
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zn- and Cd-based coordination polymers with a novel anthracene dicarboxylate ligand for highly selective detection of hydrogen peroxide†
Angela
Aleksovska
,
Peter
Lönnecke
 and
Evamarie
Hey-Hawkins
*
and
Evamarie
Hey-Hawkins
*
Leipzig University, Faculty of Chemistry and Mineralogy, Institute of Inorganic Chemistry, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de; Web: https://research.uni-leipzig.de/hh/
First published on 16th March 2020
Abstract
A one-dimensional {[Zn(L)(DMF)2]}n (1) and a three-dimensional {[Cd(L)(DMF)]·DMF}n (2) coordination polymer based on the novel anthracene derivative H2L (H2L = 4,4′-(9,10-anthracenediyl)dicinnamic acid) were obtained by solvothermal synthesis and charaterised by single-crystal and powder X-ray diffraction, thermogravimetry, and infrared spectroscopy. The anthracene derivative H2L and coordination polymers 1 and 2 were used to modify a glassy carbon electrode and as such served as an active material for detection of H2O2. Cyclic voltammograms in the potential range from 0 to −0.5 V revealed concentration-dependent cathodic current in all three cases with a lower detection limit of 200 μM. The electrode modified with compound 2 showed the best performance towards hydrogen peroxide detection. The results suggest that the development of electrodes modified with inorganic polymers based on highly conjugated ligands can serve as potential electrocatalytic materials.
1. Introduction
Over the past few decades, materials with excellent electrical and electrocatalytical properties for manufacturing of efficient electronic devices have been subjects of extensive studies.1 Therefore, conducting materials,2 metal nanoparticles,3 carbon nanotubes and nanowires,4 graphene5 and other potential electrocatalytic materials have been widely investigated. Furthermore, electrocatalytic materials for modifying electrodes for the detection of important biological and chemical compounds have received much attention.With the ubiquitous success in a wide range of applications, metal–organic frameworks (MOFs) and coordination polymers became one of the most prolific research areas of inorganic chemistry and crystal engineering in the last few decades. These crystalline materials consist of metal ions or clusters coordinated by organic, often rigid, ligands. The organic part of the frameworks or coordination polymers is usually responsible for the properties and behaviour of the framework. While a few reports on the investigation of the electrochemical behaviour of MOFs and coordination polymers6,7 and their electrochemical synthesis8 have been presented, the literature on the utilisation of these materials for modification of electrode surfaces is scarce.9
Hydrogen peroxide as the simplest peroxide and an important industrial chemical has been widely utilised in a broad variety of applications. It is a major reactive oxygen species that participates in various cellular metabolic pathways, generated by most oxidases in mitochondria. However, abnormal levels can lead to several bodily disorders such as myocardial infarction,10 atherosclerosis,11 Alzheimer's disease,12 cancer,13etc. Furthermore, it is one of the most versatile, reliable and environmentally compatible oxidising agents, opening various applications in wastewater treatment, water recycling, air pollution control, etc.14 Consequently, fast and accurate detection of H2O2 is of paramount importance in environmental and medicinal fields and has become a research hotspot.15
Among different methods for hydrogen peroxide detection, electrochemical methods have many advantages such as simplicity, portability, good selectivity and high sensitivity. Most of the modified electrodes for hydrogen peroxide detection reported in the past few decades are enzyme-based, although their high cost and their difficult immobilisation limit their widespread use. Recently, also coordination polymers and metal–organic frameworks were employed as electrode-modifying materials for hydrogen detection.16 The synergistic catalysis in these materials is based on a mechanism that includes oxidation of the metal centre and simultaneous reduction of hydrogen peroxide. Herein, we present an electrochemical sensor for hydrogen peroxide, where the organic part of the coordination polymers and the structure are responsible for the sensing activity.
Fused aromatic structures alone have been used as a platform to develop efficient electrocatalysts,17 but also as building blocks for coordination polymers and metal–organic frameworks. Literature shows several examples of employing dicarboxylate anthracene ligands as building blocks for coordination polymers, complexes and metal–organic frameworks, offering interesting luminescent and electroluminescent properties.18 Here, we present the synthesis of a novel ditopic anthracene-based ligand and its further use as a building block for a one-dimensional Zn-based and a three-dimensional Cd-based coordination polymer. Furthermore, we present a sensor based on a glassy carbon electrode modified with crystalline material of the above-mentioned compounds.
2. Experimental
2.1. General methods
9,10-Dibromoanthracene19 and 4-methoxycarboxyvinyl phenylboronic acid20 were prepared according to the literature. All other chemicals are commercially available and were used without further purification. 1H NMR and 13C NMR spectra were recorded at room temperature with a Bruker AVANCE DRX 400 spectrometer; chemical shifts are given in parts per million (ppm) at 400.13 MHz (1H NMR) and 100.63 MHz (13C NMR). Internal standard was tetramethylsilane (TMS). Melting points were determined with a Gallenkamp MPD350.BM2.5 device. The XRD patterns were recorded using a STOE STADI-P diffractometer system equipped with a sealed Cu X-ray tube and a germanium (111) monochromator crystal [λ(Cu-Kα1) = 154.060 pm], with a step size of 0.05°. The samples were measured in transmission mode in capillaries (Hilgenberg, 0.5 mm). Thermogravimetric (TG) and differential thermal analysis (DTA) were performed with a Netzsch STA 449 F1 thermoanalyser in a dynamic argon atmosphere combined with an Aeolos QMS 403 D mass spectrometer (heating rate 10 °C min−1, flow rate 25 mL min−1, aluminium oxide crucible, mass 20 mg), and temperature range from room temperature to 600 °C. Solvothermal/temperature-controlled reactions were conducted in Memmert ovens models UFE400 and UNE600. Elemental analyses were performed with a Vario El-Heraeus analyser. Infrared spectra were obtained with a Bruker TENSOR 27 (equipped with a MIRacle ZnSe ATR accessory from PIKE Technologies).2.2. Synthesis of 4,4′-(9,10-anthracenediyl)dicinnamic acid (H2L)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1). The mixture was stirred under reflux for 24 h. The organic solvents were removed, and the aqueous phase was acidified with HCl (6 M). The product precipitated as a bright yellow powder and was isolated by filtration, washed with hot water and dried in vacuum at 140 °C for several hours (2.2 g, yield 94%). M.p.: decomp. at 210 °C without melting. 1H NMR (d6-DMSO): δ = 6.81 (d, J = 16.0 Hz, 2H, vinyl H), 7.42 (m, 4H, arom. CH), 7.58 (d, J = 8.2 Hz, 4H, arom. CH), 7.75 (m, 4H, arom. CH), 7.78 (d, J = 8.2 Hz, 4H, arom. CH), 8.02 (d, J = 16.0 Hz, 2H, vinyl H), 12.12 (br s, 2H, COOH). 13C{1H} NMR (d6-DMSO): δ = 116.5 (vinyl C), 125.0 (arom. C), 125.3 (arom. C), 126.1 (arom. C), 130.5 (arom. C), 132.1 (arom. C), 132.4 (arom. C), 132.7 (arom. C), 133.9 (arom. C), 137.0 (arom. C), 144.3 (vinyl C), 171.6 (COOH). Anal. calcd (%) for C32H22O4: C, 81.69; H, 4.71; found (%): C, 81.37; H, 4.97.
:1:1). The mixture was stirred under reflux for 24 h. The organic solvents were removed, and the aqueous phase was acidified with HCl (6 M). The product precipitated as a bright yellow powder and was isolated by filtration, washed with hot water and dried in vacuum at 140 °C for several hours (2.2 g, yield 94%). M.p.: decomp. at 210 °C without melting. 1H NMR (d6-DMSO): δ = 6.81 (d, J = 16.0 Hz, 2H, vinyl H), 7.42 (m, 4H, arom. CH), 7.58 (d, J = 8.2 Hz, 4H, arom. CH), 7.75 (m, 4H, arom. CH), 7.78 (d, J = 8.2 Hz, 4H, arom. CH), 8.02 (d, J = 16.0 Hz, 2H, vinyl H), 12.12 (br s, 2H, COOH). 13C{1H} NMR (d6-DMSO): δ = 116.5 (vinyl C), 125.0 (arom. C), 125.3 (arom. C), 126.1 (arom. C), 130.5 (arom. C), 132.1 (arom. C), 132.4 (arom. C), 132.7 (arom. C), 133.9 (arom. C), 137.0 (arom. C), 144.3 (vinyl C), 171.6 (COOH). Anal. calcd (%) for C32H22O4: C, 81.69; H, 4.71; found (%): C, 81.37; H, 4.97.
Yellow needle-shaped crystals of H2L suitable for single crystal X-ray structure determination were obtained by slow evaporation of a solution of H2L in THF/DMF (molecular structure is given in Fig. 1A). Crystal structure parameters for H2L are given in Table S1 (ESI†).
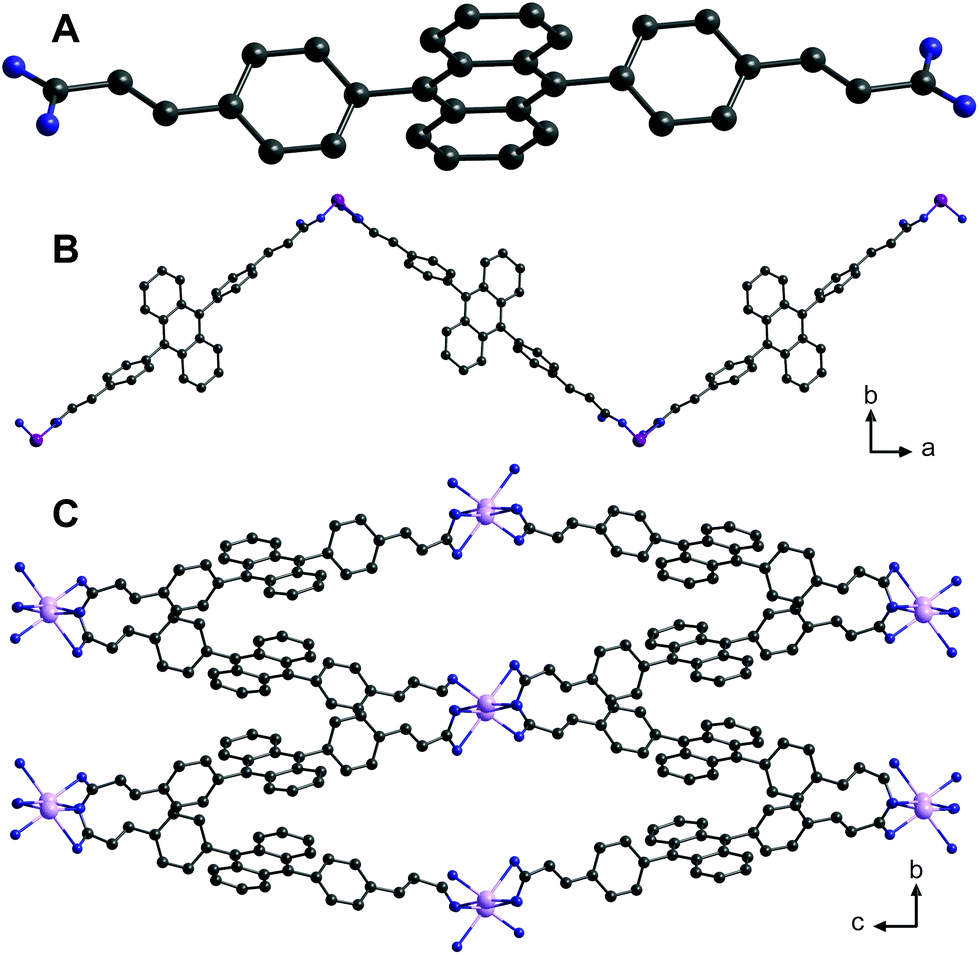 | ||
| Fig. 1 Solid-state structures of (A) H2L, (B) one-dimensional coordination polymer {[Zn(L)(DMF)2]}n (1) and (C) a three-dimensional metal–organic framework {[Cd(L)(DMF)]·DMF}n (2) based on 4,4′-(9,10-anthracenediyl)dicinnamic acid (H2L). Solvent molecules and hydrogen atoms were omitted for clarity. Colour code: C, black; O, blue; Zn, purple; Cd: pink. | ||
2.3. Synthesis of {[Zn(L)(DMF)2]}n (1)
0.012 g (0.027 mmol) H2L and 0.02 g (0.06 mmol) Zn(NO3)2·6H2O were dissolved in 2 mL DMF and a drop of HCOOH in a 4 mL vial. After 10 min of ultrasonication the vial was slowly heated to 110 °C during 4 h. After 48 h of heating at 110 °C, the mixture was cooled to room temperature by reducing the temperature by 5 °C each hour. After the reaction, yellow crystals suitable for single crystal X-ray structure determination were obtained. For further analysis the crystalline material was washed twice with methanol and dried at 200 °C. Anal. calcd (%) for C38H34N2O6Zn: C, 67.11; H, 5.04; N, 4.11; found (%): C, 67.03; H, 5.17, N, 4.01. The IR spectrum (Fig. S9) and powder XRD data (Fig. S6) are given in the ESI.†2.4. Synthesis of {[Cd(L)(DMF)]·DMF}n (2)
0.015 g (0.03 mmol) H2L, 0.02 g (0.06 mmol) Cd(NO3)2·4H2O and 0.005 g (0.04 mmol) 1,4-diazabicyclo[2.2.2]octane (DABCO) were dissolved in 2 mL DMF and a drop of HCOOH in a 4 mL vial. After 10 min of ultrasonication the vial was slowly heated to 110 °C during 4 h. After 48 h at 110 °C, the mixture was cooled to room temperature by reducing the temperature by 5 °C each hour. After the reaction, yellow crystals suitable for single crystal X-ray structure determination were obtained. Anal. calcd (%) for C38H34CdN2O6: C, 62.77; H, 4.71; N, 3.85; found (%): C, 62.79; H, 4.83, N, 3.91. The crystalline material was washed twice with methanol and dried at 200 °C; then elemental analysis was repeated. C35H26CdNO5 calculated: C, 64.38; H, 4.01; N, 2.26; found (%): C, 64.58; H, 4.13; N, 2.20. The IR spectrum (Fig. S10) and powder XRD data (Fig. S7 and S8) are given in the ESI.†2.5. Crystal structure determination
The data for H2L, 1 and 2 were collected on a Gemini diffractometer (Rigaku Oxford Diffraction) using Mo-Kα radiation (λ = 71.073 pm) and ω-scan rotation. Data reduction was performed with CrysAlis Pro21 including the program SCALE3 ABSPACK for empirical absorption correction. The structures were solved by dual-space methods with SHELXT-2014,22 and the refinement of all non-hydrogen atoms was performed with SHELXL-201823 with anisotropic thermal parameters. All hydrogen atoms are calculated on idealised positions using the riding model. Structure figures were generated with DIAMOND-4.24 CCDC 1980173 (1), 1980174 (2) and 1980175 (H2L)† contain the supplementary crystallographic data for this paper. A summary of the crystal data and relevant refinement parameters for 1, 2 and H2L are given in Table S1.† Bond lengths and angles of compounds 1 and 2 are given in Tables S2 and S3.†2.6. Cyclic voltammetry
Electrochemical experiments were conducted with a conventional three-electrode system, in which a bare glassy carbon electrode (GCE) with a diameter of 3 mm was used as working electrode and an Ag/AgCl (aq. saturated KCl) and platinum (Pt) wire were used as the reference and counter electrodes, respectively. All measurements (in the presence and absence of H2O2) were performed in 0.05 M phosphate buffer solution (PBS) at pH = 7 and with a prior nitrogen purge at a scan rate of 50 mV s−1.2.7. Preparation of the modified electrode
Three different solutions were prepared: H2L dissolved in toluene, suspension of 1 in toluene and suspension of 2 in toluene (concentration 5 mol L−1). The solution or suspension was spread on the surface of the electrode and then the electrode was dried. The resulting electrodes were immersed in solutions with different concentrations of H2O2.3. Results and discussion
3.1. Synthesis
Synthesis of H2L was achieved by a Pd-catalysed Suzuki–Miyaura coupling reaction between 9,10-dibromoanthracene and 4-methylcarboxyvinyl phenylboronic acid,19 followed by a base-catalysed hydrolysis to yield the corresponding carboxylic acid (Scheme 1).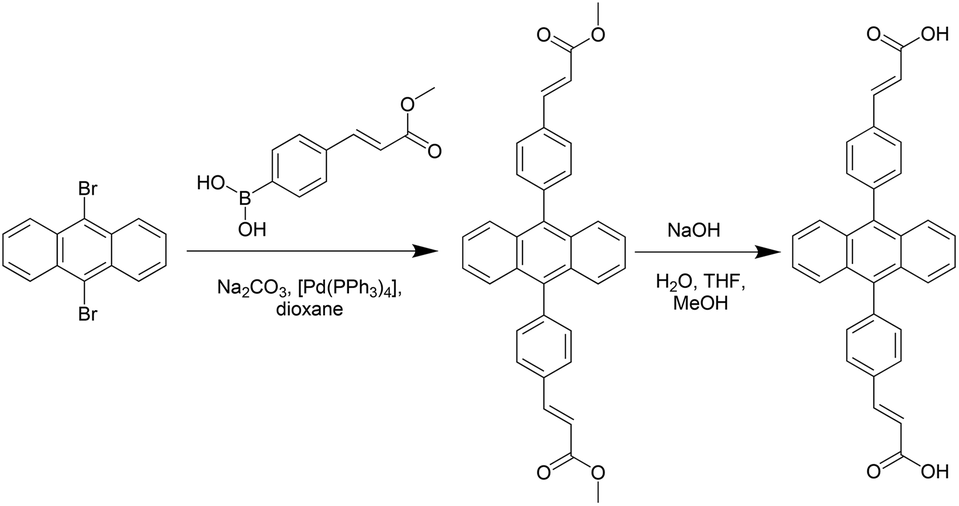 | ||
| Scheme 1 Synthesis of 4,4′-(9,10-anthracenediyl)dicinnamic acid (H2L). | ||
4,4′-(9,10-Anthracenediyl)dicinnamic acid (H2L) was then reacted with M(NO3)2·6H2O (M = Zn, Cd) in DMF and a drop of HCOOH under solvothermal conditions resulting in yellow crystals of {[Zn(L)(DMF)2]}n (1) and {[Cd(L)(DMF)]·DMF}n (2) suitable for single crystal X-ray structure determination.
3.2. Crystal structures
The single crystal X-ray structure determination of compound 1 revealed the formation of a coordination polymer consisting of (![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 01)-oriented chains (Fig. 1B).
01)-oriented chains (Fig. 1B).
Depending on the definition of a zinc–oxygen bond, each zinc(II) ion is surrounded by either two κ1- or κ2-coordinating carboxylate groups and two DMF molecules (Fig. 2). The coordination sphere is slightly distorted tetrahedral, if we consider only strong Zn–O bonds in the range of 191.7(3)–204.7(3) pm (Table S2 in ESI†). Weaker Zn–O interactions are visualised as dashed lines in Fig. 2 (Zn1–O3′: 250.9(3)pm; Zn1–O2: 295.4(3) pm).
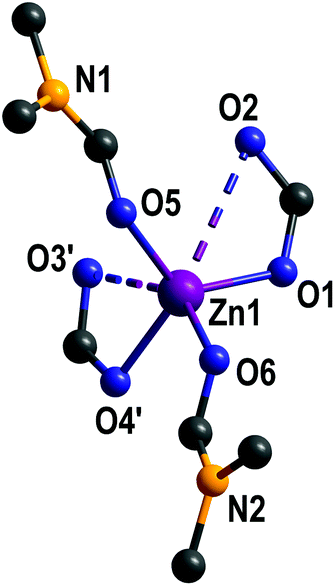 | ||
| Fig. 2 Coordination environment of zinc(II) in {[Zn(L)(DMF)2]}n (1). Hydrogen atoms and non-coordinating DMF are omitted for clarity. | ||
Compound 2 forms a three-dimensional coordination polymer (Fig. 1C). Each cadmium(II) is octahedrally coordinated by three κ1- and one κ2-coordinating carboxylate and one DMF molecule (Fig. 3). Cd–O bond lengths are in the range of 219.3–243.9 pm (Table S3 in ESI†).
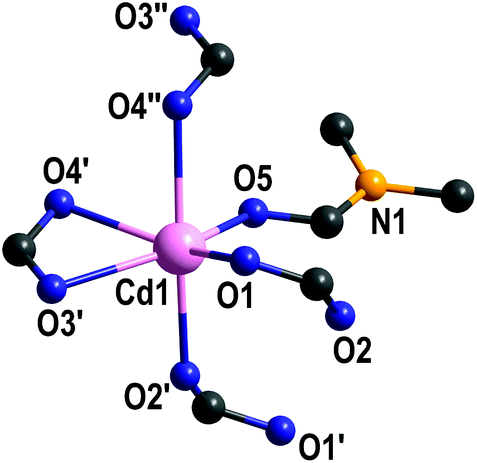 | ||
| Fig. 3 Coordination environment of cadmium(II) in {[Cd(L)(DMF)]·DMF}n (2). Hydrogen atoms and non-coordinating DMF are omitted for clarity. | ||
In 2, two octahedrally coordinated cadmium(II) ions are edge-linked via O4 and O4′, whereas the bridging carboxylate groups (O1, O2 and O1′, O2′, respectively) connect these dimers to infinite chains along (100) with alternating Cd–Cd distances of 379.3 pm and 430.6 pm (Fig. 4; top). The deprotonated organic linker L2− connects these chains to form a three-dimensional network with accessible ellipsoidal-shaped channels along the crystallographic a axis, which are filled with DMF molecules. Taking van der Waals radii into account, the dimensions of these channels are approximately 400 × 1200 pm (Fig. 1C).
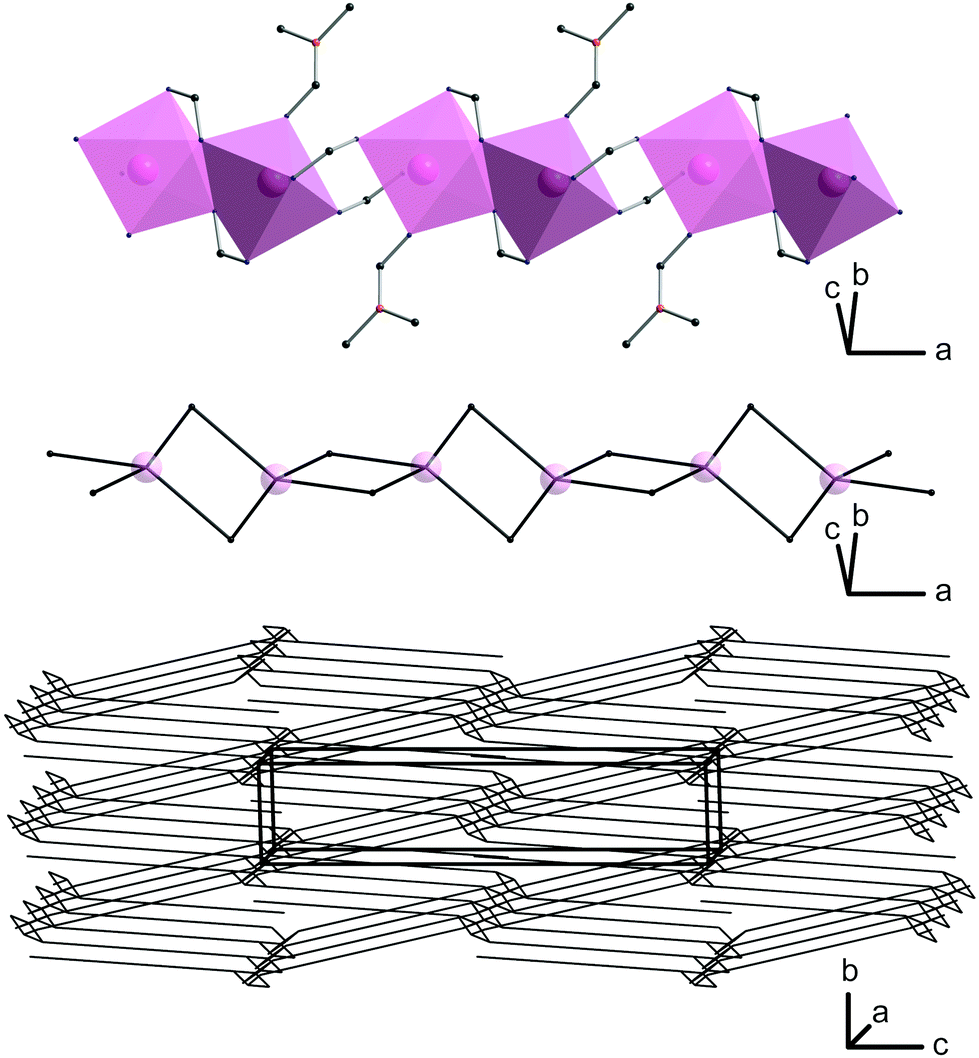 | ||
| Fig. 4 Octahedrally coordinated cadmium(II) in 2 forming chains along the a axis (top), its transformed 42-net (middle) and the resulting final dmd net-type (bottom). | ||
A more detailed topological analysis of the underlying network was performed using the cluster simplification method implemented in ToposPro.25 For a network, one-dimensional coordinating ligands like DMF are ignored, and it is no longer important, if two octahedral units are edge-linked by one or two coordinating atoms of the same linking carboxylic unit. For these reasons, the chains of octahedrally coordinated cadmium(II) ions form a fourfold meshed net along the a axis (Fig. 4; middle). The organic linker connects these units to the final three-dimensional net of dmd topology (Fig. 4; bottom) as categorised by the Reticular Chemistry Structure Resource (RCSR).
3.3. Thermogravimetric and X-ray powder diffraction (XRPD) analyses
Thermogravimetric analysis (TGA) was carried out under argon atmosphere to examine the stability of compound 1 in the temperature range from 20 to 500 °C and 2 in the temperature range from 20 to 600 °C. While the TGA curve of compound 1 shows a continuous weight loss without clearly defined steps, the TGA curve of compound 2 showed a weight loss of around 8% at 120–160 °C, which can be attributed to loss of uncoordinated DMF molecules in the pores (10.05%, theoretical mass percentage). The second weight loss around 300 °C is attributed to decomposition.The powder X-ray diffractograms of 1 and 2 (Fig. S6 and S7, ESI†) are in excellent agreement with the simulated data. According to powder XRD, elemental analysis and TG-DTA, the framework of compound 2 stays intact after removal of the solvent.
3.4. Electrochemical response towards H2O2
In order to examine the electrochemical response towards H2O2 of compounds 1 and 2, cyclic voltammetry studies in the presence and absence of H2O2 were performed as follows: (A) at a bare glassy carbon (GC) electrode, (B) GC electrode modified with H2L, (C) GC electrode modified with compound 1, and (D) GC electrode modified with compound 2. All cyclic voltammograms were recorded in a phosphate buffer solution (pH = 7) at a scan rate of 50 mV s−1, in a potential range between 0 and −0.5 V. In the absence of H2O2, the results clearly reveal no significant reduction peak in the selected potential window, while in the presence of H2O2 a significant reduction peak is observed in all four cases (Fig. 5).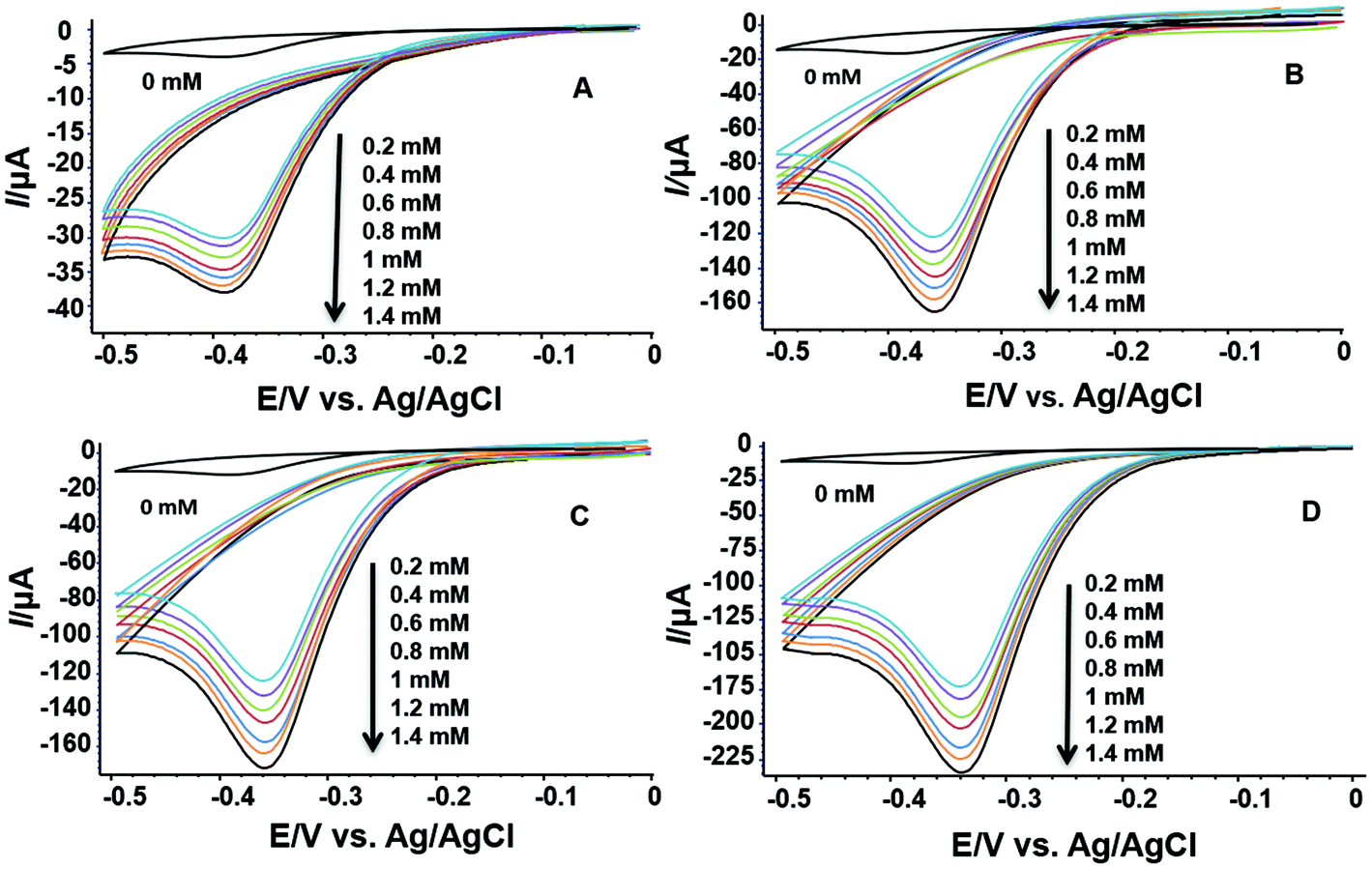 | ||
| Fig. 5 Electrochemical reduction of H2O2 (various concentrations) on (A) bare GC electrode, (B) GC electrode modified with H2L, (C) GC electrode modified with compound 1, and (D) GC electrode modified with compound 2. | ||
Furthermore, increasing the concentration of H2O2 evidently leads to increasing voltammetric response (Fig. 5). Since the amount of active material on the GC electrode was constant during the measurements, the reduction peak is clearly dependent only on the concentration of H2O2 in the electrochemical cell. This effect is most pronounced in the case where the electrode is modified with compound 2 (Fig. 5D), where the reduction peak current is six times higher than the one of the bare glassy carbon electrode (Fig. 5A).
As can be seen from Fig. 6, the reduction peak potential in the case where the electrode is modified with H2L is −0.360 V, showing greater tendency towards reduction of H2O2 than the electrode itself, thus indicating that H2L itself exhibits catalytic activity (Fig. 6B). In the case where the GC electrode is modified with compound 1 (Fig. 6C), the reduction peak potential is 0.358 V showing similar catalytic activity as H2L. However, it is evident that compound 2 shows significantly higher electrocatalytic activity with a reduction peak potential of −0.338 V. A plausible explanation for this would be that whereas the conjugated ligand (L2−) clearly acts as a driving force, the structure and the electron density around Cd2+ also play an important role.
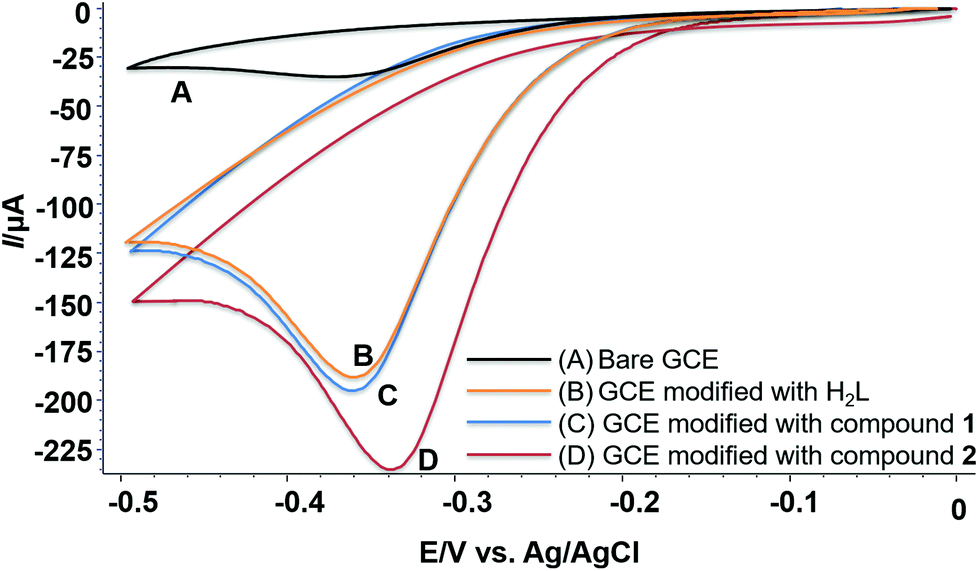 | ||
| Fig. 6 Voltammetric response of (A) bare GC electrode; (B) GC electrode modified with H2L, (C) GC electrode modified with compound 1, and (D) GC electrode modified with compound 2 in 1.4 mM H2O2. | ||
The electrochemical stability of the electrode modified with compounds 1 or 2 was confirmed by consecutive CV measurements in various concentrations of aqueous H2O2. Additionally, experiments were repeated after keeping the modified electrodes in water in the refrigerator for two weeks and showed only a decrease of around 2–3% in the current in all concentrations of H2O2. After consecutive measurements, powder X-ray diffraction was repeated and revealed an unaltered morphology of the CPs.
All three cases of modified GC electrodes showed a good linear relationship between the reduction current and H2O2 concentration in the range of 0.2 to 1.4 mM (Fig. 7). The values for R2 indicate that the regression equation is applicable for the determination of unknown H2O2 concentrations. These findings corroborate the suitable employment of electrodes modified with inorganic polymers based on highly conjugated linkers in sensing electron-transfer reactions.
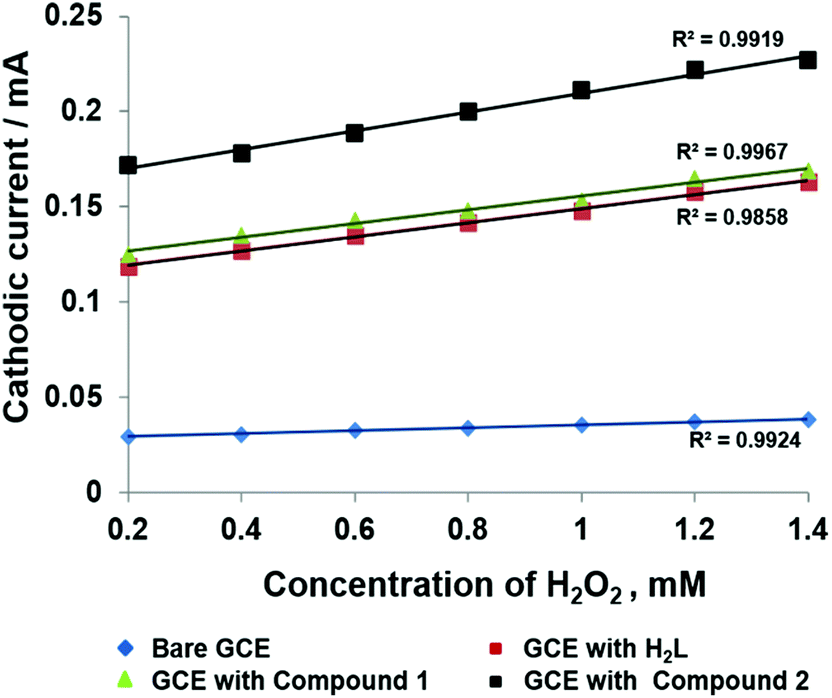 | ||
| Fig. 7 Relationship between cathodic current and H2O2 concentration in the four cases mentioned above (Fig. 5). | ||
3. Conclusions
In summary, this paper reports the synthesis and characterisation of a Zn-based one-dimensional coordination polymer 1 and Cd-based metal–organic framework 2 based on a novel anthracene dicarboxylate ligand. The porosity of compound 2 stays intact after removal of solvent molecules in the pores. Compounds 1 and 2 as well as H2L were tested for their electrocatalytic activity towards H2O2. Easy preparation of glassy carbon electrodes modified with these compounds that can act as non-enzymatic amperometric sensor for detection of H2O2 is presented. The electrode modified with compound 2 showed the most pronounced response towards H2O2 reduction. The cathodic peak potentials indicate that the structure plays a crucial role in the catalytic performance. The linear concentration range of H2O2 from 0.2 mM to 1.4 mM follows a linear increase in reduction current in all three cases, providing strong evidence that fused and highly conjugated systems incorporated in inorganic polymers can serve as an excellent means for sensing electron-transfer reactions using modified electrodes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the DAAD (doctoral grant to A.A., GSSP) and the Graduate School BuildMoNa.Notes and references
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252 CrossRef CAS; X. Chia and M. Pumera, Nat. Catal., 2018, 1, 909 CrossRef; H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Qiao, Chem. Rev., 2018, 118, 6337 CrossRef PubMed; Z. Seh, J. Kibsgaard, C. Dickens, I. Chorkendorff, J. Norskov and T. Jaramillo, Science, 2017, 355, 146 CrossRef PubMed.
- Q. Zhou and G. Shi, J. Am. Chem. Soc., 2016, 138, 2868 CrossRef CAS PubMed; R. Kingsborough and T. Swager, Chem. Mater., 2000, 12, 872 CrossRef; A. Malinauskas, Synth. Met., 1999, 107, 75 CrossRef.
- M. Montiel, F. Vidal-Iglesias, V. Montiel and J. Gullón, Curr. Opin. Electrochem., 2017, 1, 34 CrossRef CAS; L. Wang, Z. Zeng, C. Ma, Y. Liu, M. Giroux, M. Chi, J. Jin, J. Greeley and C. Wang, Nano Lett., 2017, 17, 3391 CrossRef PubMed.
- Y. Liu, J. Zhang, Y. Cheng and S. Jiang, ACS Omega, 2018, 3, 667 CrossRef CAS PubMed; W. Wang, F. Lv, B. Lei, S. Wan, M. Luo and S. Guo, Adv. Mater., 2018, 28, 10117 CrossRef PubMed; F. Mauench, Catalysts, 2018, 8, 597 CrossRef.
- D. Higgins, P. Zamani, A. Yu and Z. Chen, Energy Environ. Sci., 2016, 9, 357 RSC; M. Liu, R. Zhang and W. Chen, Chem. Rev., 2014, 114, 5117 CrossRef CAS PubMed.
- A. Doménech, H. García, M. Doménech-Carbó and F. Llabrés-i-Xamena, J. Phys. Chem. C, 2007, 111, 13701 CrossRef CAS; A. Morozan and F. Jaouen, Energy Environ. Sci., 2012, 5, 9269 RSC; L. Liu, Y. Zhou, S. Liu and M. Xu, ChemElectroChem, 2018, 5, 6 CrossRef; H. Tabbassum, W. Guo, W. Meng, R. Zhao, Q. Wang and R. Zou, Adv. Energy Mater., 2017, 7, 1601679 Search PubMed.
- G. Givaja, P. Amo-Ochoa, C. J. Gomez-Garcıa and F. Zamora, Chem. Soc. Rev., 2012, 41, 115 RSC.
- H. Al-Kutubi, J. Gascon, E. Sudholter and L. Rassaei, ChemElectroChem, 2015, 2, 462 CrossRef CAS; K. Pirzadeh, A. Ghoreyshi, M. Rahimnejad and M. Mohammadi, Korean J. Chem. Eng., 2018, 35, 974 CrossRef.
- Z. Peng, Z. Jiang, X. Huang and Y. Li, RSC Adv., 2016, 6, 13742 RSC; H. Valizadeh, J. Tashkhourian and A. Abbaspour, Microchim. Acta, 2019, 186, 455 CrossRef CAS PubMed; X. Feng, K. Zhang, M. A. Hempenius and G. J. Vancso, RSC Adv., 2015, 5, 106355 RSC.
- C. Liu, Y. Liu and Z. Yang, Neurosci. Lett., 2014, 560, 112 CrossRef CAS PubMed.
- M. R. Kopaei, M. Setorki, M. Doudi, A. Baradaran and H. Nasri, Int. J. Prev. Med., 2014, 5, 927 Search PubMed.
- N. G. Milton, Drugs Aging, 2004, 21, 81 CrossRef CAS PubMed.
- N. A. Burns, I. M. Ahmad, Y. Zhu, L. W. Oberley and D. R. Spitz, Biochem. J., 2009, 418, 29 CrossRef PubMed.
- W. Chen, S. Cai, Q. Ren, W. Wen and Y. Zhao, Analyst, 2012, 137, 49 RSC.
- H. Liu, L. Weng and C. Yang, Microchim. Acta, 2017, 184, 1267 CrossRef CAS.
- N. Lopa, M. Rahman, F. Ahmed, S. Sutradhar, T. Ryu and W. Kim, Electrochim. Acta, 2018, 274, 49 CrossRef CAS; Y. Fu, J. Dai, Y. Ge, Y. Zhang, H. Ke and W. Zhang, Molecules, 2018, 23, 2552 CrossRef PubMed.
- J. Mahmood, M. Anjum and J. Baek, Adv. Mater., 2018, 31, 1805062 CrossRef PubMed.
- K. Darzinezhad, M. M. Amini, E. Mohajerani, M. Armaghan, T. Knedal, A. Abareghi and C. Janiak, Dalton Trans., 2019, 48, 3695 RSC; J. Wang, T. Hu and X. Bu, CystEngComm, 2011, 13, 5152 RSC.
- A. Slodek, M. Filapek, E. Schab-Balcherzak, M. Grucela, S. Kotowicz, H. Janeczek, K. Smolarek, S. Mackowski, J. Malecki, A. Jedrzejowska, G. Szafraniec-Gorol, A. Chrobok, B. Marcol, S. Krompiec and M. Matussek, Eur. J. Org. Chem., 2016, 4020 CrossRef CAS.
- R. Sekiya, S. Nishikiori and K. Ogura, Inorg. Chem., 2006, 23, 9233 CrossRef PubMed.
- CrysAlis Pro: Data collection and data reduction software package, Rigaku Oxford Diffraction Search PubMed.
- G. M. Sheldrick, SHELXT, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- K. Brandenburg, DIAMOND 4: Crystal Impact GbR, Bonn, Germany Search PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Powder X-ray diffraction patterns, thermogravimetric analysis, IR spectra, UV-Vis spectra. CCDC 1980173–1980175. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/D0DT00333F |
| This journal is © The Royal Society of Chemistry 2020 |