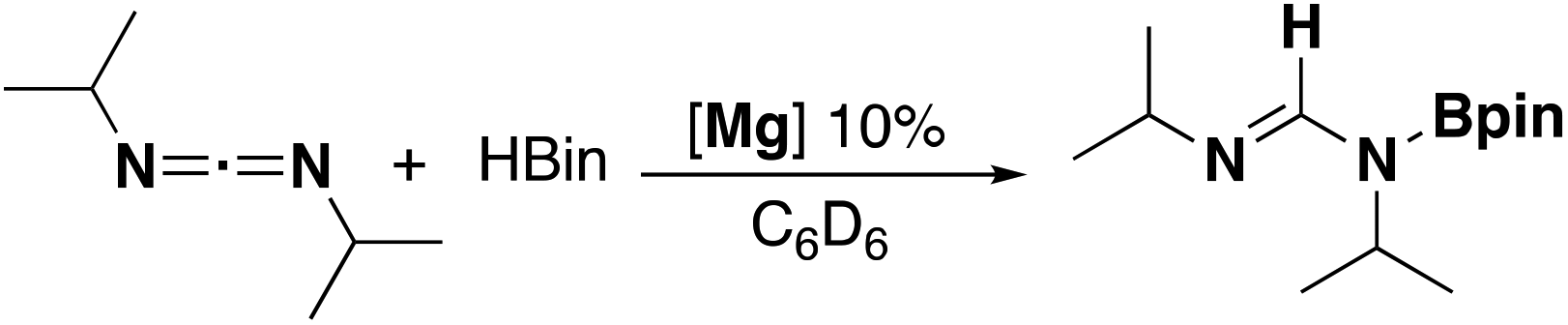DOI:
10.1039/D0DT02523B
(Paper)
Dalton Trans., 2020,
49, 11354-11360
Magnesium hydrides bearing sterically demanding amidinate ligands: synthesis, reactivity and catalytic application†
Received
17th July 2020
, Accepted 3rd August 2020
First published on 4th August 2020
Abstract
The synthesis and characterisation of a series of magnesium complexes bearing sterically demanding amidinate ligands is reported; this includes magneisum amides (1a and 1b), hydrides (3a and 3b) and alkyl complexes (2b). The solid and solution state behaviour of the complexes has been investigated using single crystal X-ray diffraction and NMR spectroscopy, revealing the magnesium hydrides to exist as dimers in the solid state, dispite the sterically demanding ligand systems and showing a degree of monomeric character in solution. The stoichiometric and catalytic activity of the amidinate complexes were investigated, with the complexes found to efficiently mediate both the hydroamination of N,N′-diisopropylcarbodiimide and the Tishchenko reaction. The metal hydrides are highly reactive towards coordinating substrates, showing a significant increase in catalytic rate compared with more ubiquitous β-diketiminate magnesium hydrides.
Introduction
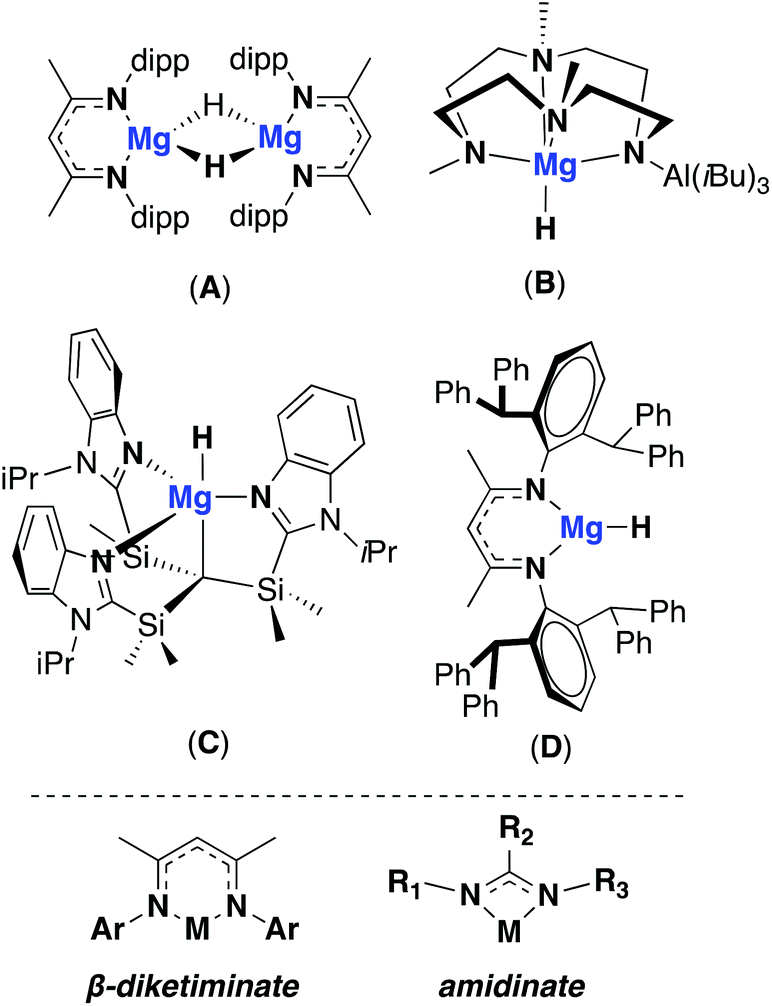
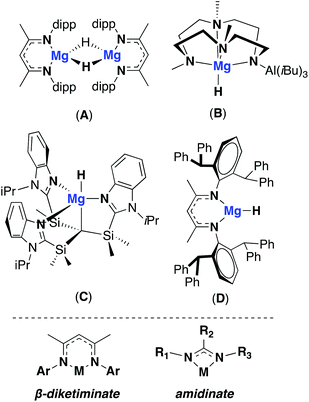
There is a growing interest in replacing expensive transitions metal based catalysts with cheap, Earth abundant and non-toxic main-group metals. Over recent years, alkaline earth metals have been shown to be adept in catalysing a host of transformations such as the inter and intra hydrofunctionalisation of unsaturated C–C bonds and dehydrogenative coupling reactions.1 Magnesium based systems have shown a lot of promise as catalysts, due to their ease of synthesis and relative stability towards Schlenk equilibrium versus heavier Group 2 metals. In hydroboration, hydroamination and hydrosilation reactions, magnesium hydrides are often identified as the active catalytic species. Despite their common use and application in catalysis, examples of discrete, stable and isolatable Mg–H species remain relatively rare.2,3 Two-coordinate β-diketiminate ligands have been shown to support magnesium hydrides, such as A, which is stable towards Schlenk redistribution at room temperature and exists as a dimer.4A has been widely employed in catalysis, either used directly or formed in situ, for reactions such as the hydroboration of aldehydes and ketones and the reduction of CO with phenylsilane.5,6 The isolation of terminal magnesium hydride species has also been possible through the use of ligand systems with high coordination numbers (Fig. 1: B and C).7,8 These compounds have been shown to be catalytically active, for instance C has is a competent catalyst for both olefin hydrosilylation and the hydroboration of carbodiimides.7 Sterics have been used to favour the formation of monomeric, low-coordinate compounds leading to the isolation of a ‘super bulky’ β-diketiminate magnesium hydride complex, D. However, the increased steric profile rendered the complex relatively unreactive, even towards water.9 In contrast to β-diketiminate ligands, the two-coordinate amidinate ligand framework has a significantly different steric profile, with the 4-membered ring directing the nitrogen substituents further from the metal centre. Amidinates also offer greater potential for structural modification due to the modular way in which they can be synthesised. Both symmetric and asymmetric ligand systems can be easily accessed, as well as modification of the bridgehead carbon substituent (guanidinate where bridgehead = NR2). Recently, examples of guanidinate and amidinate ligand frameworks bearing ‘super bulky’ aryl groups have been developed. In 2014 Fortier and co-workers reported the synthesis of a super bulky guanidinate ligand and it's coordination with group 1 metals, as well as with iron to stabilise a terminal Fe(IV) nitride species ((Ar*N)2RCH where Ar* = 2,6-bis(diphenylmethyl)-4-tert-butylphenyl and R = NC(tBu)2).10,11 Both Jones and Harder have more recently developed amidinate analogues with bulky 2,6-(diphenylmethyl) substituents, coordinating them to magnesium and strontium amides.12,13 Using an asymmetric ligand, where R1 = 2,6-(diphenylmethyl) and R2 = 2,6-(diisopropyl), Jones was also able to isolate both magnesium and strontium hydrides. These represent the first examples of group 2 hydrides stabilised by amidinate (or guanidinate) ligands. The reactivity of this new class of metal hydride complex has yet to be investigated, but the more strained and open coordination environment at the metal centre present interesting distinctions from the more ubiquitous β-diketiminate systems. Therefore diversifying this family of magnesium hydrides and exploring their potential in catalysis is of interest. Herein, the synthesis and reactivity of two novel magnesium hydride complexes stabilised by a symmetric and asymmetric amidinate ligand is discussed.
 |
| | Fig. 1 Top: Examples of monomeric and dimeric magnesium hydrides used in catalysis. Bottom: general structure of β-diketiminate and amidinate ligands. | |
Results and discussion
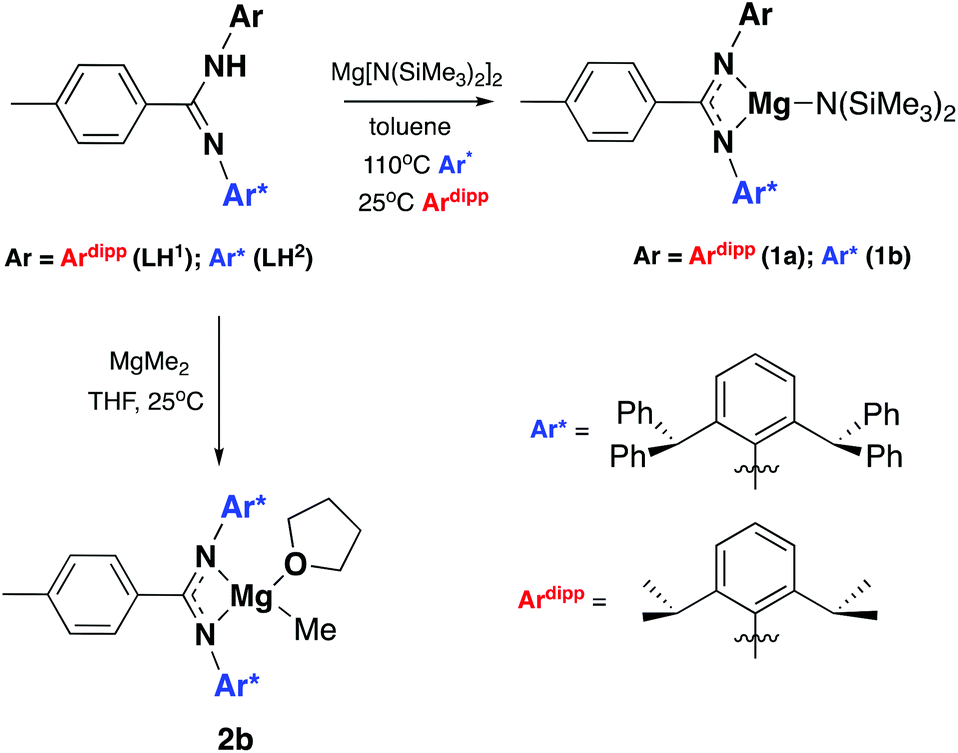
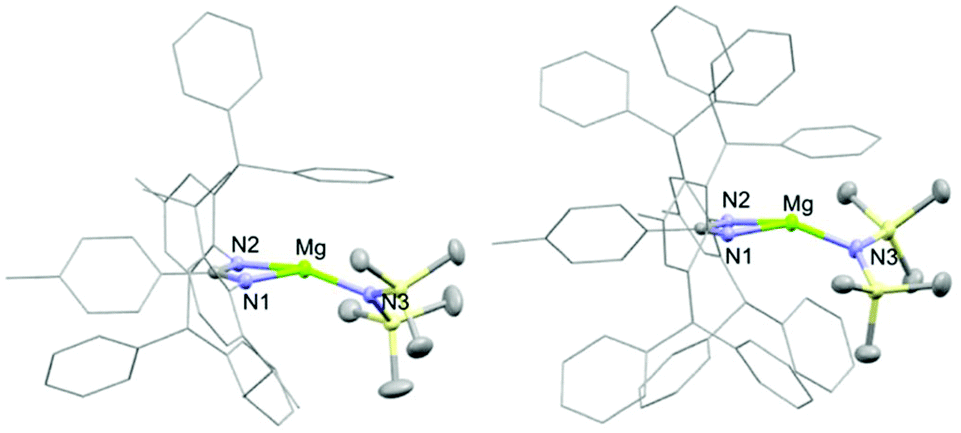
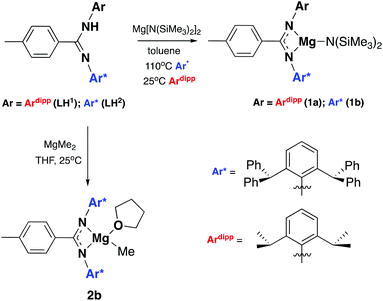
Two ligand systems were chosen to be the focus of this study, LH1 and LH2, both of which were synthesised by modifying literature procedures (Fig. S1†). Magnesium amide complexes 1a and 1b were then prepared by reaction of pro-ligand LH1 (1a) or LH2 (1b) with one equivalent of the magnesium precursor Mg[N(SiMe3)2]2 in toluene (Scheme 1). Compound 1a formed after stirring at room temperature for 5 h, whereas 1b required heating at 110 °C for 3 days. Signals in the 1H NMR spectrum at δ 0.02 and δ −0.15 ppm (1a and 1b) corresponding to eighteen protons (–N(SiMe3)2) confirm the formation of the monoligated product. Crystals suitable for single crystal X-ray diffraction were isolated and the solid state structures of both 1a and 1b show the expected three-coordinate magnesium centre, with key bond lengths and angles shown in Fig. 2. In both compounds there are significant deviations from linearity, with the amide group bent away from the N–Mg–N plane by 154.8° (1a) and 151.7° (1b). This is due to additional stabilising interactions between one of the ligand based aryl groups with the magnesium centre (Mg⋯Ar; 1a: 2.840(1); 1b: 2.767(2)). Other magnesium precursors were also explored; reaction of LH2 with MgMe2 in toluene was hindered by the limited solubility of the magnesium precursor, however in tetrahydrofuran (THF) formation of the THF adduct (2b) was facile.
 |
| | Scheme 1 Synthetic procedure for the formation of compounds 1a, 1b and 2b. | |
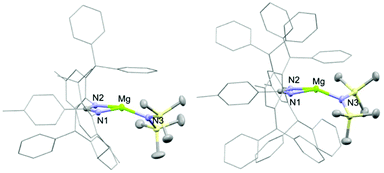 |
| | Fig. 2 Solid state structure of 1a and 1b. Selected bond lengths (Å) and angles (°). 1a: Mg–N1 2.088(1); Mg–N2 2.072(1); Mg–N3 1.967(1); N1–Mg–N2 64.7; N1–C–N2 113.3. 1b: Mg–N1 2.115(12); Mg–N2 2.087(2); Mg–N3 1.967(3); N1–Mg–N2 64.3; N1–C–N2 113.2. | |
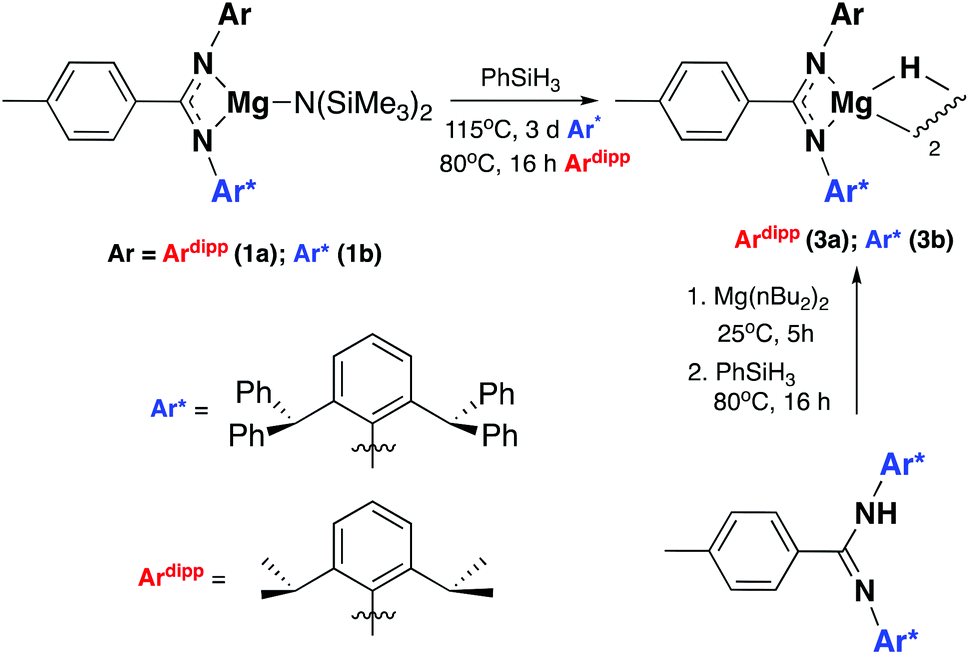
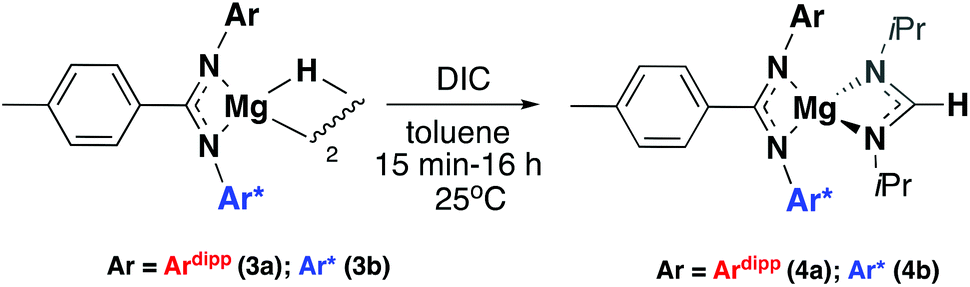
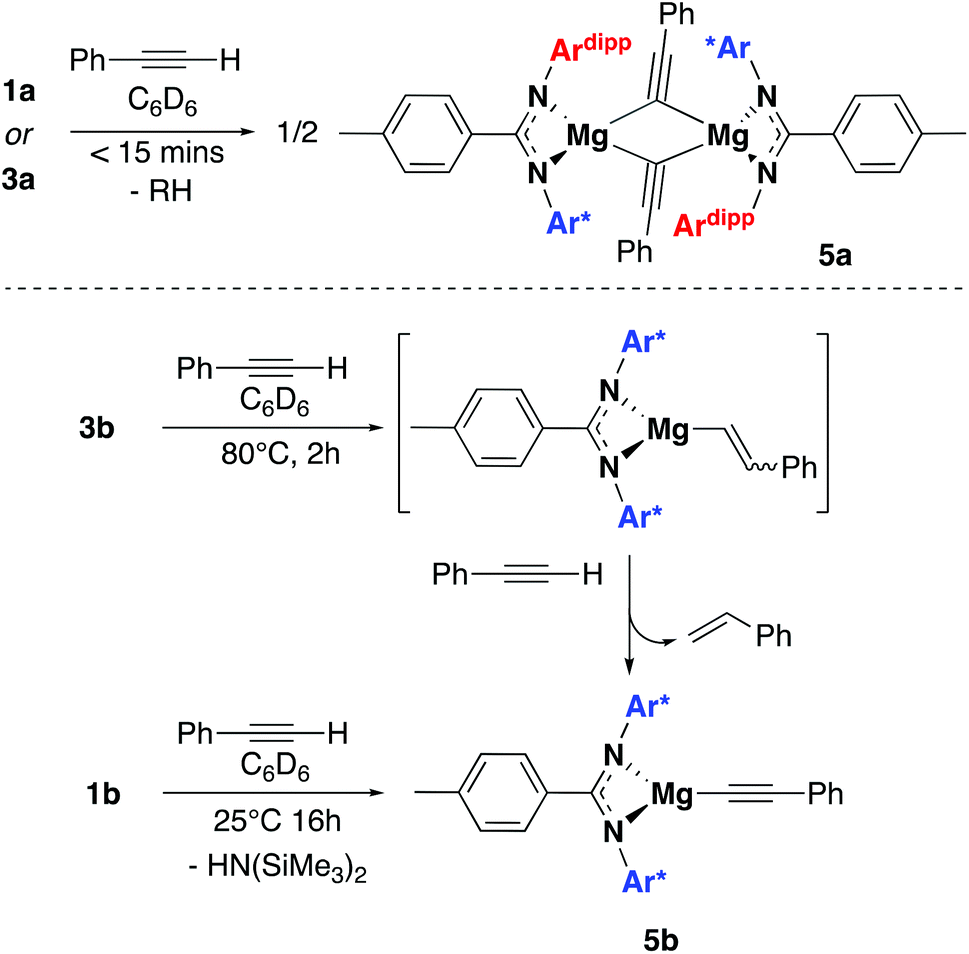
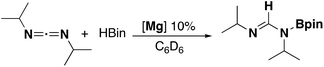
Compounds 1a and 1b were subsequently reacted with phenylsilane in toluene at 80 °C for 16 hours and at 115 °C for three days, respectively (Scheme 2). The 1H NMR spectra reveal distinctive signals at δ 3.64 ppm (3a) and δ 5.03 ppm (3b) which correspond to the newly formed magnesium hydrides (Mg–H). The Mg–H signal in 3a matches well with a closely related complex reported by Jones and co-workers.13 However, the Mg–H NMR shift of 3b is significantly further downfield, with other examples of downfield (>δ 4.5 ppm) Mg–H signals relating to structurally characterised examples of terminal magnesium hydrides (Fig. S2†).4,7–9,13–22 Compound 3b can also be made using a more facile synthetic route, reacting LH2 with dibutyl magnesium to form L2Mg(nBu), which was not isolated but further reacted with phenylsilane under milder conditions (80 °C, 16 hours) to form 3b in good yield (58%). Attempts to react 2b with phenylsilane under a variety of conditions failed to yield any magnesium hydride product. Crystals of 3a and 3b suitable for single crystal XRD were isolated from concentrated C6D6/hexane or C6D6 solutions, however both samples repeatedly co-crystallised with a hydroxide impurity (modelled as 3a 31%, 3b 24%, see ESI†). This occurred despite negligible hydroxide content in the sample (3a 3% in 1H NMR; 3b none observed in 1H NMR‡), indicating that minor hydroxide impurities crystallise out of solution preferentially. Nevertheless, the structures clearly show that both compounds are dimeric in the solid state, in contrast to the β-diketiminate magnesium hydride bearing the same nitrogen substituents (2,6-(diphenylmethyl)), which was monomeric.9 Whilst the presence of the hydroxide prevents comparison of the Mg–H bond lengths, it is still possible to compare the structures of 3a and 3b. 3b is significantly more contorted than 3a, with the two N–Mg–N planes twisted at approximately 49° from one another. The hydride/hydroxide ligands lay in a third orientation (Fig. S14†), whereas in 3a they are approximately orthogonal to the N–Mg–N plane. The dimeric structure of 3b is somewhat surprising given it's markedly different Mg-H NMR shift. Therefore, in order to determine if 3b maintained its dimeric structure in solution, a Diffusion Ordered NMR spectroscopy (DOSY) experiment was conducted and the diffusion coefficient and hydrodynamic radius compared with 1b (0.01 M in C6D6, 1b: 4.64 × 10−10 m2 s−1, 1950 Å3; 3b: 4.04 × 10−10 m2 s−1, 2950 Å3, Table S1†). Compound 3b is approximately 1.5 times the volume of 1b, which due to the sterically demanding amide co-ligand can only exist as a monomer. Conversely, 3a has only a slightly larger solution volume than 1a (1a: 4.95 × 10−10 m2 s−1, 1600 Å3; 3a: 4.72 × 10−10 m2 s−1, 1850 Å3). This suggests 3b has significant dimeric character in solution, whereas 3a appears to tend toward a monomer. This may be related to the asymmetric ligand rendering the dimer less stable. The stable formation of 3b is in contrast to attempts to isolate the analogous strontium hydride compound utilising a very similar ligand system (iPr versus Me).12 Here, the transient formation of the Sr–H species is proposed, followed by rapid deprotonation of an acidic methine proton (CHPh2) in the ligand framework to yield an intramolecular Ph2 (aryl)C− anion. Isolation of this new series of low-coordinate magnesium amide and magnesium hydride complexes presents the opportunity to investigate their stoichiometric and catalytic reactivity. Addition of one equivalent of N,N′-diisopropylcarbodiimide to 3a in C6D6 led to the facile formation of the Mg–H insertion product 4a (Scheme 3). The same reaction with compound 3b proceeded slower, but after 16 hours at room temperature the distinctive loss of the hydride resonance was observed along with the formation of a new singlet at 7.86 ppm corresponding to the formamidinate methine proton (4b, Fig. S3†). Both compounds 4a and 4b could be characterised in situ, however their solution stability was poor. Upon standing in a J. Youngs NMR tube over 2 days, a significant amount of decomposition was observed in both samples, in addition to liberation of the protonated ligand.§ Despite their relative instability, the reactivity of 3a and 3b towards N,N′-diisopropylcarbodiimide suggests they may be active catalysts for carbodiimide functionalisation. In 2016, Hill and co-workers reported the first example of the catalytic hydroboration of a carbodiimides.23 These reactions employed a β-diketiminate magnesium butyl complex as a pre-catalyst and propose magnesium hydride A to be the active catalytic species. Compounds 3a and 3b were found to catalyse the hydroboration of N,N′-diisopropylcarbodiimide with HBpin (4,4,5,5-tetramethyl-1,3,2-dioxaborolane) with surprisingly good efficiencies (Table 1). At 60 °C using 3a, 86% conversion to the singly reduced hydroboration product was achieved after 4 hours. This was reduced to 1.5 h when the reaction temperature was increased to 80 °C, approximately four times faster than the related β-diketiminate system. Compound 3a was also found to catalyse the reaction at room temperature, albeit slowly, with 19% conversion after 24 h. Compound 3b was significantly slower, requiring 5 h at 80 °C to reach high conversion, in line with its reduced activity towards N,N′-diisopropylcarbodiimide. The reduced reactivity of 3b relates to the increased sterics of 3bversus3a, in addition to higher retention of dimeric character in solution, rendering the magnesium–hydride bond less accessible towards substrates. In all cases, addition of a second equivalent of HBpin did not lead to formation of the doubly reduced product. The related amide compounds 1a and 1b were also shown
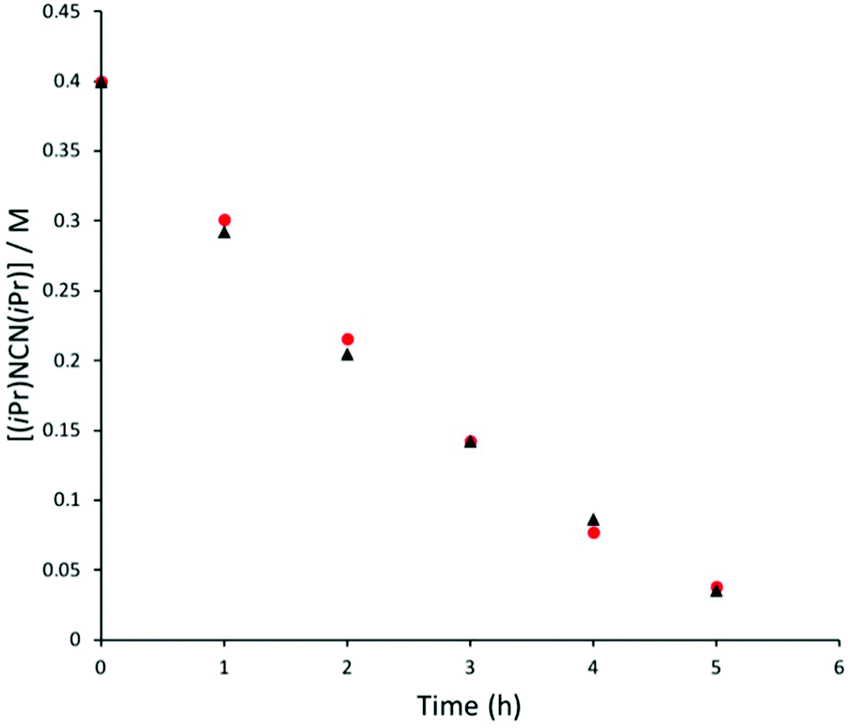
to be efficient pre-catalysts towards the hydroboration reaction, with identical reactivities to their hydride analogues. More detailed kinetic analysis was conducted at 80 °C, with 1H NMR spectra collected at regular time intervals during the reaction (Fig. 3). The rate of consumption of N,N′-diisopropylcarbodiimide and HBpin was consistent over time, with the rate not dropping significantly as the reaction progressed. The same reactivity profile was observed for 1a–b and 3a–b and the data did not fit pseudo first- or second-order reaction kinetics (Fig. S7 and 8†).
 |
| | Scheme 2 Synthetic procedure for the formation of compounds 3a and 3b. | |
 |
| | Scheme 3 Synthetic procedure for the formation of compounds 4a and 4b. | |
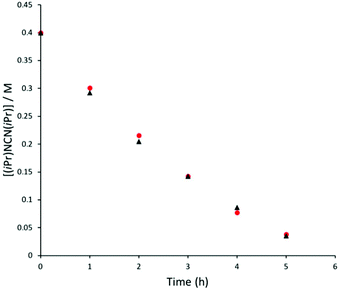 |
| | Fig. 3 Plot of [(iPr)NCN(iPr)] versus time for 1b (black triangles) and 3b (red circles). | |
Table 1 Catalytic hydroboration of N,N′-diisopropylcarbodiimide
|
|
| Complex |
t (h) |
T (°C) |
Conversion (%) |
| Reaction conditions: [HBpin] = [DIC] = 0.4 M in C6D6. Conversions determined by 1H NMR spectroscopy. |
|
1a
|
24 |
25 |
13 |
|
1a
|
1.5 |
80 |
90 |
|
3a
|
24 |
25 |
19 |
|
3a
|
4 |
60 |
86 |
|
3a
|
1.5 |
80 |
86 |
|
1b
|
5 |
80 |
91 |
|
3b
|
24 |
60 |
80 |
|
3b
|
5 |
80 |
90 |
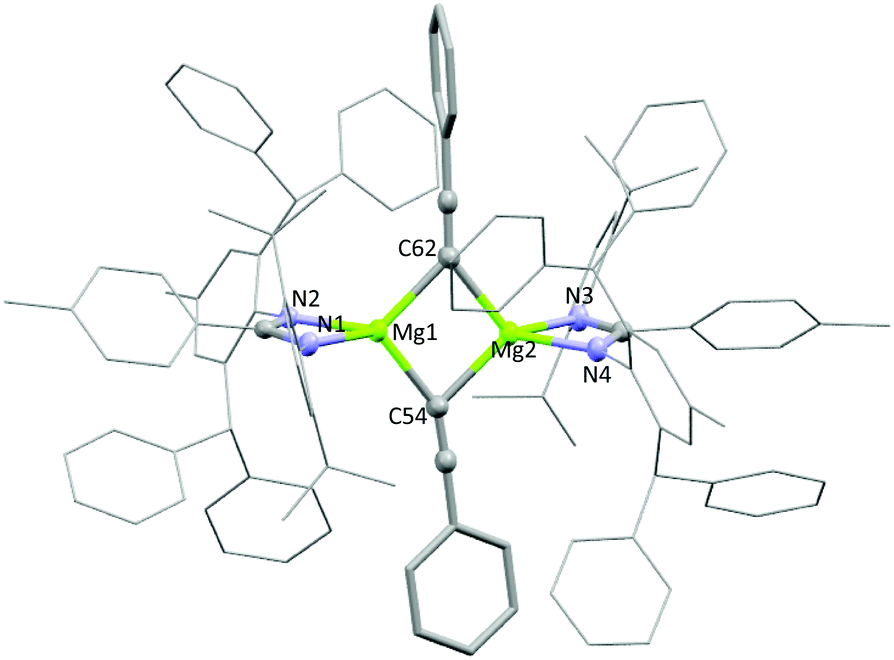
The reactivity of 3a and 3b towards terminal alkenes was also tested. Recently, a variety of alkene substrates have been shown to insert into the Mg–H bond of A, at high temperature on the day timescale.24 However, even with prolonged heating, no reaction was observed for 3a or 3b with either 1-hexene or styrene. Conversely, 1a and 3a were found to undergo facile reaction with the terminal alkyne phenylacetylene. Reaction of one equivalent of phenylacetylene with 1a in benzene-d6 led to the formation of dehydrogenation product 5a in less than 15 minutes. The characteristic Mg–N(SiMe3)2 peak at −0.03 was seen to disappear, and bis(trimethylsilyl)amine was observed to form. Reaction of 3a with one equivalent of phenylacetylene also saw the formation of 5a, with loss of the magnesium hydride signal and dihydrogen formation observed. An intermediate species was also seen to form during the reaction but it could not be identified (Fig. S4†). Crystals of 5a formed readily from a saturated benzene-d6 solution and were analysed by single crystal X-ray diffraction. The complex crystallised as a dimer in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, with the two N–Mg–N fragments slightly distorted from one another in one plane and the phenyl acetylides ligands lying orthogonal (Fig. 4). The phenyl acetylides lie slightly asymmetrically between the two magnesium centres, with an Mg1–C62
space group, with the two N–Mg–N fragments slightly distorted from one another in one plane and the phenyl acetylides ligands lying orthogonal (Fig. 4). The phenyl acetylides lie slightly asymmetrically between the two magnesium centres, with an Mg1–C62![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond angle of 133.4° and a Mg2–C62C bond angle of 143.6°. A series of related compounds based on β-diketiminate magnesium and calcium complexes has been previously reported by Hill and co-workers, however these show significantly more distorted acetylides units.25 As with 4a, all attempts to isolate a sample of 5a led to rapid product decomposition indicating 5a has poor stability. It is likely that the steric influence of the ligand in 5a prevents additional stabilising interactions between the alkyne groups and the magnesium centres. Compounds 1b and 3b also react with phenylacetylene, though the reaction is slower than with 1a and 3a. Reaction of 1b with a slight excess of phenylacetylene in benzene-d6 led to the slow formation of 5b over 16 hours at room temperature. Again the characteristic loss of the Mg–N(SiMe3)2 peak at −0.15 was observed with concomitant formation of bis(trimethylsilyl)amine. Reaction of 3b under the same conditions showed negligible product formation, however heating for two hours at 80 °C led to the complete and clean formation of compound 5b. Analysis by 1H NMR also showed the concomitant formation of styrene, as confirmed against an independent sample, and unlike reaction with 3a no liberation of dihydrogen was observed (Fig. S5†). This strongly indicates that the formation of 5b goes via a magnesium alkenyl intermediate, formed through insertion of the alkyne into the magnesium hydride bond (Scheme 4). This intermediate can then react with a second equivalent of phenylacetyene, forming 5b and liberating styrene. However, no intermediate species were detected by 1H NMR during the course of the reaction.¶ Addition of excess phenylacetylene to 5a or 5b in benzene-d6 did not lead to the formation of any stoichiometric or catalytic dimerization products and at elevated temperatures only ligand protonation was observed. Hill and co-workers also observe that β-diketiminate magnesium species were deprotonated in the presence of excess phenylacetylene at elevated temperatures.25
C bond angle of 133.4° and a Mg2–C62C bond angle of 143.6°. A series of related compounds based on β-diketiminate magnesium and calcium complexes has been previously reported by Hill and co-workers, however these show significantly more distorted acetylides units.25 As with 4a, all attempts to isolate a sample of 5a led to rapid product decomposition indicating 5a has poor stability. It is likely that the steric influence of the ligand in 5a prevents additional stabilising interactions between the alkyne groups and the magnesium centres. Compounds 1b and 3b also react with phenylacetylene, though the reaction is slower than with 1a and 3a. Reaction of 1b with a slight excess of phenylacetylene in benzene-d6 led to the slow formation of 5b over 16 hours at room temperature. Again the characteristic loss of the Mg–N(SiMe3)2 peak at −0.15 was observed with concomitant formation of bis(trimethylsilyl)amine. Reaction of 3b under the same conditions showed negligible product formation, however heating for two hours at 80 °C led to the complete and clean formation of compound 5b. Analysis by 1H NMR also showed the concomitant formation of styrene, as confirmed against an independent sample, and unlike reaction with 3a no liberation of dihydrogen was observed (Fig. S5†). This strongly indicates that the formation of 5b goes via a magnesium alkenyl intermediate, formed through insertion of the alkyne into the magnesium hydride bond (Scheme 4). This intermediate can then react with a second equivalent of phenylacetyene, forming 5b and liberating styrene. However, no intermediate species were detected by 1H NMR during the course of the reaction.¶ Addition of excess phenylacetylene to 5a or 5b in benzene-d6 did not lead to the formation of any stoichiometric or catalytic dimerization products and at elevated temperatures only ligand protonation was observed. Hill and co-workers also observe that β-diketiminate magnesium species were deprotonated in the presence of excess phenylacetylene at elevated temperatures.25
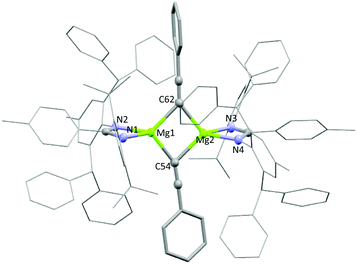 |
| | Fig. 4 Solid state structure of compound 5a. Selected bond lengths (Å) and angles (°): Mg1–N1 2.042(1); Mg1–N2 2.054(1); Mg1–C54 2.168(2), Mg1–C62 2.190(2); N1–Mg1–N2 65.8; C54–Mg1–C62 97.1. | |
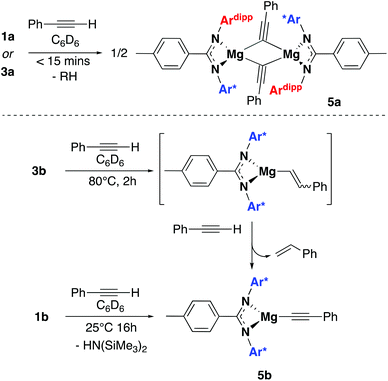 |
| | Scheme 4 Top: Synthetic procedure for the formation of 5a. Bottom: two synthetic routes to 5b. | |
Last year, Rueping and co-workers reported the first example of the magnesium catalysed hydroboration of a series of terminal and internal alkynes.26 Dibutyl magnesium (MgBu2, 10 mol%) was found to form exclusively the E-product in high yields after 18 h at 60 °C. Conversion was seen to be lower in the presence of β-diketiminate ligands. The isolated compounds 1a, 1b and 3b were all found to catalyse the hydroboration of terminal alkynes, in all cases yielding the same anti-Markovnikov E-product. Using 1a and 1b (10 mol% in benzene-d6), a small amount of conversion was observed at 25 °C after 4 days, 6% and 26% respectively. However, at 80 °C high conversions were reached within 16 h (86 and 88% after 16 hours). The magnesium hydrides 3a and 3b were found to catalyse the hydroboration reactions at near identical rates (Table SX†). When analysing the 1H NMR spectrum of the catalytic mixture of 3b after 16 hours at 80 °C free ligand was observed, suggesting loss of the ligand during catalysis. Though this is not clearly observed is the other compounds, the minimal difference in rate observed between the four compounds bearing two different ligand systems, suggests that the ligand does not remain bound during catalysis and that the true active species is a magnesium centred organometallic generated in situ through reaction with phenylacetylene and HBpin. Previous observations regarding ligand protonation in the presence of excess phenylacetylene support this argument. Comparable rates with Rueping's system suggests the presence of a common catalytically active species. Recent work by Thomas and co-workers has highlighted hidden boron containing species to be active catalysts in ‘metal catalysed’ alkene and alkyne hydroboration reactions.27 The nature of the active catalyst in this and related systems is the subject of ongoing investigation.
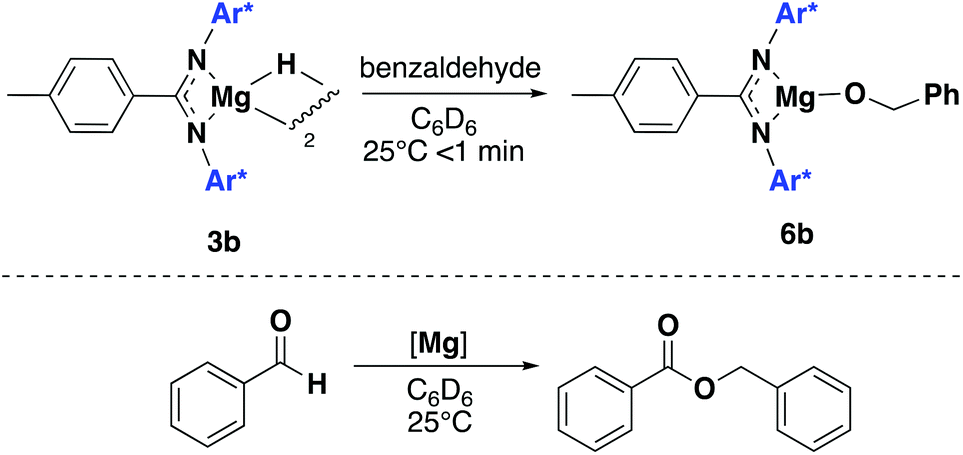
Attempts to isolate alkoxides derivatives of LH1 and LH2 were also investigated. However, the reaction of a variety of alcohols with 1a and 1b only led to protonolysis of the ligand. 3b was instead reacted with benzaldehyde: addition of 1 equivalent of benzaldehyde to 3b in C6D6 led to the facile formation of the alkoxide product 6b (Scheme 5, top). In the presence of an excess of benzaldehyde a dark orange solution was seen to form with analysis by 1H NMR spectroscopy revealing the formation of the Tishchenko dimerization product, benzyl benzoate (Scheme 5, bottom). At 1mol% catalyst loading 3b catalysed a 1 M solution of benzaldehyde in C6D6 to 72% conversion in 10 minutes. After 1 hour this has increased to 89% conversion. Coles and co-workers have previously shown related magnesium amides and alkoxides to be efficient catalysts for the Tishchenko reaction, but this is the first example of a magnesium hydride compound being used as a pre-catalyst.28,29 Compound 1b was also found to be catalytically active towards the dimerization of benzaldehyde, with only trace amounts of starting material remaining after 10 minutes under the same reaction conditions which is significantly faster than the previously reported magnesium amides (95% yield after 3 h). Interestingly, upon addition of benzaldehyde to 1b, the solution remained colourless, suggesting both 1b and 3b proceed via different catalytic intermediates. Attempts to react both 1a, 3a and magnesium hydride A with benzaldehyde led some initial unidentified reaction occurring, but no benzyl benzoate formation was observed.
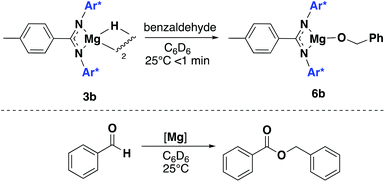 |
| | Scheme 5 Top: synthetic procedure for the formation of 6b. Bottom: tischenko reaction catalysed by 1b and 3b. | |
Conclusions
A series of novel magnesium amidinate complexes has been synthesised and fully characterised, including two new rare examples of stable magnesium hydrides (3a and 3b). The catalytic activity of these complexes has been investigated, with compounds 1a–b and 3a–b shown to efficiently mediate the hydroboration of N,N′-diisopropylcarbodiimide. 1a and 3b are also extremely proficient at catalysing the Tishchenko reaction (100 equivalents < 10 min) and 1a–b/3a–b all react stoichiometrically with phenylacetylene. Comparing this reactivity with other magnesium hydrides draws mixed conclusions. 1a and 3b catalyse the hydroboration of N,N′-diisopropylcarbodiimide significantly faster than the Mg–H dimer A, but unlike A they show no reactivity towards alkenes. The monomeric Mg–H B can catalyse both the hydroboration of carbodiimides and the hydrosilation of styrene at room temperature. The rapid reactivity of 3b towards the Tishchenko reaction is the first example of a Mg–H acting as a pre-catalyst, indeed 3a and A do not lead to any aldehyde dimerization product. This curious pattern in reactivity warrants further comment. The reduced reactivity of 1b/3bversus1a/3a is undoubtably due to the increase in steric bulk, which rather than rendering complexes 1b/3b monomeric restricts access of the substrate to the metal centre. In fact, DOSY experiments indicate that the less sterically hindered 3a has more monomeric character in solution than 3b. The steric bulk of these amidinate complexes appears to protect the dimeric magnesium hydride core more efficiently than the 2,6-diisopropylphenyl β-diketiminate ligand in A, hence no reactivity is observed with alkenes. However, when reacted with coordinating substrates, such as N,N′-diisopropylcarbodiimide or benzaldehyde, a marked increase in reactivity is observed versus the β-diketiminate system, suggesting that once aggregation is broken the Mg centre is more accessible. The 4-membered versus 6-membered ligand–metal coordination environment facilitates this increased reactivity, however, some experimental observations also suggest that the more strained ring system makes the complexes less stable. Overall, these results highlight the importance of easy access of the reactive Mg–H core and suggest that monomeric magnesium hydrides, such as B, will continue to display superior catalytic capability than complexes with more aggregated structures.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The Ramsay Memorial Trust and UCL Chemistry are thanked for their financial support. Prof. Claire Carmalt is thanked for her support and mentorship. Dr Jeremy Cockcroft and Richard Kong are thanked for useful XRD discussions.
Notes and references
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- D. Mukherjee and J. Okuda, Angew. Chem., Int. Ed., 2018, 57, 1458–1473 CrossRef PubMed.
- S. Harder, Chem. Commun., 2012, 48, 11165–11177 RSC.
- S. J. Bonyhady, S. P. Green, C. Jones, S. Nembenna and A. Stasch, Angew. Chem., Int. Ed., 2009, 48, 2973–2977 CrossRef CAS PubMed.
- M. Arrowsmith, T. J. Hadlington, M. S. Hill and G. Kociok-Köhn, Chem. Commun., 2012, 48, 4567–4564 RSC.
- M. D. Anker, M. S. Hill, J. P. Lowe and M. F. Mahon, Angew. Chem., 2015, 127, 10147–10149 CrossRef.
- M. Rauch, S. Ruccolo and G. Parkin, J. Am. Chem. Soc., 2017, 139, 13264–13267 CrossRef CAS PubMed.
- S. Schnitzler, T. P. Spaniol, L. Maron and J. Okuda, Chem. – Eur. J., 2015, 21, 11330–11334 CrossRef CAS.
- M. Arrowsmith, B. Maitland, G. Kociok-Köhn, A. Stasch, C. Jones and M. S. Hill, Inorg. Chem., 2014, 53, 10543–10552 CrossRef CAS PubMed.
- A. K. Maity, S. Fortier, L. Griego and A. J. Metta-Magaña, Inorg. Chem., 2014, 53, 8155–8164 CrossRef CAS PubMed.
- A. K. Maity, J. Murillo, A. J. Metta-Magaña, B. Pinter and S. Fortier, J. Am. Chem. Soc., 2017, 139, 15691–15700 CrossRef CAS PubMed.
- B. Freitag, J. Pahl, C. Färber and S. Harder, Organometallics, 2018, 37, 469–475 CrossRef CAS.
- C. N. de Bruin-Dickason, T. Sutcliffe, C. A. Lamsfus, G. B. Deacon, L. Maron and C. Jones, Chem. Commun., 2018, 54, 786–789 RSC.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem. – Eur. J., 2009, 16, 938–955 CrossRef.
- R. Lalrempuia, A. Stasch and C. Jones, Chem. – Asian J., 2014, 10, 447–454 CrossRef.
- H. Xie, X. Hua, B. Liu, C. Wu and D. Cui, J. Organomet. Chem., 2015, 798, 335–340 CrossRef CAS.
- L. Fohlmeister and A. Stasch, Chem. – Eur. J., 2016, 22, 10235–10246 CrossRef CAS PubMed.
- L. E. Lemmerz, D. Mukherjee, T. P. Spaniol, A. Wong, G. Ménard, L. Maron and J. Okuda, Chem. Commun., 2019, 55, 3199–3202 RSC.
- L. Garcia, M. F. Mahon and M. S. Hill, Organometallics, 2019, 38, 3778–3785 CrossRef CAS.
- D. J. Gallagher, K. W. Henderson, A. R. Kennedy, C. T. O'Hara, R. E. Mulvey and R. B. Rowlings, Chem. Commun., 2002, 376–377 RSC.
- M. Wiesinger, B. Maitland, H. Elsen, J. Pahl and S. Harder, Eur. J. Inorg. Chem., 2019, 2019, 4433–4439 CrossRef CAS.
- S. Schnitzler, P. Cui, T. P. Spaniol and J. Okuda, Dalton Trans., 2017, 46, 1761–1765 RSC.
- C. Weetman, M. S. Hill and M. F. Mahon, Chem. – Eur. J., 2016, 22, 7158–7162 CrossRef CAS.
- L. Garcia, C. Dinoi, M. F. Mahon, L. Maron and M. S. Hill, Chem. Sci., 2019, 10, 8108–8118 RSC.
- M. Arrowsmith, M. R. Crimmin, M. S. Hill, S. L. Lomas, D. J. MacDougall and M. F. Mahon, Organometallics, 2013, 32, 4961–4972 CrossRef CAS.
- M. Magre, B. Maity, A. Falconnet, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 7025–7029 CrossRef CAS.
- A. D. Bage, T. A. Hunt and S. P. Thomas, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS.
- B. M. Day, W. Knowelden and M. P. Coles, Dalton Trans., 2012, 41, 10930–10934 RSC.
- R. J. Schwamm, B. M. Day, N. E. Mansfield, W. Knowelden, P. B. Hitchcock and M. P. Coles, Dalton Trans., 2014, 43, 14302–14314 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2016829–2016833. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02523b |
| ‡ Crystals of 3b were grown from a concentrated C6D6 solution in a sealed YT NMR tube which did not indicate the presence of any Mg–OH side product in the 1H NMR spectrum. |
| § These reactions have been attempted multiple times with additional drying measures put in place, but always yielded decomposition products. The unstable molecule may be reacting intramolecularly or with solvent. |
| ¶ Repeated attempts to collect single crystal data on 5b were unsuccessful due weakly diffracting samples. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a