
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle crystal growth of water-soluble metal complexes with the help of the nano-crystallization method†
Ricardo
Alvarez
,
Philipp P.
Nievergelt
 ,
Ekaterina
Slyshkina
,
Peter
Müller
,
Roger
Alberto
and
Bernhard
Spingler
*
,
Ekaterina
Slyshkina
,
Peter
Müller
,
Roger
Alberto
and
Bernhard
Spingler
*
Department of Chemistry, University of Zurich, 8057 Zurich, Switzerland. E-mail: spingler@chem.uzh.ch
First published on 24th June 2020
Abstract
The Nano-Crystallization method has been extensively tested for the growth of single crystals of cationic coordination compounds, which are soluble in water. All three studied, diverse metal complexes could be crystallized with the help of pipetting robots commonly used for the crystallization of proteins and the anion small molecule screen. It was furthermore possible to obtain for each positively charged complex several structures with different anions. In one case, together with literature data, a total of six salts with different anions could be assembled, which allowed an investigation of the influence of the counterions on the inter-cation metal-to-metal distance. The Nano-Crystallization method can be recommended for the single crystal growth of cationic coordination complexes, which are stable in water and have an aqueous solubility of at least 2 mM. This is the first publication dedicated solely to the single crystal growth of coordination complexes.
1. Introduction
In most cases, setting up the crystallization of a small molecule is a manual process. If done intentionally, a solution of the compound is prepared and then the conditions are changed in order to reach supersaturation, which is hopefully followed by crystallization.1 While there are dedicated publications that deal exclusively with the crystallization of organic compounds,2 there are no publications to the best of our knowledge that are solely dedicated to the growth of single crystals of coordination metal complexes (see however the following references for the (single) crystal growth of MOFs from aqueous solutions,3 for the high-throughput synthesis of crystalline MOF materials4 and for the measurement of microcrystals of reactive coordination complexes5). This is really surprising, as 57% of the entries in the Cambridge Structure Database6 contain a metal ion. We have recently developed a crystallization screen consisting of 77 different anions and have successfully applied the screen for the crystallization of six out of seven very diverse organic cations, which were all water soluble to at least a concentration of 2 mM.7 The 96 different solutions of the screen are either mixed with the help of a pipetting robot7a or just with a cheap multi-channel pipette under oil.7b Vapor diffusion of water from the crystallization drops either in the bigger reservoir7a or through the oil into the air,7b induces the growth of the crystals. The first coordination complex crystallized by this technique has just been reported.8With coordination metal complex cations, the counteranion has an influence upon various structural aspects such as dimensionality, topology and nuclearity, and other parameters dependent on the structure, such as the magnetic behavior of cobalt(II),9 copper(II),10 zinc(II),11 silver(I),12 cadmium(II),13 mercury(II)14 and dysprosium(III)15 coordination complexes. It is a common feature of these studies that they are limited to simple anions, whose metal salts are already commercially available and which serve as starting materials for the complex formation. Bugris et al. described the calcium(II) complexes of two similar polyhydroxy carboxylates, gluconate and isosaccharinate.16 These two structures display very different shortest metal to metal distances of 3.7312(2) Å and 5.734(1) Å respectively, because in the first case a μ-oxo bridge formed by the carboxylate was present but not in the latter.
In the course of this study, we wanted to evaluate systematically the crystallization behavior of selected cationic coordination complexes using our newly developed Nano-Crystallization technique.7a For this purpose, we chose the racemic chloride salts of cobalt(III) tris(bipyridine) (1)17 and ruthenium(II) tris(4,4′-dimethyl-2,2′-bipyridine) (2)18 as well as cobalt(II)(6,6′′-(ethene-1,1-diyl)di-2,2′-bipyridine)(bromide)2 (3) as test candidates (Fig. 1). While complex 3 was expected to allow direct coordination of some exchanged anions to the metal center, this direct coordination mode was assumed to be unlikely in the case of the coordinatively saturated metal centers in 1 and 2. Cobalt(III) tris(bipyridine) and ruthenium(II) tris(bipyridine) complexes are important in many light-to-energy applications like dye-sensitized solar cells (DSSC).19 DSSCs use a wide band-gap semiconductor, which is sensitized to visible light by an adsorbed dye molecule and an electrolyte containing a redox mediator.20 Cobalt(III) tris(2,2′-bipyridine) complexes are used as high-performing dye-regenerating mediators in DSSC. Compared to iodide, the traditional mediator used in DSSC, cobalt complexes are noncorrosive to the metal cathodes and tend to compete less with the dye for visible light and most importantly have synthetically tunable redox potentials.19a Ruthenium(II) polypyridine complexes and their derivatives have been tested extensively as photosensitizers in DSSC.20a,21 Another area of use for ruthenium(II) tris-bipyridine complexes is the excited-state proton-coupled electron transfer (PCET), in which two ruthenium(II) tris-bipyridine cations are connected by a salt bridge to facilitate the proton transfer.19b So far, only two crystal structures have been reported that contain the cobalt(III) tris(2,2′-bipyridine) cation with a simple anion.22 Analogously, only two reported crystal structures describe the cationic ruthenium(II) tris(4,4′-dimethyl-2,2′-bipyridine) moiety.23 Facile generation of single crystals for these complex classes would be of utmost importance for many research directions.
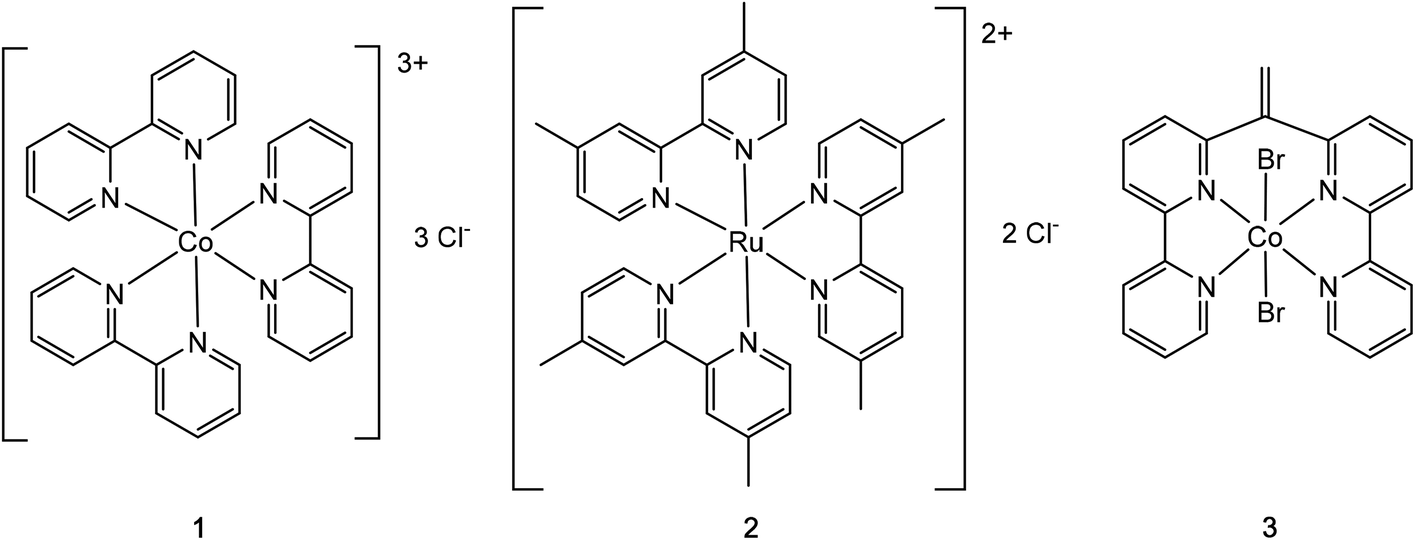 | ||
| Fig. 1 The tested coordination complexes for single crystal growth by the anion exchange Nano-Crystallization method: [Co(III)(2,2′-bipyridine)3](chloride)3 (1), [Ru(II)(4,4′-dimethyl-2,2′-bipyridine)3](chloride)2 (2), and Co(II)(6,6′′-(ethene-1,1-diyl)di-2,2′-bipyridine)(bromide)2 (3). | ||
2. Experimental
2.1. General chemical procedures
All reactions were done using standard laboratory glassware. Ruthenium(III) trichloride hydrate was purchased from Johnson Matthey, 4,4′-dimethyl-2,2′-bipyridyl (≥99%) from Acros Organics, pentaamminechlorocobalt(III) chloride from Sigma Aldrich, 2,2′-bipyridyl from TCI Chemicals, acetone (≥99.8%) from Merck, dimethyl sulfoxide (DMSO, ≥99.5%), dichloromethane (DCM, ≥99%), ether (≥99%) from Sigma Aldrich, ethanol (≥99.8%) from VWR Chemicals. All chemicals were used without further purification. Solvents were of p.a. grade. Aluminum oxide 90 (basic) for column chromatography activity level 1 purchased from Macherey-Nagel was used for purification. cis-Ru(DMSO)4Cl2 was synthesized on a 2 mmol scale according to the literature in a yield of 69%.24 The IR spectrum was recorded on a SpectrumTwo FT-IR Spectrometer (Perkin-Elmer) equipped with a Specac Golden Gate™ ATR accessory for neat samples. The frequencies are reported in wavenumbers (cm−1).
Chromatography: Merck ALOX 60 (40–63 μm) with the indicated solvent system. Thin layer chromatography (TLC): Merck TLC plates silica gel 60 on aluminum with the indicated solvent system; the spots were visualized by UV light (254 and 366 nm). 1H-NMR spectra were recorded on a Bruker AV-400 (400 MHz), the coupling constants are given in Hz. High-resolution electrospray mass spectra (HR-ESI-MS) were recorded on a maXis QTOF-MS instrument (Bruker Daltonics GmbH, Bremen, Germany). The samples were dissolved in a suitable solvent (e.g. MeOH) at a concentration of ca. 50 μg ml−1 and analyzed via continuous flow injection (2 μL min−1). The mass spectrometer was operated in the positive electrospray ionization mode at 4000 V capillary voltage, −500 V endplate offset, with a N2 nebulizer pressure of 0.8 bar and dry gas flow of 4 L min−1 at 180 °C. MS acquisitions were performed in the mass range from m/z 50 to 2000 at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 resolution (full width at half maximum) and 1.0 Hz spectra rate. The mass analyzer was calibrated between m/z 118 and 2721 using an Agilent ESI-L low concentration tuning mix solution (Agilent, USA) at a resolution of 20000 and a mass accuracy below 2 ppm. All solvents used were purchased in best LC-MS qualities. Elemental analyses were acquired on a LECO Truespec CHNS(O)-microanalyser. The used HPLC system was a VWR Hitachi Chromaster with a diode array detector 5430 and a ReproShell C18, 2.6 μm, 75 × 4.6 mm column (Dr Maisch GmbH, Germany), which was kept at 40 °C. The used gradient was 95% A (water with 0.1% formic acid) and 5% B (acetonitrile) changing linearly within 5.8 minutes to 100% B, which was maintained for one minute. The aqueous solubilities of 1 and 2 were determined by gravimetry and HPLC respectively. For 1, 50 μL of a saturated solution of 1 were mixed together with 150 μL of water in order to dilute the solution enough for the lyophilization process. After lyophilization, the remaining weight of 1 was determined. In order to estimate the errors of this procedure, a similar experiment was done with 125 μL of a saturated sodium chloride solution. Based upon this and taking the concentration25 and the density26 of the saturated sodium chloride solution at 22 °C into account, the error of the gravimetric procedure was estimated to be 2%. For 2, an aqueous saturated solution was diluted 540-fold. Its concentration was determined by comparing it with a calibration line, which was generated by 10-fold diluting an aqueous solution of 1.34 mg of 2 in 1 ml of water. Five different volumes between 1 and 20 μl of the 10-fold diluted solution of 2 were used to create the calibration line after manual integration. The error of the slope of the calibration line was calculated to be 0.5%. The HPLC procedure could not be applied to compound 1, as it eluted too early together with the injection peak on our system.
000 resolution (full width at half maximum) and 1.0 Hz spectra rate. The mass analyzer was calibrated between m/z 118 and 2721 using an Agilent ESI-L low concentration tuning mix solution (Agilent, USA) at a resolution of 20000 and a mass accuracy below 2 ppm. All solvents used were purchased in best LC-MS qualities. Elemental analyses were acquired on a LECO Truespec CHNS(O)-microanalyser. The used HPLC system was a VWR Hitachi Chromaster with a diode array detector 5430 and a ReproShell C18, 2.6 μm, 75 × 4.6 mm column (Dr Maisch GmbH, Germany), which was kept at 40 °C. The used gradient was 95% A (water with 0.1% formic acid) and 5% B (acetonitrile) changing linearly within 5.8 minutes to 100% B, which was maintained for one minute. The aqueous solubilities of 1 and 2 were determined by gravimetry and HPLC respectively. For 1, 50 μL of a saturated solution of 1 were mixed together with 150 μL of water in order to dilute the solution enough for the lyophilization process. After lyophilization, the remaining weight of 1 was determined. In order to estimate the errors of this procedure, a similar experiment was done with 125 μL of a saturated sodium chloride solution. Based upon this and taking the concentration25 and the density26 of the saturated sodium chloride solution at 22 °C into account, the error of the gravimetric procedure was estimated to be 2%. For 2, an aqueous saturated solution was diluted 540-fold. Its concentration was determined by comparing it with a calibration line, which was generated by 10-fold diluting an aqueous solution of 1.34 mg of 2 in 1 ml of water. Five different volumes between 1 and 20 μl of the 10-fold diluted solution of 2 were used to create the calibration line after manual integration. The error of the slope of the calibration line was calculated to be 0.5%. The HPLC procedure could not be applied to compound 1, as it eluted too early together with the injection peak on our system.
2.2. Synthesis of the starting coordination complexes 1–3
:17). 1H-NMR (D2O): 8.82 (dd, J = 7.7, 1.0, 2 H); 8.53 (td, J = 7.8, 1.2, 2 H); 7.79–7.75 (m, 2 H); 7.45 (d, J = 6.0, 2 H). HR-ESI(+)-MS (H2O): calc. for [C30H24CoN6]2+: 263.56910, exp.: 263.56917 (100%, Δ = 0.26 ppm); calc. for [C20H16CoN4]2+: 185.53480, exp.: 185.53461 (28%, Δ = 0.98 ppm), calc. for [C30H24CoN6]3+: 175.71260, exp.: 175.71252 (22%, Δ = 0.47 ppm). Anal. calcd for [Co(bpy)3]Cl3·5.5H2O: C, 49.16; H, 4.81; N, 11.47. Found: C, 48.74; H, 4.67; N, 11.93. The measured powder XRD pattern of 1 is shown in Fig. S3.† The aqueous solubility of 1 was determined by gravimetry to be 461 ± 9 mM.
24 (0.64 g, 1.33 mmol) and 4,4′-dimethyl-2,2′-bipyridine (0.74 g, 4.02 mmol) were added to 25 mL of a 10% solution of water in ethylene glycol in a 100 mL round bottom flask equipped with a reflux condenser under the exclusion of light in N2 atmosphere and heated to reflux (oil bath, 120 °C) for 1 hour. After cooling to room temperature, the orange solution was diluted with water (30 mL) and then a saturated solution of ammonium hexafluorophosphate salt (50 mL) was added until complete precipitation.18a The orange solid was filtered off and dried under reduced pressure to yield the hexafluorophosphate salt of 2 (97%, 1.22 g, 1.29 mmol) as orange crystals. IR (neat): 3595w, 2928w, 1620m, 1552w, 1479w, 1446w, 1304w, 1241w, 1038w, 988m, 972m, 821s. [Ru(II)(4,4′-dimethyl-2,2′-bipyridine)3]Cl2 (2) was synthesized as the tetrahydrate from the hexafluorophosphate salt on a 1.3 mmol scale according to the literature in a yield of 68%.18a Anal. calc. for C36H36N6RuCl2(H2O)4 (796.76): C 54.27, H 5.57, N 10.55, found: C 54.19, H 5.59, N 10.42. The aqueous solubility of 2 was determined by HPLC to be 309.4 ± 1.3 mM.
:1) to afford 60.2 mg (61%) of the pure product as a white solid. 1H NMR: (400 MHz, CD3CN): 8.66 (ddd, J = 4.8, 1.7, 0.9, 2H); 8.41–8.34 (m, 4H); 7.89 (t, J = 7.9, 2H); 7.82 (td, J = 7.6, 1.9, 2H); 7.53 (dd, J = 7.7, 0.9, 2H); 7.36 (ddd, J = 7.5, 4.7, 1.2, 2H); 6.25 (s, 2H). HR-ESI(+)-MS: calc. for [C22H17N4]+: 337.14477, exp.: 337.14430.
To a solution of 6,6′′-(ethene-1,1-diyl)di-2,2′-bipyridine (296 mg, 0.748 mmol, 1.0 eq.) in 12 mL acetonitrile was added CoBr2 (155 mg, 0.711 mmol, 0.95 eq.) in one portion and the resulting red suspension was stirred for 3 hours at RT. The red precipitate was collected on a P3 glass filter frit and dried under HV for several hours to obtain 316 mg (76%) of the desired product 3 as a red solid. HR-ESI-MS: calc. for [M − 2Br]2+: 197.53480, exp.: 197.53501. Anal. calcd for [Co(bpy2CCH2)Br2]·1.5H2O (%): C, 45.39; H, 3.29; N, 9.62. Found: C, 45.44; H, 3.26; N, 9.52.
2.3. Crystallographic procedures
The crystallization experiments were done as previously described7a using a Gryphon LCP nano-drop handler from Art Robbins Instruments in ARI Intelli-Plates 96-3 LVR. 500 nL of a stock solution of the cation to be crystallized (90% maximal saturation in water) were mixed with 500 nL of the stock solutions of the sodium and potassium salts and equilibrated against 75 μL of the stock solution of the same sodium or potassium salt via vapor diffusion. The screen contains 77 different sodium or potassium salts with a total of 96 different conditions and each coordination complex was tested for crystallization under those 96 conditions. The plates were incubated for 5–16 days at 20 °C. The Rock Imager 1000 took a picture of each well with normal light (immediately after setting up the plate and then after 2, 5, 10 and 16 days) and cross polarized light (immediately after setting up the plate and then after 5, 10 and 16 days).Crystallographic data were collected at 160.0(1) K (with the exception of 100.00(11) K for 2b) on a Rigaku OD XtaLAB Synergy Dualflex diffractometer equipped with a Pilatus 200 K detector and both PhotonJet Mo Kα and Cu Kα sources. Suitable crystals were covered with oil (Infineum V8512, formerly known as Paratone N), placed on a nylon loop that is mounted on a CrystalCap Magnetic™ pin (Hampton Research) and immediately transferred to the diffractometer. The program suite CrysAlisPro was used for data collection, numerical and multi-scan absorption correction as well as data reduction.28 Each structure was solved with direct methods using ShelxT29 and was refined by full-matrix least-squares methods on F2 with SHELXL-201430 using the OLEX2 GUI.31 Graphical output was produced with the help of the program Mercury32 and the calculations of the geometry were done with the program Platon.33 For 1a, a 7 keV threshold was applied for the hybrid pixel Pilatus detector34 in order to minimize the X-ray fluorescence caused by the cobalt complex being irradiated with copper radiation.35 Additionally, the crystals of 1a were twinned by a 180° rotation around the a-axis. The twin ratio was 69:31 and the two merged twin components were handled with the HKLF5 command in ShelxL. For 1c, two complexes plus 15 water molecules and additional a half-occupied sodium chloride were found in the asymmetric unit, which corresponds to the unit cell (space group P1). Several water molecules were half occupied; their hydrogen atoms could not be found. One half occupied water molecule and a half-occupied sodium are located on the same position. Several restraints for the hydrogen atoms had to be used. For 2a, the squeeze routine within Platon had to be used.36 For 4c, unexpectedly the spherical absorption correction was much better in terms of I/σ(I) and Rint than the Gaussian or the numerical absorption correction. CCDC entries 1986517–1986525† contain the supplementary crystallographic data for this paper. Powder X-ray diffraction (PXRD) patterns were recorded on a STOE STADI P diffractometer equipped with a DECTRIS MYTHEN 1K detector in transmission mode using a Ge monochromator for Mo radiation (0.70930 Å).37 Background correction of the recorded PXRD pattern was done with help of the software QUALX2.0.38 Simulated PXRD pattern based on the single crystal analyses were generated with the help of the program Mercury.32
3. Results & discussion
The three chosen cationic (or cationic after anion dissociation), water-soluble metal complexes (1–3) were all subjected to the same high throughput screening Nano-Crystallization method,7a in which 500 nl of a 90% saturated aqueous solution of the complexes were mixed with 500 nl of an aqueous sodium salt solution containing the desired anion, which was added by a pipetting robot. Subsequently, this mixture was equilibrated by vapor diffusion against an excess of the same sodium salt solution. This procedure was repeated for each coordination complex with 95 different anion salt solutions to finally yield a completely filled 96-well crystallization plate with 96 different crystallization trials starting from one metal complex solution. The plates were incubated for 5–16 days at 20 °C in an imaging system. The Rock Imager 1000 took a picture of each well with normal light and cross polarized light. In the following, the results will be discussed for each obtained individual coordination complex moiety.3.1 Crystallization of the cobalt(III) tris(2,2′-bipyridine) triscationic unit
Starting from cobalt(III) tris(2,2′-bipyridine) chloride, single crystals were obtained after mixing with either a 0.7 M solution of sodium dicyanamide, a 2.27 M solution of disodium DL-malate or a 2.92 M solution of disodium L-malate. All crystallographic data are summarized in Table S1.†The aqueous mixture of cobalt(III) tris(2,2′-bipyridine) trichloride and sodium dicyanamide yielded crystals of cobalt(II) tris(2,2′-bipyridine) dicyanamide chloride·tetrahydrate (1a) in the hexagonal space group P6/mcc (Fig. 2). A 7 keV threshold was applied for the hybrid pixel Pilatus detector34 in order to minimize the X-ray fluorescence caused by the cobalt complex being irradiated with copper radiation.35 The cobalt center in 1a was assigned without any doubt to be a Co(II), because the Co–N bond length in 1a is 2.1259(16) Å. This value is perfectly in line with the range of Co(II)–N bond lengths between 2.117(8) and 2.141(7) Å in [Co(II)(bpy)3]Cl2.39 On the other side, [Co(III)(bpy)3]Cl3 was reported to have Co–N bond lengths between 1.928(3)–1.939(3) Å.22b Synthetic experiments on a 0.4 mL scale revealed that the yield of this Co(II) complex is less than 10%. We postulate that the used dicyanamide anion is responsible for this reduction. The starting material cobalt(III) tris(2,2′-bipyridine) chloride (1) was analysed by elemental analysis and PXRD, but no signs for a cobalt(II) impurity could be found in 1.
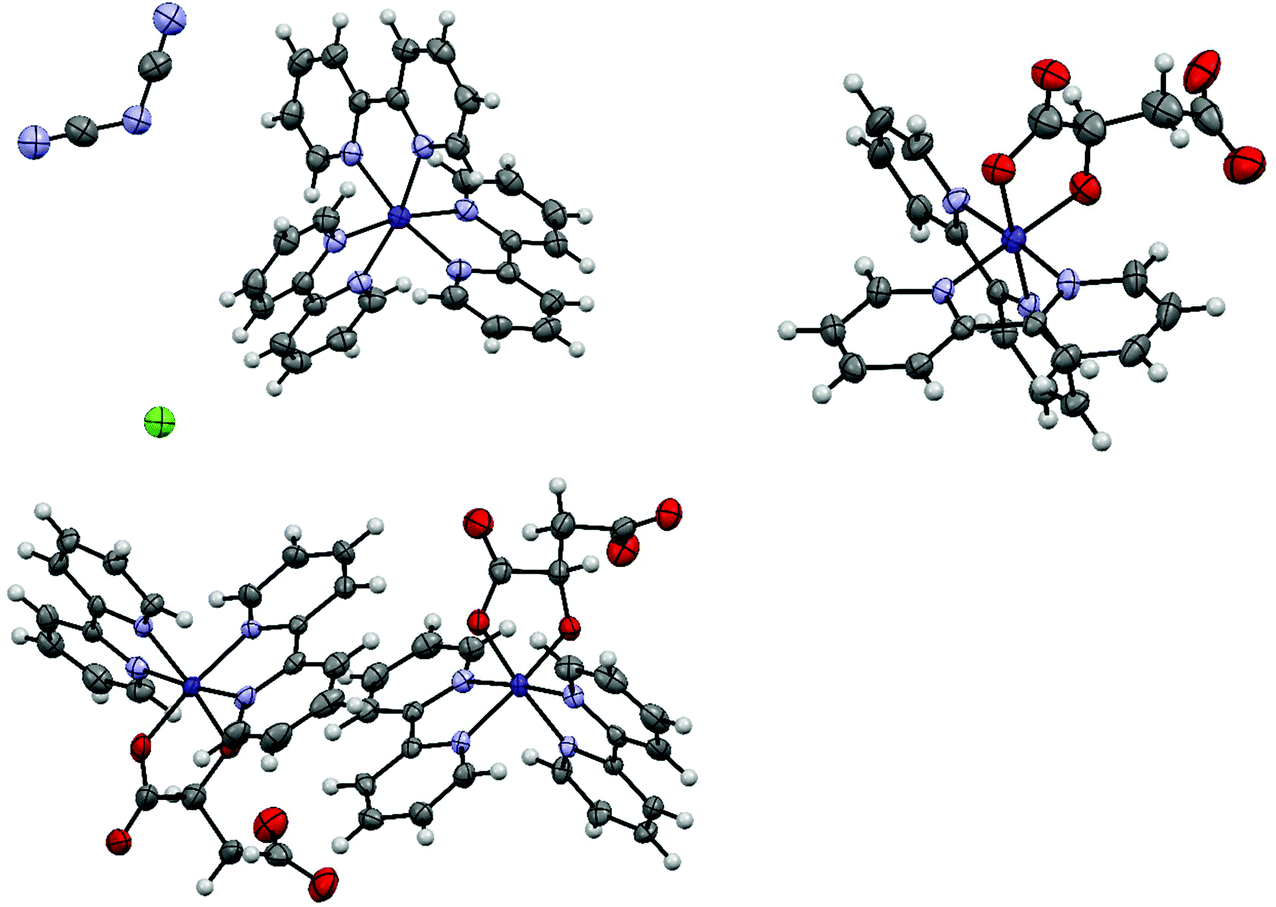 | ||
| Fig. 2 Displacement ellipsoid representation of 1a (top left), 1b (top right) and 1c (lower left). Ellipsoids are drawn at 50% probability. Disordered parts and water molecules are omitted for clarity. | ||
Single, red crystals of cobalt(III) bis(2,2′-bipyridine) DL-malate·decahydrate (1b) were obtained by vapor diffusion of a mixture of a 90% saturated solution of complex 1 and a 2.27 M solution of disodium DL-malate against a reservoir of a 2.27 M solution of disodium DL-malate. During crystallization a ligand exchange took place, whereby one 2,2′-bipyridine was replaced by a D- or L-malate anion. The resulting cobalt(III) bis(2,2′-bipyridine) DL-malate crystallized in the monoclinic space group I2/a (Fig. 2). The malate ligand coordinates as a bidentate trianion to the cobalt to yield a neutral complex overall. The substitution of one 2,2′-bipyridyl (bpy) (or the related 1,10-phenanthroline, phen) ligand within a cobalt(III)(bpy)3 or cobalt(III)(phen)3 complex by a dianionic α-hydroxycarboxylate has been reported before.40
Single, red crystals of cobalt(III) bis(2,2′-bipyridine) L-malate·9.25(H2O)·0.25(NaCl) (1c) were obtained by vapor diffusion of a mixture of a 90% saturated solution of 1 and a 2.92 M solution of disodium L-malate against a reservoir of a 2.92 M solution of disodium L-malate. Again, during crystallization a ligand exchange took place, whereby one 2,2′-bipyridine was replaced by one bidentate L-malate trianion. The resulting cobalt(III) bis(2,2′-bipyridine) L-malate crystallized in the triclinic, chiral space group P1. Both delta and lambda forms of cobalt(III) bis(2,2′-bipyridine) L-malate are present in the crystal in a 1:1 ratio.
In the crystallization trials leading to the red complexes 1b and 1c, additionally yellow crystals could be seen and were confirmed by single crystal analysis to be the already published crystal structure of starting material 1.22b Microscope images of the two crystallization trials leading to compounds 1 as well as 1b and 1c respectively are shown in Fig. S6 and S8.†
3.2 Crystallization of the ruthenium(II) tris(4,4′-dimethyl-2,2′-bipyridine) dicationic unit
Single crystals containing the ruthenium(II) tris(4,4′-dimethyl-2,2′-bipyridine) dication were obtained from a mixture of a 90% saturated aqueous solution of ruthenium(II) tris(4,4′-dimethyl-2,2′-bipyridine) chloride and either a 0.73 M solution of disodium fumarate or a 1.57 M solution of sodium saccharinate. All crystallographic data are summarized in Table S2.†The asymmetric unit of ruthenium(II) tris(4,4′-dimethyl-2,2′-bipyridine)·(hydrogenfumarate)·(hemifumarate)·(tetrahydrate) 2a consists of one ruthenium(II) tris(4,4′-dimethyl-2,2′-bipyridine) dication, one hydrogenfumarate anion, half a fumarate dianion and four water molecules (Fig. 3). The asymmetric unit in the case of 2b consists of one ruthenium(II) tris(4,4′-dimethyl-2,2′-bipyridine) dication, two saccharinate anions and four water molecules. Compound 2b was also synthesized on a macroscopic (0.13 mmol) scale from the chloride salt 2 with the help of an anion exchange column in a yield of 84% (see Chapter S5†).
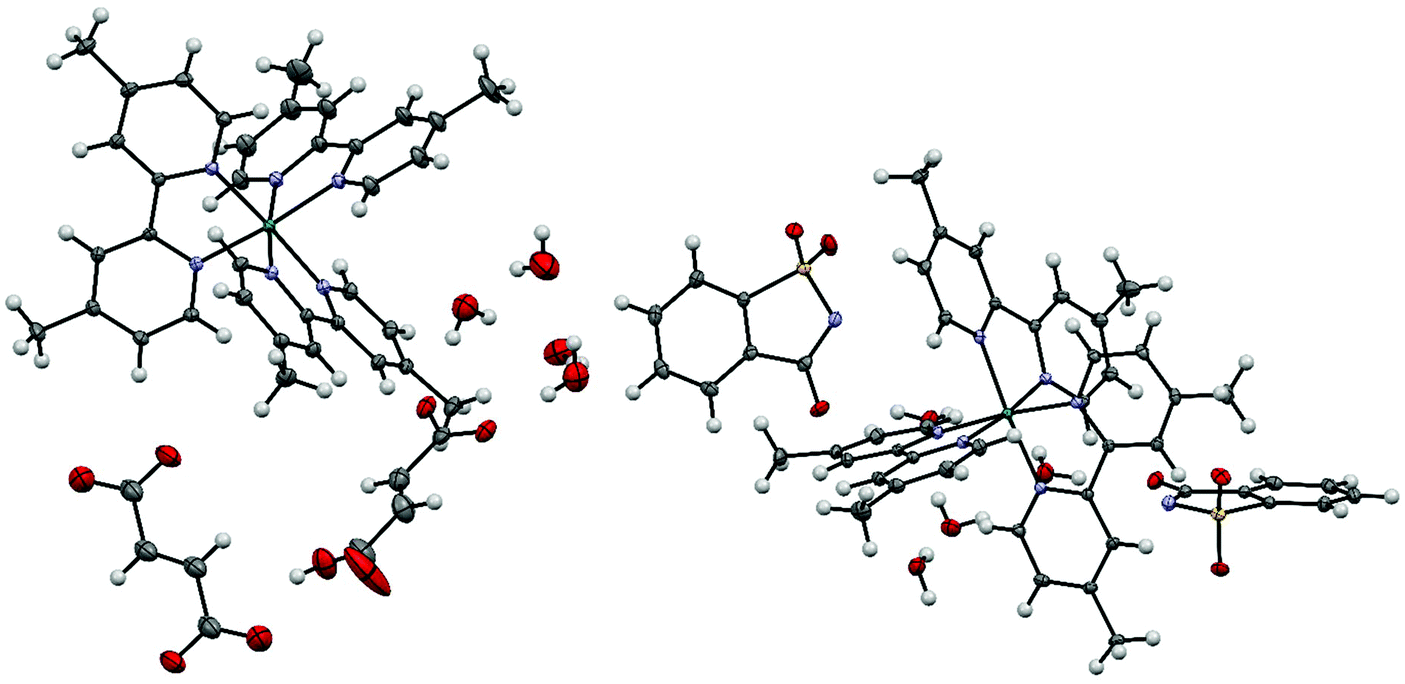 | ||
| Fig. 3 Displacement ellipsoid representation of 2a (left) and 2b (right). Ellipsoids are drawn at 50% probability. An inversion center symmetry operation has been applied to form the complete fumarate anion. The smaller ellipsoids of 2b, compared with the other structures, are caused by the lower measurement temperature of 100 K instead of 160 K. | ||
3.3 Crystallization of the Co(II)(6,6′′-(ethene-1,1-diyl)di-2,2′-bipyridine)(bromide)2 (3) complex
The starting complex for the crystallizations was always cobalt(II) (6,6′′-(ethene-1,1-diyl)di-2,2′-bipyridine)(bromide)2 (3). We expected that one or two bromide ligands might dissociate, thereby yielding a cationic complex, which might be a suitable candidate for our Anion screen. Unexpectedly, all obtained crystal structures contained the cobalt(II) bis(2,2′-bipyrid-6′-yl)ketone dication. It seems that, when complex (3) is dissolved in water, its double bond is oxidatively cleaved under aerobic conditions, resulting in the carbonyl group at the bridging position of the products 4. The hypothesis is supported by various publications, in which cobalt complexes are reported to catalyze the oxidative cleavage of terminal double bonds by molecular oxygen.41 Single crystals containing the cobalt(II) bis(2,2′-bipyrid-6′-yl)ketone dication were obtained from a mixture of a 90% saturated aqueous solution of Co(II)(6,6′′-(ethene-1,1-diyl)di-2,2′-bipyridine)(bromide)2 and the same volume of either a 0.15 M solution of sodium p-toluenesulfonate (4a), a 0.725 M solution of disodium fumarate (4b), a 2 M solution of sodium tetrafluoroborate (4c) or a 0.06 M solution of disodium terephthalate (4d). All crystallographic data are summarized in Table S3.†The asymmetric unit of 4a contains a cobalt(II) bis(2,2′-bipyrid-6′-yl)ketone dication with an octahedral metal center coordinated by one p-toluenesulfonate anion and a water molecule. The remaining charge is compensated by a second free p-toluenesulfonate anion; additionally, there are four water molecules in the asymmetric unit (Fig. 4).
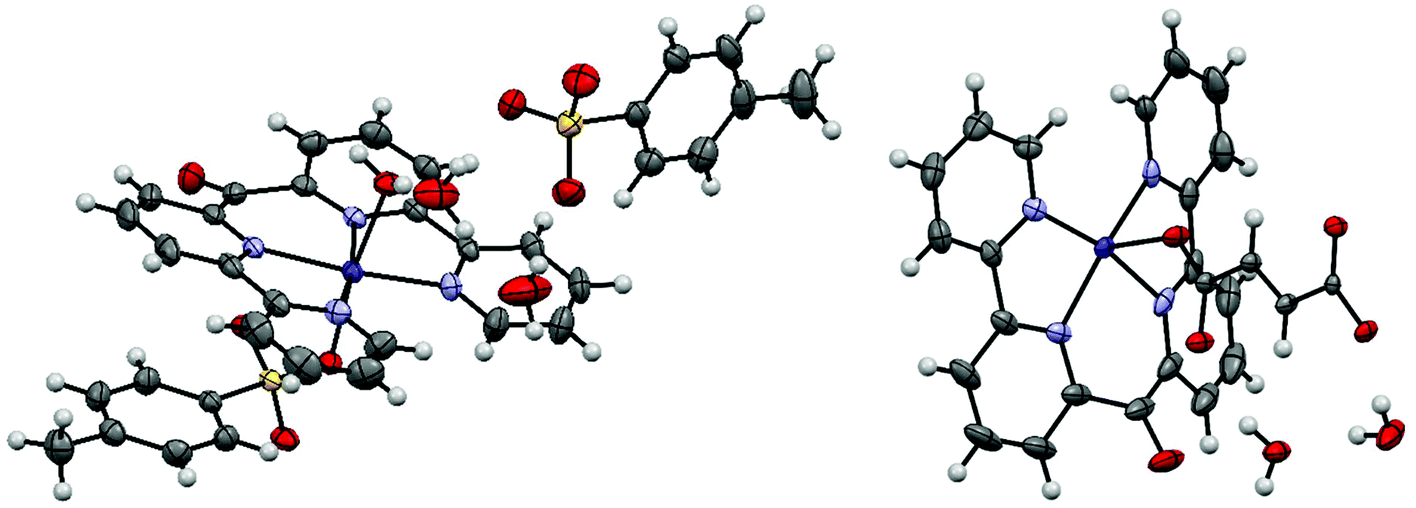 | ||
| Fig. 4 Displacement ellipsoid representation of 4a (left) and 4b (right), ellipsoids drawn at 50% probability. One disordered water molecule in 4a is omitted for clarity. | ||
The asymmetric unit of 4b contains a cobalt(II) bis(2,2′-bipyrid-6′-yl)ketone dication, one fumarate dianion and 2.25 water molecules (Fig. 4). The octahedral cobalt center is coordinated by the tetradendate ligand and two fumarate anions, where the latter bridge adjacent cobalt centers to form an infinite 1D-chain. The asymmetric unit of 4c consists of one cobalt(II) bis(2,2′-bipyrid-6′-yl)ketone dication, two tetrafluoroborate anions and two water ligands (Fig. 5). The octahedral cobalt is coordinated by the ligand and two axially coordinated water molecules.
 | ||
| Fig. 5 Displacement ellipsoid representation of 4c (left) and 4d (right). Ellipsoids are drawn at 50% probability. For 4d, the complete terephthalate ligand, which is created by a center of inversion, is shown for clarity. The free terephthalate dianion is omitted for clarity. | ||
The asymmetric unit of 4d consists of one cobalt(II) bis(2,2′-bipyrid-6′-yl)ketone dication with only half of the coordinated terephthalate ligand, one coordinated and three free water molecules and another half of a free terephthalate dianion (Fig. 5). Each terephthalate species sits on a center of inversion.
3.4 Variation of metal-to-metal distances caused by the presence of the different anions in six crystal structures containing the cobalt(II) bis(2,2′-bipyrid-6′-yl)ketone dication
The modulation of the metal-to-metal distance is of great importance for the magnetic properties in certain supramolecular molecular metal complexes.42 Therefore, we asked ourselves, whether the Nano-Crystallization method could generate a diverse collection of coordination compounds that contain the same cationic metal complex with different anions. Each salt would be expected to have a different minimal metal-to-metal distance. Including this work, there are six crystal structures known that contain cobalt(II) complexes with the bis(2,2′-bipyrid-6′-yl)ketone ligand. This unique assembly of six coordination complexes, all having a N4O2 coordination environment, allows to study the influence of the introduced anion on the metal-to-metal distances (Table 1, see Table S4† for an extended listing of metal-to-metal distances). From the data, it is clear that the shortest metal-to-metal distance does not correlate with the vdW volume of the anions. The shortest cobalt-to-cobalt distance among all six studied compounds, in 4d, is present between neighboring complexes, which are not bridged by any ligand. The second shortest distance, in 4a, is between two cobalt centers, which are bridged by an aqua ligand, which makes one hydrogen bond to a κO:κO′ bridging sulfonate group of the tosylate anion, that is coordinating to the next cobalt center.| Compound | Anion | Shortest cobalt-to-cobalt distance | Axial metal coordination | Anion vdW volume [Å3] | Ref. |
|---|---|---|---|---|---|
| 4d | Terephthalate | 6.8324(3) | Terephthalate bridged two cobalt metals, on the other side water that is bridged by one terephthalate to another water bound to Co | 133 | This work |
| 4a | Tosylate | 7.3475(3) | Tosylate on one side, water on the other | 141 | This work |
| 4c | Tetrafluoroborate | 7.9998(8) | Water molecules on both sides | 54 | This work |
| Perchlorate | 8.0982(9) | Water molecules on both sides | 57 | 44 | |
| 4b | Fumarate | 8.1800(5) | Fumarate anions on both sides | 88 | This work |
| Triflate | 8.2128(7) | Triflate on both sides | 85 | 27a |
4. Conclusion
For all three tested metal coordination complexes, the Nano-Crystallization method was able to generate novel single crystal structures with different anions, in which at least one of the original halide anions was replaced. This method can be recommended for cationic coordination complexes, which are stable in water and have an aqueous solubility of at least 2 mM. The shortest metal-to-metal distance was found to be purely dependent upon the interaction of the anion within the crystalline lattice and not upon the vdW size of the anion (Table 1).Conflicts of interest
The University of Zurich has a license agreement with Molecular Dimensions Ltd, which states that Molecular Dimension Ltd has the right to sell the Anion Screen.Acknowledgements
We thank the University of Zurich and the R'Equip programme of the Swiss National Science Foundation (project no. 206021_164018) for financial support. We thank Esmael Balaghi for measuring the PXRD, Viviane Grange for measuring the elemental analyses, Franziska Rahn and Matthias Berchtold for help with the HPLC measurements and Prof. Dr Anthony Linden for a critical reading of the manuscript.References
- (a) B. Spingler, S. Schnidrig, T. Todorova and F. Wild, CrystEngComm, 2012, 14, 751–757 RSC; (b) P. P. Nievergelt and B. Spingler, CrystEngComm, 2017, 19, 142–147 RSC.
- (a) H.-H. Tung, E. L. Paul, M. Midler and J. A. McCauley, Crystallization of Organic Compounds: An Industrial Perspective, John Wiley & Sons, Inc., 2009 CrossRef; (b) R. J. Davey, S. L. Schroeder and J. H. ter Horst, Angew. Chem., Int. Ed., 2013, 52, 2166–2179 CrossRef CAS PubMed; (c) Y. Tamura, H. Takezawa, Y. Domoto and M. Fujita, Chem. Lett., 2018, 47, 617–619 CrossRef CAS; (d) N. Wada, R. D. Kersten, T. Iwai, S. Lee, F. Sakurai, T. Kikuchi, D. Fujita, M. Fujita and J. K. Weng, Angew. Chem., Int. Ed., 2018, 57, 3671–3675 CrossRef CAS PubMed; (e) A. Tyler, R. Ragbirsingh, C. McMonagle, P. Waddell, S. Heaps, J. Steed, P. Thaw, M. Hall and M. Probert, ChemRxiv, 2019, 11366054 Search PubMed.
- (a) I. A. Ibarra, P. A. Bayliss, E. Pérez, S. Yang, A. J. Blake, H. Nowell, D. R. Allan, M. Poliakoff and M. Schröder, Green Chem., 2012, 14, 117–122 RSC; (b) P. A. Bayliss, I. A. Ibarra, E. Pérez, S. Yang, C. C. Tang, M. Poliakoff and M. Schröder, Green Chem., 2014, 16, 3796–3802 RSC; (c) B. Saccoccia, A. M. Bohnsack, N. W. Waggoner, K. H. Cho, J. S. Lee, D.-Y. Hong, V. M. Lynch, J.-S. Chang and S. M. Humphrey, Angew. Chem., Int. Ed., 2015, 54, 5394–5398 CrossRef CAS PubMed; (d) K. Sumida, K. Liang, J. Reboul, I. A. Ibarra, S. Furukawa and P. Falcaro, Chem. Mater., 2017, 29, 2626–2645 CrossRef CAS.
- (a) E. Biemmi, S. Christian, N. Stock and T. Bein, Microporous Mesoporous Mater., 2009, 117, 111–117 CrossRef CAS; (b) P. J. Kitson, R. J. Marshall, D. Long, R. S. Forgan and L. Cronin, Angew. Chem., Int. Ed., 2014, 53, 12723–12728 CrossRef CAS PubMed; (c) M. L. Kelty, W. Morris, A. T. Gallagher, J. S. Anderson, K. A. Brown, C. A. Mirkin and T. D. Harris, Chem. Commun., 2016, 52, 7854–7857 RSC; (d) S. M. Moosavi, A. Chidambaram, L. Talirz, M. Haranczyk, K. C. Stylianou and B. Smit, Nat. Commun., 2019, 10, 539 CrossRef CAS PubMed.
- C. G. Jones, M. Asay, L. J. Kim, J. F. Kleinsasser, A. Saha, T. J. Fulton, K. R. Berkley, D. Cascio, A. G. Malyutin, M. P. Conley, B. M. Stoltz, V. Lavallo, J. A. Rodríguez and H. M. Nelson, ACS Cent. Sci., 2019, 5, 1507–1513 CrossRef CAS PubMed.
- R. Taylor and P. A. Wood, Chem. Rev., 2019, 119, 9427–9477 CrossRef CAS PubMed.
- (a) P. P. Nievergelt, M. Babor, J. Čejka and B. Spingler, Chem. Sci., 2018, 9, 3716–3722 RSC; (b) M. Babor, P. P. Nievergelt, J. Čejka, V. Zvoníček and B. Spingler, IUCrJ, 2019, 6, 145–151 CrossRef CAS PubMed.
- R. Bolliger, A. Frei, H. Braband, G. Meola, B. Spingler and R. Alberto, Chem. – Eur. J., 2019, 25, 7101–7104 CrossRef CAS PubMed.
- W.-H. Jiang, H.-Z. Zhang, G.-F. Hou, D.-S. Ma, B. Liu and Y.-H. Yu, RSC Adv., 2017, 7, 45641–45651 RSC.
- (a) P. Diaz, J. Benet-Buchholz, R. Vilar and A. J. P. White, Inorg. Chem., 2006, 45, 1617–1626 CrossRef CAS PubMed; (b) I. A. Koval, M. Sgobba, M. Huisman, M. Lüken, E. Saint-Aman, P. Gamez, B. Krebs and J. Reedijk, Inorg. Chim. Acta, 2006, 359, 4071–4078 CrossRef CAS; (c) J. Suárez-Varela, A. J. Mota, H. Aouryaghal, J. Cano, A. Rodriguez-Diéguez, D. Luneau and E. Colacio, Inorg. Chem., 2008, 47, 8143–8158 CrossRef PubMed; (d) P. Mukherjee, M. G. B. Drew, M. Estrader, C. Diaz and A. Ghosh, Inorg. Chim. Acta, 2008, 361, 161–172 CrossRef CAS; (e) L. Botana, J. Ruiz, J. M. Seco, A. J. Mota, A. Rodríguez-Diéguez, R. Sillanpää and E. Colacio, Dalton Trans., 2011, 40, 12462–12471 RSC; (f) R. A. Polunin, K. S. Gavrilenko, M. A. Kiskin, I. L. Eremenko, V. M. Novotortsev and S. V. Kolotilov, Russ. J. Coord. Chem., 2016, 42, 487–493 CrossRef CAS.
- (a) J. Fielden, P. T. Gunning, D.-L. Long, M. Nutley, A. Ellern, P. Kögerler and L. Cronin, Polyhedron, 2006, 25, 3474–3480 CrossRef CAS; (b) R. Mondal, T. Basu, D. Sadhukhan, T. Chattopadhyay and M. K. Bhunia, Cryst. Growth Des., 2009, 9, 1095–1105 CrossRef CAS.
- (a) A. N. Khlobystov, A. J. Blake, N. R. Champness, D. A. Lemenovskii, A. G. Majouga, N. V. Zyk and M. Schröder, Coord. Chem. Rev., 2001, 222, 155–192 CrossRef CAS; (b) X. Li, Y. Gong, H. Zhao and R. Wang, Inorg. Chem., 2014, 53, 12127–12134 CrossRef CAS PubMed.
- X.-R. Meng, X.-J. Wu, D.-W. Li, H.-W. Hou and Y.-T. Fan, Polyhedron, 2010, 29, 2619–2628 CrossRef CAS.
- Y. Mu, Y. Zhao, H. Xu, H. Hou and Y. Fan, J. Mol. Struct., 2009, 935, 144–150 CrossRef CAS.
- P. Chen, M. Zhang, W. Sun, H. Li, L. Zhao and P. Yan, CrystEngComm, 2015, 17, 5066–5073 RSC.
- V. Bugris, C. Dudás, B. Kutus, V. Harmat, K. Csankó, S. Brockhauser, I. Pálinkó, P. Turner and P. Sipos, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2018, 74, 598–609 CrossRef CAS.
- N. Maki, Bull. Chem. Soc. Jpn., 1969, 42, 2275–2281 CrossRef CAS.
- (a) J. Lacour, C. Goujon-Ginglinger, S. Torche-Haldimann and J. J. Jodry, Angew. Chem., Int. Ed., 2000, 39, 3695–3697 CrossRef CAS; (b) A. M. Todd, A. N. Swinburne, A. E. Goeta and J. W. Steed, New J. Chem., 2013, 37, 89–96 RSC.
- (a) J. T. Kirner and C. M. Elliott, J. Phys. Chem. C, 2015, 119, 17502–17514 CrossRef CAS; (b) O. S. Wenger, Coord. Chem. Rev., 2015, 282, 150–158 CrossRef.
- (a) S. Aghazada and M. K. Nazeeruddin, Inorganics, 2018, 6, 52 CrossRef; (b) A. Colombo, C. Dragonetti, A. Valore, C. Coluccini, N. Manfredi and A. Abbotto, Polyhedron, 2014, 82, 50–56 CrossRef CAS.
- S. Aghazada, I. Zimmermann, Y. M. Ren, P. Wang and M. K. Nazeeruddin, ChemistrySelect, 2018, 3, 1585–1592 CrossRef CAS.
- (a) M. Du, X.-J. Zhao and H. Cai, Z. Kristallogr. – New Cryst. Struct., 2004, 219, 463–465 CAS; (b) W. Liu, W. Xu, J.-L. Lin and H.-Z. Xie, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1586–m1586 CrossRef CAS PubMed.
- (a) T. J. Rutherford, P. A. Pellegrini, J. Aldrich-Wright, P. C. Junk and F. R. Keene, Eur. J. Inorg. Chem., 1998, 1677–1688 CrossRef CAS; (b) J. Karges, O. Blacque, P. Goldner, H. Chao and G. Gasser, Eur. J. Inorg. Chem., 2019, 2019, 3704–3712 CrossRef CAS.
- I. Bratsos, E. Alessio, M. E. Ringenberg and T. B. Rauchfuss, Inorg. Synth., 2010, 35, 148–152 CAS.
- R. W. Potter II and M. A. Clynne, J. Res. U.S. Geol. Surv., 1978, 6, 701–705 Search PubMed.
- V. L. Thurmond, R. W. Potter II and M. A. Clynne, The densities of saturated solutions of NaCl and KCl from 10 °C to 105 °C, U. S. Geological Survey, 1984 Search PubMed.
- (a) S. Schnidrig, C. Bachmann, P. Müller, N. Weder, B. Spingler, E. Joliat-Wick, M. Mosberger, J. Windisch, R. Alberto and B. Probst, ChemSusChem, 2017, 10, 4570–4580 CrossRef CAS PubMed; (b) H. Nierengarten, J. Rojo, E. Leize, J.-M. Lehn and A. Van Dorsselaer, Eur. J. Inorg. Chem., 2002, 573–579 CrossRef CAS.
- Rigaku Oxford Diffraction, CrysAlisPro Software system 1.171.40, Rigaku Corporation, 2018 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, L. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- (a) P. Kraft, A. Bergamaschi, C. Broennimann, R. Dinapoli, E. F. Eikenberry, B. Henrich, I. Johnson, A. Mozzanica, C. M. Schlepütz, P. R. Willmott and B. Schmitt, J. Synchrotron Radiat., 2009, 16, 368–375 CrossRef CAS PubMed; (b) C. Brönnimann and P. Trüb, in Synchrotron Light Sources and Free-Electron Lasers: Accelerator Physics, Instrumentation and Science Applications, ed. E. J. Jaeschke, S. Khan, J. R. Schneider and J. B. Hastings, Springer International Publishing, Cham, 2016, pp. 995–1027 Search PubMed; (c) A. Förster, S. Brandstetter and C. Schulze-Briese, Philos. Trans. R. Soc., A, 2019, 377, 20180241 CrossRef PubMed.
- L. Prieto, M. Neuburger, B. Spingler and F. Zelder, Org. Lett., 2016, 18, 5292–5295 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- S. E. Balaghi, C. A. Triana and G. R. Patzke, ACS Catal., 2020, 10, 2074–2087 CrossRef CAS.
- A. Altomare, N. Corriero, C. Cuocci, A. Falcicchio, A. Moliterni and R. Rizzi, J. Appl. Crystallogr., 2015, 48, 598–603 CrossRef CAS.
- D. J. Szalda, C. Creutz, D. Mahajan and N. Sutin, Inorg. Chem., 1983, 22, 2372–2379 CrossRef CAS.
- (a) R. A. Haines and D. W. Bailey, Inorg. Chem., 1975, 14, 1310–1312 CrossRef CAS; (b) A. Tatehata, Inorg. Chem., 1976, 15, 2086–2090 CrossRef CAS.
- (a) R. S. Drago, B. B. Corden and C. W. Barnes, J. Am. Chem. Soc., 1986, 108, 2453–2454 CrossRef CAS PubMed; (b) Y. H. Lin, I. D. Williams and P. Li, Appl. Catal., A, 1997, 150, 221–229 CrossRef CAS; (c) X. Zhou and H. Ji, Chin. J. Chem., 2012, 30, 2103–2108 CrossRef CAS; (d) B. Biannic and J. J. Bozell, Org. Lett., 2013, 15, 2730–2733 CrossRef CAS PubMed.
- (a) M. Atzori, A. Serpe, P. Deplano, J. A. Schlueter and M. L. Mercuri, Inorg. Chem. Front., 2015, 2, 108–115 RSC; (b) V. V. Lukov, I. N. Shcherbakov, S. I. Levchenkov, Y. P. Tupolova, L. D. Popov, I. V. Pankov and S. V. Posokhova, Russ. J. Coord. Chem., 2019, 45, 163–187 CrossRef CAS.
- ChemAxon Ltd., MarvinSketch 20.6, 2020 Search PubMed.
- J. C. Knight, A. J. Amoroso, P. G. Edwards, R. Prabaharan and N. Singh, Dalton Trans., 2010, 39, 8925–8936 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description and crystallographic data for 1a–1c, 2a, 2b, 4a–4d, in CIF format, additional structural figures and experimental characterizations. CCDC 1986517–1986525. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt01236j |
| This journal is © The Royal Society of Chemistry 2020 |