
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and rhodium complexes of macrocyclic PNP and PONOP pincer ligands†
Thomas M.
Hood
 ,
Matthew R.
Gyton
and
Adrian B.
Chaplin
*
,
Matthew R.
Gyton
and
Adrian B.
Chaplin
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: a.b.chaplin@warwick.ac.uk
First published on 8th January 2020
Abstract
The synthesis of macrocyclic variants of commonly employed phosphine-based pincer ligands derived from lutidine (PNP-14) and 2,6-dihydroxypyridine (PONOP-14) is described, where the P-donors are trans-substituted with a tetradecamethylene linker. This was accomplished using an eight-step procedure involving borane protection, ring-closing olefin metathesis, chromatographic separation from the cis-substituted diastereomers, and borane deprotection. The rhodium coordination chemistry of these ligands has been explored, aided by the facile synthesis of 2,2′-biphenyl (biph) adducts [Rh(PNP-14)(biph)][BArF4] and [Rh(PONOP-14)(biph)][BArF4] (ArF = 3,5-(CF3)2C6H3). Subsequent hydrogenolysis enabled generation of dihydrogen, ethylene and carbonyl derivatives; notably the ν(CO) bands of the carbonyl complexes provide a means to compare the donor properties of the new pincer ligands with established acyclic congeners.
Introduction
Phosphine-based pincers are an important ligand class in organometallic chemistry and catalysis, enabling a diverse variety of metal-based reactivity.1 Their ability to support reactive metal fragments is often exploited in the literature, with notable examples including a σ-methane complex,2 alkane dehydrogenation catalysts,3 and complexes capable of enacting the activation of C(sp3)–F bonds.4 Although mer-tridentate donor geometries are in principle highly tuneable and adaptable ligand scaffolds, the majority of phosphine-based pincers employed in the literature feature homoleptic aryl and alkyl phosphine donors, exemplified in the case of lutidine- and 2,6-dihydroxypyridine-derived variants by PNP-tBu and PONOP-tBu (Chart 1).5,6 Motivated by the potential to exploit additional reaction control though their unique steric profile, use in the construction of interlocked assemblies, and as an extension of our related work with NHC-based pincer ligands,7,8 we became interested in developing the chemistry of macrocyclic phosphine-based pincers. We herein describe the racemic synthesis of the first macrocyclic pincers PNP-14 and PONOP-14, where the chiral P-donors are trans-substituted with a tetradecamethylene linker, and some representative complexes with rhodium.9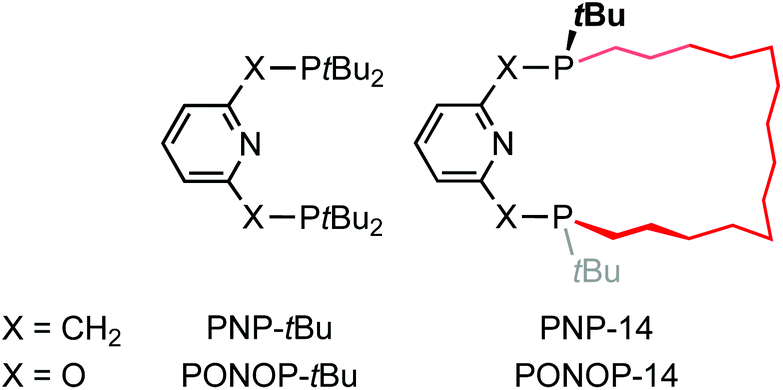 | ||
| Chart 1 | ||
Results and discussion
Preparation of borane protected ligands
PNP-14·2BH3 (trans-1a) and PONOP-14·2BH3 (trans-1b) were prepared from commercially available tert-butyldichlorophosphine using the seven-step synthesis outlined in Scheme 1. Amination of the starting material,10 enabled selective mono-alkylation (2, δ31P 73.3) and following treatment with HCl chloro-tert-butyl-octen-7-yl-phosphine 3 (δ31P 128.7) was obtained in 92% yield over three steps. Substitution of 3 by nucleophiles derived from the deprotonation of 2,6-dihydroxypyridine hydrochloride or 2,6-lutidine affords acyclic 4a (δ31P 33.7) and 4b (δ31P 144.7) as inseparable mixtures of diastereomers in 55% and 72% yield, respectively, after borane protection at −78 °C and purification by chromatography. Thereafter, olefin metathesis of 4a/b under dilute conditions (<4 mmol L−1) using Grubbs' 1st generation catalyst generated the corresponding macrocycles (cis-5a/b, δ31P 33.8/144.8; trans-5a/b, δ31P 34.0/143.4). The component diastereomers of 5a/b were separated using column chromatography and subsequently hydrogenated using Wilkinson's catalyst to produce the saturated derivatives (cis-1a/b, δ31P 33.3/145.1; trans-1a/b, δ31P 33.9/144.1). In this way trans-1a/b were obtained as analytically pure racemates, in practically useful overall yields of 14/22%, with their configurations confirmed by single crystal X-ray diffraction (Fig. 1).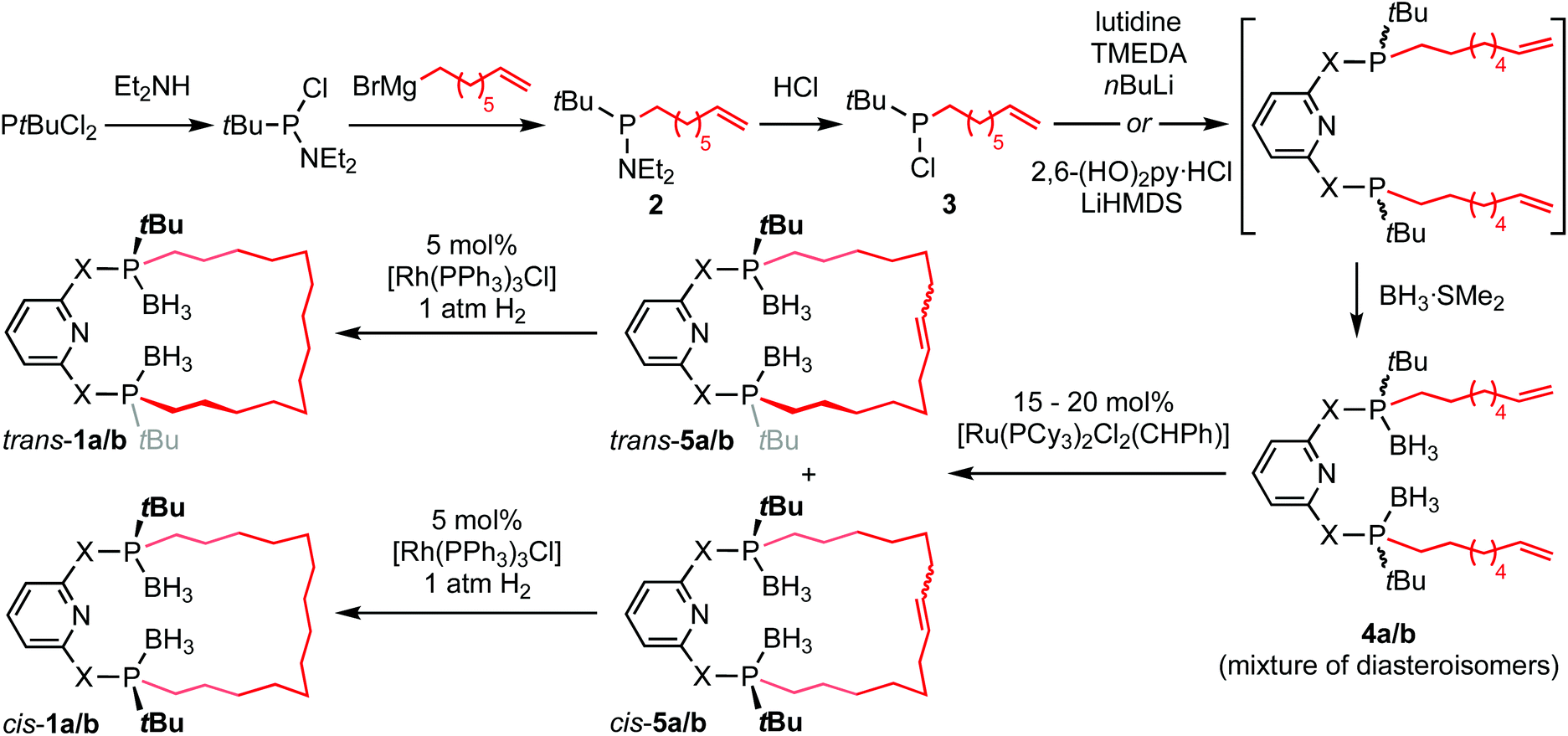 | ||
| Scheme 1 Preparation of PNP-14·2BH3 (trans-1a) and PONOP-14·2BH3 (trans-1b). | ||
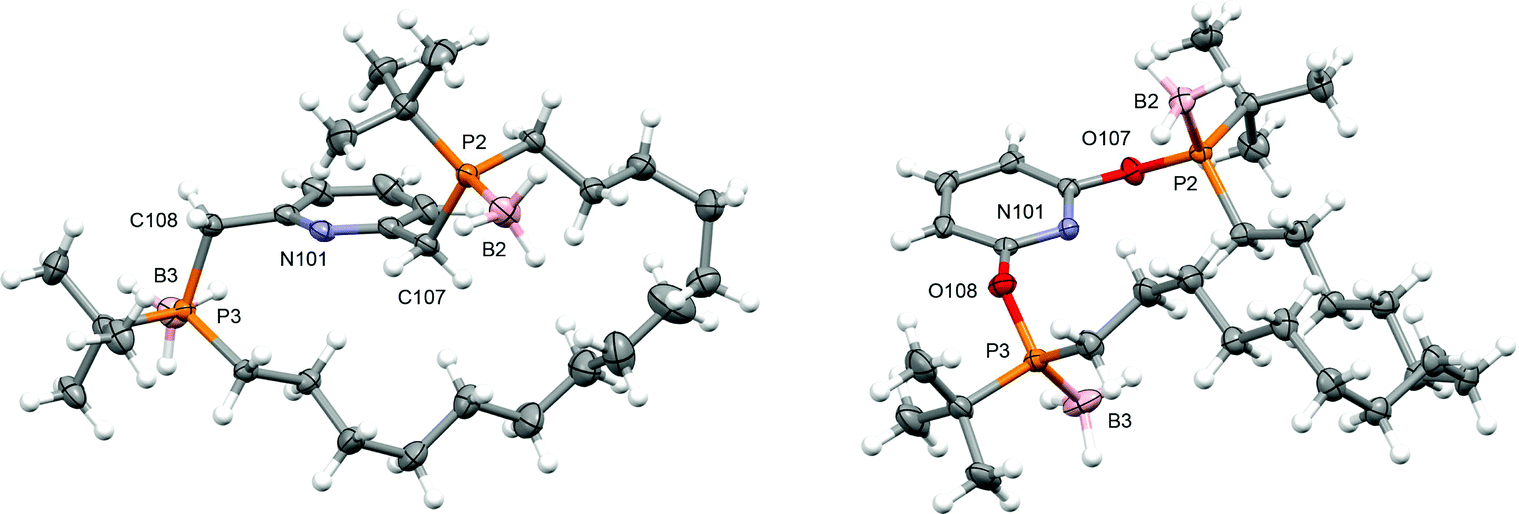 | ||
| Fig. 1 Solid-state structures of trans-1a (left) and trans-1b (right). Thermal ellipsoids drawn at 50% probability; hexane solvent (trans-1b) omitted for clarity. Selected bond lengths (Å): trans-1a, P2–B2, 1.918(2), P3–B3, 1.922(2); trans-5b, P2–B2, 1.903(3), P3–B3, 1.898(3). | ||
Deprotection
Deprotection of phosphine–boranes is commonly achieved by reactions with excess amine.11 Gratifyingly, treatment of trans-1a with neat Et2NH at 85 °C resulted in complete conversion to the free-base PNP-14 (δ31P 4.5) within 36 h, which was subsequently isolated in quantitative yield on removal of volatiles. Reactions between trans-1b and Et2NH under a range of conditions were, however, characterised by a significant degree of ligand decomposition that we ascribe to rupture of at least one of the P–O bonds.12 Evaluation of a range of other deprotection methods13 gave similar outcomes (see ESI†) and consequently we have so far been unable to obtain pure samples of the free-base. Nevertheless, conditions under which PONOP-14 (δ31P 146.5) can be generated in situ in 69–83% purity were identified: prolonged stirring of trans-1b (3.8 mmol L−1) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 THF:Et2NH at 19 °C.
:1 THF:Et2NH at 19 °C.
Rhodium complexes
As convenient {Rh(pincer)}+ synthons, the synthesis of five coordinate derivatives [Rh(pincer)(biph)][BArF4] (pincer = PNP-14, 6a; PONOP-14, 6b; biph = 2,2′-biphenyl; ArF = 3,5-(CF3)2C6H3) were targeted (Scheme 2). Exploiting a rhodium(III) precursor first described by Jones,14 and informed by previous work in our laboratories,7,15,166a/b were obtained as analytically pure materials in good isolated yield (79/69%) using a one-pot procedure involving substitution reactions of [Rh(biph)(dtbpm)Cl] (dtbpm = bis(di-tert-butylphosphino)methane) with isolated PNP-14 or in situ generated samples of PONOP-14 in the weakly coordinating solvent fluorobenzene17 and subsequent addition of Na[BArF4] as a halide abstracting agent. Complexes 6a and 6b are characterised in solution by pairs of 31P resonances centred at δ 43.1 (1JRhP = 110 Hz)/38.4 (1JRhP = 113 Hz) and δ 191.1 (1JRhP = 110 Hz)/182.9 (1JRhP = 121 Hz), which display diagnostic trans-phosphine 2JPP coupling of 339 and 372 Hz, respectively, and indicate adoption of C1 symmetry. Whilst the acyclic congeners [Rh(pincer)(biph)][BArF4] (pincer = PNP-tBu, 6a′; PONOP-tBu, 6b′) highlight the propensity for dynamic pseudorotation of the biph ligand on the NMR timescale,15 the tetradecamethylene linker appears to preclude such fluxionality in 6a/6b.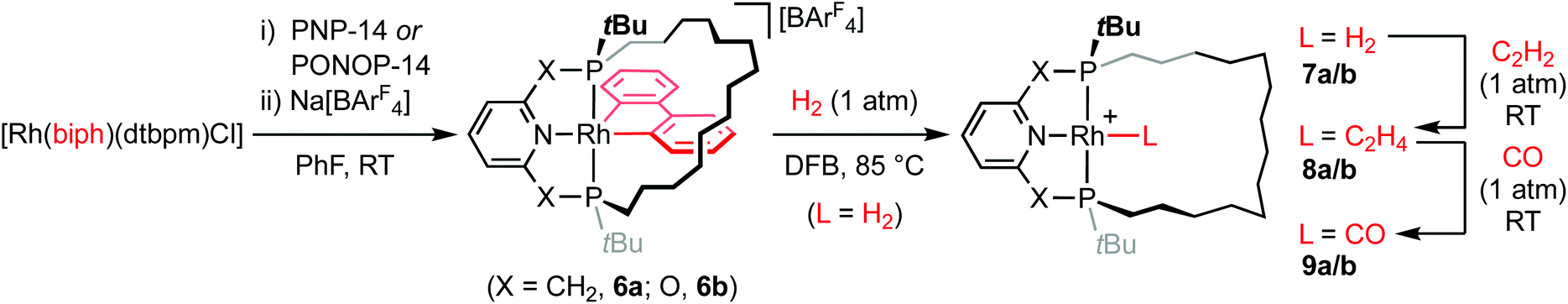 | ||
| Scheme 2 Preparation of rhodium complexes of PNP-14 and PONOP-14. | ||
The solid-state structures of 6a/6b demonstrate the adoption of distorted square pyramidal metal geometries, inferred from solution (Fig. 2). The methylene chains of the pincer ligands are skewed to one side of the basal plane, presumably to minimise steric buttressing with the biph ligand, and contorted to enable adoption of a weak γ-agostic interactions (![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif)
![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif)
![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) ⋯H–
⋯H–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif)
![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) = 3.184(2) Å 6a; 2.925(5) Å, 6b).18 Agostic interactions of comparable magnitude are observed in 6a′/6b′ and closely related rhodium 2,2′-biphenyl complexes of a NHC-based macrocyclic pincer ligand.7,15
= 3.184(2) Å 6a; 2.925(5) Å, 6b).18 Agostic interactions of comparable magnitude are observed in 6a′/6b′ and closely related rhodium 2,2′-biphenyl complexes of a NHC-based macrocyclic pincer ligand.7,15
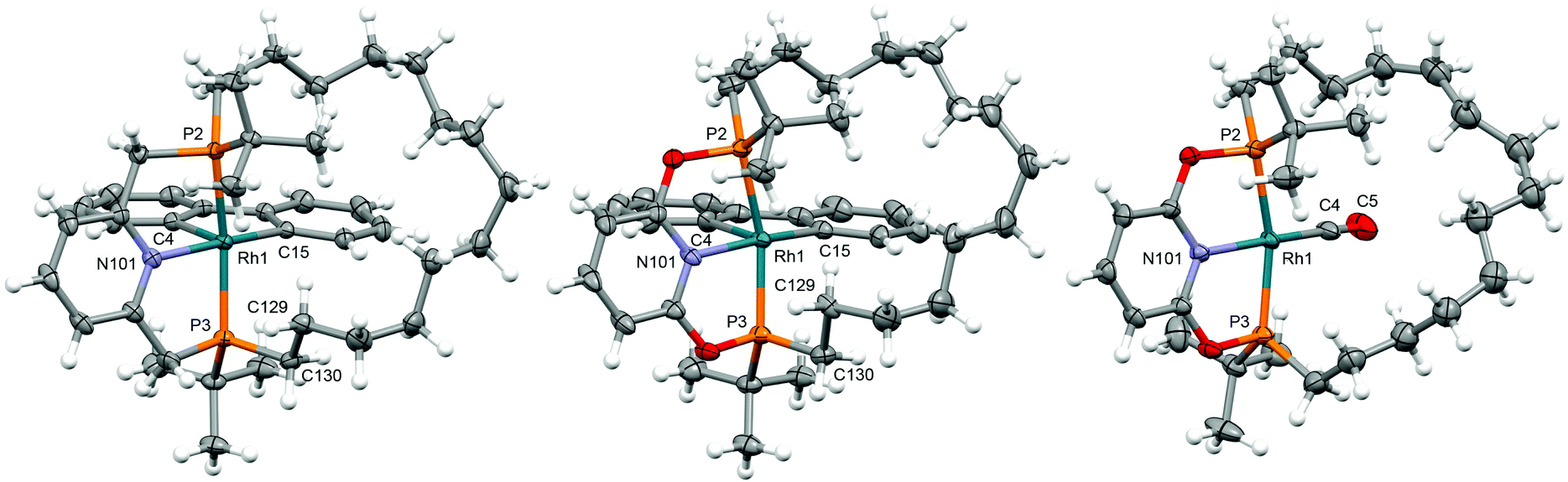 | ||
| Fig. 2 Solid-state structures of 6a (left), 6b (centre) and 9b (not unique, Z′ = 2; right). Thermal ellipsoids drawn at 50%, 30% and 30% probability, respectively; minor disordered component (9b, methylene chain) and anions omitted. Selected bond lengths (Å) and bond angles (°): 6a: Rh1–C4, 2.003(2); Rh1–C15, 2.028(2); Rh1–P2, 2.3340(4), Rh1–P3, 2.2801(4); Rh1–N101, 2.142(1); P2–Rh1–P3, 163.85(2); N101–M1–C15, 172.93(6); ⋯H–, 3.184(2); Rh1–P3–C130, 103.53(6); 6b: Rh1–C4, 2.065(5); Rh1–C15, 2.034(5); Rh1–P2, 2.330(1), Rh1–P3, 2.243(1); Rh1–N101, 2.091(4); P2–Rh1–P3, 159.89(5); N101–M1–C15, 171.2(2); ⋯H–, 2.925(5); Rh1–P3–C130, 103.0(2); 9b: Rh1–C4, 1.844(5); C4–O5, 1.141(7); Rh1–P2, 2.291(1); Rh1–P3, 2.256(1); Rh1–N101, 2.051(3); P2–Rh1–P3, 160.67(4); N101–Rh1–C4, 174.0(2); Rh11–C14, 1.846(6); C14–O15, 1.147(8); Rh11–P12, 2.288(2); Rh11–P13, 2.250(2); Rh11–N201, 2.034(4); P12–Rh11–P13, 161.16(7); N201–Rh11–C14, 172.0(3). | ||
Reaction of 6a/b with dihydrogen (1 atm) in 1,2-difluorobenzene (DFB)17 resulted in hydrogenolysis of the biph ligand and formation of 7a/b [δ31P 65.9 (1JRhP = 120 Hz)/δ31P 211.5 (1JRhP = 127 Hz)], but elevated temperature and prolonged reactions times were required for complete conversion (t = 2 days/5 days at 85 °C, Scheme 2). In both cases, no organometallic intermediates were observed during this reaction and biphenyl was the sole by-product. The spectroscopic characteristics are consistent with formulation of 7a/b as C2 symmetric rhodium(I) dihydrogen complexes, with broad 2H resonances at δ −10.76/−8.51 that exhibit short spin–lattice relaxation (T1 = 45 ± 11/48 ± 6 ms) at 298 K (600 MHz, Ar) the most diagnostic.19 Subsequent reaction in situ with ethylene (1 atm) confers the corresponding C2 symmetric π-complexes 8a/8b [δ31P 53.0 (1JRhP = 125 Hz)/δ31P 199.1 (1JRhP = 129 Hz)], with concomitant formation of ethane, in quantitative spectroscopic yield within 5 min at RT. Coordination of ethylene is substantiated by chemically inequivalent 2H signals at δ 3.70/3.52 and 3.95/3.70, and 13C resonances at δ 55.0 (1JRhC = 12 Hz) and 59.5 (1JRhC = 11 Hz), which display appreciable coupling to 103Rh, for 8a and 8b respectively. Finally, C2 symmetric carbonyl compounds 9a/b [δ31P 67.5 (1JRhP = 122 Hz)/δ31P 210.8 (1JRhP = 128 Hz)] are obtained by substitution of ethylene on reaction of 8a/b with carbon monoxide (1 atm <5 min at RT), isolated from solution in 96/72% yield overall from 6a/b and fully characterised, including in the case of 9b in the solid state by X-ray diffraction (Fig. 2). The ν(CO) bands of rhodium(I) carbonyl derivatives are diagnostic reporter groups for the donor properties of pincer ligands.20,21 Comparison of the carbonyl bands of 9a/b with those of acyclic congeners 9a/b′,15,22 recorded under the same conditions, suggests PNP-14 and PONOP-14 are marginally weaker net donors than PNP-tBu and PONOP-tBu, respectively (Table 1). By reference to IR data reported for [Rh(PNP-iPr)(CO)][BArF4] (9a′′; PNP-iPr = 2,6-(iPr2PCH2)2C5H3N) and trends established for monodentate phosphines, these minor differences are in line with changes in the phosphine/phosphinite substituents alone.20,23
Conclusions
An eight-step procedure for the synthesis of two macrocyclic phosphine-based pincer ligands, where the P-donors are trans-substituted with a tetradecamethylene linker, has been developed. These ligands are derived from lutidine (PNP-14) and 2,6-dihydroxypyridine (PONOP-14), with key steps involving borane protection, ring-closing olefin metathesis, chromatographic separation from the cis-substituted diastereomers, and borane deprotection. The final step was accomplished by borane transfer to diethylamine, but a non-trivial amount of decomposition could not be avoided in the case of the phosphinite pincer. The rhodium coordination chemistry of these ligands has been explored, with 2,2′-biphenyl (biph) complexes [Rh(PNP-14)(biph)][BArF4] and [Rh(PONOP-14)(biph)][BArF4] conveniently accessed by substitution reactions of [Rh(biph)(dtbpm)Cl] (dtbpm = bis(di-tert-butylphosphino)methane), followed by halide abstraction. These five-coordinate rhodium(III) complexes are well-defined synthons for the generation of rhodium(I) dihydrogen, ethylene and carbonyl derivatives, following hydrogenolysis of the biph ligand that serves as an ‘organometallic protecting group’. By comparison with the ν(CO) bands of rhodium(I) carbonyl adducts, determined by IR spectroscopy in CH2Cl2, PNP-14 and PONOP-14 can be considered to be marginally weaker net donors than their respective homoleptic tert-butyl substituted congeners PNP-tBu and PONOP-tBu, respectively.Experimental
General methods
All manipulations were performed under an atmosphere of argon using Schlenk and glove box techniques unless otherwise stated. Glassware was oven dried at 150 °C overnight and flame-dried under vacuum prior to use. Molecular sieves were activated by heating at 300 °C in vacuo overnight. Dihydrogen and ethylene were dried by passage through a stainless-steel column of activated 3 Å molecular sieves prior to use. Fluorobenzene and 1,2-difluorobenzene (DFB) were pre-dried over Al2O3, distilled from calcium hydride and dried twice over 3 Å molecular sieves.17 CD2Cl2 was freeze–pump–thaw degassed and dried over 3 Å molecular sieves. C6D6 was distilled from sodium and stored over 3 Å molecular sieves. THF, dioxane, diethyl ether and benzene were distilled from sodium/benzophenone and stored over 3 Å molecular sieves. Et2NH was distilled from CaH2. SiMe4 was distilled from liquid Na/K alloy and stored over a potassium mirror. Other anhydrous solvents were purchased from Acros Organics or Sigma-Aldrich, freeze–pump–thaw degassed and stored over 3 Å molecular sieves. LiHMDS was resublimed before use. nBuLi was titrated before use.24 TMEDA was distilled from sodium/benzophenone and stored over 3 Å molecular sieves. Diethylamino-tert-butyl-chlorophosphine (yield = 98%),10 BrMgC8H15,25 Wilkinson's catalyst,26 Na[BArF4],27 and [Rh(biph)(dtbpm)Cl],14 were synthesised according to published procedures. All other reagents are commercial products and were used as received. NMR spectra were recorded on Bruker spectrometers under argon at 298 K unless otherwise stated. Chemical shifts are quoted in ppm and coupling constants in Hz. NMR spectra in DFB and THF:Et2NH were recorded using an internal capillary of C6D6. ESI-MS were recorded on Bruker Maxis Plus (HR) or Agilent 6130B single Quad (LR) instruments. Infrared spectra were recorded on a Jasco FT-IR-4700 using a KBr transmission cell in CH2Cl2. Microanalyses were performed at the London Metropolitan University by Stephen Boyer.
Preparation of PNP-14·2BH3 (trans-1a) and PONOP-14·2BH3 (trans-1b)
1
H NMR (600 MHz, C6D6): δ 5.80 (ddt, 3JHH = 16.9, 3JHH = 10.2, 3JHH = 6.7, 1H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.02–5.08 (m, 1H, CHC2), 4.98–5.01 (m, 1H, CHC2), 2.90–2.97 (m, 4H, NCH2), 1.98–2.04 (m, 2H, C2CHCH2), 1.72–1.78 (m, 1H, CH2), 1.13–1.66 (m, 9H, CH2), 1.06 (d, 9H, 3JPH = 11.8, tBu), 1.00 (t, 6H, 3JHH = 7.1, NCH2C3).
CH2), 5.02–5.08 (m, 1H, CHC2), 4.98–5.01 (m, 1H, CHC2), 2.90–2.97 (m, 4H, NCH2), 1.98–2.04 (m, 2H, C2CHCH2), 1.72–1.78 (m, 1H, CH2), 1.13–1.66 (m, 9H, CH2), 1.06 (d, 9H, 3JPH = 11.8, tBu), 1.00 (t, 6H, 3JHH = 7.1, NCH2C3).
13
C{
1
H} NMR (151 MHz, C6D6): δ 139.2 (s, HCH2), 114.6 (s, CHH2), 44.5 (br, NCH2), 34.2 (s, H2CHCH2), 32.5 (d, 1JPC = 20, tBu{C}), 31.7 (d, JPC = 12, CH2), 29.5 (s, CH2), 29.4 (s, CH2), 27.6 (d, 2JPC = 16, tBu{CH3}), 26.6 (d, JPC = 18, CH2), 23.2 (d, 1JPC = 19, CH2), 15.2 (d, 4JPC = 2, NCH2H3).
31 P{ 1 H} NMR (243 MHz, C6D6): δ 73.3 (s).
1
H NMR (500 MHz, C6D6): δ 5.77 (ddt, 3JHH = 16.8, 3JHH = 10.0, 3JHH = 6.5, 1H, CCH2), 5.01–5.06 (m, 1H, CHC2), 4.97–5.01 (m, 1H, CHC2), 1.92–2.00 (m, 2H, C2CHCH2), 1.15–1.83 (m, 10H, CH2), 0.99 (d, 3JPH = 12.8, 9H, tBu).
13
C{
1
H} NMR (126 MHz, C6D6): δ 139.1 (s, HCH2), 114.6 (s, CHH2), 34.1 (s, H2CHCH2), 32.4 (d, 1JPC = 29, tBu{C}), 31.1 (d, JPC = 11, CH2), 30.7 (d, 1JPC = 36, CH2), 29.2 (s, 2 × CH2), 25.9 (d, JPC = 15, CH2), 25.5 (d, 2JPC = 17, tBu{CH3}).
31 P{ 1 H} NMR (162 MHz, C6D6): δ 128.7 (s).
1
H NMR (500 MHz, CDCl3): δ 7.55 (t, 3JHH = 7.7, 1H, py), 7.19–7.24 (m, 2H, py), 5.72–5.85 (m, 2H, CCH2), 4.95–5.01 (m, 2H, CHC2), 4.91–4.95 (m, 2H, CHC2), 3.09–3.20 (m, 4H, pyC2), 1.97–2.06 (m, 4H, C2CHCH2), 1.67–1.85 (m, 2H, CH2), 1.49–1.62 (m, 4H, CH2), 1.21–1.38 (m, 14H, CH2), 1.16 (d, 3JPH = 13.3, 7.3H, tBu), 1.12 (d, 3JPH = 13.4, 10.7H, tBu), −0.05–0.77 (m, 6H, BH3). Some peaks duplicated because of diastereomers.
13
C{
1
H} NMR (126 MHz, CDCl3): δ 154.4 (dd, 2JPC = 6, 4JPC = 1, py), 154.2 (dd, 2JPC = 5, 4JPC = 2, py), 138.97 (s, HCH2) 138.96 (s, HCH2), 136.8 (t, 4JPC = 2, py), 136.7 (t, 4JPC = 2, py), 123.3 (app t, JPC = 3, py), 123.2 (app t, JPC = 3, py), 114.5 (s, CHH2), 33.8 (s, H2CHCH2), 31.74 (d, 2JPC = 13, CH2), 31.70 (d, 2JPC = 13, CH2), 31.39 (d, 1JPC = 26, pyH2), 31.34 (d, 1JPC = 26, pyH2), 28.91 (d, 1JPC = 38, tBu{C}), 28.90 (d, 1JPC = 31, tBu{C}), 28.90 (s, CH2), 28.88 (s, CH2), 28.8 (br, CH2), 25.8 (t, 2JPC = 2, tBu{CH3}), 23.70 (s, CH2), 23.67 (s, CH2), 20.0 (d, 1JPC = 30, CH2). Some peaks duplicated because of diastereomers.
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 33.7 (vbr, fwhm = 150 Hz).
HR ESI-MS (positive ion 4 kV): 554.4366, [M + Na]+ (calcd 554.4368) m/z.
1
H NMR (500 MHz, CDCl3): δ 7.65 (t, 3JHH = 7.9, 1H, py), 6.81 (d, 3JHH = 7.9, 1.0H, py), 6.80 (d, 3JHH = 7.9, 1.0H, py), 5.80 (ddt, 3JHH = 16.9, 3JHH = 10.3, 3JHH = 6.7, 2H, CCH2), 4.96–5.02 (m, 2H, CHC2), 4.93 (d, 3JHH = 10.1, 2H, CHC2), 2.08–2.24 (m, 2H, CH2), 2.04 (app q, 3JHH = 7, 4H, C2CHCH2), 1.79–1.92 (m, 2H, CH2), 1.67–1.78 (m, 4H, CH2), 1.33–1.47 (m, 12H, CH2), 1.29 (d, 3JPH = 14.1, 9.0H, tBu), 1.29 (d, 3JHH = 14.2, 9.0H, tBu), 0.08–0.92 (m, 6H, BH3). Some peaks duplicated because of diastereomers.
13
C{
1
H} NMR (126 MHz, CDCl3): δ 158.1 (app t, JPC = 7, py), 142.09 (s, py), 142.05 (s, py), 139.07 (s, HCH2), 139.06 (s, HCH2), 114.5 (s, CHH2), 111.0 (d, 3JPC = 3, py), 110.8 (d, 3JPC = 3, py), 33.84 (s, H2CHCH2), 33.83 (s, H2CHCH2), 32.84 (d, 1JPC = 36, tBu{C}), 32.78 (d, 1JPC = 36, tBu{C}), 31.4 (s, CH2), 31.3 (s, CH2), 28.90 (s, CH2), 28.89 (s, CH2), 28.80 (s, CH2), 28.78 (s, CH2), 25.5 (d, 1JPC = 31, CH2), 25.4 (d, 1JPC = 31, CH2), 24.94 (d, 2JPC = 3, tBu{CH3}), 24.92 (d, 2JPC = 3, tBu{CH3}), 23.01, (s, CH2), 23.00 (s, CH2). Some peaks duplicated because of diastereomers.
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 144.7 (vbr, fwhm = 160 Hz).
HR ESI-MS (positive ion 4 kV): 558.3953, [M + Na]+ (calcd 558.3950) m/z.
cis-5a (RF = 0.22). Yield: 553 mg (18%).
1
H NMR (500 MHz, CDCl3): δ 7.55 (t, 3JHH = 7.7, 1H, py), 7.23 (d, 3JHH = 7.8, 2H, py), 5.27–5.41 (m, 2H, CHCH), 3.07–3.21 (m, 4H, pyC2), 1.94–2.09 (m, 4H, C2CHCH), 1.80–1.92 (m, 2H, CH2), 1.47–1.67 (m, 4H, CH2), 1.23–1.45 (m, 14H, CH2), 1.12 (d, 3JPH = 13.3, 18H, tBu), 0.02–0.82 (m, 6H, BH3).
13
C{
1
H} NMR (126 MHz, CDCl3): δ 154.5 (dd, 2JPC = 6, 4JPC = 2, py), 136.9 (t, 4JPC = 1, py), 131.1 (s, CHCH), 123.3 (app t, JPC = 3, py), 32.1 (s, H2CHCH), 31.2 (s, CH2), 31.1 (d, 1JPC = 12, pyH2), 28.9 (d, 1JPC = 31, tBu{C}), 28.7 (s, CH2), 27.5 (s, CH2), 25.8 (d, 2JPC = 2, tBu{CH3}), 23.5 (s, CH2), 19.3 (d, 1JPC = 30, CH2).
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 33.8 (vbr, fwhm = 150 Hz).
HR ESI-MS (positive ion 4 kV): 526.4051, [M + Na]+ (calcd 526.4079) m/z.
trans-5a (RF = 0.22). Yield: 840 mg (27%).
1
H NMR (500 MHz, CDCl3): δ 7.54 (t, 3JHH = 7.7, 1H, py), 7.17 (d, 3JHH = 7.8, 2H, py), 5.23–5.41 (m, 2H, CHCH), 3.07–3.20 (m, 4H, pyC2), 1.99–2.07 (m, 4H, C2CHCH), 1.78–1.92 (m, 2H, CH2), 1.54–1.71 (m, 4H, CH2), 1.28–1.51 (m, 14H, CH2), 1.16 (d, 3JPH = 13.2, 18H, tBu), −0.15–0.73 (m, 6H, BH3).
13
C{
1
H} NMR (126 MHz, CDCl3): δ 154.5 (dd, 2JPC = 5, 4JPC = 2, py), 136.7 (t, 4JPC = 2, py), 131.1 (s, CHCH), 123.2 (app t, JPC = 3, py), 31.9 (s, H2CHCH), 31.0 (d, 2JPC = 11, CH2), 30.8 (d, 1JPC = 26, pyH2), 29.0 (d, 1JPC = 31, tBu{C}), 28.6 (s, CH2), 27.2 (s, CH2), 25.9 (d, 2JPC = 2, tBu{CH3}), 23.5 (s, CH2), 19.8 (d, 1JPC = 30, CH2).
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 34.0 (vbr, fwhm = 150 Hz).
HR ESI-MS (positive ion 4 kV): 526.4054, [M + Na]+ (calcd 526.4079) m/z.
cis-5b (RF = 0.21). Yield: 520 mg (33%).
1
H NMR (500 MHz, CDCl3): δ 7.67 (t, 3JHH = 7.9, 1H, py), 6.95 (d, 3JHH = 8.0, 2H, py), 5.29–5.32 (m, 2H, CHCH), 2.12–2.24 (m, 2H, CH2), 1.95–2.08 (m, 4H, C2CHCH), 1.65–1.85 (m, 6H, CH2), 1.30–1.48 (m, 12H, CH2), 1.28 (d, 3JPH = 14.1, 18H, tBu), 0.15–0.92 (m, 6H, BH3). Data for major isomer only.
13
C{
1
H} NMR (126 MHz, CDCl3): δ 158.2 (d, 2JPC = 5, py), 142.3 (s, py), 131.0 (s, CHCH), 110.5 (d, 3JPC = 3, py), 32.8 (d, 1JPC = 37, tBu{C}), 32.1 (s, H2CHCH), 31.1 (d, 2JPC = 14, CH2), 28.7 (s, CH2), 27.7 (s, CH2), 25.5 (d, 1JPC = 31, CH2), 24.9 (d, 2JCH = 3, tBu{CH3}), 22.8 (s, CH2). Data for major isomer only.
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 144.8 (vbr, fwhm = 150 Hz).
HR ESI-MS (positive ion 4 kV): 530.3644, [M + Na]+ (calcd 530.3639) m/z.
trans-5b (RF = 0.22). Yield: 540 mg (34%).
1
H NMR (500 MHz, CDCl3): δ 7.63 (t, 3JHH = 7.8, 1H, py), 6.76 (d, 3JHH = 7.9, 2H, py), 5.29–5.33 (m, 2H, CHCH), 2.16–2.33 (m, 2H, CH2), 1.96–2.09 (m, 4H, C2CHCH), 1.83–1.92 (m, 2H, CH2), 1.32–1.46 (m, 4H, CH2), 1.32–1.46 (m, 12H, CH2), 1.28 (d, 3JPH = 14.0, 18H, tBu), 0.11–0.85 (m, 6H, BH3). Data for major isomer only.
13
C{
1
H} NMR (126 MHz, CDCl3): δ 158.2 (d, 2JPC = 6, py), 142.0 (s, py), 131.2 (s, CHCH), 110.1 (d, 3JPC = 3, py), 32.8 (d, 1JPC = 37, tBu{C}), 31.8 (s, H2CHCH), 31.2 (d, 2JPC = 14, CH2), 28.6 (s, CH2), 27.5 (s, CH2), 25.5 (d, 1JPC = 30, CH2), 24.9 (d, 2JPC = 3, tBu{CH3}), 23.4 (s, CH2). Data for major isomer only.
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 143.4 (vbr, fwhm = 180 Hz).
HR ESI-MS (positive ion 4 kV): 530.3634, [M + Na]+ (calcd 530.3639) m/z.
cis-1a (20% EtOAc in hexane, RF = 0.20).
Following the general procedure using cis-5a (80.0 mg, 0.159 mmol) and [Rh(PPh3)3Cl] (7.4 mg, 8.0 μmol) in benzene (5 mL), the product was isolated as a white solid. Yield: 73.8 mg (92%).
1
H NMR (600 MHz, CDCl3): δ 7.55 (t, 3JHH = 7.7, 1H, py), 7.32 (d, 3JHH = 7.8, 2H, py), 3.16 (app d, 2JPH = 12, 4H, pyC2), 1.71–1.82 (m, 2H, CH2), 1.47–1.60 (m, 4H, CH2), 1.21–1.39 (m, 22H, CH2), 1.12 (d, 3JPH = 13.3, 18H, tBu), 0.11–0.72 (br, 6H, BH3).
13
C{
1
H} NMR (151 MHz, CDCl3): δ 153.8 (dd, 2JPC = 4, 4JPC = 2, py), 136.6 (t, 4JPC = 2, py), 123.5 (app t, JPC = 3, py), 31.5 (d, 1JPC = 26, pyH2), 30.7 (d, 2JPC = 13, CH2), 28.9 (d, 1JPC = 31, tBu{C}), 28.0 (s, CH2), 27.87 (s, CH2), 27.85 (s, CH2), 27.8 (s, CH2), 25.7 (d, 2JPC = 2, tBu{CH3}), 22.7 (d, 3JPC = 2, CH2), 20.4 (d, 1JPC = 31, CH2).
31 P{ 1 H} NMR (243 MHz, CDCl3): δ 33.3 (vbr, fwhm = 130 Hz).
HR ESI-MS (positive ion 4 kV): 528.4204, [M + Na]+ (calcd 528.4211) m/z.
trans-1a (20% EtOAc in hexane, RF = 0.19).
Following the general procedure using trans-5a (840 mg, 1.67 mmol) and [Rh(PPh3)3Cl] (77.2 mg, 83.4 μmol) in benzene (50 mL), the product was isolated as a white solid. Yield: 818 mg (97%).
1
H NMR (500 MHz, CDCl3): δ 7.55 (t, 3JHH = 7.7, 1H, py), 7.21 (d, 3JHH = 7.8, 2H, py), 3.13–3.28 (m, 4H, pyC2), 1.75–1.86 (m, 2H, CH2), 1.52–1.68 (m, 4H, CH2) 1.38–1.50 (m, 4H, CH2), 1.26–1.35 (m, 18H, CH2), 1.10 (d, 3JPH = 13.3, 18H, tBu), 0.05–0.77 (m, 6H, BH3).
13
C{
1
H} NMR (126 MHz, CDCl3): δ 154.7 (dd, 2JPC = 6, 4JPC = 1, py), 136.8 (t, 4JPC = 2, py), 123.0 (app t, JPC = 3, py), 31.5 (d, 1JPC = 26, pyH2), 30.8 (d, 2JPC = 13, CH2), 29.1 (d, 1JPC = 31, tBu{C}), 27.91 (s, CH2), 27.89 (s, CH2), 27.74 (s, CH2), 27.71 (s, CH2), 25.9 (d, 2JPC = 2, tBu{CH3}), 22.9 (d, 3JPC = 1, CH2), 20.1 (d, 1JPC = 31, CH2).
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 33.9 (vbr, fwhm = 150 Hz).
HR ESI-MS (positive ion 4 kV): 528.4209, [M + Na]+ (calcd 528.4211) m/z.
Anal. Calcd for C29H59B2NP2 (505.37 g mol−1): C, 68.92; H, 11.77; N, 2.77; Found: C, 68.76; H 11.82; N, 2.69.
cis-1b (30% CH2Cl2 in hexane, RF = 0.19).
Following the general procedure using cis-5b (315 mg, 0.620 mmol) and [Rh(PPh3)3Cl] (27.2 mg, 29.4 μmol) in benzene (30 mL), the product was isolated as a white solid. Yield: 287 mg (91%).
1 H NMR (500 MHz, CDCl3): δ 7.67 (t, 3JHH = 7.9, 1H, py), 6.98 (d, 3JHH = 7.9, 2H, py), 2.12–2.25 (m, 2H, CH2), 1.63–1.82 (m, 6H, CH2), 1.27 (d, 3JPH = 14, 18H, tBu), 1.25–1.49 (m, 20H, CH2), 0.14–0.88 (m, 6H, BH3).
13 C{ 1 H} NMR (126 MHz, CDCl3): δ 158.2 (d, 2JPC = 5, py), 142.2 (s, py), 110.8 (d, 3JPC = 3, py), 32.9 (d, 1JPC = 36, tBu{C}), 30.6 (d, 2JPC = 13, CH2), 27.8 (s, 2 × CH2), 27.5 (s, CH2), 27.3 (s, CH2), 25.2 (d, 1JPC = 32, CH2), 24.9 (d, 2JPC = 3, tBu{CH3}), 22.1 (s, CH2).
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 145.1 (vbr, fwhm = 142 Hz).
HR ESI-MS (positive ion 4 kV): 532.3791, [M + Na]+ (calcd 532.3796) m/z.
trans-1b (30% CH2Cl2 in hexane, RF = 0.20).
Following the general procedure using trans-5b (620 mg, 1.22 mmol) and [Rh(PPh3)3Cl] (56.5 mg, 61.1 μmol) in benzene (50 mL), the product was isolated as a white. Yield: 623 mg (95%).
1 H NMR (500 MHz, CDCl3): δ 7.64 (t, 3JHH = 7.8, 1H, py), 6.81 (d, 3JHH = 7.9, 2H, py), 2.13–2.29 (m, 2H, CH2), 1.85–1.96 (m, 2H, CH2), 1.69–1.83 (m, 4H, CH2), 1.38–1.47 (m, 4H, CH2), 1.28 (d, 3JPH = 13.9, 18H, tBu), 1.23–1.37 (m, 12H, CH2), 0.11–0.99 (m, 6H, BH3).
13 C{ 1 H} NMR (126 MHz, CDCl3): δ 158.2 (d, 2JPC = 6, py), 142.1 (s, py), 110.2 (d, 3JPC = 3, py), 32.9 (d, 1JPC = 36, tBu{C}), 30.8 (d, 2JPC = 13, CH2), 27.7 (s, CH2), 27.52 (s, CH2), 27.47 (s, CH2), 26.9 (s, CH2), 25.5 (d, 1JPC = 31, CH2), 25.0 (d, 2JPC = 3, tBu{CH3}), 22.7 (s, CH2).
31 P{ 1 H} NMR (162 MHz, CDCl3): δ 144.1 (vbr, fwhm = 155 Hz).
HR ESI-MS (positive ion 4 kV): 532.3804, [M + Na]+ (calcd 532.3795) m/z.
Anal. Calcd for C27H55B2NO2P2 (509.31 g mol−1): C, 63.67; H, 10.89; N, 2.75; Found: C, 63.66; H, 11.03; N, 2.74.
Preparation of PNP-14
A solution of trans-1a in Et2NH (0.5 mL) was heated at 85 °C for 2 days within a J Young's valve NMR tube. Quantitative conversion was observed by 1H and 31P NMR spectroscopy. The volatiles were removed in vacuo to afford the product as a colourless oil, which was carried forward without further purification.
1
H NMR (500 MHz, C6D6): δ 7.08 (t, 3JHH = 7.7, 1H, py), 6.90 (d, 3JHH = 7.7, 2H, py), 3.04 (d, 2JHH = 13.0, 2H, pyC2), 2.87 (dd, 2JHH = 13.0, 2JPH = 2.9, 2H, pyC2), 1.50–1.57 (m, 2H, CH2), 1.38–1.49 (m, 8H, CH2), 1.27–1.38 (m, 18H, CH2), 1.03 (d, 3JPH = 11, 18H, tBu).
13
C{
1
H} NMR (126 MHz, C6D6): δ 160.4 (d, 2JPC = 8, py), 136.0 (s, py), 120.5 (dd, 3JPC = 6, 5JPC = 2, py), 35.5 (d, 1JPC = 24, pyH2), 30.8 (d, 1JPC = 12, tBu{C}), 28.5 (s, CH2), 28.2 (s, CH2), 28.1 (s, CH2), 28.0 (s, CH2), 27.6 (d, 2JPC = 14, tBu{CH3}), 27.3 (s, CH2), 27.1 (s, CH2), 24.4 (d, 1JPC = 20, CH2).
31 P{ 1 H} NMR (121 MHz, C6D6): δ 4.5 (s).
LR ESI-MS (positive ion, 4 kV): 532.5, [M]+ (calcd 532.3) m/z.
Preparation of PONOP-14
A solution of trans-1b (11.7 mg, 23.0 μmol) in THF (3 mL) was treated with an equal volume of Et2NH (3 mL) and the resulting solution stirred at 19 °C for 8 days. The volatiles were removed in vacuo to afford the product as a yellow oil in 65–84% purity, as determined by 31P NMR spectroscopy, which was carried forward without further purification.
31
P{
1
H} NMR (162 MHz, THF:HNEt2, selected data): δ 146.5 (s).
Preparation of [Rh(PNP-14)(biph)][BArF4] (6a)
A suspension of PNP-14 (16.1 mg, 33.7 μmol) and [Rh(biph)(dtbpm)Cl] (20.0 mg, 33.6 μmol) in PhF (0.50 mL) was stirred at ambient temperature for 16 h. Na[BArF4] (29.8 mg, 33.6 μmol) was added and the suspension stirred for a further 4 h before the volatiles were removed in vacuo. The resulting orange oil was washed with pentane (2 × 1 mL), dried in vacuo and extracted into CH2Cl2 (2 mL). The product was obtained as an orange crystalline solid by slow cooling of CH2Cl2:hexane (1:20) solution to −30 °C. Yield: 42.6 mg (79%).
1
H NMR (500 MHz, CD2Cl2): δ 7.95 (t, 3JHH = 7.9, 1H, py), 7.70–7.76 (m, 8H, ArF), 7.59–7.68 (m, 4H, 2 × py + 2 × biph), 7.56 (br, 4H, ArF), 7.48 (d, 3JHH = 7.6, 1H, biph), 7.10–7.26 (m, 2H, biph), 6.98 (t, 3JHH = 7.3, 1H, biph), 6.50 (t, 3JHH = 7.6, 1H, biph), 5.63 (d, 3JHH = 8.2, 1H, biph), 3.85–4.04 (m, 2H, pyC2), 3.51–3.76 (m, 2H, pyC2), 2.66–2.78 (m, 1H, CH2), 2.05–2.24 (m, 1H, CH2), 1.74–1.83 (m, 1H, CH2), 1.40–1.70 (m, 10H, CH2), 1.18–1.39 (m, 7H, CH2), 1.16 (d, 3JPH = 13.3, 9H, tBu), 0.96–1.09 (m, 4H, CH2), 0.66–0.87 (m, 3H, CH2), 0.51 (d, 3JPH = 15, 9H, tBu), 0.19–0.35 (m, 1H, CH2).
13
C{
1
H} NMR (126 MHz, CD2Cl2): δ 162.5 (app t, JPC = 5, py), 162.3 (q, 1JCB = 50, ArF), 162.1 (app t, JPC = 3, py), 161.7 (obscured, biph), 152.3 (d app t, 1JRhC = 44, 2JPC = 7, biph), 151.2 (s, biph), 148.9 (s, biph), 140.5 (s, py), 135.4 (s, ArF), 133.9 (s, biph), 129.6 (s, biph), 129.4 (qq, 2JFC = 32, 3JCB = 3, ArF), 128.5 (s, biph), 126.7 (s, biph), 125.2 (q, 1JFC = 272, ArF), 125.1 (s, biph), 124.1 (s, biph), 123.5 (d, 3JPC = 8, py), 123.4 (d, 3JPC = 10, py), 122.4 (s, biph), 121.9 (s, biph), 118.0 (sept, 3JFC = 4, ArF), 40.1 (d, 1JPC = 23, pyH2), 38.7 (d, 1JPC = 19, pyH2), 34.4 (dd, 1JPC = 16, 3JPC = 5, tBu{C}), 33.0 (ddd, 1JPC = 20, 3JPC = 5, 2JRhC = 2, tBu{C}), 32.0 (d, 2JPC = 14, CH2), 30.3 (s, CH2), 29.7 (s, CH2), 29.54 (s, CH2), 29.51 (s, CH2), 29.43 (d, 2JPC = 4, tBu{CH3}), 29.37 (s, CH2), 29.3 (s, CH2), 28.0 (s, CH2), 27.3 (s, CH2), 26.2 (d, 1JPC = 21, PCH2), 25.7 (s, tBu{CH3}), 25.6 (obscured, CH2), 24.9 (s, CH2), 24.6 (s, CH2), 21.0 (d app t, 1JPC = 16, J = 2, PCH2).
31 P{ 1 H} NMR (162 MHz, CD2Cl2): δ 43.1 (dd, 2JPP = 339, 1JRhP = 110, 1P), 38.4 (dd, 2JPP = 339, 1JRhP = 113, 1P).
HR ESI-MS (positive ion, 4 kV): 732.3329, [M]+ (calcd 732.3329) m/z.
Anal. Calcd for C73H73BF24NP2Rh (1596.02 g mol−1): C, 54.94; H, 4.61; N, 0.88; Found: C, 54.89; H, 4.80; N, 0.86.
Preparation of [Rh(PONOP-14)(biph)][BArF4] (6b)
A suspension of PONOP-14 (17.8 μmol, generated in situ as described above) and [Rh(biph)(dtbpm)Cl] (10.6 mg, 17.8 μmol) in PhF (0.5 mL) was stirred at ambient temperature for 16 h. Na[BArF4] (15.8 mg, 17.8 μmol) was added and the suspension stirred for a further 4 h before the volatiles were removed in vacuo. The resulting orange oil was washed with pentane (2 × 1 mL), dried in vacuo and extracted into CH2Cl2 (2 mL). The product was recrystallised by slow diffusion of hexane into a CH2Cl2 solution (1:20). Yield: 19.6 mg (69%).
1 H NMR (500 MHz, CD2Cl2): δ 8.11 (t, 3JHH = 8.2, 1H, py), 7.70–7.76 (m, 8H, ArF), 7.65 (d, 3JHH = 7.3, 1H, biph), 7.56 (br, 4H, ArF), 7.54 (obscured, 1H, biph), 7.47 (d, 3JHH = 7.2, 1H, biph), 7.16–7.21 (m, 2H, biph), 7.15 (d, 3JHH = 8.2, 1H, py), 7.10 (d, 3JHH = 8.2, 1H, py), 7.06 (t, 3JHH = 7.4, 1H, biph), 6.56 (t, 3JHH = 7.7, 1H, biph), 5.32 (d, 3JHH = 8.8, 1H, biph), 2.64–2.86 (m, 2H, CH2), 1.85–2.08 (m, 3H, CH2), 1.60–1.78 (m, 4H, CH2), 1.29 (d, 3JPH = 14.6, 9H, tBu), 1.00–1.58 (m, 13H, CH2), 0.84–0.96 (m, 1H, CH2), 0.65–0.83 (m, 3H, CH2), 0.62 (d, 3JPH = 17.4, 9H, tBu), 0.37–0.48 (m, 2H, CH2).
13 C{ 1 H} NMR (126 MHz, CD2Cl2): δ 162.7 (dd, 2JPC = 6, 4JPC = 2, py), 162.3 (q, 1JCB = 50, ArF), 161.5 (dd, 2JPC = 6, 4JPC = 2, py), 159.1 (ddd, 1JRhC = 32, 2JPC = 11, 2JPC = 5, biph), 151.9 (ddd, 1JRhC = 43, 2JPC = 9, 2JPC = 7, biph), 151.2 (s, biph), 149.1 (br, biph), 147.2 (s, py), 135.4 (s, ArF), 133.9 (s, biph), 129.4 (qq, 2JFC = 32, 3JCB = 3, ArF), 129.35 (s, biph), 128.3 (s, biph), 127.6 (biph), 126.2 (s, biph), 125.2 (q, 1JFC = 272, ArF), 125.0 (s, biph), 123.3 (s, biph), 122.5 (s, biph), 118.0 (sept, 3JFC = 4, ArF), 106.1 (d, 3JPC = 4, py), 105.7 (d, 3JPC = 5, py), 41.6 (dd, 1JPC = 9, 2JRhC = 7, tBu{C}), 38.1 (ddd, 1JPC = 17.8, 3JPC = 7, 2JPC = 3, tBu{C}), 35.8 (d, JPC = 11, CH2), 31.3 (s, CH2), 30.9 (dd, 1JPC = 15, 3JPC = 3, PCH2), 30.7 (s, CH2), 30.5 (s, CH2), 30.3 (s, CH2), 30.0 (s, CH2), 29.2 (s, CH2), 28.6 (s, CH2), 28.2 (s, CH2), 28.0 (d, JPC = 7, CH2), 27.5 (d, 2JPC = 5, tBu{CH3}), 25.0 (d app t, 1JPC = 14, J = 3, PCH2), 24.4 (d, 2JPC = 4, tBu{CH3}), 24.2 (d, JPC = 4, CH2), 23.7 (s, CH2).
31 P{ 1 H} NMR (162 MHz, CD2Cl2): δ 191.1 (dd, 2JPP = 373, 1JRhP = 110, 1P), 182.9 (dd, 2JPP = 373, 1JRhP = 121, 1P).
HR ESI-MS (positive ion, 4 kV): 736.2909, [M]+ (calcd 736.2914) m/z.
Anal. Calcd for C71H69BF24NO2P2Rh (1599.96 g mol−1): C, 53.30; H, 4.35; N, 0.88; Found: C, 53.12; H, 4.48; N, 0.86.
General procedure for in situ synthesis of dihydrogen complexes 7
A solution of 6 in DFB (0.5 mL) was freeze–pump–thaw degassed and placed under dihydrogen (1 atm) within a J Young's valve NMR tube and heated at 85 °C to afford the corresponding dihydrogen complex, which was characterised in situ under dihydrogen, and biphenyl.
1
H NMR (500 MHz, DFB, H2): δ 8.09–8.15 (m, 8H, ArF), 7.54 (t, 3JHH = 7.8, 1H, py), 7.49 (br, 4H, ArF), 7.22 (obscured 2H, py), 3.46 (dvt, 2JHH = 17.7, JPH = 4, 2H, pyC2), 3.23 (dvt, 2JHH = 17.7, JPH = 4, 2H, pyC2), 1.51–1.71 (m, 10H, CH2), 1.14–1.41 (m, 18H, CH2), 0.94 (vt, JPH = 8, 18H, tBu), −10.43 (vbr, fwhm ∼800 Hz, 2H, RhH).
13
C{
1
H} NMR (126 MHz, DFB, H2): δ 164.1 (vt, JPC = 5, py), 162.3 (q, 1JCB = 50, ArF), 140.1 (s, py), 135.4 (s, ArF), 129.6 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.2 (q, 1JFC = 272, ArF), 121.1 (vt, JPC = 5, py), 117.6 (sept, 3JFC = 4, ArF), 37.9 (vt, JPC = 9, pyH2), 32.0 (vt, JPC = 12, tBu{C}), 28.7 (vt, JPC = 4, CH2), 28.5 (s, CH2), 28.4 (s, CH2), 28.0 (s, CH2), 27.0 (s, CH2), 26.4 (vt, JPC = 3, tBu{CH3}), 24.7 (vt, JPC = 3, CH2), 20.8 (vtd, JPC = 12, 2JRhC = 2, PCH2).
31 P{ 1 H} NMR (162 MHz, DFB, H2): δ 65.9 (d, 1JRhP = 120).
1 H NMR (600 MHz, DFB, selected data under argon): δ −10.76 (vbr, fwhm = 60 Hz, T1 = 45 ± 11 ms, 2H, RhH).
1 H NMR (500 MHz, DFB, H2): δ 8.09–8.15 (m, 8H, ArF), 7.63 (t, 3JHH = 8.2, 1H, py), 7.49 (br, 4H, ArF), 6.63 (obscured 2H, py), 2.03–2.18 (m, 4H, CH2), 1.53–1.78 (m, 6H, CH2), 1.15–1.41 (m, 18H, CH2), 1.11 (vt, JPH = 8, 18H, tBu), −8.65 (vbr, fwhm = 100 Hz, 2H, RhH).
13 C{ 1 H} NMR (126 MHz, DFB, H2): δ 163.5 (br, py), 162.3 (q, 1JCB = 50, ArF), 145.9 (s, py), 135.4 (s, ArF), 129.6 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.2 (q, 1JFC = 272, ArF), 117.6 (sept, 3JFC = 4, ArF), 103.3 (vt, JPC = 3, py), 37.6 (vt, JPC = 12, tBu{C}), 29.0 (br, CH2), 28.5 (s, CH2), 28.1 (s, CH2), 28.0 (s, CH2), 27.5 (vt, JPC = 9, PCH2), 27.3 (s, CH2), 24.7 (vt, JPC = 4, tBu{CH3}), 23.9 (vt, JPC = 3, CH2).
31 P{ 1 H} NMR (162 MHz, DFB, H2): δ 211.5 (d, 1JRhP = 127).
1 H NMR (600 MHz, DFB, selected data under argon): δ −8.51 (vbr d, fwhm = 60 Hz, 1JRhH = 21, T1 = 48 ± 6 ms, 2H, RhH).
General procedure for in situ synthesis of ethylene complexes 8
A solution of 7 in DFB (0.5 mL) was freeze–pump–thaw degassed and placed under ethylene (1 atm) within a J Young's valve NMR tube to afford the corresponding ethylene complex, which was characterised in situ under ethylene. All spectra contained ethane (δ1H 0.70).
1
H NMR (500 MHz, DFB, C2H4): δ 8.09–8.15 (m, 8H, ArF), 7.51 (t, 3JHH = 8.0, 1H, py), 7.49 (br, 4H, ArF), 7.15 (obscured, 2H, py), 3.70 (br, 2H, C2H4), 3.52 (br, 2H, C2H4), 3.31 (dvt, 2JHH = 17.3, JPH = 4, 2H, pyC2), 3.22 (dvt, 2JHH = 17.4, JPH = 4, 2H, pyC2), 1.72–1.93 (m, 4H, CH2), 1.55–1.67 (m, 2H, CH2), 1.39–1.50 (m, 2H, CH2), 1.07–1.37 (m, 20H, CH2), 0.83 (vt, JPH = 7, 18H, tBu).
13
C{
1
H} NMR (126 MHz, DFB, C2H4): δ 162.9 (vt, JPH = 5, py), 162.3 (q, 1JCB = 50, ArF), 140.1 (s, py), 135.1 (s, ArF), 129.6 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.2 (q, 1JFC = 272, ArF), 120.7 (vt, JPC = 5, py), 117.6 (sept, 3JFC = 4, ArF), 55.0 (d, 1JRhC = 12, C2H4), 37.5 (vt, JPC = 8, pyH2), 32.8 (vt, JPC = 10, tBu{C}), 29.5 (vt, JPC = 4, CH2), 29.3 (s, CH2), 28.7 (s, CH2), 28.2 (s, CH2), 27.9 (s, CH2), 26.6 (vt, JPC = 3, tBu{CH3}), 24.1 (s, CH2), 21.9 (vt, JPC = 10, PCH2).
31 P{ 1 H} NMR (162 MHz, DFB, C2H4): δ 53.0 (d, 1JRhP = 125).
1 H NMR (500 MHz, DFB, C2H4): δ 8.09–8.15 (m, 8H, ArF), 7.61 (t, 3JHH = 8.1, 1H, py), 7.49 (br, 4H, ArF), 6.59 (obscured 2H, py), 3.95 (br, 2H, C2H4), 3.70 (br, 2H, C2H4), 2.19–2.29 (m, 2H, CH2), 2.05–2.16 (m, 2H, CH2), 1.74–1.86 (m, 2H, CH2), 1.46–1.60 (m, 4H, CH2), 1.02–1.45 (m, 18H, CH2), 0.96 (vt, JPH = 8, 18H, tBu).
13 C{ 1 H} NMR (126 MHz, DFB, C2H4): δ 162.8 (vt, JPC = 3, py), 162.3 (q, 1JCB = 50, ArF), 145.3 (s, py), 135.1 (s, ArF), 129.6 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.2 (q, 1JFC = 272, ArF), 117.6 (sept, 3JFC = 4, ArF), 103.2 (vt, JPC = 3, py), 59.5 (d, 1JRhC = 11, C2H4), 39.1 (vt, JPC = 10, tBu{C}), 30.0 (vt, JPC = 2, CH2), 28.7 (s, CH2), 28.6 (s, CH2), 28.5 (s, CH2), 28.4 (s, CH2), 27.3 (vtd, JPC = 8, 2JRhC = 2, PCH2), 24.9 (vt, JPC = 3, tBu{CH3}), 23.7 (s, CH2).
31 P{ 1 H} NMR (162 MHz, DFB, C2H4): δ 199.1 (d, 1JRhP = 129).
General procedure for the preparation of carbonyl complexes 9
A solution of 8 in DFB (0.5 mL) was freeze–pump–thaw degassed and placed under carbon monoxide (1 atm) within a J Young's valve NMR tube, resulting in an immediate colour change. The volatiles were removed in vacuo, and the resulting yellow solid washed and dried in vacuo.
1
H NMR (500 MHz, CD2Cl2): δ 7.79 (t, 3JHH = 7.8, 1H, py), 7.70–7.76 (m, 8H, ArF), 7.56 (br, 4H, ArF), 7.42 (d, 3JHH = 7.9, 2H, py), 3.70 (dvt, 2JHH = 17.5, JPH = 4, 2H, pyC2), 3.56 (dvt, 2JHH = 17.5, JPH = 4, 2H, pyC2), 2.02–2.09 (m, 4H, CH2), 1.78–1.98 (m, 4H, CH2), 1.63–1.75 (m, 2H, CH2), 1.49–1.63 (m, 2H, CH2), 1.21–1.49 (m, 16H, CH2), 1.13 (vt, JPH = 8, 18H, tBu).
13
C{
1
H} NMR (126 MHz, CD2Cl2): δ 194.7 (dt, 1JRhC = 70, 2JPC = 13, CO), 163.8 (vtd, JPC = 5, 2JRhC = 1, py), 162.3 (q, 1JCB = 50, ArF), 141.6 (s, py), 135.4 (s, ArF), 129.4 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.2 (q, 1JFC = 272, ArF), 122.1 (vt, JPC = 5, py), 118.0 (sept, 3JFC = 4, ArF), 38.7 (vt, JPC = 9, pyH2), 33.9 (vt, JPC = 12, tBu{C}), 30.3 (vt, JPC = 4, CH2), 29.3 (s, CH2), 28.94 (s, CH2), 28.88 (s, CH2) 28.4 (s, CH2), 27.8 (vt, JPC = 3, tBu{CH3}), 26.2 (s, CH2), 23.2 (vtd, JPC = 12, 2JRhC = 3, PCH2).
31 P{ 1 H} NMR (162 MHz, CD2Cl2): δ 67.5 (d, 1JRhP = 122).
IR (CH2Cl2): ν(CO) 1997 cm−1.
HR ESI-MS (positive ion, 4 kV): 608.2653, [M]+ (calcd 608.2652) m/z.
Anal. Calcd for C62H65BF24NOP2Rh (1471.83 g mol−1): C, 50.60; H, 4.45; N, 0.95 Found: C, 50.53; H, 4.47; N, 1.08.
1 H NMR (500 MHz, CD2Cl2): δ 7.91 (t, 3JHH = 8.2, 1H, py), 7.70–7.76 (m, 8H, ArF), 7.56 (br, 4H, ArF), 6.86 (d, 3JHH = 8.2, 2H, py), 2.40–2.60 (m, 4H, CH2), 1.76–1.98 (m, 6H, CH2), 1.52–1.65 (m, 3H, CH2), 1.11–1.48 (m, 15H, CH2), 1.29 (vt, JPH = 8, 18H, tBu).
13 C{ 1 H} NMR (126 MHz, CD2Cl2): δ 193.2 (dt, 1JRhC = 71, 2JPC = 13, CO), 163.1 (vt, JPC = 3, py), 162.3 (q, 1JCB = 50, ArF), 147.7 (s, py), 135.4 (s, ArF), 129.5 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.2 (q, 1JFC = 272, ArF), 118.0 (sept, 3JFC = 4, ArF), 104.5 (vt, JPC = 3, py), 39.9 (vtd, JPC = 11, 2JRhC = 2, tBu{C}), 30.9 (vt, JPC = 2, CH2), 29.5 (vtd, JPC = 9, 2JRhC = 3, PCH2), 29.3 (s, CH2), 29.1 (s, 2 × CH2), 28.8 (s, CH2), 26.1 (vt, JPC = 4, tBu{CH3}), 25.1 (vt, JPC = 2, CH2).
31 P{ 1 H} NMR (162 MHz, CD2Cl2): δ 210.8 (d, 1JRhP = 128).
IR (CH2Cl2): ν(CO) 2020 cm−1.
HR ESI-MS (positive ion, 4 kV): 612.2228, [M]+ (calcd 612.2237) m/z.
Anal. Calcd for C60H61BF24NO3P2Rh (1475.78 g mol−1): C, 48.83; H, 4.17; N, 0.95 Found: C, 48.91; H, 4.26; N, 1.02.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council (ERC, grant agreement 637313) and Royal Society (UF100592, UF150675, A. B. C.) for financial support. High-resolution mass-spectrometry data were collected using instruments purchased through support from Advantage West Midlands and the European Regional Development Fund. Crystallographic data were collected using an instrument that received funding from the ERC under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 637313).References
- (a) Pincer Compounds: Chemistry and Applications, ed. D. Morales-Morales, Elsevier, 2018, vol. 1 Search PubMed; (b) E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC; (c) R. E. Andrew, L. González-Sebastián and A. B. Chaplin, Dalton Trans., 2016, 45, 1299–1305 RSC; (d) The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications, ed. G. van Koten and R. A. Gossage, Topics in Organometallic Chemistry, Springer, 2016, vol. 45 Search PubMed; (e) Pincer and Pincer-Type Complexes: Applications in Organic Synthesis and Catalysis, ed. K. J. Szabó and O. F. Wendt, Wiley-VCH, 2014 Search PubMed; (f) Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein; Topics in Organometallic Chemistry, Springer, 2013, vol. 40 Search PubMed; (g) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed; (h) M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS.
- W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553–556 CrossRef CAS PubMed.
- A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS PubMed.
- J. Choi, D. Y. Wang, S. Kundu, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Science, 2011, 332, 1545–1548 CrossRef CAS PubMed.
- (a) E. M. Pelczar, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Organometallics, 2008, 27, 5759–5767 CrossRef CAS; (b) D. Hermann, M. Gandelman, H. Rozenberg, L. J. W. Shimon and D. Milstein, Organometallics, 2002, 21, 812–818 CrossRef CAS.
- W. H. Bernskoetter, S. K. Hanson, S. K. Buzak, Z. Davis, P. S. White, R. Swartz, K. I. Goldberg and M. Brookhart, J. Am. Chem. Soc., 2009, 131, 8603–8613 CrossRef CAS PubMed.
- M. R. Gyton, B. Leforestier and A. B. Chaplin, Organometallics, 2018, 37, 3963–3971 CrossRef CAS PubMed.
- (a) C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Angew. Chem., Int. Ed., 2018, 57, 12003–11200 CrossRef CAS PubMed; (b) S. L. Apps, R. E. Alflatt, B. Leforestier, C. M. Storey and A. B. Chaplin, Polyhedron, 2018, 143, 57–61 CrossRef CAS; (c) R. E. Andrew, C. M. Storey and A. B. Chaplin, Dalton Trans., 2016, 45, 8937–8944 RSC; (d) R. E. Andrew, D. W. Ferdani, C. A. Ohlin and A. B. Chaplin, Organometallics, 2015, 34, 913–917 CrossRef CAS; (e) R. E. Andrew and A. B. Chaplin, Inorg. Chem., 2015, 54, 312–322 CrossRef CAS PubMed; (f) R. E. Andrew and A. B. Chaplin, Dalton Trans., 2014, 43, 1413–1423 RSC.
- The synthesis and coordination chemistry of analogous ortho-xylene- and resorcinol-derived macrocyclic pincer ligands will be described in a following contribution.
- D. Dakternieks and R. Di Giacomo, Phosphorus Sulfur Relat. Elem., 1985, 24, 217–224 CrossRef CAS.
- (a) T. Imamoto, T. Kusumoto, N. Suzuki and K. Sato, J. Am. Chem. Soc., 1985, 107, 5301–5303 CrossRef CAS; (b) G. C. Lloyd-Jones and N. P. Taylor, Chem. – Eur. J., 2015, 21, 5423–5428 CrossRef CAS PubMed.
- A. P. T. Athanasopoulos, PhD Thesis, University of Waterloo, 2009.
- (a) M. Van Overschelde, E. Vervecken, S. G. Modha, S. Cogen, E. Van der Eycken and J. Van der Eycken, Tetrahedron, 2009, 65, 6410–6415 CrossRef CAS; (b) K. Jouvin, R. Veillard, C. Theunissen, C. Alayrac, A.-C. Gaumont and G. Evano, Org. Lett., 2013, 15, 4592–4595 CrossRef CAS PubMed.
- C. N. Iverson and W. D. Jones, Organometallics, 2001, 20, 5745–5750 CrossRef CAS.
- T. M. Hood, B. Leforestier, M. R. Gyton and A. B. Chaplin, Inorg. Chem., 2019, 58, 7593–7601 CrossRef CAS PubMed.
- (a) J. Emerson-King, I. Prokes and A. B. Chaplin, Chem. – Eur. J., 2019, 25, 6317–6319 CrossRef CAS PubMed; (b) R. C. Knighton, J. Emerson-King, J. P. Rourke, C. A. Ohlin and A. B. Chaplin, Chem. – Eur. J., 2018, 24, 4927–4938 CrossRef CAS PubMed.
- S. D. Pike, M. R. Crimmin and A. B. Chaplin, Chem. Commun., 2017, 53, 3615–3633 RSC.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
- (a) G. J. Kubas, Metal Dihydrogen and o-Bond Complexes, Kluwer Academic/Plenum Publishers, New York, 2001 Search PubMed; (b) R. H. Crabtree, Chem. Rev., 2016, 116, 8750–8769 CrossRef CAS PubMed.
- G. L. Parker, S. Lau, B. Leforestier and A. B. Chaplin, Eur. J. Inorg. Chem., 2019, 3791–3798 CrossRef CAS PubMed.
- J. J. Davidson, J. C. DeMott, C. Douvris, C. M. Fafard, N. Bhuvanesh, C.-H. Chen, D. E. Herbert, C.-I. Lee, B. J. McCulloch, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2015, 54, 2916–2935 CrossRef CAS PubMed.
- M. R. Gyton, T. M. Hood and A. B. Chaplin, Dalton Trans., 2019, 48, 2877–2880 RSC.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- T. R. Hoye, B. M. Eklov and M. Voloshin, Org. Lett., 2004, 6, 2567–2570 CrossRef CAS PubMed.
- Y. Chen, T. P. Clark, B. A. Jazdzewski, S. B. Klamo and T. T. Wenzel, Polyhedron, 2014, 84, 32–36 CrossRef CAS.
- J. A. Osborn, G. Wilkinson and J. J. Mrowca, Inorg. Synth., 1990, 28, 77–79 CAS.
- (a) A. J. Martínez-Martínez and A. S. Weller, Dalton Trans., 2019, 48, 3551–3554 RSC; (b) W. E. Buschmann, J. S. Miller, K. Bowman-James and C. N. Miller, Inorg. Synth., 2002, 33, 83–91 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, IR and ESI-MS spectra of new compounds, and selected reactions (PDF). Primary NMR data (MNOVA). CCDC 1966918–1966922. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt04474d |
| This journal is © The Royal Society of Chemistry 2020 |