
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric bis-PNP pincer complexes of zirconium and hafnium – a measure of hemilability†
Celia
Idelson
a,
Leah
Webster
a,
Tobias
Krämer
 *b and
F. Mark
Chadwick
*a
*b and
F. Mark
Chadwick
*a
aDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, White City, Shepherds Bush, London, W12 0BZ, UK. E-mail: m.chadwick@imperial.ac.uk
bDepartment of Chemistry, Maynooth University, Maynooth, Co. Kildare, Ireland. E-mail: tobias.kraemer@mu.ie
First published on 10th November 2020
Abstract
Asymmetrically-bound pyrrolide-based bis-PNP pincer complexes of zirconium and hafnium have been formed. The [κ2-PNPPh][κ3-PNPPh]MCl2 species are in direct contrast to previous zirconium PNP pincer complexes. The pincer ligands are fluxional in their binding and the energy barrier for exchange has been approximated using VT-NMR spectroscopy and the result validated by DFT calculations.
Pincer ligands, tridentate ligands where a central ligating moiety is flanked by two others, occupy a privileged position in coordination chemistry. Their modular nature allows a chemist to fine-tune both the steric and electronic properties of the metal centre.1 Such design has allowed the development of active catalysts for a vast array of transformations. When first used by Van Koten in 1989 the term pincer ligand strictly applied to an ‘LXL’ ligand, where a central anionic carbon atom was flanked by two donors (which would force a meridianal geometry).2 This definition has been broadened to include several different central heteroatom donors including O, Si and N.1 For the last, an archetypical motif is that of PNP, where a central pyridine is flanked by methylene phosphines (Fig. 1, top). The non-innocence of this ‘LLL’ ligand has been exploited to accomplish difficult bond activations.3 A somewhat less frequently deployed PNP pincer ligand is one where the pyridine ring is replaced by a pyrrole ring,4,5 giving a formally anionic ‘LXL’ ligand ([PNPR], Fig. 1, bottom). What we found particularly interesting is that, for both PNP congeners, the phosphorus centres are usually further substituted with alkyl substituents rather than aryl groups. Since its first synthesis in 2012 the phenyl substituted pyrrolide-PNP pincer ligand has been relatively underused, especially given both the commercial availability of the precursors and its varied reactivity.6–14
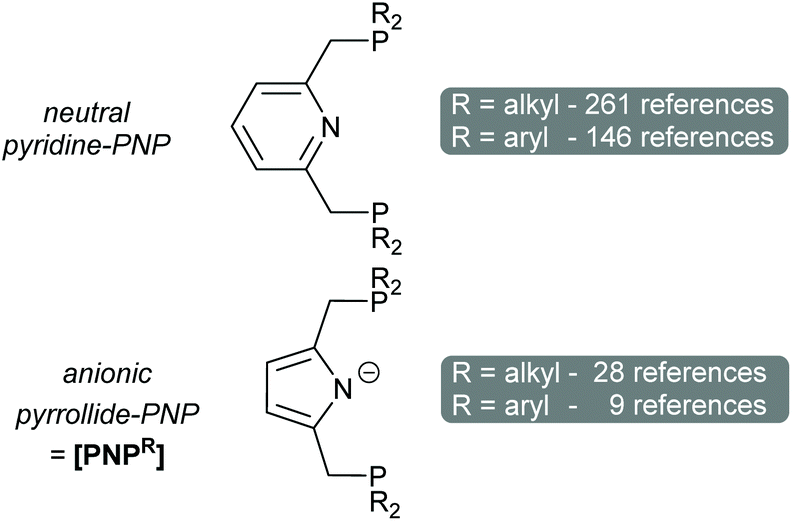 | ||
| Fig. 1 The general structures of pyridine-based and pyrrolide-based PNP ligands. Emboldened [PNPR] refers exclusively to the anionic pyrrolide-PNP, where R indicates the substituents on the phosphine. | ||
Our interest was also piqued by the recent resurgence of early transition metal pincer chemistry. Initially pioneered by Fryzuk and others from the 1970s onwards,15 it remains remarkably underdeveloped when compared with the later transition metals. However recent work has shown rich chemistry within early transition metal pincer complexes which are capable of facilitating extremely challenging transformations, for example the activation of methane.16
With the pyrrolide-based [PNPR] ligand (where R represents the substituents on the phosphines), the formally anionic pyrrolide should bind strongly, however the phosphorus centres will form comparatively weaker interactions with early transition metals versus later transition metals. This provides the tantalising prospect of hemilability. Early transition metal [PNPR] complexes have exclusively had alkyl substituents in the past. Sc[PNPR] and Y[PNPR] (R = Cy, tBu) have been shown to facilitate C–H activation, hydrosilylation and hydrogenation of olefins and support reactive methylidene complexes.17–19 Recently Nishibayashi and co-workers have shown that Ti[PNPtBu], V[PNPtBu] and Zr[PNPtBu] complexes are able to catalyse dinitrogen reduction.20,21 In order to explore the steric and electronic profile of pyrrolide-based PNP ligands we wished to investigate the synthesis and properties of the phenylated derivative [PNPPh] (Scheme 1).
 | ||
| Scheme 1 The synthesis of [PNPPh]2MCl2, M = Zr, Hf. (i) nBuLi in hexane and toluene, −78 °C then RT for 1 hour, THF extraction. (ii) ZrCl4·2THF/HfCl4·2THF. | ||
The synthesis of [PNPPh]H and its subsequent lithiation has been previously reported and is analogous to the alkylated congeners.12,17,20–29 A variety of lithium bases have been used in the literature but in our hands the conditions outlined in Scheme 1 and the ESI† worked best. The lithium salt could be isolated from an Et2O solution in very good yield (86%), but even after prolonged exposure to dynamic vacuum (16 h, <1 × 10−2 mbar) one equivalent of Et2O remained (this was not found to affect onward reactivity). Dimeric {[PNPPh]Li·THF}2 was crystallised by layering a THF solution with pentane. The structure is shown in Fig. 2.
 | ||
| Fig. 2 The single crystal X-ray structure of {[PNPPh]Li·THF}2. Thermal probability ellipsoids at 50%, hydrogen and non-ipso phenyl carbons omitted for clarity. Symmetry related labels marked with an apostrophe. Selected bond distances (Å): Li(1)–N(1)′ 2.532(5), Li(1)–C(1)′ 2.573(6), Li(1)–C(2)′ 2.544(6), Li(1)–C(3)′ 2.453(6), Li(1)–C(4)′ 2.441(5), Li(1)–N(1) 2.042(4), Li(1)–O(1) 1.957(5), Li(1)–P(1) 2.867(4). | ||
A dimeric structure of {Li[PNPiPr]}2 has been previously reported. In {Li[PNPiPr]}2 the Li bridges through the pyrrole nitrogens,25 but in {[PNPPh]Li·THF}2 it preferentially binds through a π-interaction with the pyrrolide ring. This π-interaction is significantly slipped towards an η3 coordination. This may be induced by the presence of the THF donor. It is unclear whether the dimer remains in solution with both 31P{1H} and 7Li{1H} NMR showing broad singlets and no coupling resolved.
Anticipating a [PNPPh]ZrCl3 stoichiometry in the product, initial reactions of [PNPPh]Li·Et2O with ZrCl4·2THF were carried out in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. However these reactions would give broad 31P{1H} NMR spectra and complex 1H NMR spectra. The synthesis was repeated with a 2:1 excess of [PNPPh]Li·Et2O (to ZrCl4·2THF). Despite the presumed excess of ligand in the reaction mixture, after one hour over 95% of the [PNPPh]Li·Et2O had been consumed. Subsequent work-up yielded the product in good yield (74% with respect to ZrCl4·2THF). Single crystals could be grown by layering a saturated THF solution with pentane and showed the structure to be an asymmetrical bis-pincer complex: [κ2-PNPPh][κ3-PNPPh]ZrCl2, [PNPPh]2ZrCl2, Fig. 3. Asymmetric binding of two pincer ligands is extremely rare.30 An analogous synthesis furnished the hafnium derivative, [PNPPh]2HfCl2, and crystals suitable for X-ray diffraction could be grown similarly (see ESI†). This complex is the first time a pyrrolide-PNP motif has been ligated onto hafnium. We have seen no evidence for a mono-[PNPPh] complex, even when ZrCl4·2THF is in excess and the Li salt is added slowly and at low temperature.
:1 ratio. However these reactions would give broad 31P{1H} NMR spectra and complex 1H NMR spectra. The synthesis was repeated with a 2:1 excess of [PNPPh]Li·Et2O (to ZrCl4·2THF). Despite the presumed excess of ligand in the reaction mixture, after one hour over 95% of the [PNPPh]Li·Et2O had been consumed. Subsequent work-up yielded the product in good yield (74% with respect to ZrCl4·2THF). Single crystals could be grown by layering a saturated THF solution with pentane and showed the structure to be an asymmetrical bis-pincer complex: [κ2-PNPPh][κ3-PNPPh]ZrCl2, [PNPPh]2ZrCl2, Fig. 3. Asymmetric binding of two pincer ligands is extremely rare.30 An analogous synthesis furnished the hafnium derivative, [PNPPh]2HfCl2, and crystals suitable for X-ray diffraction could be grown similarly (see ESI†). This complex is the first time a pyrrolide-PNP motif has been ligated onto hafnium. We have seen no evidence for a mono-[PNPPh] complex, even when ZrCl4·2THF is in excess and the Li salt is added slowly and at low temperature.
 | ||
| Fig. 3 The single crystal X-ray structure of [PNPPh]2ZrCl2. Thermal probability ellipsoids at 50%, hydrogen and non-ipso phenyl carbons omitted for clarity. Selected bond distances (Å) and angles (°): Zr(1)–Cl(1) 2.4412(10), Zr(1)–Cl(2) 2.4193(11), Zr(1)–N(1) 2.260(3), Zr(1)–N(2) 2.264(3), Zr(1)–P(1) 2.7935(10), Zr(1)–P(2) 2.8267(10), Zr(1)–P(3) 2.8390(11), Zr(1)–P(4) 4.836(1), Cl(1)–Zr(1)–Cl(2) 170.50(4), P(2)–Zr(1)–N(1) 67.06(9), N(1)–Zr(1)–P(1) 67.96 (9), P(1)–Zr(1)–N(2) 81.51(9), N(2)–Zr(1)–P(3) 67.97(9), P(2)–Zr(1)–P(3) 81.20(3). ∑(equatorial angles) 365.7°. | ||
The zirconium and hafnium complexes have very similar structures. Both crystallise in P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with near identical unit cells. Such similarity is common. The metal centre adopts a near-perfect pentagonal bipyramidal geometry (Cl(1)–Zr(1)–Cl(2): 170.50(4)°; ∑(equatorial angles) = 365.7°). One phosphorus centre is clearly pendant. Of the remaining phosphorus centres two, P(2) and P(3), are trans to the bisector of an N–Zr–P angle and one, P(1), is trans to the P–Zr–P angle bisector. These differing phosphorus environments are reflected in the low temperature 31P{1H} NMR studies. The individual bond lengths are similar to the comparable ones in Nishibayashi's bis(tert-butyl)phosphinopyrrolide complex {[PNPtBu]ZrCl2(μ-Cl)}2.21 Though unsymmetrical binding in a PNP pincer ligand is unprecedented κ2-[PNP] binding has been observed before in the homoleptic [κ2-PNPtBu]2Mn complex and in a DMAP substituted Sc complex.17,22 No significant disorder is observed in the crystal structure of either [PNPPh]2MCl2 species, implying the structures to be static in crystallographic conditions.
with near identical unit cells. Such similarity is common. The metal centre adopts a near-perfect pentagonal bipyramidal geometry (Cl(1)–Zr(1)–Cl(2): 170.50(4)°; ∑(equatorial angles) = 365.7°). One phosphorus centre is clearly pendant. Of the remaining phosphorus centres two, P(2) and P(3), are trans to the bisector of an N–Zr–P angle and one, P(1), is trans to the P–Zr–P angle bisector. These differing phosphorus environments are reflected in the low temperature 31P{1H} NMR studies. The individual bond lengths are similar to the comparable ones in Nishibayashi's bis(tert-butyl)phosphinopyrrolide complex {[PNPtBu]ZrCl2(μ-Cl)}2.21 Though unsymmetrical binding in a PNP pincer ligand is unprecedented κ2-[PNP] binding has been observed before in the homoleptic [κ2-PNPtBu]2Mn complex and in a DMAP substituted Sc complex.17,22 No significant disorder is observed in the crystal structure of either [PNPPh]2MCl2 species, implying the structures to be static in crystallographic conditions.
We were curious to investigate whether the structure in the solid-state reflects the behaviour in solution. Pincer ligands often have the dissociation of a single arm of the ligand inferred in mechanistic pathways and our system could allow us to investigate experimentally the energetics of this.31 Variable temperature NMR studies were therefore undertaken (Fig. 4).
 | ||
| Fig. 4 The variable temperature 31P{1H} NMR of [PNPPh]2MCl2 (M = Zr, Hf) in d8-toluene. Red spectrum is measured at 298 K, blue spectrum is measured at 203 K. For spectra at 10 K increments see ESI.† | ||
At room temperature a single 31P{1H} environment is observed, indicating fluxionality in the structure. However, on cooling to 203 K the 31P{1H} resolves into four clear environments. For both Zr and Hf congeners a high-field sharp singlet is observed at approximately δ −15.0, assigned as the free pendant phosphine ([PNPPh]H has a single 31P{1H} NMR resonance at δ −16.0). For [PNPPh]2ZrCl2 the remaining resonances resolve into a doublet at δ 16.3, and two remaining signals are overlapping at δ 15.0. In the case of [PNPPh]2HfCl2 the resonances resolve more clearly. Two of the low-field resonances appear as doublet of doublets [presumably corresponding to P(1) and P(3)] and a well-defined triplet [P(2)]. Assignments are proposed since P(2) is effectively identically trans to P(1) and P(3), therefore extremely similar coupling is expected.
Upon slowly warming the sample coalescence phenomena for both samples were observed. Coalescence of all resonances occurs simultaneously; however it is unlikely that direct exchange of the pendant phosphorus with all three others is degenerate. A more likely alternative is that P4 exchange with P2 and P3 is of similar energy, which could undergo sequential exchange with either P1 or P4. Using this model the exchange can be modelled as single-site exchange, and therefore a ΔG‡ = +11.3 kcal mol−1 can be estimated for [PNP]2ZrCl2 from the variable temperature 31P{1H} NMR (coalescence temperature 249 K, for [PNP]2HfCl2 coalescence temperature 270 K, ΔG‡ = + 12.1 kcal mol−1). These values agree well with those calculated using DFT (vide infra). The somewhat elevated value for the hafnium congener could arise from the stronger Hf–P bond strength and implies a dissociative pathway for exchange.
In order to elucidate plausible mechanisms for the fluxional ligand exchange process, we performed DFT calculations using a truncated model complex in which Ph groups were replaced by Me groups. Despite the differences in electronic and steric properties of the resulting phosphines, we observe no qualitative effect on the proposed ligand exchange pathways that would alter our conclusions. A comparison of the geometries of the starting complex [PNPPh]2ZrCl2 and its truncated variant [PNPMe]2ZrCl2 optimised at the BP86/SDD/6-31G(d,p) level of theory, reveals that both give satisfactory agreement with the experimental counterpart (see ESI†). Ligand exchange occurs via a dissociative mechanisms, involving initial decoordination of one phosphine arm followed by coordination of P4. In the case of P1/P4 exchange, the rate-determining step is associated with P1 decoordination (ΔG‡ = + 11.7 kcal mol−1), while for the P2/P4 and P4/P3 exchange processes their respective overall barriers of ∼16.5 kcal mol−1 represent rate determining steps that are associated with a small rearrangement of the pyrrolide ring (Fig. 5). This rearrangement shifts the pyrrolide ring onto the vacant coordination site initially coordinated by the leaving phosphine. We note that we here report energies uncorrected for London dispersion effects. Inclusion of D3 correction across a large range of tested functionals adds up to 10 kcal mol−1 to barrier heights (see ESI†).
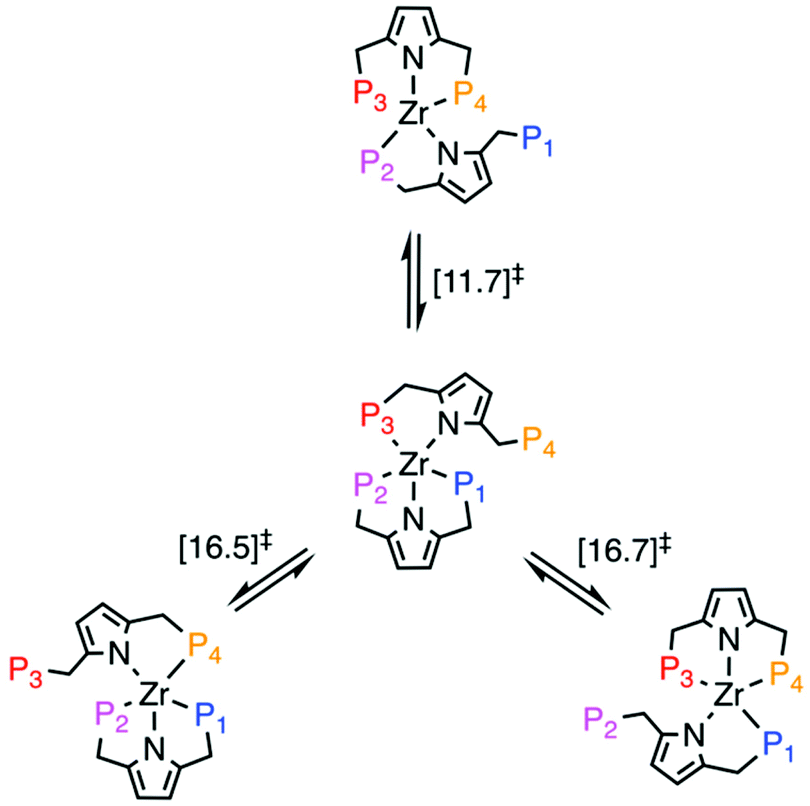 | ||
| Fig. 5 Summary of ligand exchange pathways for [PNPMe]2ZrCl2. Free Energies (BP86/def2-TZVP corrected for toluene solvent) of the rate-determining step in each pathway are given in kcal mol−1. | ||
Obtaining accurate ligand binding energies remains a challenge for quantum chemical methods.32 In particular, the use of polarisable continuum models often restricts the accuracy of predictions of dissociative processes. This is due to them falling short of correctly describing intermolecular solute-solvent interactions on one hand, and both intramolecular dispersive interactions and intermolecular solvent–solvent interactions on the other. It has been argued that the solvent can significantly attenuate inter- and intramolecular dispersive interactions.33,34 Nonetheless, we believe that the suggested mechanism for the ligand exchange is plausible and provides a rationale for the experimentally observed temperature-dependence of the NMR resonances.
In conclusion we have synthesised the first asymmetric bis-pincer complexes in good yield. We have shown that by simple alterations of the modular pincer ligand framework interesting binding modes can be found. We have investigated the fluxionality in these complexes and experimentally quantified the energetics of the processes involved. We also synthesised the first pyrrolide-PNP complex of hafnium.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- G. van Koten, Pure Appl. Chem., 1989, 61, 1681–1694 CAS.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS.
- D. J. Chadwick, in Chemistry of Heterocyclic Compounds, John Wiley & Sons, Ltd, 1990, vol. 48, pp. 1–103 Search PubMed.
- C. V. Thompson and Z. J. Tonzetich, in Advances in Organometallic Chemistry, Academic Press Inc., 2020, vol. 74, pp. 153–240 Search PubMed.
- N. Grüger, H. Wadepohl and L. H. Gade, Dalton Trans., 2012, 41, 14028 RSC.
- S. Kumar, G. Mani, S. Mondal and P. K. Chattaraj, Inorg. Chem., 2012, 51, 12527–12539 CrossRef CAS.
- G. T. Venkanna, T. V. M. Ramos, H. D. Arman and Z. J. Tonzetich, Inorg. Chem., 2012, 51, 12789–12795 CrossRef CAS.
- G. T. Venkanna, S. Tammineni, H. D. Arman and Z. J. Tonzetich, Organometallics, 2013, 32, 4656–4663 CrossRef CAS.
- S. Kumar, G. Mani, D. Dutta and S. Mishra, Inorg. Chem., 2014, 53, 700–709 CrossRef CAS.
- G. T. Venkanna, H. D. Arman and Z. J. Tonzetich, ACS Catal., 2014, 4, 2941–2950 CrossRef CAS.
- S. Kuriyama, K. Arashiba, K. Nakajima, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, Eur. J. Inorg. Chem., 2016, 2016, 4856–4861 CrossRef.
- S. Kumar, O. Jana, V. Subramaniyan and G. Mani, Inorg. Chim. Acta, 2018, 480, 113–119 CrossRef CAS.
- J. Madera, M. Slattery, H. D. Arman and Z. J. Tonzetich, Inorg. Chim. Acta, 2020, 504, 119457 CrossRef CAS.
- R. J. Burford, A. Yeo and M. D. Fryzuk, Coord. Chem. Rev., 2017, 334, 84–99 CrossRef CAS.
- T. Kurogi, J. Won, B. Park, O. S. Trofymchuk, P. J. Carroll, M.-H. Baik and D. J. Mindiola, Chem. Sci., 2018, 9, 3376–3385 RSC.
- D. S. Levine, T. D. Tilley and R. A. Andersen, Organometallics, 2015, 34, 4647–4655 CrossRef CAS.
- D. S. Levine, T. D. Tilley and R. A. Andersen, Organometallics, 2017, 36, 80–88 CrossRef CAS.
- D. S. Levine, T. D. Tilley and R. A. Andersen, Chem. Commun., 2017, 53, 11881–11884 RSC.
- Y. Sekiguchi, K. Arashiba, H. Tanaka, A. Eizawa, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2018, 57, 9064–9068 CrossRef CAS.
- Y. Sekiguchi, F. Meng, H. Tanaka, A. Eizawa, K. Arashiba, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Dalton Trans., 2018, 47, 11322–11326 RSC.
- A. L. Narro, H. D. Arman and Z. J. Tonzetich, Organometallics, 2019, 38, 1741–1749 CrossRef CAS.
- R. Kawakami, S. Kuriyama, H. Tanaka, K. Arashiba, A. Konomi, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Chem. Commun., 2019, 55, 14886–14889 RSC.
- J. A. Kessler and V. M. Iluc, Inorg. Chem., 2014, 53, 12360–12371 CrossRef CAS.
- M. Kreye, M. Freytag, P. G. Jones, P. G. Williard, W. H. Bernskoetter and M. D. Walter, Chem. Commun., 2015, 51, 2946–2949 RSC.
- S. Kuriyama, K. Arashiba, H. Tanaka, Y. Matsuo, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2016, 55, 14291–14295 CAS.
- N. Ehrlich, M. Kreye, D. Baabe, P. Schweyen, M. Freytag, P. G. Jones and M. D. Walter, Inorg. Chem., 2017, 56, 8415–8422 CAS.
- N. Ehrlich, D. Baabe, M. Freytag, P. G. Jones and M. D. Walter, Polyhedron, 2018, 143, 83–93 CrossRef CAS.
- Y. Sekiguchi, S. Kuriyama, A. Eizawa, K. Arashiba, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2017, 53, 12040–12043 RSC.
- P. Dani, T. Karlen, R. A. Gossage, W. J. J. Smeets, A. L. Spek and G. Van Koten, J. Am. Chem. Soc., 1997, 119, 11317–11318 CrossRef CAS.
- P. M. Pérez García, P. Ren, R. Scopelliti and X. Hu, ACS Catal., 2015, 5, 1164–1171 CrossRef.
- T. Husch, L. Freitag and M. Reiher, J. Chem. Theory Comput., 2018, 14, 2456–2468 CrossRef CAS.
- R. Pollice, M. Bot, I. J. Kobylianskii, I. Shenderovich and P. Chen, J. Am. Chem. Soc., 2017, 139, 13126–13140 CrossRef CAS.
- L. Yang, C. Adam, G. S. Nichol and S. L. Cockroft, Nat. Chem., 2013, 5, 1006–1010 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, variable temperature 1H NMR spectra, and further crystallographic details. CCDC 2023865–2023867. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03544k |
| This journal is © The Royal Society of Chemistry 2020 |