolyte. | ||
Based on the polarization curves of water electrolysis in 1.0 M KOH (Fig. S36–S38, ESI†), Ni–P–B/paper‖Ni–P–B/paper, Ni–P–B/cloth‖Ni–P–B/cloth and Ni–P–B/PU‖Ni–P–B/PU systems demand cell voltages of only 1.796 V, 1.776 V and 1.781 V to afford 200 mA cm−2, respectively, which are superior to the corresponding Pt–C and IrO2 couple for overall water splitting as well as most of the traditional bifunctional electrodes with metal substrates (Fig. 5d). These results clearly illustrate the great potential of the strategy on the development of practical and cost efficient electrodes for overall water splitting using insulating substrates.
Conclusions
In summary, we have developed a family of robust catalytic electrodes by depositing conductive catalysts on insulating substrates of paper, cloth and sponge, which bring dramatic advantages of high performance, low cost, lightweight, eco-friendliness, flexibility, and simple and scalable fabrication. One of the electrolyzers assembled by a Ni–P–B/paper electrode which can survive at large current density of 1000 mA cm−2 for over 240 h, delivered a current density of 50 mA cm−2 at a cell voltage of only 1.661 V for overall water splitting. The remarkable performance of the catalytic electrodes may be attributed to a combination of high conductivity, active electrocatalyst and favourable structure. Further advances in the degradation performance could promote the practical application of the catalytic electrodes. Moreover, a ring Ni–P–B/paper electrode that enables in situ generation and collection of H2 and O2 products has been constructed by a filter paper substrate. This work may open scientific and engineering opportunities in the development of cost-effective, efficient and more eco-friendly electrodes for a wide variety of applications.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (51571063, 51672049, U1732268, 51727801, 51871060 and 61874128), Science and Technology Committee of Shanghai (17ZR1402500), the National Thousand Young Talents Program, China Postdoctoral Science Foundation (KLH2021056), Chinese-Austrian Cooperative R&D Project (GJHZ201950), and Key Project of Shanghai Polytechnic University (XXKZD1602).References
- Q. Zhu and Q. Xu, Energy Environ. Sci., 2015, 8, 478 RSC.
- S. Anantharaj, K. Karthick, S. Sam Sankar, K. Sangeetha and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- A. Sahasrabudhe, H. Dixit, R. Majee and S. Bhattacharyya, Nat. Commun., 2018, 9, 2014 CrossRef.
- D. Wang and D. Astruc, Chem. Soc. Rev., 2017, 46, 816–854 RSC.
- G. Li, G. R. Blake and T. M. Palstra, Chem. Soc. Rev., 2017, 46, 1693–1706 RSC.
- X. Wang, W. Li, D. Xiong, D. Y. Petrovykh and L. Liu, Adv. Funct. Mater., 2016, 26, 4067–4077 CrossRef CAS.
- Z. Zhao, D. E. Schipper, A. P. Leitner, H. Thirumalai, J. H. Chen, L. Xie, F. Qin, M. K. Alam, L. C. Grabow, S. Chen, D. Wang, Z. Ren, Z. Wang, K. H. Whitmire and J. Bao, Nano Energy, 2017, 39, 444–453 CrossRef CAS.
- T. Liu, D. Liu, F. Qu, D. Wang, L. Zhang, R. Ge, S. Hao, Y. Ma, G. Du, A. M. Asiri, L. Chen and X. Sun, Adv. Energy Mater., 2017, 7, 1700020 CrossRef.
- C. Niether, S. Faure, A. Bordet, J. Deseure, M. Chatenet, J. Carrey, B. Chaudret and A. Rouet, Nat. Energy, 2018, 3, 476–483 CrossRef CAS.
- T. Liu, K. Wang, G. Du, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2016, 4, 13053–13057 RSC.
- M. Xie, X. Xiong, L. Yang, X. Shi, A. M. Asiri and X. Sun, Chem. Commun., 2018, 54, 2300–2303 RSC.
- W. Hao, R. Wu, R. Zhang, Y. Ha, Z. Chen, L. Wang, Y. Yang, X. Ma, D. Sun, F. Fang and Y. Guo, Adv. Energy Mater., 2018, 1801372 CrossRef.
- C. Tang, R. Zhang, W. Lu, L. He, X. Jiang, A. M. Asiri and X. Sun, Adv. Mater., 2017, 29, 1700805 CrossRef PubMed.
- Y. Ge, P. Dong, S. R. Craig, P. M. Ajayan, M. Ye and J. Shen, Adv. Energy Mater., 2018, 1800484 CrossRef.
- C. You, Y. Ji, Z. Liu, X. Xiong and X. Sun, ACS Sustainable Chem. Eng., 2018, 6, 1527–1531 CrossRef CAS.
- X. Zhao, P. Pachfule, S. Li, J. R. J. Simke, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2018, 57, 8921–8926 CrossRef CAS PubMed.
- Y. Ha, L. Shi, Z. Chen and R. Wu, Adv. Sci., 2019, 1900272 CrossRef PubMed.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef CAS PubMed.
- Z. Chen, R. Wu, Y. Liu, Y. Ha, Y. Guo, D. Sun, M. Liu and F. Fang, Adv. Mater., 2018, 30, 1802011 CrossRef PubMed.
- P. Z. Zhang, F. R. Zhou, W. J. Zeng, H. B. Su, G. Li, J. H. Gao, R. Sun and C. P. Wong, ACS Nano, 2016, 10, 1273–1282 CrossRef PubMed.
- Y. Z. Wang, X. Lu, Z. J. Ma, C. Xie and Z. J. Zheng, Chem. Soc. Rev., 2018, 47, 4611 RSC.
- F. Han, Y. Wang, G. P. Zhang and C. P. Wong, J. Mater. Chem. C, 2018, 6, 8135 RSC.
- N. B. Sanjeev, K. Bhardwaj, R. Kaur, J. Mehta and A. L. Sharma, J. Mater. Chem. A, 2018, 6, 14992 RSC.
- B. Qiu, M. Xing and J. Zhang, Chem. Soc. Rev., 2018, 47, 2165–2216 RSC.
- S. Carenco, D. Portehault, C. Boissiere, N. Mezailles and C. Sanchez, Chem. Rev., 2013, 113, 7981–8065 CrossRef CAS PubMed.
- D. W. Shin, M. D. Barnes, K. Walsh, D. Dimov, P. Tian, A. I. S. Neves, C. D. Wright, S. M. Yu, J. B. Yoo, S. Russo and M. F. Craciun, Adv. Mater., 2018, 30, 1802953 CrossRef PubMed.
- S. Gupta, N. Patel, A. Miotello and D. C. Kothari, J. Power Sources, 2015, 279, 620–625 CrossRef CAS.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6, 1502313 CrossRef.
- S. Gupta, N. Patel, R. Fernandes, S. Hanchate, A. Miotello and D. C. Kothari, Electrochim. Acta, 2017, 232, 64–71 CrossRef CAS.
- P. Zhang, M. Wang, Y. Yang, T. Yao, H. Han and L. Sun, Nano Energy, 2016, 19, 98–107 CrossRef CAS.
- M. M. Madrigal, E. Armelin, F. Sanzcde and C. Aleman, Polym. Chem., 2014, 5, 1248 RSC.
- Y. Uchida, S. Nakandakari, K. Kawahara, S. Yamasaki, M. Mitsuhara and H. Ago, ACS Nano, 2018, 8, 03055 Search PubMed.
- W. Wu, Nanoscale, 2017, 9, 7342–7372 RSC.
- S. Escolastico, S. C. Soli, C. Kjolseth and J. M. Serra, ACS Appl. Mater. Interfaces, 2017, 9, 35749–35756 CrossRef CAS PubMed.
- C. Z. Cai, A. Khan, L. Q. Wang and W. D. Li, ACS Appl. Mater. Interfaces, 2018, 10, 28754–28763 CrossRef PubMed.
- Y. H. Liang, A. M. Asiri and Y. Q. He, Nanotechnology, 2016, 27, 12LT01 CrossRef PubMed.
- S. P. Lee, J. Mol. Catal. A: Chem., 2000, 152, 213–223 CrossRef CAS.
- W. L. Wang, D. H. Xiong and L. F. Liu, J. Mater. Chem. A, 2016, 4, 5639 RSC.
- L. F. Stern, F. Song and X. L. Hu, Energy Environ. Sci., 2015, 8, 2347 RSC.
- P. Li, Z. Jin and D. Xiao, J. Mater. Chem. A, 2014, 2, 18420–18427 RSC.
- L. Yuan, X. Xiao, T. Ding, J. Zhong, X. Zhang, Y. Shen, B. Hu, Y. Huang, J. Zhou and Z. L. Wang, Angew. Chem., Int. Ed., 2012, 51, 4934–4938 CrossRef CAS PubMed.
- L. Hu, M. Pasta, F. L. Mantia, L. Cui, S. Jeong, H. D. Deshazer, J. W. Choi, S. M. Han and Y. Cui, Nano Lett., 2010, 10, 708–714 CrossRef CAS PubMed.
- Z. Zhao, H. Wu, H. He, X. Xu and Y. Jin, Adv. Funct. Mater., 2014, 24, 4698–4705 CrossRef CAS.
- I. K. Mishra, J. Y. Sun, F. Qin, K. Dahal and J. M. Bao, Energy Environ. Sci., 2018, 11, 2246 RSC.
- J. Masa, I. Sinev, H. Mistry, E. Ventosa, M. de la Mata, J. Arbiol, M. Muhler, B. Roldan Cuenya and W. Schuhmann, Adv. Energy Mater., 2017, 7, 1700381 CrossRef.
- X. Wang, W. Li, D. Xiong, D. Y. Petrovykh and L. Liu, Adv. Funct. Mater., 2016, 26, 4066 CrossRef.
- L. A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 RSC.
- W. Yuan, X. Zhao, W. Hao, J. Li, L. Wang, X. Ma and Y. Guo, ChemElectroChem, 2018, 6, 764–770 CrossRef.
- J. Luo, G. Su, Y. L. Tang, H. Q. Liu, F. Y. Tian and D. Lia, J. Mater. Chem. A, 2017, 5, 14865–14872 RSC.
- W. B. Lu, L. S. Xie, C. Tang, D. N. Liu, S. Hao, F. L. Qu, Y. M. Gu Du, A. M. Asiri and X. P. Sun, Small, 2017, 13, 1700805 CrossRef PubMed.
- P. Prashanth, W. Menezes, S. Loos, C. W. Florian Bunschei-Bruns, M. Schwarze and X. H. Deng, Energy Environ. Sci., 2018, 11, 1287–1298 RSC.
- J. L. Zheng, T. Liu, S. J. Liu, C. B. Wang and L. Guo, Nanoscale, 2017, 9, 4409 RSC.
- L. Peng, X. Zheng, L. Li, L. Zhang, N. Yang, K. Xiong, H. Chen, J. Li and Z. Wei, Appl. Catal., B, 2019, 245, 122–129 CrossRef CAS.
- L. P. Zheng, L. Li, N. Yang, Y. J. Yang, J. Li, J. C. Wang and Z. Wei, Chem. Sci., 2018, 9, 1822 RSC.
- J. Wang, S. Mao, Z. Liu, Z. Wei, H. Wang, Y. Chen and Y. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 7139–7147 CrossRef CAS PubMed.
- T. Liu, A. Li, C. Wang, W. Zhou, S. Liu and L. Guo, Adv. Mater., 2018, 1803590 CrossRef PubMed.
- P. W. Menezes, C. Panda, S. Loos, F. Bunschei-Bruns, C. Walter, M. Schwarze, X. Deng, H. Dau and M. Driess, Energy Environ. Sci., 2018, 11, 1287 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee00839j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |