
Unique vapochromism of a paddlewheel-type dirhodium complex accompanied by dynamic structural and phase transitions†

Abstract
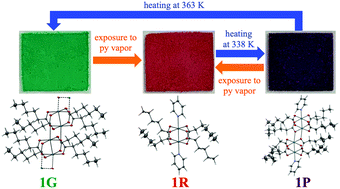
The one-dimensional coordination polymer [Rh2(HA)4]n (1G; HA = hexanoate) exhibits a drastic vapochromic color change from green to red upon exposure to pyridine (py) vapor. Heating the red discrete complex [Rh2(HA)4(py)2] (1R) at 338 K affords the purple discrete tetrarhodium complex [Rh2(HA)4(py)]2 (1P), which is an intermediate species in the vapochromic transformation of 1G to 1R. The obtained complexes 1G, 1R, and 1P differ not only in their color in the solid state, but also in their temperature-dependent phase transition properties.
 Please wait while we load your content...
Please wait while we load your content...

