
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and group 9 complexes of macrocyclic PCP and POCOP pincer ligands†
Baptiste
Leforestier
 ,
Matthew R.
Gyton
and
Adrian B.
Chaplin
*
,
Matthew R.
Gyton
and
Adrian B.
Chaplin
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: a.b.chaplin@warwick.ac.uk
First published on 7th February 2020
Abstract
The synthesis of macrocyclic variants of commonly employed phosphine-based pincer (pro)ligands derived from meta-xylene (PCP-14) and resorcinol (POCOP-14) is described, where the P-donors are trans-substituted with a tetradecamethylene linker. The former was accomplished using a seven-step asymmetric procedure involving (−)-cis-1-amino-2-indanol as a chiral auxiliary and ring-closing olefin metathesis. A related, but non-diastereoselective route was employed for the latter, which consequently necessitated chromatographic separation from the cis-substituted by-product. The proligands are readily metalated and homologous series of MI(CO) and MIIICl2(CO) derivatives (M = Rh, Ir) have been isolated and fully characterised in solution and the solid state. Metal hydride complexes are generated during the synthesis of the former and have been characterised in situ using NMR spectroscopy.
Introduction
Conferring thermal stability whilst permitting a broad range of metal-based reactivity, the application of mer-tridentate “pincer” ligands in organometallic chemistry and homogeneous catalysis has had a profound impact.1 Phosphine-based examples bearing a central aryl donor were first reported by Shaw in the late 1970s,2 but these archetypical PCP ligands continue to attract contemporary interest, with iridium derivatives noteworthy for their ability to catalyse the dehydrogenation of alkanes.3 The predictable and modular composition of pincer ligands enables the steric and electronic properties of metal derivatives to be tuned through changes to the constituent donor groups, their substituents or the backbone configuration itself, and these adaptions have been extensively explored over the intervening four decades.4 Motivated by the potential to exploit additional reaction control though their unique steric profile and their use in the construction of interlocked assemblies, we have become interested in exploring the chemistry of macrocyclic pincer ligands, with our initial efforts devoted to NHC-based CNC ligands.5 We herein describe the synthesis and coordination chemistry of meta-xylene- and resorcinol-derived macrocyclic (pro)ligands PCP-14 and POCOP-14, where the chiral P-donors are trans-substituted with a tetradecamethylene linker (Chart 1). The preparation of analogous lutidine- and 2,6-dihydroxypyridine-derived PNP-14 and PONOP-14 ligands was described in a preceding contribution.6 | ||
| Chart 1 | ||
Results and discussion
Proligand synthesis
Circumventing problems associated with the generation of the unwanted cis-substituted diastereoisomer, the proligand PCP-14′ was prepared using an adapted asymmetric procedure developed by Riera and Verdaguer that employs (−)-cis-1-amino-2-indanol as a chiral auxiliary and ring-closing olefin metathesis (Scheme 1).7 The known oxazaphospholidine borane 1 (δ31P 167.9) was first stereoselectively ring-opened by treatment with octen-7-yl-magnesium bromide, affording 2 (δ31P 76.5). The auxiliary was then removed by acidolysis using an excess of H2SO4 in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH–water to generate phosphinous acid borane 3 (δ31P 125.6), which was subsequently converted into phosphine borane 4 (δ31P 22.2, 1JPH = 352 Hz) by reduction of the mesylate with tetrabutylammonium borohydride and isolated in 65% overall yield from 1. Isolated 4 is unstable at room temperature, presumably due to a propensity to undergo hydrophosphination,8 and is best converted directly into the corresponding phosphide by deprotonation with nBuLi at −78 °C. Reaction thereafter with 1,3-bis(bromomethyl)benzene afforded 5 (δ31P 32.3), which was cyclised by portion-wise treatment with Grubbs’ 1st generation catalyst (totalling 10 mol%) under dilute conditions in CH2Cl2 (5 mmol L−1) to afford the corresponding unsaturated macrocycle 6 (δ31P 31.3–33.4). Subsequent hydrogenation using Wilkinson's catalyst (producing PCP-14′·2BH37, δ31P 32.5) and deprotection by borane transfer in neat diethylamine afforded the proligand (δ31P 3.2) as the (R,R)-diastereomer with a respectable overall yield of 48% and >95% ee, as determined by 1H and 31P NMR spectroscopic analysis of the oxide derivative using the chiral shift reagent (−)-3,5-dinitro-N-(1-phenylethyl)benzamide (see ESI†).9
:1 MeOH–water to generate phosphinous acid borane 3 (δ31P 125.6), which was subsequently converted into phosphine borane 4 (δ31P 22.2, 1JPH = 352 Hz) by reduction of the mesylate with tetrabutylammonium borohydride and isolated in 65% overall yield from 1. Isolated 4 is unstable at room temperature, presumably due to a propensity to undergo hydrophosphination,8 and is best converted directly into the corresponding phosphide by deprotonation with nBuLi at −78 °C. Reaction thereafter with 1,3-bis(bromomethyl)benzene afforded 5 (δ31P 32.3), which was cyclised by portion-wise treatment with Grubbs’ 1st generation catalyst (totalling 10 mol%) under dilute conditions in CH2Cl2 (5 mmol L−1) to afford the corresponding unsaturated macrocycle 6 (δ31P 31.3–33.4). Subsequent hydrogenation using Wilkinson's catalyst (producing PCP-14′·2BH37, δ31P 32.5) and deprotection by borane transfer in neat diethylamine afforded the proligand (δ31P 3.2) as the (R,R)-diastereomer with a respectable overall yield of 48% and >95% ee, as determined by 1H and 31P NMR spectroscopic analysis of the oxide derivative using the chiral shift reagent (−)-3,5-dinitro-N-(1-phenylethyl)benzamide (see ESI†).9
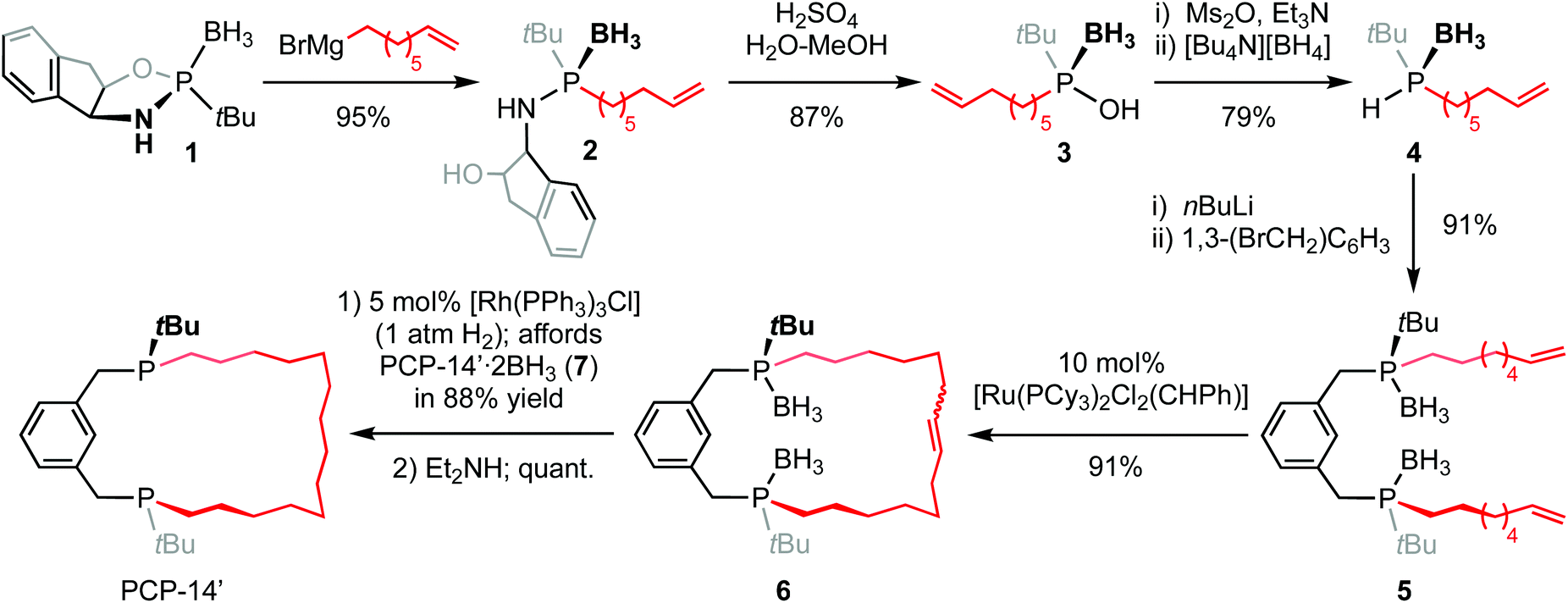 | ||
| Scheme 1 Asymmetric synthesis of PCP-14′. | ||
Attempts to develop an asymmetric procedure for the preparation of the proligand POCOP-14′ starting from enantiopure 3, involving reaction of the mesylate with disodium resorcinolate or nucleophilic aromatic substitution of 1,3-difluorobenzene by the conjugate base, were unsuccessful (see ESI† for details). Likewise, attempted conversion of 2 to the corresponding borane protected chlorodialkylphosphine by acidolysis with HCl was unsuccessful. Instead the five step racemic synthesis outlined in Scheme 2, adapted from our work with PNP-14 and PONOP-14 and also involving ring-closing olefin metathesis,6 was employed with the borane protected (saturated) macrocycle 12 proving to be the most conducive to resolution of the two diastereoisomers by column chromatography (cis-12, δ31P 143.7; trans-12, δ31P 143.8) and enabling determination of their respective configurations by single crystal X-ray diffraction (Fig. 1). In this way POCOP-14′ was obtained as an analytically pure racemate in 30% overall yield (δ31P 141.6). Use of the chiral shift reagent (−)-3,5-dinitro-N-(1-phenylethyl)benzamide enabled the component phosphine oxide enantiomers to be resolved by 31P NMR spectroscopy (see ESI†).9
 | ||
| Scheme 2 Preparation of POCOP-14′. | ||
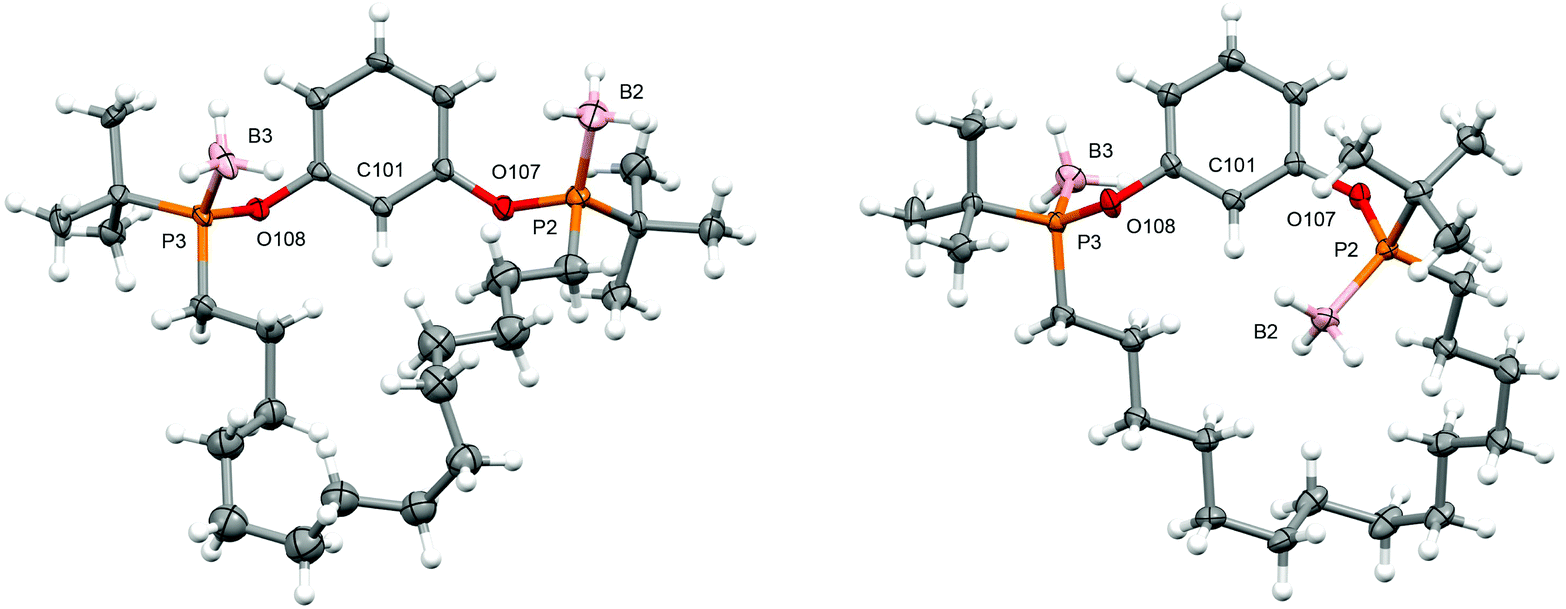 | ||
| Fig. 1 Solid-state structures of cis-12 (left) and trans-12 (right; not unique, Z′ = 2). Thermal ellipsoids drawn at 30% and 50% probability, respectively; minor disordered components omitted (cis-12; tBu groups and methylene chain). Selected bond lengths (Å): cis-12, P2–B2, 1.915(8); P3–B3, 1.896(7); P2–O107, 1.617(4); P3–O108, 1.630(4); trans-12, B2–P2, 1.908(2); B3–P3, 1.905(2); P2–O107, 1.6323(9); P3–O108, 1.6270(9); B12–P12, 1.901(2); B13–P13, 1.904(2); P12–O207, 1.6284(10); P13–O208, 1.6274(10). | ||
Rhodium complexes
With the new macrocyclic proligands in hand, the synthesis of rhodium derivatives was targeted (Scheme 3). Using an adapted literature procedure,10 rhodium dihydrogen complexes [Rh(pincer)(H2)] (pincer = PCP-14, 13a; POCOP-14, 13b) were prepared in quantitative spectroscopic yield using a one-pot procedure involving metalation of the proligands with [Rh(COE)2Cl]2 (COE = cyclooctene) in toluene at elevated temperature and subsequent treatment with KOtBu at 120 °C under an atmosphere of dihydrogen. Complexes 13a/b were characterised in situ and are notable for the adoption of C2 symmetry, 31P resonances at δ 70.4 (1JRhP = 153 Hz)/198.4 (1JRhP = 165 Hz) displaying large coupling to 103Rh, and broad 2H hydride signals at δ −4.36/−2.87 (298 K), which exhibit fast spin–lattice relaxation (T1 = 74 ± 4/42 ± 2 ms at 298 K; T1 = 117 ± 15/89 ± 11 ms at 200 K; 600 MHz, Ar) consistent with the assignment as dihydrogen complexes.11,12 The formulation of 13a/b as σ-complexes is further substantiated by their instability to vacuum and onward reactivity with CO (1 atm), conferring C2 symmetric Rh(I) carbonyl complexes [Rh(pincer)(CO)] (pincer = PCP-14, 14a; POCOP-14, 14b), which are characterised by 31P resonances at δ 75.0 (1JRhP = 146 Hz)/201.6 (1JRhP = 156 Hz) with 1JRhP coupling constants of similar magnitude. Complexes 14a/b were thereafter oxidised to the corresponding C2 symmetric Rh(III) carbonyl complexes 15a/b by reaction with PhICl2, as evidenced by 31P resonances at δ 64.9 (1JRhP = 87 Hz)/181.0 (1JRhP = 92 Hz) with the expected reductions in 1JRhP values, isolated as analytically pure materials in 80/48% overall yield, following purification by silica chromatography (CH2Cl2–pentane), and fully characterised (Fig. 2, Table 1).13 Analytically pure samples of very alkane soluble 14a/b were secured from the reaction of 15a/b with 3 equivalents iPrMgCl·LiCl in THF and subsequent recrystallisation from SiMe4 in 85/94% yield; as confirmed by combustion analysis.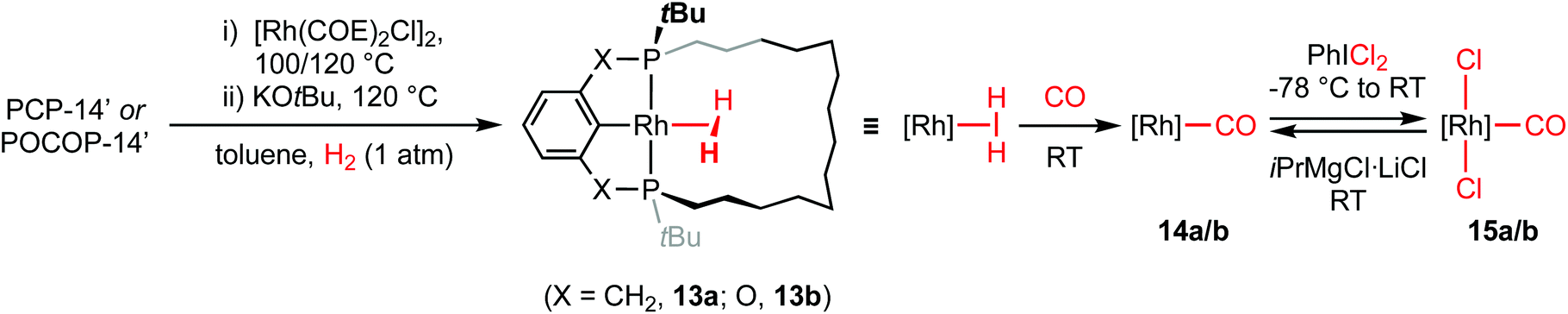 | ||
| Scheme 3 Synthesis of rhodium PCP-14 and POCOP-14 complexes 13–15. | ||
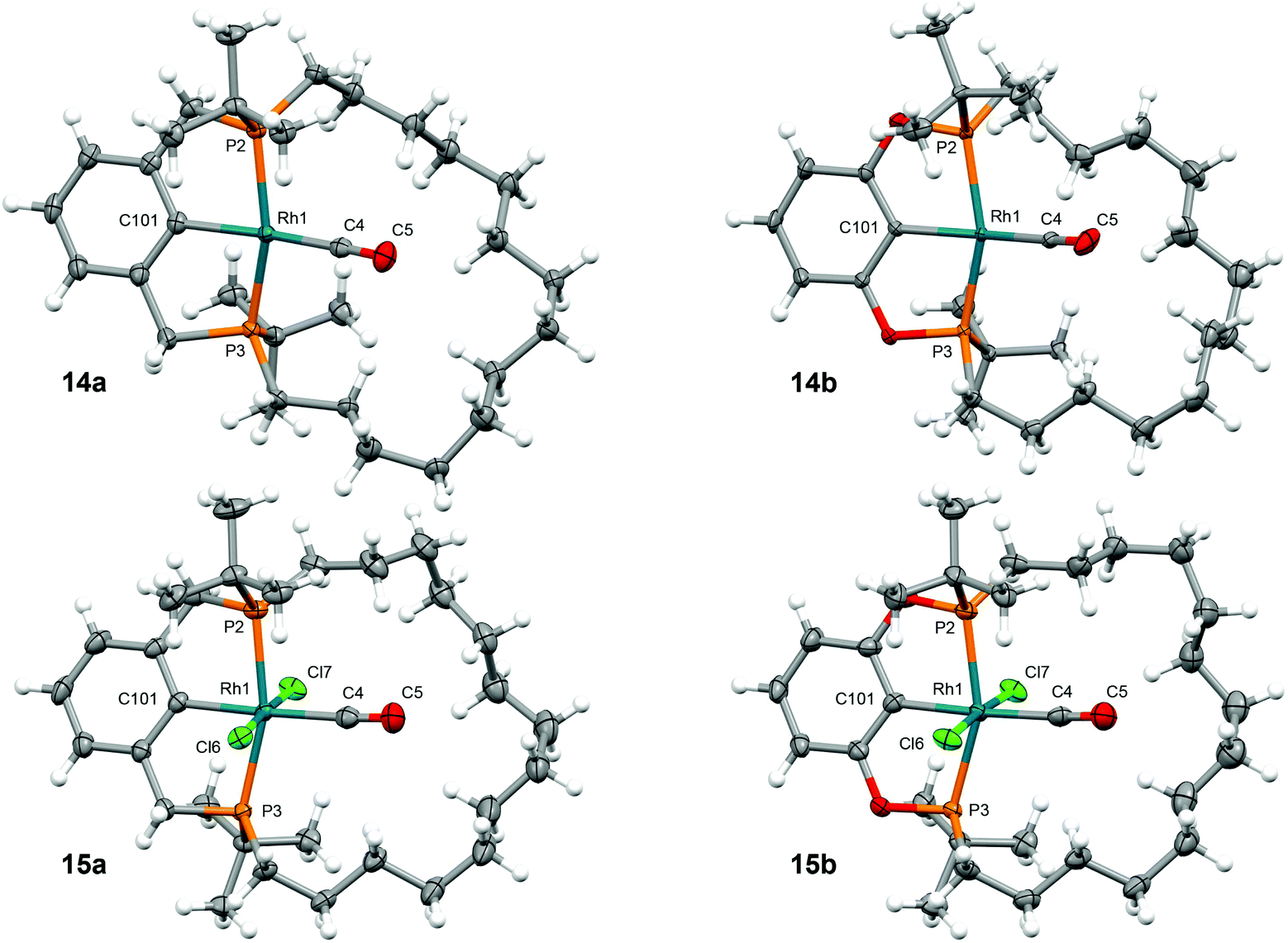 | ||
| Fig. 2 Solid-state structures of 14 and 15. Thermal ellipsoids drawn at 50% probability; minor disordered components omitted (15a/b; methylene chains). Selected metrics provided in Table 1. | ||
| M = Rh | M = Ir | |||||||
|---|---|---|---|---|---|---|---|---|
| 14a | 14b | 15a | 15b | 18a | 18b | 19a | 19b | |
| a Angle between the least-squares mean planes of the aryl group and the MP2C(aryl) atoms. | ||||||||
| M1–P2 | 2.2844(6) | 2.2704(15) | 2.3615(7) | 2.3324(12) | 2.2817(7) | 2.2694(11) | 2.3637(9) | 2.336(2) |
| M1–P3 | 2.2731(6) | 2.2856(13) | 2.3938(6) | 2.3768(11) | 2.2760(7) | 2.2798(10) | 2.3922(8) | 2.377(2) |
| M1–C4 | 1.865(3) | 1.879(6) | 2.005(3) | 1.996(6) | 1.862(3) | 1.879(4) | 1.940(3) | 1.926(9) |
| M1–C101 | 2.083(3) | 2.037(6) | 2.061(3) | 2.018(5) | 2.085(3) | 2.048(4) | 2.089(3) | 2.034(10) |
| M1–Cl6 | — | — | 2.3656(5) | 2.3616(12) | — | — | 2.3828(7) | 2.380(2) |
| M1–Cl7 | — | — | 2.3591(6) | 2.3521(12) | — | — | 2.3793(8) | 2.371(2) |
| P2–M1–P3 | 160.83(2) | 155.98(5) | 162.17(2) | 156.80(5) | 161.16(2) | 156.21(3) | 160.81(3) | 156.18(9) |
| C101–M1–C4 | 171.38(10) | 165.3(3) | 177.22(11) | 174.6(2) | 171.91(12) | 165.6(2) | 177.24(14) | 174.9(4) |
| Cl6–M1–Cl7 | — | — | 176.92(2) | 176.32(5) | — | — | 178.81(3) | 177.37(9) |
| Aryl twista | 14.30(6) | 6.7(2) | 18.52(7) | 12.34(13) | 13.95(8) | 6.49(14) | 17.93(9) | 11.3(3) |
The solid-state structures of 14a/b have been determined by single crystal X-ray diffraction and demonstrate the adoption of distorted square planar metal coordination geometries, with the tetradecamethylene linker of the pincer ligands considerably skewed to one side of the coordination plane conferring overall C1 symmetry and inducing an appreciable deviation of the C101–Rh1–C4 angles from linearity, especially in the case of the phosphinite pincer (171.38(10)°, 14a; 165.3(3)°, 14b). The methylene chains of the macrocycles are also non-symmetrically positioned in the Rh(III) congeners, but in this case contortion is counteracted by buttressing with the ancillary chloride ligands and more ideal C101–Rh1–C4 angles are observed (177.22(11)°, 15a; 174.6(2)°, 15b). These asymmetric configurations are not retained in solution, where time averaged C2 symmetry is observed at 298 K (500–600 MHz) consistent with the tetradecamethylene linker being sufficiently large and flexible to accommodate the carbonyl ligand within the annulus of the macrocycle. The more obtuse P2–Rh1–P3 bite angles and rigid backbone conformations observed in the complexes of POCOP-14 in the solid state, compared to those of PCP-14, are fully in line with expectations for these pincer architectures.14 This homologous series of complexes showcases the effect of increased steric crowding on the latter, where C2 symmetric twisting of the central aryl donor is considerably more pronounced in the octahedral Rh(III) congeners. Finally, the observed differences in Rh–P, Rh–CO, and Rh–aryl contacts for the Rh(I) and Rh(III) congeners are consistent with the nature of associated donors (vide infra for further discussion on the carbonyl ligand).
Iridium complexes
In a similar manner to that employed for the rhodium analogues, iridium tetrahydride complexes [Ir(pincer)H4] (pincer = PCP-14, 16a; POCOP-14, 16b) were prepared using a one-pot procedure involving metalation of the macrocyclic proligands with [Ir(COE)2Cl]2 (16a) or [Ir(COD)Cl]2 (16b, COD = cyclooctadiene) in toluene at elevated temperature and subsequent treatment with KOtBu at 120 °C under an atmosphere of dihydrogen (Scheme 4). Complexes 16a/b were characterised in situ and are notable for the adoption of C2 symmetry, 31P resonances at δ 49.9/165.0, and relatively sharp triplet 4H hydride signals at δ −8.99 (2JPH = 9.8 Hz)/−8.26 (2JPH = 9.9 Hz) at 298 K that only broaden slightly on cooling to 200 K, with moderate spin–lattice relaxation (T1 = 300 ± 10/194 ± 4 ms at 298 K; T1 = 626 ± 15/242 ± 15 ms at 200 K; 600 MHz, Ar). These data are consistent with fluxional hydride ligands, where dynamic interconversion between classical tetrahydride and dihydride dihydrogen formulations occurs on the NMR timescale.11,15 Evidencing the presence of the latter, reaction with CO (1 atm) resulted in immediate substitution of one equivalent of dihydrogen and formation of cis-17a/b [δ1H(hydride) −10.66, −11.59; δ31P 51.0, 45.8 (2JPP = 286 Hz)/δ1H(hydride) −9.82, −10.75; δ31P 160.4, 154.6 (2JPP = 305 Hz)]. Prolonged thermolysis at 120 °C under CO (1 atm) in the presence of tert-butylethylene (TBE) followed by removal of CO resulted in the formation of C2 symmetric Ir(I) carbonyl complexes [Ir(pincer)(CO)] (pincer = PCP-14, δ31P 68.1, 18a; POCOP-14, δ31P 186.7, 18b). Transient formation of trans-17a/b [δ1H(hydride) −9.65; δ31P 54.1/δ1H(hydride) −9.76; δ31P 167.3] and generation of [Ir(pincer)(CO)2] (pincer = PCP-14, δ31P 51.6; POCOP-14, δ31P 164.8) were noted during this transformation. Complexes 18a/b were thereafter oxidised to the corresponding C2 symmetric Ir(III) carbonyl complexes 19a/b (δ31P 33.8/144.4) by reaction with PhICl2 and isolated as analytically pure and air-stable materials in 51/73% overall yield, following purification by silica chromatography (CH2Cl2–hexane), and fully characterised. Analytically pure samples of very alkane soluble 18a/b were secured from the reaction of 19a/b with 10 equivalents of KC8 in pentane and subsequent recrystallisation from neohexane/SiMe4 in 84/79% yield; as confirmed by combustion analysis.16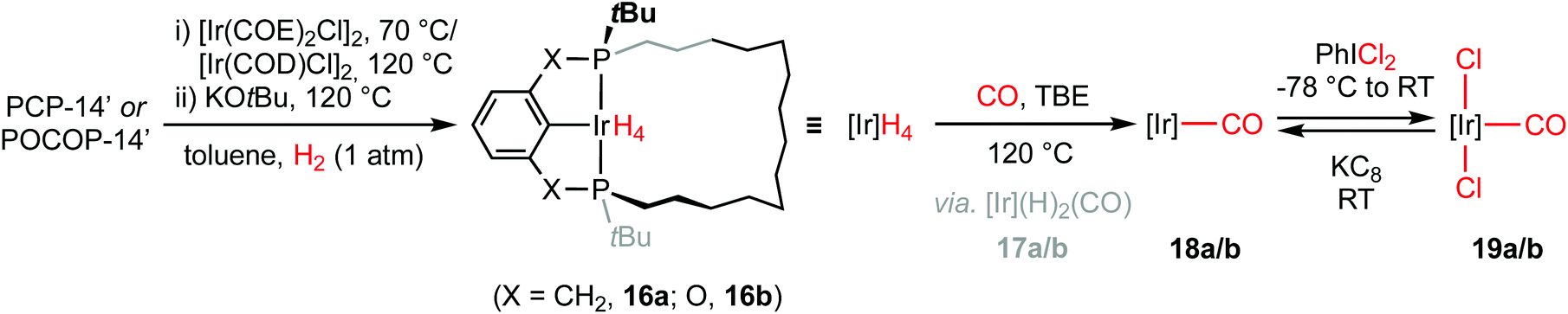 | ||
| Scheme 4 Synthesis of iridium PCP-14 and POCOP-14 complexes 16–19. | ||
The solid-state structures of 18a/b and 19a/b have all been determined by single crystal X-ray diffraction and are isomorphous to the corresponding rhodium congeners 14a/b and 15a/b, respectively (Table 1).
Carbonyl stretching frequencies
The ν(CO) bands of metal carbonyl derivatives are established reporter groups for donor properties of ligands.17 In the case of pincer ligands, group 9 examples have an established track record in the literature.18 The carbonyl stretching frequencies of 14, 15, 18 and 19 and those of known acyclic pincer complexes [M(pincer)(CO)] (M = Rh, Ir; pincer = 2,6-(tBu2PCH2)2C6H3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PCP-tBu, 2,6-(tBu2PO)2C6H3POCOP-tBu) have all been determined by IR spectroscopy under the same conditions and compiled in Table 2. These data suggest that PCP-14 and POCOP-14 are marginally weaker donors than PCP-tBu and POCOP-tBu, respectively, consistent with similar findings for the neutral PNP and PONOP analogues and attributed to changes in the phosphine/phosphinite substituents alone.6 Other trends apparent in the IR data associated with the extent of π-backbonding increasing in the order Ir > Rh, M(I) > M(III) and PCP > POCOP are fully in line with expectation and reinforced by similar trends in the X-ray derived metrics (Table 1) and NMR data (see Experimental section).
PCP-tBu, 2,6-(tBu2PO)2C6H3POCOP-tBu) have all been determined by IR spectroscopy under the same conditions and compiled in Table 2. These data suggest that PCP-14 and POCOP-14 are marginally weaker donors than PCP-tBu and POCOP-tBu, respectively, consistent with similar findings for the neutral PNP and PONOP analogues and attributed to changes in the phosphine/phosphinite substituents alone.6 Other trends apparent in the IR data associated with the extent of π-backbonding increasing in the order Ir > Rh, M(I) > M(III) and PCP > POCOP are fully in line with expectation and reinforced by similar trends in the X-ray derived metrics (Table 1) and NMR data (see Experimental section).
| Macrocyclic | Acyclic | |
|---|---|---|
| [Rh(PCP)(CO)] | 1939 | 1933 |
| [Rh(POCOP)(CO)] | 1958 | 1954 |
| [Rh(PCP)Cl2(CO)] | 2069 | — |
| [Rh(POCOP)Cl2(CO)] | 2083 | — |
| [Ir(PCP)(CO)] | 1925 | 1918 |
| [Ir(POCOP)(CO)] | 1943 | 1939 |
| [Ir(PCP)Cl2(CO)] | 2034 | — |
| [Ir(POCOP)Cl2(CO)] | 2049 | — |
Conclusions
Procedures that enable the preparation of two phosphine-based macrocyclic pincer (pro)ligands PCP-14 and POCOP-14, where the chiral P-donors are trans-substituted with a tetradecamethylene linker, have been developed. The synthesis of meta-xylene-based proligand PCP-14′ was achieved using a seven-step asymmetric protocol involving (−)-cis-1-amino-2-indanol as a chiral auxiliary and ring-closing olefin metathesis, affording the (R,R)-diastereomer in 48% overall yield and >95% ee. Whilst we were unable to apply this methodology to the synthesis of resorcinol-based POCOP-14′, this proligand was obtained as analytically pure racemate in 30% overall yield using a non-diastereoselective route, where separation from the unwanted cis-substituted by-product was achieved by chromatography. Rhodium and iridium complexes of these new macrocyclic pincer ligands have been obtained through metalation of the proligands with dimeric rhodium(I) and iridum(I) halide precursors and subsequent dehydrohalogenation under a dihydrogen atmosphere. The resulting hydride complexes were characterised in situ by NMR spectroscopy and reacted on to generate isolatable MI(CO) and MIIICl2(CO) derivatives that have been extensively characterised in solution and the solid state, including all eight complexes by single crystal X-ray diffraction. By comparison of the ν(CO) bands of the rhodium(I) and iridum(I) carbonyl adducts, determined by IR spectroscopy in toluene, PCP-14 and POCOP-14 can be considered to be marginally weaker net donors than their respective homoleptic tert-butyl substituted congeners PCP-tBu and POCOP-tBu, respectively. Work exploring the organometallic chemistry of these novel macrocyclic pincer ligands is ongoing in our laboratories and will be reported in due course.Experimental
General methods
All manipulations were performed under an atmosphere of argon using Schlenk and glove box techniques unless otherwise stated. Hydrogen was dried by passage through a stainless-steel column of activated 3 Å molecular sieves. Carbon monoxide was used as received. Glassware was oven-dried at 150 °C overnight and flame-dried under vacuum prior to use. Molecular sieves were activated by heating at 300 °C in vacuo overnight. CD2Cl2 was freeze–pump–thaw degassed and dried over 3 Å molecular sieves. Toluene-d8 and C6D6 were dried over sodium overnight, distilled, freeze–pump–thaw degassed and stored over 3 Å molecular sieves. THF was distilled from sodium and benzophenone and stored over 3 Å molecular sieves. Et2NH was distilled from CaH2. tert-Butylethylene was freeze–pump–thaw degassed and stored over 3 Å molecular sieves. SiMe4 was distilled from liquid Na/K alloy and stored over a potassium mirror. Neohexane (2,2-dimethylbutane) was distilled from liquid Na/K alloy and stored over 3 Å molecular sieves. Other anhydrous solvents were purchased from Acros Organics or Sigma-Aldrich, freeze–pump–thaw degassed and stored over 3 Å molecular sieves. The concentration of nBuLi was titrated by 1H NMR spectroscopy before use.19 Oxazaphospholidine 1,7 MgBr(C8H15),208,6 PhICl2,21 [Rh(PPh3)3Cl],22 [Rh(COE)2Cl]2,23 [Ir(COD)Cl]2,24 [Ir(COE)2Cl]2,24 [M(pincer)(CO)] (M = Rh, Ir; pincer = PCP-tBu, POCOP-tBu)2,14d,25 were synthesised according to published procedures. NMR spectra were recorded on Bruker spectrometers under argon at 298 K unless otherwise stated. Chemical shifts are quoted in ppm and coupling constants in Hz.26 NMR spectra in toluene-d0 were recorded using an internal capillary of C6D6. HR ESI-MS were recorded on a Bruker MaXis mass spectrometer. IR spectra were recorded on a Bruker Alpha Platinum ATR FT-IR spectrometer at RT (for full details see ESI†). Microanalyses were performed at the London Metropolitan University by Stephen Boyer in all cases, except for samples of 14a/b and 18a/b, which were analysed by Elemental Microanalysis Ltd.Synthesis of PCP-14′
1
H NMR (500 MHz, CDCl3): δ 7.35–7.42 (m, 1H, Ar), 7.23–7.28 (m, 3H, Ar), 5.81 (ddt, 3JHH = 16.9, 10.1, 6.5, 1H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) CH2), 5.01 (app dq, 3JHH = 16.9, JHH = 2, 1H, CHC2), 4.95 (ddt, 3JHH = 10.1, 2JHH = 2.2, 4JHH = 1.2, 1H, CHC2), 4.77 (app td, J = 10, 3JHH = 4.5, 1H, NC), 4.51 (app q, 3JHH = 5, 1H, OC), 3.10 (dd, 2JHH = 16.6, 3JHH = 4.7, 1H, OCHC2), 2.90 (d, 2JHH = 16.6, 1H, OCHC2), 2.11 (d, 3JHH = 10.0, NH), 2.05 (app q, 3JHH = 7, 2H, C2CHCH2), 1.81 (d, 3JHH = 4.4, 1H, OH), 1.56–1.83 (m, 4H, CH2), 1.30–1.49 (m, 6H, CH2), 1.26 (d, 3JPH = 13.6, 9H, tBu), 0.55 (partially collapsed quartet, fwhm = 310 Hz, 3H, BH3).
CH2), 5.01 (app dq, 3JHH = 16.9, JHH = 2, 1H, CHC2), 4.95 (ddt, 3JHH = 10.1, 2JHH = 2.2, 4JHH = 1.2, 1H, CHC2), 4.77 (app td, J = 10, 3JHH = 4.5, 1H, NC), 4.51 (app q, 3JHH = 5, 1H, OC), 3.10 (dd, 2JHH = 16.6, 3JHH = 4.7, 1H, OCHC2), 2.90 (d, 2JHH = 16.6, 1H, OCHC2), 2.11 (d, 3JHH = 10.0, NH), 2.05 (app q, 3JHH = 7, 2H, C2CHCH2), 1.81 (d, 3JHH = 4.4, 1H, OH), 1.56–1.83 (m, 4H, CH2), 1.30–1.49 (m, 6H, CH2), 1.26 (d, 3JPH = 13.6, 9H, tBu), 0.55 (partially collapsed quartet, fwhm = 310 Hz, 3H, BH3).
13
C{
1
H} NMR (126 MHz, CDCl3): δ 142.9 (d, 3JPC = 7, Ar{C}), 139.9 (s, Ar{C}), 139.1 (s, ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) HCH2), 128.1 (s, Ar), 127.1 (s, Ar), 125.6 (s, Ar), 124.3 (s, Ar), 114.5 (s, CHH2), 74.8 (s, OCH), 62.3 (d, 2JPC = 3, NCH), 39.4 (s, OCHH2), 33.8 (s, H2CHCH2), 31.9 (d, 1JPC = 39, tBu{C}), 31.5 (d, 2JPC = 13, CH2), 28.9 (s, CH2), 28.8 (s, CH2), 25.1 (d, 2JPC = 2, tBu{CH3}), 24.1 (d, 1JPC = 36, PCH2), 23.0 (s, CH2).
HCH2), 128.1 (s, Ar), 127.1 (s, Ar), 125.6 (s, Ar), 124.3 (s, Ar), 114.5 (s, CHH2), 74.8 (s, OCH), 62.3 (d, 2JPC = 3, NCH), 39.4 (s, OCHH2), 33.8 (s, H2CHCH2), 31.9 (d, 1JPC = 39, tBu{C}), 31.5 (d, 2JPC = 13, CH2), 28.9 (s, CH2), 28.8 (s, CH2), 25.1 (d, 2JPC = 2, tBu{CH3}), 24.1 (d, 1JPC = 36, PCH2), 23.0 (s, CH2).
31 P{ 1 H} NMR (121 MHz, CDCl3): δ 76.5 (partially collapsed quartet, fwhm = 200 Hz).
HR ESI-MS (positive ion, 4 kV): 384.2603, [M + Na]+ (calcd 384.2602) m/z.
:45 mL). The reaction mixture was heated at 70 °C for 4 days, with periodic monitoring by TLC. The mixture was allowed to cool to RT, exposed to air, and CH2Cl2 (40 mL) and saturated aqueous NaCl (20 mL) added. The aqueous phase was extracted with CH2Cl2 (3 × 15 mL) and the combined organic extracts dried (MgSO4), filtered, and concentrated under reduced pressure to give a colourless opaque oil. The crude material was dissolved in a minimal amount of CH2Cl2 and run through a short silica plug, eluting with CH2Cl2 (Rf = 0.6). The volatiles were removed under reduced pressure to afford 3 as a colourless oil. Yield: 2.51 g (87%).
1
H NMR (500 MHz, toluene-d8): δ 5.75 (ddt, 3JHH = 17.0, 10.2, 6.7, 1H, CCH2), 5.02 (br d, 3JHH = 17.1, 1H, CHC2), 4.97 (br d, 3JHH = 10.1, 1H, CHC2), 3.14 (br, 1H, OH), 1.95 (app q, 3JHH = 7, 2H, C2CHCH2), 1.60–1.75 (m, 1H, CH2), 1.39–1.56 (m, 2H, CH2), 1.03–1.38 (m, 7H, CH2), 0.95 (d, 3JPH = 13.7, 9H, tBu), 0.63–1.30 (obscured, 3H, BH3).
13
C{
1
H} NMR (126 MHz, toluene-d8): δ 139.1 (s, HCH2), 114.6 (s, CHH2), 34.2 (s, H2CHCH2), 31.5 (d, 2JPC = 12, CH2), 31.0 (d, 1JPC = 38, tBu{C}), 29.24 (s, CH2), 29.15 (s, CH2), 24.5 (d, 1JPC = 35, PCH2), 24.0 (d, 2JPC = 3, tBu{CH3}), 22.8 (s, CH2).
31 P{ 1 H} NMR (121 MHz, toluene-d8): δ 125.6 (partially collapsed quartet, fwhm = 200 Hz).
1
H NMR (500 MHz, CDCl3): δ 5.80 (ddt, 3JHH = 17.0, 10.0, 6.5, 1H, CCH2), 4.99 (app dq, 3JHH = 17.0, JHH = 2, 1H, CHC2), 4.94 (ddt, 3JHH = 10.0, 2JHH = 2.3, 4JHH = 1.2, 1H, CHC2), 4.23 (dm, 1JPH = 350.6, 1H, PH), 2.04 (app q, 3JHH = 7, 2H, C2CHCH2), 1.65–1.90 (m, 2H, CH2), 1.45–1.60 (m, 2H, CH2), 1.24–1.40 (m, 6H, CH2), 1.21 (d, 3JPH = 14.2, 9H, tBu), 0.46 (partially collapsed quartet, fwhm = 330, 3H, BH3).
13
C{
1
H} NMR (126 MHz CDCl3): δ 139.0 (s, HCH2), 114.5 (s, CHH2), 33.8 (s, CH2), 30.9 (d, 2JPC = 11, CH2), 28.8 (s, CH2), 28.7 (s, CH2), 26.9 (d, 2JPC = 2, tBu{CH3}), 26.8 (d, 1JPC = 35, tBu{C}), 25.0 (d, 3JPC = 2, CH2), 17.6 (d, 1JPC = 32, PCH2).
31 P NMR (202 MHz, CDCl3): δ 22.2 (br d, 1JPH = 352).
31 P{ 1 H} NMR (202 MHz, CDCl3): δ 22.2 (partially collapsed quartet, fwhm = 165 Hz).
1
H NMR (300 MHz, CDCl3): δ 7.12–7.24 (m, 4H, Ar), 5.78 (ddt, 3JHH = 16.9, 10.2, 6.8, 2H, CCH2), 4.88–5.03 (m, 4H, CHC2), 2.85–3.10 (m, 4H, PCH2), 2.01 (app q, 3JHH = 7.0, 4H, C2CHCH2), 1.10–1.65 (m, 24H, CH2), 1.17 (d, 3JPH = 13.1, 18H, tBu), 0.44 (partially collapsed quartet, fwhm = 270 Hz, 6H, BH3).
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 32.3 (partially collapsed quartet, fwhm = 140 Hz).
HR ESI-MS (positive ion, 4 kV): 553.4469, [M + Na]+ (calcd 553.4416) m/z.
1
H NMR (500 MHz, CDCl3): δ 7.14–7.25 (m, 4H, Ar), 5.21–5.29 (m, 0.6H, CHCH minor), 5.21–5.29 (m, 1.4H, CHCH major), 3.02 (dd, 2JHH = 13.8, 2JPH = 9.0, 2H, ArC2), 2.92 (app t, 2J = 14, 1.4H, ArC2 major), 2.92 (app t, 2J = 14, 1.4H, ArC2 major), 2.91 (app t, 2J = 14, 0.6H, ArC2 minor), 1.88–2.05 (m, 4H, CH2), 0.93–1.70 (m, 20H, CH2), 1.21 (d, 3JPH = 13.0, 5.4H, tBu minor), 1.20 (d, 3JPH = 13.0, 12.6H, tBu major), 0.45 (partially collapsed quartet, fwhm = 270 Hz, 6H, BH3).
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 31.3–33.4 (br m).
HR ESI-MS (positive ion, 4 kV): 525.4105, [M + Na]+ (calcd 525.4102) m/z.
1
H NMR (600 MHz, CDCl3): δ 7.25 (s, 1H, Ar{2-CH}), 7.22 (t, 3JHH = 7.6, 1H, Ar), 7.17 (d, 3JHH = 7.6, 2H, Ar), 3.03 (dd, 2JHH = 14.0, 2JPH = 9.9, 2H, ArC2), 3.03 (app t, 2J = 14, 2H, ArC2), 1.50–1.61 (m, 4H, PCH2), 1.40–1.50 (m, 2H, CH2), 1.13–1.35 (m, 22H, CH2), 1.17 (d, 3JPH = 13.1, 18H, tBu), 0.46 (partially collapsed quartet, fwhm = 270 Hz, 6H, BH3).
13
C{
1
H} NMR (151 MHz, CDCl3): δ 133.9 (dd, 2JPC = 5, 4JPC = 2, Ar{C}), 132.0 (t, 3JPC = 4, Ar{2-CH}), 128.7 (dd, 3JPC = 4, 5JPC = 3, Ar), 128.5 (t, 4JPC = 2, Ar), 30.8 (d, 2JPC = 12, CH2), 28.8 (d, 1JPC = 31, tBu{C}), 28.7 (d, 1JPC = 27, ArH2), 27.8 (s, CH2), 27.6 (s, 2 × CH2), 27.4 (s, CH2), 25.8 (d, 2JPC = 1, tBu{CH3}), 23.3 (s, CH2), 20.2 (d, 1JPC = 31, PCH2).
31 P{ 1 H} NMR (243 MHz, CDCl3): δ 32.5 (partially collapsed quartet, fwhm = 155 Hz).
HR ESI-MS (positive ion, 4 kV): 527.4250, [M + Na]+ (calcd 527.4259) m/z.
Anal. Calcd for C30H60B2P2 (508.32 g mol−1): C, 71.44; H, 11.99; found: C, 71.37; H, 12.10.
1
H NMR (600 MHz, toluene-d8): δ 7.34 (s, 1H, Ar{2-CH}), 7.06 (br, 3H, Ar), 2.75 (d, 2JHH = 13.5, 2H, ArC2), 2.51 (dd, 2JHH = 13.5, 2JPH = 3.0, 2H, ArC2), 1.10–1.50 (m, 28H, CH2), 0.98 (d, 3JPH = 10.9, 18H, tBu).
13
C{
1
H} NMR (151 MHz, toluene-d8): δ 140.2 (d, 2JPC = 8, Ar{C}), 130.9 (t, 3JPC = 7, Ar{2-CH}), 128.5 (s, Ar), 127.1 (dd, 3JPC = 7, 5JPC = 2, Ar), 32.6 (d, 1JPC = 23, ArH2), 30.8 (d, 2JPC = 12, CH2), 28.6 (s, CH2), 28.3 (s, CH2), 28.23 (d, 1JPC = 15, tBu{C}), 28.21 (s, CH2), 28.1 (s, CH2), 27.5 (d, 2JPC = 13, tBu{CH3}), 27.3 (s, CH2), 24.4 (d, 1JPC = 20, PCH2).
31 P{ 1 H} NMR (243 MHz, toluene-d8): δ 3.2 (s).
1
H NMR (500 MHz, CD2Cl2): δ 7.37 (s, 1H, Ar), 7.23–7.28 (m, 1H, Ar), 7.21 (d, 3JHH = 7.6, 2H, Ar), 3.12 (app t, 2J = 14, 2H, ArC2), 3.02 (dd, 2JHH = 14.5, 2JPH = 10.1, 2H, ArC2), 1.61–1.76 (m, 2H, PCH2), 1.50–1.61 (m, 2H, PCH2), 1.39–1.50 (m, 2H, CH2), 1.10–1.50 (m, 22H, CH2), 0.98 (d, 3JPH = 10.9, 18H, tBu).
13
C{
1
H} NMR (126 MHz, CD2Cl2): δ 134.2 (dd, 2JPC = 8, 4JPC = 2, Ar{C}), 131.9 (t, 3JPC = 5, Ar{2-CH}), 128.8 (t, 4JPC = 2, Ar), 128.6 (app t, JPC = 3, Ar), 33.2 (d, 1JPC = 65, tBu{C}), 32.9 (d, 1JPC = 55, ArH2), 30.9 (d, 2JPC = 12, CH2), 28.3 (s, CH2), 28.2 (s, CH2), 28.14 (s, CH2), 28.08 (s, CH2), 24.9 (s, tBu{CH3}), 24.4 (d, 1JPC = 62, PCH2), 22.0 (d, 3JPC = 5, CH2).
31 P{ 1 H} NMR (162 MHz, CD2Cl2): δ 52.2 (s).
HR ESI-MS (positive ion, 4 kV): 531.3490, [M + Na]+ (calcd 531.3491) m/z.
Preparation of POCOP-14′
:1 mixture of diastereoisomers) as a colourless oil on drying. Yield: 16.1 g (95%).
1
H NMR (300 MHz, C6D6): δ 7.43 (s, 1H, Ar{2-CH}), 7.04 (t, 3JHH = 8.0, 1H, Ar), 6.95 (d, 3JHH = 8.4, 2H, Ar), 5.76 (ddt, 3JHH = 16.8, 11.8, 6.7, 2H, CCH2), 4.95–5.10 (m, 4H, CHC2), 1.95 (app q, 3JHH = 7, 4H, C2CHCH2), 1.75–1.90 (m, 2H, CH2), 1.61 (app sex, J = 8, 4H, CH2), 1.15–1.40 (m, 14H, CH2), 1.04 (d, 3JPH = 12.0, 18H, tBu).
13
C{
1
H} NMR (75 MHz, C6D6): δ 161.0 (d, 2JPH = 9.0, Ar{C}), 139.2 (s, HCH2), 130.2 (s, Ar), 114.6 (s, CHH2), 112.4 (d, 3JPH = 11, Ar), 109.6 (t, 3JPH = 12, Ar{2-CH}), 34.2 (s, H2CHCH), 32.7 (d, 1JPH = 16, tBu{C}), 31.5 (d, 2JPH = 12, CH2), 29.3 (s, CH2), 29.2 (s, CH2), 29.2 (d, 1JPH = 24, PCH2), 25.9 (d, 3JPH = 16, CH2), 25.4 (d, 2JPH = 16, tBu{CH3})
31 P{ 1 H} NMR (121 MHz, C6D6): δ 142.40 (s, 1P), 142.36 (s, 1P).
HR ESI-MS (positive ion, 4 kV): 561.3231, [M + 2O + Na]+ (calcd 561.3233) m/z.
:1 mixture of diastereoisomers) as a colourless oil. Yield: 12.9 g (79%).
1
H NMR (600 MHz, CDCl3): δ 7.19 (t, 3JHH = 8.2, 1H, Ar), 6.99 (app q, J = 2, 1H, Ar{2-CH}), 6.90 (app dt, 3JHH = 8.4, J = 2, 2H, Ar), 5.79 (app ddt, 3JHH = 16.9, 10.1, 6.7, 2H, CCH2), 4.99 (app dq, 3JHH = 17.0, JHH = 2, 2H, CHC2), 4.93 (app dq, 3JHH = 10.2, JHH = 2, 2H, CHC2), 2.04 (app q, 3JHH = 7, 4H, C2CHCH2), 1.92–2.01 (m, 2H, PCH2), 1.68–1.79 (m, 4H, CH2), 1.55–1.65 (m, 2H, CH2), 1.28–1.44 (m, 12H, CH2), 1.26 (d, 3JPH = 14.0, 18H, tBu), 0.53 (partially collapsed quartet, fwhm = 300 Hz, 6H, BH3).
13
C{
1
H} NMR (151 MHz, CDCl3): δ 154.2 (d, 2JPC = 6, Ar{C}), 139.1 (s, HCH2), 129.6 (s, Ar), 117.0 (d, 3JPC = 3, Ar), 114.5 (s, CHH2), 114.1 (t, 3JPC = 3.0, Ar{2-CH}), 33.8 (s, H2CHCH2), 32.9 (d, 1JPC = 37, tBu{C}), 31.4 (d, 2JPC = 13.0, CH2), 28.9 (d, 4JPC = 3, CH2), 28.7 (s, CH2), 25.4 (d, 1JPC = 33, PCH2), 24.9 (d, 2JPC = 3, tBu{CH3}), 22.8 (s, CH2).
31 P{ 1 H} NMR (121 MHz, CDCl3): δ 143.7 (partially collapsed quartet, fwhm = 165 Hz).
HR ESI-MS (positive ion, 4 kV): 557.4000, [M + Na]+ (calcd 557.3990) m/z.
:1 mixture of diastereoisomers) as a colourless oil. Yield: 832 mg (94%).
1
H NMR (500 MHz, CDCl3): δ 7.20 (t, 3JHH = 8.3, 0.5H, Ar cis), 7.19 (t, 3JHH = 8.3, 0.5H, Ar trans), 7.03 (dd, 3JHH = 8.3, 4JHH = 2.4, 1H, Ar cis), 6.97 (t, 4JHH = 2.3, 0.5H, Ar{2-CH} trans), 6.89 (dd, 3JHH = 8.3, 4JHH = 2.3, 1H, Ar trans), 6.74 (t, 4JHH = 2.4, 0.5H, Ar{2-CH} cis), 5.26–5.36 (m, 2H, CHCH), 1.93–2.10 (m, 6H, CH2), 1.61–1.81 (m, 4H, CH2), 1.24–1.60 (m, 14H, CH2), 1.27 (d, 3JPH = 14.0, 18H, tBu), 0.55 (partially collapsed quartet, fwhm = 300 Hz, 6H, BH3).
13
C{
1
H} NMR (126 MHz, CDCl3): δ 154.4 (d, 2JPC = 6, Ar{C} cis), 154.1 (d, 2JPC = 6, Ar{C} trans), 130.9 (s, CHCH trans), 130.8 (s, CHCH cis), 129.7 (s, Ar cis), 129.5 (s, Ar trans), 117.0 (d, 3JPC = 4, Ar trans), 116.4 (d, 3JPC = 3, Ar cis), 114.4 (t, 3JPC = 3, Ar{2-CH} trans), 113.3 (t, 3JPC = 4, Ar{2-CH} cis), 32.8 (d, 1JPC = 37, tBu{C} cis), 32.7 (d, 1JPC = 38, tBu{C} trans), 32.10 (s, H2CHCH trans), 32.06 (s, H2CHCH cis), 31.6 (d, 2JPC = 13, CH2trans), 31.2 (d, 2JPC = 13, CH2cis), 28.8 (s, CH2trans), 28.7 (s, CH2cis), 28.1 (s, CH2trans), 27.9 (s, CH2cis), 25.5 (d, 1JPC = 33, PCH2cis), 25.4 (d, 1JPC = 33, PCH2trans), 24.9 (d, 2JPC = 3, tBu{CH3} cis), 24.8 (d, 2JPC = 3, tBu{CH3} trans), 23.0 (d, 3JPC = 2, CH2trans), 22.7 (d, 3JPC = 2, CH2cis). Data for major alkene isomer only.
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 142.0–144.6 (br m).
HR ESI-MS (positive ion, 4 kV): 529.3689, [M + Na]+ (calcd 529.3687) m/z.
POCOP-14′·2BH3 (trans-12, Rf = 0.45). Yield: 951 mg (43%, colourless oil which slowly crystallised upon standing).
1 H NMR (600 MHz, CDCl3): δ 7.19 (t, 3JHH = 8.2, 1H, Ar), 7.03 (t, 4JHH = 2.5, 1H, Ar{2-CH}), 6.93 (dd, 3JHH = 8.2, 4JHH = 2.5, 2H, Ar), 1.94–2.04 (m, 2H, PCH2), 1.67–1.76 (m, 4H, CH2), 1.50–1.61 (m, 2H, CH2), 1.36–1.44 (m, 4H, CH2), 1.22–1.34 (m, 16H, CH2), 1.26 (d, 3JPH = 13.8, 18H, tBu), 0.58 (partially collapsed quartet, fwhm = 300 Hz, 6H, BH3).
13 C{ 1 H} NMR (151 MHz, CDCl3): δ 154.2 (d, 2JPC = 6, Ar{C}), 129.6 (s, Ar), 116.9 (d, 3JPC = 3, Ar), 114.1 (t, 3JPC = 3, Ar{2-CH}), 32.8 (d, 1JPC = 37, tBu{C}), 30.7 (d, 2JPC = 12, CH2), 28.1 (s, CH2), 28.0 (s, CH2), 27.6 (s, CH2), 27.5 (s, CH2), 25.1 (d, 1JPC = 33, PCH2), 24.8 (d, 3JPC = 3, tBu{CH3}), 22.3 (s, CH2).
31 P{ 1 H} NMR (243 MHz, CDCl3): δ 143.8 (partially collapsed quartet, fwhm = 165 Hz).
HR ESI-MS (positive ion, 4 kV): 531.3840, [M + Na]+ (calcd 531.3844) m/z.28
Anal. Calcd for C28H56B2O2P2 (508.32 g mol−1): C, 66.16; H, 11.10; found: C, 66.09; H, 11.26.28
cis-12 (Rf = 0.42). Yield: 984 mg (45%, white crystalline solid).
1 H NMR (500 MHz, CDCl3): δ 7.20 (t, 3JHH = 8.3, 1H, Ar), 7.03 (dd, 3JHH = 8.3, 4JHH = 2.3, 2H, Ar), 6.82 (t, 4JHH = 2.5, 1H, Ar{2-CH}), 1.90–2.06 (m, 2H, PCH2), 1.67–1.81 (m, 4H, CH2), 1.53–1.66 (m, 2H, CH2), 1.20–1.50 (m, 20H, CH2), 1.26 (d, 3JPH = 14.0, 18H, tBu), 0.61 (partially collapsed quartet, fwhm = 285 Hz, 6H, BH3).
13 C{ 1 H} NMR (126 MHz, CDCl3): δ 154.4 (d, 2JPC = 6, Ar{C}), 129.7 (s, Ar), 116.6 (d, 3JPC = 3, Ar), 113.4 (t, 3JPC = 4, Ar{2-CH}), 32.9 (d, 1JPC = 37, tBu{C}), 30.5 (d, 2JPC = 12, CH2), 28.2 (s, CH2), 27.9 (s, CH2), 27.8 (s, CH2), 27.6 (s, CH2), 25.2 (d, 1JPC = 33, PCH2), 24.9 (d, 3JPC = 3, tBu{CH3}), 22.0 (d, 3JPC = 2, CH2).
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 143.7 (br, fwhm = 175 Hz).
HR ESI-MS (positive ion, 4 kV): 531.3840, [M + Na]+ (calcd 531.3844) m/z.28
Anal. Calcd for C28H56B2O2P2 (508.32 g mol−1): C, 66.16; H, 11.10; found: C, 66.09; H, 11.26.28
1 H NMR (500 MHz, toluene-d8): δ 7.30 (app p, J = 2, 1H, Ar{2-CH}), 6.97 (t, 3JHH = 8.1, 1H, Ar), 6.87 (app dt, 3JHH = 8.2, J = 2, 2H, Ar), 1.84 (dtd, 2JHH = 14.4, 3JHH = 7.3, 2JPH = 3.2, 2H, PCH2), 1.54–1.67 (m, 4H, CH2), 1.15–1.50 (m, 22H, CH2), 1.00 (d, 3JPH = 12.1, 18H, tBu).
13 C{ 1 H} NMR (126 MHz, toluene-d8): δ 160.9 (d, 2JPC = 9, Ar{C}), 129.9 (s, Ar), 112.2 (d, 3JPC = 11, Ar), 109.2 (t, 3JPC = 12, Ar{2-CH}), 32.5 (d, 1JPC = 16, tBu{C}), 31.1 (d, 2JPC = 11, CH2), 29.0 (d, 1JPC = 24, PCH2), 28.9 (s, CH2), 28.5 (s, CH2), 28.2 (s, CH2), 28.0 (s, CH2), 25.5 (d, 3JPC = 15, CH2), 25.3 (d, 2JPC = 15, tBu{CH3}).
31 P{ 1 H} NMR (162 MHz, toluene-d8): δ 141.6 (s).
1 H NMR (500 MHz, toluene-d8): δ 7.21 (app p, J = 2, 1H, Ar{2-CH}), 6.97 (t, 3JHH = 7.9, 1H, Ar), 6.91 (app dt, 3JHH = 7.9, J = 2, 2H, Ar), 1.85 (dtd, 2JHH = 14.2, 3JHH = 7.4, 2JPH = 3.1, 1H, PCH2), 1.58–1.68 (m, 4H, CH2), 1.41–1.52 (m, 2H, CH2), 1.16–1.40 (m, 20H, CH2), 1.01 (d, 3JPH = 12.0, 18H, tBu).
13 C{ 1 H} NMR (126 MHz, toluene-d8): δ 160.8 (d, 2JPC = 9, Ar{C}), 129.9 (s, Ar), 111.7 (d, 3JPC = 13, Ar), 109.4 (t, 3JPC = 11, Ar{2-CH}), 32.5 (d, 1JPC = 16, tBu{C}), 30.9 (d, 2JPC = 11, CH2), 29.1 (d, 1JPC = 24, PCH2), 28.9 (s, CH2), 28.5 (s, CH2), 28.2 (s, CH2), 28.1 (s, CH2), 25.4 (d, 3JPC = 15, CH2), 25.3 (d, 3JPC = 15, tBu{CH3}).
31 P{ 1 H} NMR (162 MHz, C6D6): δ 140.5 (s).
1 H NMR (500 MHz, CD2Cl2): δ 7.23 (t, 3JHH = 8.3, 1H, Ar), 7.19 (t, 4JHH = 2.4, 1H, Ar{2-CH}), 7.04 (dd, 3JHH = 8.3, 4JHH = 7.1, 2H, Ar), 1.75–1.91 (m, 4H, CH2), 1.16–1.73 (m, 24H, CH2), 1.22 (d, 3JPH = 15.5, 18H, tBu).
13 C{ 1 H} NMR (126 MHz, CD2Cl2): δ 153.6 (d, 2JPC = 10, Ar{C}), 130.2 (s, Ar), 116.4 (d, 3JPC = 4, Ar), 113.3 (t, 3JPC = 4, Ar{2-CH}), 33.9 (d, 1JPC = 91, tBu{C}), 30.9 (d, 2JPC = 14, CH2), 28.5 (s, CH2), 28.3 (s, CH2), 28.0 (s, CH2), 27.7 (s, CH2), 24.6 (s, tBu{CH3}), 24.4 (d, 1JPC = 83, PCH2), 21.9 (d, 3JPC = 6, CH2).
31 P{ 1 H} NMR (162 MHz, CD2Cl2): δ 64.1 (s).
HR ESI-MS (positive ion, 4 kV): 535.3078, [M + Na]+ (calcd 535.3077) m/z.
Synthesis of rhodium complexes of PCP-14
A solution of PCP-14′ (118.2 mg, 248 μmol) and [Rh(COE)2Cl]2 (88.9 mg, 124 μmol) in toluene (5 mL) within a reaction flask fitted with a J Young's value was freeze–pump–thaw-degassed and placed under dihydrogen (1 atm), sealed, and heated at 100 °C for 18 h. The solution was cooled to RT and cannula transferred under dihydrogen into a reaction flask fitted with a J Young's value and charged with KOtBu (34.0 mg, 303 μmol). The flask was then sealed and heated at 120 °C for 2 h to generate 13a, which was characterised in situ. The solution was freeze–pump–thaw degassed and placed under carbon monoxide (1 atm) resulting in immediate formation of 14a, which was isolated by removal of the volatiles in vacuo and extraction of the residue with hexane. Crude 14a obtained in this way was dissolved in toluene (5 mL) and then slowly added to a suspension of PhICl2 (68.7 mg, 250 μmol) in toluene (2 mL) at −78 °C. The reaction mixture was allowed to warm to RT overnight, the volatiles removed in vacuo, and the resulting residue extracted with hexane to afford the crude product, which was purified by alumina chromatography in air (20% CH2Cl2 in hexane, Rf = 0.55) to afford analytically pure 15a. Yield: 136.4 mg (80%). Complex 15a is stable in the solid state, but partial loss of CO was observed in solution over time.
1
H NMR (500 MHz, toluene-d8, H2): δ 7.12 (d, 3JHH = 7.4, 2H, Ar), 7.05 (t, 3JHH = 7.4, 1H, Ar), 3.08 (vt, JPH = 3.9, 4H, ArC2), 1.87–2.00 (m, 2H, CH2), 1.74–1.84 (m, 2H, CH2), 1.17–1.67 (m, 24H, CH2), 1.01 (vt, JPH = 6.5, 18H, tBu), −4.36 (br, 2H, RhH).
13
C{
1
H} NMR (126 MHz, toluene-d8, H2): δ 176.3 (dt, 1JRhC = 40, 2JPC = 6, Ar{CRh}), 151.6 (vtd, JPC = 12, 2JRhC = 3, Ar{C}), 124.1 (s, Ar), 120.7 (vt, JPC = 10, Ar), 39.9 (vtd, JPC = 10, 2JRhC = 4, ArH2), 31.9 (vt, JPC = 11, tBu{C}), 29.5 (vt, JPC = 4, CH2), 29.2 (s, CH2), 29.0 (s, CH2), 28.6 (s, CH2), 28.4 (vt, JPC = 3, tBu{CH3}), 27.7 (s, CH2), 25.6 (vt, JPC = 4, CH2), 22.9 (vt, JPC = 8, PCH2).
31 P{ 1 H} NMR (162 MHz, toluene-d8, H2): δ 70.4 (d, 1JRhP = 153).
1 H NMR (600 MHz, toluene-d8, Ar, selected data): δ −4.36 (br d, 1JRhH = 17.9, 2H, RhH, T1 = 74 ± 4 ms).
1 H NMR (600 MHz, toluene-d8, Ar, 200 K, selected data): δ −4.26 (br, 2H, RhH, T1 = 117 ± 15 ms).
[Rh(PCP-14)Cl2(CO)] (15a)
1 H NMR (600 MHz, toluene-d8, CO): δ 6.95 (t, 3JHH = 7.4, 1H, Ar), 6.90 (d, 3JHH = 7.4, 2H, Ar) 3.71 (dvt, 2JHH = 15.6, JPH = 4.8, 2H, ArC2), 2.96–3.04 (m, 2H, PCH2), 2.91 (dvt, 2JHH = 15.6, JPH = 4.1, 2H, ArC2), 1.84–1.95 (m, 2H, CH2), 1.25–1.78 (m, 24H, CH2), 1.16 (vt, JPH = 6.9, 18H, tBu).
13
C{
1
H} NMR (151 MHz, toluene-d8, CO): δ 189.1 (dt, 1JRhC = 39, 2JPC = 7, CO), 163.7 (d, 1JRhC = 21, Ar{CRh}), 147.4 (vt, JPC = 8, Ar{C}), 125.6 (s, Ar), 122.6 (vt, JPC = 9, Ar), 38.1 (vt, JPC = 14, ArH2), 34.9 (vt, JPC = 10, tBu{C}), 30.9 (vt, JPC = 5, CH2), 29.2 (s, CH2), 29.1 (s, CH2), 28.9 (s, CH2), 28.6 (s, CH2), 27.6 (s, tBu{CH3}), 26.4 (s, CH2), 22.3 (vt, JPC = 11, PCH2).
31 P{ 1 H} NMR (243 MHz, toluene-d8, CO): δ 64.9 (d, 1JRhP = 87).
IR (toluene): ν(CO) 2069 cm−1.
Anal. Calcd for C31H53Cl2OP2Rh (677.52 g mol−1): C, 54.96; H, 7.89; found: C, 54.99; H, 8.06.
Synthesis of rhodium complexes of POCOP-14
A solution of POCOP-14′ (217 mg, 452 μmol) and [Rh(COE)2Cl]2 (163 mg, 227 μmol) in toluene (5 mL) within a reaction flask fitted with a J Young's value was freeze–pump–thaw-degassed and placed under dihydrogen (1 atm), sealed, and heated at 120 °C for 3 days. The solution was cooled to RT and cannula transferred under dihydrogen into a reaction flask fitted with a J Young's value and charged with KOtBu (61.1 mg, 545 μmol). The flask was then sealed and heated at 120 °C for 1 h to generate 13b, which was characterised in situ. The solution was freeze–pump–thaw degassed and placed under carbon monoxide (1 atm) resulting in immediate formation of 14b, which was isolated by removal of the volatiles in vacuo and extraction of the residue with hexane. Crude 14b obtained in this way was dissolved in toluene (5 mL) and then slowly added to a suspension of PhICl2 (125 mg, 455 μmol) in toluene (2 mL) at −78 °C. The reaction mixture was allowed to warm to RT overnight, the volatiles removed in vacuo, and the resulting residue extracted with hexane to afford the crude product, which was purified by alumina chromatography in air (20% CH2Cl2 in hexane, Rf = 0.45) to afford analytically pure 15b. Yield: 149.3 mg (48%). Complex 15b is stable in the solid state, but partial loss of CO was observed in solution over time.1 H NMR (500 MHz, toluene-d8, H2): δ 6.94 (t, 3JHH = 7.9, 1H, Ar), 6.76 (d, 3JHH = 7.9, 2H, Ar), 1.93–2.04 (m, 4H, CH2), 1.83–1.92 (m, 2H, PCH2), 1.17–1.67 (m, 22H, CH2), 1.15 (vt, JPH = 7.1, 18H, tBu), −2.87 (br d, 1JRhH = 18.8, 2H, RhH).
13 C{ 1 H} NMR (126 MHz, toluene-d8, H2): δ 167.3 (vt, JPC = 10, Ar{C}), 143.1 (dt, 1JRhC = 35, 2JPC = 10, Ar{CRh}), 126.7 (s, Ar), 104.8 (vt, JPC = 7, Ar), 36.7 (vtd, JPC = 12, 2JRhC = 2, tBu{C}), 29.0 (vt, JPC = 2, CH2), 28.9 (s, CH2), 28.7 (s, CH2), 28.6 (s, CH2), 28.1 (vtd, JPC = 9, 2JRhC = 2, PCH2), 27.7 (s, CH2), 26.8 (vt, JPC = 4, tBu{CH3}), 25.0 (vt, JPC = 4, CH2).
31 P{ 1 H} NMR (162 MHz, toluene-d8, H2): δ 198.4 (d, 1JRhP = 165).
1 H NMR (600 MHz, toluene-d8, Ar, selected data): δ −2.87 (br d, 1JRhH = 18.9, 2H, RhH, T1 = 42 ± 2 ms).
1 H NMR (600 MHz, toluene-d8, Ar, 200 K, selected data): δ −2.72 (br, 2H, RhH, T1 = 89 ± 11 ms).
[Rh(POCOP-14)Cl2(CO)] (15b)
1 H NMR (500 MHz, toluene-d8, CO): δ 6.80 (t, 3JHH = 7.9, 1H, Ar), 6.61 (d, 3JHH = 7.9, 2H, Ar), 3.63 (app dp, 2JHH = 14.1, J = 7, 2H, PCH2), 1.87–1.94 (m, 2H, CH2), 1.78–1.87 (m, 2H, PCH2), 1.29 (vt, JPH = 7.6, 18H, tBu), 1.11–1.72 (m, 22H, CH2).13 C{ 1 H} NMR (126 MHz, toluene-d8, CO): δ 187.2 (dt, 1JRhC = 41, 2JPC = 5, CO), 163.6 (vt, JPC = 6, Ar{C}), 136.1 (dt, 1JRhC = 22, 2JPC = 6, Ar{CRh}), 128.4 (s, Ar), 107.6 (vt, JPC = 6, Ar), 41.5 (vt, JPC = 11, tBu{C}), 30.7 (vt, JPC = 6, CH2), 29.5 (s, CH2), 29.4 (s, CH2), 29.2 (s, CH2), 28.2 (s, CH2), 26.5 (s, tBu{CH3}), 24.1 (vt, JPC = 3, CH2), 23.8 (vt, JPC = 13, PCH2).
31 P{ 1 H} NMR (162 MHz, toluene-d8, CO): δ 181.0 (d, 1JRhP = 92).
IR (toluene): ν(CO) 2083 cm−1.
Anal. Calcd for C29H49Cl2O3P2Rh (681.46 g mol−1): C, 51.11; H, 7.25; found: C, 51.09; H, 7.31.
Synthesis of iridium complexes of PCP-14
A solution of PCP-14′ (57.6 mg, 121 μmol) and [Ir(COE)2Cl]2 (54.1 mg, 60.4 μmol) in toluene (5 mL) within a reaction flask fitted with a J Young's value was freeze–pump–thaw-degassed and placed under dihydrogen (1 atm), sealed, and heated at 70 °C for 18 h. The solution was cooled to RT and cannula transferred under dihydrogen into a reaction flask fitted with a J Young's value and charged with KOtBu (16.3 mg, 145 μmol). The flask was then sealed and heated at 120 °C for 18 h to generate 16a, which was characterised in situ. The solution was freeze–pump–thaw degassed and placed under carbon monoxide (1 atm) resulting in immediate formation of cis-17a, which was characterised in situ. To this solution tert-butylethylene (78 μL, 0.60 mmol) was added and the reaction mixture heated under carbon monoxide (1 atm, sealed flask) at 120 °C for 18 h, resulting in partial conversion to trans-17a but ultimately affording [Ir(PCP-14)(CO)2] (δ31P 51.6). The solution was freeze–pump–thaw degassed and placed under argon to generate 18a, filtered, and slowly added to a suspension of PhICl2 (33.3 mg, 121 μmol) in toluene (1 mL) at −78 °C. The reaction mixture was allowed to warm to RT overnight, the volatiles removed in vacuo, and the resulting residue extracted with hexane to afford the crude product, which was purified by silica chromatography in air (40% CH2Cl2 in hexane, Rf = 0.43) to afford analytically pure 19a. Yield: 76.9 mg (51%).
1
H NMR (600 MHz, toluene-d8, H2): δ 6.99–7.03 (m, 3H, Ar), 3.50 (dvt, 2JHH = 16.1, JPH = 3.6, 2H, ArC2), 2.89 (dvt, 2JHH = 16.6, JPH = 4.6, 2H, ArC2), 1.78–1.88 (m, 2H, PCH2), 1.67–1.77 (m, 2H, CH2), 1.53–1.63 (m, 2H, CH2), 1.27–1.53 (m, 22H, CH2), 0.94 (vt, JPH = 6.8, 18H, tBu), −8.99 (t, 2JPH = 9.8, 4H, IrH).
13
C{
1
H} NMR (151 MHz, toluene-d8, H2): δ 151.7 (s, Ar{CIr}), 147.7 (vt, JPC = 8, Ar{C}), 123.3 (s, Ar), 120.1 (vt, JPC = 8, Ar), 46.1 (vt, JPC = 17, ArH2), 30.0 (vt, JPC = 4, CH2), 29.7 (s, CH2), 29.3 (vt, JPC = 15, tBu{C}), 29.1 (s, CH2), 28.9 (s, CH2), 28.2 (s, CH2), 27.4 (br, CH2), 27.2 (vt, JPC = 15, PCH2), 25.7 (vt, JPC = 3, tBu{CH3}).
31 P{ 1 H} NMR (243 MHz, toluene-d8, H2): δ 49.9 (s).
1 H NMR (600 MHz, toluene-d8, Ar, selected data): δ −8.99 (t, 2JPH = 9.8, 4H, IrH, T1 = 300 ± 10 ms).
1 H NMR (600 MHz, toluene-d8, Ar, 200 K, selected data): δ −8.95 (br, 4H, IrH, T1 = 626 ± 15 ms).
1
H NMR (500 MHz, toluene-d8): δ 6.99–7.04 (m, 3H, Ar), 3.50–3.65 (m, 2H, ArC2), 2.98 (dd, 2JHH = 16.0, 2JPH = 7.3, 1H, ArC2), 2.86 (dd, 2JHH = 16.8, 2JPH = 8.8, 1H, ArC2), 2.06–2.16 (m, 1H, PCH2), 1.92–2.04 (m, 2H, CH2), 1.14–1.91 (m, 25H, CH2), 0.96 (d, 2JPH = 13.3, 9H, tBu), 0.89 (d, 2JPH = 13.6, 9H, tBu), −10.66 (ddd, 2JPH = 23.4, 2JPH = 10.5, 2JHH = 3.0, 1H, IrH), −11.59 (app td, 2JPH = 12, 2JHH = 3.0, 1H, IrH).
13
C{
1
H} NMR (126 MHz, toluene-d8): δ 183.3 (m, CO), 155.1 (br, Ar{CIr}), 148.8 (dd, 2JPC = 10, JPC = 4, Ar{C}), 148.1 (dd, 2JPC = 9, JPC = 4, Ar{C}), 123.5 (s, Ar), 120.0 (d, 3JPC = 14, Ar), 119.8 (d, 3JPC = 16, Ar), 48.4 (d, 1JPC = 35, ArH2), 46.2 (d, 1JPC = 35, ArH2), 31.2 (dd, 1JPC = 24, 3JPC = 3, tBu{C}), 30.5 (dd, 1JPC = 27, 3JPC = 5, tBu{C}), 30.2 (d, 2JPC = 10, CH2), 29.9 (s, CH2), 29.7 (d, 2JPC = 9, CH2), 29.4 (s, CH2), 29.3 (s, CH2), 29.1 (s, CH2), 28.8 (s, CH2), 28.5 (s, CH2), 28.3 (s, CH2), 28.2 (s, CH2), 27.9 (dd, 1JPC = 29, 3JPC = 4, PCH2), 27.7 (s, CH2), 27.3 (dd, 1JPC = 24, 3JPC = 3, PCH2), 27.2 (s, CH2), 26.2 (d, 2JPC = 4, tBu{CH3}), 26.1 (d, 2JPC = 4, tBu{CH3}).
31 P{ 1 H} NMR (162 MHz, toluene-d8): δ 51.0 (d, 2JPP = 286, 1P), 45.8 (d, 2JPP = 286, 1P).
trans-[Ir(PCP-14)(H)2(CO)] (trans-17a)
1 H NMR (400 MHz, toluene-d0, CO, selected data): δ 1.01 (vt, 2JPH = 7.1, 18H, tBu), −9.65 (t, 2JPH = 14.4, 2H, IrH).31 P{ 1 H} NMR (162 MHz, toluene-d0, CO): δ 54.1 (s).
[Ir(PCP-14)Cl2(CO)] (19a)
1 H NMR (500 MHz, toluene-d8): δ 6.94–7.00 (m, 3H, Ar), 3.64 (dvt, 2JHH = 15.8, JPH = 5.0, 2H, ArC2), 3.05 (dvt, 2JHH = 15.8, JPH = 3.8, 2H, ArC2), 2.91–3.01 (m, 2H, PCH2), 1.81–1.97 (m, 2H, CH2), 1.02–1.81 (m, 24H, CH2), 1.13 (br, 18H, tBu).
13
C{
1
H} NMR (126 MHz, toluene-d8): δ 175.7 (t, 2JPC = 5, CO), 155.3 (s, Ar{CIr}), 148.5 (vt, JPC = 8, Ar{C}), 126.0 (s, Ar), 121.6 (vt, JPC = 8, Ar), 38.9 (vt, JPC = 17, ArH2), 34.3 (vt, 1JPC = 13, tBu{C}), 30.8 (vt, JPC = 5, CH2), 29.2 (s, CH2), 29.1 (s, CH2), 29.0 (s, CH2), 28.6 (s, CH2), 27.5 (s, tBu{CH3}), 26.4 (s, CH2), 20.6 (vt, JPC = 13, PCH2).
31 P{ 1 H} NMR (162 MHz, toluene-d8): δ 33.8 (s).
IR (toluene): ν(CO) 2034 cm−1.
Anal. Calcd for C31H53Cl2IrOP2 (766.83 g mol−1): C, 48.56; H, 6.97; found: C, 48.66; H, 6.92.
Synthesis of iridium complexes of POCOP-14
A solution of POCOP-14′ (69.1 mg, 144 μmol) and [Ir(COD)Cl]2 (53.1 mg, 79.1 μmol) in toluene (5 mL) within a reaction flask fitted with a J Young's value was freeze–pump–thaw-degassed and placed under dihydrogen (1 atm), sealed, and heated at 100 °C for 3 days. The solution was cooled to RT and cannula transferred under dihydrogen into a reaction flask fitted with a J Young's value and charged with KOtBu (19.4 mg, 173 μmol). The flask was then sealed and heated at 120 °C for 2 h to generate 16b, which was characterised in situ. The solution was freeze–pump–thaw degassed and placed under carbon monoxide (1 atm) resulting in immediate formation of cis-17b, which was characterised in situ. To this solution tert-butylethylene (93 μL, 0.72 mmol) was added and the reaction mixture heated under carbon monoxide (1 atm, sealed flask) at 120 °C for 18 h, resulting in partial conversion to trans-17b but ultimately affording [Ir(POCOP-14)(CO)2] (δ31P 164.8, fwhm = 127 Hz). The solution was freeze–pump–thaw degassed and placed under argon to generate 18b, filtered, and slowly added to a suspension of PhICl2 (39.5 mg, 144 μmol) in toluene (2 mL) at −78 °C. The reaction mixture was allowed to warm to RT overnight, the volatiles removed in vacuo, and the resulting residue extracted with hexane to afford the crude product, which was purified by silica chromatography in air (20% CH2Cl2 in hexane, Rf = 0.28) to afford analytically pure 19b. Yield: 107.9 mg (73%).1 H NMR (500 MHz, toluene-d0, H2, selected data): δ 6.67 (d, 3JHH = 7.9, 2H, Ar), 1.07 (vt, JPH = 7.5, 18H, tBu), −8.26 (t, 2JPH = 9.9, 4H, IrH).
31 P{ 1 H} NMR (162 MHz, toluene-d0, H2): δ 165.0 (s).
1 H NMR (600 MHz, toluene-d0, Ar, selected data): δ −8.26 (t, 2JPH = 10.0, 4H, IrH, T1 = 194 ± 4 ms).
1 H NMR (600 MHz, toluene-d0, Ar, 200 K, selected data): δ −8.10 (br, 4H, IrH, T1 = 242 ± 15 ms).
1 H NMR (500 MHz, toluene-d8): δ 6.86 (t, 3JHH = 7.9, 1H, Ar), 6.70 (d, 3JHH = 7.9, 1H, Ar), 6.70 (d, 3JHH = 7.9, 1H, Ar), 2.68–2.79 (m, 1H, PCH2), 2.30–2.46 (m, 3H, CH2), 1.85–1.97 (m, 1H, PCH2), 1.12–1.70 (m, 23H, CH2), 1.08 (d, 2JPH = 14.8, 9H, tBu), 1.04 (d, 2JPH = 15.0, 9H, tBu), −9.82 (app t, 2JPH = 9, 1H, IrH), −10.75 (dd, 2JPH = 21.8, 2JPH = 14.7, 1H, IrH).
13 C{ 1 H} NMR (126 MHz, toluene-d8): δ 180.6 (m, CO), 163.2 (dd, 2JPC = 6, JPC = 4, Ar{C}), 162.8 (dd, 2JPC = 7, JPC = 4, Ar{C}), 126.2 (s, Ar), 123.0 (app t, 2JPC = 6, Ar{CIr}), 104.8 (d, 3JPC = 12, Ar), 104.7 (d, 3JPC = 16, Ar), 37.2 (dd, 1JPC = 28, 3JPC = 7, tBu{C}), 37.0 (dd, 1JPC = 27, 3JPC = 6, tBu{C}), 34.6 (dd, 1JPC = 30, 3JPC = 5, PCH2), 33.0 (dd, 1JPC = 27, 3JPC = 5, PCH2), 30.3 (s, CH2), 29.8 (s, CH2), 29.7 (d, JPC = 6, CH2), 29.5 (s, CH2), 29.16 (s, CH2), 29.15 (s, CH2), 28.9 (s, CH2), 28.6 (s, CH2), 28.5 (s, CH2), 27.7 (d, JPC = 6, CH2), 25.7 (d, JPC = 5, CH2), 25.2 (app vt, JPC = 5, tBu{CH3}), 25.1 (d, JPC = 6, CH2).
31 P{ 1 H} NMR (162 MHz, toluene-d8): δ 160.4 (d, 2JPP = 305, 1P), 154.6 (d, 2JPP = 305, 1P).
trans-[Ir(POCOP-14)(H)2(CO)] (trans-17b)
1 H NMR (400 MHz, toluene-d0, CO, selected data): δ 1.10 (vt, JPH = 7.6, 18H, tBu), −9.76 (t, 2JPH = 16.5, 2H, IrH).31 P{ 1 H} NMR (162 MHz, toluene-d0, CO): δ 167.3 (s).
[Ir(POCOP-14)Cl2(CO)] (19b)
1 H NMR (500 MHz, toluene-d8): δ 6.80 (t, 3JHH = 8.0, 1H, Ar), 6.64 (d, 3JHH = 8.0, 2H, Ar), 3.57 (app dp, 2JHH = 14.3, J = 7, 2H, PCH2), 1.85–1.99 (m, 2H, CH2), 1.72–1.83 (m, 2H, PCH2), 1.26 (vt, JPH = 7.7, 18H, tBu), 1.11–1.71 (m, 22H, CH2).
13
C{
1
H} NMR (126 MHz, toluene-d8): δ 175.6 (t, 2JPC = 3, CO), 164.4 (vt, JPC = 6, Ar{C}), 129 (obsc, Ar{CIr}), 128.9 (s, Ar), 106.8 (vt, JPC = 6, Ar), 41.0 (vt, JPC = 15, tBu{C}), 30.3 (vt, JPC = 6, PCH2H2), 29.5 (s, CH2), 29.4 (s, CH2), 29.1 (s, CH2), 28.3 (s, CH2), 26.5 (s, tBu{CH3}), 24.2 (vt, JPC = 2, CH2), 22.0 (vt, JPC = 16, PCH2).
31 P{ 1 H} NMR (121 MHz, toluene-d8): δ 144.4 (s).
IR (toluene): ν(CO) 2049 cm−1.
Anal. Calcd for C29H49Cl2IrO3P2 (770.77 g mol−1): C, 45.19; H, 6.41; found: C, 44.95; H, 6.30.
General procedure for the preparation of Rh(I) carbonyls
To a solution of 15 (ca. 30 μmol) in THF (2 mL) at RT was added iPrMgCl·LiCl (1.3 M in THF, 3 equivalents). The resulting solution was stirred at RT for 1 h, excess MeOH added and then reduced to dryness in vacuo. The residue was extracted with pentane to afford the crude product on removal of volatiles, which was subsequently recrystallised by slow evaporation of SiMe4.
1
H NMR (600 MHz, toluene-d8): δ 7.06 (d, 3JHH = 7.3, 2H, Ar), 7.02 (t, 3JHH = 7.3, 1H, Ar) 3.19 (dvt, 2JHH = 16.3, JPH = 3.4, 2H, ArC2), 3.15 (dvt, 2JHH = 16.5, JPH = 4.2, 2H, ArC2), 2.10–2.01 (m, 2H, CH2), 1.77–1.36 (m, 26H, CH2), 1.00 (vt, JPH = 6.6, 18H, tBu).
13
C{
1
H} NMR (151 MHz, toluene-d8): δ 201.5 (dvt, 1JRhC = 57, 2JPC = 12, CO), 179.4 (dt, 1JPC = 29, 2JPC = 7, Ar{CRh}), 152.2 (vt, JPC = 12, Ar{C}), 125.7 (s, Ar), 120.5 (vt, JPC = 9, Ar), 41.3 (vt, JPC = 12, ArH2), 32.7 (vt, JPC = 11, tBu{C}), 30.5 (vt, JPC = 4, CH2), 29.4 (s, CH2), 29.2 (s, CH2), 29.0 (s, CH2), 28.52 (vt, JPC = 4, tBu{CH3}), 28.47 (s, CH2), 26.4 (vt, JPC = 4, CH2), 24.2 (vt, 1JPC = 9, PCH2).
31 P{ 1 H} NMR (243 MHz, toluene-d8): δ 75.0 (d, 1JRhP = 146).
IR (toluene): ν(CO) 1939 cm−1.
Anal. Calcd for C31H53OP2Rh (606.62 g mol−1): C, 61.38; H, 8.81; found: C, 61.34; H, 8.74.
1 H NMR (600 MHz, toluene-d8): δ 6.91 (t, 3JHH = 7.9, 1H, Ar), 6.70 (d, 3JHH = 7.9, 2H, Ar), 2.00–2.06 (m, 4H, PCH2), 1.73–1.85 (m, 4H, CH2), 1.26–1.60 (m, 20H, CH2), 1.15 (vt, JPH = 7.2, 18H, tBu).
13 C{ 1 H} NMR (151 MHz, toluene-d8): δ 201.2 (dt, 1JRhC = 60, 2JPC = 10, CO), 167.7 (vt, JPC = 9, Ar{C}), 145.5 (dt, 1JRhC = 26, 2JPC = 10, Ar{CRh}), 128.8 (obsc, Ar), 104.8 (vt, JPC = 7, Ar), 38.1 (vt, 1JPC = 12, tBu{C}), 30.6 (br, CH2), 29.4 (s, CH2), 29.3 (s, CH2), 29.24 (vt, JPC = 9, PCH2), 29.16 (s, CH2), 28.8 (s, CH2), 26.8 (vt, JPC = 4, tBu{CH3}), 25.8 (vt, JPC = 4, CH2).
31 P{ 1 H} NMR (243 MHz, toluene-d8): δ 201.6 (d, 1JRhP = 156).
IR (toluene): ν(CO) 1958 cm−1.
Anal. Calcd for C29H49O3P2Rh (610.56 g mol−1): C, 57.05; H, 8.09; found: C, 57.14; H, 8.07.
General procedure for the preparation of Ir(I) carbonyl complexes
A solution of 19 (ca. 20 μmol) in pentane (2 mL) was added to a flask charged with KC8 (10 equivalents) and stirred at RT for 2 days. The solution was filtered, reduced to dryness in vacuo, and the analytically pure product obtained following recrystallisation.16
1
H NMR (500 MHz, toluene-d8): δ 7.14 (d, 3JHH = 7.5, 2H, Ar), 7.02 (t, 3JHH = 7.5, 1H, Ar), 3.35 (dvt, 2JHH = 16.4, JPH = 4.0, 2H, ArC2), 3.09 (dvt, 2JHH = 16.4, JPH = 3.8, 2H, ArC2), 2.03–2.19 (m, 2H, CH2), 1.90–1.78 (m, 2H, PCH2), 1.76–1.34 (m, 24H, CH2), 1.01 (vt, JPH = 6.7, 18H, tBu).
13
C{
1
H} NMR (126 MHz, toluene-d8): δ 198.6 (t, 2JPC = 7, CO), 181.3 (t, 2JPC = 4, Ar{CIr}), 153.8 (vt, JPC = 11, Ar{C}), 126.1 (s, Ar), 120.2 (vt, JPC = 8, Ar), 41.7 (vt, JPC = 15, ArH2), 33.5 (vt, JPC = 14, tBu{C}), 30.5 (vt, JPC = 5, CH2), 29.5 (s, CH2), 29.3 (s, CH2), 29.0 (s, CH2), 28.5 (s, CH2), 28.2 (vt, JPC = 3, tBu{CH3}), 26.3 (vt, JPC = 3, CH2), 24.2 (vt, JPC = 13, PCH2).
31 P{ 1 H} NMR (162 MHz, toluene-d8): δ 68.1 (s).
IR (toluene): ν(CO) 1925 cm−1.
Anal. Calcd for C31H53IrOP2 (695.93 g mol−1): C, 53.50; H, 7.68; found: C, 53.24; H, 7.76.
1 H NMR (500 MHz, toluene-d8): δ 6.87 (t, 3JHH = 7.9, 1H, Ar), 6.74 (d, 3JHH = 7.9, 2H, Ar), 2.03–2.19 (m, 4H, CH2), 1.73–1.87 (m, 4H, CH2), 1.23–1.66 (m, 20H, CH2), 1.15 (vt, JPH = 7.4, 18H, tBu).
13 C{ 1 H} NMR (126 MHz, toluene-d8): δ 200.5 (t, 2JPC = 5, CO), 168.3 (vt, JPC = 8, Ar{C}), 148.7 (t, 2JPC = 9, Ar{CIr}), 129.5 (s, Ar), 104.4 (vt, JPC = 6, Ar), 39.5 (vt, JPC = 16, tBu{C}), 30.8 (vt, JPC = 2, CH2), 29.6 (vt, JPC = 14, PCH2), 29.4 (s, CH2), 29.3 (s, CH2), 29.2 (s, CH2), 28.9 (s, CH2), 26.7 (vt, JPC = 3, tBu{CH3}), 26.0 (vt, JPC = 3, CH2).
31 P{ 1 H} NMR (162 MHz, toluene-d8): δ 186.7 (s).
IR (toluene): ν(CO) 1943 cm−1.
Anal. Calcd for C29H49IrO3P2 (699.87 g mol−1): C, 49.77; H, 7.06; found: C, 49.85; H 7.01.
Crystallographic details
Data were collected on a Rigaku Oxford Diffraction SuperNova AtlasS2 CCD diffractometer using graphite monochromated Mo Kα (λ = 0.71073 Å) or CuKα (λ = 1.54184 Å) radiation and an Oxford Cryosystems N-HeliX low temperature device [150(2) K]. Data were collected and reduced using CrysAlisPro and refined using SHELXL,29 through the Olex2 interface.30 Full details about the collection, solution, and refinement are documented in CIF format, which have been deposited with the Cambridge Crystallographic Data Centre under CCDC 1972811–1972820.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council (ERC, grant agreement 637313) and Royal Society (UF100592, UF150675, A. B. C.) for financial support. High-resolution mass-spectrometry data were collected using instruments purchased through support from Advantage West Midlands and the European Regional Development Fund. Crystallographic data were collected using an instrument that received funding from the ERC under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 637313).References
- (a) Pincer Compounds: Chemistry and Applications, ed. D. Morales-Morales, Elsevier, 2018, vol. 1 Search PubMed; (b) S. Murugesan and K. Kirchner, Dalton Trans., 2016, 45, 416–439 RSC; (c) R. E. Andrew, L. González-Sebastián and A. B. Chaplin, Dalton Trans., 2016, 45, 1299–1305 RSC; (d) The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications, in Topics in Organometallic Chemistry, ed. G. van Koten and R. A. Gossage, Springer, 2016; vol. 45 Search PubMed; (e) M. Asay and D. Morales-Morales, Dalton Trans., 2015, 44, 17432–17447 RSC; (f) S. Werkmeister, J. Neumann, K. Junge and M. Beller, Chem. – Eur. J., 2015, 21, 12226–12250 CrossRef PubMed; (g) Pincer and Pincer-Type Complexes: Applications in Organic Synthesis and Catalysis, ed. K. J. Szabó and O. F. Wendt, Wiley-VCH, 2014 Search PubMed; (h) Organometallic Pincer Chemistry; ed. G. van Koten and D. Milstein, Topics in Organometallic Chemistry, Springer, 2013, vol. 40 Search PubMed; (i) M. Albrecht and M. M. Lindner, Dalton Trans., 2011, 40, 8733–8744 RSC; (j) J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832–8846 CrossRef; (k) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef; (l) M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef.
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020–1024 RSC.
- (a) A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef; (b) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- (a) M. R. Gyton, B. Leforestier and A. B. Chaplin, Organometallics, 2018, 37, 3963–3971 CrossRef CAS; (b) C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Angew. Chem., Int. Ed., 2018, 57, 12003–11200 CrossRef CAS PubMed; (c) S. L. Apps, R. E. Alflatt, B. Leforestier, C. M. Storey and A. B. Chaplin, Polyhedron, 2018, 143, 57–61 CrossRef CAS; (d) R. E. Andrew, C. M. Storey and A. B. Chaplin, Dalton Trans., 2016, 45, 8937–8944 RSC; (e) R. E. Andrew, D. W. Ferdani, C. A. Ohlin and A. B. Chaplin, Organometallics, 2015, 34, 913–917 CrossRef CAS; (f) R. E. Andrew and A. B. Chaplin, Inorg. Chem., 2015, 54, 312–322 CrossRef CAS; (g) R. E. Andrew and A. B. Chaplin, Dalton Trans., 2014, 43, 1413–1423 RSC.
- T. M. Hood, M. R. Gyton and A. B. Chaplin, Dalton Trans., 2020 10.1039/c9dt04474d.
- (a) E. Salomó, S. Orgué, A. Riera and X. Verdaguer, Synthesis, 2016, 48, 2659–2663 CrossRef; (b) S. Orgué, A. Flores-Gaspar, M. Biosca, O. Pàmies, M. Diéguez, A. Riera and X. Verdaguer, Chem. Commun., 2015, 51, 17548–17551 RSC; (c) T. León, A. Riera and X. Verdaguer, J. Am. Chem. Soc., 2011, 133, 5740–5743 CrossRef.
- D. Mimeau, O. Delacroix and A.-C. Gaumont, Chem. Commun., 2003, 2928–2929 RSC.
- Z. Pakulski, O. M. Demchuk, R. Kwiatosz, P. W. Osiński, W. Świerczyńska and K. M. Pietrusiewicz, Tetrahedron: Asymmetry, 2003, 14, 1459–1462 CrossRef CAS.
- K. Zhu, P. D. Achord, X. Zhang, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2004, 126, 13044–13053 CrossRef CAS.
- (a) G. J. Kubas, Metal Dihydrogen and σ-Bond Complexes, Kluwer Academic Publishers, 2001 CrossRef; (b) R. H. Crabtree, Chem. Rev., 2016, 116, 8750–8769 CrossRef CAS.
- (a) M. Findlater, K. M. Schultz, W. H. Bernskoetter, A. Cartwright-Sykes, D. M. Heinekey and M. Brookhart, Inorg. Chem., 2012, 51, 4672–4678 CrossRef CAS; (b) K.-W. Huang, J. H. Han, C. B. Musgrave and E. Fujita, Organometallics, 2007, 26, 508–513 CrossRef CAS.
- It is interesting to note that the PC2 resonances of the MIIICl2(CO) derivatives are appreciably diastereotopic, giving rise to downfield 2H signals at ca. δ 3.0 (PCP-14) and 3.6 (POCOP-14) presumably as a consequence of diamagnetic anisotropy induced by close proximity to the chloride ligands. See for example: A. M. Camp, M. R. Kita, J. Grajeda, P. S. White, D. A. Dickie and A. J. M. Miller, Inorg. Chem., 2017, 56, 11141–11150 CrossRef CAS.
- For structures of [M(pincer)(CO)] (M = Rh, Ir; pincer = PCP-tBu, POCOP-tBu) see: (a) M. Montag, I. Efremenko, R. Cohen, L. J. W. Shimon, G. Leitus, Y. Diskin-Posner, Y. Ben-David, H. Salem, J. M. L. Martin and D. Milstein, Chem. – Eur. J., 2010, 16, 328–353 CrossRef CAS; (b) S. A. Kuklin, A. M. Sheloumov, F. M. Dolgushin, M. G. Ezernitskaya, A. S. Peregudov, P. V. Petrovskii and A. A. Koridze, Organometallics, 2006, 25, 5466–5476 CrossRef CAS; (c) K.-W. Huang, D. C. Grills, J. H. Han, D. J. Szalda and E. Fujita, Inorg. Chim. Acta, 2008, 361, 3327–3331 CrossRef CAS; (d) D. Morales-Morales, R. Redón, Z. Wang, D. W. Lee, C. Yung, K. Magnuson and C. M. Jensen, Can. J. Chem., 2001, 79, 823–829 CrossRef CAS.
- T. J. Hebden, K. I. Goldberg, D. M. Heinekey, X. Zhang, T. J. Emge, A. S. Goldman and K. Krogh-Jespersen, Inorg. Chem., 2010, 49, 1733–1742 CrossRef CAS.
- Reduction of 19a/b can also be achieved by reaction with iPrMgCl·LiCl (3 equiv.) in THF. Whilst 18a/b are formed in quantitative spectroscopic yield within 4 h at RT, they decompose on treatment with protic solvents that are needed to quench the excess Grignard reagent.
- For NHC and phosphine ligands see: (a) H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS; (b) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef; (c) R. A. Kelly III, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef; (d) C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS; (e) C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2953–2956 CrossRef CAS.
- (a) G. L. Parker, S. Lau, B. Leforestier and A. B. Chaplin, Eur. J. Inorg. Chem., 2019, 3791–3798 CrossRef CAS; (b) L. Maser, C. Schneider, L. Vondung, L. Alig and R. Langer, J. Am. Chem. Soc., 2019, 141, 7596–7760 CrossRef CAS; (c) J. J. Davidson, J. C. DeMott, C. Douvris, C. M. Fafard, N. Bhuvanesh, C.-H. Chen, D. E. Herbert, C.-I. Lee, B. J. McCulloch, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2015, 54, 2916–2935 CrossRef CAS.
- T. R. Hoye, B. M. Eklov and M. Voloshin, Org. Lett., 2004, 6, 2567–2570 CrossRef CAS.
- Y. Chen, T. P. Clark, B. A. Jazdzewski, S. B. Klamo and T. T. Wenzel, Polyhedron, 2014, 84, 32–36 CrossRef CAS.
- M. S. Carle, G. K. Shimokura and G. K. Murphy, Eur. J. Org. Chem., 2016, 3930–3933 CrossRef CAS.
- J. A. Osborn, G. Wilkinson and J. J. Mrowca, Inorg. Synth., 1990, 28, 77–79 CAS.
- A. van der Ent, A. L. Onderdelinden and R. A. Schunn, Inorg. Synth., 1990, 28, 90–92 CAS.
- J. L. Herde, J. C. Lambert, C. V. Senoff and M. A. Cushing, Inorg. Synth., 1974, 15, 18–20 CAS.
- (a) S. Nemeh, C. Jensen, E. Binamira-Soriaga and W. C. Kaska, Organometallics, 1983, 2, 1442–1447 CrossRef CAS; (b) I. Göttker-Schnetmann, P. S. White and M. Brookhart, Organometallics, 2004, 23, 1766–1776 CrossRef; (c) A. V. Polezhaev, S. A. Kuklin, D. M. Ivanov, P. V. Petrovskii, F. M. Dolgushin, M. G. Ezernitskaya and A. A. Koridze, Russ. Chem. Bull., 2009, 58, 1847–1854 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- C. R. Hilliard, N. Bhuvanesh, J. A. Gladysz and J. Blümel, Dalton Trans., 2012, 41, 1742–1754 RSC.
- Data for a mixture of the cis- and trans-diastereoisomers.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details; NMR, IR and ESI-MS spectra of new compounds, and selected reactions (PDF); primary NMR data (MNOVA). CCDC 1972811–1972820. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt04835a |
| This journal is © The Royal Society of Chemistry 2020 |