
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of large-pore zeolites from chiral structure-directing agents with two L-prolinol units†
Luis
Gómez-Hortigüela
 *a,
Álvaro
Mayoral
bcd,
Haining
Liu
a,
Laura
Sierra
a,
Laura
Vaquerizo
a,
Cristina
Mompeán
a and
Joaquín
Pérez-Pariente
a
*a,
Álvaro
Mayoral
bcd,
Haining
Liu
a,
Laura
Sierra
a,
Laura
Vaquerizo
a,
Cristina
Mompeán
a and
Joaquín
Pérez-Pariente
a
aInstituto de Catálisis y Petroleoquímica, ICP-CSIC, C/Marie Curie 2, 28049, Madrid, Spain. E-mail: lhortiguela@icp.csic.es
bInstitute of Materials Science of Aragon (ICMA), CSIC-University of Zaragoza, 12, Calle de Pedro Cerbuna, 50009 Zaragoza, Spain
cLaboratorio de Microscopias Avanzadas (LMA), University of Zaragoza, Spain
dCenter for High-resolution Electron Microscopy (CħEM), School of Physical Science and Technology, ShanghaiTech University, 393 Middle Huaxia Road, Pudong, Shanghai 201210, China
First published on 23rd June 2020
Abstract
In this work, we perform an in-depth experimental and computational study about the structure-directing effect of two new chiral organic quaternary ammonium dications bearing two N-methyl-prolinol units linked by a xylene spacer in para or meta relative orientation, displaying four enantiopure stereogenic centers in (S) configuration. Synthesis results show that the para-xylene derivative is an efficient structure-directing agent, promoting the crystallization of ZSM-12 (in pure-silica composition), beta zeolite (as pure-silica, or in the presence of Al or Ge), and a mixture of polymorphs C, A and B of zeolite beta (in the presence of Ge). In contrast, the meta-xylene derivative showed a much poorer structure-directing activity, yielding only amorphous materials unless Ge is present in the gel, where beta and polymorph C (together with A and B) zeolites crystallized. Molecular simulations showed that the para-xylene dication displays a cylindrical shape suitable for confining in zeolite pores, while the meta-xylene derivative has an angular shape that shifts from the typical dimensions required for 12MR zeolite channels. Despite enantio-purity of the para-xylene dication with (S,S,S,S) configuration, no enrichment in polymorph A of the zeolite beta samples obtained was observed by Transmission Electron Microscopy. With the aid of molecular simulations, the failure in transferring chirality to the zeolite is explained by the loose fit of this SDA in the large-pores of zeolite beta, and a lack of close geometrical fit with the chiral element of polymorph A, as evidenced by the very similar interaction of the cation with the two enantiomorphic space groups of polymorph A. Nevertheless, the molecular-level knowledge gained in this work can provide insights for the future design of more efficient SDAs towards the synthesis of chiral zeolites.
1. Introduction
Zeolite materials have three-dimensional frameworks with pores and cavities of molecular dimensions that provide a confinement effect to guest species occluded within their framework, promoting their characteristic shape-selective and molecular sieve properties.1,2 Combined with their cationic-exchange and catalytic properties, this has stimulated the use of zeolite materials in a wide range of applications in the chemical industry, especially as catalysts for different types of reactions with high selectivity towards desired products.3,4Confinement effects associated to zeolite microporous frameworks enable the discrimination between guest species (sorbates, reactants, transition states or products) with small steric differences.5 This has been widely exploited in catalytic uses, but it is also essential during the synthesis of these materials where guest extra-framework species are confined within the zeolite pores and cavities during the crystallization process.6 In particular, the addition of organic cations with particular geometric properties (size and shape) to the zeolite synthesis gels has enabled to gain control on the zeolite porous architecture that crystallizes through host–guest chemistry and, in turn, confinement effects. These organic species, which are usually referred as structure-directing agents (SDA), direct the crystallization process towards a particular framework type through a geometric relationship between the size and shape of the organic species and that of the porosity of the zeolite framework.7–10
One of the greatest challenges in zeolite science is the development of enantiomerically pure chiral zeolites or at least enriched in one of the two enantiomorphic crystals.11–13 These chiral zeolites should be able to perform enantioselective operations, both in adsorption and catalysis processes, because of an asymmetric confinement of guest species in the chiral pores and/or cavities.14–18 Indeed, several chiral zeolite frameworks do actually exist.12,19–23 However, they usually crystallize as mixtures of crystals with the two handednesses, either as racemic mixtures of enantiopure crystals (like STW, that can crystallize in P6122 or P6522 enantiomorphic space groups)24 or as intergrown polymorphs of chiral frameworks, like zeolite beta where polymorph A is chiral (and can crystallize in P4122 or P4322 space groups).19 Once again, confinement in a restricted space is crucial for developing enantio-discriminating properties, not only for potential applications but also during crystallization in the presence of organic SDAs. In this context, the usual host–guest geometrical relationship between organic SDAs and zeolite frameworks provides a straightforward tool to promote the crystallization of chiral zeolite frameworks through the use of chiral organic species as SDAs.11,25 For this imprint of chirality to occur, a true template effect, in the sense of establishing a close geometrical relationship between the guest molecular shape and the host framework walls, should be established. Although this strategy has been used for very long, only very recently a single successful example of enantio-enrichment of a chiral zeolite (STW) through the use of a rationally-designed chiral organic SDA has been reported.26 In this context, an in-depth knowledge at molecular level of the structure-directing role played by chiral SDAs during crystallization of zeolite frameworks is vital for successfully promoting a transfer of chirality from organic guests to the zeolite host framework.
In recent years, we have successfully studied several organic SDAs prepared from chiral precursors derived from the chiral pool, on the one hand from chiral alkaloids (1R,2S)-ephedrine and (1S,2S)-pseudoephedrine,27–32 and on the other from L-prolinol (derived from L-proline amino acid).33–38L-Prolinol is a useful chiral precursor since it provides a rigid ring with an N atom that can be quaternized with two different alkyl substituents, providing an additional stereogenic centre. Interestingly, a careful selection of the synthesis protocol during the alkylation reactions enables a preferential attack by one particular side of the molecule, leading to enantiopure stereogenic N atoms in addition to the enantiopure C atoms of the original L-prolinol units.33 In previous works, we studied the structure-directing effect of N-methyl-N-benzyl-prolinol, and observed the crystallization of several zeolite materials, including MTW,33 MWW38 and FER frameworks,36 as a function of the synthesis conditions. Another report with similar but smaller prolinol derivatives with methyl and ethyl substituents showed the production of layered precursors of CDO zeolite.39
In order to maximize the transfer of chirality to zeolite frameworks, it is essential to adapt the chiral dimension of the organic SDAs, which is expressed at a molecular level, to that of the zeolite frameworks, which is usually expressed at a long-range level in the form of helicoidal channels. Based upon these grounds, in this work we build new SDAs based on two L-prolinol units linked by a xylene ring in different positions, resulting in large SDAs with four enantiopure stereogenic centres that should enhance the asymmetry of the organic species (Scheme 1). In order to understand from a molecular level the structure-directing activity of these SDAs, we perform a combined experimental and computational study of the zeolite materials obtained.
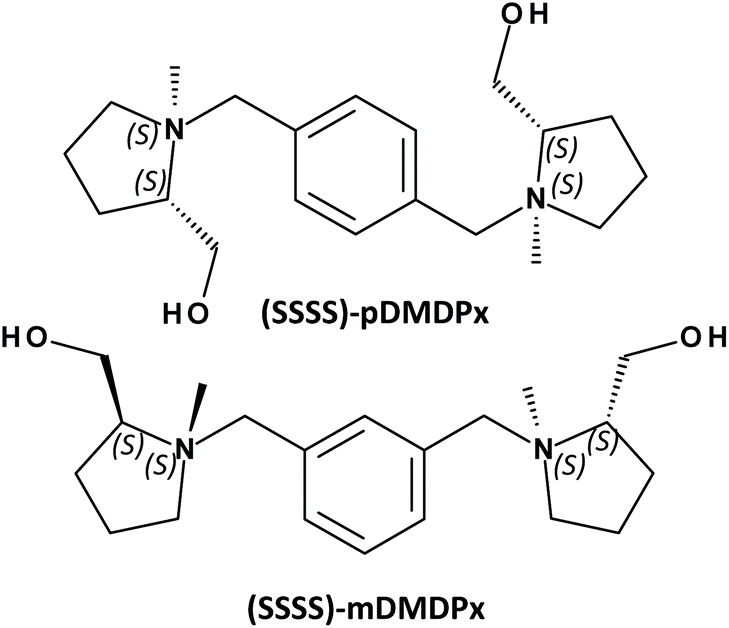 | ||
| Scheme 1 Molecular structure of the SDAs used in this work. | ||
2. Experimental
2.1 Synthesis of the organic structure directing agents
The organic SDAs, 1,1′-(1,X-phenylenebis(methylene))bis(2-(hydroxymethyl)-1-methylpyrrolidin-1-ium (where ‘X’ is ‘4’ for the para-substituted and ‘3’ for the meta-substituted derivatives, and will be referred as ‘pDMDPx’ and ‘mDMDPx’, respectively, see Scheme 1), were prepared through alkylation reactions from the chiral precursor L-prolinol. Quaternization of the N atoms with different substituents (methyl and xylene derivatives) raises new stereogenic centres on N, which could display (S) or (R) configuration, while the absolute configuration of the stereogenic C atoms in L-prolinol units invariably remains as (S). Therefore, in principle two types of diastereoisomers could be produced, with prolinol units in (S,S) or (S,R) configuration (where the first refers to C and the second to N stereogenic centres). The order of alkylation reactions is crucial in order to obtain pure diastereoisomers. In a previous work, we observed that for N-benzyl-N-methyl-prolinol cation, initial alkylation with the bulky aromatic derivative followed by methylation led to pure (S,S)-derivatives, while inversion of the order of alkylation reactions (first methyl and second benzyl groups) yielded a mixture of (S,S) and (S,R) isomers.33 The same strategy has been followed here in order to obtain pure (S,S,S,S)-isomers (in this case the organic cations contain two L-prolinol units, see Scheme 1).Synthesis of pDMDPx and mDMDPx was carried out by alkylation of (S)-2-pyrrolidinemethanol (L-prolinol) with α,α′-dibromo-para-xylene or α,α′-dibromo-meta-xylene, respectively. In a typical synthesis of pDMDPx, 10.00 g of (S)-2-pyrrolidinemethanol were carefully added to a cooled solution of 13.05 g of α,α′-dibromo-para-xylene in 350 mL of acetonitrile with 20.50 g of potassium carbonate (careful, exothermic reaction). The reaction mixture was kept refluxing overnight, after which the inorganic solids were removed by filtration, and the solvent was rotoevaporated, yielding a yellowish solid (14.30 g, yield 95%). 13C NMR (CDCl3): 23.4; 27.8; 54.5; 58.3; 61.9; 64.3; 128.7; 138.2.
13.70 g of this solid were dissolved in 300 mL of cooled acetonitrile, and 12.80 g of methyl iodide were added dropwise (careful, exothermic reaction). The mixture was kept at room temperature for 5 days, after which the solvent was rotoevaporated, and the obtained yellow oil was washed with diethyl ether. The resulting product was 1,1′-(1,4-phenylenebis(methylene))bis(2-(hydroxymethyl)-1-methyl-pyrrolidin-1-ium iodide (pDMDPx+I−) (20.70 g, yield 91%). 13C NMR (D2O): 18.9; 23.5; 42.5; 58.8; 64.3; 67.2; 74.5; 130.4; 133.5.
In order to confirm the production of the (S,S,S,S)-isomer, the organic synthesis was also carried out by reverting the order of alkylation, first adding a methyl group to L-prolinol through the Leucart reaction, and then adding the dibromo-para-xylene derivative. In this case, we obtained a product where 13C NMR signals corresponding to the C atoms directly attached to N were doubled (Fig. S1†), evidencing the presence of the two isomers, (S,R,S,R) and (S,S,S,S), and confirming the isomeric purity of our original product.
The corresponding derivative in meta-position, (1,1′-(1,3-phenylenebis(methylene))bis(2-(hydroxymethyl)-1-methyl-pyrrolidin-1-ium iodide (mDMDPx+I−), was obtained in the same way using the α,α′-dibromo-meta-xylene derivative. 13C NMR (D2O): 18.9; 23.5; 42.4; 58.8; 64.3; 67.3; 74.6; 129.1; 130.1; 134.9; 136.8.
The hydroxide forms of the quaternary ammonium iodide salts were obtained by ion exchange with an anionic resin (Amberlite IRN-78; exchange capacity, 4 meq g−1; Supelco), and the hydroxide solutions were concentrated to ∼30 wt%.
2.2 Synthesis of zeolite materials
Zeolite materials were prepared by hydrothermal method using pDMDPx and mDMDPx as SDA under different synthesis conditions in fluoride medium. The molar composition of the synthesis gels was 0.25R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(1 − x)SiO2:xGeO2:yAl2O3:0.5HF:wH2O, where R stands for the organic SDA. Pure-silicate, aluminosilicate and germanosilicate materials were prepared with different compositions, as explained in the corresponding section. In a typical preparation, the corresponding amounts of the organic hydroxide and GeO2 were mixed and stirred for 30 minutes, after which tetraethylorthosilicate (TEOS) and aluminium isopropoxide were added and stirred until all the ethanol coming up from the hydrolysis of TEOS and the required amount of water to achieve the desired composition were evaporated. HF (48%) was then added and manually stirred (with the help of a spatula) until a homogenous thick gel was obtained. The gels were introduced into 60 ml Teflon lined stainless steel autoclaves and heated statically at different temperatures under autogenous pressure for selected periods of time. The resulting solids were separated by filtration, thoroughly washed with ethanol and water and dried at room temperature overnight.
:(1 − x)SiO2:xGeO2:yAl2O3:0.5HF:wH2O, where R stands for the organic SDA. Pure-silicate, aluminosilicate and germanosilicate materials were prepared with different compositions, as explained in the corresponding section. In a typical preparation, the corresponding amounts of the organic hydroxide and GeO2 were mixed and stirred for 30 minutes, after which tetraethylorthosilicate (TEOS) and aluminium isopropoxide were added and stirred until all the ethanol coming up from the hydrolysis of TEOS and the required amount of water to achieve the desired composition were evaporated. HF (48%) was then added and manually stirred (with the help of a spatula) until a homogenous thick gel was obtained. The gels were introduced into 60 ml Teflon lined stainless steel autoclaves and heated statically at different temperatures under autogenous pressure for selected periods of time. The resulting solids were separated by filtration, thoroughly washed with ethanol and water and dried at room temperature overnight.
2.3 Characterization of zeolite materials
The obtained solids were characterized by powder X-Ray Diffraction (XRD), using a Philips X'PERT diffractometer with CuKα radiation with a Ni filter. Thermogravimetric analyses (TGA) were registered using a PerkinElmer TGA7 instrument (heating rate = 20 °C min−1) under air flow. Liquid NMR spectra were recorded with a Bruker Avance III-HD Nanobay 300 MHz spectrometer, using a 5 mm HBO 1H/X probe. Solid State MAS-NMR spectra of the solid samples were recorded with a Bruker AV 400 WB spectrometer, using a BL7 probe. 1H to 13C Cross-Polarization spectra were recorded using π/2 rad pulses of 2.75 μs for 1H, a contact time of 3 ms and a recycle delay of 4 s. The spectra were recorded while spinning the samples at ca. 11.2 kHz.Electron microscopy analyses were carried out in a cold FEG JEOL GrandARM 300 operated at 300 kV. The microscope was equipped with a double spherical aberration (Cs) corrector from JEOL Company. Images were recorded under low-dose conditions to minimize the electron beam damage using an annular dark field detector (ADF). Prior to observation, the samples were deeply crushed using mortar and pestle dispersed in ethanol and few drops of the suspension were placed onto holey carbon copper grids.
2.4 Computational details
In order to understand the structure-directing role of the two chiral SDAs and the effect of their molecular structure, molecular simulations based on a combination of molecular mechanics (Dreiding forcefield) and quantum mechanics (DFT) were carried out. Calculations of the stability of different conformers in vacuum were performed at ab initio level with the CASTEP code,40 using DFT + D and plane waves (with an energy cut-off of 571.4 eV), and the PBE functional (including the Grimme dispersion term).41 Molecular structures of the organic cations and their interaction with the different zeolite frameworks were simulated using the Dreiding force-field42 and an atomic charge distribution obtained by the ESP charge calculation method at DFT + D level; our results showed that such Dreiding-ESP model reproduced well the conformational energy landscape of the organic cations studied at DFT + D level. The atomic coordinates of the different zeolite frameworks were kept fixed during all the calculations. Different supercell systems were built as a function of the particular framework type; details will be given in the corresponding section. The organic cations were manually loaded in different conformations and orientations, and the most stable system was obtained through simulated annealing calculations. Interaction energies were calculated by subtracting the energy of the cations in vacuum to the total energy of the system; all energies are expressed in kcal mol−1 of SDA.Calculation of the NMR chemical shielding of the different isomers was carried out with the gauge-including projector augmented-wave method (GIPAW) developed by Pickard and Mauri,43 as implemented in the CASTEP code, using a σref value of 176 ppm, the same as in our previous works.29,32,44
The conformational behaviour of the SDA cations in water was studied by NVT Molecular Dynamics simulations, in the same way as reported in our previous work.35 8 SDA cations, 16 Cl− anions (for charge-compensation) and 160 water molecules were included in the simulation cell, and 10 ns of MD simulations in NVT ensemble were run at 423 K.
3. Experimental results
3.1 Pure-silicate materials
Table 1 summarizes the phases obtained using the two SDAs in pure-silica compositions under different crystallization conditions, and Fig. 1 shows the XRD patterns of some of the crystalline products obtained. The use of the para-derivative (pDMDPx) led to highly crystalline MTW materials with 0.25R and 5H2O molar composition, both at 130 and 150 °C crystallization temperatures (Fig. 1A). A decrease of the water content to 3 led to a mixture of zeolite beta (BEA) and MTW at 130 °C, while an increase of the temperature to 175 °C prevented the formation of zeolite beta. A further decrease of the water content to 1 resulted in zeolite beta as the only crystalline phase at 130 °C, although this was accompanied with some amorphous material (Fig. 1B); this provides a new example of the so-called Villaescusa's rule45 where an increase of the SDA concentration tends to promote the crystallization of more open frameworks. Worth is noting that BEA and MTW frameworks share the mtw composite building unit. In line with this, a decrease of the SDA concentration (to 0.125) favoured the crystallization of MTW instead of beta (Table 1). In sharp contrast, no crystalline products were obtained when the meta-xylene derivative (mDMDPx) was used as SDA under similar crystallization conditions; even under the more favourable conditions for BEA of low water contents, no crystalline products were observed, evidencing a poorer structure-directing ability of mDMDPx, at least in pure-silica composition.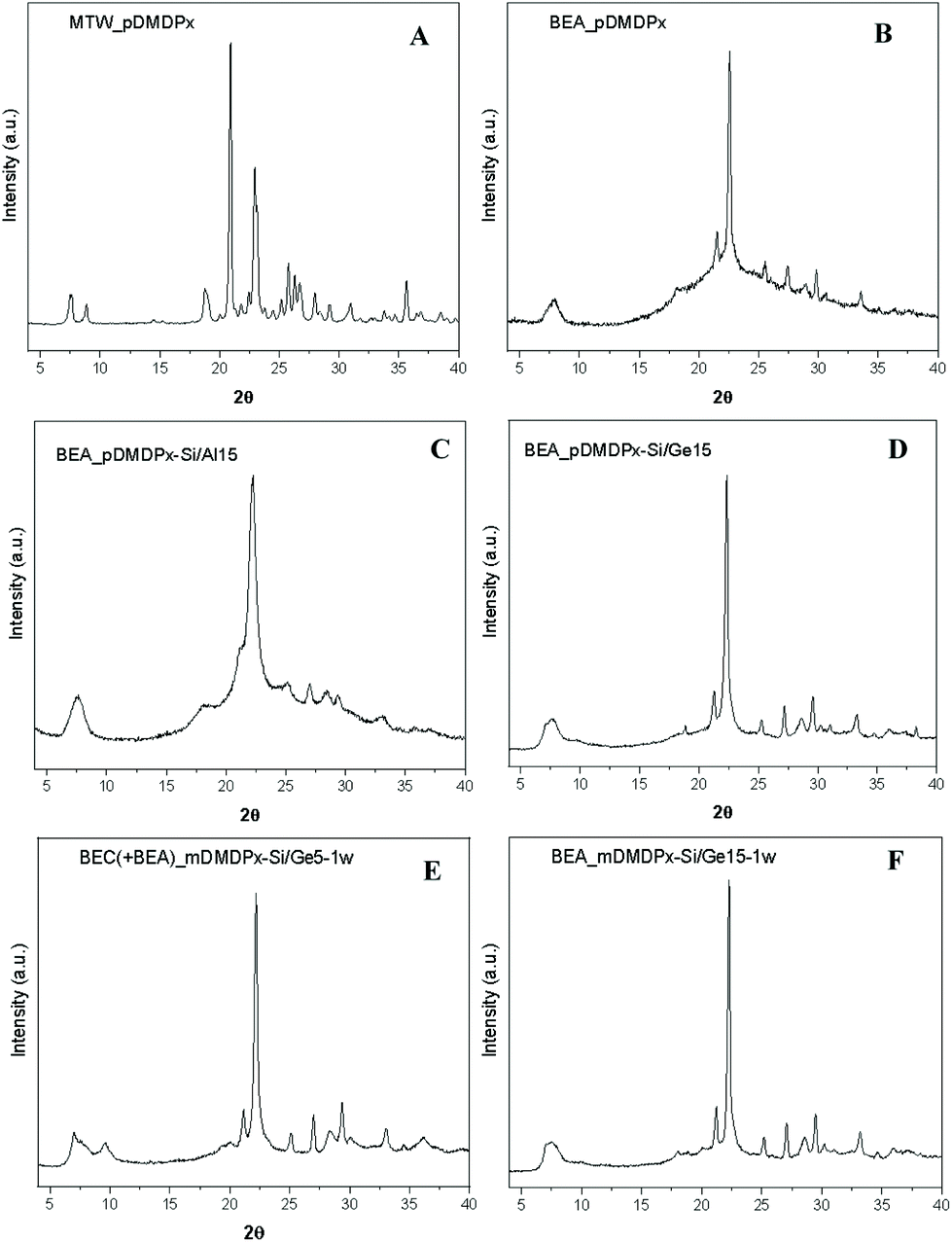 | ||
| Fig. 1 XRD patterns of (top): pure-silica materials obtained with pDMDPx as SDA with 5 (A: MTW, 10 days at 150 °C) or 1 (B: BEA, 7 days at 130 °C) H2O molar compositions. Middle: BEA materials obtained with pDMDPx in the presence of Al (C, Si/Al 15, 14 days at 150 °C) or Ge (D, Si/Ge 15, 7 days at 150 °C). Bottom: BEC/BEA materials obtained with mDMDPx in the presence of Ge, with Si/Ge ratios of 5 (E, 1H2O, 6 days at 150 °C) or 15 (F, 2.7H2O, 6 days at 150 °C), with low water contents. | ||
| SDA | R | H2O | Temperature (°C) | Time (days) | Phase |
|---|---|---|---|---|---|
| pDMDPx | 0.25 | 5.0 | 130 | 10 | MTW |
| 150 | 5 | MTW | |||
| 150 | 10 | MTW | |||
| 3.0 | 130 | 10 | BEA + MTW | ||
| 175 | 10 | MTW + Am | |||
| 1.0 | 130 | 7 | BEA | ||
| 0.125 | 1.0 | 130 | 7 | MTW + Am | |
| mDMDPx | 0.25 | 5.0 | 130 | 10 | Am |
| 150 | 5 | Am | |||
| 150 | 10 | Am | |||
| 2.0 | 130 | 6 | Am | ||
| 150 | 6 | Am | |||
| 180 | 6 | Am | |||
3.2 Aluminosilicate materials
We next studied the synthesis of zeolite materials in the presence of Al (Table 2). Using pDMDPx as SDA with a Si/Al ratio of 15 under standard conditions (0.25R and 5H2O molar compositions), zeolite beta was obtained both at 130 and 150 °C (Fig. 1C). Zeolite beta is an intergrowth of related polymorphs A and B, with the former being chiral. Hence, this result was interesting since chiral polymorph A could be potentially enriched by the use of our chiral SDA. Indeed, much effort has been applied in recent years in order to enrich zeolite beta in polymorph A, which has been partially accomplished by using special synthesis conditions.46–52 Besides, theoretical studies based on molecular simulations have showed that polymorph A could be obtained by using particular SDAs designed with specific geometric features.53 In our case, no hints of a potential enrichment in polymorph A was observed by looking at the profile of the first diffraction peak at around 7.5° 2θ (enrichment in polymorph A should enhance peaks at 7.0, 7.7 and 9.7°).| SDA | Si/Al | R | H2O | Temperature (°C) | Time (days) | Product |
|---|---|---|---|---|---|---|
| pDMDPx | 15 | 0.25 | 5.0 | 130 | 10 | BEA |
| 150 | 5 | BEA | ||||
| 150 | 14 | BEA | ||||
| 15 | 0.125 | 5.0 | 130 | 14 | Am | |
| 150 | 14 | Am | ||||
| 15 | 0.25* | 5.0 | 130 | 13 | Am | |
| 150 | 6 | Am | ||||
| 150 | 13 | Am | ||||
| 5 | 0.25 | 5.0 | 130 | 13 | Am | |
| 150 | 5 | Am | ||||
| 150 | 13 | Am | ||||
| 22 | 0.25 | 5.0 | 130 | 13 | BEA | |
| 30 | 0.25 | 5.0 | 130 | 13 | BEA | |
| 30 | 0.25 | 3.0 | 130 | 9 | BEA | |
| 40 | 0.25 | 5.0 | 130 | 13 | Am | |
| mDMDPx | 15 | 0.25 | 5.0 | 130 | 10 | Am |
| 150 | 5 | Am | ||||
| 150 | 10 | Am | ||||
| 15 | 0.25 | 2.2 | 150 | 6 | Am | |
In order to limit the crystallization of zeolite beta in an attempt to promote the crystallization of polymorph A, the SDA concentration was reduced (0.125); however, this prevented the formation of zeolite beta (Table 2). Similarly, an increase of the HF concentration also impeded the crystallization of zeolite beta. An increase of the amount of Al (Si/Al = 5) resulted in no crystalline products. In contrast, under the typical SDA and H2O contents (0.25 and 5 molar compositions, respectively), zeolite beta was able to crystallize up to a Si/Al ratio of 30, but a further reduction of the Al content (Si/Al = 40) prevented its formation.
In line with the previous result for pure-silicate composition, the conditions that promoted the crystallization of highly crystalline zeolite beta with pDMDPx did not result in any crystalline material when the SDA was the meta-derivative (mDMDPx), further evidencing the poor structure-directing ability of this cation.
3.3 Germanosilicate materials
Table 3 shows our synthesis results with different Ge contents in the gel. When using the more efficient pDMDPx SDA, the incorporation of Ge in high amounts (Si/Ge ratios of 1 and 5) resulted in the crystallization of polymorph C (BEC) of zeolite beta, possibly mixed with polymorphs A and B. The formation of BEC is typical in the presence of Ge due to the occurrence of D4R units under this stacking configuration of the beta sheets. At high temperatures (175 °C), BEC zeolite was accompanied by minor amounts of clathrasil AST, where pDMDPx cannot be accommodated, suggesting that at this temperature the SDA starts to degrade, and the resulting fragments direct the formation of AST. Interestingly, when the Ge content was decreased to a Si/Ge ratio of 15, zeolite beta crystallized, though probably with a minor amount of BEC (Fig. 1D). Similarly, pure and highly crystalline zeolite beta was formed at a higher Si/Ge ratio of 30, while in the absence of Ge, MTW zeolite was obtained under the same conditions (Table 1). This clearly indicates that the incorporation of even a small amount of Ge tends to favour the crystallization of zeolite beta, even though it does not contain D4Rs (though we cannot discard a minor presence of BEC present in this sample, as will be shown below). We note that these zeolite beta samples obtained in the presence of Ge show very subtle increases of the diffraction peaks at 7.0 and 9.7° 2θ (Fig. 1D); however, this could correspond either to polymorph A or C. Electron microscopy will be used below in order to distinguish the occurrence of both polymorphs.| SDA | Si/Ge | H2O | Temperature (°C) | Time (days) | Product |
|---|---|---|---|---|---|
| pDMDPx | 1 | 5.0 | 150 | 7 | BEC |
| 150 | 14 | BEC | |||
| 175 | 7 | BEC + AST | |||
| 5 | 5.0 | 150 | 7 | BEC | |
| 150 | 14 | BEC | |||
| 175 | 7 | BEC | |||
| 5 | 3.0 | 150 | 7 | BEC | |
| 175 | 7 | BEC + AST | |||
| 15 | 5.0 | 150 | 7 | BEA* | |
| 175 | 7 | BEA* | |||
| 30 | 5.0 | 130 | 10 | BEA | |
| 150 | 7 | BEA | |||
| 150 | 14 | BEA | |||
| mDMDPx | 5 | 5.0 | 130 | 11 | BEC |
| 30 | 5.0 | 130 | 11 | BEA | |
| 40 | 5.0 | 130 | 10 | BEA | |
| 30 | 3.0 | 130 | 11 | BEA | |
| 15 | 2.7 | 130 | 6 | BEA* | |
| 150 | 6 | BEA* | |||
| 30 | 2.0 | 130 | 6 | Am | |
| 150 | 6 | BEA | |||
| 5 | 1.0 | 150 | 6 | BEC + BEA | |
We then analysed the structure-directing ability of mDMDPx. In contrast to previous cases where no crystalline products were observed with this SDA, polymorph C of zeolite beta (together with some minor amounts of polymorphs A and B) was obtained with a Si/Ge ratio of 5 with high water content (H2O = 5), while an increase of the concentration (to 1H2O) resulted in the crystallization of a mixture of the three beta polymorphs (Fig. 1E). As with pDMDPx, an increase of the Si/Ge ratio beyond 15 results in the crystallization of zeolite beta (BEA), being more crystalline the samples obtained with a lower water content (Fig. 1F). Hence, only in the presence of Ge did the less efficient mDMDPx SDA enabled the crystallization of zeolite materials related to beta.
3.4 Characterization of zeolites
We then characterized by TGA and 13C CP MAS NMR the organic content of the most interesting samples. TGA of MTW_pDMDPx material (Fig. S2†) showed no desorption of water, as typically in pure-silica materials, and a total weight loss of 14.7%. BEA materials showed much higher amounts of volatile compounds, with 1–2% of water, and 24.2% (BEA_pDMDPx-Si/Al15), 21.6% (BEA_pDMDPx-Si/Ge15), and 22.0% (BEA_mDMDPx-Si/Ge15) of volatile compounds desorbing in the 120–900 °C temperature range. Worth is noting that the intensity of desorption in the second step is higher for the BEA sample containing Al, which is an evidence of the presence of Al and the formation of acid sites during calcination.The chemical integrity of the SDAs occluded within the different zeolite materials was studied by 13C CP MAS NMR of the solid samples (Fig. 2). All the zeolite materials obtained with pDMDPx (left) showed several bands corresponding to the different C atoms of the pristine cation (as represented by the iodide salt in D2O solution), confirming the integral incorporation of the cation in the solids. Nevertheless, some additional bands with lower intensity between 115–120 ppm and at ∼30 ppm reveal the presence of minor amounts of other organic fragments in certain samples, probably as a result of a partial decomposition; this is particularly true in zeolite beta obtained in the presence of Al and in polymorph C (blue and green lines). Similarly, 13C NMR spectra of the solids obtained with mDMDPx also showed the presence of all the bands characteristic of the cation (Fig. 2-right), again evidencing the integral incorporation of this cation within the zeolite beta-related materials. The same bands corresponding to degradation products are also observed in these samples, especially in BEA. Overall, these results demonstrate the incorporation of pDMDPx and mDMDPx in the zeolite frameworks.
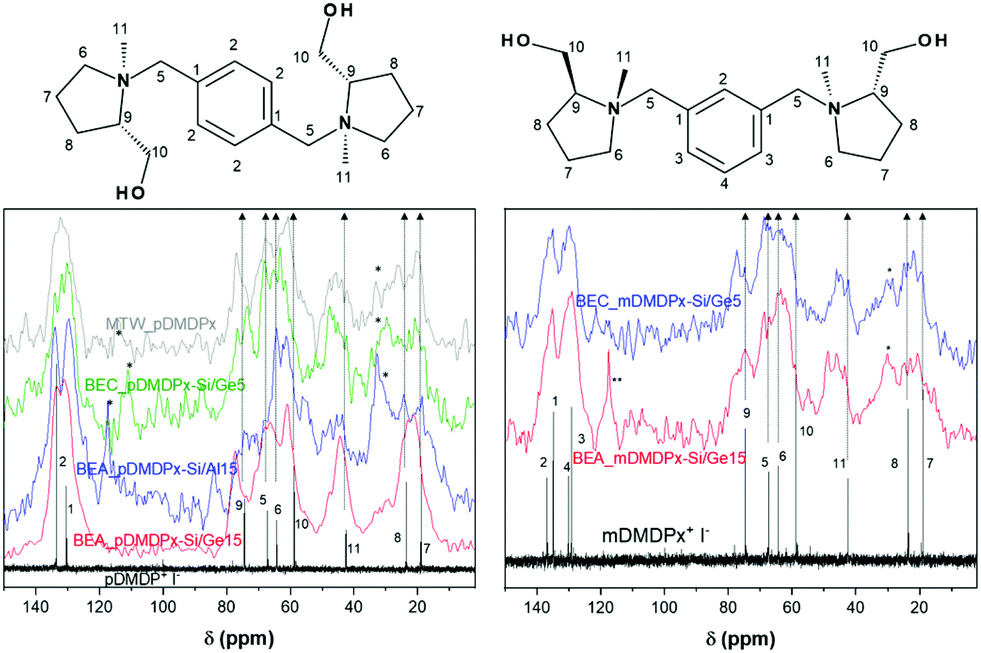 | ||
| Fig. 2 13C CP MAS NMR spectra of zeolite materials obtained with pDMDPx (left) and mDMDPx (right); the corresponding liquid 13C NMR spectra of the iodide salts in D2O are also shown for comparison (black lines). ‘*’ indicates minor decomposition products. | ||
The BET surface area and porosity of the sample of zeolite beta crystallized from gels with Si/Al = 15 by using pDMDPx were determined in order to compare with those of zeolite beta samples synthesized from conventional templates. The micropore volume was 0.17 cm3 g−1 and the total surface 621 m2 g−1, of which 408 m2 g−1 corresponds to micropores and 213 m2 g−1 to external surface area. These results are comparable to those of Al-containing beta zeolites prepared from a conventional template, namely tetraethylammonium hydroxide, taking into account the well-known observed trend for the micropore volume of zeolite beta and other zeolites to decrease as the external surface area increases, being the relative proportion of micropore-to-external surface a function of crystal size and specific synthesis parameters.54,55
3.5 Electron Microscopy characterization of the polymorphs
Although XRD results showed no evidence of enrichment in the chiral polymorph A, we wanted to study the occurrence of the different polymorphs (A, B or C) by Spherical aberration corrected (Cs-corrected) Scanning Transmission Electron Microscopy (STEM) using an annular dark field detector (ADF). As previously mentioned, polymorph A (PA) can crystallize in the P4122 and P4322 enantiomorphic space groups, depending on the handedness, while polymorph B (PB) belongs to the monoclinic C2/m. On the other hand, polymorph C is tetragonal with the space group: P42/mmc. To differentiate the three structures, observations need to be performed perpendicular to the largest pores, the 12 MRs, along ‘b’ axis. On this orientation, PA layers are stacked in a zig-zag manner, ABABA…; in PB they are shifted by 1/3 along the observation plane, obtaining a sequence ABCABC…, while for polymorph C there is no shift on the layer stacking sequence, being AAAA….Selected samples obtained with pDMDPx were studied by electron microscopy with the intention of obtaining some insights of the materials formed. In the case of Al-containing beta zeolite (obtained after 14 days at 150 °C with a Si/Al ratio of 15), Fig. 3a presents the high-resolution Cs-corrected STEM-ADF image of a typical crystallite, which already proves the excellent crystallinity of the sample and where the existence of the structural defects is evidenced as a consequence of the intergrowth of both polymorphs of zeolite beta, polymorphs A and B. The Electron diffraction (ED) pattern (inset) obtained from the same crystal exhibits well-defined spots together with diffuse lines along c* axis produced as a consequence of the mixture of both polymorphs. Such types of images allowed the calculation of the relationship of both polymorphs on different zeolite crystallites, obtaining a ratio for this Al-beta sample of 40% of polymorph A and 60% of B. The stacking sequence is marked showing the zig-zag pillaring of PA in yellow, while the regions of polymorph B are marked by straight red lines.
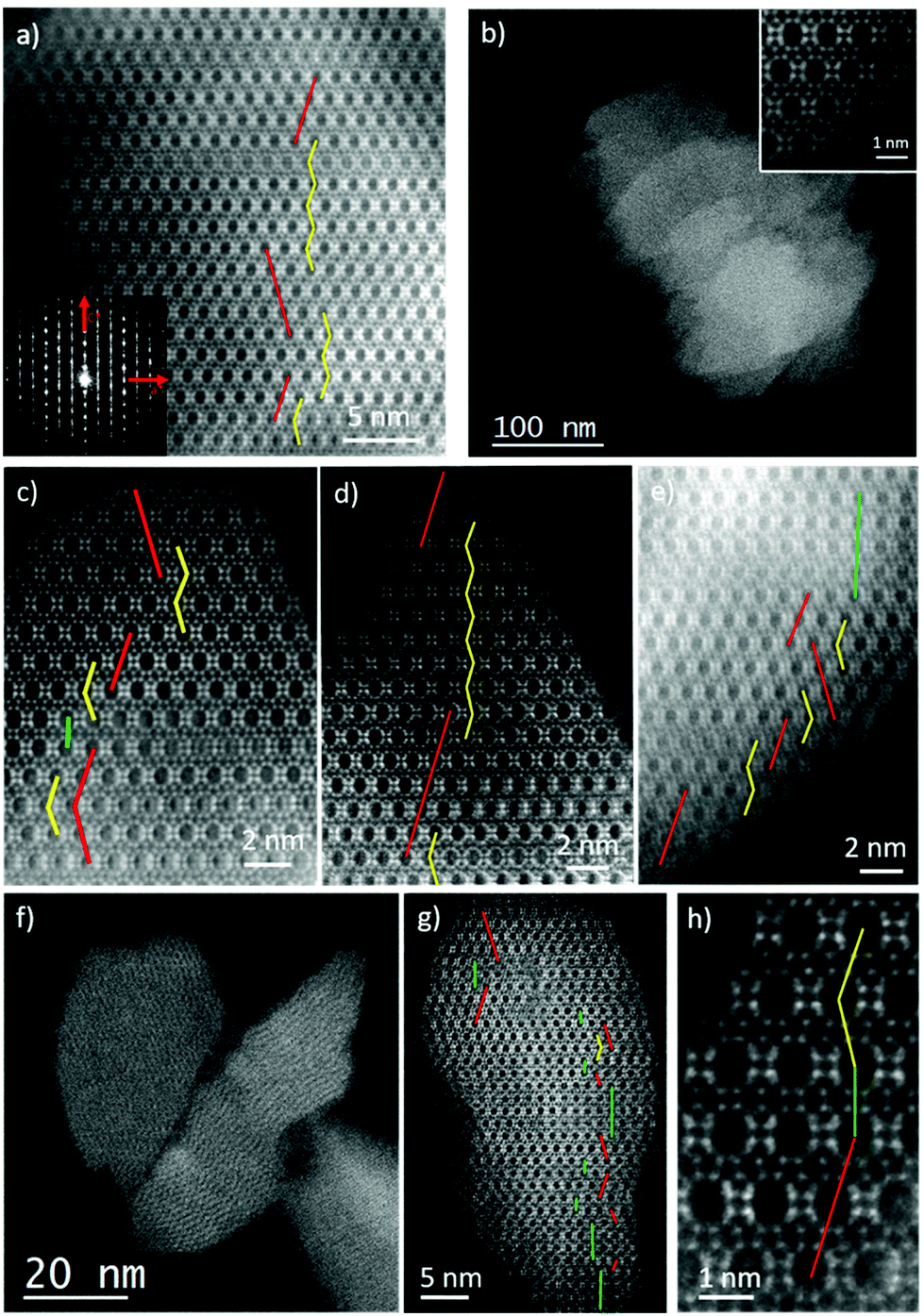 | ||
| Fig. 3 Cs-corrected STEM-ADF analysis of several zeolite beta materials obtained with pDMDPx. (a) High-magnification image of zeolite beta with Si/Al = 15. The ED is shown inset. (b) Low-magnification micrograph of several crystallites of zeolite beta with Si/Ge = 30 (atomic resolution observation is shown inset), and (c) close observation with the different polymorphs observed. (d) and (e) High-magnification images of the material with Si/Ge = 15. (f) Low-magnification data of the material obtained with Si/Ge = 5, (g) high resolution image of a zeolite nanocrystal with the identification of the polymorphs marked, and (h) atomic observation of the 3 polymorphs. | ||
In the case of Ge-containing materials obtained in the presence of pDMDPx, samples prepared with different Ge amounts (with 5 H2O molar ratio, Table 3) were analysed. For materials with a low Ge content (Si/Ge = 30), an excellent crystallinity was observed, displaying the typical morphology for zeolite beta, as shown in Fig. 3b. This image allows the observation of several zeolite crystallites with truncated octahedral shaped particles of few hundreds of nm (the atomic resolution data of the framework, again along ‘b’ axis, is shown in the inset). As for the previous case, the stacking sequence can be also followed if the data is sufficiently good to distinguish the different polymorphs (Fig. 3c). In this sample, a mixture of 50% of polymorph B, 49% of A and 1% of C was observed (Fig. 3c uses the same colour code to denote the polymorphs, including green lines for polymorph C).
Very similar results were also obtained when the amount of Ge was increased to a Si/Ge of 15 (Fig. 3d and e), obtaining a product with very good crystallinity where the predominant polymorphs were A and B (Fig. 3d). In this case, polymorph A was found to be 42% of the sample, while B was 54% with a slight increment of polymorph C (3%) (Fig. 3e). In conclusion, no evidence of an enrichment in polymorph A is observed in the Ge-containing beta materials, while minor amounts of C-stacking sequences are observed, evidencing that the slightly increased peaks at 7 and 9.7° observed previously in the XRD patterns (Fig. 1D) are due to minor amounts of polymorph C.
Increasing the amount of Ge to Si/Ge = 5 made a significant influence on the structure and morphology of the materials obtained. Fig. 3f shows the low magnification image of few particles representative of this material. The first difference in comparison with the previous zeolites is the particle size, which was much smaller, obtaining crystallites of sizes under 100 nm. The atomic resolution observation is depicted in Fig. 3g, corresponding to a zeolite nanoparticle of ∼50 nm with the different polymorphs marked. Interestingly, the increase of polymorph C is evident in this micrograph, reaching up to 18%, being 44% and 38% for A and B, respectively. A closer look of the occurrence of the three polymorphs is displayed in Fig. 3h, showing how the three polymorphs stack onto each other. From these observations, we conclude that Ge has a big influence on the final material obtained, where high Ge contents increases the amount of polymorph C while reducing the crystal size.
In terms of the relative crystallinity of the materials, no significant differences were observed for beta zeolites obtained with Si/Al = 15, Si/Ge = 30 or Si/Ge = 15, presenting in every case excellent crystallinities without the observation of amorphous phases. Despite no amorphous material was visualized for Si/Ge = 5 either, the crystallinity was not as good as for the others, although this aspect could be related to the small size of the particles obtained and maybe the entire structure was not completely formed along the particle. Nevertheless, most of the particles observed displayed a reasonable well-defined framework.
4. Computational results
4.1 Characterization of the pDMDPx diastereomeric configuration
First, we wanted to confirm the diastereomeric configuration of the organic pDMDPx cation obtained. As explained below, the order of addition of the alkylation agents did lead to a pure isomer (by first addition of xylene and second of methyl group) or to a mixture of isomers (first methyl and second xylene group). In order to verify the diastereomeric configuration of our pure isomer, we calculated by DFT the theoretical 13C chemical shifts of the (S,S,S,S) and (S,R,S,R) isomers (in their most stable conformation in aqueous solution, as explained below), and compared with the experimental results (Fig. S1†). The theoretical and experimental results are shown in Table 4; in the experimental data, ‘1’ corresponds to the isomer produced pure (whose shifts are obtained from Fig. S1-top†), and ‘2’ corresponds to the other one (whose bands are those in Fig. S1-bottom† that are not present in Fig. S1-top†). Theoretical NMR calculations show that C11, C6, C5 and C9 chemical shifts are those that differ mostly depending on the isomer, as expected since these are directly attached to N whose absolute configuration varies. Theoretical results show that change from (S) to (R) configuration of N involves a downfield shift of C11 and C9, and an upfield shift of C6 and C5. Interestingly, change from ‘1’ to ‘2’ isomer in the experimental data involves exactly the same type of shifts (in qualitative terms) of the bands associated to those C atoms (downfield in C11, C9, and upfield in C6, C5), clearly confirming that the pure pDMDPx isomer that we obtained had (S,S,S,S) configuration, as expected because of steric reasons (and the same for mDMDPx).| Isomer | C7 | C8 | C11 | C10 | C6 | C5 | C9 | C1 | C2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Theor. | SSSS | 21.4 | 25.4 | 38.7 | 68.3 | 70.3 | 76.2 | 84.9 | 138.0 | 140.0 |
| SRSR | 22.2 | 25.8 | 53.4 | 67.8 | 66.0 | 58.3 | 86.4 | 136.8 | 140.4 | |
| Exper. | 1 | 18.9 | 23.5 | 42.5 | 58.8 | 64.3 | 67.2 | 74.5 | 130.4 | 133.5 |
| 2 | 19.3 | 23.8 | 49.6 | 58.0 | 58.4 | 61.9 | 78.1 | 130.0 | 133.4 |
4.2 Characterization of the conformational space of pDMDPx and mDMDPx
Next, we characterized the conformational behaviour of the two SDAs, and their geometrical properties as a function of the aromatic substitutional position. The main torsional angle that defines the conformational space is C11–N–C5–C1, which defines three different conformations with angles of 60, 180 and −60° (for each ring). Besides, the two L-prolinol units can be at opposite or the same side of the aromatic ring; Fig. 4 shows the molecular shape of the different conformers with the two prolinol units in opposite sides. Different force-field/charge models were tried; we found that Dreiding and a DFT atomic charge distribution (ESP method) reproduced well the relative energies calculated at DFT level in vacuum (Table S1†), and hence we used this method for our simulations.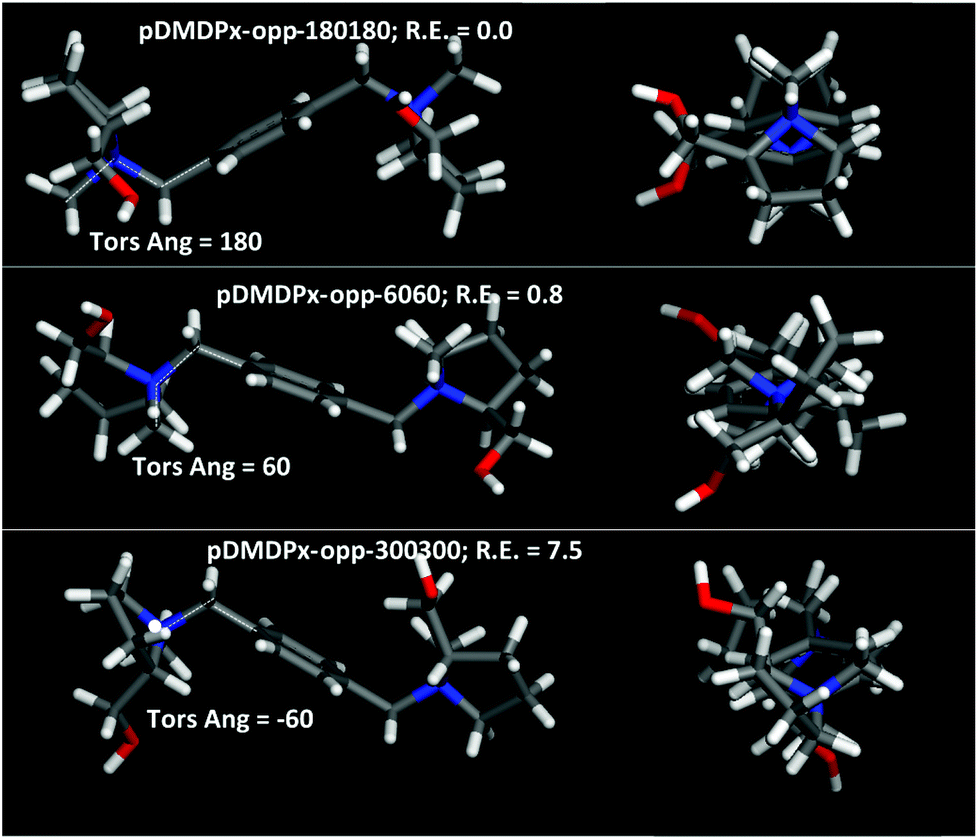 | ||
| Fig. 4 Molecular structure of the three conformers of pDMDPx with prolinol units in opposite sides; relative energies (calculated with Dreiding method) are also reported, as well as C11–N–C5–C1 torsion angle. | ||
Energy results for pDMDPx showed that the most stable conformation in vacuum was for a C11–N–C5–C1 torsional angle of ∼180°, where methyl C11 and phenyl ring are in anti configuration (labelled as pDMDPx-opp-180180), followed closely by conformers with torsional angles of ∼60°, where they are in syn configuration, having a relative energy of 0.8 kcal mol−1 (labelled as pDMDPx-opp-6060) (both cases with prolinol units in opposite sides) (Fig. 4-top and middle and Table S1†). Similar results were found for mDMDPx, with the same two most stable conformers with relative energies of 0.0 and 0.2 kcal mol−1, respectively (Fig. S3†).
Due to the similar stability of these two conformations (opp-180180 and opp-6060), we studied their relative occurrence in aqueous solution in order to analyse which one will be responsible for the structure-direction of zeolite materials. SDA cations were initially arranged in the most stable conformation in vacuum (opp-180180), and were then allowed to relax in the presence of water. Fig. S4† shows the torsional angle distribution at the beginning (0–0.5 ns time interval, left) and at the end of the simulation (7.5–10 ns, right) (top: pDMDPx; bottom: mDMDPx). We can clearly observe that both cations tend to shift from opp180180 towards opp-6060 conformations in aqueous solution, despite the former being slightly more stable in vacuum. These MD results suggest that the opp-6060 conformations will dominate in aqueous solution and, consequently, the molecular shape of these conformers will determine the porosity of the resulting zeolite materials.
We then analysed the geometrical properties of the two cations in their most stable conformations (opp-6060) by running 1 ns of MD NVT simulations (at 25 °C) in vacuum, and calculated the molecular shadow lengths during these MD simulations (Fig. S5†). Results showed that pDMDPx displays a roughly cylindrical shape, with two similar dimensions centred at 7.3 and 8.5 Å and a large molecular axis of 16.5 Å, while mDMDPx displays a more elliptical shape with dimensions of 7.3 and 9.6 Å, and a slightly shorter molecular main axis of 15.2 Å.
4.3 Structure-directing effect towards MTW
We then studied the incorporation of both cations within the one-dimensional channels of the MTW framework. First, we considered the incorporation of one SDA in 1 × 6 × 1 MTW supercells. The elliptical dimensions of the MTW channels forced the incorporation of pDMDPx in the opp-6060 conformation (opt-180180 did not fit within the MTW channels). Indeed, the geometric characteristics of pDMDPx with a cylindrical shape were suitable for being confined within the MTW channels, where three subsequent side-pockets of the channels were occupied by the three rings of pDMDPx, giving an interaction energy of −175.5 kcal mol−1 (Fig. 5-top). Similarly, energy results showed that the mDMDPx dication could be also allocated in a similar position, developing an even higher interaction energy of −204.7 kcal mol−1 (Fig. 5-middle). However, we know from experiments that this cation did not direct the formation of the MTW structure. This could be caused by a number of factors; one likely would be due to kinetic reasons since the geometric properties of the cation in vacuum (Fig. S5†), with a larger length of 9.6 Å, do not fit within the MTW channel dimensions, and consequently the SDA cation would have to alter its most stable conformation to be confined within MTW. Indeed, the conformational energy penalty of mDMDPx to display the conformation adopted within the MTW framework (calculated as the energy difference between the conformation in MTW and the most stable one in vacuum) is more than twice higher than that of pDMDPx, evidencing a kinetic barrier for mDMDPx to get occluded in the MTW channels, even despite developing a strong interaction energy with the framework once confined.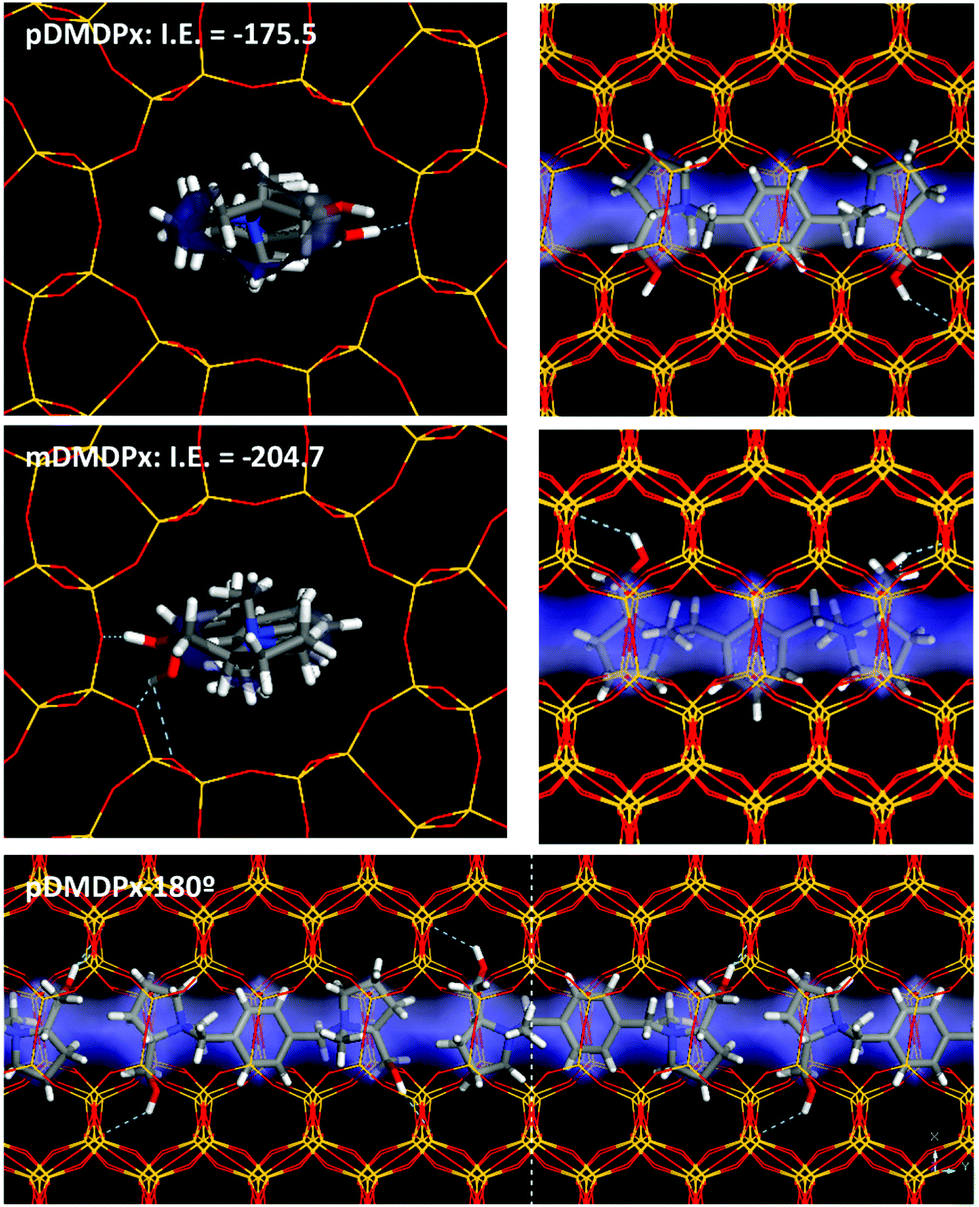 | ||
| Fig. 5 Location of pDMDPx (top) and mDMDPx (middle) within MTW framework, and interaction energy (1SDA per supercell); bottom: packing of pDMDPx in the MTW channels, with 180° rotation. Free volume of the MTW channels is displayed as a transparent blue surface. | ||
Finally, packing of pDMDPx cations along the MTW channels can take place with adjacent dications in the same orientation, or rotated consecutively by 180° (Fig. S6†). Energy results showed a higher stability for the latter case by 3.1 kcal mol−1 SDA because of a better packing of the pyrrolidinium rings (Fig. 5-bottom), giving a packing value of 1.33 SDAs per u.c.
4.4 Structure-directing effect towards BEA
Next, we analysed the host–guest chemistry of these chiral cations within zeolite beta. Polymorphs A and B of zeolite beta differ in the stacking sequence of bea sheets along the ‘c’ axis, with an invariable rotation of 90° (or −90°) in polymorph A, and an alternated rotation of + and −90° in polymorph B. Therefore, if a preferred rotation (+ or −90°) in the presence of these cations is met, this should favour the corresponding polymorph A (in P4122 or P4322 space group), while if no preference is found, a mixture of polymorph A (in the two space groups) and B should be observed. Hence, in an attempt to understand the potential chiral influence of the SDAs into the crystallization of the chiral polymorph A, we calculated the interaction energy of these SDAs in the two enantiomorphic groups of polymorph A.We started by loading one SDA in 4 × 1 × 1 BEA supercells, i.e. neglecting packing interactions. After an extensive search by simulated annealing calculations, we found one particularly stable location for pDMDPx along the BEA channels of both enantiomorphic polymorphs (Fig. 6); pDMDPx dications site aligned with the [100] (or the equivalent [010]) channels, with the bulkier prolinol units spanning consecutive channel intersections, displaying the opp-6060 conformation. Despite enantiopurity of pDMDPx, we found a very similar stability of the SDA in the two chiral enantiomorphic polymorphs, with interaction energies of −163.85 and −164.00 kcal mol−1 in P4122 and P4322 frameworks, respectively; such energy difference was very similar when energies were averaged from MD simulations (<0.3 kcal mol−1). This lack of chiral recognition might be associated to the chirality of BEA being imposed by the stacking sequence of bea sheets along the ‘c’ axis, while pDMDPx SDAs tend to site aligned with the [100] and [010] channels, thus not expressing its chirality in the appropriate orientation for the BEA framework to feel it. Therefore, these SDAs should not impose any particular stacking sequence with +90 or −90° rotation of sheets along ‘c’, and therefore a random sequence would be expected, resulting in the crystallization of zeolite beta with the typical mixture of polymorphs A (in the two space groups) and B. This is in line with our experimental observations where no particular enrichment of this zeolite beta was observed by XRD or STEM.
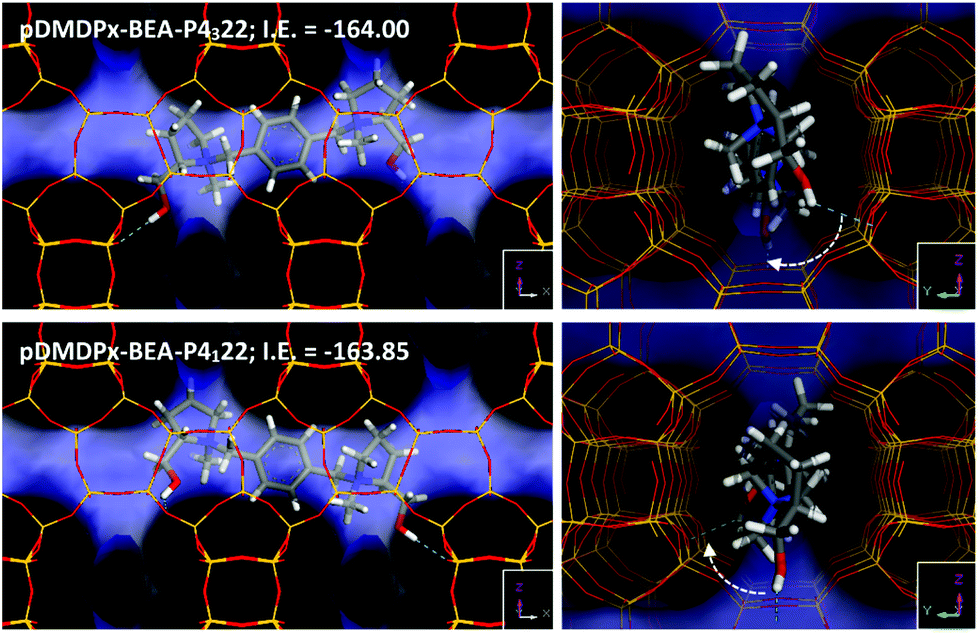 | ||
| Fig. 6 Location of pDMDPx in chiral BEA polymorphs P4322 (top) and P4122 (bottom); free volume of BEA channels is displayed as the blue surface. Right views show the asymmetric orientation of the organic hydroxyl groups with angles around 45° consecutively rotated in clockwise direction. | ||
However, worth is noting that the location of hydroxyl groups in the dication follows always (in the two enantiomorphic frameworks of BEA) an asymmetric orientation where they rotate ∼45° in clockwise direction, regardless of the polymorph (Fig. 6-right); this particular orientation is imposed by the handedness of (S,S,S,S)-pDMDPx. If one assumes that the location of Al (which generates a negative framework charge) could be at some extent directed by these polar hydroxyl groups through the formation of H-bonds, an asymmetric orientation of these active sites could be expected, what could potentially lead to asymmetric catalysts. At present, this is just a mere hypothesis, but work is in progress in order to study this issue.
We next studied the packing of pDMDPx along the [100] (or [010]) channels in an attempt to analyse if a potential asymmetric packing arrangement could lead to an enantio-differentiation of polymorphs. Imposing the condition of maximum load of organic cations while preventing too short contacts, we estimated that 3 SDAs could be loaded in 4 u.c. along each channel. Such packing arrangement gives a total SDA loading of 3 SDAs per u.c. (there are 4 channels per u.c.), corresponding to an organic content of 21%, which is in line with the values obtained by TGA in the different zeolite beta materials. In this fully-loaded systems, we found interaction energies of −190.91 and −190.98 kcal mol−1 of SDA for pDMDPx in P4122 and P4322 frameworks, respectively, being extremely similar, as occurred in the absence of packing interactions; Fig. 7 (top) shows the final arrangement of pDMDPx cations in P4322 framework. These energy results evidence that the packing of pDMDPx cations in the BEA channels does not impose any asymmetric arrangement. Indeed, RDFs (during MD simulations) between H atoms of the SDAs and framework O atoms (Fig. 7-bottom) show a very similar profile for the two diastereomeric pairs, evidencing that the host–guest fitting in the fully-loaded systems is very similar for (S,S,S,S)-pDMDPx in the two BEA chiral frameworks. Again, this would explain the non-prevalence of a particular stacking sequence of bea sheets in the presence of this cation. The lack of such host–guest chiral recognition might be associated to the looseness of the host–guest fit, given the high porosity and large size of BEA channels and the limited geometric fit with the molecular shape of pDMDPx, as indicated by the large distance between framework O and pDMDPx H atoms (higher than 3 Å, Fig. 7-bottom).
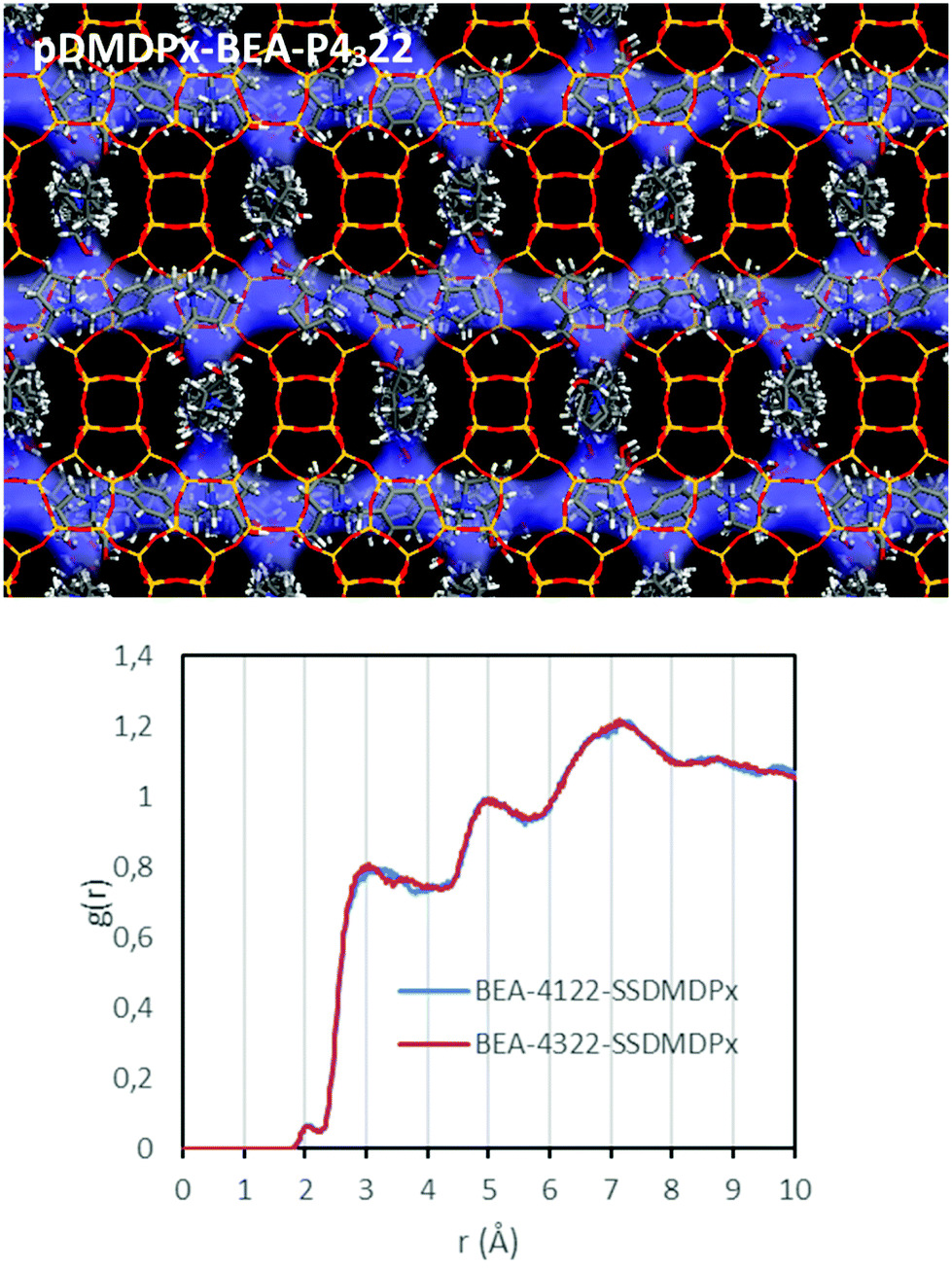 | ||
| Fig. 7 Top: most stable location of pDMDPx in chiral polymorph A with P4322 space group under a packing of 3 SDAs per u.c. Bottom: RDF between H atoms of pDMDPx and framework O atoms of BEA during MD simulations in P4122 (blue) and P4322 frameworks (red). | ||
Despite having 4 enantiomerically-pure stereogenic centres, (S,S,S,S)-pDMDPx does not display enantioselective discrimination properties between the two enantiomorphic BEA chiral frameworks. This might be associated to the small size of the substituents attached to the stereogenic centres (C and N, with methyl and hydroxymethyl groups, respectively), which promotes small asymmetric shapes that are not sufficient to be imprinted on the stacking of bea sheets due to the loose host–guest fit. For this reason, we studied the effect of having larger substituents attached to N; in particular, we analysed the interaction energy difference in the two frameworks of a related cation but where the methyl substituents attached to N were replaced by ethyl groups (pDEDPx). In this case, the larger size of ethyl groups involved that these pointed to the perpendicular adjacent channels along the ‘c’ axis (which determines the stacking sequence) (Fig. S7†). As a consequence, the interaction energy difference of (S,S,S,S)-pDEDPx dications in P4122 and P4322 frameworks notably increased from 0.15 (for pDMDPx) to 1.12 kcal mol−1. Although this evidences the validity of this argument, the energy difference between the two chiral polymorphs is still not very high. In any case, the arrangement of these SDAs with ethyl groups partially filling the perpendicular adjacent channels (Fig. S7†) could potentially disrupt an efficient loading of those channels, what might prevent an effective crystallization of zeolite beta with this SDA. Anyhow, unfortunately attempts to experimentally synthesize this cation failed because of the higher steric hindrance of the ethyl substituent.
We finally studied the incorporation of mDMDPx in the two BEA enantiomorphic frameworks. In this case, two different stable orientations of the SDAs were found. In the most stable orientation, mDMDPx cations locate in the intersections spanning perpendicular [100] and [010] channels, roughly aligned with the ‘c’ axis, which is a consequence of the angular shape of this cation (compared with the linear shape of pDMDPx) (Fig. 8-top). In principle, this orientation would be interesting since the stacking sequence of bea sheets along ‘c’ determines the chirality of the resulting polymorph. However, again we observed a negligible difference in the interaction energy of mDMDPx with the two chiral frameworks P4122 and P4322, being −213.42 and −213.27 kcal mol−1, respectively (giving an enantio-differentiation energy of 0.15 kcal mol−1). Once again, this might be a consequence of the small size of the asymmetric methyl and hydroxymethyl substituents. Nevertheless, in this case we also observed another stable orientation where mDMDPx located aligned with the [100] (or [010]) channels (Fig. 8-bottom), giving interaction energies of −207.31 and −208.20 kcal mol−1 for P4122 and P4322, respectively; note that the energy difference in this case is slightly higher (0.89 kcal mol−1), though still low. Nevertheless, we note that the conformational energy penalty for this orientation of mDMDPx aligned with BEA channels is higher than in the previous orientation. In any case, once again we did not find any evidence for an enrichment of polymorph A in this zeolite. Replacement of methyl by ethyl substituents did result in an increase of the enantio-discrimination energy (to 1.51 kcal mol−1) (Fig. S8†), although in this case a very low interaction energy was found (−80.72 and −79.21 kcal mol−1 for P4122 and P4322, respectively), evidencing a poor fit of this cation in BEA.
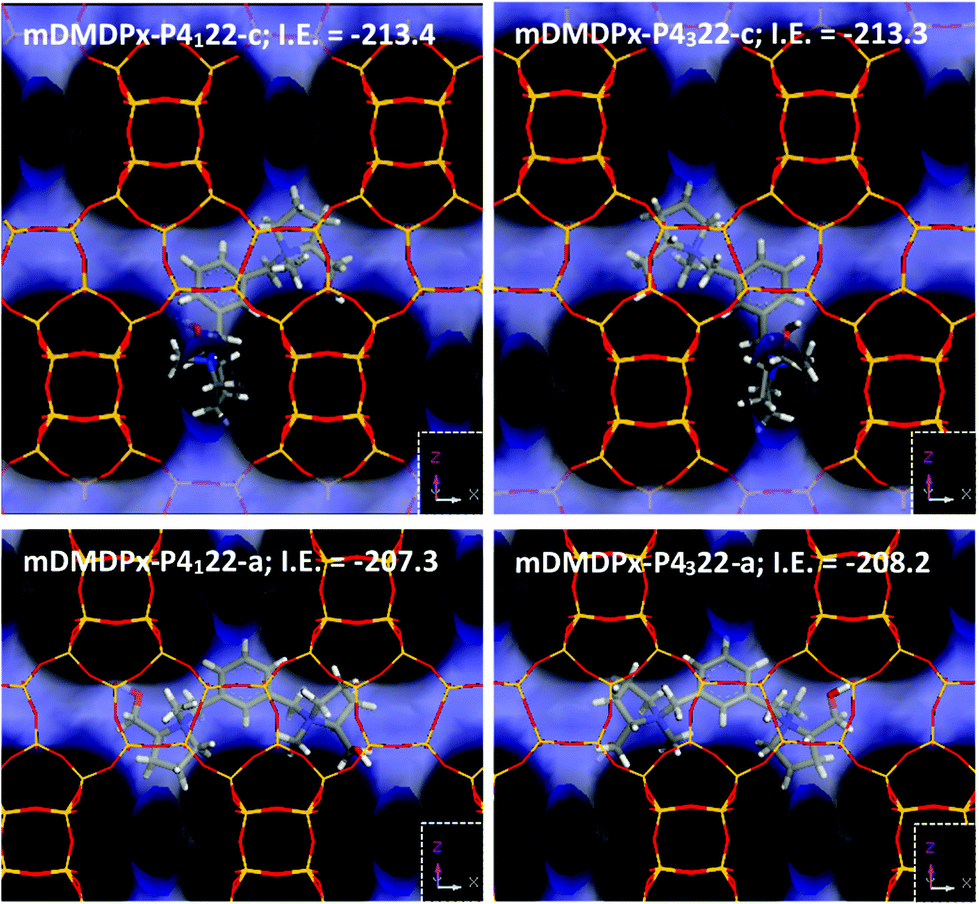 | ||
| Fig. 8 Location of mDMDPx in chiral BEA polymorph A with P4122 (left) and P4322 (right) space groups in the two possible orientations, along ‘c’ (top) or aligned with ‘a’ (or ‘b’) channels (bottom). | ||
We note that the crystallization of zeolite materials in the presence of mDMDPx invariably required the presence of Ge. Therefore, it seems that the main structure-directing agent in this system was provided by Ge rather than by mDMDPx, which might act more as a space-filling agent. The same as for the MTW framework, the low structure-directing efficiency of mDMDPx might be associated to the angular shape of this cation, which shifts away from the cylindrical shape typical of pore-based zeolites.
For the sake of completeness, we also studied the most stable location of these cations in polymorph C (BEC) (Fig. S9†). Similar orientations of the SDAs were observed, with pDMDPx aligned with the [100] (or [010]) channels, and mDMDPx spanning the two types of channels along ‘c’ axis. On the other hand, the interaction energies of each SDA were also very similar as those for BEA.
5. Conclusions
In this work, we have performed a thorough study about the structure-directing effect of two chiral SDAs based on two L-prolinol quaternary ammonium units linked by a xylene ring in para- or meta-position. The synthesis protocol of the chiral SDAs ensures the production of pure enantiomers with a unique diastereomeric configuration, with the four resulting stereogenic centres in (S) absolute configuration. Our synthesis results indicate that the para-SDA is much more efficient in directing the crystallization of large-pore zeolites, being able to promote the formation of MTW in pure-silica composition, zeolite beta in the presence of Al, and mixtures of polymorphs A, B and C of zeolite beta in the presence of Ge. In contrast, the meta-derived SDA is a very poor SDA, only allowing the production of crystalline zeolite materials in the presence of Ge, probably acting more as a pore-filling agent than a true structure-director, and being Ge the actual structure-directing agent. A conformational study of the two SDAs suggests that the worse structure-directing efficiency of the meta-derivative is a consequence of its angular shape that fits worse in the tubular channels of zeolite frameworks; in contrast, the para-derivative displays a more cylindrical shape suitable to be confined in zeolite pores.Because of the presence of the chiral polymorph A in zeolite beta, we analysed the stability of our enantiomerically-pure para-xylene SDA in the two enantiomorphic space groups, yielding a similar interaction energy in both cases. This might be associated to the large size of the BEA zeolite channels and the loose fit of the SDA, which prevents an effective transfer of the asymmetric nature of the chiral SDA to the stacking of beta sheets along the ‘c’ direction that determines the BEA chirality. As a consequence, zeolite beta obtained in this work seems to have the usual proportion of A and B polymorphs. Nevertheless, our results suggest that larger substituents associated to the stereogenic centres should result in larger energy differences between the chiral polymorphs, providing clues about the future design of new chiral SDAs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been partially financed by the Spanish State Research Agency (Agencia Española de Investigación, AEI) and the European Regional Development Fund (Fondo Europeo de Desarrollo Regional, FEDER) through the Projects MAT2016-77496-R (AEI/FEDER, UE) and PID2019-107968RB-I00, and by CSIC through the Projects 201780I043 and 2019AEP074. Secretaría General Adjunta de Informática-CSIC is acknowledged for running the calculations, and BIOVIA for providing the computational software. AM acknowledges the Spanish Ministry of Science through the Ramon y Cajal program (RYC2018-024561-I), to the Natural National Science Foundation of China (NFSC-21850410448, NFSC-21835002) and to The Centre for High-resolution Electron Microscopy (CħEM), supported by SPST of ShanghaiTech University under contract No. EM02161943. C. Márquez-Álvarez is acknowledged for his help with the N2 isotherm data. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- J. Cejka, A. Corma and S. I. Zones, Zeolites and catalysis: synthesis reactions and applications, Wiley-VCH Verlag GmbH & Co. KGaA, 2010 Search PubMed.
- M. E. Davis, Ordered porous materials for emerging applications, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- E. T. C. Vogt and B. M. Weckhuysen, Fluid catalytic cracking: recent developments on the grand old lady of zeolite catalysis, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC.
- S. Abate, K. Barbera, G. Centi, P. Lanzafame and S. Perathoner, Disruptive catalysis by zeolites, Catal. Sci. Technol., 2016, 6, 2485–2250 RSC.
- B. Smit and T. L. M. Maesen, Towards a molecular understanding of shape selectivity, Nature, 2008, 451, 671–678 CrossRef CAS PubMed.
- A. W. Burton and S. I. Zones, Organic molecules in zeolite synthesis: their preparation and structure-directing effects, Stud. Surf. Sci. Catal., 2007, 168, 137–179 CrossRef CAS.
- Y. Kubota, M. M. Helmkamp, S. I. Zones and M. E. Davis, Properties of organic cations that lead to the structure-direction of high-silica molecular sieves, Microporous Mater., 1996, 6, 213–229 CrossRef CAS.
- M. Moliner, F. Rey and A. Corma, Towards the rational design of efficient organic structure-directing agents for zeolite synthesis, Angew. Chem., Int. Ed., 2013, 52, 13880–13889 CrossRef CAS PubMed.
- C. M. Lew, T. M. Davis and S. Elomari, Synthesis of new molecular sieves using novel structure-directing agents (Chapter 2), in Verified syntheses of zeolitic materials, ed. S. Mintova, Synthesis Commission of the International Zeolite Association, 3rd revised edn, 2016, pp. 29–35. ISBN: 978-0-692-68539-6 Search PubMed.
- L. Gómez-Hortigüela and M. A. Camblor, Introduction to the Zeolite Structure-Directing Phenomenon by Organic Species: General Aspects, in Insights into the Chemistry of Organic Structure-Directing Agents in the Synthesis of Zeolitic Materials, ed. L. Gómez-Hortigüela, Structure and Bonding, Springer, Cham, 2017, vol. 175 Search PubMed.
- M. E. Davis, Reflections on Routes to Enantioselective Solid Catalysts, Top. Catal., 2003, 25, 3–7 CrossRef CAS.
- J. Yu and R. Xu, Chiral zeolitic materials: structural insights and synthetic challenges, J. Mater. Chem., 2008, 18, 4021–4030 RSC.
- M. E. Davis, A thirty-year journey to the creation of the first enaniomerically enriched molecular sieve, ACS Catal., 2018, 8, 10082–10088 CrossRef CAS.
- T. S. van Erp, T. P. Caremans, D. Dubbeldam, A. Martín-Calvo, S. Calero and J. A. Martens, Enantioselective adsorption in achiral zeolites, Angew. Chem., Int. Ed., 2010, 49, 3010–3013 CrossRef CAS PubMed.
- J. M. Castillo, T. J. H. Vlugt, D. Dubbeldam, S. Hamad and S. Calero, Performance of Chiral Zeolites for Enantiomeric Separation Revealed by Molecular Simulation, J. Phys. Chem. C, 2010, 114, 22207–22213 CrossRef CAS.
- R. E. Morris and X. H. Bu, Induction of chiral porous solids containing only achiral building blocks, Nat. Chem., 2010, 2, 353–361 CrossRef CAS PubMed.
- D. Dubbledam, S. Calero and T. J. H. Vlugt, Exploring new methods and materials for enantioselective separations and catalysis, Mol. Simul., 2014, 40, 585–598 CrossRef.
- K. D. M. Harris and S. J. M. Thomas, Selected Thoughts on Chiral Crystals, Chiral Surfaces, and Asymmetric Heterogeneous Catalysis, ChemCatChem, 2009, 1, 223–231 CrossRef CAS.
- M. M. J. Treacy and J. M. Newsam, 2 New 3-dimensional 12-ring zeolite frameworks of which zeolite beta is a disordered intergrowth, Nature, 1988, 332, 249–251 CrossRef CAS.
- N. Rajic, N. Z. Logar and V. Kaucic, A novel open framework zincophosphate - synthesis and characterization, Zeolites, 1995, 15, 672–678 CrossRef CAS.
- J. Sun, C. Bonneau, A. Cantín, A. Corma, M. J. Díaz-Cabañas, M. Moliner, D. Zhang, M. Li and X. Zou, The ITQ-37 mesoporous chiral zeolite, Nature, 2009, 458, 1154–1157 CrossRef CAS PubMed.
- A. Rojas and M. A. Camblor, A Pure Silica Chiral Polymorph with Helical Pores, Angew. Chem., Int. Ed., 2012, 51, 3854–3856 CrossRef CAS PubMed.
- L. Q. Tang, L. Shi, C. Bonneau, J. L. Sun, H. J. Yue, A. Ojuva, B. L. Lee, M. Kritikos, R. G. Bell, Z. Bacsik, J. Mink and X. D. Zou, A zeolite family with chiral and achiral structures built from the same building layer, Nat. Mater., 2008, 7, 381–385 CrossRef CAS PubMed.
- A. Rojas, O. Arteaga, B. Kahr and M. A. Camblor, Synthesis, structure, and optical activity of HPM-1, a pure silica chiral zeolite, J. Am. Chem. Soc., 2013, 135, 11975–11984 CrossRef CAS PubMed.
- L. Gómez-Hortigüela and B. Bernardo-Maestro, Chiral Organic Structure-Directing Agents, in Insights into the Chemistry of Organic Structure-Directing Agents in the Synthesis of Zeolitic Materials, ed. L. Gómez-Hortigüela, Structure and Bonding, Springer, Cham, 2017, vol. 175 Search PubMed.
- S. K. Brand, J. E. Schmidt, M. W. Deem, F. Daeyaert, Y. Ma, O. Terasaki, M. Orazov and M. E. Davis, Enantiomerically enriched, polycrystalline molecular sieves, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5101–5106 CrossRef CAS PubMed.
- B. Bernardo-Maestro, F. López-Arbeloa, J. Pérez-Pariente and L. Gómez-Hortigüela, Comparison of the Structure-Directing Effect of Ephedrine and Pseudoephedrine during Crystallization of Nanoporous Aluminophosphates, Microporous Mesoporous Mater., 2017, 254, 211–224 CrossRef CAS.
- L. Gómez-Hortigüela, T. Álvaro-Muñoz, B. Bernardo-Maestro and J. Pérez-Pariente, Towards chiral distributions of dopants in microporous frameworks: helicoidal supramolecular arrangement of (1R,2S)-ephedrine and transfer of chirality, Phys. Chem. Chem. Phys., 2015, 17, 348–357 RSC.
- B. Bernardo-Maestro, P. Gálvez, D. González, F. López-Arbeloa, J. Pérez-Pariente and L. Gómez-Hortigüela, Conformational Space of (1R,2S)-Dimethyl-Ephedrinium and (1S,2S)-Dimethyl-Pseudoephedrinium in the Synthesis of Nanoporous Aluminophosphates, J. Phys. Chem. C, 2018, 122, 20377–20390 CrossRef CAS.
- P. Gálvez, B. Bernardo-Maestro, E. Vos, I. Díaz, F. López-Arbeloa, J. Pérez-Pariente and L. Gómez-Hortigüela, ICP-2: A New Hybrid Organo-Inorganic Ferrierite Precursor with Expanded Layers Stabilized by π–π Stacking Interactions, J. Phys. Chem. C, 2017, 121, 24114–24127 CrossRef.
- B. Bernardo-Maestro, F. López-Arbeloa, J. Pérez-Pariente and L. Gómez-Hortigüela, Supramolecular chemistry controlled by conformational space during structure-direction of nanoporous materials: self-assembly of ephedrine and pseudoephedrine, J. Phys. Chem. C, 2015, 119, 28214–28225 CrossRef CAS.
- D. Nieto, J. Pérez-Pariente, E. Toran, F. López-Arbeloa and L. Gómez-Hortigüela, Conformational Sieving Effect of Organic Structure-Directing Agents during the Synthesis of Zeolitic Materials, Microporous Mesoporous Mater., 2019, 287, 56–64 CrossRef CAS.
- R. García, L. Gómez-Hortigüela, F. Sánchez and J. Pérez-Pariente, Diasteroselective structure directing effect of (1S,2S)-2-Hydroxymethyl-1-benzyl-1-methylpyrrolidinium in the synthesis of ZSM-12, Chem. Mater., 2010, 22, 2276–2286 CrossRef.
- L. Gómez-Hortigüela, F. Corà, C. R. A. Catlow and J. Pérez-Pariente, Computational study of a chiral supramolecular arrangement of organic structure directing molecules for the AFI structure, Phys. Chem. Chem. Phys., 2006, 8, 486–493 RSC.
- L. Gómez-Hortigüela, S. Hamad, F. López-Arbeloa, A. B. Pinar, J. Pérez-Pariente and F. Corà, Molecular Insights into the Self-Aggregation of Aromatic Molecules in the Synthesis of Nanoporous Aluminophosphates: A Multilevel Approach, J. Am. Chem. Soc., 2009, 131, 16509–16524 CrossRef PubMed.
- R. García, L. Gómez-Hortigüela, F. Sánchez and J. Pérez-Pariente, Structure-direction of chiral 2-hydroxymethyl-1-benzyl-1-methylpyrrolidinium in the cotemplated synthesis of ferrierite: fundaments of diastereo-recognition from non-chiral microporous structures, Microporous Mesoporous Mater., 2011, 146, 57–68 CrossRef.
- A. B. Pinar, L. Gómez-Hortigüela, L. B. McCusker and J. Pérez-Pariente, Synthesis of Zn−containing microporous aluminophosphate with the STA−1 structure, Dalton Trans., 2011, 40, 8125–8131 RSC.
- R. García, L. Gómez-Hortigüela and J. Pérez-Pariente, Study of the structure directing effect of the chiral cation (1S, 2S)-2-hydroxymethyl-1-benzyl-1-methylpyrrolidinium in aluminosilicate preparations in the presence of co-structure directing agents, Catal. Today, 2012, 179, 16–26 CrossRef.
- R. Martínez-Franco, C. Paris, J. Martínez-Triguero, M. Moliner and A. Corma, Direct synthesis of the aluminosilicate form of the small pore CDO zeolite with novel OSDAs and the expanded polymorphs, Microporous Mesoporous Mater., 2017, 246, 147–157 CrossRef.
- M. J. Frisch, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, DREIDING: a generic force field for molecular simulations, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- C. J. Pickard and F. Mauri, All-electron magnetic response with pseudopotentials: NMR chemical shifts, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- P. Lu, L. Gómez-Hortigüela, L. Xu and M. A. Camblor, Synthesis of STW zeolite using imidazolium-based dications of varying length, J. Mater. Chem. A, 2018, 6, 1485–1495 RSC.
- L. A. Villaescusa and M. A. Camblor, The fluoride route to new zeolites, Recent Res. Dev. Chem., 2003, 1, 93–141 CAS.
- F. Taborda, T. Willhammar, Z. Wang, C. Montes and X. Zou, Synthesis and characterization of pure silica zeolite beta obtained by an aging-drying method, Microporous Mesoporous Mater., 2011, 143, 196–205 CrossRef CAS.
- M. Tong, D. Zhang, W. Fan, J. Xu, L. Zhu, W. Guo, W. Yan, J. Yu, S. Qiu, J. Wang, F. Deng and R. Xu, Synthesis of chiral polymorph A-enriched zeolite beta with an extremely concentrated fluoride route, Sci. Rep., 2015, 5, 11521 CrossRef PubMed.
- T. Lu, R. Xu and W. Yan, Co-templated synthesis of polymorph A-enriched zeolite beta, Microporous Mesoporous Mater., 2016, 226, 19–24 CrossRef CAS.
- T. Lu, L. Zhu, X. Wang, W. Yan, W. Shi and R. Xu, A green route for the crystallization of a chiral polymorph A-enriched zeolite beta, Inorg. Chem. Front., 2018, 5, 802–805 RSC.
- M. Tong, D. Zhang, L. Zhu, J. Xu, F. Deng, R. Xu and W. Yan, An elaborate structure investigation of the chiral polymorph A-enriched zeolite beta, CrystEngComm, 2016, 18, 1782–1789 RSC.
- T. Lu, L. Zhu, X. Wang, W. Yan, W. Shi and R. Xu, Identification of the key factor promoting the enrichment of chiral polymorph A in zeolite beta and the synthesis of chiral polymorph A highly enriched zeolite beta, Inorg. Chem. Front., 2018, 5, 1640–1645 RSC.
- T. Lu, W. Yan and R. Xu, Chiral zeolite beta: Structure, synthesis, and application, Inorg. Chem. Front., 2019, 6, 2209–2210 RSC.
- F. Daeyaert and M. W. Deem, Design of organic structure directing agents for polymorph A zeolite beta, J. Mater. Chem. A, 2019, 7, 9854–9866 RSC.
- M. A. Camblor, A. Corma and S. Valencia, Characterization of nanocrystalline zeolite Beta, Microporous Mesoporous Mater., 1998, 25, 59–74 CrossRef CAS.
- N. Suárez, J. Pérez-Pariente, F. Mondragón and A. Moreno, Generation of hierarchical porosity in beta zeolite by post-synthesis treatment with the cetyltrimethylammonium cationic surfactant under alkaline conditions, Microporous Mesoporous Mater., 2019, 280, 144–150 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1, Additional liquid 13C NMR, Fig. S2, Thermogravimetric analyses, Table S1, Relative energies, Fig. S3, Additional pictures of molecular structure, Fig. S4, Torsion angle distributions, Fig. S5, Geometric properties of SDAs, Fig. S6, Packing of SDAs in MTW, Fig. S7 and S8, Location of alternative SDAs, Fig. S9, Location of SDAs in BEC. See DOI: 10.1039/d0dt01834a |
| This journal is © The Royal Society of Chemistry 2020 |