
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactions of Rh(PNP) pincer complexes with terminal alkynes: homocoupling through a ring or not at all†
Thomas M.
Hood
 and
Adrian B.
Chaplin
*
and
Adrian B.
Chaplin
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: a.b.chaplin@warwick.ac.uk
First published on 4th November 2020
Abstract
Through use of a bespoke macrocyclic variant, we demonstrate a novel approach for tuning the reactivity of rhodium PNP pincer complexes that enables formation of conjugated enynes from terminal alkynes, rather than vinylidene derivates. This concept is illustrated using tert-butylacetylene as the substrate and rationalised by a ring-induced switch in mechanism.
The transition metal-mediated coupling of terminal alkynes into conjugated enynes is an attractive and atom-economic method for the preparation of conjugated enynes.1,2 Whilst this is a conceptually simple reaction, the formal addition of the C(sp)–H bond of one alkyne across the C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond of another is a process that can and often does result in mixtures of different 1,3-enyne isomers by virtue of head-to-tail (gem-) and/or head-to-head coupling (E- and Z-). In this context, the application of rigid mer-tridentate “pincer” ligands is particularly notable, with a number of systems capable of producing one enyne isomer with high fidelity.3,4 With regards to the work presented herein, the underlying mechanisms of these reactions invoke distinct pathways involving either alkyne insertion into a M–H bond (“hydrometallation”) or formation of a metal vinylidene intermediate (“vinylidene”; Scheme 1).1
C bond of another is a process that can and often does result in mixtures of different 1,3-enyne isomers by virtue of head-to-tail (gem-) and/or head-to-head coupling (E- and Z-). In this context, the application of rigid mer-tridentate “pincer” ligands is particularly notable, with a number of systems capable of producing one enyne isomer with high fidelity.3,4 With regards to the work presented herein, the underlying mechanisms of these reactions invoke distinct pathways involving either alkyne insertion into a M–H bond (“hydrometallation”) or formation of a metal vinylidene intermediate (“vinylidene”; Scheme 1).1
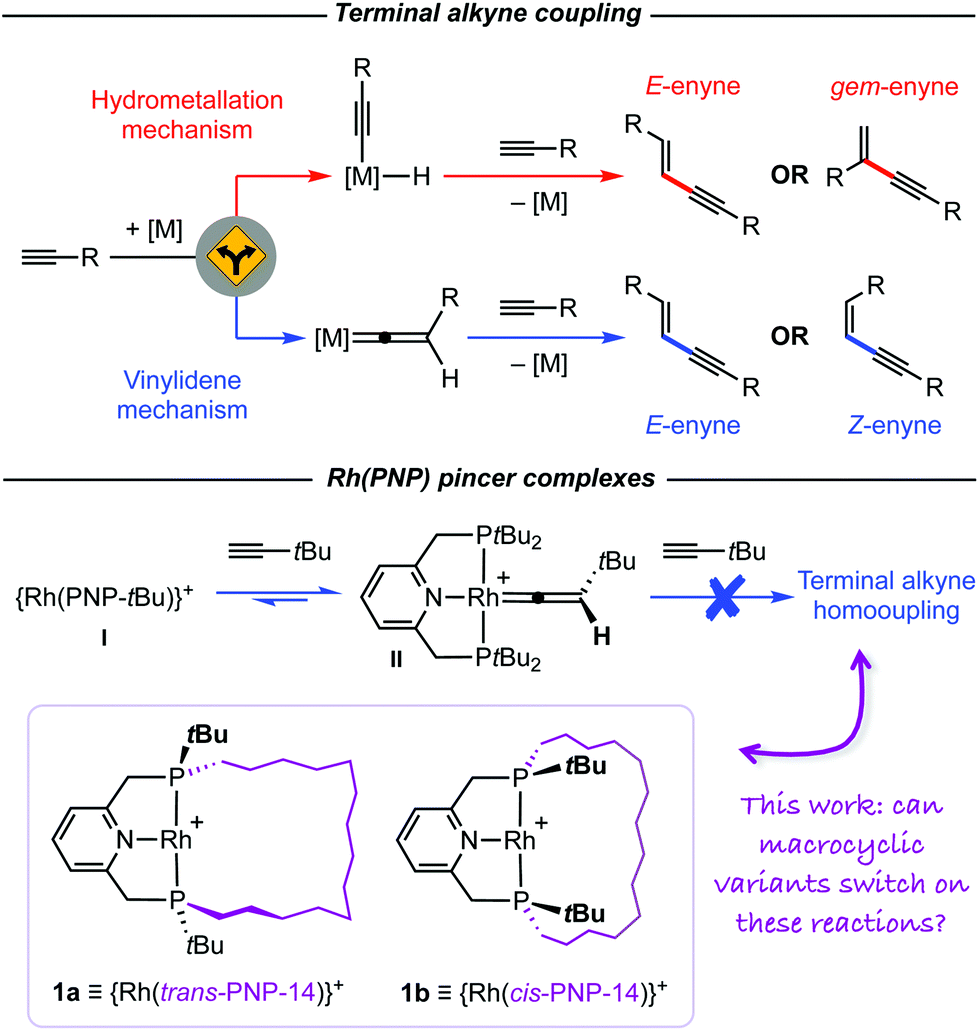 | ||
| Scheme 1 Terminal alkyne coupling reactions promoted by rhodium pincer complexes. | ||
As part of our work exploring the chemistry of phosphine-based pincer complexes of rhodium,5,6 we recently discovered that reaction of complex I with tert-butylacetylene resulted in the reversible formation of the vinylidene derivative II (Scheme 1).7 The corresponding alkynyl hydride was not observed, but species of this nature are established intermediates in alkyne/vinylidene tautomerisation reactions of rhodium(I) complexes.8,9 Whilst this complex is in principle an intermediate in the generation of tBuCCCHCHtBu via the vinylidene mechanism, in the presence of excess tert-butylacetylene we can confirm no homocoupling occurs, even upon prolonged heating at 80 °C in the weakly coordinating solvent 1,2-difluorobenzene (DFB).10 Having previously noted interesting effects when terminal alkyne coupling reactions are performed through the annulus of a macrocyclic ancillary ligand,4 we speculated that use of an appropriately designed PNP variant could destabilise the formation of vinylidene derivatives relative to the corresponding alkynyl hydride, and in doing so “switch on” the capacity to promote terminal alkyne homocoupling reactions. We herein present work evaluating this hypothesis using reactive rhodium(I) fragments 1, featuring PNP pincer ligands with P-donors that are either trans- or cis-substituted with a tetradecamethylene linker (Scheme 1).5 The linker traverses the coordination plane in 1a, counteracting formation of a vinylidene derivative, but is skewed to one side in 1b. The latter therefore represents a strictly isoelectronic control for the former (vide infra).
Emulating the method used in the synthesis of II, substitution of [Rh(COD)2][BArF4] (COD = 1,5-cyclooctadiene, ArF = 3,5-(CF3)2C6H3) in DFB was chosen to access the organometallic chemistry of the target pincer complexes 1a and 1b.7 Coordination of the macrocyclic pincer ligands is rapid and quantitative at RT, conferring [Rh(PNP-14)(η2-COD)]+ (1a′δ31P 57.4, 45.9, 2JPP = 312 Hz, 1JRhP = 131, 138 Hz resp.; 1b′δ31P 46.8, 1JRhP = 134 Hz) as the exclusive rhodium derivatives in solution by 1H and 31P NMR spectroscopy. Generation of [{Rh(PNP-14)}2(μ2–η2:η2-COD)]2+1′′ under equilibrium is also implied, as these dications are ultimately the products obtained upon crystallisation in both cases (see ESI† for solid-state structures). Going forward, generation of 1′in situ proved most expedient and addition of excess HCCtBu (2.5 equiv.) at RT afforded the corresponding vinylidene derivatives [Rh(PNP-14)(CCHtBu)]+2 in quantitative spectroscopic yield (Scheme 2), but under disparate timeframes. Complex 2a was formed within 5 min, but 2b required 42 h; indicating more strongly bound COD in this case. The formation of 2 are marked by distinctive deep green (2a)/blue (2b) colours in solution and exhibit 31P resonances at δ 58.6, 50.8 (2a, 2JPP = 312 Hz) and 50.1 (2b) that are coupled to 103Rh (1JRhP = 137–142 Hz). The vinylidene 13C resonances were located in both cases (2aδ 323.7; 2bδ 328.9) and are in good agreement with that of II (δ 317.5) and, moreover, other related rhodium precedents.7,9,11
 | ||
| Scheme 2 Terminal alkyne coupling reactions promoted by rhodium pincer complexes. Reactions in DFB at RT unless otherwise stated. Solid-state structures of 3a (not unique, Z′ = 2) and 2b: thermal ellipsoids drawn at 30% probability, minor disordered components (1 × tBu group, 3a; methylene chain, 2b), anions, and most hydrogen atoms omitted. Selected bond lengths (Å) and angles (°): 3a, Rh1–P2, 2.316(2); Rh1–P3, 2.323(2); Rh1–N101, 2.080(7); P2–Rh1–P3, 161.16(9); Rh1–Cnt(C2,C3), 2.042(5); C2–C3, 1.258(10); C2–C4, 1.431(10); C4–C5, 1.319(10); N101–Rh1–Cnt(C2,C3), 178.6(3); C2–C4–C5, 125.5(7); py-Rh–CC twist, 59.6(5); 2b, Rh1–P2, 2.2801(15); Rh1–P3, 2.2698(14); Rh1–N101, 2.116(5); Rh1–C4, 1.822(6); C4–C5, 1.311(9); C5–C6, 1.491(9); P2–Rh1–P3, 166.73(6); N101–Rh1–C4, 177.8(2); Rh1–C4–C5, 177.5(6); C4–C4–C6, 127.0(7). Cnt = centroid. | ||
In line with the hypothesis, 2a is characterised by low solution stability and we have so far been unsuccessful in isolating it from solution. In the presence of an excess of terminal alkyne, however, slow conversion into interpenetrated E-enyne complex 3a was observed in situ by NMR spectroscopy at RT (Scheme 2). This product was more expediently obtained by heating the reaction at 80 °C for 16 h, isolated in 87% yield, and fully characterised (δ31P 56.6, 51.0, 2JPP = 393 Hz, 1JRhP = 133, 129 Hz resp.; Rh-alkyne, 2.042(5) Å). For comparison, treatment of 1a′ with independently synthesised E-tBuCCCHCHtBu in DFB did not afford 3a, even upon heating at 80 °C,12 indicating that it can only result from homocoupling directly through the ring. Emulating II, 2b exhibits excellent solution-phase stability, was readily isolated in the solid state (90%), and showed no onward reaction with terminal alkyne (1.5 equiv.) upon prolonged thermolysis at 80 °C (16 h; Scheme 2). Inspection of the solid-state structure of 2b corroborates the formation of the vinylidene, with the Rh![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1.822(6) Å) and CC bonds (1.319(9) Å) in good agreement with those of II, and demonstrates the disposition of the methylene strap to one side of the complex; distinctly remote from the vinylidene, with all the RhCCHtBu{
C (1.822(6) Å) and CC bonds (1.319(9) Å) in good agreement with those of II, and demonstrates the disposition of the methylene strap to one side of the complex; distinctly remote from the vinylidene, with all the RhCCHtBu{![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3}⋯H2 contacts over 4 Å.
H3}⋯H2 contacts over 4 Å.
To gain deeper insight into the mechanism associated with the formation of 3a, isotope-labelling experiments were conducted. Heating 2a with excess DCCtBu (10 equiv.) in DFB at 80 °C resulted in extensive D incorporation into both positions of the enyne core of the product (totalling 83% D), indicating that reversible vinylidene formation is fast relative to its onward reactivity (ca. 2 × faster). Under the same conditions 54% D incorporation in the vinylidene was observed for 2b, consistent with slower retro-migration than in 2a. Supporting this assertion, the irreversible reaction of 2b with CO forming [Rh(cis-PNP-14)(CO)]+4b and liberating HCCtBu is appreciably slower than the equivalent reaction of 2a with CO, which likewise affords [Rh(trans-PNP-14)(CO)]+4a and HCCtBu.5 Incidentally, both carbonyl derivatives are characterised by ν(CO) bands at 1997 cm−1, as expected for ligands with equivalent donor properties,13 and slightly red-shifted to that of [Rh(PNP-tBu)(CO)]+ (1990 cm−1).5,14
Based on the observations presented herein – in particular the absence of onward reactivity of II and 2b, requirement for C–C bond formation to occur through the ring, and more facile retro-migration of the vinylidene in 2a compared to 2b – the production of 3a is best reconciled by a hydrometallation mechanism involving steady state formation of a rhodium(III) alkynyl hydride and not a vinylidene mechanism (Scheme 2); as hypothesised. More generally, this work showcases an unconventional approach for tuning the reactivity of pincer ligands15,16 and provides new insight into how terminal alkyne coupling reactions can be controlled.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council (ERC, grant agreement 637313) and Royal Society (UF100592, UF150675, A. B. C.) for financial support.Notes and references
- Q. Liang, K. Hayashi and D. Song, ACS Catal., 2020, 10, 4895–4905 CrossRef CAS; B. M. Trost and J. T. Masters, Chem. Soc. Rev., 2016, 45, 2212–2238 RSC; Y. Zhou, Y. Zhang and J. Wang, Org. Biomol. Chem., 2016, 14, 6638–6650 RSC.
- B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS; B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef.
- For representative examples see: N. Gorgas, B. Stöger, L. F. Veiros and K. Kirchner, ACS Catal., 2018, 8, 7973–7982 CrossRef CAS; N. Gorgas, L. G. Alves, B. Stöger, A. M. Martins, L. F. Veiros and K. Kirchner, J. Am. Chem. Soc., 2017, 139, 8130–8133 CrossRef; O. Rivada-Wheelaghan, S. Chakraborty, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 6942–6945 RSC; G. Kleinhans, G. Guisado-Barrios, D. C. Liles, G. Bertrand and D. I. Bezuidenhout, Chem. Commun., 2016, 52, 3504–3507 CrossRef; J. Alós, T. Bolaño, M. A. Esteruelas, M. Oliván, E. Oñate and M. Valencia, Inorg. Chem., 2014, 53, 1195–1209 CrossRef; C. J. Pell and O. V. Ozerov, ACS Catal., 2014, 4, 3470–3480 RSC; R. Ghosh, X. Zhang, P. Achord, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2007, 129, 853–866 Search PubMed; W. Weng, C. Guo, R. Çelenligil-Çetin, B. M. Foxman and O. V. Ozerov, Chem. Commun., 2006, 254, 197–199 Search PubMed.
- C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Chem. – Eur. J. DOI:10.1002/chem.202002962; C. M. Storey, A. Kalpokas, M. R. Gyton, T. Krämer and A. B. Chaplin, Chem. Sci., 2020, 11, 2051–2057 RSC; C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Angew. Chem., Int. Ed., 2018, 57, 12003–12006 CrossRef CAS.
- T. M. Hood, M. R. Gyton and A. B. Chaplin, Dalton Trans., 2020, 49, 2077–2086 RSC.
- B. Leforestier, M. R. Gyton and A. B. Chaplin, Angew. Chem., Int. Ed. DOI:10.1002/anie.202009546; B. Leforestier, M. R. Gyton and A. B. Chaplin, Dalton Trans., 2020, 49, 2087–2101 RSC; M. R. Gyton, B. Leforestier and A. B. Chaplin, Angew. Chem., Int. Ed., 2019, 58, 15295–15298 CrossRef CAS; T. M. Hood, B. Leforestier, M. R. Gyton and A. B. Chaplin, Inorg. Chem., 2019, 58, 7593–7601 CrossRef.
- M. R. Gyton, T. M. Hood and A. B. Chaplin, Dalton Trans., 2019, 48, 2877–2880 RSC.
- J. M. Lynam, Chem. – Eur. J., 2010, 16, 8238–8247 CrossRef CAS.
- M. J. Cowley, J. M. Lynam and J. M. Slattery, Dalton Trans., 2008, 4552–4554 RSC; M. Schafer, J. Wolf and H. Werner, Organometallics, 2004, 23, 5713–5728 CrossRef CAS; M. Schafer, J. Wolf and H. Werner, J. Organomet. Chem., 1995, 485, 85–100 CrossRef; M. Schafer, J. Wolf and H. Werner, J. Chem. Soc., Chem. Commun., 1991, 1341–1343 RSC; H. Werner and U. Brekau, Z. Naturforsch., 1989, 44, 1438–1446 Search PubMed; H. Werner, F. J. G. Alonso, H. Otto and J. Wolf, Z. Naturforsch., 1988, 43, 722–726 Search PubMed; F. J. G. Alonso, A. Höhn, J. Wolf, H. Otto and H. Werner, Angew. Chem., Int. Ed. Engl., 1985, 24, 406–408 CrossRef.
- S. D. Pike, M. R. Crimmin and A. B. Chaplin, Chem. Commun., 2017, 53, 3615–3633 RSC.
- R. Wiedemann, P. Steinert, M. Schaefer and H. Werner, J. Am. Chem. Soc., 1993, 115, 9864–9865 CrossRef CAS.
- In this case, a non-interpentrated enyne complex is initially formed (δ31P 44.3, 36.9, 2JPP = 408 Hz, 1JRhP = 131, 128 resp.). Extended thermolysis ultimately results in dehydrogenation of the tetradecamethylene linker alongside generation of E,E-tBuCHCHCHCHtBu from transfer hydrogenation of the enyne. Details are provided in the ESI.†.
- G. L. Parker, S. Lau, B. Leforestier and A. B. Chaplin, Eur. J. Inorg. Chem., 2019, 3791–3798 CrossRef CAS; L. Maser, C. Schneider, L. Vondung, L. Alig and R. Langer, J. Am. Chem. Soc., 2019, 141, 7596–7604 CrossRef; J. J. Davidson, J. C. DeMott, C. Douvris, C. M. Fafard, N. Bhuvanesh, C.-H. Chen, D. E. Herbert, C.-I. Lee, B. J. McCulloch, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2015, 54, 2916–2935 CrossRef.
- A. B. Chaplin and A. S. Weller, Organometallics, 2011, 30, 4466–4469 CrossRef CAS; M. Feller, E. Ben-Ari, T. Gupta, L. J. W. Shimon, G. Leitus, Y. Diskin-Posner, L. Weiner and D. Milstein, Inorg. Chem., 2007, 46, 10479–10490 CrossRef.
- This methodology can also be used to prepare the phosphinite analogue of 3a, [Rh(trans-PONOP-14)(E-tBuCCCHCHtBu)][BArF4] (δ31P 204.5, 194.5, 2JPP = 395 Hz, 1JRhP = 140, 133 resp.; Rh-alkyne, 2.061(2) Å). Details are provided in the ESI.†.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and characterisation data, including NMR spectra. CCDC 2025828–2025833. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03550e |
| This journal is © The Royal Society of Chemistry 2020 |