
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aerobic dehydrogenation of amines to nitriles catalyzed by triazolylidene ruthenium complexes with O2 as terminal oxidant†
Marta
Olivares
 ,
Pascal
Knörr
and
Martin
Albrecht
*
,
Pascal
Knörr
and
Martin
Albrecht
*
Departement für Chemie und Biochemie, Universität Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: martin.albrecht@dcb.unibe.ch
First published on 21st January 2020
Abstract
Pyridyl-substituted mesoionic triazolylidene ruthenium cymene complexes catalyze the oxidation of both aromatic and aliphatic amines to nitriles with high activity and selectivity under benign conditions using dioxygen as the terminal oxidant. Modification on the pyridyl moiety of the ligand scaffold has negligible effect on the catalytic performance, while substituents on the triazolylidene directly affect the catalytic fitness of the metal center, leading to distinct catalytic profiles. Pre-dissociation of the cymene ligand and formation of a solvento analogue further enhances the catalytic activity towards nitrile formation. Variation of reaction conditions provided valuable mechanistic insights and resulted in a highly efficient protocol for nitrile formation with maximum turnover numbers around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The turnover frequency reaches up to 400 h−1, providing one of the fastest catalytic systems known to date for this transformation.
000. The turnover frequency reaches up to 400 h−1, providing one of the fastest catalytic systems known to date for this transformation.
Introduction
Nitriles are a prominent class of organic molecules which serve as versatile intermediates in organic transformations and are included in a wide variety of natural products and biologically active compounds.1,2 Thus, nitriles are valuable synthetic building blocks in the synthesis of fine chemicals.3,4 They are prepared by conventional methods such as dehydration of amides/aldoximes,5,6 through cyanation of alkyl or aryl halides,7 or by the Sandmeyer and Schmidt-type reactions,8,9 among many others.10,11 These traditional synthetic routes to prepare nitriles proceed with low atom economy, produce stoichiometric waste, require toxic reagents like HCN, have limited selectivity and often require harsh reaction conditions.12 In recent years, a substantial amount of research has been directed towards the development of direct oxidative dehydrogenation (ODH) of primary amines to nitriles by molecular oxygen in the presence of transition metal catalysts, a pathway that starts from abundant low-value precursors and avoids toxic reagents and harsh reaction conditions.13,14 However, the ODH of primary amines to nitriles is a challenging task, as it requires the removal of four electrons and four protons and generally involves several competing pathways, such as dehydrogenation, coupling and transamination.15 Amine dehydrogenation can hence lead to a broad range of products such as oximes, imines, amides, nitriles, amine oxides and azo compounds. For this reason, the design and development of efficient and selective catalytic systems is of great importance. McWhinnie et al. first reported the conversion of amine ligands coordinated to a ruthenium(II) center to nitriles upon exposure to oxygen under ambient conditions.16,17 Later, Taube and co-workers studied the oxidation of different types of amine ligands coordinated to ruthenium centers.18,19 Pioneering studies of Tang et al. reported the first catalytic reaction for the oxidative dehydrogenation of amines with high activity but poor selectivity when using 2–3 atm of oxygen at 100 °C.20 This study triggered the development of numerous ruthenium-based homogeneous catalytic systems for the aerobic oxidation of amines.21–24 For example, James et al. reported the synthesis of a ruthenium–porphyrin complex that exhibited 100% selectivity under mild conditions (50 °C, <16 h).21 Later on Parvulescu introduced a faster novel ruthenium–terpyridyl complex that showed conversion of amines to nitriles after 2 h when using 5 atm of oxygen at 60 °C,25 while Maiti and co-workers developed new ruthenium hydrido complexes for the selective generation of nitriles and imines by varying the catalyst instead of the substrates.26 More recently, different N-chelated ruthenium complexes27–29 as well as simple [Ru(arene)Cl2]2 complexes have been demonstrated to be highly competent in amine dehydrogenation.30,31Herein we describe new ruthenium(II) complexes featuring a range of functionalized triazolylidene ligands and their catalytic activity in amine oxidation. This subclass of N-heterocyclic carbene (NHC) ligands32,33 is particularly attractive due to the strong σ-donor capability and easy synthetic accessibility through versatile “click chemistry”.34–38 Mesoionic NHCs can act as electron reservoirs, displaying a non-innocent behavior when coordinated to metal centers. Specifically in this work, we have included pendant ester, ether and aliphatic groups adjacent to the carbene carbon, as this creates opportunities to electronically modulate the catalytic properties of the metal center. Further modifications include the pyridyl moiety by incorporation of a methoxy group in different positions, as well as the ancillary ligands through replacing the p-cymene with MeCN ligands and formation of the corresponding solvento complex. Interestingly, these mesoionic triazolylidene ruthenium complexes display higher catalytic activity than other catalytic systems reported so far for the oxygen-mediated conversion of amines into the corresponding nitriles.
Results and discussion
Synthesis and characterization of a series of pyridyl–triazolylidene ruthenium complexes
The differently functionalized triazolium salts 1–3 were prepared via a previously reported procedure.39 Metalation of 1–3 and formation of the corresponding triazolylidene ruthenium(II) complexes 4–6 was achieved via a silver triazolylidene intermediate and in situ carbene transfer to [RuCl2(p-cymene)]2 in the presence of Me4NCl (Scheme 1). The chloride salt serves both as a means to remove silver upon transmetalation and to prevent scrambling of the anionic ancillary ligand in the ruthenium complex (Cl−vs. OTf−). Purification by column chromatography yielded complexes 4–6 as orange air-stable solids in 20–60% yield.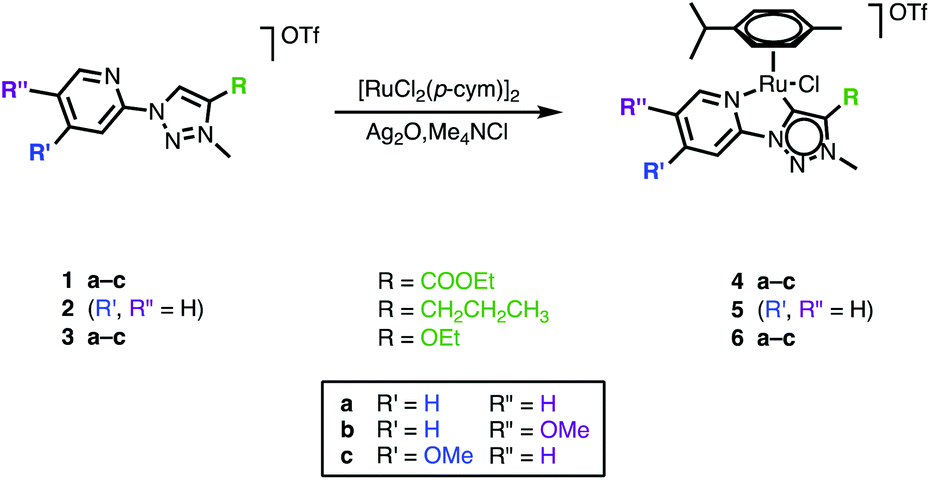 | ||
| Scheme 1 Synthesis of 1,2,3-triazolylidene ruthenium(II) complexes 4–6. | ||
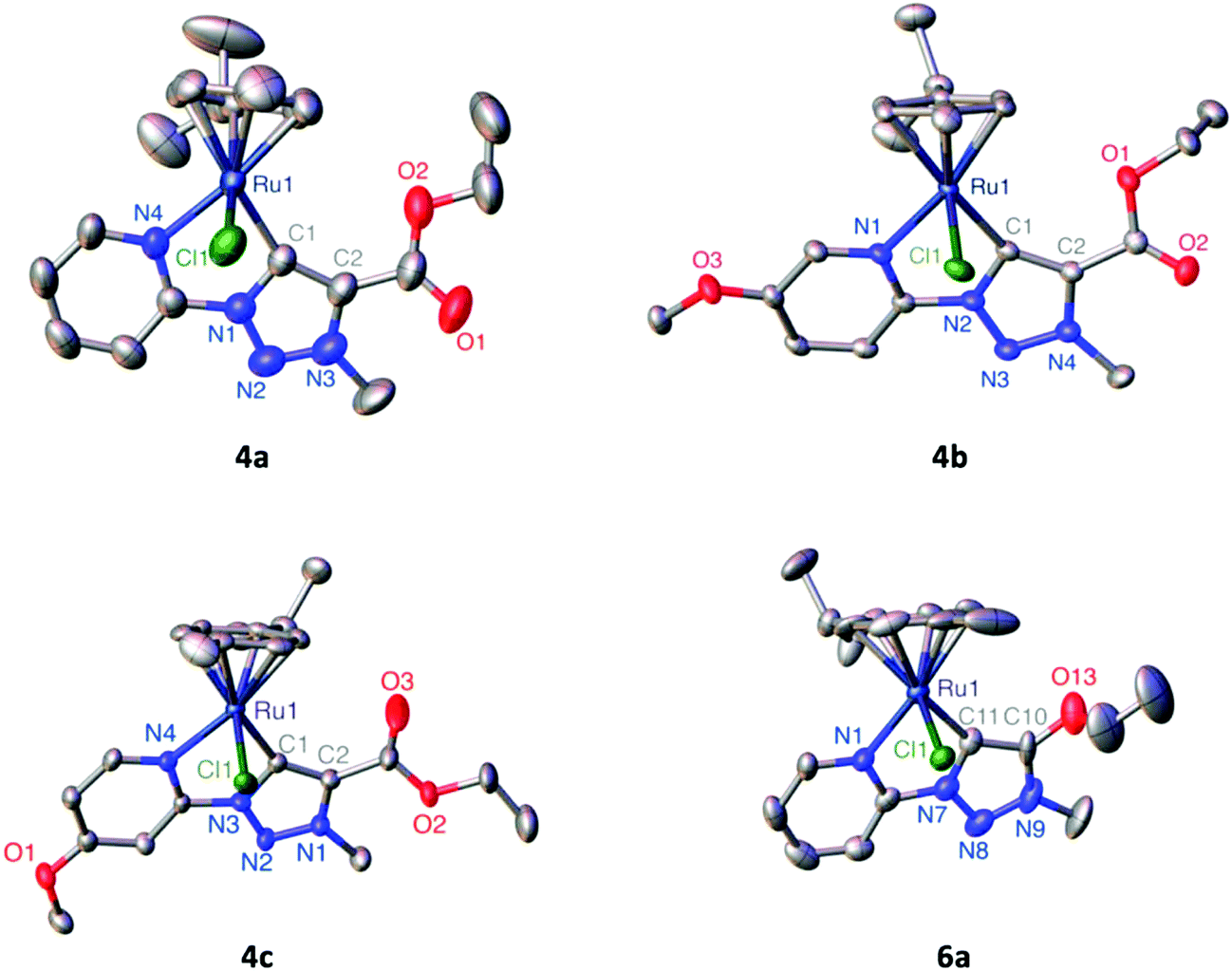 | ||
| Fig. 1 ORTEP presentation of complexes 4a–c and 6a (50% probability ellipsoids, non-coordinating OTf− anions and co-crystallized solvent molecules omitted for clarity). | ||
|
4aa |
4b |
4cb |
6ab |
|
|---|---|---|---|---|
| a Data from ref. 41. b Unit cell contains two independent complex molecules; bond lengths (Å) and angles (°) only given for one of these molecules as the second features identical bond lengths and angles within esd's. | ||||
| Ru–Npy | 2.0983(18) | 2.1024(15) | 2.108(4) | 2.113(7) |
| Ru–Ctrz | 2.049(2) | 2.0570(19) | 2.034(6) | 2.032(8) |
| Ru–Cgcym | 1.706(10) | 1.705(8) | 1.707(2) | 1.692(3) |
| Ru–Cl | 2.4004(6) | 2.4091(5) | 2.4022(13) | 2.395(2) |
| Ctrz–Ctrz | 1.399(3) | 1.392(3) | 1.399(8) | 1.387(12) |
| Ctrz–Ru–Npy | 77.92(8) | 77.60(7) | 77.7(2) | 76.4(3) |
Catalytic oxidation of amines to nitriles
The catalytic activity of the novel ruthenium complexes 4–6 was evaluated in amine oxidation using 4-methylbenzylamine as model substrate and 1,2-dichlorobenzene as solvent. An initial run with complex 4b as catalyst precursor at 5 mol% catalyst loading produced the corresponding nitrile as the major product in a high 86% yield after 3 h. The catalytic profile of the reaction reveals rapid conversion of the substrate that is almost complete within the first hour of reaction (94% conversion) to yield a mixture of benzonitrile (29%) and the corresponding imine (49%; Fig. 2). Nitriles are formed by a double-dehydrogenation of amines, while the secondary aldimine is a product from mono-dehydrogenation followed by condensation with a substrate amine. Although imine formation is favored over nitrile production in the first hour, the imine product is intermittent and disappeared over time. Consumption of the imine was accompanied with an increase of nitrile formation and the appearance of traces of tolylaldehyde, suggesting that the imine is formed as a transient species. After 3 h, all imine was consumed, and the corresponding nitrile was the main product together with low quantities (<15%) of aldehyde as a side-product. A blank reaction under identical conditions yet in the absence of complex 4b led to slow formation of small quantities of imine (15% after 22 h), yet no nitrile nor aldehyde was observed, indicating a pivotal role of the triazolylidene ruthenium complex as catalyst for amine oxidation.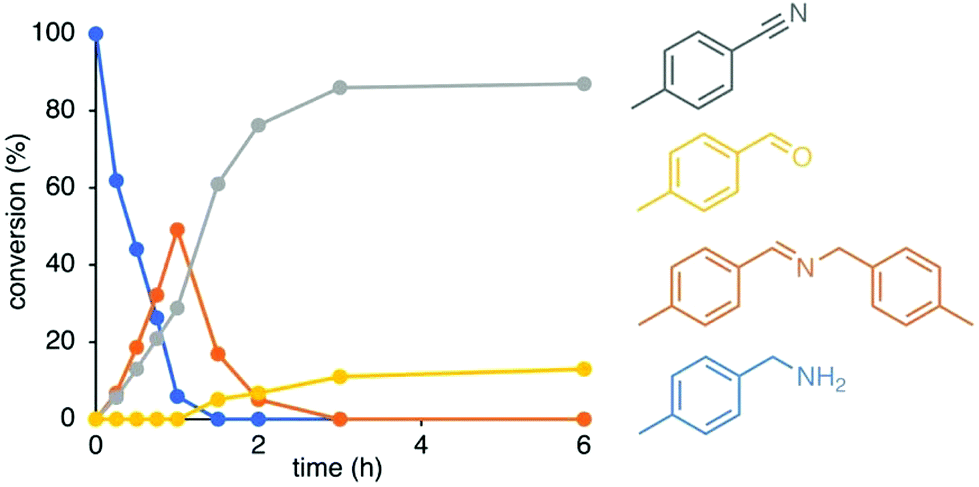 | ||
| Fig. 2 Time-dependent conversion profile for the catalytic oxidation of 4-methylbenzylamine with complex 4b. General conditions: 4-Methylbenzylamine (0.2 mmol), [Ru] (0.01 mmol, 5 mol%), 1,2-dichlorobenzene (2 mL), 150 °C. Conversions were determined by 1H NMR integration (1,3,5-trimethoxybenzene as internal standard) and are averaged over 2 runs. | ||
To better understand the catalytic profile and optimize the reactions conditions, a series of experiments were monitored under strict control of the atmosphere (Table S1†). The first set of experiments were directed towards enhancing the selectivity of the oxidation towards either nitrile or imine formation. Therefore, the reaction was run under inert atmosphere using dry solvent either in an open system with a condenser and argon atmosphere (1 bar) or in a closed Schlenk flask with a small headspace. Both experiments barely gave any products. However, the reaction proceeded well when performed under an atmosphere of air, both in an open and closed system. Under these conditions, a mixture of nitrile and imine formed within 1 h in about 30% and 50% yield, respectively, suggesting H2O rather than H2 as the potential by-product of the dehydrogenation of the amine to the aldimine. Moreover, the fact that both imine and nitrile are formed in similar ratios irrespective of whether the reactor is an open or closed system strongly suggests that the by-product does not inhibit the catalytic reaction. Further support for this conclusion was provided by the insensitivity of the reaction to H2 atmosphere or moist solvent. Thus, when a reaction was first performed for 1 h in air followed by saturation with H2, no hydrogenation of the imine or the nitrile back to the benzylamine was observed even after 1 h under H2 atmosphere. Hence, the ruthenium catalyst derived from complex 4b catalyzes the dehydrogenation of amines and imines, but not the revers, i.e. the hydrogenation of imines or nitriles. Continuous saturation of the reaction mixture with air did slow down the dehydrogenation, however the activity is substantially enhanced when the reaction was run under an atmosphere of molecular oxygen. These conditions reduced the time to completion from 3 to 1 h, and they also markedly enhanced the product selectivity to afford the nitrile in high yield (89%), with only 6% imine formed (15:1 nitrile/imine selectivity, cf. 1:1.7 selectivity in air after 1 h).
Attempts to detect the formation of water during the reaction by NMR spectroscopy were not conclusive. However, monitoring the gas phase of the catalytic reaction with anhydrous cobalt(II) chloride, as an indicator showed the characteristic color change from blue to purple only when the Ru complex was present, thus confirming formation of water as a by-product. These experiments clearly indicate that oxygen is essential for this dehydrogenation acting as the terminal oxidant. While an inert atmosphere impedes the reaction, air leads to full conversion of the amine in 1 h to an imine/nitrile mixture, and an atmosphere of oxygen further accelerates the dehydrogenation and produces the nitrile in high yield and selectivity within the same time.
The formation of high ratios of nitrile from intermittent imine/nitrile ratios implies that N-substituted aldimines are converted to nitriles. This transformation is suggested to involve a presumably non-catalyzed equilibrium between the primary and secondary aldimine III, with the former prone to undergo a second dehydrogenation to generate the nitrile product II (Scheme 2). We note that the aldehyde IV is formed in much smaller quantities than the nitrile once all amine is consumed (cf.Fig. 2), which excludes quantitative imine transformation to the aldehyde. Formation of aldehyde is surmised to be a side reaction from competition of water with ammonia in the reaction with the N-substituted imine.
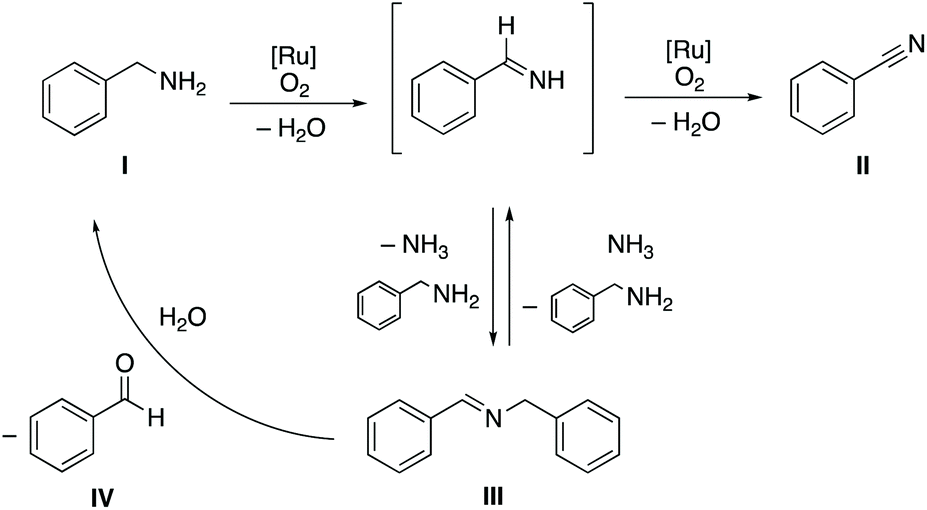 | ||
| Scheme 2 Benzylamine dehydrogenation using oxygen as terminal oxidant by ruthenium(II) complex bearing pyridyl-triazolylidene ligand. | ||
In order to evaluate the role of the pyridyl–triazolylidene ligand, the activity of complexes 4–6 with different ligand substitution patterns were compared in amine oxidation catalysis under aerobic reaction conditions. Complexes 4a–c with different substituents on the pyridyl moiety feature an essentially identical time-conversion profile with no significant difference in activity nor selectivity (Fig. S7,† entries 1–3, Table 2). This outcome suggests that the pyridyl site does not have a direct impact on the catalytic activity. However, complexes 5 and 6a bearing a propyl and an ethoxy group on the triazolylidene unit, respectively, were considerably more active at early stage. Conversions reached 90% already after 30 min and were quantitative within 1 h compared to about 50% at 30 min with complexes 4a–c (entries 4 and 5). Higher activity is also quantified by the improved turnover frequency at 50% conversion, TOF50, which reaches 33 h−1 with complex 6a containing an OEt substituent at the triazolylidene unit (cf. TOF50 = 20 h−1 for 4a). Moreover, only trace amounts of aldehyde were detectable when using complexes 5 and 6a as catalyst precursors. The higher activity of complexes 5 and 6a indicates that the electron-donating properties of the triazolylidene are relevant for imparting high catalytic activity. Moreover, complexes 5 and 6a showed formation of nitrile and imine in an equal ratio within the first 30 min of the reaction, whereas complexes 4a–c formed preferentially the imine over the nitrile at early stage (Table 2). Although the conversion of the substrate took less time and the amount of imine formed was lower when using complex 5 and 6a compared to complexes 4a–c, it is worth noting that the transformation of the initially formed imine intermediate to benzonitrile required about the same time for all complexes 4–6, suggesting a rate-limiting step for this transformation that is not metal-catalyzed. We assume that this second transformation is rate-limited by the NH3 concentration, which is required to generate the primary aldimine that is then dehydrogenated to nitrile (cf.Scheme 2).
a
| Entry | Complex | Conversion/% | Product yieldb/% | TOF50 (h−1) | ||
|---|---|---|---|---|---|---|
| 1 h | 3 h | III/II at 1 h | III/II/IV at 3 h | |||
|
a General conditions: 4-Methylbenzylamine (0.2 mmol), [Ru] (0.01 mmol, 5 mol%), 1,2-dichlorobenzene (2 mL), 150 °C, under aerobic reaction conditions (cf. entry 3, Table S1†).
b II (nitrile), III (imine), IV (aldehyde), see Scheme 2.
|
||||||
| 1 | 4a | 90 | >99% | 48/28 | 0/85/13 | 20 |
| 2 | 4b | 94 | >99% | 49/29 | 0/86/11 | 17 |
| 3 | 4c | 88 | >99% | 54/29 | 0/86/13 | 17 |
| 4 | 5 | 98 | >99% | 21/67 | 1/86/6 | 24 |
| 5 | 6a | 99 | >99% | 23/62 | 3/85/10 | 33 |
Overall, the activities for complexes 5 and 6a vary only subtly and therefore, complex 5 was selected for further optimization since its synthesis is more facile than 6a. Under molecular oxygen, complex 5 reaches full conversion already after 20 min. At this stage, the nitrile/imine ratio is almost 3:1 (67% nitrile, 24% imine; Fig. S8†), considerably higher than the 1:2 ratio observed for 4b at the same stage in air. After 2 h, the reaction is essentially complete, with 96% yield of nitrile together with 4% of aldehyde as side-product.
Since conversion of the secondary imine to the nitrile is independent of the ruthenium complex, yet limits the rate of nitrile formation once all amine is converted, we reasoned that the equilibrium between the primary and secondary imine (cf.Scheme 2) will be favorably shifted to the former if ammonia is supplied. Indeed, a catalytic run performed under oxygen in the presence of gaseous ammonia (6 mL) accelerated nitrile formation considerably. While amine conversion is not affected (20 min, Fig. S9†), the fraction of nitrile increased to 81% (cf. 67% in the absence of ammonia) and the concentration of imine was considerably reduced from 24% to 12%. After 40 min, the reaction was complete and yielded essentially quantitatively the nitrile product (<1% aldehyde detected). These optimized conditions provided an estimated TOF50 = 75 h−1, about a fourfold increase compared to the initial experiments under air and in the absence of added ammonia. Moreover, these conditions also successfully suppress the formation of aldehyde as a side-product due to the kinetically favorable reaction of the secondary imine with NH3vs. H2O, which allows for further dehydrogenation and nitrile formation. This experiment supports the mechanistic proposal and in particular the uncatalyzed re-activation of the secondary imine for nitrile formation. Controlling this equilibrium with the primary imine is critical to accomplish selective oxidation to the nitrile. In the absence of ammonia, competitive reaction of the imine with water, presumably formed as a by-product from aerobic H2 fixation, produces the aldehyde and compromises the selectivity. The shorter reaction time also indicates that while the first dehydrogenation is fast (quantitative conversion of the amine), the second dehydrogenation is impeded by competitive imine formation, i.e. the rate of dehydrogenation to form the nitrile is slower than the rate of condensation of the primary imine with amine to form the secondary imine III. This rate difference depletes the concentration of primary imine if no ammonia is present, while excess ammonia is reversing the formation of the secondary imine and eliminates the otherwise rate-limiting and uncatalyzed aminolysis of the secondary imine for nitrile production. From a process point of view, ammonia addition is unproblematic as it is removed easily due to its low boiling point.
Different primary amine substrates were oxidized by complex 5 under these optimized conditions in an atmosphere of oxygen and ammonia. Both para-substituted benzylamines with electron-donating MeO and electron-withdrawing CF3 groups were converted. Reaction rates are highly similar (Fig. 3), suggesting that the electronically sensitive benzylic C–H bond activation is not part of the turnover-limiting step. In contrast, aliphatic amines were oxidized significantly faster and time to nitrile formation was reduced to 20 min (cf. 40 min for benzylamines).
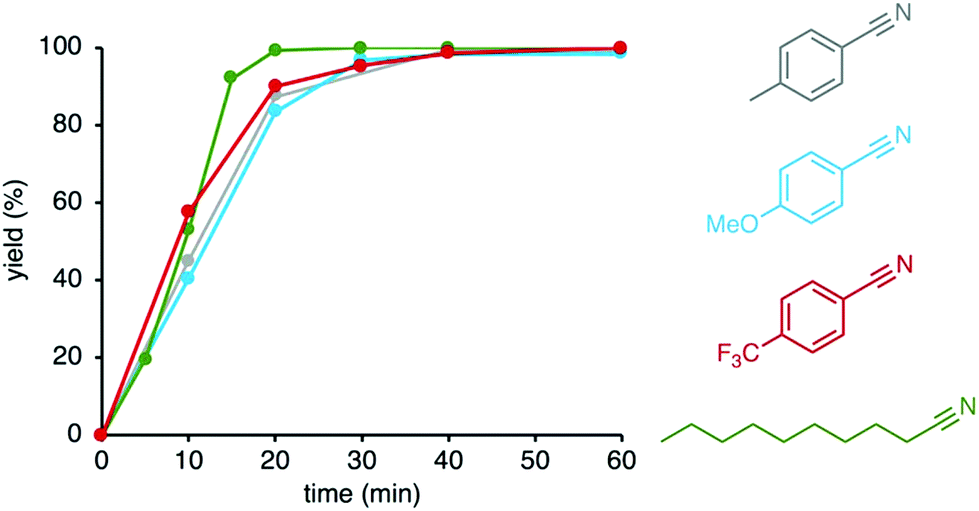 | ||
| Fig. 3 Time-dependent profiles for the catalytic oxidation towards nitrile with complex 5 under ammonia gas (6 mL) and molecular oxygen. General conditions: Amine substrate (0.2 mmol), [Ru] (0.01 mmol, 5 mol%), 1,2-dichlorobenzene (2 mL), 150 °C. Conversions were determined by 1H NMR integration (1,3,5-trimethoxybenzene as internal standard) and are averaged over 2 runs. Isolated yields between 52% (4-Me-benzonitrile) and 86% (4-MeO-benzonitrile). | ||
Mechanistic insights into the nature of the active species were obtained from 1H NMR spectroscopic analysis of the reaction mixture, which revealed the complete disappearance of the resonances due to the ruthenium-bound cymene within 2 h when the reaction was heated at 150 °C. At room temperature, the cymene ligand was stably bound to the ruthenium center over extended periods. These observations suggest a thermally induced activation of the ruthenium complexes 4–6 by cymene dissociation. In order to eliminate this potentially temperature-limiting step, the solvento complex 7 was prepared from the parent cymene analogue 5 upon reaction with AgOTf in refluxing MeCN (Scheme 3) via established procedures.41,46–50 In contrast to complex 5, the solvento complex 7 is moderately air-sensitive and gradually degrades over several days, as indicated by a color change from yellow to green. Successful ligand exchange was indicated by 1H NMR spectroscopy, which showed the absence of the cymene resonances and the appearance of two singlets attributed to coordinated MeCN in 1:2 ratio (δH = 2.54 and 2.07, respectively, CD3CN solution). The observation of only three MeCN ligands is in agreement with fast exchange of the MeCN ligand trans to the carbene with CD3CN.41,46 Unequivocal evidence for the ligand exchange and the octahedral geometry around the ruthenium center of complex 7 was obtained by X-ray diffraction studies.
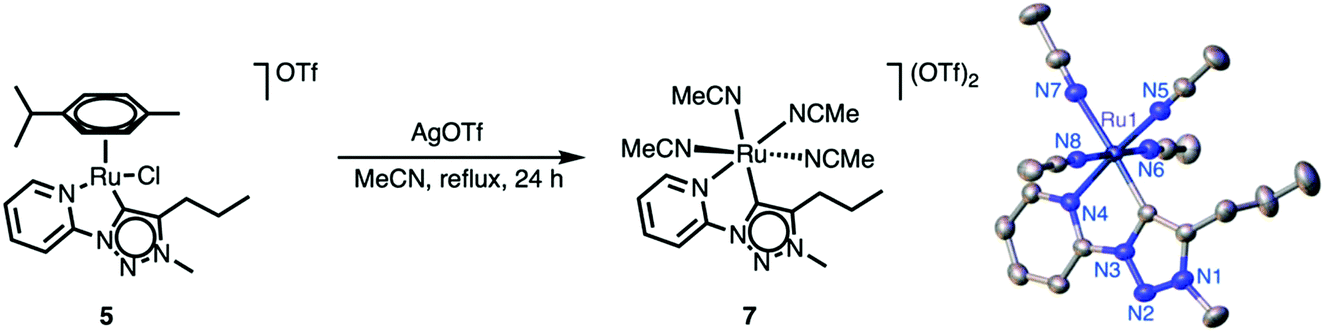 | ||
| Scheme 3 Synthesis of the solvento ruthenium complex 7 from complex 5 and ORTEP plot (50% probability ellipsoids). | ||
Catalytic amine oxidation using the solvento complex 7 as catalyst precursor was indeed considerably accelerated when compared to reactions with complex 5 under identical conditions, reaching full amine conversion already after 10 min (cf. 57% conversion with complex 5 after this time, Fig. 4a). After these initial stages, complex 5 and 7 show similar catalytic profile with formation of nitrile in 81% and 79% yield respectively, suggesting the evolution of an identical catalytic species, which supports the initial loss of cymene from complex 5. However, the solvento complex 7 showed only very modest activity at lower temperature and reached a mere 50% conversion after 3 d at 80 °C, indicating that unlike N-coordinated systems,29,51 high temperatures are needed also for the catalytic dehydrogenation and not only for cymene dissociation during catalyst activation. We note that nitrile formation was more favored when running the catalysis with complex 7 under higher concentration of ammonia gas (30 instead of 6 mL), reaching 71% nitrile and 22% imine after 10 min, and almost full conversion to nitrile (95% yield) after 20 min reaction (Fig. 4b).
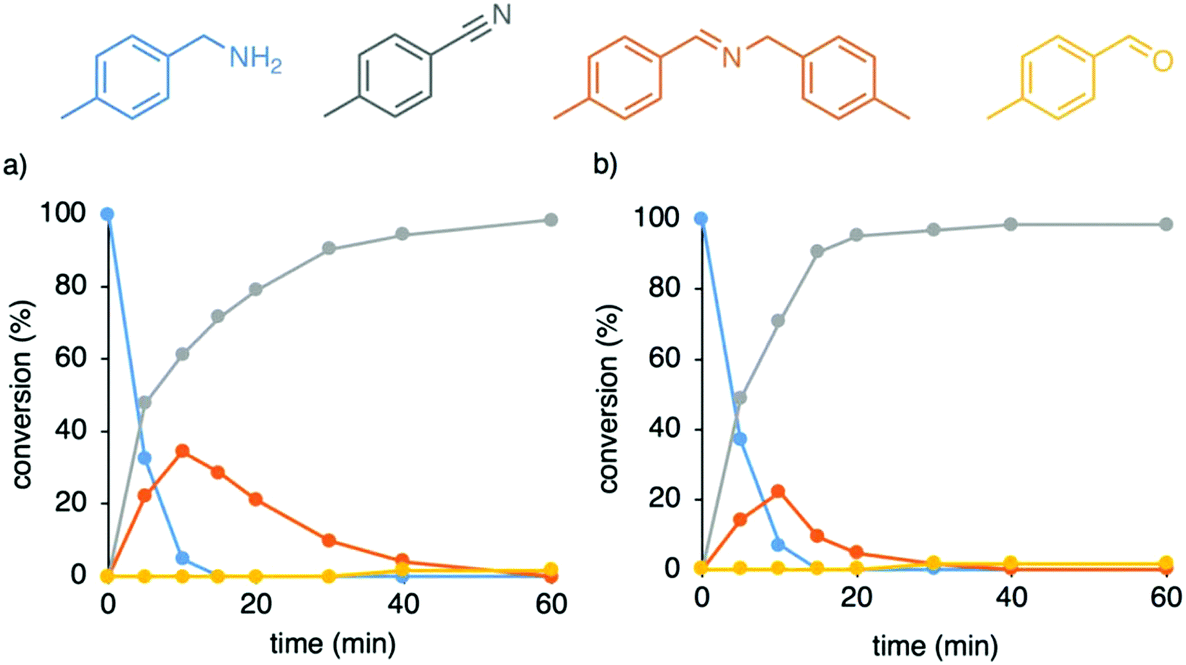 | ||
| Fig. 4 Time-dependent profile for the catalytic 4-methylbenzylamine oxidation with complex 7 under molecular oxygen and (a) 6 mL ammonia gas and (b) 30 mL ammonia gas conditions. General conditions: Amine substrate (0.2 mmol), [Ru] (0.01 mmol, 5 mol%), 1,2-dichlorobenzene (2 mL), 150 °C. Conversions were determined by 1H NMR integration (1,3,5-trimethoxybenzene as internal standard) and are averaged over 2 runs. | ||
The solvento complex 7 is also competently catalyzing the dehydrogenation of aliphatic amines, such as decylamine (Fig. S10†). After 10 min reaction, 93% of the substrate was converted and nitrile formation reached 99% after 15 min, displaying a TOFmax of about 150 h−1. Due to the high activity of complex 7, formation of the imine intermediate or aldehyde as a side-product were not detected by NMR spectroscopy, indicating an excellent selectivity with this catalyst under the applied reaction conditions.
Further studies using complex 7 were carried out to evaluate its catalytic activity at lower catalyst loading. Thus, 4-methylbenzylamine was fully converted at 0.1 mol% catalyst loading with a maximum turnover frequency TOFmax = 250 h−1. Decreasing the catalyst loading to 0.01 mol% increased the time required for full conversion to 6 days, (TON = 10000), indicating a highly robust catalytic cycle and little catalyst degradation. Under these conditions TOFmax reach a remarkable 400 h−1. The catalytic activity is comparable to that reported for simpler systems like [Ru(cym)Cl2]2.30,31 Control experiments indicate that under identical conditions (i.e. 150 °C in 1,2-dichlorobenzene), the activity of this simple complex is indeed comparable to that of complex 7, despite the different ligand set of the two ruthenium species. This similar activity might point to complete ligand loss and the formation of a heterogeneous Ru system as catalytically active species. Addition of elemental mercury to catalytic runs with complex 7 did, however, not poison the catalytically active species,52 and conversion continued at about the same rate as in the absence of Hg (Fig. S11†). These results together with the highly reversible kinetics of the dehydration reaction strongly suggest that a homogeneous catalyst is formed when using triazolylidene ruthenium complexes 4–7 as catalyst precursors.
Conclusion
A new series of 1,2,3-triazolylidene ruthenium(II) complexes were prepared. Simple modifications on the ligand through electronic modulation by introducing electron-donating methoxy substituents on the pyridyl ring (4a–c), or by altering the electronic properties on the triazolylidene heterocycle through the incorporation of different substituents on the C4-position (4a, 5, 6a–c) were evaluated in the catalytic oxidation of 4-methylbenzylamine. While the modifications on the pyridyl moiety series have no significant effects and activities are very similar, the electronic modulation on the triazolylidene however, significantly modulate the catalytic performance. Complexes bearing electron-withdrawing substituent (–COOEt) showed slower conversion of the substrate than those complexes containing electron-donating groups (–OEt or –nPr). These results suggest that the electron donating character on the triazolylidene heterocycle influences the ruthenium metal center in amine oxidation reactions.Overall, all ruthenium(II) complexes were active catalyst precursors for the oxidation of amine under aerobic conditions, achieving very good yields of nitrile product without the need of any additive (86% after 3 h). In the presence of ammonia and molecular oxygen, the catalytic activity is boosted towards formation of nitrile in quantitative yield and with exclusive selectivity towards the nitrile (99% after 40 min). Further optimization of the catalyst by removing the p-cymene ligand to give the acetonitrile solvento complex yielded a high activity catalyst which induces fast and selective amine oxidation to the corresponding nitrile (full conversion within 15 min, TOFmax ∼ 400 h−1) and tolerating catalyst loadings as low as 0.01 mol% (10000 TON), thus constituting one of the most efficient catalyst for the direct oxidation of amines to nitriles to date.
Experimental section
General
The metalation reactions were carried out under nitrogen atmosphere using standard Schlenk techniques, and all the reagents and solvents were used as obtained from commercial suppliers. Triazolium salts 1a,411b,411c,393b,39 and ruthenium complex 4a41 were prepared according to previously reported procedures. Unless specified, NMR spectra were recorded at 25 °C on Bruker spectrometers operating at 400 MHz (1H NMR) and 100 MHz (13C NMR), respectively. Chemical shifts (δ in ppm, coupling constants J in Hz) were referenced to residual solvent signals (1H, 13C). Assignments are based on homo- and heteronuclear shift correlation spectroscopy. All complexes show a quartet around 120 ppm in the 13C NMR spectrum due to the OTf counterion. Purity of the complexes has been established by NMR spectroscopy, and by elemental analysis, which were performed by the University of Bern Microanalytic Laboratory using a Thermo Scientific Flash 2000 CHNS-O elemental analyzer. High-resolution mass spectrometry was carried out with a Thermo Scientific LTQ Orbitrap XL (ESI-TOF).
Synthesis of 2-(4-propyl-1H-1,2,3-triazol-1-yl)pyridine. A solution of 2-azidopyridine (200 mg, 1.66 mmol), 1-pentyne (197 μL, 1.99 mmol), CuSO4·5H2O (83 mg, 0.33 mmol) and Cu powder (74 mg, 1.16 mmol) in tBuOH
:H2O (9:9 mL) were placed into a sealed microwave vial. The reaction was irradiated at 100 °C for 15 h. The mixture reaction was diluted with 100 mL of CH2Cl2 and extracted with NH4OH 10% sol. (2 × 100 mL), H2O (2 × 100 mL) and brine (2 × 100 mL). The organic layer was dried over MgSO4, filtered and all volatiles were evaporated under reduced pressure. The residue was washed with pentane to afford compound 1 as a white solid (285 mg, 91%). 1H NMR (400 MHz, CDCl3): δ = 8.44–8.37 (m, 1H, CpyH), 8.26 (s, 1H, CtrzH), 8.11 (d, 3JHH = 8.2 Hz, 1H, CpyH), 7.87–7.78 (m, 1H, CpyH), 7.25 (ddd, 3JHH = 7.4 Hz, 3JHH = 4.9 Hz, 4JHH = 1.0 Hz, 1H, CpyH), 2.73 (t, 3JHH = 7.4 Hz, 2H, CtrzCH2), 1.71 (h, 2H, 3JHH = 7.4 Hz, CtrzCH2CH2), 0.95 (t, 3JHH = 7.4 Hz, 3H, CH2CH3). 13C{1H} NMR (100 MHz, CDCl3): δ = 149.4 (Cpy–Ntrz), 148.7 (CtrzCH2), 148.4 (CpyH), 139.0 (CpyH), 123.2 (CpyH), 118.1 (CtrzH), 113.7 (CpyH), 27.7 (CH2CH2CH3), 22.5 (CH2CH2CH3), 13.8 (CH2CH3). HR-MS (CH3CN): m/z calculated for C10H13N4 [M + H]+ = 189.1135; found, 189.1131.
Synthesis of triazolium salt 2. To a solution of the 2-(4-propyl-triazol-1-yl)pyridine (420 mg, 2.2 mmol) in CH2Cl2 at 0 °C was added MeOTf (270 μL, 2.45 mmol) and the mixture was stirred at 0 °C for 30 min. Addition of Et2O gave a white solid (740 mg, 94%). 1H NMR (400 MHz, CDCl3): δ = 8.85 (s, 1H, CtrzH), 8.61 (d, 4JHH = 3.8 Hz, 1H, CpyH), 8.16 (d, 3JHH = 8.1 Hz, 1H, CpyH), 8.11–8.05 (m, 1H, CpyH), 7.60 (dd, 3JHH = 7.3 Hz, 4JHH = 4.9 Hz, 1H, CpyH), 4.43 (s, 3H, NCH3), 2.99 (t, 3JHH = 7.7 Hz, 2H, CtrzCH2), 1.89 (m, 2H, CtrzCH2CH2), 1.13 (t, 3JHH = 7.3 Hz, 3H, CH2CH3). 13C{1H} NMR (100 MHz, CDCl3): δ = 149.4 (CpyH), 146.7 (Cpy–Ntrz), 146.2 (CtrzCH2), 140.6 (CpyH), 127.1 (CpyH), 124.2 (CtrzH), 115.4 (CpyH), 39.9 (NCH3), 25.7 (CH2CH2CH3), 20.5 (CH2CH2CH3), 13.7 (CH2CH3). Anal. calcd for C12H15F3N4O3 (352.33): C, 40.91; H, 4.29; N, 15.90. Found: C, 41.08; H, 4.18; N, 15.77. HR-MS (CH3CN): m/z calculated for C11H15N4 [M − OTf]+ = 203.1291; found, 203.1285.
Synthesis of 2-(4-ethoxy-1H-1,2,3-triazol-1-yl)pyridine. The reaction of 2-azidopyridine (500 mg, 4.16 mmol), ethoxyacetylene (2.9 mL, 12.48 mmol), CuI (795 mg, 4.16 mmol) and 2,6-lutidine (970 μL, 8.32 mmol) in THF (27 mL) and DMSO (470 μL) was stirred under reflux for 24 h and afforded this compound as an off-white solid (608 mg, 77%). 1H NMR (300 MHz, CDCl3): δ = 8.47 (ddd, 3JHH = 4.9 Hz, 4JHH = 1.9 Hz, 5JHH = 0.9 Hz, 1H, CpyH), 7.99 (s, 1H, CtrzH), 7.89 (ddd, 3JHH = 8.3 Hz, 3JHH = 7.4 Hz, 4JHH = 1.9 Hz, 1H, CpyH), 7.53–7.46 (m, 1H, CpyH), 7.32 (ddd, 3JHH = 7.4 Hz, 4JHH = 4.9 Hz, 5JHH = 0.9 Hz, 1H, CpyH), 4.33 (q, 3JHH = 7.1 Hz, 2H, OCH2Me), 1.46 (t, 3JHH = 7.1 Hz, 3H, OCH2CH3).
Synthesis of triazolium salt 3a. To a solution of the 2-(4-ethoxy-triazol-1-yl)pyridine (607 mg, 3.2 mmol) in CH2Cl2 at 0 °C was added MeOTf (400 μL, 3.67 mmol) and the mixture was stirred at 0 °C for 30 min. Addition of Et2O gave a white precipitate (245 mg, 69%). 1H NMR (400 MHz, CDCl3): δ = 8.78 (s, 1H, CtrzH), 8.63–8.60 (m, 1H, CpyH), 8.15 (d, 3JHH = 8.2 Hz, 1H, CpyH), 8.07 (td, 3JHH = 7.4 Hz, 4JHH = 4.8 Hz, 1H, CpyH), 4.65 (q, 3JHH = 7.0 Hz, 2H, OCH2Me), 4.23 (s, 3H, NCH3), 1.60 (t, 3JHH = 7.0 Hz, 3H, OCH2CH3). 13C{1H} NMR (100 MHz, CDCl3): δ = 153.9 (Ctrz–OEt), 149.4 (CpyH), 140.5 (CpyH), 127.1 (CpyH), 115.1 (CpyH), 108.6 (CtrzH), 72.9 (OCH2Me), 36.1 (NCH3), 14.5 (OCH2CH3), (Cpy–Ntrz) n.d. HR-MS (CH3CN): m/z calculated for C10H13ON4 [M − OTf]+ = 205.1084; found, 205.1079.
Synthesis of 2-(4-ethoxy-1H-1,2,3-triazol-1-yl)-4-methoxypyridine. The reaction of 2-azido-4-methoxypyridine (500 mg, 3.33 mmol), ethoxyacetylene (2.4 mL, 9.98 mmol), CuI (635 mg, 3.33 mmol) and 2,6-lutidine (770 μL, 6.65 mmol) in THF (27 mL) and DMSO (470 μL) was stirred under reflux for 72 h and afforded this compound as an off-white solid (270 mg, 36%). 1H NMR (300 MHz, CDCl3): δ = 8.23 (d, 3JHH = 5.8 Hz, 1H, CpyH), 7.96 (s, 1H, CtrzH), 7.66 (d, 4JHH = 2.3 Hz, 1H, CpyH), 6.82 (dd, 3JHH = 5.8 Hz, 4JHH = 2.3 Hz, 1H, CpyH), 4.29 (q, 3JHH = 7.0 Hz, 2H, OCH2Me), 3.93 (s, 3H, OCH3), 1.44 (t, 3JHH = 7.0 Hz, 3H, OCH2CH3).
Synthesis of triazolium salt 3c. To a solution of the 2-(4-ethoxy-triazol-1-yl)-4-methoxypyridine (190 mg, 0.86 mmol) in CH2Cl2 at 0 °C was added MeOTf (110 μL, 0.99 mmol) and the mixture was stirred at 0 °C for 15 min. Addition of Et2O gave a white precipitate (150 mg, 45%). 1H NMR (300 MHz, CDCl3): δ = 8.78 (s, 1H, CtrzH), 8.36 (d, 3JHH = 5.8 Hz, 1H, CpyH), 7.69 (d, 4JHH = 2.2 Hz, 1H, CpyH), 7.05 (dd, 3JHH = 5.8 Hz, 4JHH = 2.2 Hz, 1H, CpyH), 4.63 (q, 3JHH = 7.0 Hz, 2H, OCH2Me), 4.23 (s, 3H, NCH3), 4.02 (s, 3H, OCH3), 1.60 (t, 3JHH = 7.0 Hz, 3H, OCH2CH3).
General procedure for the synthesis of the complexes 4–6
The corresponding triazolium salt 1–3 (1 eq.), Ag2O (2 eq.), Me4NCl (1.5 eq.) and [RuCl2(p-cymene)]2 (0.5 eq.), were suspended in CH2Cl2 and stirred under exclusion of light at 40 °C for 24–48 h. The reaction was filtered through Celite and all volatiles were evaporated under reduced pressure. The crude solid was purified by gradient column chromatography (SiO2; CH2Cl2 to CH2Cl2/acetone 10:2). Precipitation with CH2Cl2/Et2O gave the title complexes as pure orange solids.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 157.4 (Cpy–OMe), 143.2 (Cpy–Ntrz), 142.4 (CpyH), 138.9 (Ctrz–COOEt), 125.8 (CpyH), 115.2 (CpyH), 108.5, 105.1 (2 × Ccym–C), 90.2, 89.0, 87.6, 84.3 (4 × CcymH), 63.3 (OCH2Me), 57.3 (OCH3), 42.7 (NCH3), 31.3 (CHMe2), 22.6, 22.4 (2 × CH–CH3), 19.0 (Ccym–CH3), 14.4 (OCH2CH3). Anal. calcd for C23H28ClF3RuN4O6S (682.07): C, 40.50; H, 4.14; N, 8.21. Found: C, 40.35; H, 3.84; N, 7.95. HR-MS (CH3CN): m/z calculated for C22H28O3N4ClRu [M − OTf]+ = 533.0888; found, 533.0882.
O), 156.2 (CpyH), 150.9 (Cpy–Ntrz), 139.0 (Ctrz–COOEt), 114.1 (CpyH), 107.7, 105.3 (2 × Ccym–C), 100.8 (CpyH), 89.9, 88.5, 87.9, 84.1 (4 × CcymH), 63.4 (OCH2Me), 57.3 (OCH3), 42.9 (NCH3), 31.3 (CHMe2), 22.7, 22.4 (2 × CH–CH3), 19.1 (Ccym–CH3), 14.4 (OCH2CH3). Anal. calcd for C23H28ClF3RuN4O6S (682.07): C, 40.50; H, 4.14; N, 8.21. Found: C, 40.27; H, 3.75; N, 7.98. HR-MS (CH3CN): m/z calculated for C22H28O3N4ClRu [M − OTf]+ = 533.0888; found, 533.0865.
O), 157.4 (Cpy–OMe), 143.2 (Cpy–Ntrz), 142.4 (CpyH), 138.9 (Ctrz–COOEt), 125.8 (CpyH), 115.2 (CpyH), 108.5, 105.1 (2 × Ccym–C), 90.2, 89.0, 87.6, 84.3 (4 × CcymH), 63.3 (OCH2Me), 57.3 (OCH3), 42.7 (NCH3), 31.3 (CHMe2), 22.6, 22.4 (2 × CH–CH3), 19.0 (Ccym–CH3), 14.4 (OCH2CH3). Anal. calcd for C23H28ClF3RuN4O6S (682.07): C, 40.50; H, 4.14; N, 8.21. Found: C, 40.35; H, 3.84; N, 7.95. HR-MS (CH3CN): m/z calculated for C22H28O3N4ClRu [M − OTf]+ = 533.0888; found, 533.0882.
O), 156.2 (CpyH), 150.9 (Cpy–Ntrz), 139.0 (Ctrz–COOEt), 114.1 (CpyH), 107.7, 105.3 (2 × Ccym–C), 100.8 (CpyH), 89.9, 88.5, 87.9, 84.1 (4 × CcymH), 63.4 (OCH2Me), 57.3 (OCH3), 42.9 (NCH3), 31.3 (CHMe2), 22.7, 22.4 (2 × CH–CH3), 19.1 (Ccym–CH3), 14.4 (OCH2CH3). Anal. calcd for C23H28ClF3RuN4O6S (682.07): C, 40.50; H, 4.14; N, 8.21. Found: C, 40.27; H, 3.75; N, 7.98. HR-MS (CH3CN): m/z calculated for C22H28O3N4ClRu [M − OTf]+ = 533.0888; found, 533.0865.
General procedure for the 4-methylbenzylamine oxidation
In a 100 mL Schlenk flask, a mixture of complex (0.01 mmol), 4-methylbenzylamine (0.2 mmol) and 1,3,5-trimethoxybenzene (5.6 mg, 0.033 mmol; internal standard) in 1,2-dichlorobenzene (2 mL) was mixed. Then, molecular oxygen was bubbled into the solution for 5 min and 6 mL of gaseous ammonia was injected. An oxygen balloon was connected to the reaction vessel via a septum. The reaction mixture was heated to 150 °C. Aliquots were taken at specific times, diluted with CDCl3 and analyzed by 1H NMR spectroscopy. Conversions and yields were determined relative to 1,3,5-trimethoxybenzene as internal standard. Products were isolated at the end of the reaction by cooling the mixture to room temperature, and subsequent purification by column chromatography using SiO2 as stationary phase and elution first with pentane (100 mL) to remove the 1,2-dichlorobenzene, followed by elution with pentane:Et2O (either 80:20 or 90:10). The NMR spectra of the isolated products showed less than 5% of aldehyde (due to imine hydrolysis).
Crystal structure determinations
Crystal data for 4b, 4c, 6a and 7 were collected using an Oxford Diffraction SuperNova area-detector diffractometer53 with mirror optics monochromated Mo Kα radiation (λ = 0.71073 Å) and Al filtered.54 Data reduction was performed using the CrysAlisPro program. The intensities were corrected for Lorentz and polarization effects, and numerical absorption correction based on Gaussian integration over a multifaceted crystal model was applied. The structure was solved by direct methods using SHELXT,55 which revealed the positions of all not disordered non-hydrogen atoms of the title compound. The non-hydrogen atoms were refined anisotropically. All H-atoms were placed in geometrically calculated positions and refined using a riding model where each H-atom was assigned a fixed isotropic displacement parameter with a value equal to 1.2Ueq of its parent atom (1.5Ueq for the methyl groups and water). Refinement of the structure was carried out on F2 using full-matrix least-squares procedures, which minimized the function ∑w(Fo2 − Fc2)2. The weighting scheme was based on counting statistics and included a factor to downweight the intense reflections. All calculations were performed using the SHELXL-2014/7 program.56 Further crystallographic details are compiled in Tables S2–5.† Crystallographic data for all structures have been deposited with the Cambridge Crystallographic Data Centre (CCDC) as supplementary publication numbers 4b (1958588), 4c (1958592), 6a (1958633) and 7 (1958593).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge generous financial support from the European Research Council (ERC CoG 615653) and the Swiss National Science Foundation (200020_182663, R'equip projects 206021_128724 and 206021_170755).Notes and references
- D. Srimani, M. Feller, Y. Ben-David and D. Milstein, Chem. Commun., 2012, 48, 11853–11855 RSC.
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS.
- S. Yao, S. Saaby, R. G. Hazell and K. A. Jørgensen, Chem. – Eur. J., 2000, 6, 2435–2448 CrossRef CAS.
- S. I. Murahashi, Angew. Chem., Int. Ed. Engl., 1995, 34, 2443–2465 CrossRef CAS.
- K. Yamaguchi, H. Fujiwara, Y. Ogasawara, M. Kotani and N. Mizuno, Angew. Chem., Int. Ed., 2007, 46, 3922–3925 CrossRef CAS.
- K. Ishihara, Y. Furuya and H. Yamamoto, Angew. Chem., Int. Ed., 2002, 41, 2983–2986 CrossRef CAS.
- S. L. F. Cavani and N. Ballarini, Top. Catal., 2009, 52, 935–947 CrossRef.
- T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1885, 18, 1496–1500 CrossRef.
- T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1885, 18, 1492–1496 CrossRef.
- M. Lamani and K. R. Prabhu, Angew. Chem., Int. Ed., 2010, 49, 6622–6625 CrossRef CAS PubMed.
- S. Zhou, K. Junge, D. Addis, S. Das and M. Beller, Org. Lett., 2009, 11, 2461–2464 CrossRef CAS PubMed.
- R. K. Grasselli, Catal. Today, 1999, 49, 141–153 CrossRef CAS.
- M. T. Schümperli, C. Hammond and I. Hermans, ACS Catal., 2012, 2, 1108–1117 CrossRef.
- K. N. T. Tseng and N. K. Szymczak, Synlett, 2014, 25, 2385–2389 CrossRef CAS.
- S. Muthusamy, N. Kumarswamyreddy, V. Kesavan and S. Chandrasekaran, Tetrahedron Lett., 2016, 57, 5551–5559 CrossRef CAS.
- W. R. McWhinnie, J. D. Miller, J. B. Watts and D. Y. Waddan, J. Chem. Soc. D, 1971, 629–630 RSC.
- W. R. McWhinnie, J. D. Miller, J. B. Watts and D. Y. Waddan, Inorg. Chim. Acta, 1973, 7, 461–466 CrossRef CAS.
- S. E. Diamond, G. M. Tom and H. Taube, J. Am. Chem. Soc., 1975, 97, 2661–2664 CrossRef CAS.
- B. C. Lane, J. E. Lester and F. Basolo, J. Chem. Soc. D, 1971, 1618–1619 RSC.
- R. Tang, S. E. Diamond, N. Neary and F. Mares, J. Chem. Soc., Chem. Commun., 1978, 562 RSC.
- A. J. Bailey and B. R. James, Chem. Commun., 1996, 2343–2344 RSC.
- J. S. M. Samec, A. H. Éll and J. E. Bäckvall, Chem. – Eur. J., 2005, 11, 2327–2334 CrossRef CAS PubMed.
- D. L. J. Broere, Phys. Sci. Rev., 2018, 20170029 Search PubMed.
- R. Ray, A. S. Hazari, G. K. Lahiri and D. Maiti, Chem. – Asian J., 2018, 13, 2138–2148 CrossRef CAS PubMed.
- L. Cristian, S. Nica, O. D. Pavel, C. Mihailciuc, V. Almasan, S. M. Coman, C. Hardacre and V. I. Parvulescu, Catal. Sci. Technol., 2013, 3, 2646–2653 RSC.
- R. Ray, S. Chandra, V. Yadav, P. Mondal, D. Maiti and G. K. Lahiri, Chem. Commun., 2017, 53, 4006–4009 RSC.
- R. Ray, S. Chandra, V. Yadav, P. Mondal, D. Maiti and G. K. Lahiri, Chem. Commun., 2017, 53, 4006–4009 RSC.
- K. N. T. Tseng, A. M. Rizzi and N. K. Szymczak, J. Am. Chem. Soc., 2013, 135, 16352–16355 CrossRef CAS PubMed.
- I. Dutta, S. Yadav, A. Sarbajna, S. De, M. Hölscher, W. Leitner and J. K. Bera, J. Am. Chem. Soc., 2018, 140, 8662–8666 CrossRef CAS PubMed.
- M. Kannan and S. Muthaiah, Organometallics, 2019, 38, 3560–3567 CrossRef CAS.
- T. Achard, J. Egly, M. Sigrist, A. Maisse-François and S. Bellemin-Laponnaz, Chem. – Eur. J., 2019, 25, 13271–13274 CrossRef CAS PubMed.
- P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS PubMed.
- Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145–1159 RSC.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC.
- M. Olivares, M. van der Ham, C. J. M. Mdluli, V. Schmidtendorf, M. Müller-Bunz, H. Verhoeven, T. W. G. M. Li, M. Niemantsverdriet, J. W. (Hans) Hetterscheid, D. G. H. Bernhard and S. Albrecht, submitted.
- L. Suntrup, S. Hohloch and B. Sarkar, Chem. – Eur. J., 2016, 22, 18009–18018 CrossRef CAS PubMed.
- S. Sabater, H. Müller-Bunz and M. Albrecht, Organometallics, 2016, 35, 2256–2266 CrossRef CAS.
- M. Valencia, A. Pereira, H. Müller-Bunz, T. R. Belderraín, P. J. Pérez and M. Albrecht, Chem. – Eur. J., 2017, 23, 8901–8911 CrossRef CAS PubMed.
- J. A. Woods, R. Lalrempuia, A. Petronilho, N. D. McDaniel, H. Müller-Bunz, M. Albrecht and S. Bernhard, Energy Environ. Sci., 2014, 7, 2316–2328 RSC.
- M. Delgado-Rebollo, D. Canseco-Gonzalez, M. Hollering, H. Muller-Bunz and M. Albrecht, Dalton Trans., 2014, 43, 4462–4473 RSC.
- A. Bolje, S. Hohloch, D. Urankar, A. Pevec, M. Gazvoda, B. Sarkar and J. Košmrlj, Organometallics, 2014, 33, 2588–2598 CrossRef CAS.
- L. Bernet, R. Lalrempuia, W. Ghattas, H. Mueller-Bunz, L. Vigara, A. Llobet and M. Albrecht, Chem. Commun., 2011, 47, 8058–8060 RSC.
- W. Ghattas, H. Müller-Bunz and M. Albrecht, Organometallics, 2010, 29, 6782–6789 CrossRef CAS.
- V. Leigh, W. Ghattas, R. Lalrempuia, H. Müller-Bunz, M. T. Pryce and M. Albrecht, Inorg. Chem., 2013, 52, 5395–5402 CrossRef CAS PubMed.
- M. Nussbaum, O. Schuster and M. Albrecht, Chem. – Eur. J., 2013, 19, 17517–17527 CrossRef CAS PubMed.
- R. Le Lagadec, L. Rubio, L. Alexandrova, R. A. Toscano, E. V. Ivanova, R. Meškys, V. Laurinavičius, M. Pfeffer and A. D. Ryabov, J. Organomet. Chem., 2004, 689, 4820–4832 CrossRef CAS.
- A. J. Bailey and B. R. James, Chem. Commun., 1996, 20, 2343–2344 RSC.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- CrysAlisPro (Version 1.171.34.44), Oxford Diffraction Ltd., Yarnton, Oxfordshire, UK, 2010 Search PubMed.
- P. Macchi, H. B. Bürgi, A. S. Chimpri, J. Hauser and Z. Gál, J. Appl. Crystallogr., 2011, 44, 763–771 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures for ligand precursors, NMR spectra of all complexes, catalytic and crystallographic details. CCDC 1958588, 1958592, 1958593 and 1958633. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt04873a |
| This journal is © The Royal Society of Chemistry 2020 |