
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMixed-ligand lanthanide complexes supported by ditopic bis(imino-methyl)-phenol/calix[4]arene macrocycles: synthesis, structures, and luminescence properties of [Ln2(L2)(MeOH)2] (Ln = La, Eu, Tb, Yb)†
Steve
Ullmann
ab,
Peter
Hahn
a,
Parvathy
Mini
c,
Kellie L.
Tuck
 c,
Axel
Kahnt
d,
Bernd
Abel
de,
Matias E.
Gutierrez Suburu
fg,
Cristian A.
Strassert
fg and
Berthold
Kersting
*a
c,
Axel
Kahnt
d,
Bernd
Abel
de,
Matias E.
Gutierrez Suburu
fg,
Cristian A.
Strassert
fg and
Berthold
Kersting
*a
aInstitut für Anorganische Chemie, Universität Leipzig, Johannisallee 29, 04103 Leipzig, Germany. E-mail: b.kersting@uni-leipzig.de; Fax: +49(0)341-97-36199
bInstitut für Nichtklassiche Chemie e.V., Permoserstraße. 15, D-04318 Leipzig, Germany
cSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia
dLeibniz Institute of Surface Engineering (IOM), Department of Functional Surfaces, Permoserstr. 15, D-04318 Leipzig, Germany
eWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, Linnéstraße 2, D-04103 Leipzig, Germany
fInstitut für Anorganische und Analytische Chemie, CiMIC, SoN - Westfälische Wilhelms-Universität Münster, Corrensstraße 28/30, 48149 Münster, Germany
gCeNTech - Westfälische Wilhelms-Universität Münster, Heisenbergstraße 11, 48149 Münster, Germany
First published on 28th July 2020
Abstract
The lanthanide binding ability of a macrocyclic ligand H6L2 comprising two bis(iminomethyl)phenol and two calix[4]arene units has been studied. H6L2 is a ditopic ligand which provides dinuclear neutral complexes of composition [Ln2(L2)(MeOH)2] (Ln = La (1), Eu (2), Tb (3), and Yb (4)) in very good yield. X-ray crystal structure analyses for 2 and 3 show that (L2)6− accommodates two seven coordinated lanthanide ions in a distorted monocapped trigonal prismatic/octahedral coordination environment. UV-vis spectroscopic titrations performed with La3+, Eu3+, Tb3+ and Yb3+ ions in mixed MeOH/CH2Cl2 solution (I = 0.01 M NBu4PF6) reveal that a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (metal:ligand) stoichiometry is present in solution, with log K11 and K21 values ranging from 5.25 to 6.64. The ratio α = K11/K21 of the stepwise formation constants for the mononuclear (L2 + M = ML2, logK11) and the dinuclear complexes (ML2 + M = M2L2, logK21) was found to be invariably smaller than unity indicating that the binding of the first Ln3+ ion augments the binding of the second Ln3+ ion. The present complexes are less luminescent than other seven-coordinated Eu and Tb complexes, which can be traced to vibrational relaxation of excited EuIII and TbIII states by the coligated MeOH and H2O molecules and/or low-lying ligand-to-metal charge-transfer (LMCT) states.
:1 (metal:ligand) stoichiometry is present in solution, with log K11 and K21 values ranging from 5.25 to 6.64. The ratio α = K11/K21 of the stepwise formation constants for the mononuclear (L2 + M = ML2, logK11) and the dinuclear complexes (ML2 + M = M2L2, logK21) was found to be invariably smaller than unity indicating that the binding of the first Ln3+ ion augments the binding of the second Ln3+ ion. The present complexes are less luminescent than other seven-coordinated Eu and Tb complexes, which can be traced to vibrational relaxation of excited EuIII and TbIII states by the coligated MeOH and H2O molecules and/or low-lying ligand-to-metal charge-transfer (LMCT) states.
Introduction
The development of ligand systems that form both stable and luminescent lanthanide complexes is an active research area due to their many potential applications,1 particularly the optical detection and sensing of biologically relevant or environmentally harmful molecules and ions.2–4 The calix[4]arenes5–8 with their hard O4 donor set have turned out to be versatile supporting ligands that form stable complexes with a range of trivalent lanthanide ions.9–17 Calixarenes bearing pendant chromophores can display an antenna effect, and several luminescent lanthanide calixarene complexes have already been investigated.18–23Calix[4]arene bearing pendant Schiff-base moieties have proved to be versatile ligands for transition metals.24 We have investigated previously the lanthanide coordination chemistry of the hybrid calix[4]arene-Schiff-base ligand H4L1 (Fig. 1). This monotopic ligand supports dinuclear lanthanide complexes of composition [HNEt3][Ln2(HL1)(L1)] with Ln = Sm3+, Eu3+, Gd3+, and Tb3+,25 where two monomeric [LnL1] units dimerize via phenolato bridges from the calix[4]arenes. The coordinated salicylaldimines exhibit a triplet-state energy of 22000 cm−1,25 and act as antennas enabling sensitization of the EuIII and TbIII emission when irradiated in the solid state (λex = 311 nm). We have also reported a macrocyclic ligand H6L2 comprising of fluorescent bis(iminomethyl)phenol and calix[4]arene head units, which enabled the optical detection of Zn2+ ions among a series of biologically relevant metal ions in solution.26 In the presence of Zn2+ ions the fluorescent intensity output increased, presumably due to coordination of the Zn2+ ions reducing the PET quenching mechanism of the imino functional groups. The observations made with H4L1 prompted us to investigate the lanthanide coordination chemistry of H6L2. Of particular interest to us was whether the macrocyclic structure would form stable Ln3+ complexes and whether the bis(iminomethyl)phenolate units could function as antennae and enable excitation of the lanthanide ion, noting that the ligand H6L2 displayed strong absorption bands at 346 nm and 447 nm.26 To our knowledge, no lanthanide complexes of H6L2 have been reported so far only transition metal complexes have been investigated.27–29 This paper demonstrates that H6L2 behaves as a ditopic ligand that supports dinuclear macrocyclic lanthanide complexes of the type [Eu2(L2)(L′)2] (Ln = La, Eu, Tb, Yb, L′ = solvent). Their structures and properties are compared with those of the previously published [HNEt3][Ln2(HL1)(L1)] complexes.
 | ||
| Fig. 1 Structures of the hybrid Schiff-base/calix[4]arene ligands H4L1 and H6L2. | ||
Results and discussion
Synthesis and characterization of compounds
The macrocyclic ligand H6L2 was prepared by a [2 + 2] Schiff-base condensation reaction between 25,27-bis(aminoethoxy)-26,28-dihydroxy-calix[4]arene and 4-tert-butyl-2,6-diformylphenol according to literature procedures.26,30 Reaction of H6L2 with La(NO3)3 and NEt3 as a base, in a 1:2.2:5 molar ratio in MeOH/CH2Cl2, results in immediate formation of a yellow precipitate of composition [La2(L2)(MeOH)2] (1, where (L2)6− represents the fully deprotonated form of H6L2) in a very good (81%) yield (Scheme 1). The analogous Eu3+ (2), Tb3+ (3), and Yb3+ complexes (4) were also synthesized in this manner. Attempts to prepare mononuclear complexes (utilizing a 1:1 stoichiometric ratio) were unsuccessful, and only low yields of the dinuclear complexes could be isolated, most likely due to a lower solubility of the dinuclear complex species, but other reasons cannot be ruled out. All complexes are air-stable both in the solid and solution state. They are moderately soluble in polar aprotic solvents such as CHCl3, CH2Cl2, MeCN, and THF, but do not dissolve in MeOH or EtOH. The guest solvate molecules hosted in the cavities of the calix[4]arenes are lost upon standing in air. The complexes gave satisfactory elemental analysis and were further characterized by ESI mass spectrometry, FTIR and UV-vis spectroscopy, and X-ray crystallography (for 2 and 3).
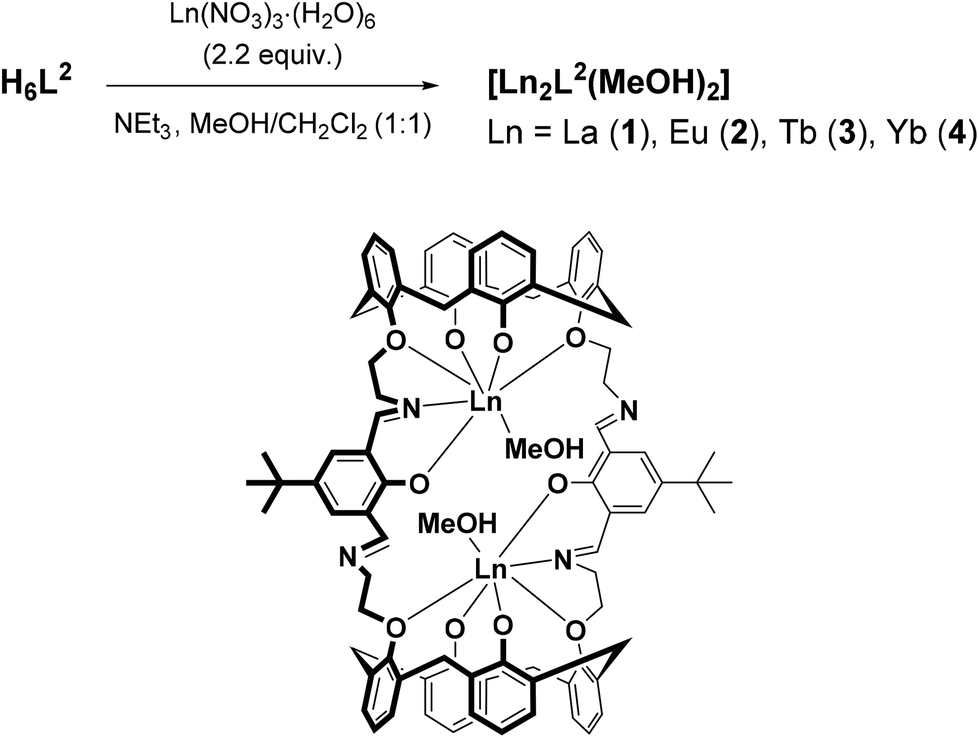 | ||
| Scheme 1 Synthesis of the complexes 1–4. | ||
Selected analytical data for H6L2 and the complexes 1–4 are listed in Table 1. FT-IR spectra of powdered samples were recorded in the ATR mode. As can be seen (Fig. S1, S3, S5 and S7†), the OAr–H stretches that were seen in the spectra of the free ligand at 3400 cm−1 are absent in the spectra of complexes 1–4, indicative of Ln3+-bound phenolate functions. Coordination of the phenolate groups is further supported by the red shifted ν(C–Ophenol) stretches which occurs at 1340 cm−1 for the free ligand (Fig. 2). The FT-IR spectra of the complexes feature also pronounced differences in the ν(ArO–Cether) stretching region (1050–950 cm−1), which imply the coordination of the phenol ether O donors as well.31 All complexes exhibit a band around 1635 cm−1 of medium intensity that can be attributed to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) (imine) stretching vibrations, a typical wavelength value for imine functional groups that are coordinated to Ln3+ ions.
N) (imine) stretching vibrations, a typical wavelength value for imine functional groups that are coordinated to Ln3+ ions.
 | ||
| Fig. 2 ATR-FT-IR spectra in the 1400–950 cm−1 region of H6L2 and complexes 1–4. | ||
| Compound |
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1
/cm−1 |
λ max/nm (ε/M−1 cm−1)b |
|---|---|---|
| a ATR mode. b CH2Cl2/MeOH (1/1 v/v), 2 × 10−5 M. | ||
| H6L2 | 1640 (CN) |
226 (70395) (π–π*, calixarene) |
| 1340 (C–O) | 245 (sh, 55435) (π–π*, calixarene) |
|
| 275 (sh, 25651) (π–π*, calixarene) |
||
| 346 (10735) (π–π*, PhCHNR) |
||
| 446 (3721) (π –π*, PhCHNR) |
||
| 1 (La) | 1632 (CN) |
231 (87976) (π–π*, calixarene–OLn) |
| 1322 (C–O) | 250 (59730) (π–π*, calixarene–OLn) |
|
| 304 (13306) (π–π*, PhCHNR–Ln) |
||
| 410 (14224) (π–π*, PhCHNR–Ln) |
||
| 2 (Eu) | 1635 (CN) |
231 (87092) (π–π*, calixarene–OLn) |
| 1317 (C–O) | 254 (sh, 57697) (π–π*, calixarene–OLn) |
|
| 302 (sh, 15985) (π–π*, PhCHNR–Ln) |
||
| 409 (15339) (π–π*, PhCHNR–Ln) |
||
| 3 (Tb) | 1636 (CN) |
227 (107414) (π–π*, calixarene–OLn) |
| 1318 (C–O) | 251 (69553) (π–π*, calixarene–OLn) |
|
| 303 (18343) (π–π*, PhCHNR–Ln) |
||
| 409 (16976) (π–π*, PhCHNR–Ln) |
||
| 4 (Yb) | 1634 (CN) |
230 (72147) (π–π*, calixarene–OLn) |
| 1330 (C–O) | 257 (42642) (π–π*, calixarene–OLn) |
|
| 307 (7994) (π–π*, PhCHNR–Ln) |
||
| 417 (11100) (π–π*, PhCHNR–Ln) |
||
The ESI-MS data confirms the presence of dinuclear lanthanide complexes of the type [Ln2(L2)(L′)2] with two exchangeable coligands L′, in agreement with the X-ray crystallographic findings detailed below. However, the large and neutral charged dinuclear complexes are hard to vaporize leading to strong fragmentation and only weak intensity for the molecular ion peaks in the respective mass spectra (Fig. S2, S4, S6 and S8†).
Crystallographic characterization
In order to determine the structures of the complexes, single-crystals (for 2 and 3) were grown and subjected to X-ray crystallographic analyses. Single crystals of [Eu2(L2)(MeOH)2]·4CH2Cl2·MeOH (2·4CH2Cl2·MeOH), obtained from a mixed MeOH/CH2Cl2 system, were subjected to an X-ray crystallographic analysis. The crystal structure is composed of dinuclear [Eu2(L2)(MeOH)2] complexes (Fig. 3), and CH2Cl2 and MeOH solvate molecules. Some of the CH2Cl2 and MeOH solvate molecules were observed to occupy the voids in the structure. Each calixarene cavity is also occupied by one CH2Cl2 molecule, which is held in place by CH⋯π interactions with the aromatic rings of the calixarene units as illustrated in Fig. 4. Selected metrical data are listed in Table 2 and Fig. 3 and 4. | ||
| Fig. 3 Single-crystal X-ray diffraction structure of [Eu2(L2)(MeOH)2]·4CH2Cl2·MeOH (2·4CH2Cl2·MeOH) (ball-and-stick representation). The CH2Cl2 and MeOH solvate molecules are omitted for clarity. Dashed lines refer to intramolecular hydrogen bonding interactions (O11⋯N4 = 2.71, O12⋯N2 = 2.64). | ||
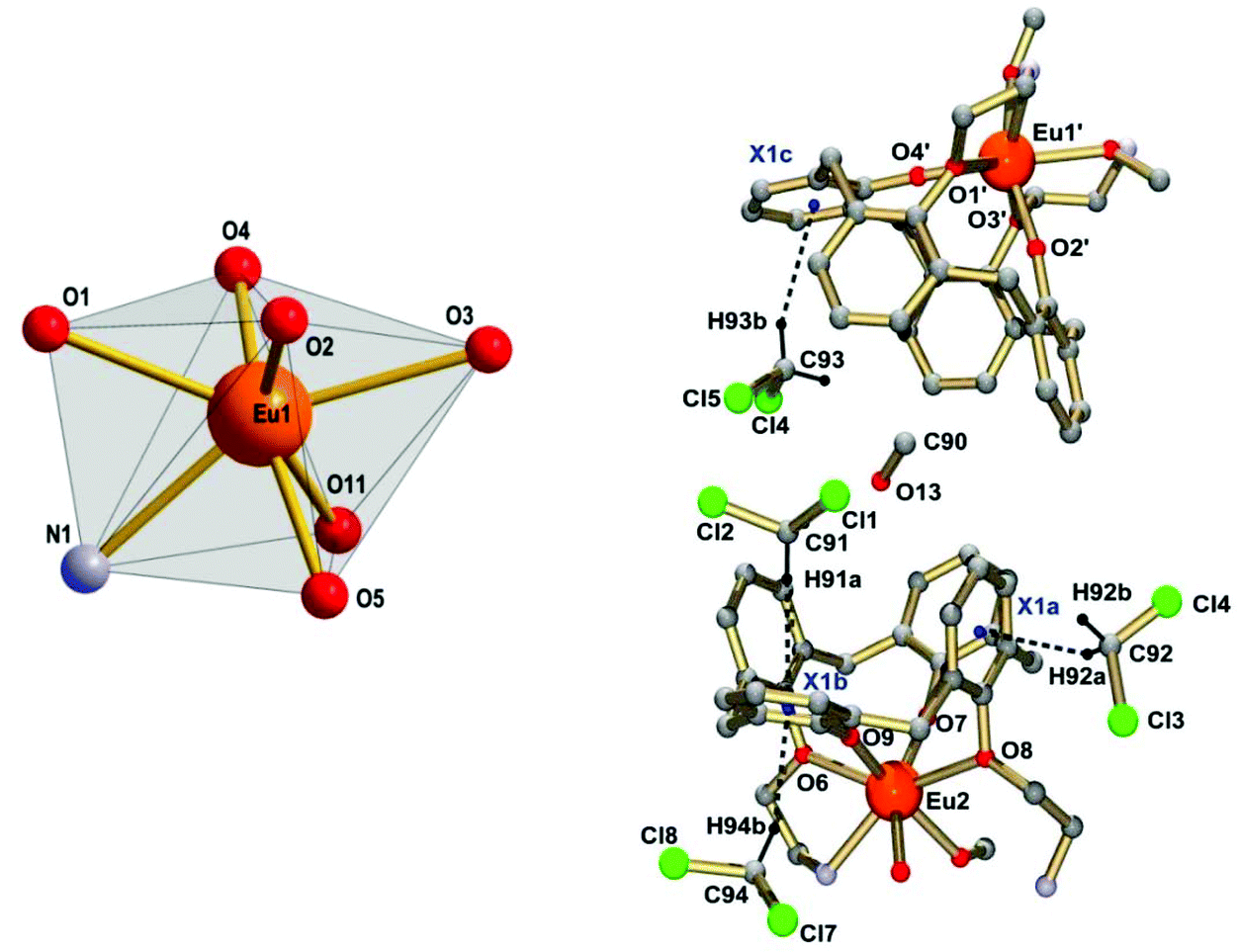 | ||
| Fig. 4 Left: Immediate coordination environment of the Eu3+ ions in 2·4CH2Cl2·MeOH. Right: Representation of the intermolecular CH⋯π interactions between the CH2Cl2 solvate molecules and the calix[4]arene units of the ligand. Parts of the complexes have been omitted for clarity. Symmetry code used to generate equivalent atoms: −x, 0.5 + y, 0.5 − z (′). Selected distances/Å: C92⋯X1a (center of ring) 3.566, C91⋯X1b 3.696, C94⋯X1b 3.450, C93⋯X1c 3.771. | ||
| M | 2·4CH2Cl2·MeOH (M = Eu) | 3·xH2O (M = Tb) |
|---|---|---|
| M1–O1 | 2.528(3) | 2.482(2) |
| M1–O2 | 2.164(3) | 2.095(3) |
| M1–O3 | 2.576(3) | 2.505(3) |
| M1–O4 | 2.141(3) | 2.089(3) |
| M1–O5 | 2.254(3) | 2.189(2) |
| M1–O11 | 2.457(3) | 2.322(3) |
| M1–N1 | 2.516(4) | 2.442(3) |
| M2–O6 | 2.498(3) | 2.487(2) |
| M2–O7 | 2.180(3) | 2.110(2) |
| M2–O8 | 2.561(3) | 2.439(3) |
| M2–O9 | 2.143(4) | 2.068(3) |
| M2–O10 | 2.266(3) | 2.205(3) |
| M2–O12 | 2.398(3) | 2.324(2) |
| M2–N2 | 2.503(4) | 2.402(3) |
| M1⋯M2 | 9.297(1) | 9.291(1) |
The complex 2·4CH2Cl2·MeOH has pseudo-C2 symmetry. The Eu3+ ions are seven-coordinate by four calix[4]arene O atoms and the imine N and O atoms of the appended Schiff base unit. A MeOH ligand completes the coordination polyhedron, which is best described as intermediate between distorted mono-capped trigonal prismatic or mono-capped octahedral, two of the most common coordination geometries for seven coordinate lanthanide complexes.32,33 The Eu–O bond lengths range from 2.14 to 2.59 Å. The hard phenolate functions of the calixarenes form the shortest Eu–O bonds (mean value 2.15 Å), followed by slightly longer (weaker) Eu–O iminophenolate linkages at 2.26 Å. The weaker donor ability of the latter can be traced to mesomeric effects induced by the imine functions, which reduce the electron density on the O donors through delocalization into the aromatic π-system. Indeed, the stronger electron delocalization is also reflected in a decrease of the C–O bond length from 1.32 Å for the calixarene moiety to a value of 1.29 Å in the iminophenol (Schiff-base) units. The MeOH and phenol ether O atoms have much weaker donor ability, implicated by the very long Eu–O bond distances of 2.43 and 2.54 Å, respectively. The Eu–N bonds are also quite long at 2.51 Å. The structure is further stabilized by intramolecular hydrogen bonds between the MeOH ligands and the non-coordinating imine functions.34–36 The intramolecular Eu⋯Eu distance in 2 is 9.29(3) Å, and interestingly the intermolecular Eu⋯Eu distance is shorter, 6.34(3) Å.
Crystals of [Tb2(L2)(MeOH)(H2O)]·(H2O)x (3·(H2O)x, x ∼ 10) obtained from a mixed CH2Cl2/MeOH solution are monoclinic, space group P21/n. The asymmetric unit contains a neutral, dinuclear [Tb2(L2)(MeOH)(H2O)] complex and several water solvate molecules. Most of the latter were heavily disordered and the corresponding electron density was removed with the SQUEEZE routine implemented in Platon.37 The MeOH and H2O molecules bonded to the Tb3+ atoms, on the other hand, were not disordered and were included in the structure refinement. Fig. 5 shows the structure of the dinuclear terbium complex 3. Table 2 lists selected bonds lengths. The atom labeling utilized for 2 was adapted for 3 to aid structural comparisons.
 | ||
| Fig. 5 Single-crystal X-ray diffraction structure of the dinuclear [Tb2(L2)(MeOH)(H2O)] complex in crystals of 3·(H2O)x, x ∼ 10 (ball-and-stick representation). Dashed lines refer to intramolecular hydrogen bonding interactions (O11⋯N4 = 2.75 Å, O11⋯O13 2.78 Å, O12⋯N2 = 2.62 Å). | ||
The structure of this complex is very similar to that of the europium complex 2. The macrocycle wraps around the two Tb3+ ions in the same fashion, but in contrast to 2, the coordination spheres of the Tb atoms are completed by either one methanol (Tb2) or one water ligand (Tb1). As in 2, the Tb-bound coligands are hydrogen bonded to the imine-nitrogen atoms of the supporting ligand, but the H2O ligand hydrogen bonds also to a water solvate molecule (O11⋯O13). Likewise, three sets of Tb–O distances can be distinguished. However, the corresponding metal–ligand bond lengths in 2 and 3 differ by ca. 0.05 Å, as one might expect in view of the smaller ionic radius of Tb3+.38 The intramolecular Tb⋯Tb distance in 3 is 9.290(1) Å, again about 3 Å longer than the shortest intermolecular Tb⋯Tb distance (7.65 Å). The packing of the [Ln2(L2)(MeOH)2] complexes in 2 and 3 is very similar, and appears not to be influenced by the type of co-crystallized solvate molecules.
The binding mode of H6L2 seen in the present lanthanide complexes differs significantly from that observed in the Zn and Ni complexes [Zn(L2)(py)2] and [Ni(L2)], the only other complexes, whose crystal structures were reported so far with this large macrocyclic ligand system.26 In both cases, H6L2 is a tetradentate ligand, binding the divalent 3d ions via two bidentate iminomethylphenolate functions in a square-planar (Ni2+) or octahedral (Zn2+) coordination geometry, respectively. The present lanthanide structures clearly demonstrate the potential of H6L2 in providing additional coordinating modes in which the calix[4]arene moieties act as additional binding sites.
Spectroscopic and photophysical properties
Electronic absorption spectra of the complexes 1–4 were recorded in a mixed CH2Cl2/MeOH (1:1 v/v) solution, as this was the solvent system in which they had the best solubility. In case of the free ligand, there are two intense absorption bands at 346 nm and 446 nm attributable to transitions into 1(π–π*) states of the o,o′-bis(iminomethyl)phenol chromophore.39 The intense band at 226 nm and the shoulders at 245 nm and 275 nm can be attributed to transitions into 1(π–π*) states of the aromatic rings of the calix[4]arene moiety.40
The UV-vis spectrum of the Eu complex 2 in CH2Cl2/MeOH differs from that of the free ligand in the same solvent system. Complex 2 exhibits two well-resolved absorption bands at 231 nm and 409 nm, both with higher intensity than the absorptions of H6L2. Two shoulders are observed at 254 nm and 302 nm. The two high-energy features below 300 nm region are associated with 1(π–π*) states within the aromatic rings of the Eu-bound calix[4]arene units.40 The broad band at 409 nm, on the other hand, is assigned to an intraligand transition to a 1(π–π*) state involving the bis(iminomethyl)phenolate chromophore units. The electronic absorption spectral properties of the other complexes are very similar to those of the Eu complex. Again all spectra are dominated by the intraligand absorptions.
Calix[4]arenes18,41,42 and iminomethyl-phenolate ligands43 are known to act as antennas for sensitization of lanthanide luminescence. Also, lanthanide complexes with odd coordination numbers, in particular seven-coordinated structures, have the potential to be strong luminescent materials.44–47 On these grounds, the Eu (2) and Tb (3) complexes were studied by luminescence spectroscopy. Although 2 is non-emissive in the solution (CH3CN or CH2Cl2/MeOH), a red emission can be detected by the naked eye when solid 2 (neat powder) is irradiated at 77 K (see graphical abstract). Fig. 6 displays the photoluminescence spectrum (λex = 361 nm) of a transparent poly-methylmethacrylate (PMMA) thin film containing 4 wt% of the Eu complex 2.48 The luminescence spectrum of a PMMA film containing 4 wt% of the complex [HNEt3][Eu2(HL1)(L1)] is also shown in Fig. 6 for comparative purposes. Photoluminescence spectra of complex 2 were also recorded as a neat powder or in a frozen CH2Cl2/MeOH (1:1) matrix at 77 K and are provided in the ESI.†
 | ||
| Fig. 6 Photoluminescence spectra of [Eu2(L2)(MeOH)2] (2) and [HNEt3][Eu2(HL1)(L1)] at 77 K (PMMA thin films doped with 4 wt% 2 or [HNEt3][Eu2(HL1)(L1)], thickness ∼0.2 mm). The excitation wavelength is 361 nm. The transitions above 575 nm start from the 5D0 state. The spectra are normalized with respect to the 5D0 → 7F1 transition. | ||
The luminescence spectrum of 2 in PMMA matrix is characteristic and will be discussed. The spectrum displays several broad features attributed to the characteristic 5D0 → 7FJ transitions (J = 0–6) from the excited 5D0 state of the Eu3+ ion.49 The 5D0 → 7F1 transition splits into three components (590 nm, 592 nm, 598 nm) and the 5D0 → 7F2 into two components (615 nm, 627 nm) as a consequence of the crystal-field splitting in the ground 7F1 and 7F2 manifolds, as observed in other seven-coordinate Eu(III) salicylaldimine complexes.46,49 The features at 650 nm and 750 nm, attributed to the 5D0 → 7F3 and 5D0 → 7F4 transitions, respectively, are also split by the crystal field. The feature at 581 nm is very broad and can be assigned as the 5D0 → 7F0 transition, considering that immobilization in a PMMA matrix causes inhomogeneous line broadening.49 In most Eu complexes, the 5D0 → 7F0 transition is weak; however, when the symmetry is low it may gain intensity, even higher intensity than the 5D0 → 7F1 band.50 This observation is in agreement with the low local site symmetry of the coordination environment in 2 (shape factors = 1.60 (ESI) for the capped trigonal prismatic geometry and 1.62 for the capped octahedral coordination environment vide infra).51 Along with the corresponding photoluminescence profile, the excitation spectra of a thin PMMA film of complex 2 at 77 K monitored at 615 nm is shown in Fig. S17.† A broad absorption band from 300 to 400 nm attributable to the transition into the 1(π–π*) state of the bis(iminomethyl)phenolato ligand is clearly discernable, showing that it can function as an antenna for sensitizing the Eu3+ ion.
Fig. 6 also displays the photoluminescence spectrum of the dinuclear europium(III) complex [HNEt3][Eu2(HL1)(L1)]. The spectrum is very similar to that of 2 in terms of the number and energies of the emission bands. The ratio R of the intensities of the 5D0 → 7F2 and 5D0 → 7F1 transitions is 10.3 in 2 and 10.1 in [Eu2(HL1)L1]− (R = 10.3 vs. 10.1). The similar spectral features can be traced to the similar coordination environments of the lanthanide ions in [Eu2(L2)(MeOH)2] and in [Eu2(L1)(HL1)]− as illustrated in Fig. 7. Thus, the changes in the coordination environments that occur upon going from [Eu(HL1)(L1)]− to [Eu2(L2)(MeOH)2] concern only the organic residues on two of the seven donor atoms. That is, the phenolate donors (OaH and Ob) in [Eu(HL1)(L1)]− become phenolether Oc and methanol OdH donors in 2. Notice that both coordination environments include also one O–H donor, each of which is involved in an intramolecular hydrogen bonding interaction. The similarity in the coordination environments is also illustrated by the shape factors, which provide a measure for the deviation from the ideal coordination environments. As already stated, the coordination geometry of the seven-coordinate Eu3+ ions in both 2 and [Eu2(L2)(MeOH)2] is intermediate between the capped octahedral and the capped trigonal prismatic coordination environments, with the calculated shape factors differing by only ΔS ∼ 0.9 (capped octahedron: S = 1.60 for 2, 2.30 for [Eu2(L1)(HL1)]−, capped trigonal prism: S = 1.61 (2), S = 2.49 for ([Eu2(L1)(HL1)]−)). The other possible coordination geometries reveal much larger deviations of the shape factor from the ideal values. The similarity of the luminescence spectra of the two seven-coordinate Eu complexes is not surprising in view of the structural similarities.
 | ||
| Fig. 7 Comparison of the immediate coordination environments in [HNEt3][Eu2(HL1)(L1)]25 (left) and [Eu2(L2)(MeOH)2] (right) with major changes highlighted. The phenolate O atoms (OaH, Ob) in the left structure become phenolether (Oc) and MeOH (Od) donors in the right structure. Parts of the structures were omitted for clarity. | ||
It was of interest to determine the quantum efficiencies for comparison with literature reporting that Eu3+ complexes with seven-coordinate coordination geometries exhibit high intrinsic quantum yields (Φff) and large radiative rate constants (kr).52,53 Time resolved photoluminescence decay measurements on the PMMA thin film doped with complex 2 revealed upon excitation at 361 nm an emission lifetime τobs of 330 μs at 77 K for the 5D0 level. The emission lifetime for [HNEt3][Eu2(HL1)(L1)] (τobs = 300 μs) under the same conditions is only slightly shorter than that for 2, again in good agreement with the similar coordination environments. Table 3 gathers selected photophysical parameters (kr, knr, Φff) for the two compounds along with the shape symmetry factors (average values for the two most ideal coordination environments). Complex 2 exhibits a slightly larger intrinsic quantum yield (Φff = 28%) than [HNEt3][Eu2(HL1)(L1)] (Φff = 17%) as a consequence of a larger radiative rate constant (kr = 0.857 × 103 s−1versus 0.560 × 103 s−1) and a smaller non-radiative rate constant knr (2.219 × 103 s−1). To our knowledge the photophysical parameters of only few seven-coordinated Eu3+ Schiff-base complexes have been reported. Thomas et al. reported a seven coordinated Eu3+ complex supported by TREN-(3,5-di-tert-butylsalicylidene)3.33 As for the present case the luminescence of this complex is only observed at low temperature upon enforcement of a delay time of 50 μs together with a residual emission from the ligand state, attributable to the presence of low-lying ligand-to-metal charge-transfer (LMCT) states.54 The observations made for the present Eu3+ compounds are in good agreement with the reported trend.
| Compound | T/K | Matrix | τ obs/μs |
τ
rc/μs |
k
rc/s−1 |
k
nrc/s−1 |
Φ
ffd/% |
Symmetry factor for Ln3+ ione |
|---|---|---|---|---|---|---|---|---|
| a λ ex = 310 nm (Perkin Elmer Fluorescence Spectrometer FL6500). b λ ex = 360 nm. c Radiative rate constant (kr) for EuIII complexes (in PMMA matrix) were estimated as follows: kr = 1/τr = AMD,0·n3 (Itot/IMD), where AMD,0 = 14.65 s−1 (spontaneous emission probability for the 5D0 → 7F1 transition in vacuum), n = 1.5 (refractive index), and Itot/IMD being the ratio of the total area of the corrected Eu3+ emission spectrum to the integrated intensity of the 5D0 → 7F1 transition; for the non radiative rate constant: knr = 1/τobs − 1/τr.46 Radiative rate constant (kr) for TbIII complexes (in PMMA matrix) were calculated assuming that the emission lifetime (τobs) at 77 K is the radiative emission lifetime (τr).55 d Intrinsic emission quantum yield: Φff = kr/(kr + knr) = τobs/τr. e Mean value of all S factors in this structure. | ||||||||
| [HNEt3][Eu2(HL1)(L1)] | 77a | PMMA thin film | 304 ± 9a | 1790 ± 90 | 560 ± 30 | 2700 ± 200 | 17 ± 1 | 2.40 |
| [Eu2(L2)(MeOH)2] (2) | 77 | PMMA thin film | 325 ± 9b | 1170 ± 60 | 860 ± 50 | 2200 ± 200 | 28 ± 2 | 1.62 |
| [HNEt3][Tb2(HL1)(L1)] | 298 | PMMA thin film | 175 ± 9a | 972 ± 3 | 1029 ± 3 | 4700 ± 300 | 18 ± 1 | 2.40 |
| 77 | PMMA thin film | 972 ± 3a | 972 ± 3 | 1029 ± 3 | <6 | 100 < Φff > 99 | ||
| [Tb2(L2)(MeOH)2] (3) | 298 | PMMA thin film | 186 ± 2b | 1099 ± 2 | 910 ± 2 | 4480 ± 60 | 17 ± 1 | 1.42 |
| 77 | PMMA thin film | 1099 ± 2b | 1099 ± 2 | 910 ± 2 | <3 | 100 < Φff > 99 | ||
Emission spectra of complex 2 (neat powder or CH2Cl2/MeOH 1:1 frozen glassy matrix) were also recorded at 77 K (utilizing a single photon counting photoluminescence spectrometer FluoTime 300 from PicoQuant). The spectra and photophysical parameters are similar to those recorded in PMMA matrix (Fig. S18 and S25†).
Fig. 8 displays photoluminescence spectra of PMMA thin films doped with the Tb3+ complexes [HNEt3][Tb2(HL1)(L1)] and [Tb2(L2)(MeOH)2] (3) at 298 K and 77 K. The 77 K spectrum of 3 shows four transitions centered at 480 nm, 550 nm, 580 nm, and 625 nm, which correspond to the 5D4 → 7FJ (J = 6, 5, 4, 3) transitions from the excited 5D4 state of the Tb3+ ion. Of these, the “green” 5D4 → 7F5 transition has the highest intensity. All signals are split by the crystal field. The spectrum of [HNEt3][Tb2(HL1)(L1)] is very similar to that of 3 with four transitions split by the crystal field at similar wavelengths. The similar spectroscopic features can again be traced to the similar coordination environments of the seven-coordinate Tb3+ ions. Thus, the symmetry shape factors differ again only slightly (capped octahedron: S = 1.63 for 3, 2.30 for [Tb2(L1)(HL1)]−, capped trigonal prism: S = 1.22 (3), S = 2.49 for ([Tb2(L1)(HL1)]−)), as observed for the above pair of Eu3+ complexes. The excitation spectrum of a thin PMMA film of complex 3 at 298 K monitored at 545 nm is shown in Fig. S17.† A broad absorption band occurs at 380 nm attributable to the transition into the 1(π–π*) state of the bis(iminomethyl)phenolato ligand, showing that it can function as an antenna for sensitizing the Tb3+ ion.
 | ||
| Fig. 8 Photoluminescence spectra of [HNEt3][Tb2(HL1)(L1)] and [Tb2(L2)(MeOH)2] (3) immobilized in a PMMA thin film (4 wt% 3, thickness ∼0.2 mm) at 77 K and 298 K. The excitation wavelength is 361 nm. The spectra are normalized with respect to the 5D4 → 7F6 transition. | ||
Time resolved photoluminescence decay measurements on the PMMA thin film doped with Tb3+ complex 3 revealed upon excitation at 361 nm an emission lifetime τobs of 1100 μs at 77 K for the emitting 5D4 level. The emission lifetime for [HNEt3][Tb2(HL1)(L1)] under the same conditions was found to be similar (907 μs), again in good agreement with the similar coordination environments. The photophysical parameters (kr, knr, Φff) for the two Tb3+ complexes (in PMMA matrix) are listed in Table 3. At room temperature, the value of Φff is 18% for complex 3 and 17% for [HNEt3][Tb2(HL1)(L1)]. There are not many seven-coordinated Tb3+ Schiff-base complexes whose photophysical parameters have been reported. Hasegawa and co-workers have reported the photophysical properties of [Tb(tmh)3(tppo)], where tmh = tetramethylheptanedionato and tppo = triphenylphosphine oxide.46 This mono-capped octahedral Tb3+ complex has a similar emission lifetime (840 ms) and kr value (1040 s−1) but a much smaller knr value (149 s−1) such that the value of Φff is much higher (88% vs. 18% for 3). It should be mentioned that Φff decreases significantly with increasing temperature. This decrease in intensity is attributed to O–H-overtone-mediated vibrational relaxation of the excited state of the metal ion by the coligated MeOH and H2O, although quenching due to low-lying ligand-to-metal charge-transfer (LMCT) states cannot be ruled out.33 Emission spectra of complex 2 (neat powder or CH2Cl2/MeOH 1:1 frozen glassy matrix) were also recorded at 77 K (utilizing the spectrometer from PicoQuant mentioned above). The spectra and photophysical parameters are similar to those recorded in PMMA matrix (Fig. S22 and S28,†Table 4).
| Compound | T/K | λ exc/nm | Form |
τ
avb/μs |
Φ L/% |
k
rc/s−1 |
k
nrd/103 s−1 |
|---|---|---|---|---|---|---|---|
| λ em/nm | |||||||
| a Data recorded on a PicoQuant FluoTime300 photoluminescence spectrometer and an integrating sphere from Hamamatsu. This experimental setup cannot measure these quantities for samples embedded in a PMMA matrix. b Amplitude-weighted average lifetime. The decay components and their relative amplitudes can be found along with the fitted decays in the ESI.† c Average radiative rate constant calculated using the equation kr = ΦL/τav. d Average radiationless deactivation rate constant calculated using the equation knr = (1 − ΦL)/τav. e The upper limit of the ΦL is used for the calculation. f The upper and the lower limits of ΦL are used to calculate knr. g Data recorded on a Varian Cary-Eclipse fluorescence spectrometer. | |||||||
| 2 | 77 | 360 | CH2Cl2/MeOHa | 402 ± 6 | 3 ± 2 | 80 ± 50 | 2.4 ± 0.9 |
| 610 | (1:1) glassy matrix |
||||||
| 77 | 360 | Powdera | 143 ± 4 | <2 | <140e | 6.98 > knr > 6.84f | |
| 607 | |||||||
| 3 | 77 | 360 | CH2Cl2/MeOHa | 900 ± 30 | 19 ± 2 | 280 ± 40 | 1.19 ± 0.07 |
| 543 | (1:1) glassy matrix |
||||||
| 77 | 360 | Powdera | 30 ± 2 | <2 | <670e | 3.49 > knr > 3.42f | |
| 550 | |||||||
| 298 | 292 | DMSO/HEPESg | 526 | ||||
| 550 | |||||||
Unlike 2, complex 3 was found to be emissive in the solution state, albeit very weakly. Fig. 9 displays the photoluminescence spectrum of a 50 μM solution of complex 3 in H2O/DMSO (95/5 v/v) solution containing 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer. As can be seen, the spectrum is similar to that recorded for the neat powder or the PMMA thin film, exhibiting four well-separated transitions centered at 490 nm, 545 nm, 582 nm, and 620 nm. The lifetime of 3 was found to be significantly longer (τ = 526 μs) under these conditions. This increase in luminescence lifetime presumably relates to substitution of the Tb-bound MeOH co-ligands by the anionic sulfonate groups from the HEPES buffer, as replacement of MeOH by RSO3− (or DMSO) ligands is expected to prevent vibrational quenching through the loss of the O–H oscillators.56,57 It is not clear whether the Tb3+ complexes retain their seven-coordinate structures. Nevertheless, this finding suggests that these complexes may be interesting to investigate as receptors for anionic sulfate ions.
 | ||
| Fig. 9 Emission spectrum of a 5 × 10−5 M solution of complex 3 in H2O/DMSO (95/5 v/v, HEPES-buffer, 1 × 10–2 M concentration, pH 7.4). Excitation wavelength λex 292 nm. | ||
Spectrophotometric titrations
UV-vis spectrophotometric titrations were performed in order to determine the speciation in solution and the binding affinity of H3L2 for the La3+, Eu3+, Tb3+ and Yb3+ ions. The titrations were carried out in a batch mode in CH2Cl2/MeOH (1:1 v/v) solution where all compounds had the best solubility. Measurements were performed at constant ionic strength (10–2 M nBu4NPF6) and pH value (2 × 10–4 M NEt3 buffer) as done in previous titrations.
Fig. 10 shows the spectral changes that occur upon addition of a solution of Eu(NO3)3·6H2O to a solution of H6L2, which is assumed to be deprotonated under our experimental conditions, in the 190 nm–700 nm spectral region. The other titration data are shown in the ESI (Fig. S9, S12 and S14†). As can be seen, the intensity of the bands/shoulder (sh) at 231 nm, 271 (sh) nm, 345 nm, and 420 nm for the ligand decreases steadily with increasing Eu3+ concentration, and new bands for [Eu2(L2)(MeOH)2] develop with maxima at 231 nm and 409 nm. The spectrophotometric titration of H6L2 with the other lanthanide salts displayed a similar evolution of absorbance values (ESI†).
 | ||
| Fig. 10 Spectrophotometric titration of H6L2 with Eu(NO3)3·6H2O in a MeOH/CH2Cl2 (1:1 v/v) solvent mixture (2 × 10–5 M concentration) at constant ionic strength (1 × 10–2 M N(n-Bu4)PF6, T = 298 K) in the presence of 2 × 10–4 M NEt3. The green curve refers to a final molar ratio of Eu3+/(L2)6− = 5.0. The inset shows the evolution of selected absorbance values versus the [Eu3+]/[(L2)6−] molar ratio. The solid lines are drawn to illustrate the consecutive complex formations from 1:1 to a 2:1 molar ratio. | ||
The mole ratio method was applied to determine the metal to ligand ratio.58 The inset of Fig. 10 shows a plot of two selected absorbance values versus molar ratio [Eu3+]/[(L2)6−]. This ratio increases steadily up to a molar ratio of about 2 and then remains constant. This behavior clearly confirms that the 2:1 (metal:ligand) stoichiometry found for the solid state is also maintained in the solution phase.
The titration data were analyzed by nonlinear least-squares refinements. The data could only be fitted by assuming a speciation model that involved the deprotonated ligand, a mononuclear 1:1 (M:L2) complex and a dinuclear 2:1 (M:L2) complex. Apparent stability constants of logK11 = 6.01(7) for the mononuclear and logK21 = 6.04(6) for the dinuclear complex were determined. The data for the other lanthanide ions are listed in Table 5. Overall, the stability constants show a strong affinity of (L2)6− towards lanthanide ions.
| La3+ | Eu3+ | Tb3+ | Yb3+ | |
|---|---|---|---|---|
|
a Solvent: CH2Cl2/MeOH (1:1 v/v), cBase = 2 × 10–4 M NEt3, I = 0.01 M N(n-Bu)4PF6, T = 298 K.
|
||||
| logK11 |
5.42(5) | 6.01(7) | 6.56(6) | 5.25(8) |
| logK21 |
6.07(6) | 6.04(6) | 6.64(5) | 6.33(8) |
| α | 0.06 | 0.23 | 0.21 | 0.02 |
The interaction parameter (α)59 for the stepwise formation of a homotropic 2:1 M2:L2 complex according to eqn (1) provides an insight for the cooperativity effect between the formation of 1:1 and 2:1 complexes.60 If α = 1 (i.e. K11 = 4K21), the stepwise 2:1 binding is noncooperative; if α < 1 (i.e. K11 < 4K21), the stepwise 1:2 binding is positively cooperative; if α > 1 (i.e. K11 > 4K21) the stepwise 1:2 binding is negatively cooperative.61 For the present complexes the parameter α was found invariably smaller than 1, indicative of positive cooperativity, that is the binding of the first Ln3+ ion augments the binding of the second Ln3+ ion. This may be traced to mechanical coupling of the two binding sites by the rather stiff bis(iminomethyl)phenol functions. Cooperative binding of lanthanide ions by multitopic receptors is rare, but is of importance for the design of selective receptors and also the targeted construction of supramolecular structures.62,63
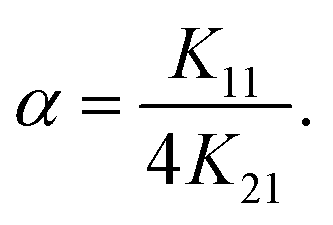 | (1) |
Few studies have reported solution thermodynamic data for f-element calixarene complexes.64–69 We have investigated the Ln3+ binding ability of the related salicylaldimine functionalized calix[4]arene ligand (L1)4− in MeOH. The logK11 value for this complex of 6.0 compares well with that determined for the present complexes.
Conclusion
The coordination behaviour of the macrocyclic ligand H6L2 comprising two calix[4]arene head units linked by two bis(iminomethyl)phenol-chromophores towards selected lanthanide ions (La3+, Eu3+, Tb3+, Yb3+) has been investigated both in solution and solid state. H6L2 was found to be an effective dinucleating ligand for the Ln3+ ions forming mixed-ligand complexes of composition [Ln2(L2)(MeOH)2] with one exchangeable coligand per seven-coordinated Ln site. A 2:1 complex stoichiometry is also present in solution state as suggested by ESI MS and spectroscopic titration. The two binding pockets of H6L2 have similar affinities for the Ln3+ ions, with the stepwise binding constants logK11 and logK21 in a narrow 5.2 to 6.6 range. The interaction parameter α = K11/4K21 is invariably smaller than unity indicating that the binding of the first Ln3+ ion augments the binding of the second Ln3+ ion. The investigation of the photophysical properties clearly show that the bis(iminomethyl)phenolate units in H6L2 can sensitize luminescence of the coordinated Eu3+ and Tb3+ ions. However, the external MeOH coligands in 2 and 3 provide non-emissive relaxation pathways, which render the complexes less luminescent than other 7-coordinate Eu3+ and Tb3+ compounds supported by enolate ligands. On the other hand, replacement of the MeOH by RSO3− ligands appears to prevent vibrational quenching through the loss of the O–H oscillators, and it could be interesting to investigate them as displacement receptors for anionic sulfonate or other S-based ions.
Experimental section
Materials and methods
The ligand H6L2 was prepared as described in the literature.26,30 All reagents and solvents were commercial grade and used without further purification. Melting points were determined with an Electrothermal IA9000 series instrument using open glass capillaries and are uncorrected. Elemental analyses were carried out on a VARIO EL elemental analyzer (Elementar Analysensysteme GmbH, Hanau). Mass spectra were obtained using the positive ion electrospray ionization modus (ESI) on a Bruker Daltronics ESQUIRE 3000 Plus ITMS or Impact II UHR Qq-TOF instrument. Infrared spectra (4000–640 cm−1) were recorded at 2 cm−1 resolution on a Bruker TENSOR 27 FT-IR spectrometer using a MIRacle ZnSe ATR accessory from PIKE Technologies. Solution absorption spectra were collected on a Jasco V-670 UV–vis–NIR device using 1 cm quartz cells (Hellma).Synthesis and analysis of compounds
= 670 (w), 714 (w), 758 (m), 810 (w), 838 (w), 863 (w), 913 (w), 950 (w), 1011 (w), 1039 (w), 1084 (m), 1109 (w), 1126 (w), 1156 (w), 1190 (w), 1222 (w), 1244 (w), 1299 (m), 1322 (w), 1364 (w), 1394 (w), 1425 (w), 1452 (s), 1530 (w), 1591 (w), 1632 (m), 1650 (m), 2874 (w), 2914 (w), 2954 (w), 3019 (w), 3057 (w), 3450 cm−1 (br); UV-vis (CH2Cl2/MeOH, 1/1 v/v): λmax (ε) = 231 (87976), 250 (sh) (59730), 304 (sh) (13306), 410 (14224) nm (M−1 cm−1); ESI-MS(+): m/z: 1633.386 ([M + H+]+, calc. 1633.425), 1696.387 ([M + 2MeOH]+, calc. 1696.469); elemental analysis calcd (%) for C88H82La2N4O10·2CH3OH: C 63.68, H 5.34, N 3.30; found: C 63.83, H 5.49, N 3.33.
= 673 (w), 695 (w), 714 (w), 746 (m), 754 (m), 784 (w), 805 (w), 832 (w), 867 (w), 899 (w), 931 (w), 955 (w), 980 (w), 1006 (w), 1033 (w), 1075 (w), 1085 (w), 1124 (w), 1152 (w), 1187 (w), 1224 (w), 1246 (m), 1266 (w), 1289 (s), 1317 (w), 1402 (w), 1426 (w), 1449 (s), 1547 (w), 1590 (w), 1635 (m), 2839 (w), 2879 (w), 2915 (w), 2998 (w), 3020 (w), 3053 (w), 3508 (br), 3586 cm−1 (w); UV-vis (CH2Cl2/MeOH, 1/1 v/v): λmax (ε) = 231 (87092), 254 (sh) (57697), 302 (sh) (15985), 409 (15339) nm (M−1 cm−1); ESI-MS(+): m/z: 1677.469 ([M + H2O + H+]+, calc. 1677.461), 1722.452 ([M + 2MeOH]+, calc. 1722.495); elemental analysis calcd (%) for C88H82Eu2N4O10·2CH3OH: C 62.72, H 5.26, N 3.25; found: C 62.84, H 5.44, N 3.26.
= 676 (w), 695 (w), 716 (w), 745 (m), 806 (w), 833 (w), 867 (w), 900 (w), 934 (w), 960 (w), 984 (w), 1033 (w), 1076 (w), 1085 (w), 1123 (w), 1152 (w), 1188 (w), 1224 (w), 1246 (w), 1268 (w), 1289 (m), 1319 (m), 1401 (w), 1427 (m), 1449 (s), 1548 (w), 1591 (w), 1636 (m), 2841 (w), 2882 (w), 2913 (w), 2998 (w), 3018 (w), 3054 (w), 3601 cm−1 (w); UV-vis (CH2Cl2/MeOH, 1/1: v/v): λmax (ε) = 227 (107414), 251 (69553), 303 (sh) (18343), 409 (16976) nm (M−1 cm−1); ESI-MS(+): m/z: 1657.521 ([M − CH3]+, calc. 1657.430), 1705.500 ([M + MeOH + H+]+, calc. 1705.488), 1737.440 ([M + 2MeOH + H+]+, calc. 1737.506); elemental analysis calcd (%) for C88H82N4O10Tb2·2CH3OH: C 62.21, H 5.22, N 3.22; found: C 62.36, H 5.55, N 3.18.
= 668 (w), 718 (w), 756 (m), 800 (w), 812 (w), 834 (w), 871 (w), 911 (w), 999 (w), 1036 (w), 1082 (m), 1108 (w), 1126 (w), 1155 (w), 1188 (w), 1222 (w), 1245 (w), 1304 (m), 1330 (m), 1363 (w), 1396 (w), 1428 (w), 1456 (s), 1536 (m), 1589 (w), 1634 (m), 1653 (m), 2839 (w), 2872 (w), 2913 (w), 2956 (w), 3021 (w), 3055 cm−1 (w); UV-vis (CH2Cl2/MeOH, 1/1 v/v): λmax (ε) = 230 (72147), 257 (sh) (42642), 307 (sh) (7994), 417 (11100), 456 (sh) (7020) nm (M−1 cm−1); ESI-MS(+): m/z: 1703.445 ([M + H+]+, calc. 1703.476), 1764.440 ([M + 2MeOH]+, calc. 1764.521); elemental analysis calcd (%) for 88H82N4O10Yb2·2CH3OH: C 61.22, H 5.14, N 3.17; found: C 61.42, H 5.27, N 3.19.
Crystallography
Single crystals of [Eu2(L2)(MeOH)2]·4CH2Cl2·MeOH (2·4CH2Cl2·MeOH) and [Tb2(L2)(MeOH)(H2O)]·(H2O)x (3·(H2O)x, x ∼ 10) suitable for X-ray crystallographic analyses were selected and mounted on the tip of a glass fibre using perfluoropolyether oil. The data sets were collected at 180(2) K using a STOE STADIVARI diffractometer using graphite monochromated Cu-Kα radiation (λ = 1.54186 Å). The data were processed with the programs XAREA.70 Structures were solved by direct methods71 and refined by full-matrix least-squares techniques on the basis of all data against F2 using SHELXL-2018/3.72 PLATON was used to search for higher symmetry.37 All non-hydrogen atoms were refined anisotropically. Graphics were produced with Ortep3 for Windows and PovRAY.925 reflections measured, 17529 unique, 10619 with I > 2σ(I). Final R1 = 0.0431, wR2 = 0.1074 (I > 2σ(I)), 1109 parameters/0 restraints, min./max. residual electron density = −1.422 /0.895 e Å−3. The hydrogen atoms of the Eu-bound MeOH ligands were located from final Fourier maps and were refined isotropically.
989 reflections measured, 17592 unique, 14074 with I > 2σ(I). Final R1 = 0.0405, wR2 = 0.1125 (I > 2σ(I)), 979 parameters/0 restraints, min./max. residual electron density = −1.159 /−0.891 e Å−3. The O–H hydrogen atom of the Tb-bound H2O and MeOH ligands were located from final Fourier maps and were refined isotropically. The H2O solvate molecules in this structure were found to be heavily disordered and all attempts to model this disorder failed. The corresponding electron density was removed from the structure (and the corresponding F0) with the SQUEEZE algorithm implemented in the PLATON program suite. The SQUEEZE routine revealed total potential solvent area of 2500 Å3 (400e−) per unit cell corresponding to approximately ten H2O molecules per formula unit.
CCDC 2005666 (2) and 2005669 (3)† contain the supplementary crystallographic data for this paper.
Synthesis of PMMA thin films
PMMA (0.30 g) was dissolved in CH2Cl2 (1.5 mL) and stirred for 10 min. A solution of the lanthanide complex (V = 1 mL, 8 × 10–3 M) in CH2Cl2 was added to the PMMA solution. The resulting mixture was spread on a Petri dish (d = 5 cm) and the solvent was allowed to evaporate in open air over night.Photophysical characterization
Steady state fluorescence excitation and emission spectra and luminescence lifetime measurements for PMMA thin films of [HNEt3][Eu2(HL1)(L1)], [HNEt3][Tb2(HL1)(L1)], 2, and 3 were recorded on a PerkinElmer FL 6500 fluorescence spectrophotometer using precision cells for powder samples. Lifetime analysis was performed using the commercial PerkinElmer software. The luminescence emission spectrum and lifetime measurement of an aqueous solution of complex 3 was recorded at 23 °C using a Varian Cary-Eclipse fluorescence spectrophotometer set to phosphorescence mode. A quartz cell with a path length of 10 mm and a volume of 400 μL was used, the instrument excitation and emission slit widths were both set at 5 nm.Steady-state excitation and emission spectra of compounds 2 and 3 (neat powder or frozen CH2Cl2/MeOH (1:1) matrix at 77 K) were recorded on a FluoTime300 spectrometer from PicoQuant equipped with a 300 W ozone-free Xe lamp (250–900 nm), a 10 W Xe flash-lamp (250–900 nm, pulse width <10 μs) with repetition rates of 0.1–300 Hz, an excitation monochromator (Czerny-Turner 2.7 nm mm−1 dispersion, 1200 grooves per mm, blazed at 300 nm), diode lasers (pulse width <80 ps) operated by a computer-controlled laser driver PDL-820 (repetition rate up to 80 MHz, burst mode for slow and weak decays), two emission monochromators (Czerny-Turner, selectable gratings blazed at 500 nm with 2.7 nm mm−1 dispersion and 1200 grooves per mm, or blazed at 1250 nm with 5.4 nm mm−1 dispersion and 600 grooves per mm), Glan–Thompson polarizers for excitation (Xe-lamps) and emission, a Peltier-thermostatized sample holder from Quantum Northwest (−40 °C–105 °C), and two detectors, namely a PMA Hybrid 40 (transit time spread FWHM < 120 ps, 300–720 nm) and a R5509-42 NIR-photomultiplier tube (transit time spread FWHM 1.5 ns, 300–1400 nm) with external cooling (−80 °C) from Hamamatsu. Steady-state and fluorescence lifetimes were recorded in TCSPC or MCS mode by a PicoHarp 300 (minimum base resolution 4 ps). Emission and excitation spectra were corrected for source intensity (lamp and grating) by standard correction curves. Lifetime analysis was performed using the commercial FluoFit software. The quality of the fit was assessed by minimizing the reduced chi squared function (χ2) and visual inspection of the weighted residuals and their autocorrelation.
Luminescence quantum yields (ΦL) for 2 and 3 (neat powder or frozen CH2Cl2/MeOH (1:1) matrix at 77 K) were measured with a Hamamatsu Photonics absolute PL quantum yield measurement system (C9920-02) equipped with a L9799-01 CW Xenon light source (150 W), monochromator, C7473 photonic multi-channel analyzer, integrating sphere and employing U6039-05 PLQY measurement software (Hamamatsu Photonics, Ltd, Shizuoka, Japan). All solvents used were of spectrometric grade (Uvasol®).
Spectrophotometric titrations
Stock solutions of ligands and metal salts were obtained by dissolving carefully weighed samples in HPLC-grade solvent mixtures. Batch titrations were carried out with quartz cells (Hellma 110-QS, 1 cm optical path length) containing a stock solution of H6L2 (1 mL) with (n-Bu)4NPF6 as supporting electrolyte (0.01 M) and constant concentrations (2 × 10−5 M) at 298 K. For each experiment, 19 solutions were prepared by adding aliquots of 0–200 μL of the metals salt solutions (Ln(NO3)3·6H2O, Ln = La, Eu, Yb, 10−3 M) with an Eppendorf micropipette (volume range 10–100 μL and 100–1000 μL; 0.71–0.10% error). After stirring for 12 h, UV–vis absorption spectra were collected in the 190–700 nm range at uniform data point intervals of 1 nm with a double-beam V-670 (Jasco) spectrophotometer. The multiwavelength data sets were analyzed by a nonlinear least-squares procedure implemented in the Hyperquad2008 v1.1.33 software.73,74 Goodness-of-fit parameters were assessed by the overall standard deviation (σ), the visual inspection of the residuals, and by the physical meaning of the calculated electronic absorption spectra. Unless otherwise noted, the reported uncertainties correspond to the standard deviation of the refined parameters that were returned by the software.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are thankful to Prof. Dr H. Krautscheid for providing facilities for X-ray crystallographic measurements. This work was supported by the Deutsche Forschungsgemeinschaft (DFG Priority Program, “Light Controlled Reactivity of Metal Complexes”) and the DAAD “Towards Luminescent Hydrosulfide Probes”. Matias E. Gutierrez Suburu acknowledges the DAAD for a doctoral fellowship. Cristian A. Strassert gratefully acknowledges the DFG (Cluster of Excellence Cells in Motion EXC 1003, Priority Program, “Light Controlled Reactivity of Metal Complexes”) for financial support.References
- J.-C. G. Bünzli and S. Eliseeva, Basics of Lanthanide Photophysics, in Lanthanide Luminescence, ed. P. Hänninen and H. Härmä, Springer Berlin Heidelberg, Berlin, 2011, vol. 7, pp. 1–45 Search PubMed.
- A. E. V. Gorden, J. D. Xu, K. N. Raymond and P. Durbin, Chem. Rev., 2003, 103, 4207–4282 CrossRef CAS PubMed.
- S. J. Butler and D. Parker, Chem. Soc. Rev., 2013, 42, 1652–1666 RSC.
- M. L. Aulsebrook, B. Graham, M. R. Grace and K. L. Tuck, Coord. Chem. Rev., 2018, 375, 191–220 CrossRef CAS.
- D. Gutsche, Calixarenes: An Introduction, RSC Publishing, Cambridge, 2nd edn, 2008 Search PubMed.
- V. Böhmer, Angew. Chem., 1995, 107, 785–818 ( Angew. Chem., Int. Ed. Engl. , 1995 , 34 , 713–745 ) CrossRef.
- R. Kumar, Y. Jung and J. S. Kim, in Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M.-X. Wang, Springer, 2016 Search PubMed.
- L. Baldini, F. Sansone, A. Casnati and R. Ungaro, Calixarenes in Molecular Recognition, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, John Wiley & Sons, vol. 3, 2012 Search PubMed.
- D. Semeril and D. Matt, Coord. Chem. Rev., 2014, 279, 58–95 CrossRef CAS.
- P. D. Beer and E. J. Hayes, Coord. Chem. Rev., 2003, 240, 167–189 CrossRef CAS.
- C. Redshaw, Coord. Chem. Rev., 2003, 244, 45–70 CrossRef CAS.
- M. A. McKervey, F. Arnaud-Neu and M.-J. Schwing-Weill, in Comprehensive Supramolecular Chemistry, ed. G. Gokel, Pergamon, Oxford, 1996 Search PubMed.
- J. M. Harrowfield, M. I. Ogden and A. H. White, J. Chem. Soc., Dalton Trans., 1991, 2625–2632 RSC.
- P. D. Beer, M. G. B. Drew, M. Kan, P. B. Leeson, M. I. Ogden and G. Williams, Inorg. Chem., 1996, 35, 2202–2211 CrossRef CAS PubMed.
- A. Casnati, L. Baldini, F. Sansone, R. Ungaro, N. Armaroli, D. Pompei and F. Barigelletti, Supramol. Chem., 2002, 14, 281–289 CrossRef CAS.
- G. Guillemot, B. Castellano, T. Prangé, E. Solari and C. Floriani, Inorg. Chem., 2007, 46, 5152–5154 CrossRef CAS PubMed.
- G. B. Deacon, M. G. Gardiner, P. C. Junk, J. P. Townley and J. Wang, Organometallics, 2012, 31, 3857–3864 CrossRef CAS.
- N. Sabbatini, M. Guardigli, A. Mecati, V. Balzani, R. Ungaro, E. Ghidini, A. Casnati and A. Pochini, J. Chem. Soc., Chem. Commun., 1990, 878–879 RSC.
- N. Sato, I. Yoshida and S. Shinkai, Chem. Lett., 1993, 621–624 CAS.
- F. J. Steemers, W. Verboom, D. N. Reinhoudt, E. B. van der Tol and J. W. Verhoeven, J. Am. Chem. Soc., 1995, 117, 9408–9414 CrossRef CAS.
- B. W. Ennis, S. Muzzioli, B. L. Reid, D. D'Alessio, S. Stagni, D. H. Brown, M. I. Ogden and M. Massi, Dalton Trans., 2013, 42, 6894–6901 RSC.
- D. D'Alessio, S. Muzzioli, B. W. Skelton, S. Stagni, M. Massi and M. I. Ogden, Dalton Trans., 2012, 41, 4736–4739 RSC.
- M. Massi and M. I. Ogden, Materials, 2017, 10, 1369–1381 CrossRef PubMed.
- (a) R. Joseph and C. P. Rao, Chem. Rev., 2011, 111, 4658–4702 CrossRef CAS PubMed; (b) S. A. Ansari, P. K. Mohapatra, W. Verboom, Z. Zhang, P. D. Dau, J. K. Gibson and L. Rao, Dalton Trans., 2015, 44, 6416–6422 RSC.
- S. Ullmann, P. Hahn, L. Blömer, A. Mehnert, C. Laube, B. Abel and B. Kersting, Dalton Trans., 2019, 48, 3893–3905 RSC.
- S. Ullmann, R. Schnorr, M. Handke, C. Laube, B. Abel, J. Matysik, M. Findeisen, R. Rüger, T. Heine and B. Kersting, Chem. – Eur. J., 2017, 23, 3824–3827 CrossRef CAS PubMed.
- (a) R. K. Mahajan, I. Kaur, V. Sharma and M. Kumar, Sensors, 2002, 2, 417–423 CrossRef CAS; (b) R. K. Mahajan, I. Kaur and M. Kumar, Sens. Actuators, B, 2003, 91, 26–31 CrossRef CAS; (c) R. K. Mahajan, M. Kumar, V. Sharma (nee Bhalla) and I. Kaur, Analyst, 2001, 126, 505–507 RSC; (d) R. K. Mahajan, I. Kaur, R. Kaur, V. Bhalla and M. Kumar, Bull. Chem. Soc. Jpn., 2005, 78, 1635–1640 CrossRef CAS.
- (a) M. Kumar, R. K. Mahajan, V. Sharma, H. Singh, N. Sharma and I. Kaur, Tetrahedron Lett., 2001, 42, 5315–5318 CrossRef CAS; (b) M. Kumar, V. Sharma (nee Bhalla) and J. N. Babu, Tetrahedron, 2003, 59, 3267–3273 CrossRef CAS.
- A. Ali, R. Joseph, B. Mahieu and C. P. Rao, Polyhedron, 2010, 29, 1035–1040 CrossRef CAS.
- W.-C. Zhang and Z.-T. Huang, Synthesis, 1997, 1073–1076 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, John Wiley & Sons, Inc., Hoboken, New Jersey, 6th edn, 2009 Search PubMed.
- S. Dasari and A. K. Patra, Dalton Trans., 2015, 44, 19844–19855 RSC.
- J. K. Molloy, C. Philouze, L. F. D. Imbert, O. Jarjayes and F. Thomas, Dalton Trans., 2018, 47, 10742–10751 RSC.
- I. D. Brown, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 24–31 CrossRef.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, UK, 1997 Search PubMed.
- T. Steiner, Angew. Chem., 2002, 114, 50–80 ( Angew. Chem., Int. Ed. , 2002 , 41 , 48–76 ) CrossRef.
- A. L. Spek, PLATON - A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2000 Search PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- M. La Deda, M. Ghedini, I. Aiello and A. Grisolia, Chem. Lett., 2004, 33, 1060–1061 CrossRef CAS.
- L. J. Charbonnière, C. Balsiger, K. J. Schenk and J.-C. G. Bünzli, J. Chem. Soc., Dalton Trans., 1998, 505–510 RSC.
- D. D'Alessio, S. Muzzioli, B. W. Skelton, S. Stagni, M. Massi and M. I. Ogden, Dalton Trans., 2012, 41, 4736–4739 RSC.
- R. Z. H. Phe, B. W. Skelton, M. Massi and M. I. Ogden, Eur. J. Inorg. Chem., 2020, 94–1010 CrossRef CAS.
- R. D. Archer, H. Chen and L. C. Thompson, Inorg. Chem., 1998, 37, 2089–2095 CrossRef CAS.
- Z. Ahmed and K. Iftikhar, J. Phys. Chem. A, 2013, 117, 11183–11201 CrossRef CAS PubMed.
- S. V. Eliseeva, O. X. Kotova, F. Gumy, S. N. Semenov, V. G. Kessler, L. S. Lepnev, J.-C. G. Bünzli and N. P. Kuzmina, J. Phys. Chem. A, 2008, 112, 3614–3626 CrossRef CAS PubMed.
- K. Yanagisawa, T. Nakanishi, Y. Kitagawa, T. Seki, T. Akama, M. Kobayashi, T. Taketsugu, H. Ito, K. Fushimi and Y. Hasegawa, Eur. J. Inorg. Chem., 2015, 4769–4774 CrossRef CAS.
- P. P. Ferreira da Rosa, Y. Kitagawa and Y. Hasegawa, Coord. Chem. Rev., 2020, 406, 213153 CrossRef CAS.
- The PMMA films were utilized in place of the neat powders to protect the compounds from atmospheric moisture.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- C. C. Bryden and C. N. Reiley, Anal. Chem., 1982, 54, 610–615 CrossRef CAS.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE version 2.1, University of Barcelona, Barcelona, 2013 Search PubMed.
- Y. Hasegawa, M. Yamamuro, Y. Wada, N. Kanehisa, Y. Kai and S. Yanagida, J. Phys. Chem. A, 2003, 107, 1697–1702 CrossRef CAS.
- S. Biju, R. O. Freire, Y. K. Eom, R. Scopelliti, J.-C. G. Bünzli and H. K. Kim, Inorg. Chem., 2014, 53, 8407–8417 CrossRef CAS.
- S. Wang, B. Zhang, Y. Hou, C. Du and Y. Wu, J. Mater. Chem., 2013, 1, 406–409 CAS.
- L. Prodi, M. Maestri, R. Ziessel and V. Balzani, Inorg. Chem., 1991, 30, 3798–3802 CrossRef CAS.
- J.-C. G. Bünzli and M. M. Vuckovic, Inorg. Chim. Acta, 1983, 73, 53–61 CrossRef.
- S. Comby and J.-C. G. Bünzli, Lanthanide Near-Infrared Luminescence in Molecular Probes and Devices, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner, Jr., J.-C. G. Bünzli and V. K. Pecharsky, Elsevier B.V., vol. 37, 2007 Search PubMed.
- J. H. Yoe and A. E. Harvey, J. Am. Chem. Soc., 1948, 70, 648–654 CrossRef CAS.
- K. A. Connors, A. Paulson and D. Toledo-Velasquez, J. Org. Chem., 1988, 53, 2023–2026 CrossRef CAS.
- K. A. Connors, Measurement of Cyclodextrin Complex Stability Constants, in Comprehensive Supramolecular Chemistry, ed. J. L. D. Atwood, D. MacNicol, F. Vögtle and J.-M. Lehn, Pergamon, Oxford, 1996, vol. 3, pp. 205–241 Search PubMed.
- E. Chekmeneva, C. A. Hunter, M. J. Packer and S. M. Turega, J. Am. Chem. Soc., 2008, 130, 17718–17725 CrossRef CAS PubMed.
- P. J. Huang, M. Vazin and J. Liu, Biochemistry, 2016, 55, 2518–2525 CrossRef CAS PubMed.
- X.-Z. Li, L.-P. Zhou, L.-L. Yan, Y.-M. Dong, Z.-L. Bai, X.-Q. Sun, J. Diwu, S. Wang, J.-C. Bünzli and Q.-F. Sun, Nat. Commun., 2018, 9, 547 CrossRef PubMed.
- P. Di Bernardo, A. Melchior, M. Tolazzi and P. L. Zanonato, Coord. Chem. Rev., 2012, 256, 328–351 CrossRef CAS.
- A. F. Danil de Namor and O. Jafou, J. Phys. Chem. B, 2001, 105, 8018–8027 CrossRef CAS.
- P. M. Marcos, J. R. Ascenso, M. A. P. Segurado, P. J. Cragg, S. Michel, V. Hubscher-Bruder and F. Arnaud-Neu, Supramol. Chem., 2011, 23, 93–101 CrossRef CAS.
- P. M. Marcos, J. D. Fonseca, C. S. Proença, J. R. Ascenso, R. J. Bernardino, J. Kulesza and M. Bochenska, Supramol. Chem., 2016, 28, 367–376 CrossRef CAS.
- A. F. Danil de Namor, K. Baron, S. Chahine and O. Jafou, J. Phys. Chem. A, 2004, 108, 1082–1089 CrossRef CAS.
- N. E. Borisova, A. A. Kostin, E. A. Eroshkina, M. D. Reshetova, K. A. Lyssenko, E. N. Spodine and L. N. Puntus, Eur. J. Inorg. Chem., 2014, 2014, 2219–2229 CrossRef CAS.
- Stoe & Cie, X-AREA and X-RED 32; V1.35, Stoe & Cie, Darmstadt, Germany, 2006 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, (2015), SHELXL-97, “Crystal Structure refinement with SHELXL”, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, Hyperquad2008, Protonic Software, Leeds, West Yorkshire, UK, 2008 Search PubMed.
- (a) P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed; (b) P. Gans, A. Sabatini and A. Vacca, Annu. Chim., 1999, 89, 45–49 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2005666 and 2005669. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02303e |
| This journal is © The Royal Society of Chemistry 2020 |