
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDinuclear Tb and Dy complexes supported by hybrid Schiff-base/calixarene ligands: synthesis, structures and magnetic properties†
Peter
Hahn
a,
Steve
Ullmann
a,
Jennifer
Klose
a,
Yan
Peng
b,
Annie K.
Powell
 b and
Berthold
Kersting
*a
b and
Berthold
Kersting
*a
aInstitut für Anorganische Chemie, Universität Leipzig, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: b.kersting@uni-leipzig.de; Fax: +49/(0)341-97-36199
bInstitute of Inorganic Chemistry, Karlsruhe Institute of Technology, Engesserstrasse 15, D-76131 Karlsruhe, Germany. E-mail: annie.powell@kit.edu
First published on 17th July 2020
Abstract
The synthesis of the new lanthanide complexes [HNEt3][Dy2(HL1)(L1)] (5), and [Ln2(L2)2] (Ln = TbIII (7), DyIII (8)) supported by the hybrid Schiff-base/calix[4]arene ligands H4L1 (25-[2-((2-methylphenol)imino)ethoxy]-26,27,28-trihydroxy-calix[4]arene) and H3L2 (25-[2-((2-methylpyridine)imino)ethoxy]-26,27,28-trihydroxy-calix[4]arene) are reported. Spectroscopic data (for 5) and X-ray crystallographic analysis (for 7·4MeCN, 8·4MeCN) reveal the presence of dimeric structures, featuring doubly-bridged NO4Ln(μ-O)2LnO4N (5) or N2O3Ln(μ-O)2LnO3N2 cores (7, 8) with seven-coordinated Ln3+ ions. The magnetic properties of polycrystalline samples of 5, 7 and 8 were studied by variable temperature dc and ac magnetic susceptibility measurements. The χ′′(T) vs. T plots show no maxima in zero field, but the maxima can be detected under a 3 kOe dc field. The relaxation times τ obey the Arrhenius law above 5 K. Anisotropy barriers of ∼18 cm−1 (26 K) for 5 and ∼23 cm−1 (33 K) for 8 were determined.
Introduction
The synthesis and investigation of magnetic properties of novel types of lanthanide complexes are very important for the development of molecular based magnetic materials,1–3 and many different types of lanthanide complexes with remarkable magnetic properties have been reported in the last several years.4–7 In 2003, slow relaxation of the magnetization was observed in the mononuclear lanthanide complexes [Pc2Tb]− and [Pc2Dy]−, where Pc = phthalocyaninato, and this discovery spurred the search for other lanthanide-based single-molecule magnets (SMMs).8 Considerable efforts have been dedicated to the understanding of the mechanisms in mono- and polynuclear 4f SMMs in order to inform and direct ongoing research. It has been established that variation of the ligand field symmetry and coordination geometry, alteration of the charge and type of the donor atoms, and the type and magnitude of exchange interactions (in polynuclear systems) can have a great effect on the single ion anisotropy of the coordinated Ln3+ ions and thus the dynamics of the magnetization.9 Polydentate ligands have turned out to be ideal candidates for systematic investigations because they can be rationally designed to control the coordination environment of the lanthanide ions.10 Thus, phthalocyaninato,11 β-diketonato,12 calix[4]arenes,13 and various Schiff base ligands14,15 or mixtures thereof16 have been used extensively in synthesizing functional coordination compounds of the 4f elements. In addition, these complexes have the added advantage that they can be developed for surface deposition, and in turn for device applications.17,18Several dinuclear terbium19–24 and dysprosium complexes with SMM behaviour have been reported.16,25–32 Most are mixed-ligand complexes, with the coordination spheres often being partly completed by solvate molecules or the anions. We previously reported a hybrid Schiff-base/calixarene ligand H4L1 (Fig. 1) that supports homoleptic lanthanide complexes [HNEt3][Ln2(HL1)(L1)] without additional coligands.33 Hybrid ligands of this sort are known to complex first-row transition metals readily, but their lanthanide chemistry, particularly their magnetic properties,13,34 remains largely unexplored.35–42 In order to investigate the effect of donor atom variations on the structures and properties of such lanthanide calix[4]arene complexes, we have now focused on the coordination properties of the ligand H3L2, bearing a pyridyl-aldiminato podand arm in place of a salicylaldimine unit.
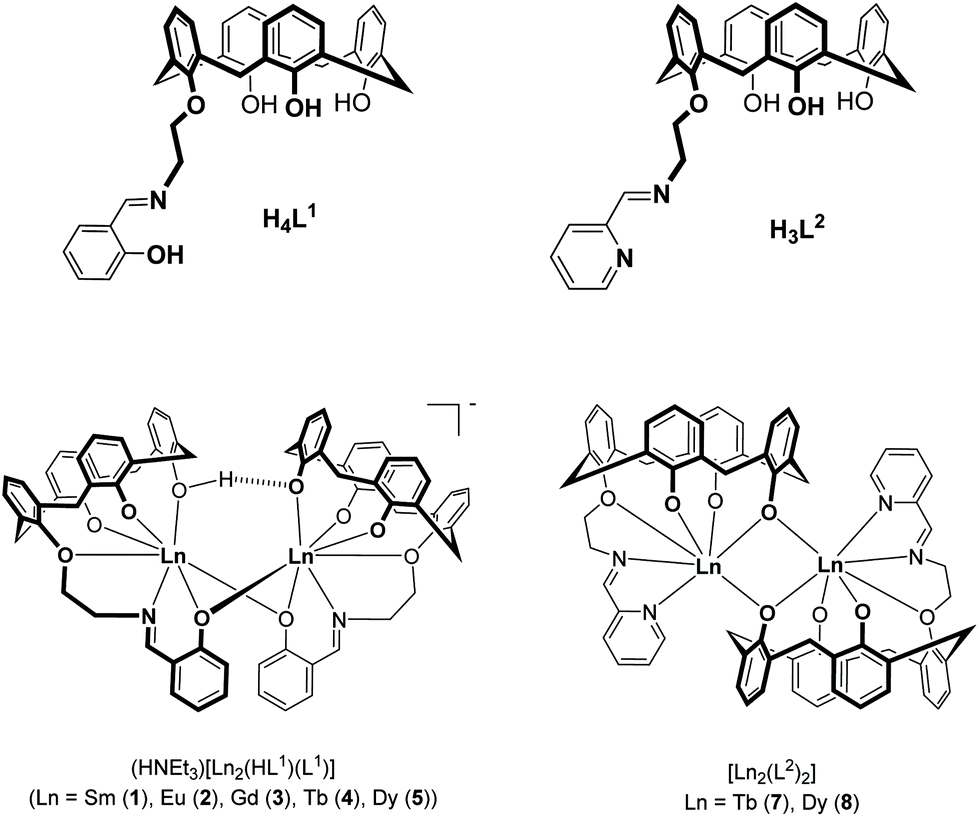 | ||
| Fig. 1 Structures of hybrid Schiff-base/calix[4]arene ligands H4L1 and H3L2 and their lanthanide complexes 1–5, 7, and 8. The complexes 1–4 have been reported previously.33 | ||
Herein, we report the synthesis, structures and magnetic properties of the corresponding TbIII and DyIII complexes 7 and 8 (Fig. 1). Their properties are compared with those of complexes 4 and 5. The contrasting responses to dynamic magnetic fields (in an applied DC field) are discussed in terms of the different coordination environments and geometries.
Results and discussion
Synthesis and spectroscopic characterization
The new ligand H3L2 was obtained in 74% yield by a Schiff-base condensation between 25-(aminoethoxy)-26,27,28-trihydroxy-calix[4]arene 6 and pyridine-2-carbaldehyde as illustrated in eqn (1).43 H3L2 is soluble in aprotic solvents such as CH2Cl2, CHCl3, DMSO, MeCN and insoluble in alcohols. H3L2 was characterized by IR, 1H and 13C NMR spectroscopy and electrospray ionization mass spectrometry (ESI-MS). The NMR spectra show one set of signals. 2D NMR experiments were performed to correctly assign the chemical shifts of hydrogen and carbon atoms (ESI†). The data are in accordance with the formulation of H3L2 and the “cone”-conformation of the calix[4]arene unit in H3L2.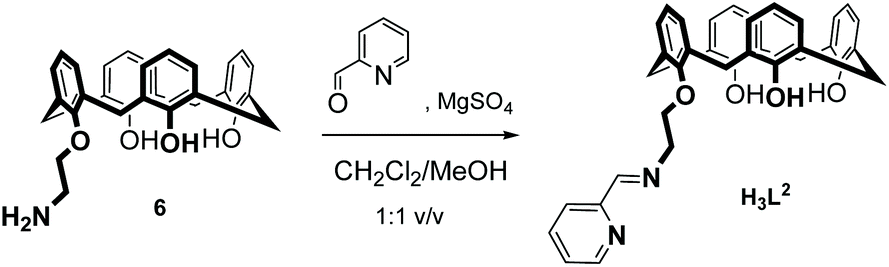 | (1) |
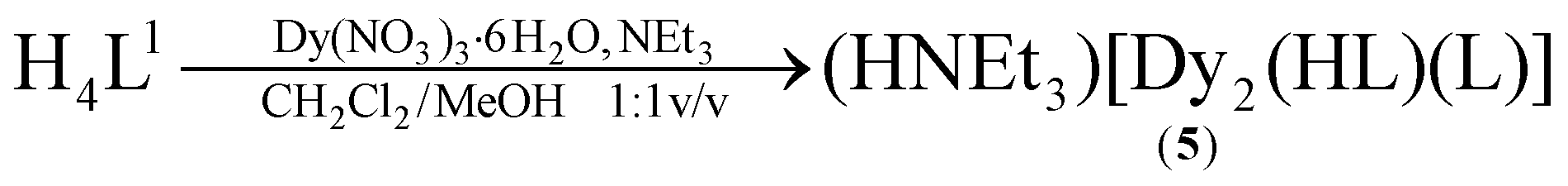 | (2) |
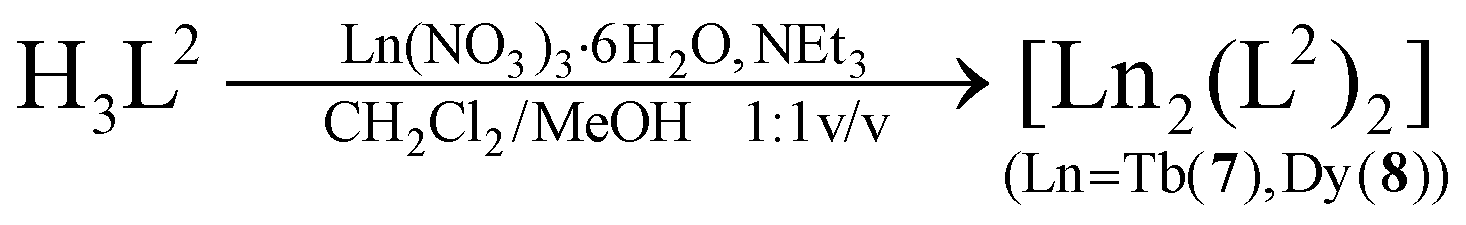 | (3) |
The syntheses of the SmIII2, GdIII2, EuIII2 and TbIII2 complexes 1–4 have been reported previously.33 The corresponding DyIII complex 5 was synthesized in an analogous fashion (eqn (2)). Similarly, reaction of H3L2 with terbium(III) nitrate hexahydrate or dysprosium(III) nitrate hexahydrate and NEt3 as a base in a mixed CH2Cl2/MeOH solution at room temperature, followed by extraction from MeCN affords [Ln2(L2)2]·4MeCN complexes (7·4MeCN, 8·4MeCN) as yellow crystals in 85–87% yield (eqn (3)). The compounds are air stable, have little solubility in common organic solvents, and lose solvate molecules upon standing in air. All new complexes were characterized by elemental analysis, mass spectrometry, and IR spectroscopy, and additionally by X-ray crystallography (for 7·4MeCN, 8·4MeCN). Table 1 lists selected analytical data.
| Compound | ESI-MSa | IRb/cm−1 | Ref. | |||
|---|---|---|---|---|---|---|
| ν(OH) |
ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) N) |
ν(C–O) | ||||
| a Concentration of solutions were ∼1.0 × 10−3 M. b MeCN solution. c ESI(−) mode. d ESI(+) mode. | ||||||
| H4L1 | 570.3c | [H3L1]− | 3635 | 1635 | 1338 | 33 |
| 3500 | ||||||
| 3320 | ||||||
| 5 (Dy) | 1461.25c | [Dy2(HL1)(L1)]− | 3439 | 1635 | 1327 | This work |
| 730.12c | [Dy(L1)]− | |||||
| H3L2 | 557.2d | [H5L2]+ | 3250 | 1651 | 1377 | This work |
| 7 (Tb) | 1447.29d | [Tb2(L2)2 + Na+]+ | — | 1652 | 1331 | This work |
| 1463.24d | [Tb2(L2)2 + K+]+ | |||||
| 713.16d | [Tb(L2) + H+]+ | |||||
| 8 (Dy) | 1433.22d | [Dy2(L2)2 + Na+]+ | — | 1655 | 1333 | This work |
| 717.1d | [Dy(L2) + H+]+ | |||||
A negative ESI-MS spectrum of a solution of 5 (10–3 M) in MeCN displays a molecular ion peak at m/z = 1461.25 with the correct isotopic peak pattern for a dimeric [Dy2(HL1)(L1)]− anion (ESI). A signal at m/z = 730.12 for a monomeric [Dy(L1)]− species is also observed. Similar peaks were observed for complexes 1–4.33 Complexes 7 and 8 were investigated by ESI MS in MeCN (concentration ∼10−3 M) using the positive mode. Under these conditions, molecular ion peaks with the correct isotopic peak pattern for dimeric [Ln2(L2)2 + H+]+, [Ln2(L2)2 + Na+]+ or [Ln2(L2)2 + K+]+ cations are detected (Fig. S7–S10†). Signals for monomeric [Ln(L2) + H+]+ species are also observed, but all attempts to isolate monomeric complexes failed so far. The dimerisation is believed to be driven by the coordination requirements of the lanthanide ions. As in H3L1, the hexadentate N2O4 ligand H3L2 cannot saturate the coordination sphere of the lanthanide ions alone and so dimerization via bridging phenolate atoms takes place to increase the coordination number. Peaks characteristic for host guest complexes with intracavity MeCN molecules could not be detected.
The FTIR spectrum of 5 is similar to those of 1–4, with a strong band at 1635 cm−1 for the ν(CN) stretching frequency. A broad band at 3439 cm−1 is attributed to the O–H stretching vibration. The ν(C–O) stretching frequency observed for H4L1 at 1338 cm−1 is shifted to 1327 cm−1, indicative of the coordination of the pendant phenol ether moiety.44
The most characteristic bands in the FTIR-spectrum of H3L2 are the bands at 1651cm−1 for the imine group (ν(CN)) and a broad feature at 3250 cm−1 for the O–H stretches of the calix[4]arene moiety. The latter band is not seen in the IR spectra of the [Ln2(L2)2] complexes in agreement with the triply deprotonated form of H3L2. The stretching vibration for the imine group is observed at 1651 cm−1 for H3L2, 1652 cm−1 for 7 and 1655 cm−1 for 8, respectively. The intensity of the ν(CN) band decreases significantly upon coordination of the lanthanide ions.
X-ray crystallography
We have not been able to grow single-crystals of the DyIII2 complex 5. However, on the basis of the spectroscopic data, this complex is believed to be isostructural with the SmIII2 (1) and GdIII2 complexes (3), both of which have been crystallographically characterized.33Single crystals of [Tb2(L2)2]·4MeCN (7·4MeCN) obtained by extraction from MeCN were found to be suitable for X-ray crystallographic analyses. The crystal structure determination revealed the presence of dimeric [Tb2(L2)2]·4MeCN complexes (Fig. 2). There are two heavily disordered MeCN solvent molecules, which were removed from the structure using the Squeeze routine implemented in Platon.45 Two well-defined MeCN molecules are situated in the calix[4]arene cavities as in other calix[4]arene structures.46
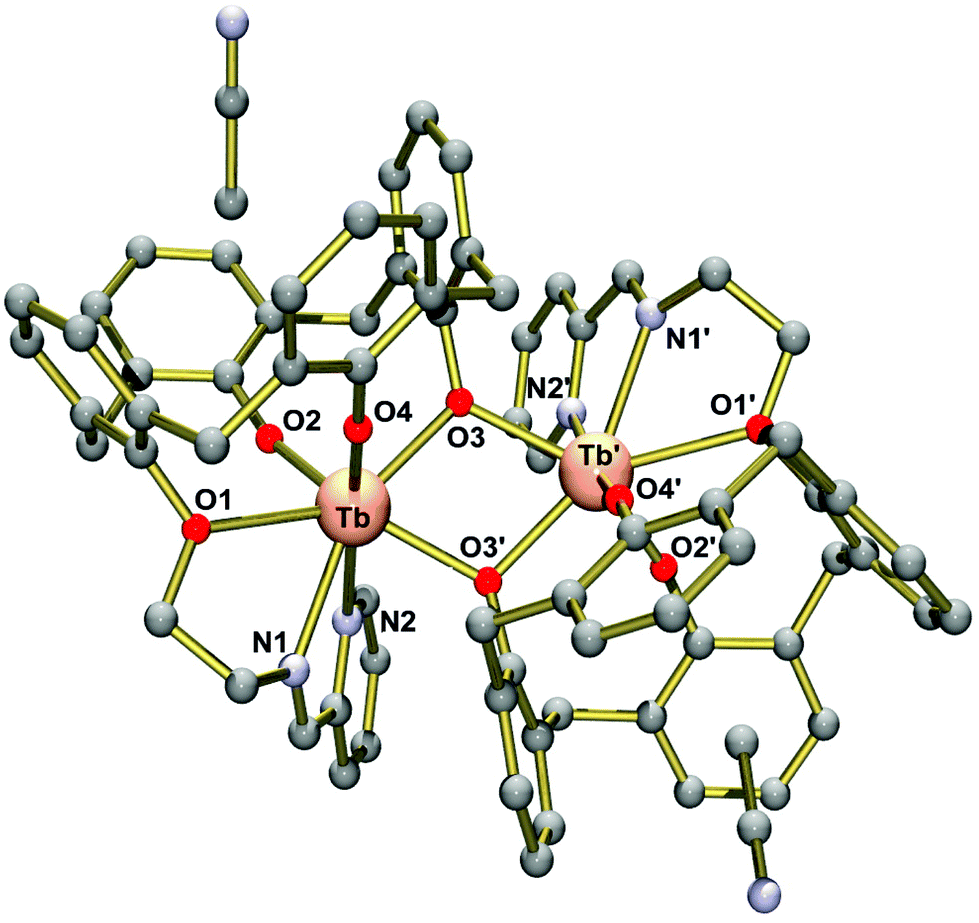 | ||
| Fig. 2 Molecular structure of the neutral [Tb2(L2)2] complex in crystals of 7·4MeCN (Ball and stick representation). The disordered MeCN solvate molecules are omitted for clarity. Symmetry code used to generate equivalent atoms 0.5 − x, −0.5 + y, −z. | ||
Complex 7 exhibits crystallographically imposed C2 symmetry, with the C2 axis passing through the centre of a central four-membered Tb2O2 ring. Two [TbIII(L2)] units are joined by two phenolato bridges at a Tb⋯Tb distance of 3.8011(8) Å. This structure is strikingly different from that seen in 1–5. In the latter, dimerization occurs via phenolato groups from the pendant salicylaldimine units, while [TbIII(L2)] monomers are linked via phenolato groups from the capping calix[4]arene entities. In 1–5, one Ln3+ ion adopts a capped octahedral geometry and the other is a capped trigonal prism as indicated by the SHAPE algorithm (ESI).47 Conversely, in 7, both Ln3+ ions are surrounded in a distorted capped trigonal prismatic fashion by five calix[4]arene O, one imine N and one pyridyl N atom. Also the Ln2O2 cores in the Ci symmetric complexes 1–5 are bent, while the Tb2O2 core in 7 is almost flat, with the TbO2 planes intersecting at 12.5°. It should be noted that the dimerization mode of the calix[4]arene units in 7 is reminiscent of that seen in lanthanide complexes supported by unmodified calixarene ligands. The complexes [{Eu(4-tert-butyl-calix(4)arene)(DMF)2}]48 and [{Nd(calix[4]H)(thf)}2]49 represent two typical examples. There are other dimeric Ln2 complexes containing phenolato bridges, but the coordination numbers tend to be higher than in the present case.50–52 Significant interactions between the complexes are not observed due to the steric bulk of the supporting ligand. Thus, the closest intermolecular Tb⋯Tb distance is at 9.145 Å.
Table 2 lists selected bond lengths and angles. The Tb–O bond lengths vary considerably from 2.136(5) Å to 2.605(5) Å. The Tb–O bonds involving the non-bridging phenolato groups (O2, O4) are significantly shorter than those involving the bridging phenolato groups (O3, O3′) (mean value 2.33 Å), as in other seven-coordinate TbIII calix[4]arene complexes.13,53 The phenol ether O atom (O1) forms the longest Tb–O bond at 2.61 Å. The Tb–N bonds are also quite long, with the Tb–Nimine bond length at 2.536(7) Å being somewhat shorter than the Tb–Npyridine bond 2.691(7) Å. The Tb–O–Tb angles of 109.0° are normal for phenolato-bridged Ln2 complexes (Fig. 3).10
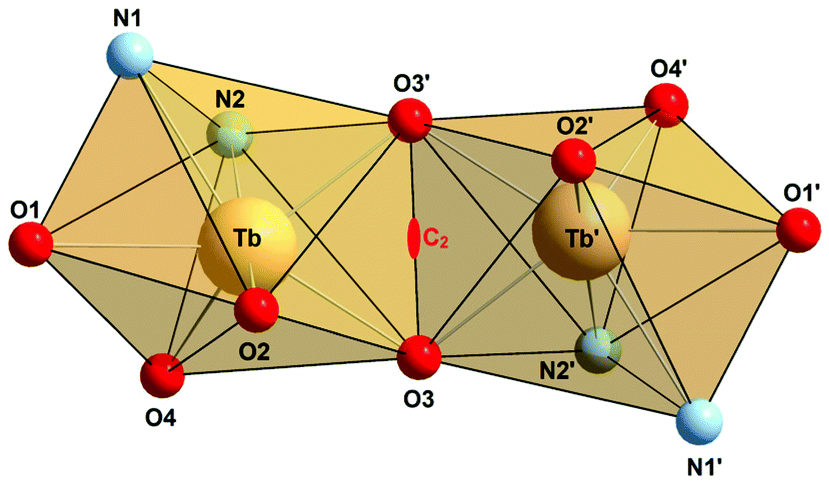 | ||
| Fig. 3 Polyhedral representation of the dinuclear N2O3Tb(μ-O)2TbO3N2 core in 7·4MeCN highlighting the mono-capped trigonal prismatic coordination geometry. The C2 axis is indicated. Symmetry code used to generate equivalent atoms 0.5 − x, −0.5 + y, −z. | ||
| [Tb2(L2)2]·4MeCN | [Dy2(L2)2]·4MeCN | |
|---|---|---|
| M–O1 | 2.605(5) | 2.608(6) |
| M–O2 | 2.136(5) | 2.134(5) |
| M–O3 | 2.323(5) | 2.311(5) |
| M–O3′ | 2.345(4) | 2.343(5) |
| M–O4 | 2.109(5) | 2.106(6) |
| M–N1 | 2.536(7) | 2.509(7) |
| M–N2 | 2.691(7) | 2.675(8) |
| M⋯M′ | 3.8011(8) | 3.7819(9) |
| M–O3–M | 109.03(18) | 108.7(2) |
The structure of the dysprosium complex [Dy2(L2)2]·4MeCN (8·4MeCN) is isomorphous with 7·4MeCN (Fig. S13†), having slightly shorter Dy–O and Dy–N distances (Table 2), in agreement with the smaller ion radius of Dy3+.54 The intramolecular Dy⋯Dy distance is at 3.7819(9) Å. Taken together, the two ligands behave similarly in the sense that they both support phenolato-bridged Ln2 complexes, but the fashion in which this is individually achieved is strikingly different for the two cases.
Magnetic properties
The lanthanide complexes 5, 7, and 8 were further studied by variable temperature direct and alternating current (dc and ac) magnetic susceptibility measurements using a SQUID-Magnetometer (MPMS Quantum Design) in order to probe their magnetic properties. All samples were desolvated at 80 °C in vacuum to remove the enclathrated solvate and intracavity MeCN molecules. The magnetic properties of compounds 1–4 have been reported previously.33Plots of χMT versus T for 5, 7, and 8 are displayed in Fig. 4. At room temperature, the observed χMT values are 30.79 cm3 K mol−1, 25.12 cm3 K mol−1 and 30.49 cm3 K mol−1 for complexes 5, 7, and 8. The values for the Dy complexes 5 and 8 are slightly larger than the theoretical value of 28.34 cm3 K mol−1 for two non-interacting Dy3+ ions (6H15/2, S = 5/2, L = 5, g = 4/3, χT = 14.17 cm3 K mol−1). In both cases, the χMT values decrease slightly upon decreasing the temperature reaching 21.54 cm3 K mol−1 (5) and 21.17 cm3 K mol−1 (8) at 4 K, respectively. This decrease is attributed to depopulation of the Stark sublevels and/or the magnetic anisotropy of the Dy3+ ions, although contributions from antiferromagnetic exchange interactions cannot be ruled out. In the GdIII complex 3, for example, a weak antiferromagnetic exchange interaction is present, J = −0.065 cm−1.33
 | ||
| Fig. 4 Temperature dependence of the χMT product (at 500 Oe) for the dinuclear complexes 5, 7 and 8, χm being the molar susceptibility per dinuclear complex defined as M/H. | ||
The observed χMT value of the TbIII complex 7 is also slightly higher than the expected value of 23.60 cm3 K mol−1 for two non-interacting Tb3+ ions (7F6, S = 3, L = 3, g = 3/2, χT = 11.82 cm3 K mol−1). As observed for 5 and 8, the χMT values decrease with decreasing temperature reaching 17.17 cm3 K mol−1 at 4 K. A similar behaviour was observed for the TbIII complex 4. Tb3+ is known to exhibit significant magnetic anisotropy, and fitting of susceptibility data is therefore difficult.55
The field dependence of the magnetization for complexes 5, 7 and 8 was studied below 8 K in order to see whether magnetic anisotropy is present. Indeed, in all cases the magnetization values increase relatively rapidly at low fields and then linearly at higher field but without a clear saturation (Fig. S14–S16†). Thus, the magnetization values of 11.81μB (5), 9.93μB (7) or 11.92μB for 8 (at 40 kOe and 2 K) are significantly lower than the theoretical value of 18μB or 20μB for dinuclear TbIII and DyIII complexes. Moreover, the iso-field lines are not superposed on each other as would be expected for an isotropic system with a well-defined ground state. These observations clearly reflect the presence of significant magnetic anisotropy and/or low lying excited states.56,57
Variable temperature ac measurements were performed for 5, 7, and 8 in order to study their response to dynamic magnetic fields. Under a zero dc field, however, none of the complexes revealed frequency dependent in-phase (χ′) or out-of-phase (χ′′) signals, ruling out the presence of true SMM behaviour.4 This behaviour was also observed for the TbIII complex 4 and is in striking contrast to many other phenolato-bridged structures, which are true SMMs (Table 3). Inspection of the data in Table 3 shows that the known phenolato-bridged Dy2 and Tb2 SMM systems exhibit coordination numbers 8 and 9 with a more pronounced axial symmetry, a factor which is known to enhance the anisotropy.5 Our complexes (i.e. the coordination polyhedra) lack axial symmetry and magnetic anisotropy, and this observation is in good agreement with the reported trend.15
| Complex | CN | Donor set | Coordination geometry | Magnetic parametersaUeff /K (t0/s) | Ref. |
|---|---|---|---|---|---|
| a HL = 8-hydroxyquinaldine. b H3L = N′-(2-hydroxy-3-methoxy-5-nitrobenzylidene)-2-(hydroxyamino)propanehydrazide. c H2L = 3,5-dichloro-4-hydroxy benzoic acid; phen: 1,10-phenanthroline; H2bfbpen: N,N′-bis-(2-hydroxy-5-fluoro-benzyl)-N,N′-bis-(pyridin-2-ylmethyl)ethylenediamine; H2bcbpen: N,N′-bis-(2-hydroxy-5-chloro-benzyl)-N,N′-bis-(pyridin-2-ylmethyl)ethylenediamine. d H2L = N1,N3-bis(4-chlorosalicyladehyde) diethylenetriamine; H2hmi: (2-hydroxy-3-methoxyphenyl)methylene (isonicotino)hydrazine; H3api: 2-(2-hydroxyphenyl)-1,3-bis[4-(2-hydroxyphenyl)-3-azabut-3-enyl]-1,3-imidazoline; dbm: 1,3-diphenyl-1,3-propanedione. e HL = 2-[[(4-methoxy-phenyl)imino]methyl]-8-hydroxy-quinoline. f HL = 2-[[(4-ethoxyphenyl)imino]methyl]-8-hydroxyquinoline; H2valdien: N1,N3-bis(3-methoxysalicylidene) diethylenetriamine. | |||||
| [Tb2(HLa)4(NO3)6] | 9 | O9 | Distorted spherical capped square antiprism | Not determined | 20 |
| [NHEt3]2[Tb2-(μ-NO3)2(NO3)2(HL)2]b | 9 | O7N2 | Distorted muffin like | 34 (1.1 × 10−8) | 22 |
| [NHEt3]2[Dy2-(μ-NO3)2(NO3)2(HL)2]b | 9 | O7N2 | Distorted muffin-like | 80 (2.2 × 10−6) | 22 |
| [Dy(Lc)(HLc)(phen)] | 8 | O6N2 | Triangular dodecahedron | 160 (9 × 10−8) | 25 |
| [Dy2(bfbpen)2(H2O)2](I)2 | 8 | O4N4 | Square antiprism | 16.9 (3.04 × 10−6) | 26 |
| [DyIII2(bcbpen)2(H2O)2](I)2·0.5H2O | 8 | O4N4 | Square antiprism | 49.2 (6.79 × 10−7) | 26 |
| [Dy2L2(O2CPh)2]·2MeOHd | 8 | O5N3 | Distorted triangular dodecahedron | 47.5 (6.42 × 10−6) | 31 |
| [Dy2L2{(2-NO2)O2CPh}2]d | 8 | O5N3 | Distorted triangular dodecahedron | 67.6 (2.52 × 10−7) | 31 |
| [Dy2L2{(2-OH)O2CPh}2] MeOH·MeCNd | 8 | O5N3 | Distorted triangular dodecahedron | 51.6 (7.50 × 10−6) | 31 |
| [Dy2(hmi)2(NO3)2(MeOH)2] | 8 | O7N1 | Not determined | 56 (3 × 10−7) | 58 |
| [Dy2(api)2] | 8 | O4N4 | Square antiprism | 25.8 (6.79 × 10−6) | 59 |
| [Dy(dbm)2(L)]2e | 8 | O6N2 | Distorted dodecahedron | 34.5 (1.54 × 10−6) | 60 |
| [Dy(dbm)2(L)]2f | 8 | O6N2 | Distorted dodecahedron | 67.6 (6.1 × 10−7) | 9 |
| [Dy2(valdien)2(NO3)2] | 8 | O5N3 | Square antiprism; dodecahedral | 76 (6.04 × 10−7) | 9 |
| [HNEt3][Dy2(HL1)(L1)] 5 | 7 | O6N1 | Capped trigonal prism, capped octahedron | 21.9 (4.73 × 10−5) | This work |
| [Dy2(L2)2] 8 | 7 | O5N2 | Capped trigonal prism | 32.9 (9.01 × 10−6) | This work |
Variable temperature ac measurements were also performed in an applied DC field of 3 kOe to suppress quantum tunneling of the magnetization. Under these conditions, frequency dependent in-phase and out-of-phase signals could be observed, but only for the Dy complexes (Fig. 5, S17 and S18†).
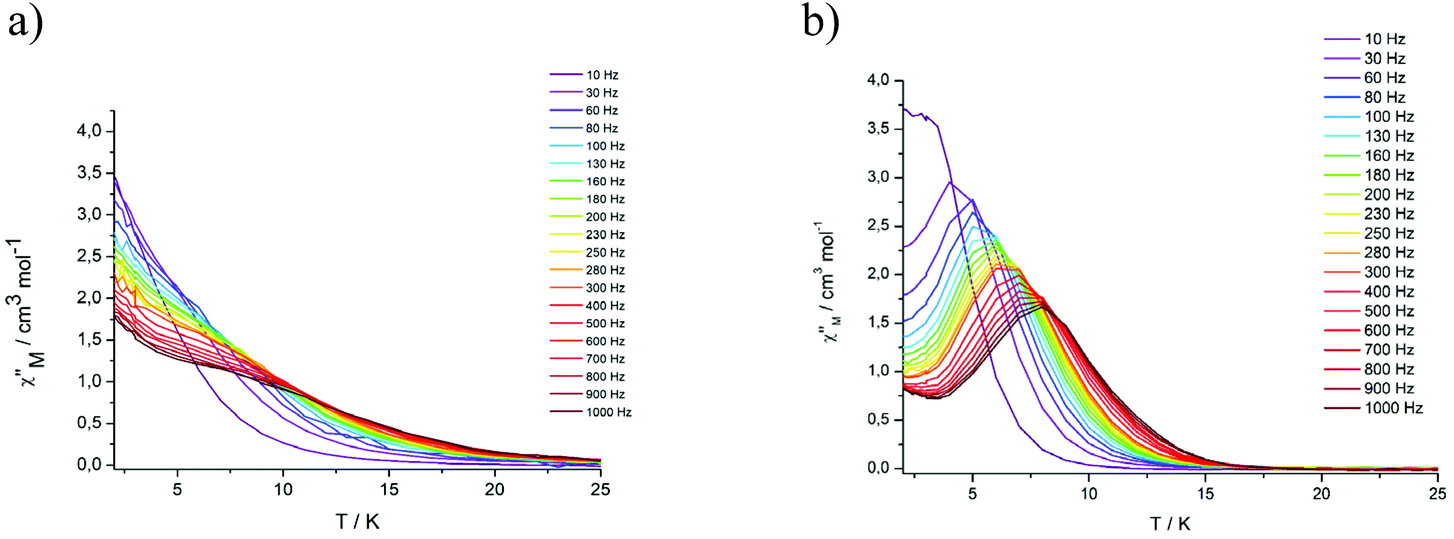 | ||
| Fig. 5 Frequency dependence of the out-of-phase (χ′′) ac susceptibility from 2 to 25 K under an applied static field (3000 Oe) at indicated frequencies for DyIII2 complexes 5 (a) and 8 (b). | ||
Complex 5 displays only a broad shoulder between 6 and 10 K at 1000 Hz. For complex 8, a well developed peak is clearly discernible with the maximum located at 8 K at 1000 Hz. In both cases, an enhancement of the peak intensity is indicated as the quantum tunneling is reduced with decreasing temperature. The relaxation time τ of complex 8 derived from the χ′′ peaks follow the Arrhenius law (Fig. S22†) with τ0 = 9.01 × 10−6 s and the effective anisotropy barrier is determined to be ΔE/KB = 32.9 K. The corresponding values for the Dy2 complex 5 are significantly smaller ΔE/KB = 26.2 K and τ0 = 3.11 × 10−5 s, attributable to the changes in the coordination environments, as detailed above. The values for 5 and 8 are otherwise in a typical range seen for other Dy2 complexes displaying SMM behaviour in a dc field.21,26,61
Conclusion
In summary, a new monofunctionalized hybrid Schiff-base calix[4]arene ligand H3L2, bearing a pyridyl-aldiminato podand arm has been synthesized and its ability to form dimeric complexes [Ln2(L2)2] with Ln = Tb3+ (7) and Dy3+ (8) has been demonstrated. The dinuclear complex [HNEt3][Dy2(HL1)(L1)] (5), where a salicylaldimine podand arm is installed at the calix[4]arene moiety, has also been successfully isolated and characterized. Crystallographic studies show that the structures supported by H4L1 and H3L2 differ greatly. In 7 and 8, [Ln(L2)] monomers are linked via phenolato groups from the capping calix[4]arene, while in 5 dimerization occurs via phenolato groups from the pendant salicylaldimine units. Although both Dy compounds require applied static fields to allow for observation of their SMM, the impact of the change in the coordination environments on the dynamic magnetism is significant.Experimental section
Materials and methods
Precursor compound 6 and the ligand H4L1 were prepared as previously described.33 All other chemicals and solvents were obtained from commercial suppliers in reagent grade and were used without further purification. Melting points were determined in open-glass capillaries and are uncorrected. Mass spectra were obtained using the positive or negative electrospray ionization modus (ESI) on a Bruker Daltronics ESQUIRE 3000 Plus ITMS or Impact II UHR Qq-TOF instrument. 1H and 13C NMR spectra were recorded on a Bruker FOURIER 300 or a Bruker AVANCE DRX 400 spectrometer at 298 K. Chemical shifts refer to solvent signals. Mid (4000–400 cm−1) infrared spectra at 2 cm−1 resolution were recorded on a Bruker TENSOR 27 (equipped with a MIRacle ZnSe ATR accessory from PIKE Technologies) FT-IR spectrometer. Elemental analyses were performed on a vario EL elemental analyzer (Elementar Analysensysteme GmbH, Hanau).Synthesis of compounds
CH, C31). 13C{1H} NMR (100 MHz, CD2Cl2, see Fig. S1† for labeling scheme): δ 31.85 (Ar–CH2–Ar, C2/20); 32.11 (Ar–CH2–Ar, C8/14); 61.13 (CH2–N, C30); 76.54 (O–CH2, C29); 121.06 (para CAr, C5/17); 121.76 (Cpy, C33); 122.08 (para CAr, C11); 125.44 (Cpy, C35); 126.33 (para CAr, C23); 128.74 (ortho CAr, C7/15); 128.83 (ortho CAr, C3/19); 128.99 (meta CAr, C4/18); 129.12 (meta CAr, C6/16); 129.20 (ortho CAr, C9/13); 129.25 (meta CAr, C10/12); 129.85 (meta CAr, C22/24); 134.87 (ortho CAr, C1/21); 137.00 (Cpy, C34); 149.87 (CAr–OH, C27); 149.91 (Cpy, C36); 151.66 (CAr–OH, C26/28); 152.23 (CAr–OH, C25); 155.28 (Cpy, C32); 165.86 (CN, C31). ATR-IR (ZnSe) ν/cm−1 = 3285 (s), 2927 (m), 1651 (m), 1592 (m), 1465 (s), 1439 (s), 1375 (m), 1246 (s), 1193 (s), 1084 (w), 1044 (m), 921 (w), 822 (w), 786 (w), 768 (w), 751 (s). m/z (ESI+, MeCN): C36H32N2O4 (556.24) [M + H+]+ calcd: 557.24; found 557.2; [M + Na+]+ calcd: 579.23; found: 579.2. Elemental analysis for C36H32N2O4·H2O (556.66 + 18.02) calc. C 75.24, N 4.87, H 5.96%; found. C 75.11, N 4.85, H 5.62%.
X-ray crystallography
Single crystals of [Tb2(L2)2]·4MeCN (7·4MeCN) and [Dy2(L2)2]·4MeCN (8·4MeCN) were obtained from acetonitrile. Suitable specimens were selected and mounted on the tip of a glass needle using perfluoropolyether oil. The data sets were collected at 180(2) K using a STOE Stadivari diffractometer equipped with graphite monochromated Cu-Kα radiation (λ = 1.54186 Å). The data were processed with the program XAREA.62 The structure was solved by direct methods63 and refined by full-matrix least-squares techniques on the basis of all data against F2 using SHELXL-2014/7.64 PLATON was used to search for higher symmetry.45 Two of the four MeCN solvate molecules in the structures of 7·4MeCN and 8·4MeCN were found to be heavily disordered and all attempts to model this disorder failed. The corresponding electron density was removed from the structures (and the corresponding F0) with the Squeeze algorithm implemented in the Platon program suite. The SQUEEZE routine revealed total potential solvent area of 150 Å3 per unit cell for 7·4MeCN and 8·4MeCN corresponding to approximately two MeCN solvate molecules per formula unit. All non-hydrogen atoms were refined anisotropically. Graphics were produced with Ortep3 for Windows and PovRAY.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 266 reflections collected, 6114 unique. Final R1 [F2 > 2σ(F2)] = 0.0719, wR2 (all data) = 0.1835.
509 reflections collected, 6070 unique. Final R1 [F2 > 2σ(F2)] = 0.0713, wR2 (all data) = 0.2067.
266 reflections collected, 6114 unique. Final R1 [F2 > 2σ(F2)] = 0.0719, wR2 (all data) = 0.1835.
509 reflections collected, 6070 unique. Final R1 [F2 > 2σ(F2)] = 0.0713, wR2 (all data) = 0.2067.
CCDC 2011007 (7) and 2011008 (8)† contain the supplementary crystallographic data for this paper.
SQUID magnetometry
Magnetic susceptibility measurements were carried out on a MPMS Quantum Design SQUID-magnetometer in the temperature range of 2–300 K. DC measurements were performed from 2–300 K in an applied external field of 0.5 T. AC measurements were collected in a 3.0 Oe ac field at various frequencies (10–1000 Hz) with an applied 3 kOe dc field.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are thankful to Prof. Dr H. Krautscheid for providing facilities for X-ray crystallographic measurements. Financial support from the German Federal Ministry of Education and Research (BMBF – project 02NUK046C, “FENABIUM”) is gratefully acknowledged. YP and AKP gratefully acknowledge funding from the DFG SFB/TRR88 “3Met”.References
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, 2006 Search PubMed.
- S. Hill, R. S. Edwards, N. Aliaga-Alcalde and G. Christou, Science, 2003, 302, 1015–1018 CrossRef CAS PubMed.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC.
- R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- J. Long, F. Habib, P.-H. Lin, I. Korobkov, G. Enright, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 5319–5328 CrossRef CAS PubMed.
- P.-H. Lin, W.-B. Sun, M.-F. Yu, G.-M. Li, P.-F. Yan and M. Murugesu, Chem. Commun., 2011, 47, 10993–10995 RSC.
- S. Takamatsu, T. Ishikawa, S.-Y. Koshihara and N. Ishikawa, Inorg. Chem., 2007, 46, 7250–7252 CrossRef CAS PubMed.
- S.-D. Jiang, B.-W. Wang, G. Su, Z.-M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 49, 7448–7451 CrossRef CAS PubMed.
- F. Gao, L. Cui, Y. Song, Y.-Z. Li and J.-L. Zuo, Inorg. Chem., 2014, 53, 562–567 CrossRef CAS PubMed.
- F. Luan, T. Liu, P. Yan, X. Zou, Y. Li and G. Li, Inorg. Chem., 2015, 54, 3485–3490 CrossRef CAS PubMed.
- M. Ren, Z.-L. Xu, S.-S. Bao, T.-T. Wang, Z.-H. Zheng, R. A. S. Ferreira, L.-M. Zheng and L. D. Carlos, Dalton Trans., 2016, 45, 2974–2982 RSC.
- K. R. Vignesh, D. I. Alexandropoulos, B. S. Dolinar and K. R. Dunbar, Dalton Trans., 2019, 48, 2872–2876 RSC.
- R. Barhoumi, A. Amokrane, S. Klyatskaya, M. Boero, M. Ruben and J.-P. Bucher, Nanoscale, 2019, 11, 21167–21179 RSC.
- M. Studniarek, C. Wäckerlin, A. Singha, R. Baltic, K. Diller, F. Donati, S. Rusponi, H. Brune, Y. Lan, S. Klyatskaya, M. Ruben, A. P. Seitsonen and J. Dreiser, Adv. Sci., 2019, 6, 1901736 CrossRef CAS PubMed.
- R. J. Holmberg, M. A. Polovkova, A. G. Martynov, Y. G. Gorbunova and M. Murugesu, Dalton Trans., 2016, 45, 9320–9327 RSC.
- L. Mandal, S. Biswas, G. Cosquer, Y. Shen and M. Yamashita, Dalton Trans., 2018, 47, 17493–17499 RSC.
- T. Morita, M. Damjanovic, K. Katoh, Y. Kitagawa, N. Yasuda, Y. Lan, W. Wernsdorfer, B. K. Breedlove, M. Enders and M. Yamashita, J. Am. Chem. Soc., 2018, 140, 2995–3007 CrossRef CAS PubMed.
- P. Kalita, J. Goura, J. M. H. Martinez, E. Colacio and V. Chandrasekhar, Eur. J. Inorg. Chem., 2019, 212–220 CrossRef CAS.
- G. Velkos, D. S. Krylov, K. Kirkpatrick, L. Spree, V. Dubrovin, B. Büchner, S. M. Avdoshenko, V. Bezmelnitsyn, S. Davis, P. Faust, J. Duchamp, H. C. Dorn and A. A. Popov, Angew. Chem., Int. Ed., 2019, 58, 5891–5896 CrossRef CAS PubMed.
- K. Katoh, B. K. Breedlove and M. Yamashita, Chem. Sci., 2016, 7, 4329–4340 RSC.
- J.-W. Zhang, Y. Man, W.-H. Liu, B.-Q. Liu and Y.-P. Dong, Dalton Trans., 2019, 48, 2560–2563 RSC.
- X. Ma, B. Chen, Y.-Q. Zhang, J. Yang, Q. Shi, Y. Ma and X. Liu, Dalton Trans., 2019, 48, 12622–12631 RSC.
- S. Biswas, L. Mandal, Y. Shen and M. Yamashita, Dalton Trans., 2019, 48, 14096–14102 RSC.
- M. Li, H. Wu, Z. Xia, V. Montigaud, O. Cador, B. Le Guennic, H. Ke, W. Wang, G. Xie and S. Chen, Chem. Commun., 2019, 55, 14661–14664 RSC.
- G. Huang, X. Yi, F. Gendron, B. Le Guennic, T. Guizouarn, C. Daiguebonne, G. Calvez, Y. Suffren, O. Guillou and K. Bernot, Dalton Trans., 2019, 48, 16053–16061 RSC.
- Y.-X. Zhang, X.-Y. Cheng, Y.-T. Tang, Y.-H. Zhang, S.-C. Wang, H.-Y. Wei and Z.-L. Wu, Polyhedron, 2019, 166, 23–27 CrossRef CAS.
- Y. Ge, D. Li, G. Wang, Y. Cui, M. S. Najib, Y. Li and B.-L. Wang, Chem. – Asian J., 2019, 14, 2846–2852 CAS.
- P. Liu, C. Li and W.-J. Yuan, Inorg. Chim. Acta, 2019, 490, 1–5 CrossRef CAS.
- S. Ullmann, P. Hahn, L. Blömer, A. Mehnert, C. Laube, B. Abel and B. Kersting, Dalton Trans., 2019, 48, 3893–3905 RSC.
- M. Massi and M. I. Ogden, Materials, 2017, 10, 1369–1381 CrossRef PubMed.
- R. K. Pathak, A. G. Dikundwar, T. N. Guru Row and C. P. Rao, Chem. Commun., 2010, 46, 4345–4347 RSC.
- D. D'Alessio, S. Muzzioli, B. W. Skelton, S. Stagni, M. Massi and M. I. Ogden, Dalton Trans., 2012, 41, 4736–4739 RSC.
- R. K. Pathak, V. K. Hinge, M. Mondal and C. P. Rao, J. Org. Chem., 2011, 76, 10039–10049 CrossRef CAS PubMed.
- R. Joseph, J. P. Chinta and C. P. Rao, Inorg. Chem., 2011, 50, 7050–7058 CrossRef CAS PubMed.
- V. V. S. Mummidivarapu, V. K. Hinge and C. P. Rao, Dalton Trans., 2015, 44, 1130–1141 RSC.
- R. K. Pathak, V. K. Hinge, A. Rai, D. Panda and C. P. Rao, Inorg. Chem., 2012, 51, 4994–5005 CrossRef CAS PubMed.
- S. Ullmann, R. Schnorr, M. Handke, C. Laube, B. Abel, J. Matysik, M. Findeisen, R. Rüger, T. Heine and B. Kersting, Chem. – Eur. J., 2017, 23, 3824–3827 CrossRef CAS PubMed.
- S. Ullmann, R. Schnorr, C. Laube, B. Abel and B. Kersting, Dalton Trans., 2018, 47, 5801–5811 RSC.
- S. Bhagat and A. K. Chakraborti, J. Org. Chem., 2007, 72, 1263–1270 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, John Wiley & Sons, Inc., Hoboken, New Jersey, 6th edn, 2009 Search PubMed.
- A. L. Spek, PLATON – A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2000 Search PubMed.
- W. E. I. Xu, R. J. Puddephatt and C. S. Frampton, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 277–290 CrossRef CAS.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE version 2.1, University of Barcelona, Barcelona, 2013 Search PubMed.
- B. M. Furphy, J. M. Harrowfield, M. I. Ogden, B. W. Skelton, A. H. White and F. R. Wilner, J. Chem. Soc., Dalton Trans., 1989, 478, 2217–2221 RSC.
- G. B. Deacon, M. G. Gardiner, P. C. Junk, J. P. Townley and J. Wang, Organometallics, 2012, 31, 3857–3864 CrossRef CAS.
- L. Zou, L. Zhao, P. Chen, Y.-N. Guo, Y. Guo, Y.-H. Li and J. Tang, Dalton Trans., 2012, 41, 2966–2971 RSC.
- Y.-N. Guo, G.-F. Xu, W. Wernsdorfer, L. Ungur, Y. Guo, J. Tang, H.-J. Zhang, L. F. Chibotaru and A. K. Powell, J. Am. Chem. Soc., 2011, 133, 11948–11951 CrossRef CAS PubMed.
- Y.-N. Guo;, X.-H. Chen, S. Xue and J. Tang, Inorg. Chem., 2011, 50, 9705–9713 CrossRef PubMed.
- G. L. Nealon, M. Mocerino, M. I. Ogden and B. W. Skelton, J. Inclusion Phenom. Macrocyclic Chem., 2009, 65, 25–30 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- P.-H. Lin, M. Leclère, J. Long, T. J. Burchell, I. Korobkov, R. Clérac and M. Murugesu, Dalton Trans., 2010, 39, 5698–5704 RSC.
- L. Zhang, S. Lu, C. Zhang, C. Du and H. Hou, CrystEngComm, 2015, 846–855 RSC.
- S. Osa, T. Kido, N. Matsumoto, N. Re, A. Pochaba and J. Mrozinski, J. Am. Chem. Soc., 2004, 126, 420–421 CrossRef CAS PubMed.
- P.-H. Lin, T. J. Burchell, R. Clerac and M. Murugesu, Angew. Chem., Int. Ed., 2008, 47, 8848–8851 CrossRef CAS PubMed.
- M. Nematirad, W. J. Gee, S. K. Langley, N. F. Chilton, B. Moubaraki, K. S. Murray and S. R. Batten, Dalton Trans., 2012, 41, 13711–13715 RSC.
- W.-M. Wang, W.-Z. Qiao, H.-X. Zhang, S.-Y. Wang, Y.-Y. Nie, H.-M. Chen, Z. Lui, H.-L. Gao, J.-Z. Cui and B. Zhao, Dalton Trans., 2016, 45, 8182–8191 RSC.
- W. R. Reed, M. A. Dunstan, R. W. Gable, W. Phonsri, K. S. Murray, R. A. Mole and C. Boskovic, Dalton Trans., 2019, 48, 15635–15645 RSC.
- Stoe & Cie, X-AREA and X-RED 32, V1.35, Stoe & Cie, Darmstadt, Germany, 2006 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, SHELXL-97, Computer program for crystal structure refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2011007 and 2011008. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02209h |
| This journal is © The Royal Society of Chemistry 2020 |