DOI:
10.1039/D0DT03315D
(Paper)
Dalton Trans., 2020,
49, 17183-17193
Bifunctional Fe(II) spin crossover-complexes based on ω-(1H-tetrazol-1-yl) carboxylic acids†
Received
23rd September 2020
, Accepted 26th October 2020
First published on 26th October 2020
Abstract
To increase the supramolecular cooperativity in Fe(II) spin crossover materials based on N1-substituted tetrazoles, a series of ω-(1H-tetrazol-1-yl) carboxylic acids with chain-lengths of C2–C4 were synthesized. Structural characterization confirmed the formation of a strong hydrogen-bond network, responsible for enhanced cooperativity in the materials and thus largely complete spin-state transitions for the ligands with chain lenghts of C2 and C4. To complement the structural and magnetic investigation, electronic spectroscopy was used to investigate the spin-state transition. An initial attempt to utilize the bifunctional coordination ability of the ω-(1H-tetrazol-1-yl) carboxylic acids for preparation of mixed-metallic 3d–4f coordination polymers resulted in a novel one-dimensional gadolinium-oxo chain system with the ω-(1H-tetrazol-1-yl) carboxylic acid acting as μ2-η2:η1 chelating–bridging ligand.
Introduction
First row transition metals with 3d4–3d7 electron configurations allow for population of their 3d-orbitals with either a maximum, or a minimum of paired electrons. Depending on the coordinative environment around the metal centre, the energetic difference between these electron configurations (high-spin (HS) and low-spin (LS) state) may be small enough to be governed by an external stimulus such as temperature,1,2 pressure,1–3 light4 or external electric field,5–8 among others.1 This phenomenon was first observed in the 1930s by Italian researchers, working on iron(III) N,N-dialkyldithiocarbamates and has since been known as spin crossover (SCO), or spin state transition.9–11
The intrinsic correlation between the bistability of SCO materials and several of their physical properties makes them appealing candidates for novel miniaturized sensors,12,13 memory/storage devices,14 and a key technology in spintronics.15 Today, SCO materials are primarily investigated by an academic bottom-up approach, resulting in highly specialized materials for devices on a nanoscale. Molecular actuators,16 their utilization in quenching and affecting luminescence and fluorescence,17,18 their sensing capacity in form of porous switchable materials,12,19 or their combination with chirality20–22 have recently been reported.
Since the occurrence of SCO under ambient conditions is very sensitive to the ligand field, it is strongly affected by the nature of the ligand system, the presence or absence of solvates23 and counter anions applied.24 (Substituted) azole ligands, including N1-substituted tetrazoles, are a promising platform for systematic development of SCO-materials.1
Homoleptic hexa-coordinated Fe(II)-complexes based on N1-substituted tetrazoles are often spin switchable, as coordination of Fe(II) by tetrazoles results in an appropriate ligand field strength. In this case, Fe(II) switches between a paramagnetic HS state with S = 2 and a diamagnetic LS state with S = 0.
In previous work, we have emphasized fine-tuning of the ligands’ geometry and flexibility, shaping cooperativity and transition temperature on a molecular scale,25,26 as well as post functionalization of the ligand backbone.27 Therefore, in this work, a carboxylic acid moiety has been introduced to the ligand system, allowing for the investigation of the SCO in [Fe(ω-(1H-tetrazol-1-yl) carboxylic acid)6]2+-cations. Alkylcarboxylic acid substituted tetrazoles provide three useful features: On a molecular level the COOH-group should establish a network of hydrogen bonds, increasing cooperativity and governing the abruptness of the spin state transition,28–30 the COOH-group may act as an additional ligand allowing for the formation of multi-metallic networks with a second transition metal, or rare-earth element and, finally, the COOH termination of the ligand may allow for deposition on oxide surfaces.
In this contribution, we report the ω-(1H-tetrazol-1-yl) carboxylic acids with C2–C4 alkyl chains and the magnetic, structural and spectroscopic characterization of their Fe(II)–SCO complexes, as well as an initial attempt to prepare a multi-metallic 3d–4f coordination polymer.
Results and discussion
Synthesis
The spin crossover behaviour of Fe(II)–N1-alkyl-substituted tetrazole complexes has been shown to be governed by the length of the N1-alkyl substituent and the choice of weakly coordinating anion.24,31 Therefore, in the current study, the length of the alkyl-skeleton was varied from C2–C4 (see Scheme 1) and BF4− and ClO4− were used as weakly coordinating tetrahedral anions of comparable volume (BF4− 49 Å3; ClO4− 54 Å3).
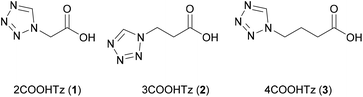 |
| | Scheme 1 ω-(1H-Tetrazol-1-yl) carboxylic acids used in this study, the number before the name displaying the chain-length. | |
The ligands 2–4COOHTz (1–3) were prepared from the corresponding amino-acids based on the Franke-tetrazole synthesis,32 in analogy to the reported synthesis of (1H-tetrazol-1-yl)-2-acetic acid (2).33–35 Treatment of the resulting N1-alkyl-substituted tetrazoles with FeX2·6H2O (X = BF4−, ClO4−) in acetonitrile (MeCN) followed by subsequent washing with THF results in the desired complexes. 1H NMR analysis of the bulk samples of all prepared complexes shows that only [Fe(2COOHTz)6]2+ incorporated solvent molecules, for both X = BF4−and ClO4− amounting to 2 MeCN per Fe2+ center.
Magnetic properties
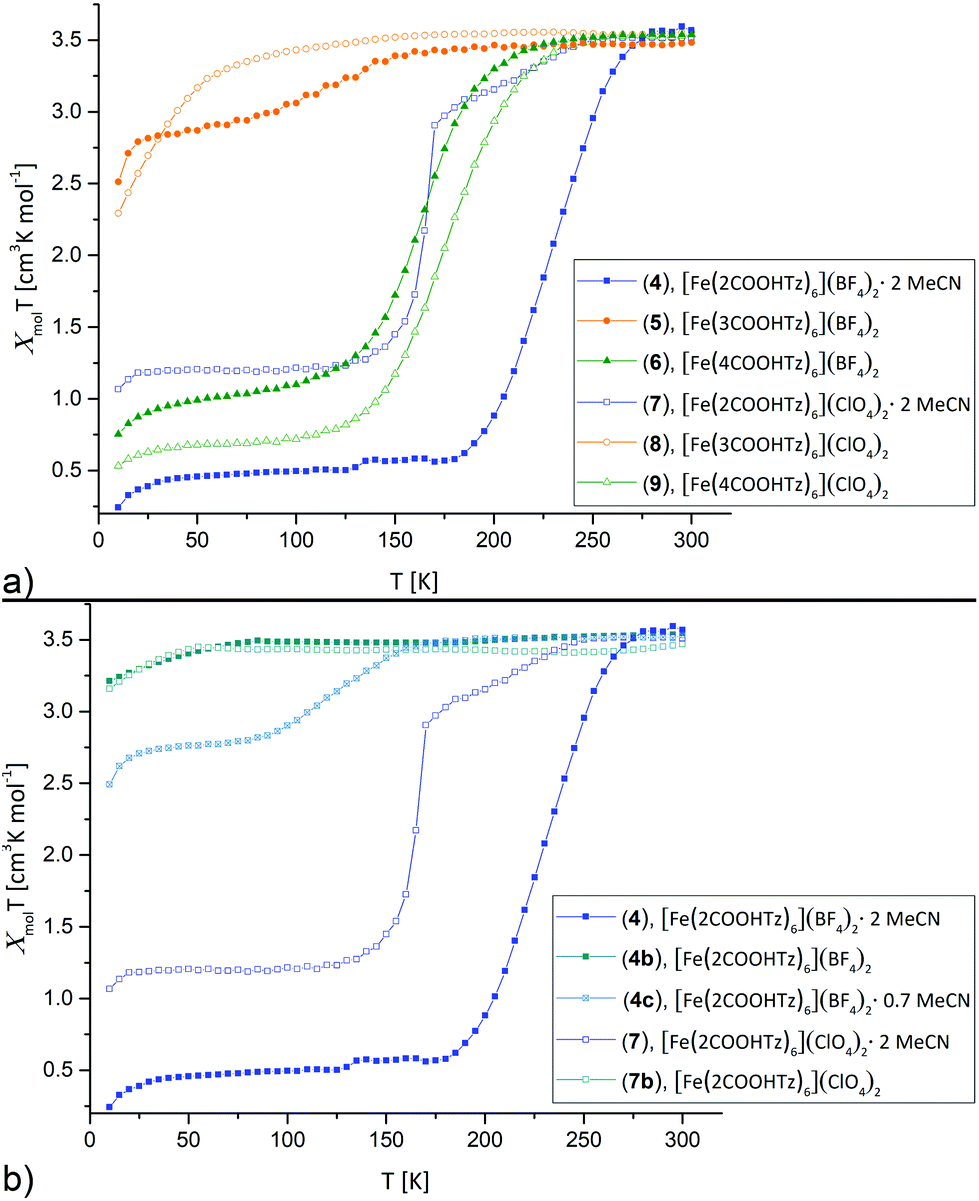
The dependence of molar magnetic susceptibility on temperature (10 to 300 K) was investigated for all six Fe(II)-complexes (Fig. 1a). The resulting χmolT curves show three distinct tendencies: partial, incomplete spin crossover (5), complete spin crossover (4, 6, 7 and 9) and no spin crossover (8). All changes in χmolT below 50 K may be attributed to zero-field splitting, expected for residual HS Fe(II).
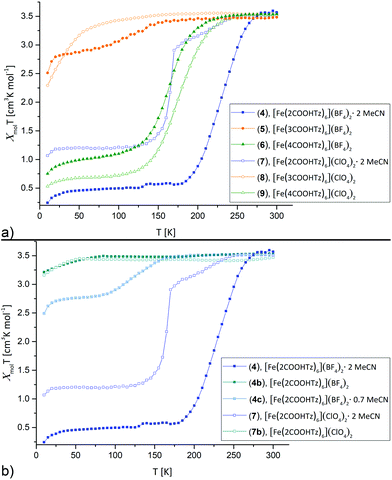 |
| | Fig. 1 Thermal variation of the molar magnetic susceptibility χmolT for [Fe(nCOOHTz)6]X2, n = 2–4, X = BF4−, ClO4− in the solid state between 10 K–300 K. Cooling and heating curves were found identical for all materials, so only the cooling data are displayed. (a) Overview on all materials. (b) Comparison for the solvated, partially solvated and desolvated samples of 4 and 7. | |
χ
mol
T decreases for 5 gradually below 170 K from 3.51 cm3 K mol−1 at 300 K to 2.86 cm3 K mol−1 at 50 K. This seems to correspond to ∼25% of Fe(II) sites in a LS state. [Fe(2COOHTz)6](BF4)2·2 MeCN (4) shows the highest T1/2 of the series with 227 K and decreases to a full LS-state below 180 K. [Fe(4COOHTz)6](BF4)2 (6) has a T1/2 of 163 K, the spin-state transition taking place gradually from below 235 K to a final χmolT of 0.98 cm3 K mol−1 at 50 K. This corresponds to ∼72% of LS-state.
Within the ClO4− compounds, [Fe(2COOHTz)6](ClO4)2·2 MeCN (7) reveals a two-step transition, featuring a kink at 170 K. Below 245 K χmolT of 7 decreases gradually to 2.9 cm3 K mol−1 at 170 K, pointing to a first HS–LS transition of ∼18% of the Fe(II) sites. A sharp decrease of χmolT to a constant value of 1.2 cm3 K mol−1 below 115 K is attributed to further 50% of the Fe(II) sites undergoing the HS–LS transition. [Fe(3COOHTz)6](ClO4)2 (8) remains all HS-state. [Fe(4COOHTz)6](ClO4)2 (9) shows by far the completest HS–LS transition with a T1/2 of 175 K and a final χmolT of 0.66 cm3 K mol−1 at 50 K, corresponding to only ∼18% of Fe(II)-centres remaining in the HS state.
The spin state transition in the BF4− and ClO4− complexes of 2COOHTz, both incorporating two molecules acetonitrile, are overly sensitive towards loss of the solvate. 4 partially loses the solvate already during drying in vacuum during the synthesis, or on shelf storage. This resulted in an intermediate with 0.7 MeCN molecules incorporated (4c, determined by 1H NMR) and on subsequent drying in vacuum in a non-solvate (4b), shown in Fig. 1b. For 0.7 MeCN molecules incorporated in 4c the spin state transition is shifted to lower temperatures with T1/2 at 132 K and a final χmolT of 2.75 cm3 K mol−1, corresponding to a ∼25% LS-state. The completely desolvated compound 4b is spin crossover inactive, remaining in the HS-state for all temperatures. Similarly, the desolvated ClO4−-complex 7b shows no spin crossover anymore (Fig. 1b). This demonstrates, how crucial the incorporated solvate molecules are in this case additional to the H-bonding network.
All in all, the differing picture obtained from this comparison of the SCO-behaviour is not only caused by the weak-coordinating anion (as obvious by comparing 4vs.7, or 6vs.9), but rather attributed to a combination of different effects. Additional to the impact of the anion, as discussed in the case of 2COOHTz the 2 MeCN solvate makes the game. Furthermore, there is the variation in the length of the alkyl-spacer, which for 4COOHTz seems to be a good fit between establishing cooperativity and preventing too much motional freedom in form of a shock-absorber effect,24,31 which was evidenced in the past for increasing chain-lengths. Missing the structural characterization of the 3COOHTz-compounds, the differing SCO-behaviour is probably related to the odd number of C-atoms, which already for unsubstituted alkyl-tetrazoles were found challenging in the past.24,31
Crystallographic analysis
Single crystals suitable for determination of the molecular structure could be grown for 4, 6, 7 and 9 by vapour-diffusion of diethyl ether (Et2O) into a concentrated MeCN-solution of the corresponding compounds. No suitable crystals of 5 and 8 could be obtained by similar means. Single crystal diffraction data were collected at 100 and 200 K for 4, 6, 7 and 9 in order to determine both low- and high-spin molecular structures. In addition variable temperature P-XRD patterns were acquired for 4 and 7 from 100–300 K.
[Fe(2COOHTz)6](BF4)2·2 MeCN (4).
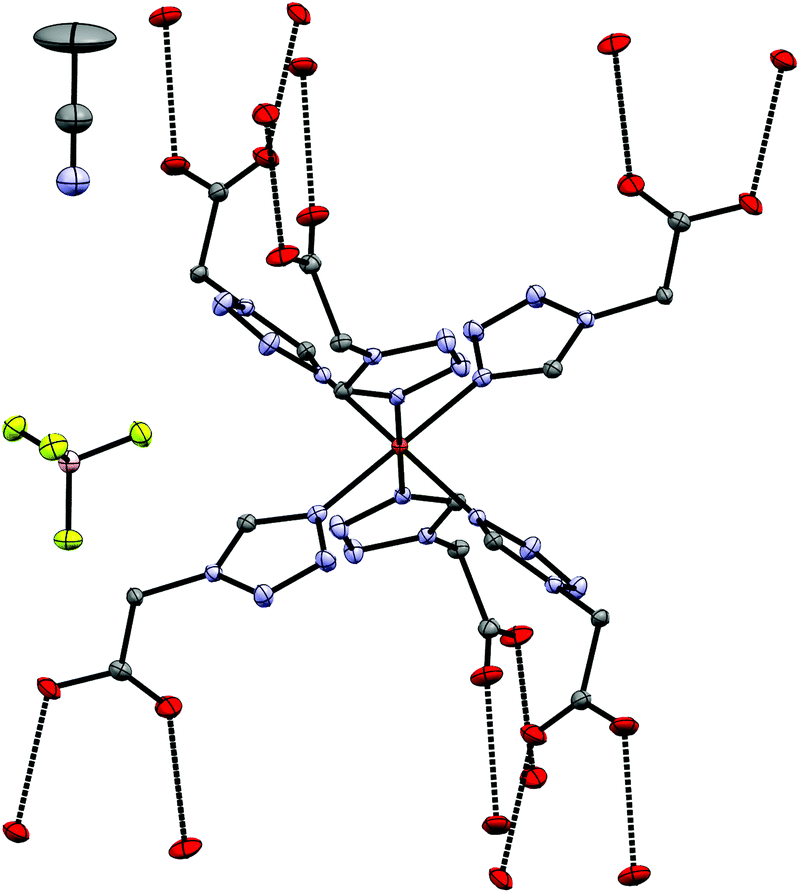
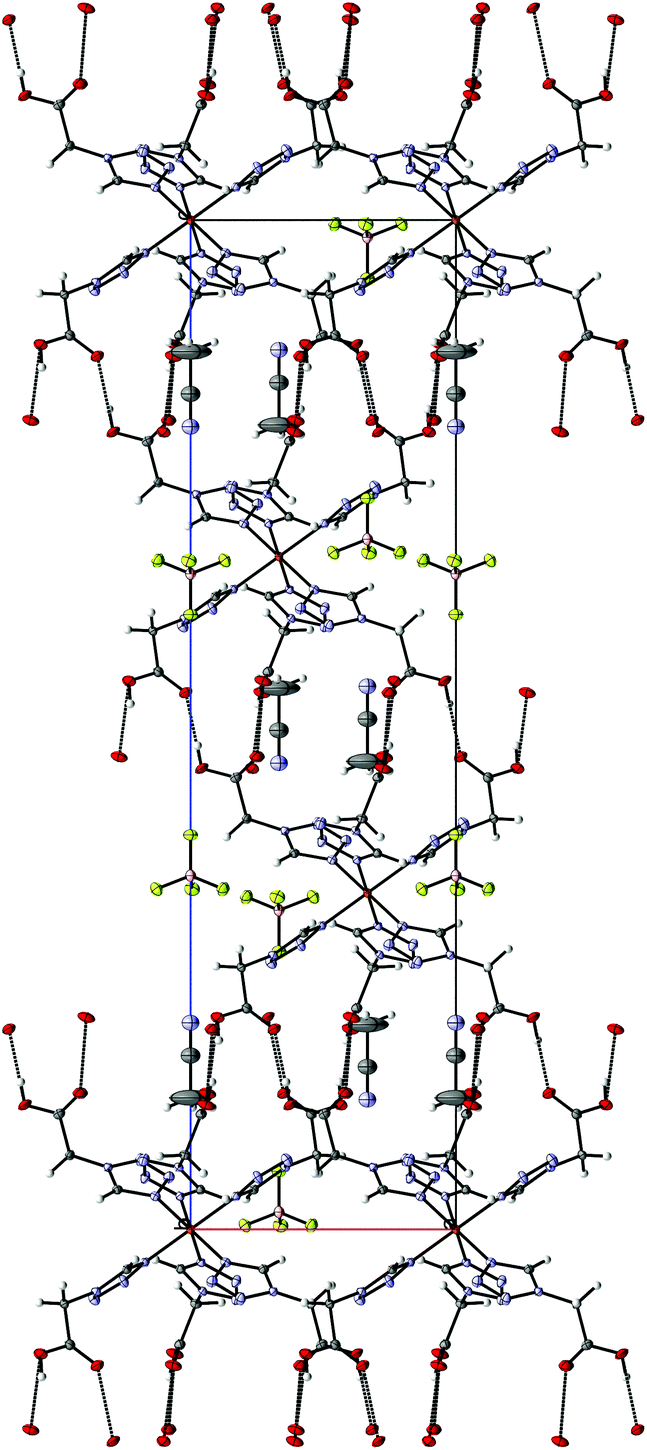
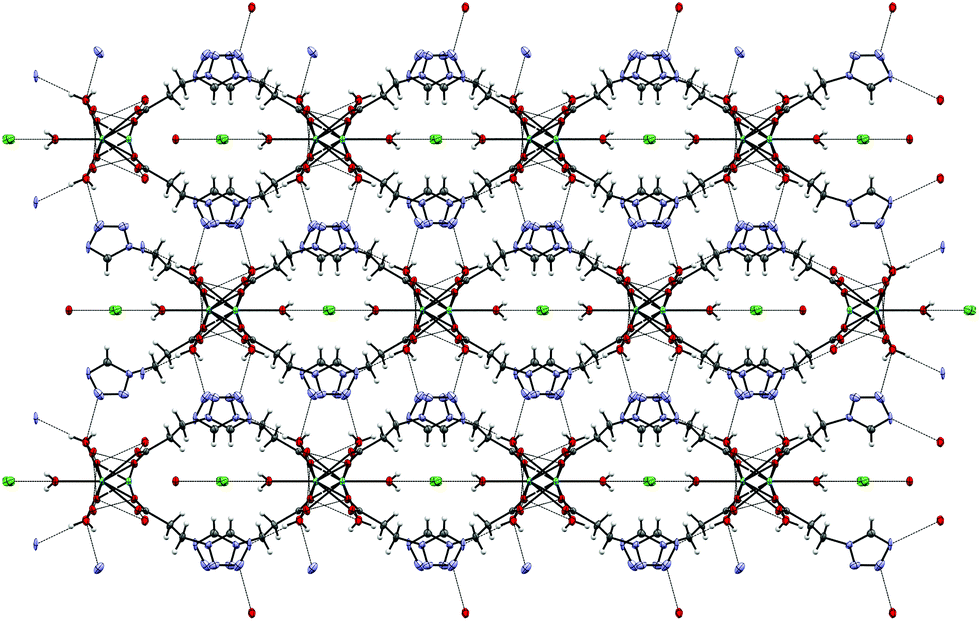
4 crystallizes as MeCN solvate in the trigonal R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group and this symmetry is retained both at 100 K (low-spin, see Fig. 2) and 200 K (high-spin). The iron-centre is octahedrally surrounded by six 2COOHTz-ligands coordinating via the exo N4–nitrogen. The Fe–N bond lengths for all six ligands are equal (1.987 Å), typical for a Fe(II) LS-state. At 200 K the distance increases ∼9% to 2.176 Å, characteristic for the Fe(II) HS-state (see Table S1†).1 The packing in the crystal is governed – as predicted for a COOH-group – by an H-bond network composed of infinite C(5) type chains between the COOH-groups. Viewed along the b axis, the [Fe(2COOHTz)6]2+-cations stack in layers, with three of the ligands pointing upwards and three downwards (see Fig. 2) to the adjacent layer. This arrangement is further stabilised by the above-mentioned H-bonds (see Fig. 3). The Fe-atoms are located on the Wyckoff-position 3a, alternating with an acetonitrile, an apex-up and an apex-down BF4− group. The BF4−groups interact strongly with the [Fe(2COOHTz)6]2+-cations through C–H⋯F-bonds with both the tetrazolic CH and CH2-group (see Fig. 4) in both the high- and low-spin states.
space group and this symmetry is retained both at 100 K (low-spin, see Fig. 2) and 200 K (high-spin). The iron-centre is octahedrally surrounded by six 2COOHTz-ligands coordinating via the exo N4–nitrogen. The Fe–N bond lengths for all six ligands are equal (1.987 Å), typical for a Fe(II) LS-state. At 200 K the distance increases ∼9% to 2.176 Å, characteristic for the Fe(II) HS-state (see Table S1†).1 The packing in the crystal is governed – as predicted for a COOH-group – by an H-bond network composed of infinite C(5) type chains between the COOH-groups. Viewed along the b axis, the [Fe(2COOHTz)6]2+-cations stack in layers, with three of the ligands pointing upwards and three downwards (see Fig. 2) to the adjacent layer. This arrangement is further stabilised by the above-mentioned H-bonds (see Fig. 3). The Fe-atoms are located on the Wyckoff-position 3a, alternating with an acetonitrile, an apex-up and an apex-down BF4− group. The BF4−groups interact strongly with the [Fe(2COOHTz)6]2+-cations through C–H⋯F-bonds with both the tetrazolic CH and CH2-group (see Fig. 4) in both the high- and low-spin states.
 |
| | Fig. 2 Molecular structure of 4 at 100 K in the LS-state showing H-bond contacts at the carboxylic acid groups; H-atoms omitted for clarity. | |
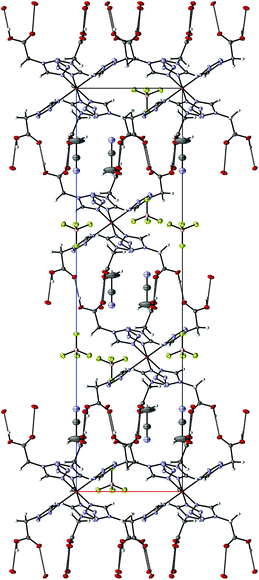 |
| | Fig. 3 H-Bonding network in 4 at 100 K, seen along the b-axis. | |
 |
| | Fig. 4 H⋯F bonds between the tetrazolic CH and the CH2-groups in 4 at 100 K. | |
[Fe(2COOHTz)6](ClO4)2·2 MeCN (7).
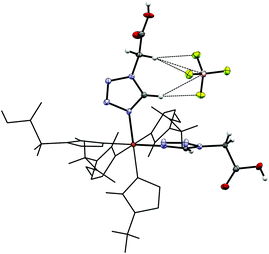
7 crystallizes isotypically to 4 with slightly larger cell parameters (see Table 1) as an MeCN solvate in the trigonal R space group. In 7 the Fe–N distances (2.004 Å, 100 K, LS-state; 2.184 Å, 200 K, HS-state, see Table S2†) are slightly longer than in 4, but still compar able and characteristic for Fe(II) in the corresponding spin-state. Also, in 7 both at 100 K and 200 K the ClO4−-anion is not disordered and stabilized through interaction with the [Fe(2COOHTz)6]2+-cation (see Fig. 5).
 |
| | Fig. 5 Hydrogen-bonds between the tetrazolic CH and the ClO4−-anion, as well as short interactions with the CH2-group in 7 at 100 K. | |
Table 1 Crystallographic parameters for the crystal structures of 4, 6, 7 and 9
| |
4·MeCN, HS |
4·MeCN, LS |
7·MeCN, HS |
7·MeCN, LS |
| Formula |
C22H30B2F8FeN26O12 |
C22H30B2F8FeN26O12 |
C22H30Cl2FeN26O20 |
C22H30Cl2FeN26O20 |
| Weight [g mol−1] |
1080.19 |
1080.19 |
1105.47 |
1105.47 |
|
T [K] |
200 |
100 |
200 |
100 |
| Colour |
White |
Pink |
White |
Pink |
| Shape |
Platelet |
Platelet |
Platelet |
Platelet |
| Crystal system |
Trigonal |
Trigonal |
Trigonal |
Trigonal |
| Space group |
R |
R |
R |
R |
|
a [Å] |
10.6588(7) |
10.4735(5) |
10.747(3) |
10.587(2) |
|
b [Å] |
10.6588(7) |
10.4735(5) |
10.747(3) |
10.587(2) |
|
c [Å] |
34.838(3) |
34.480(2) |
34.689(9) |
34.314(7) |
|
α [°] |
90 |
90 |
90 |
90 |
|
β [°] |
90 |
90 |
90 |
90 |
|
γ [°] |
120 |
120 |
120 |
120 |
|
V [Å3] |
3427.7(5) |
3275.6(4) |
3470(2) |
3330.8(14) |
|
Z
|
3 |
3 |
3 |
3 |
|
ρ
calc. [g cm−3] |
1.570 |
1.643 |
1.587 |
1.653 |
|
μ [mm−1] |
0.445 |
0.466 |
0.543 |
0.565 |
| Measured refl's. |
22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 250 250 |
21166 |
27060 |
26274 |
| Indep't refl's |
1573 |
1495 |
1935 |
1859 |
| Refl's I ≥ 2σ(I) |
1421 |
1374 |
1389 |
1444 |
|
R
int
|
0.0308 |
0.0309 |
0.0952 |
0.0798 |
| GooF |
1.068 |
1.072 |
1.071 |
1.115 |
| wR2 |
0.0804 |
0.0673 |
0.1449 |
0.1329 |
|
R
1
|
0.0318 |
0.0271 |
0.0569 |
0.0550 |
| CCDC† |
2024140
|
2024142
|
2024146
|
2024147
|
| |
|
|
6·Et2O, HS |
6·Et2O, LS |
9·Et2O, HS |
9·Et2O, LS |
| Formula |
C34H58B2F8FeN24O13 |
C34H58B2F8FeN24O13 |
C34H58Cl2FeN24O21 |
C34H58Cl2FeN24O21 |
| Weight [g mol−1] |
1240.45 |
1240.45 |
1265.79 |
1265.79 |
|
T [K] |
200 |
100 |
200 |
100 |
| Colour |
White |
Pink |
White |
Pink |
| Shape |
Platelet |
Platelet |
Platelet |
Platelet |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
|
a [Å] |
10.630(3) |
10.530(3) |
10.6262(13) |
10.6120(12) |
|
b [Å] |
10.971(3) |
10.576(3) |
11.0192(13) |
10.6186(12) |
|
c [Å] |
14.235(4) |
13.836(4) |
27.044(3) |
26.596(3) |
|
α [°] |
74.796(6) |
75.400(8) |
96.080(4) |
95.237(3) |
|
β [°] |
71.624(6) |
73.122(7) |
93.160(4) |
95.276(3) |
|
γ [°] |
63.563(6) |
64.974(7) |
116.571(3) |
114.758(3) |
|
V [Å3] |
1396.1(6) |
1321.2(7) |
2797.5(6) |
2681.9(5) |
|
z
|
1 |
1 |
2 |
2 |
|
ρ
calc. [g cm−3] |
1.512 |
1.665 |
1.503 |
1.567 |
|
μ [mm−1] |
0.377 |
0.396 |
0.459 |
0.479 |
| Measured refl's. |
32025 |
28095 |
61136 |
53754 |
| Indep't refl's |
6916 |
6513 |
13866 |
13356 |
| Refl's I ≥ 2σ(I) |
2942 |
2733 |
6407 |
6404 |
|
R
int
|
0.1497 |
0.2107 |
0.0757 |
0.0644 |
| GooF |
1.014 |
1.002 |
1.051 |
1.010 |
| wR2 |
0.3257 |
0.1728 |
0.2197 |
0.1016 |
|
R
1
|
0.1327 |
0.0910 |
0.0874 |
0.0596 |
| CCDC† |
2024143
|
2024141
|
2024144
|
2024145
|
Variable temperature P-XRD of bulk (4) and (7).
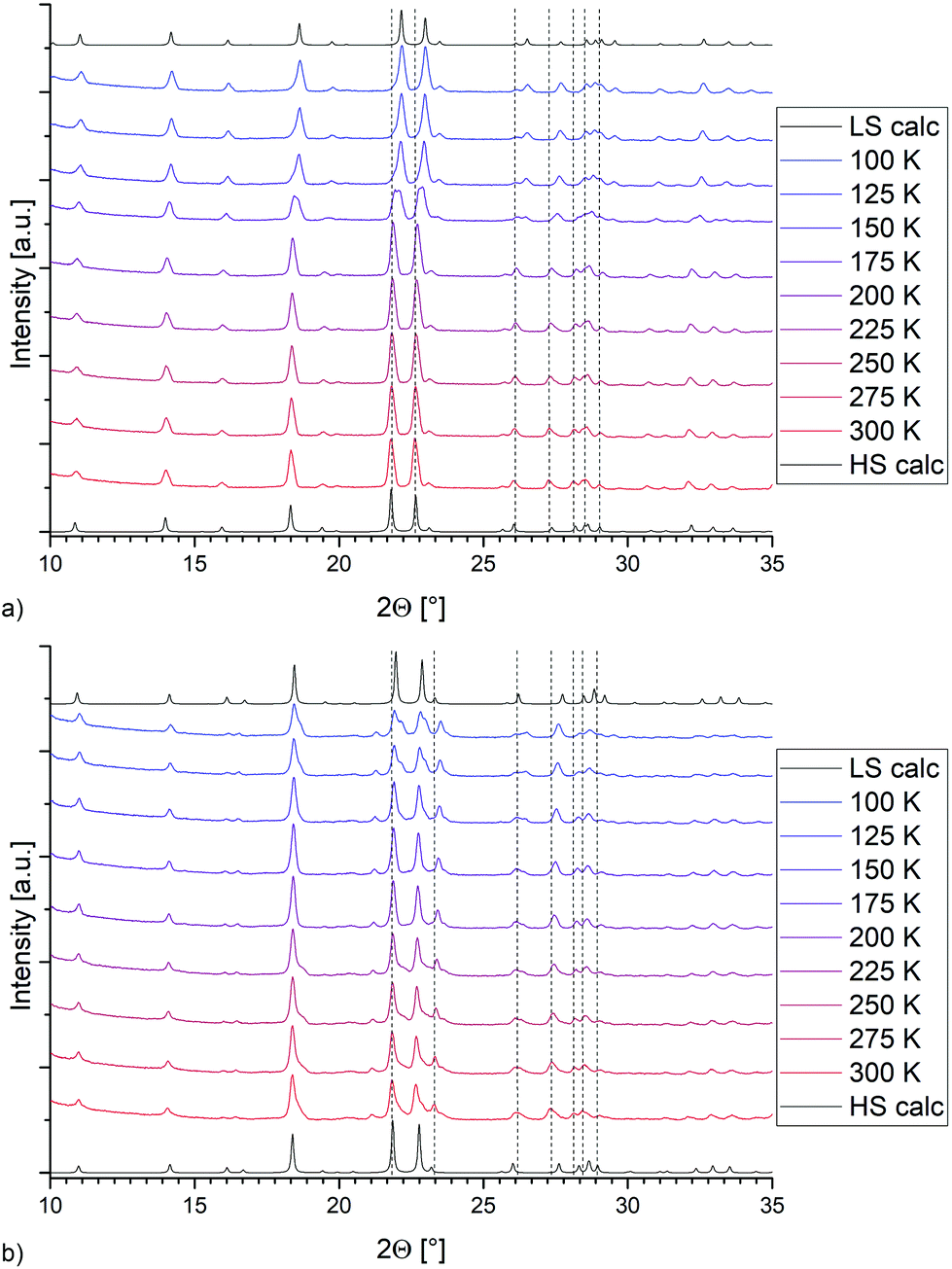
Magnetic measurements of bulk 4 and 7 show an incomplete spin transition, whereas the Fe–N bond distances obtained from the single crystal data suggest a largely complete transition. Therefore, variable temperature P-XRD was used to follow any structural changes of the bulk material during the SCO (see Fig. 6). For both compounds, the calculated diffractograms obtained from the HS and LS molecular structures are in reasonable agreement with the powder-patterns of the bulk material. For both 4 (Fig. 6a) and 7 (Fig. 6b) the temperature dependent PXRD measurements show more pronounced changes in the powder pattern (dotted lines as a guide) than to those just expectable from thermal contraction. On this basis it seems clear that both bulk and single crystalline samples are the same compound.
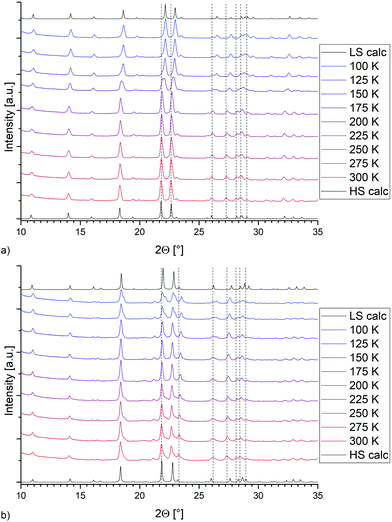 |
| | Fig. 6 Temperature-dependent P-XRD data for (a) [Fe(2COOHTz)6](BF4)2·2MeCN (4) and (b) [Fe(2COOHTz)6](ClO4)2·2MeCN (7). | |
[Fe(4COOHTz)6](BF4)2·Et2O (6).
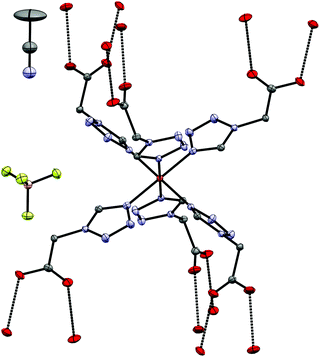
6 crystallizes as an Et2O solvate, as a result of vapour-diffusion crystallization, in the triclinic P space group. The structure was determined at 100 K (LS-state) and 200 K (HS-state). The asymmetric unit (see Fig. 7) includes the Fe-centre with three 4COOHTz ligands, one crystallographic independent BF4−-anion, as well as half of the Et2O-solvate. The Fe–N distances of the three coordinated ligands are slightly different (1.961 Å (N4), 1.979 Å (N4a) and 1.989 Å (N4b), see Table S3†). In the HS-state at 200 K the Fe–N distances increase to 2.185 Å (N4), 2.182 Å (N4a) and 2.185 Å (N4b). As for 4 and 7, the supramolecular structure is governed by an H-bond network with the COOH-groups forming a three-dimensional network of R22 (8) type ring motifs joining layers of [Fe(4COOHTz)6]2+-cations (see Fig. 8). The voids between the ligand alkyl-chains are occupied by Et2O, which also interacts with the COOH-groups via a single H-bond. Comparing the supramolecular arrangement of 4/7 to 6 it becomes evident, that in this case only due to the longer alkyl-chains Et2O could be incorporated as solvate molecule. In the former case the voids between the cationic Fe-layers would not have been large enough to fit the Et2O-molecules. Whereas at 100 K the BF4−-anions are stabilized by the H⋯F bonds similar to 4 preventing a potential disorder, at 200 K in the HS-state those interactions are no longer strong enough, resulting in an apex up-down disorder of the BF4−-groups.
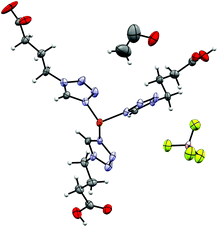 |
| | Fig. 7 Asymmetric unit of [Fe(4COOHTz)6](BF4)2·Et2O (6) at 100 K. | |
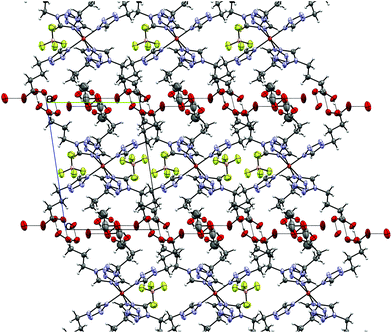 |
| | Fig. 8 Supramolecular H-bonding network in 6 at 100 K, seen along the a-axis. | |
Structure of [Fe(4COOHTz)6](ClO4)2·Et2O (9).
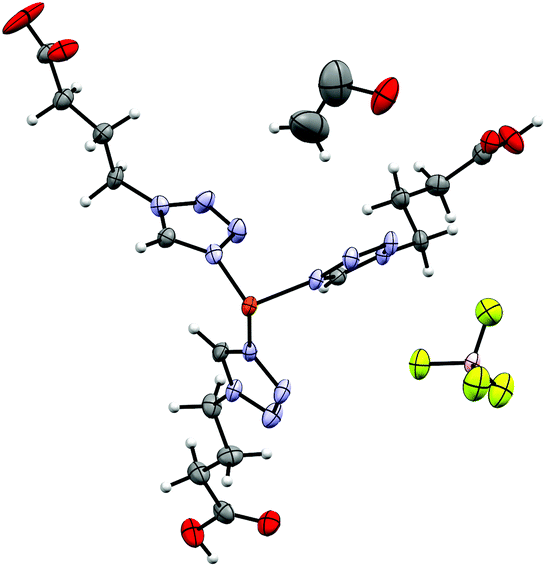
9 crystallizes as Et2O solvate like 6 in the triclinic P space group but has two crystallographic independent Fe-centres and two ClO4−-anions in the asymmetric unit. Both Fe1 and Fe2 are surrounded by each three independent 4COOHTz-ligands, which do not interact with each other via H-bonds. The solvate molecule is located in the cavity formed by the ligands, stabilized by an H-bond (see Fig. 9). At 100 K, all Fe–N bond lengths (1.984 Å Fe1–N4, 1.987 Å Fe1–N4a, 1.982 Å Fe1–N4b, 1.987 Å Fe2N4c, 1.987 Å Fe2–N4d, 1.984 Å Fe2–N4e) are characteristic for Fe(II) in its low-spin state. In contrast, at 200 K the bonds extend to typical HS distances (2.181 Å Fe1–N4, 2.178 Å Fe1–N4a, 2.173 Å Fe1–N4b, 2.174 Å Fe2N4c, 2.185 Å Fe2–N4d, 2.178 Å Fe2–N4e, see Table S4†). In 9 the [Fe(4COOHTz)6]2+-cations form layers connected through the H-bond network to each other and the ClO4−-anions.
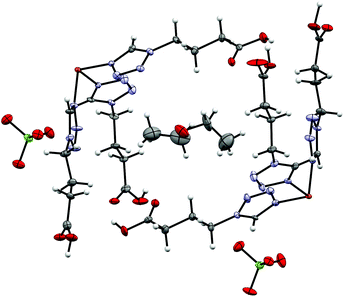 |
| | Fig. 9 Asymmetric unit of [Fe(4COOHTz)6](ClO4)2·Et2O (9) at 100 K. | |
The relevant crystallographic parameters for all Fe-containing structures are given in Table 1.
Spectroscopic characterization
MIR and FIR spectroscopy.
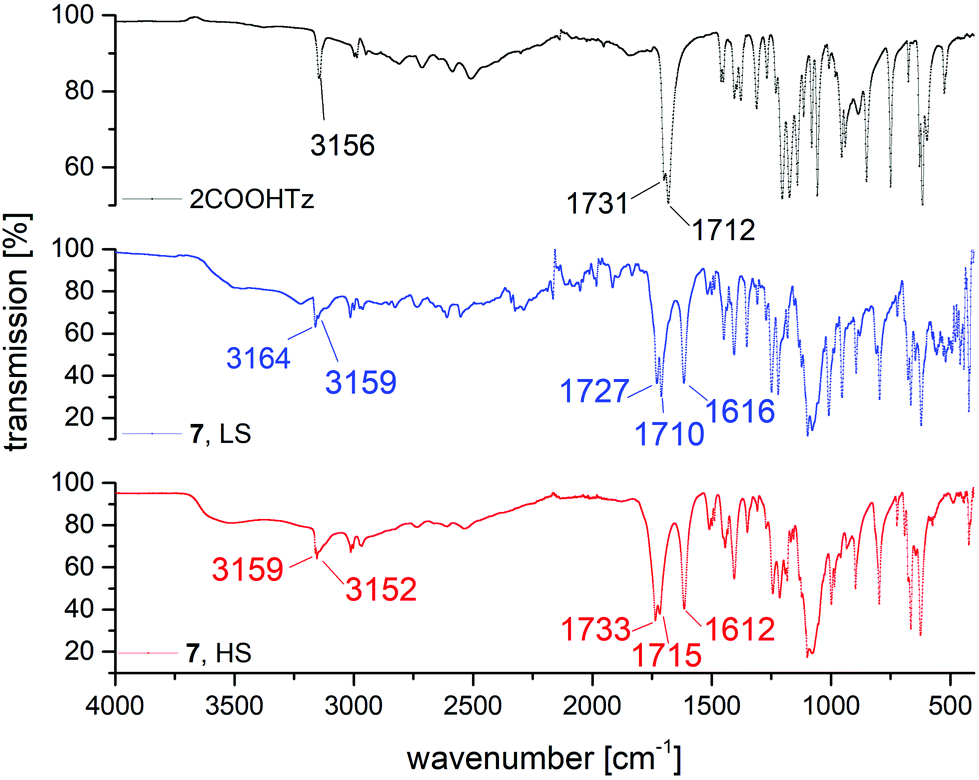
For homoleptic Fe(II)-tetrazole-complexes IR-spectroscopy allows for confirmation of successful SCO-complex formation, as well as gaining insight into the spin state present, since the tetrazolic CH-vibration is sensitive to the N4 coordination environment.36 In Fig. 10, the IR spectra of 2COOHTz (1) and its ClO4−-complex (7) in the HS-state (red) and LS-state (blue) are compared. At 3156 cm−1 the free ligand shows its tetrazolic CH-vibration, which upon successful coordination is split and shifted to 3159 cm−1 and 3152 cm−1. On cooling to the LS-state, at 100 K the CH-vibration is shifted to 3164 cm−1 and 3159 cm−1, respectively. Around 1600 cm−1 the νN2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N3 stretching vibration of the tetrazole after coordination is similarly affected by the spin-state transition, shifting from 1612 cm−1 to 1616 cm−1 in the LS-state. The change of the carbonyl-vibration at 1731 cm−1 on coordination and afterwards during SCO may be attributed to interaction within the H-bond network, transferring the volume work of the spin-state transition. The broad absorption around 1030 cm−1 corresponds to the B–F stretching vibrations of the BF4−-anion. The CN-absorption of the MeCN solvate in 7 is very weak, and is only observable in the LS-spectrum of 7 in Fig. 10 around 2250 cm−1.
N3 stretching vibration of the tetrazole after coordination is similarly affected by the spin-state transition, shifting from 1612 cm−1 to 1616 cm−1 in the LS-state. The change of the carbonyl-vibration at 1731 cm−1 on coordination and afterwards during SCO may be attributed to interaction within the H-bond network, transferring the volume work of the spin-state transition. The broad absorption around 1030 cm−1 corresponds to the B–F stretching vibrations of the BF4−-anion. The CN-absorption of the MeCN solvate in 7 is very weak, and is only observable in the LS-spectrum of 7 in Fig. 10 around 2250 cm−1.
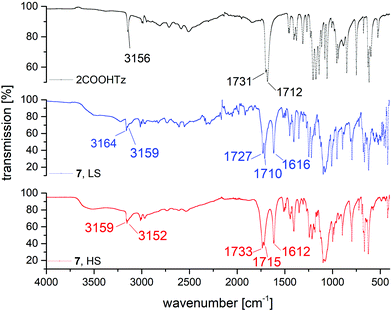 |
| | Fig. 10 Comparison of MIR spectra for 2COOHTz (1), [Fe(2COOHTz)6](ClO4)2·2 MeCN (7) in the HS-state (red) and LS-state (blue). | |
In the FIR-region of the spectra, both spin-states show the characteristic displacement of the Fe-centre towards the centroid of the trigonal faces of the N6-coordination octahedron.
Those vibrations are nearly entirely decoupled from other vibrational modes and therefore indicative of spin state. For the HS-state, this vibration is found at 226 cm−1, for the LS-state the characteristic vibrations are located at 470 and 443 cm−1 (see Fig. 11).
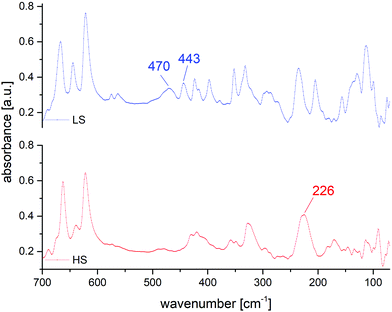 |
| | Fig. 11 Comparison of FIR spectra for [Fe(2COOHTz)6](ClO4)2·2MeCN (7) in the HS-state (red) and LS-state (blue). | |
UV-VIS/NIR spectroscopy.
Electronic transitions are also affected by the spin state transition. For homoleptic Fe-tetrazole SCO complexes the HS-state absorbs around 850 nm in the near-infrared, causing a broad and often weak absorption in the ligand field spectrum. This 5T2 → 5E transition weakens with cooling and spin-state transition, giving rise to two absorptions in the visible region of the spectrum at 545 nm and 386 nm, responsible for the pink colour of the LS-state. These absorption bands correspond to the 1A1 → 1T1 transition, as well as the 1A1 → 1T2 transition. From the maxima observed in the HS and the LS state, a ligand-field strength 10 Dq can be estimated.37 In the case of 7, measured in the solid state, a value of 1.65 Dq is obtained (Fig. 12).
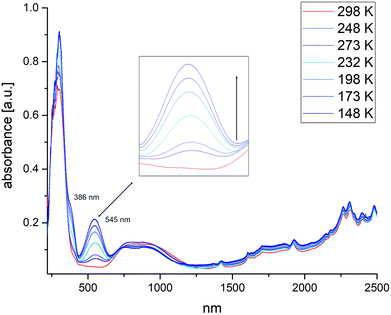 |
| | Fig. 12 Temperature-dependent UV-VIS/NIR spectra of [Fe(2COOHTz)6](ClO4)2·2MeCN (7). Inset: Increase of the absorption corresponding to the 1A1 → 1T1 transition on cooling and increasing number of LS Fe-centres. | |
Towards a mixed-metallic 3d–4f coordination polymer
The functionalization of alkyl-tetrazoles with a COOH-group introduces an H-bond donor–acceptor system thereby enhancing supramolecular cooperativity of SCO. To take advantage of the coordination ability of the COOH-group and the bifunctional ligand, we attempted to prepare a multi-metallic 3d–4f coordination polymer. Coronado et al. have successfully applied such an approach to the formation of a mixed Fe(III)–Fe(II) SCO compound using a COOH-functionalized ligand.38
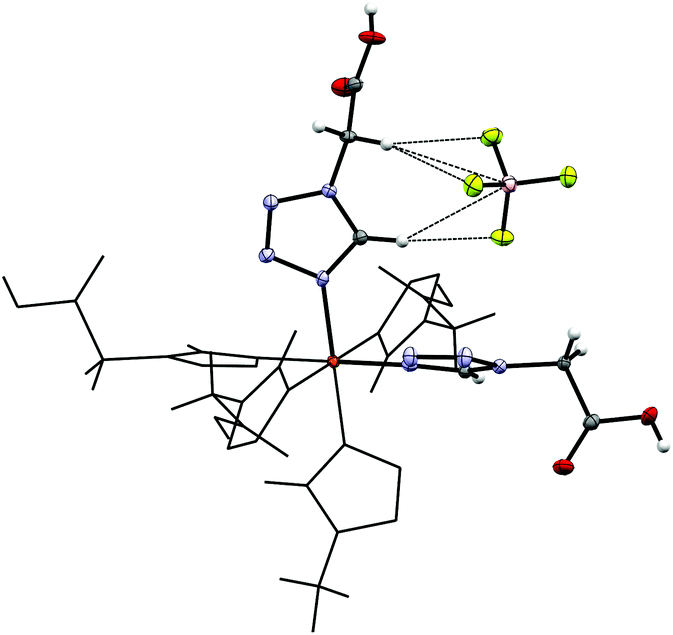
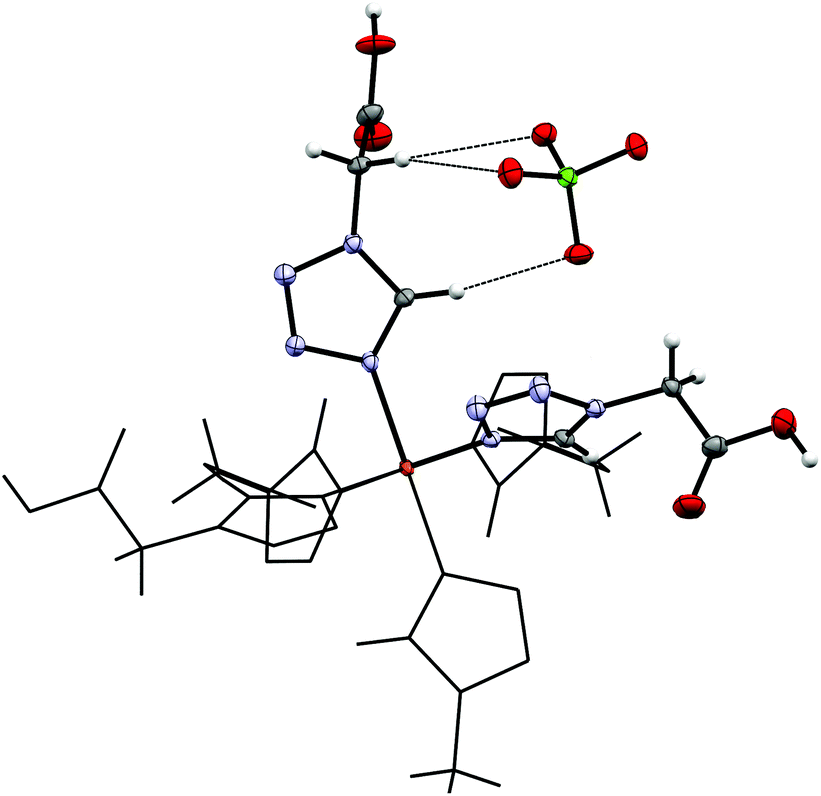
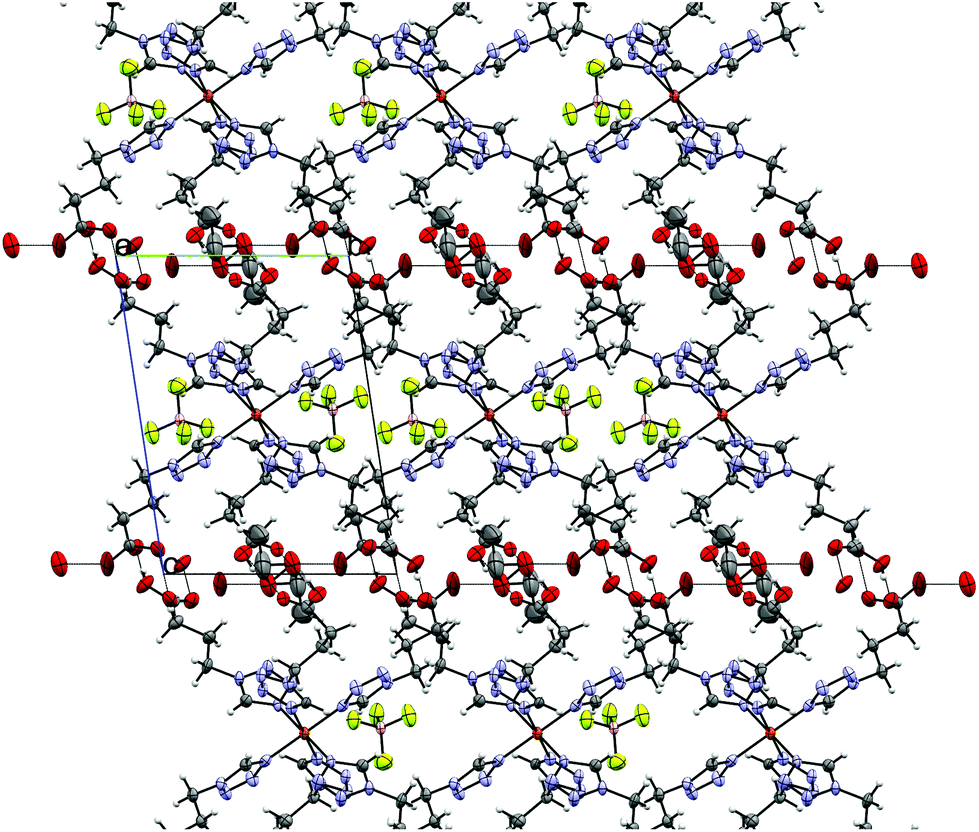
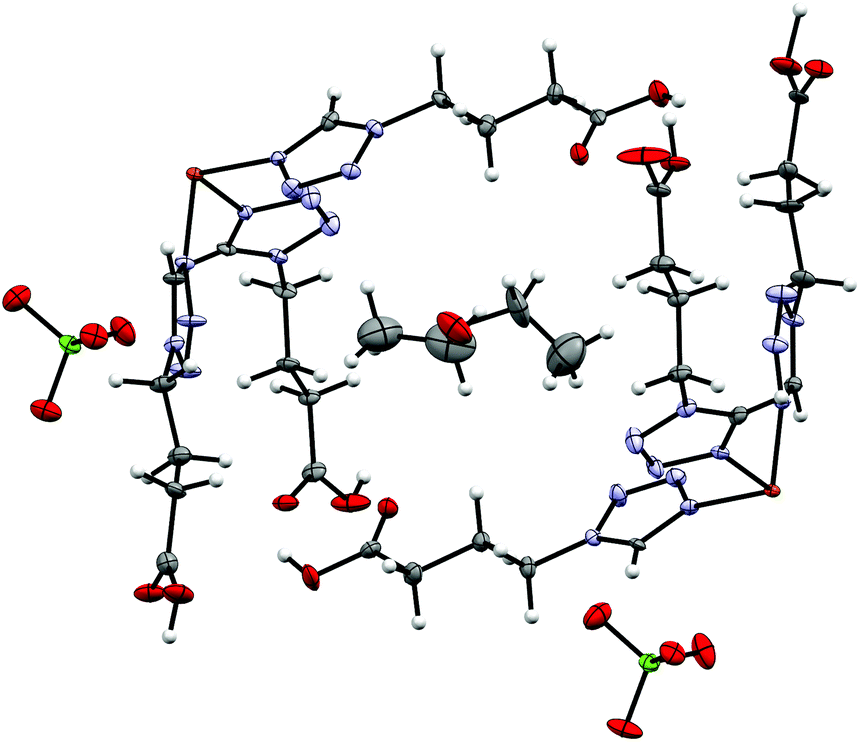
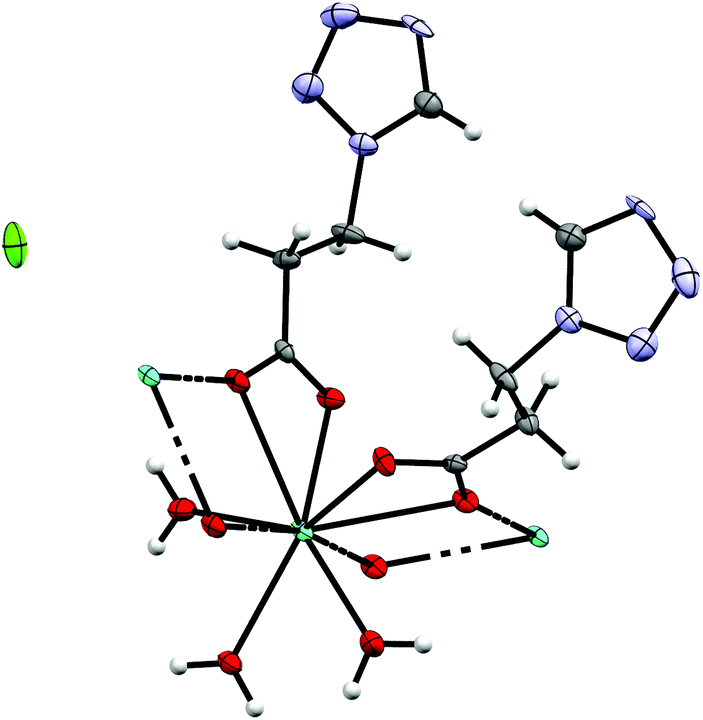
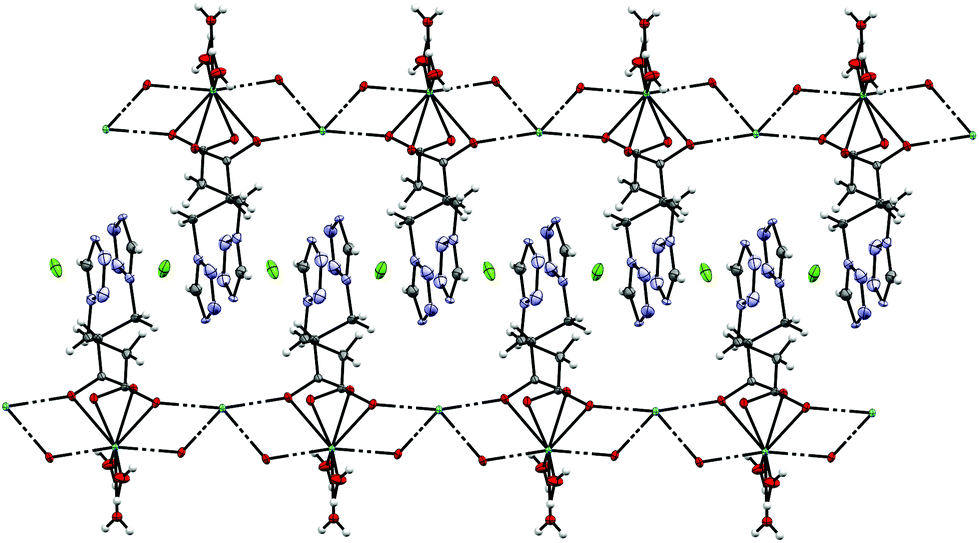
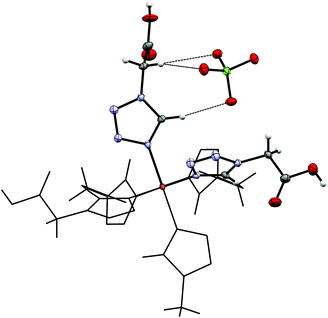
By reacting 3COOHTz (2) with Fe(ClO4)2·6H2O and GdCl3·6H2O in aqueous MeOH and subsequent evaporation a slightly yellow oil was obtained, which did not be solidify. After a few weeks at 4 °C a few colourless crystals had formed, allowing for determination of the structure by X-ray diffraction. Instead of a mixed-metallic coordination polymer, [Gd(3COOTz)2(H2O)3]Cl (10) had crystallized in the monoclinic C2/c space group. During the reaction/crystallization process the COOH-groups were deprotonated and the resulting carboxylate groups act as chelating, bridging ligands to the gadolinium-atoms in a μ2-η2:η1 fashion (see Fig. 13). Each gadolinium atom is coordinated nine-fold by oxygen-donors as the centre of a monocapped square antiprismatic coordination polyhedron. Due to the μ2-η2:η1 coordination of the carboxylate-ligands one-dimensional chains are formed, in which the Gd-atom is coordinated by four 3COOTz-ligands: two of them coordinate as bidentate, and one 3COOTz-ligand of each adjacent Gd-centre on both sides coordinates in an η1-fashion (see Fig. 14). H2O molecules occupy the remaining coordination sites (3). The η1-interaction of the carboxylate-ligands with the adjacent Gd-atoms is remarkable, as the Gd–O-bond is the shortest of all three: the η1 Gd–O interaction is 2.350 Å (Gd–O1), whereas the η2 Gd–O bonds are with 2.432 Å (Gd–O2) and 2.685 Å (Gd–O1) 3.48% and 14.3% longer. The tetrazoles establish channels parallel to the gadolinium-oxo chains. Their tetrazolic CH groups are directed to the inner of the voids, the N2 and N3 atoms of the ring interacting with the H2O ligands of the nearby gadolinium-oxo chain. In the voids the Cl−-anions are stabilized by a zig-zag H-bond structure, originating from the H2O-ligands at the Gd-centre (see Fig. 15). The crystallographic parameters for 10 are given in Table 2.
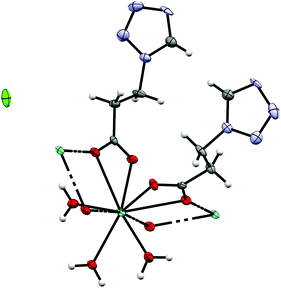 |
| | Fig. 13 μ2-η2:η1 coordination pattern of the carboxylate-ligands in [Gd(3COOTz)2(H2O)3]Cl (10). | |
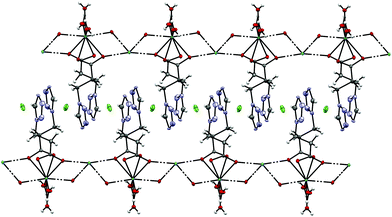 |
| | Fig. 14 One-dimensional chains in [Gd(3COOTz)2(H2O)3]Cl (10) with Cl−-atoms parallel to the c-axis, seen along the a-axis. | |
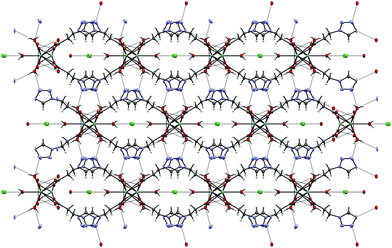 |
| | Fig. 15 Cl−-Atoms located in the voids formed by the tetrazole rings stabilized by H-bonds to the H2O ligands in the molecular structure of 10, view along c-axis. | |
Table 2 Crystallographic parameters for 10
| |
10
|
| Formula |
C8H16ClGdN8O7 |
| Weight [g mol−1] |
528.96 |
|
T [K] |
100 |
| Colour |
Clear colourless |
| Shape |
Plate |
| Crystal system |
Monoclinic |
| Space group |
C2/c |
|
a [Å] |
18.0057(4) |
|
b [Å] |
11.2323(6) |
|
c [Å] |
8.0169(9) |
|
α [°] |
90 |
|
β [°] |
91.838(3) |
|
γ [°] |
90 |
|
V [Å3] |
1620.5(2) |
|
z
|
4 |
|
r
calc. [g cm−3] |
2.1674 |
|
m [mm−1] |
4.311 |
| Measured refl's |
9983 |
| Indep't refl's |
2927 |
| Refl's I ≥ 2s(I) |
2505 |
|
R
int
|
0.0618 |
| GooF |
1.67 |
| wR2 |
0.0367 |
|
R
1
|
0.0331 |
| CCDC† |
2024832
|
Fig. 16 shows a comparison between the MIR spectrum of 2 and a single crystal of [Gd(3COOTz)2(H2O)3]Cl. Whereas the tetrazolic CH-band is only slightly shifted after the coordination to the Gd3+, the deprotonated and coordinated carbonyl-vibration is shifted by 20 cm−1. The three coordinated H2O-ligands appear in the MIR, resulting the OH-stretching mode at 3346 cm−1 and the OH-scissoring vibration at 1672 cm−1.
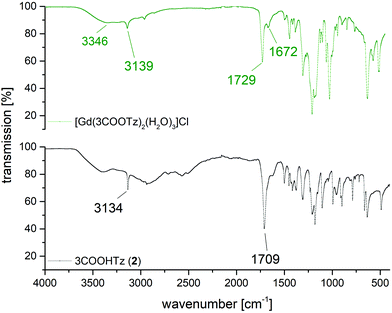 |
| | Fig. 16 Comparison of MIR spectra of 3COOHTz (2) and [Gd(3COOTz)2(H2O)3]Cl (10). | |
Experimental
Materials and methods
All operations involving Fe(II) were carried out under inert gas atmosphere (argon 5.0). The glassware used was oven dried at 120 °C before use for at least 2 hours. All solvents for the complexation reactions were dried before use and stored over molecular sieve 3 Å under argon.39 Unless otherwise stated, all starting materials were commercially obtained and used without further purification. All NMR spectra were recorded in dry deuterated solvents on a Bruker Avance UltraShield 400 MHz. Chemical shifts are reported in ppm; 1H and 13C shifts are referenced against the residual solvent resonance. For the measurement of MIR and FIR spectra, a PerkinElmer Spectrum 400, fitted with a coolable/heatable PIKE Gladi ATR unit was used within the range of 4000–100 cm−1. Solid state UV/Vis/NIR spectra were recorded with a PerkinElmer Lambda 900 spectrophotometer between 300 and 1600 nm in diffuse reflectance against BaSO4. A Harrick coolable/heatable powder sample holder in “Praying Mantis” configuration was used. Melting points were determined by differential scanning calorimetry, using a Netzsch STA 449 C Jupiter® with heating rates of 2 K min−1, see Fig. S1–S3.† The magnetic moment of the Fe(II) complexes was measured using a Physical Property Measurement System (PPMS®) by Quantum Design. The experimental setup consisted of a vibrating sample magnetometer attachment (VSM), bearing a brass sample holder with a quartz-glass powder container. The magnetic moment was determined in an external field of 1 T in the range of 10 K to 300 K, measuring all 5 K with a previous thermal stabilization of 5 minutes. Variable temperature mid-range (4000–450 cm−1) infrared spectra were recorded by the ATR technique on a PerkinElmer Spectrum 400, fitted with a coolable/heatable PIKE Gladi ATR Unit.25 Single crystals were attached to a glass fiber by using perfluorinated oil and were mounted on a Bruker KAPPA APEX II diffractometer equipped with a CCD detector with Mo Kα radiation (Incoatec Microfocus Source IμS: 30 W, multilayer mirror, λ = 0.71073 Å). For all measurements data were reduced to intensity values by using SAINT Plus,40 and an absorption correction was applied by using the multi scan method implemented by SADABS.40 For the iron(II) complexes, protons were placed at calculated positions and refined as riding on the parent C atoms. All non-H atoms were refined with anisotropic displacement parameters. For 2024832 (10), a Bruker KAPPA APEX II diffractometer equipped with a CCD detector was used and data were collected at 100 K. The powder X-ray diffraction measurements were carried out on a PANalytical X'Pert Pro diffractometer in Bragg–Brentano geometry using Cu Kα1,2 radiation filtered with a BBHD mirror and an X'Celerator linear detector. For in situ experiments below ambient temperature an Oxford PheniX Cryochamber from Oxford Cryosystems was used. The powder sample were mounted on a copper sample holder on top of a background-free silicon support. The sample chamber was evacuated, and all measurements were carried out under vacuum. The actual sample temperature is directly monitored by a thermocouple on the sample holder. The diffractograms were evaluated using the PANalytical program suite HighScorePlus, correcting for the background.
Synthesis of ligands
Tetrazoles and their derivatives – especially in combination with perchlorates – are potentially shock sensitive or explosive compounds and should, therefore, be handled with great care and with the appropriate safety precautions!
2-(1H-Tetrazol-1-yl)acetic acid (2COOHTz) (1).
25 g (333 mmol, 1 eq.) glycine and 32.4 g (499.6 mmol, 1.5 eq.) NaN3 were suspended in 97 ml (516.2 mmol, 1.55 eq.) triethylorthoformate and 250 ml acetic acid. The suspension was heated for 18 h at 95 °C, filtrated and evaporated to dryness. After extracting the residue with CH2Cl2, the solvent was removed, and the resulting yellow oil refluxed for 6 h in concentrated hydrochloric acid. After removal of all volatiles the oily residue was adsorbed on SiO2 and was further purified by column chromatography (MeCN:CH2Cl2 = 3:2). After evaporation, 1 was obtained as beige crystalline material. Yield: 7.9 g, 18.5%; MP: 130 °C; νCH(Tz) 3156, νCO 1731/1712 cm−1; 1H NMR (400 MHz, DMSO-d6) δ = 13.43 (s, 1H, COOH), 9.38 (s, 1H, Tz), 5.43 (s, 2H, CH2) ppm; 13C{1H} NMR (101 MHz, DMSO-d6) δ = 167.94 (COOH), 144.99 (Tz), 48.52 (CH2) ppm.
3-(1H-Tetrazol-1-yl)propanoic acid (3COOHTz) (2).
25 g (280.6 mmol, 1 eq.) β-alanine and 27.4 g (420.9 mmol, 1.5 eq.) NaN3 were suspended in 81.7 ml (434.9 mmol, 1.55 eq.) triethylorthoformate and 250 ml acetic acid. The suspension was heated for 18 h at 95 °C, filtrated and evaporated to dryness. After extracting the residue with CH2Cl2, the solvent was removed, and the resulting yellow oil refluxed for 6 h in concentrated hydrochloric acid. After removal of all volatiles the oily residue was adsorbed on SiO2 and was further purified by column chromatography (MeCN:CH2Cl2 = 3:2). After evaporation, 3COOHTz was obtained as off-white solid. Yield: 9.1 g, 22.8%; MP: 125 °C; νCH(Tz) 3134, νCO 1709 cm−1; 1H NMR (400 MHz, DMSO-d6) δ = 12.34 (s, 1H, COOH), 9.37 (s, 1H, Tz), 4.64 (t, J = 6.6 Hz, 2H, CH2), 2.98 (t, J = 6.7 Hz, 2H, CH2) ppm; 13C{1H} NMR (101 MHz, DMSO-d6) δ = 171.72 (COOH), 144.30 (Tz), 43.74 (CH2), 33.49 (CH2).
4-(1H-Tetrazol-1-yl)butanoic acid (4COOHTz) (3).
25 g (242.4 mmol, 1 eq.) 4-aminobutanoic acid and 23.6 g (363.7 mmol, 1.5 eq.) NaN3 were suspended in 70.6 ml (375.8 mmol, 1.55 eq.) triethylorthoformate and 250 ml acetic acid. The suspension was heated for 18 h at 95 °C, filtrated and evaporated to dryness. After extracting the residue with CH2Cl2, the solvent was removed, and the resulting yellow oil refluxed for 6 h in concentrated hydrochloric acid. After removal of all volatiles the oily residue crystallized in the freezer overnight. The yellow solid was adsorbed on SiO2 and further purified by column chromatography (MeCN:CH2Cl2 = 3:2) After evaporation, 4COOHTz was obtained as off-white solid. Yield: 11.24 g, 29.7%; MP: 86 °C; νCH(Tz) 3114, νCO 1714 cm−1; 1H NMR (400 MHz, DMSO-d6) δ = 12.21 (s, 1H, COOH), 9.41 (s, 1H, Tz), 4.48 (t, J = 7.1, 2H, CH2), 2.27 (t, J = 7.3, 2H, CH2), 2.06 (p, J = 7.2, 2H, CH2) ppm; 13C{1H} NMR (101 MHz, DMSO-d6) δ = 173.52 (COOH), 143.99 (Tz), 46.91 (CH2), 30.37 (CH2), 24.72 (CH2).
Synthesis of Fe(II)-complexes
[Fe(2COOHTz)6][BF4]2·2MeCN (4).
Fe(BF4)2·6H2O (100 mg, 296.3 μmol, 1 eq.) and 1 (231.5 mg, 1.81 mmol, 6.1 eq.) were dissolved in 10 ml MeCN and stirred at 50 °C for 18 h. After evaporation of the solvent, the residual oil was triturated in 10 ml THF for 1 h to remove the excess of ligand. The precipitated material was separated, washed with further THF (10 ml) and dried in a stream of N2. Yield: 98.8 mg, 33.4%; νCH(Tz) 3138, νCO 1744 cm−1.
[Fe(3COOHTz)6][BF4]2 (5).
Same procedure as for 4. 5 was isolated as off-white, sticky solid. Yield: 75.7 mg, 23.6%; νCH(Tz) 3135, νCO 1714 cm−1.
[Fe(4COOHTz)6][BF4]2 (6).
Same procedure as for 4. 6 was isolated as white solid. Yield: 122 mg, 35.3%; νCH(Tz) 3144, νCO 1733 cm−1.
[Fe(2COOHTz)6][ClO4]2·2MeCN (7).
Fe(ClO4)2·6H2O (100 mg, 275.6 μmol, 1 eq.) was dissolved under Ar together with a tiny amount of ascorbic acid in 2 ml MeCN and added by filtration through a syringe filter (PTFE, 0.45 μm) to a degassed solution of 1 (215.4 mg, 1.68 mmol, 6.1 eq.) in 8 ml MeCN. The resulting mixture was stirred at 50 °C for 18 h. After evaporation of the solvent, the residual oil was triturated in 10 ml THF for 1 h to remove the excess of ligand. The precipitated material was separated, washed with further THF (10 ml) and dried in a stream of N2. Yield: 51.5 mg, 18.2%; νCH(Tz) 3159/3152, νCO 1733/1715 cm−1.
[Fe(3COOHTz)6][ClO4]2 (8).
Same procedure as for 7. 8 was isolated as white sticky solid. Yield: 127.1 mg, 41.6%; νCH(Tz) 3137, νCO 1740/1715 cm−1.
[Fe(4COOHTz)6][ClO4]2 (9).
Same procedure as for 7. 9 was isolated as white solid. Yield: 118.9 mg, 36.2%; νCH(Tz) 3134, νCO 1728 cm−1.
[Gd(3COOTz)2(H2O)3]Cl (10).
Fe(ClO4)2·6H2O (42.55 mg, 117.27 μmol, 1 eq.) was dissolved under Ar together with a tiny amount of ascorbic acid in 2 ml MeOH and added by filtration through a syringe filter (PTFE, 0.45 μm) to a degassed solution of 2 (100 mg, 703.64 mmol, 6 eq.) in 8 ml of a 1:1 mixture MeOH:H2O. The resulting mixture was stirred at 50 °C for 10 minutes, then 43.6 mg (117.27 μmol, 1 eq.) GdCl3·6H2O was added. The mixture was further stirred for 6 h at 50 °C. After evaporation of the solvent, the residual slightly yellow oil was kept at 50 °C for 6 h under vacuum and afterwards left for 3 weeks at 4 °C. A few tiny colourless crystals formed.
Conclusions
Fe(II)–SCO complexes with bifunctional ω-(1H-tetrazol-1-yl) carboxylic acids allow formation of supramolecular hydrogen bonding networks that enhance the cooperativity of the material. Furthermore, the COOH-group can act as anchor-group for deposition on oxidic surfaces, or for formation of multi-metallic coordination polymers. Three ligands bearing a COOH-group with alkyl-chain lengths of C2–C4 were synthesized and coordinated to Fe(II) with BF4− and ClO4− as weak-coordinating anions. Complete and sharp spin state transitions were observed in the cases of 4, 6, 7 and 9. An initial attempt to prepare a 3d–4f mixed-metallic coordination polymer with Gd3+ resulted in the unexpected formation of one-dimensional Gd-oxo chains with the 3COOHTz-ligand having been deprotonated and coordinating to the Gd3+-atoms in a chelating–bridging μ2-η2:η1 fashion. Future work will focus on a more suitable synthetic approach to realise a coordination on both ligand functional groups.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge financial support of the Austrian Science Fund (FWF Der Wissenschaftsfond) project P 31076-N28. The X-Ray center (XRC) of the Vienna University of Technology provided access to the powder X-Ray diffractometer.
References
-
P. Gütlich and H. A. Goodwin, Topics in Current Chemistry, Spin Crossover in Transition Metal Compounds I–III, Springer Berlin Heidelberg, 2004 Search PubMed.
- P. Gütlich, Eur. J. Inorg. Chem., 2013, 581–591, DOI:10.1002/ejic.201300092.
- H. J. Shepherd, C. Bartual-Murgui, G. Molnar, J. A. Real, M. C. Munoz, L. Salmon and A. Bousseksou, New J. Chem., 2011, 35, 1205–1210 RSC.
- S. Decurtins, P. Gutlich, C. P. Kohler, H. Spiering and A. Hauser, Chem. Phys. Lett., 1984, 105, 1–4 CrossRef CAS.
- A. C. Aragonès, D. Aravena, J. I. Cerdá, Z. Acís-Castillo, H. Li, J. A. Real, F. Sanz, J. Hihath, E. Ruiz and I. Díez-Pérez, Nano Lett., 2016, 16, 218–226 CrossRef.
- C. Lefter, V. Davesne, L. Salmon, G. Molnár, P. Demont, A. Rotaru and A. Bousseksou, Magnetochemistry, 2016, 2, 18 CrossRef.
- C. Lefter, S. Rat, J. S. Costa, M. D. Manrique-Juárez, C. M. Quintero, L. Salmon, I. Séguy, T. Leichle, L. Nicu, P. Demont, A. Rotaru, G. Molnár and A. Bousseksou, Adv. Mater., 2016, 28, 7508–7514 CrossRef CAS.
- C. Lefter, R. Tan, J. Dugay, S. Tricard, G. Molnár, L. Salmon, J. Carrey, W. Nicolazzi, A. Rotaru and A. Bousseksou, Chem. Phys. Lett., 2016, 644, 138–141 CrossRef CAS.
- L. Cambi and A. Cagnasso, Atti R. Accad. Naz. Lincei, Mem. Cl. Sci. Fis., Mat. Nat., 1931, 13, 809 CAS.
- L. Cambi and L. Szegö, Ber. Dtsch. Chem. Ges. (A and B), 1933, 66, 656–661 CrossRef.
- L. Cambi and L. Szegö, Ber. Dtsch. Chem. Ges. (A and B), 1931, 64, 2591–2598 CrossRef.
- M. Ohba, K. Yoneda and S. Kitagawa, CrystEngComm, 2010, 12, 159–165 RSC.
- C. J. Kepert, Chem. Commun., 2006, 695, 10.1039/b515713g.
- A. Holovchenko, J. Dugay, M. Giménez-Marqués, R. Torres-Cavanillas, E. Coronado and H. S. J. van der Zant, Adv. Mater., 2016, 28, 7228–7233 CrossRef CAS.
- J. Dugay, M. Gimenez-Marques, T. Kozlova, H. W. Zandbergen, E. Coronado and H. S. J. van der Zant, Adv. Mater., 2015, 27, 1288–1293 CrossRef CAS.
- H. J. Shepherd, I. Y. A. Gural’skiy, C. M. Quintero, S. Tricard, L. Salmon, G. Molnár and A. Bousseksou, Nat. Commun., 2013, 4, 2607 CrossRef.
- M. Matsuda, H. Isozaki and H. Tajima, Thin Solid Films, 2008, 517, 1465–1467 CrossRef CAS.
- C. Lochenie, K. Schötz, F. Panzer, H. Kurz, B. Maier, F. Puchtler, S. Agarwal, A. Köhler and B. Weber, J. Am. Chem. Soc., 2018, 140, 700–709 CrossRef CAS.
- C. Bartual-Murgui, A. Akou, C. Thibault, G. Molnar, C. Vieu, L. Salmon and A. Bousseksou, J. Mater. Chem. C, 2015, 3, 1277–1285 RSC.
- W.-Q. Gao, Y.-S. Meng, C.-H. Liu, Y. Pan, T. Liu and Y.-Y. Zhu, Dalton Trans., 2019, 48, 6323–6327 RSC.
- J. Ru, F. Yu, P.-P. Shi, C.-Q. Jiao, C.-H. Li, R.-G. Xiong, T. Liu, M. Kurmoo and J.-L. Zuo, Eur. J. Inorg. Chem., 2017, 2017, 3144–3149 CrossRef CAS.
- K. E. Burrows, S. E. McGrath, R. Kulmaczewski, O. Cespedes, S. A. Barrett and M. A. Halcrow, Chem. – Eur. J., 2017, 23, 9067–9075 CrossRef CAS.
- I. A. Gass, S. Tewary, G. Rajaraman, M. Asadi, D. W. Lupton, B. Moubaraki, G. Chastanet, J.-F. Létard and K. S. Murray, Inorg. Chem., 2014, 53, 5055–5066 CrossRef CAS.
- A. Absmeier, M. Bartel, C. Carbonera, G. N. L. Jameson, F. Werner, M. Reissner, A. Caneschi, J. F. Letard and W. Linert, Eur. J. Inorg. Chem., 2007, 3047–3054, DOI:10.1002/ejic.200601096..
- M. Muttenthaler, M. Bartel, P. Weinberger, G. Hilscher and W. Linert, J. Mol. Struct., 2005, 741, 159–169 CrossRef CAS.
- M. Seifried, C. Knoll, G. Giester, J. M. Welch, D. Müller and P. Weinberger, Eur. J. Org. Chem., 2017, 2017, 2416–2424 CrossRef CAS.
- M. Seifried, C. Knoll, G. Giester, M. Reissner, D. Müller and P. Weinberger, Magnetochemistry, 2016, 2, 1–13 CrossRef CAS.
- M. Nihei, Y. Yanai, I. J. Hsu, Y. Sekine and H. Oshio, Angew. Chem., Int. Ed., 2017, 56, 591–594 CrossRef CAS.
- C. Lochenie, J. Heinz, W. Milius and B. Weber, Dalton Trans., 2015, 44, 18065–18077 RSC.
- W. Bauer, C. Lochenie and B. Weber, Dalton Trans., 2014, 43, 1990–1999 RSC.
- A. Absmeier, M. Bartel, C. Carbonera, G. N. L. Jameson, P. Weinberger, A. Caneschi, K. Mereiter, J. F. Letard and W. Linert, Chem. – Eur. J., 2006, 12, 2235–2243 CrossRef CAS.
- P. L. Franke and W. L. Groeneveld, Transition Met. Chem., 1981, 6, 54–56 CrossRef CAS.
-
T. Kamiya and Y. Saito, US3767667A, 1971.
-
T. Kamiya and Y. Saito, DE2147023A1, 1971.
- O. H. Jústiz, R. Fernández-Lafuente, J. M. Guisán, P. Negri, G. Pagani, M. Pregnolato and M. Terreni, J. Org. Chem., 1997, 62, 9099–9106 CrossRef.
- D. Müller, C. Knoll, M. Seifried and P. Weinberger, Vib. Spectrosc., 2016, 86, 198–205 CrossRef.
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed. Engl., 1994, 33, 2024–2054 CrossRef.
- A. Abhervé, M. J. Recio-Carretero, M. López-Jordà, J. M. Clemente-Juan, J. Canet-Ferrer, A. Cantarero, M. Clemente-León and E. Coronado, Inorg. Chem., 2016, 55, 9361–9367 CrossRef.
-
D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, 4th ed., Butterworth-Heinemann, Oxford, 1997 Search PubMed.
-
I. Bruker, Bruker AXS Inc., Madison, Wisconsin, USA, 2012.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2024140 (4, HS), 2024142 (4, LS), 2024146 (6, HS), 2024147 (6, LS), 2024143 (7, HS), 2024141 (7, LS), 2024144 (9, HS), 2024145 (9, LS) and 2024832 (10). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03315d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Jan M.
Welch
b,
Gerald
Giester
c,
Berthold
Stöger
d,
Werner
Artner
d,
Michael
Reissner
e,
Danny
Müller
a,
Jan M.
Welch
b,
Gerald
Giester
c,
Berthold
Stöger
d,
Werner
Artner
d,
Michael
Reissner
e,
Danny
Müller