
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe reactivity of molecular oxygen and reactive oxygen species with [FeFe] hydrogenase biomimetics: reversibility and the role of the second coordination sphere†
Vincent C.-C.
Wang
 *a,
Charlène
Esmieu
b,
Holly J.
Redman
b,
Gustav
Berggren
b and
Leif
Hammarström
*a
*a,
Charlène
Esmieu
b,
Holly J.
Redman
b,
Gustav
Berggren
b and
Leif
Hammarström
*a
aProgram of Physical Chemistry, Department of Chemistry – Ångström Laboratory, Uppsala University, Uppsala 75120, Sweden. E-mail: leif.hammarstrom@kemi.uu.se
bProgram of Molecular Biomimetics, Department of Chemistry – Ångström Laboratory, Uppsala University, Uppsala 75120, Sweden
First published on 12th December 2019
Abstract
The development of oxygen-tolerant H2-evolving catalysts plays a vital role for a future H2 economy. For example, the [FeFe] hydrogenase enzymes are excellent catalyst for H2 evolution but rapidly become inactivated in the presence of O2. The mechanistic details of the enzyme's inactivation by molecular oxygen still remain unclear. Here, two H2-evolving diiron complexes [Fe2(μ-SCH2NHCH2S)(CO)6] (1adt) and [Fe2(μ-SCH2CH2CH2S)(CO)6] (2pdt), inspired by the active site of [FeFe] hydrogenase, were investigated for their reactivity with molecular oxygen and reactive oxygen species. A one-electron reduced and oxygenated 1adt species was identified and characterized spectroscopically, which can be directly generated by reacting with molecular oxygen and chemical reductants at room temperature but it is unstable and gradually decomposes. Interestingly, the whole process is reversible and the addition of protons can facilitate the deoxygenation process and prevent further degradation at room temperature. This new identification of intermediate species serves as a model for studying the reversible inactivation and degradation of oxygen-sensitive [FeFe] hydrogenases by O2, and provides chemical precedence for such processes. In comparison, the complex lacking the nitrogen bridgehead, 2pdt, exhibits reduced reactivity towards O2 in the presence of reductants, highlighting that the importance of the second coordination sphere on modulating the oxygenation processes. These results provide new directions to design molecular electrocatalysts for proton reduction operated at ambient conditions and the re-engineering of [FeFe] hydrogenases for improving oxygen tolerance.
Introduction
In the past decade, the development of noble-metal-free complexes for H2 evolution has been a very active research field. A variety of metal complexes inspired by hydrogenases, both functional1 or structural2 mimics, has been prepared to generate synthetic catalysts for H+/H2 interconversion and to understand key feature for catalysis. However, most of these molecular complexes are oxygen-sensitive under catalytic turnover conditions. For example, Ni-bis(diphosphine) complexes developed by the Dubois group3,4 and diiron complexes based on the active site of [FeFe] hydrogenases,5,6 have been shown to be incompatible with air-operated conditions. Therefore, it is important to understand the damage processes by molecular oxygen during H2 evolution, in order to provide new ideas for re-engineering of catalysts to achieve improved oxygen-tolerance. To gain further insight into this chemistry, we have probed two biomimetic molecular electrocatalysts, Fe2(adt)(CO)6 (adt = azadithiolate), 1adt and Fe2(pdt)(CO)6 (pdt = propanedithiolate), 2pdt (Fig. 1),5–9 and investigated their reactivity with molecular oxygen. Both complexes are structurally similar to the diiron subsite of the catalytic cofactor of oxygen-sensitive [FeFe] hydrogenases (the “H-cluster” as shown in Fig. 1).10–13 However, they differ in the nature of the bridgehead atom in the dithiolato ligand, and thus provide insight into the influence of proton relays for the reactivity of the low valence metal center towards O2.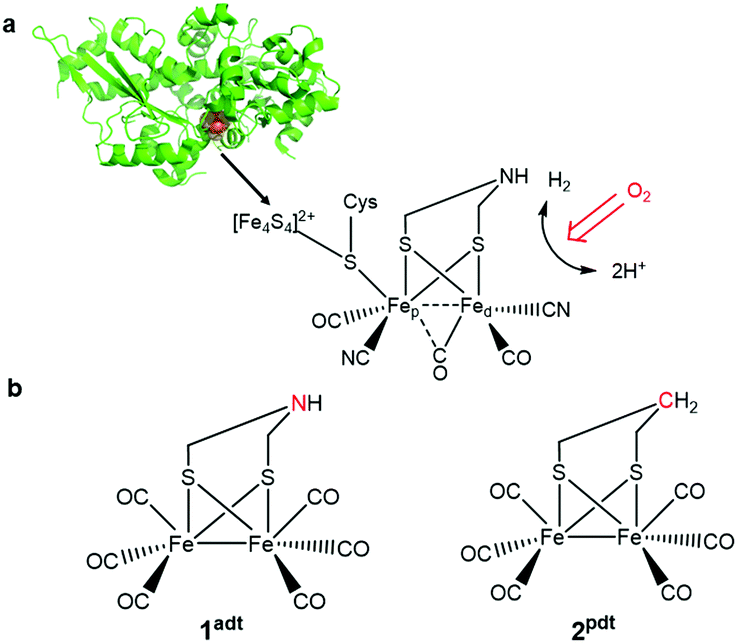 | ||
| Fig. 1 (a) A schematic representation of the [FeFe] hydrogenase from Chlamydomonas reinhardtii (Cr HydA1, PDBID: 3LX4) and the H-cluster. (b) The two biomimetic complexes, 1adt and 2pdt, used to investigate reactivity towards O2 and ROS. | ||
The reactivity of analogues of 1adt and 2pdt towards O2 under electrocatalytic conditions has previously been reported.5,6 However, under such conditions, it is challenging to characterize the true species that attack molecular catalysts and the corresponding engaged state from catalysts. For example, a variety of reactive oxygen species (ROS) can be generated directly from the electrode as suggested in eqn (1)–(3) before proton reduction reaction takes place (eqn (4) and Fig. S1†).
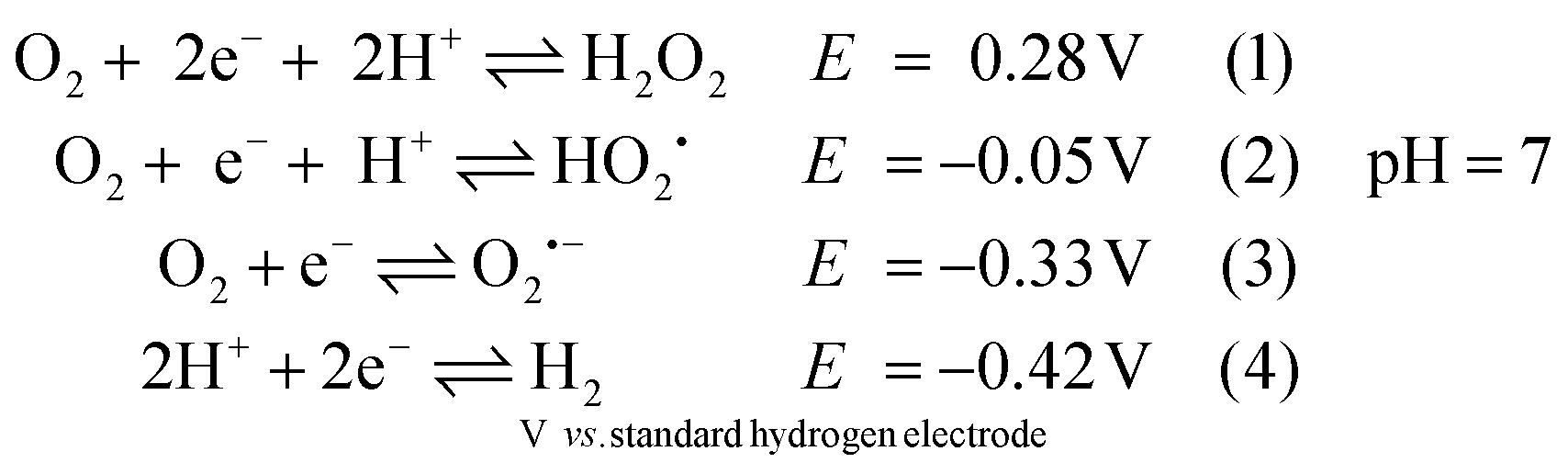
Herein, we report on spectroscopic investigations of the reactivity of these two complexes towards molecular oxygen. More specifically, the distinct vibrational absorption bands for carbonyl groups (νCO) from 1adt and 2pdt allow to monitor changes in the electronic and geometric structure of the diiron center by IR spectroscopy. Chemical reductants were employed to address the influence of the redox states on the reaction. This alternative approach enabled the generation of one-electron reduced and oxygenated 1adt and 2pdt by adding O2 in combination with reductants that have a more positive reduction potential than those of the respective diiron complexes (FeIFeI/FeIFe0). The whole process for 1adt is reversible, although the reduced and oxygenated species is unstable at room temperature. The addition of protons prevents further damage and facilitates the deoxygenation process to regenerate the starting material. The chemical process may be biologically relevant to the reactivity of [FeFe] hydrogenase towards molecular oxygen and represent a general oxygenation and reactivation scheme for this class of complexes.
Results and discussion
Reactivity of 1adt and 2pdt towards O2 under reducing conditions
The reactivity of 1adt and 2pdt with molecular oxygen was probed in the absence and presence of chemical reductants, i.e. bis(benzene)chromium (Cr(bz)2), cobaltocene (CoCp2) and bis(pentamethylcyclopentadienyl)cobalt (CoCp2*). The reduction potential for these reductants are respectively E = −1.15 V (all potentials reported herein are against Fc/Fc+), E = −1.33 V and E = −1.94 V, compared to the reduction potential of −1.6 V for 1adt and 2pdt (Fig. S1 and Table S1†). Recently, some reductants, such as CoCp2 and CoCp2* have been applied to study H2-evolving mechanism with related diiron complexes via transient spectroscopy.14,15 As expected in light of their respective reduction potentials, 1adt and 2pdt did not react with CoCp2, Cr(bz)2 or molecular oxygen in isolation. Upon addition of the chemical reductants or purging with O2, the IR spectra featured νCO bands of 1adt and 2pdt at 2074, 2033, 2000 and 1989 cm−1, which are indiscernible from the starting material (Fig. 2). In comparison, multiple new νCO bands from the two Fe complexes were observed at lower wavenumbers, between 1970 cm−1 and 1850 cm−1 (Fig. S2†), when mixing with the stronger reducing agent, CoCp2*. These features indicate the dimerization of one-electron reduced 1adt or 2pdt species as reported from earlier studies.16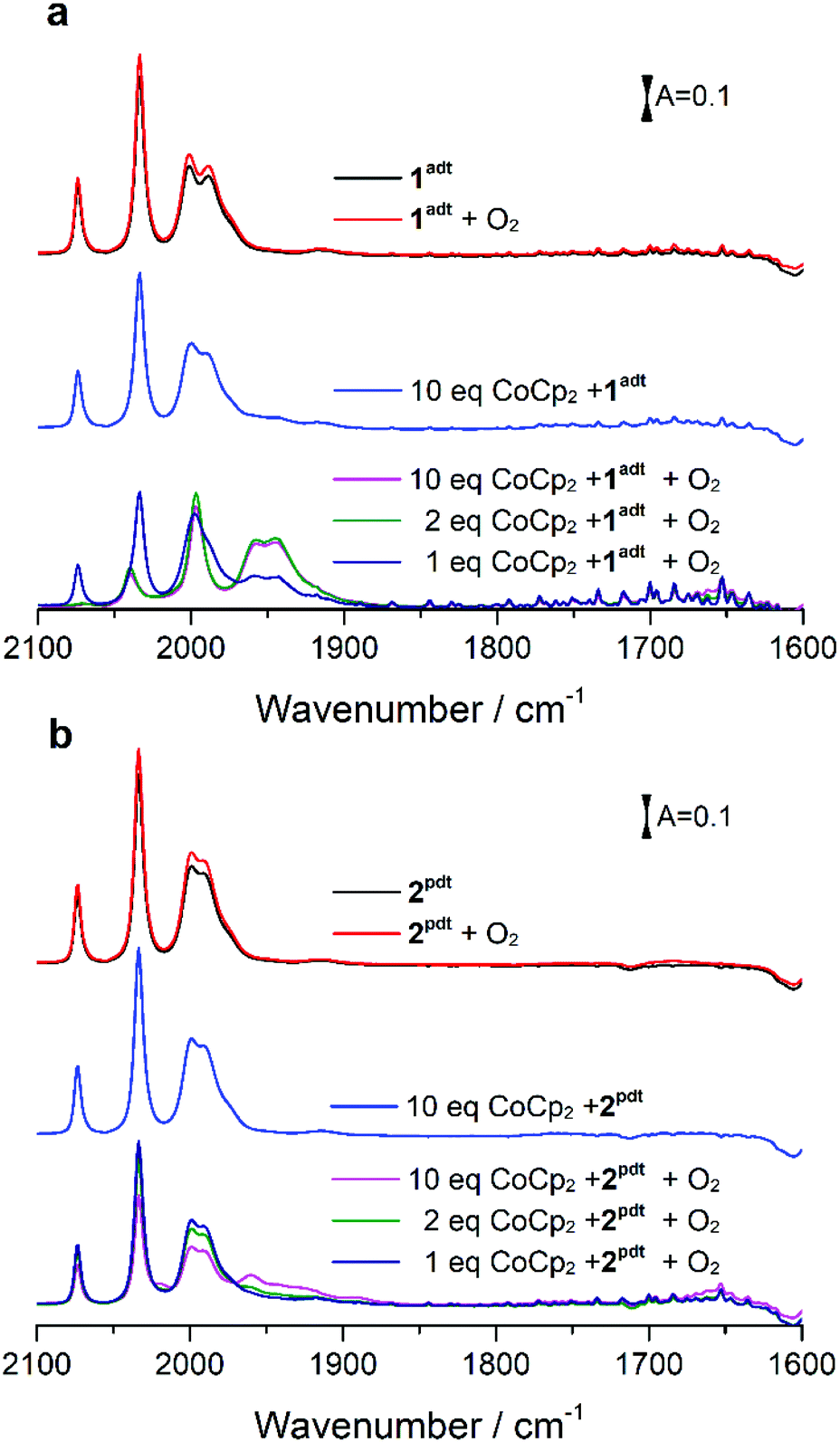 | ||
| Fig. 2 Infrared spectra obtained from a mixture of 1adt (a) (or 2pdt (b)) and cobaltocene upon exposure to the excess of O2. The complex 1adt (0.5 mM) or 2pdt (0.5 mM) was mixed with different equivalents of CoCp2 first and then molecular oxygen was added to a mixture solution. The intensity of red line is stronger than that of black line because solvent (dichloromethane, DCM) was purged away by O2 during the reaction. | ||
Unexpectedly, we noticed that two new peaks at 1957 and 1946 cm−1 appeared when a mixture of CoCp2 and 1adt was exposed to air under ambient conditions. When mixing more than two equivalents of CoCp2 (or Cr(bz)2) and 1adt with excess O2, the four bands attributed to 1adt almost disappeared and four new distinct bands for νCO were observed at lower wavenumbers, 2038, 1997, 1957 and 1946 cm−1 as shown in Fig. 2a and 3. This new spectrum is distinctly different from one-electron reduced species of 1adt reported before (Table S2†). Still, the red-shift of the four bands by ca. 35–45 cm−1 relative to the starting material 1adt suggests that the newly formed species is reduced. In control experiments, no new peaks were observed when adding oxygen-free DI water (Fig. S3†) to a mixture of CoCp2 and 1adt (or 2pdt), which exclude the formation of a protonated and reduced species. In comparison, a mixture of 2pdt and CoCp2 exhibited less reactivity toward O2. Still, new bands for νCO were observed at 2020, 1960 and 1891 cm−1 and a broad shoulder peak spanned between 1950 and 1910 cm−1 (Fig. 2b). The multiple number of new and broad peaks from 2pdt suggest at least two different species coexist after reacting a mixture of CoCp2 and 2pdt with O2 and likely dimerization occurred comparing to IR spectra from mixing CoCp2* and 2pdt. In both cases, black precipitates were observed after introducing O2 into a mixture of reductants and 1adt or 2pdt. These precipitates were also observed when adding O2 to a solution containing only CoCp2, and were thus not analyzed in detail. The newly formed IR bands from these new species gradually decayed in ca. 20 minutes under anaerobic conditions, indicating that they were unstable at room temperature. Critically, the νCO bands for as-prepared 1adt were partially recovered again, while full degradation occurred for the new species generated from 2pdt (vide infra and Fig. S18†). Thus, distinct differences were observed between 2pdt and the more biologically relevant 1adt. The latter revealed not only a more defined product in the reaction with O2, with only four distinct new νCO peaks, but also showed a high degree of reversibility, as compared to mixed products and irreversibility observed for 2pdt.
Spectroscopic characterization of transient species
The minute-scale lifetime of the transient species at room temperature allowed for a more detailed spectroscopic characterization to explore its geometric and electronic structure. The resulting product directly obtained by reacting a mixture of 1adt and Co(Cp)2 (or Cr(bz)2) with O2 showed a mass peak at m/z 401.76 amu, detected by electron-spray mass spectroscopy, assigned to [O + 1adt − H], indicated that an oxygenated species, O-1adt− was formed (Fig. S4 and S5†). Indeed, a shift of two amu units for O-1adt− was observed when 18O2 was introduced to a mixture of CoCp2 and 1adt (Fig. S6†). Similarly, a mass peak for [O + 2pdt−] at 402.6 amu was also observed (Fig. S7†). Earlier work conducted by Darensbourg and co-workers has demonstrated that meta-chloroperoxybenzoic acid (mCPBA) can oxygenate 2pdt to generate a mono-oxygenated 2pdt (O-2pdt), through an oxygen atom transfer reaction to a thiol ligand of 2pdt.17 Consequently, we explored the possibility of the oxygen atom binding to a S atom of 1adt as well. Indeed, an IR band assigned to νS![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from O-1adt− was observed at 1030 cm−1 and the peak displayed the expected isotope shift to 993 cm−1 when 18O2 was used in the oxygenation reaction (Fig. 4). The IR bands assigned to νSO are in excellent agreement with the DFT-simulated IR spectrum (1028 cm−1 for 16O and 996 cm−1 for 18O) for a one-electron reduced and oxygenated species featuring an oxygenated thiol group (SO) (Fig. 4c). Noticeably, these two peaks disappeared in ca. 25 minutes, which is in line with the disappearance of the 1946 cm−1 (νCO) peak assigned to O-1adt− (Fig. S17†).
O from O-1adt− was observed at 1030 cm−1 and the peak displayed the expected isotope shift to 993 cm−1 when 18O2 was used in the oxygenation reaction (Fig. 4). The IR bands assigned to νSO are in excellent agreement with the DFT-simulated IR spectrum (1028 cm−1 for 16O and 996 cm−1 for 18O) for a one-electron reduced and oxygenated species featuring an oxygenated thiol group (SO) (Fig. 4c). Noticeably, these two peaks disappeared in ca. 25 minutes, which is in line with the disappearance of the 1946 cm−1 (νCO) peak assigned to O-1adt− (Fig. S17†).
To characterize the electronic structure of O-1adt−, electron paramagnetic resonance (EPR) spectroscopy was employed. A new EPR signal for O-1adt− was observed at g ∼ 2.14 and 2.02 compared to EPR-silent 1adt, confirming that O-1adt− is a one-electron reduced species. Spin quantification of the double integral spectrum using Cu-EDTA as a standard, suggested that 50% of the initial 1adt complex converted to this one-electron reduced species (Fig. S8†). In the case of 2pdt, the yield of the oxygenated reduced species was limited to ca. 30%. The formation of O-1adt− also results in the appearance of a new absorption peak at 605 nm in the UV-visible spectrum (Fig. S9a†). In contrast, 1adt− shows absorption bands around 560 and 700 nm.14,15 As this new signal at 605 nm disappears with a half-life time (t1/2) of ∼3.6 minutes, which coincides with the concomitant decreasing intensity of IR peaks at 1957 and 1946 cm−1 (t1/2 avg 5.4 min, Fig. S9†), we attribute the absorption band at 605 nm to O-1adt−.
The instability of O-1adt− has so far precluded its characterization by X-ray crystallography. Nevertheless, the mass spectrometry, EPR, UV-visible and FTIR data, in combination with the DFT calculations allow us to converge on a structural model for O-1adt−. First, the μ-oxo diiron (FeIIFeII), end-on peroxide diiron (FeIIFeII) and bis(μ-oxo) diiron (FeIIIFeIII) species can be ruled out due to the observed red shift of the IR spectrum. Moreover, the observed EPR signal underscores that the formed product, O-1adt−, is paramagnetic. Three low valent (FeIFe0) and oxygenated species were further examined: ligand-based oxygenation at the bridgehead N atom or the thiolate S atom, or metal-based oxygenation producing a μ-oxo diiron state. For the first postulated structure, geometry optimization by DFT calculation methods could not be made to converge. For the low valent μ-oxo diiron state, the simulated IR spectrum based on DFT calculations revealed five νCO bands, in contrast with experimental results (Fig. S20†). In addition, no peaks between 1200 and 800 cm−1 were observed from the simulated IR spectrum of the μ-oxo diiron (FeIFe0) state. Finally, the electron-rich μ-oxo diiron (FeIFe0) center would be expected to be highly reactive at room temperature. Therefore, we assign O-1adt− to a FeIFe0 species with a retained Fe2(CO)6 core and an oxygenated ligand derived S-atom (Scheme 1). Indeed, the IR-spectrum for this species is in good agreement with the vibrational spectrum obtained from DFT calculations (Fig. 4 and Fig. S19–20†), and all aforementioned spectrometry and spectroscopic analysis.
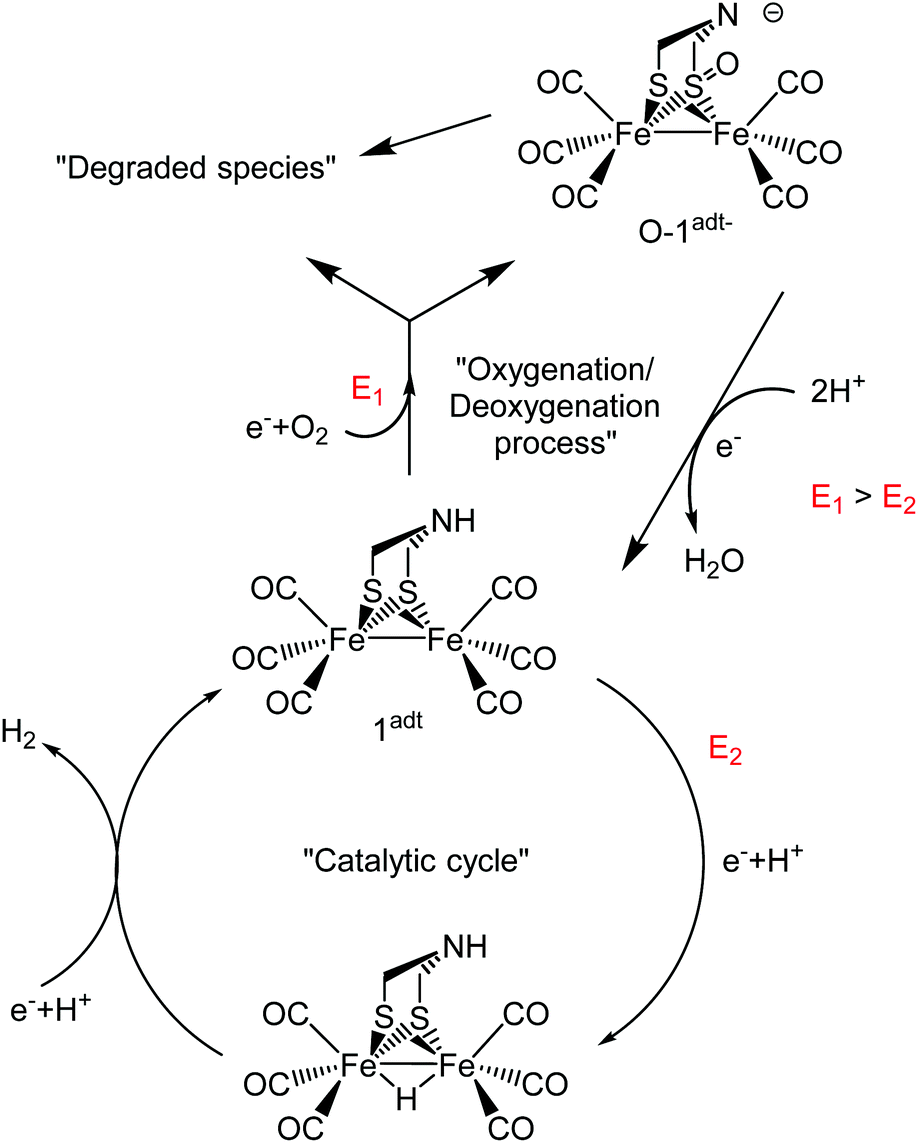 | ||
| Scheme 1 Summary of the mechanism for oxygenation/deoxygenation processes and the catalytic cycle for H2 evolution catalyzed by 1adt. The red character, E is the reduction potential. | ||
Reactivity towards O-atom transfer reagents
As described above, the reaction of mCPBA with 2pdt generates O-2pdtvia an oxygen atom transfer reaction, which allowed us to explore whether the formation of O-1adt− or O-2pdt− could be achieved via such oxygenated intermediates. The formation of O-2pdt proceeded smoothly upon mixing 2pdt with 2 eq. of mCPBA (Fig. S12†).17 In contrast, no new peaks in the CO region were observed when treating 1adt with 2 eq. of mCPBA, and larger excess of mCPBA led to diminishing νCO peaks from 1adt, with concomitant appearance of only two weak peaks at 2082 and 2040 cm−1 (Fig. S12†). The new peaks remained after 1-hour incubation for both reactions. In addition, no apparent change in νCO peaks was observed when adding CoCp2 into a mixture of 2pdt (or 1adt) and mCPBA. This is attributable to the lower reduction potential for O-2pdt, −1.58 V.17 Thus, the preparation of O-1adt or O-2pdt by mCPBA do not appear to be intermediate species during the formation of the reduced oxygenated species (O-1adt−, O-2pdt−) reported here.The role of ROS
We further examined whether these oxygenated adducts can be prepared through ROS generated from reacting CoCp2 with O2. In addition, the reactivity of ROS with these reduced and oxygenated adducts was studied as well. The reaction of O2˙− with 1adt and 2pdt respectively, resulted in the appearance of a broad shoulder band between 1980 cm−1 and 1900 cm−1 in both cases (Fig. S13†). The newly formed peaks remained for 20 hours, in contrast to the reversible formation of O-1adt−. Thus, superoxide is highly unlikely to be involved in the formation of O-1adt−. Furthermore, no new peaks were observed when mixing H2O2 with 1adt and 2pdt respectively, which is similar with the earlier report.6 However, the νCO peaks for O-2pdt− diminished after mixing H2O2 with O-2pdt−, while no apparent decrease in νCO absorption was observed for O-1adt− after mixing with H2O2 (Fig. S14†). Critically, the formation of O-1adt− was also observed using the reducing agent, Cr(bz)2 which has a more positive reduction potential (∼0.12 V, Table S1†) than the O2/O2˙−couple (Fig. 3). In combination, these results suggest that ROS is highly unlikely to be involved in the formation of neither O-1adt− nor O-2pdt−.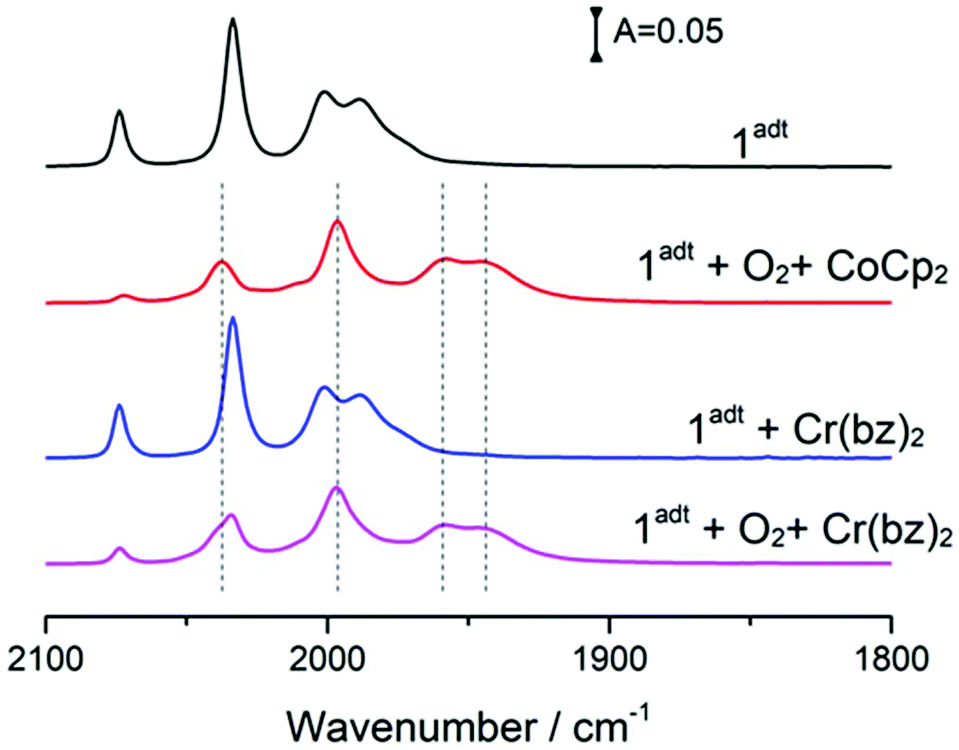 | ||
| Fig. 3 The formation of a new species, O-1adt− by mixing 1adt, O2 and reductants, CoCp2 or Cr(bz)2. The complex 1adt (0.36 mM) was first mixed with 4 eq. of CoCp2 or 2 eq. of Cr(bz)2 and then 4 eq. of O2 from O2-saturated DCM solution was added. | ||
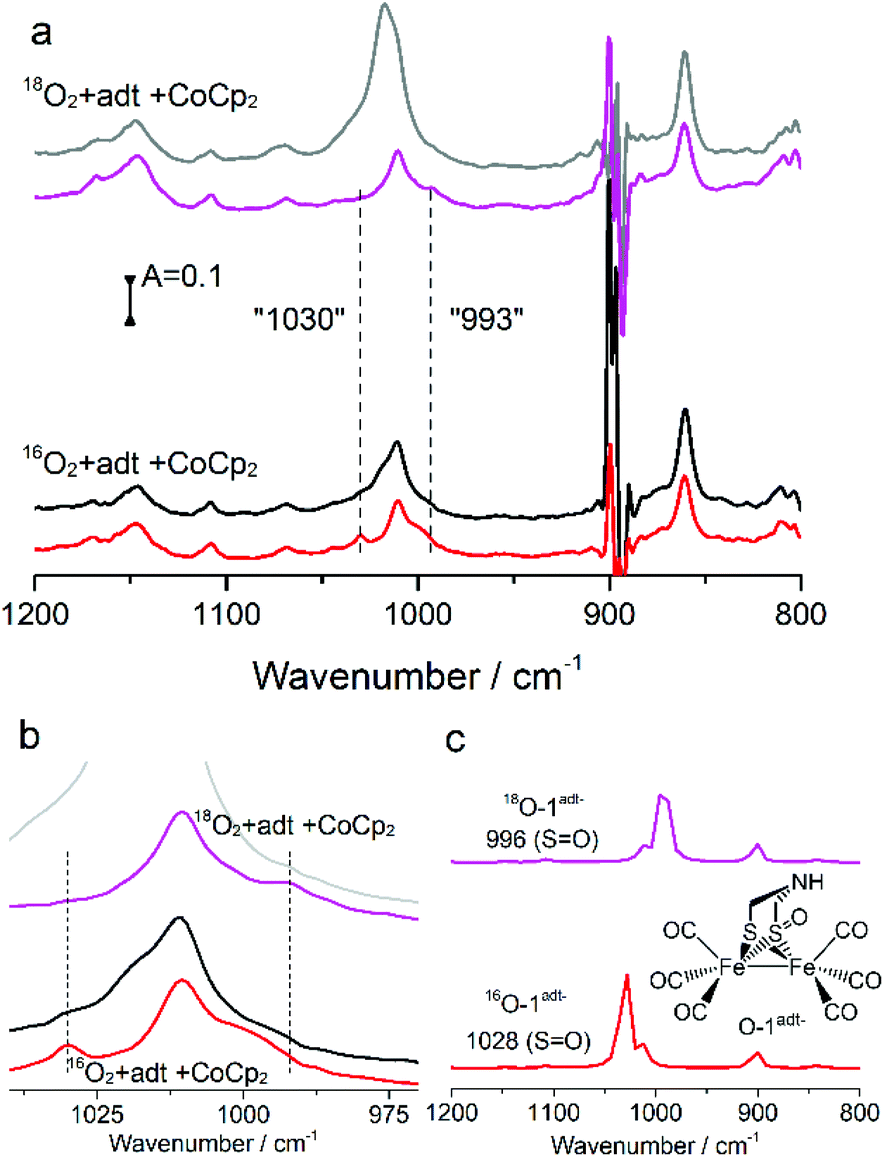 | ||
| Fig. 4 (a) The IR spectra of νSO from isotope 18O2vs.16O2 experiments. A mixture of 1adt (4.44 mM) and 5 eq. of CoCp2 reacted with 18O2 and 16O2 respectively. The IR spectra were collected at 1 min (magenta and red traces) and 25 min (grey and black traces) after addition of O2, respectively. (b) Zoom of the IR peak region for νSO. (c) Comparison of the νSO peak from experiments with that from DFT calculations based on the structure shown in the figure. | ||
Reactivity of O-1adt−
Incubation of O-1adt− under a glovebox atmosphere for 20 hours at room temperature, resulted in recovery of approximately 30% of 1adt (Fig. 5). Multiple new high-wavenumber bands were observed during the deoxygenation process from O-1adt− to 1adt, indicating the formation of intermediate species during the reaction (Fig. S9†). Additionally, the distinct IR peaks for H2O accumulated during deoxygenation (Fig. S10†). The O-1adt− species could subsequently be regenerated by addition of fresh cobaltocene and O2 as shown in Fig. 5. In comparison, much lower amount of reformed 1adt (<5%) was observed when O-1adt− samples were placed at ambient conditions, while samples were stable for 20 hours inside the glovebox at −30 °C (Fig. S11†).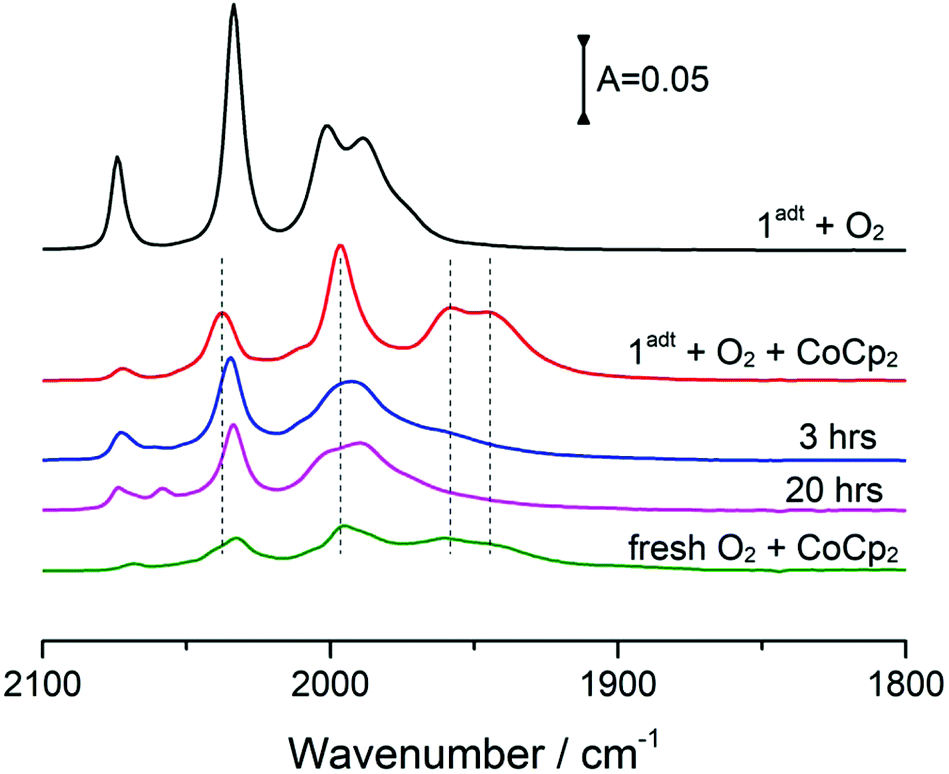 | ||
| Fig. 5 The stability and reversibility of O-1adt− at room temperature. Samples were prepared and stored in the glovebox at room temperature and covered with tin foil to prevent photodissociation. Red line: mix 4 eq. of CoCp2 and 1 eq. of 1adt (0.36 mM) with 4 eq. of O2. Blue line: 3 hours after mixing. Magenta line: 20 hours after mixing. Olive line: addition of 10 eq. fresh CoCp2 and O2 to re-generate 1adt to O-1adt−. | ||
The role of protons
The reactivity of O-1adt− and O-2pdt− towards protons was further probed with two acids, chloroacetic acid (pKa = 18.8 in acetonitrile) and trichloroacetic acid (pKa = 10.7 in acetonitrile),18 both of which have been used as proton source in electrocatalytic H2 evolution studies with the two complexes.2,7,9 It has been suggested that the bridgehead N atom of 1adt can be protonated by trichloroacetic acid but not chloroacetic acid.9 The reformed 1adt species was observed rapidly upon the addition of either of the acids with O-1adt−, as shown in Fig. 6. Compared to reformation of 1adt in the absence of acids, the deoxygenation process is faster and the yield is higher in the presence of acids (ca. 60% recovery vs. 30% in the absence of acids). Similarly, the conversion from O-2pdt− to 2pdt was also accelerated under acidic conditions (Fig. S15†). Thus, in both cases, the addition of protons facilitates the deoxygenation process and prevents further damage at room temperature. As opposed to the clear effect of acid, no apparent increase in the amount of regenerated 1adt was observed by adding excess of reduced CoCp2 to facilitate the deoxygenation process (i.e. electron source) (Fig. S16†).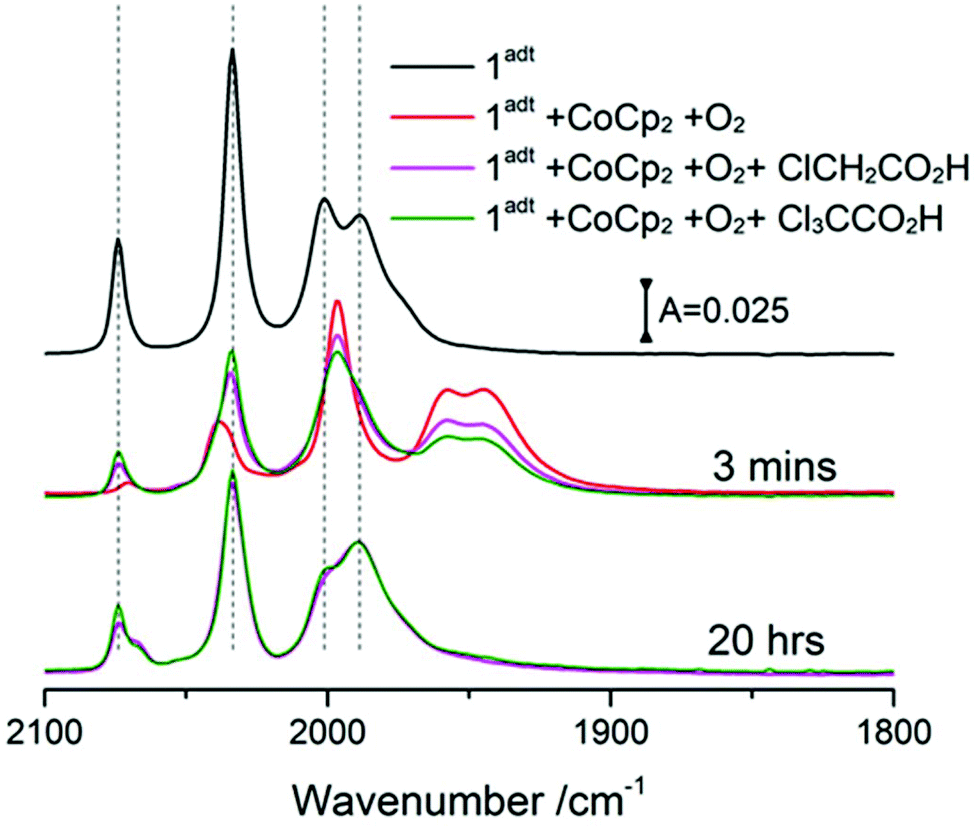 | ||
| Fig. 6 The reactivity of acids with O-1adt−. A solution with four equivalents of CoCp2 and an equivalent of 1adt was mixed with four equivalents of O2 (red line). Then two equivalents of acid against the starting concentration of 1adt (0.36 mM), chloroacetic acid (magenta color) and trichloroacetic acid (olive color), were added later respectively. The samples were stored in the glovebox at room temperature. | ||
Discussion
Herein, the one-electron reduced and oxygenated species, O-1adt− and O-2pdt−, were prepared and investigated. Recent studies by Dey, Darensbourg and co-workers have shown that similar analogues of 1adt and 2pdt attached on the electrode surface are capable of catalyzing the O2 reduction reaction (ORR) in aqueous solution, albeit with gradual degradation during electrochemical catalysis.5,6 Noticeably, the onset current for O2 reduction was observed more positive (∼0.1 V) than the first reduction potential of the studied 1adt analogues, which suggests that electron transfer can occur at the more positive potential through coupling chemical steps. This observation is firmly supported by the formation of O-1adt− through chemical reductants in the presence of molecular oxygen reported here. Since the reduction potential for FeIFeI to FeIFe0 is significantly more negative (∼ca. 0.4 V) than the reduction potential of the mildest reductant, Cr(bz)2, it is highly unlikely that reduction chemistry occurs prior to the oxygenation reaction (i.e. the chemical step). Instead, the most likely scenario is that a pre-equilibrium exists with reversible binding of O2 to 1adt or 2pdt before reduction chemistry takes place, although the association constant for the complex, [O2⋯1adt] or [O2⋯2pdt], must be very small. The formation of a complex of [O2⋯1adt] or [O2⋯2pdt] provides an anodic shift of the reduction potential of the diiron center and facilitates sequential redox and oxygenation reactions.Although the bridgehead atoms are different (i.e. N vs. C), the two diiron center complexes, 1adt and 2pdt, have a similar electronic structure. Still, the protonatable aza group in the second coordination sphere in this family of complexes generally improves their catalytic properties. Analogously, their reactivity with O2 in the presence of reductants led to different oxygenated structures and the reactivity of 2pdt was more sluggish. Noticeably, the oxygenated 2pdt complex has more νCO peaks than the four distinct peaks observed for O-1adt−, suggesting the formation of multiple products. When mCPBA was used as the oxygen donor, the oxygenated, non-reduced, adduct of 2pdt was formed, whereas 1adt was much less reactive and did not form the corresponding complex. Thus, the present results suggest that the second coordination sphere also modulates the reactivity of oxygenation. However, upon addition of protons, no apparent differences between these complexes were observed in the deoxygenation process. Both 1adt and 2pdt were rapidly regenerated after addition of acids. Water was observed as the product during the deoxygenation process of O-1adt− and attempts to detect H2O2via three various methods were not successful. Based on the oxygenation/deoxygenation processes of 1adt discovered here and the mechanism for H2 evolution suggested by earlier studies,2,9,15 a schematic catalytic cycle of 1adt for H2 evolution in the presence of molecular oxygen is summarized in Scheme 1.
As pointed out in the introduction, the formation of ROS, expected to irreversibly damage the catalyst, is thermodynamically more favorable than proton reduction. Nevertheless, examples where proton reduction occurs under ambient conditions, have been reported. In nature, [Fe4S3]-containing [NiFe] hydrogenases have been shown to catalyze H2 oxidation in the presence of O2. The oxygen-tolerant property from this subset of hydrogenases is generally ascribed to the unique [Fe4S3] cluster that accommodates three oxidation states and acts as electron reservoirs for the reduction of O2 to harmless water instead of the formation of ROS.19,20 Therefore, one way to prevent such oxygen-induced damage is to develop bifunctional catalysts that can catalyze ORR to water. Cobalt diimine-dioxime, one of few known oxygen-tolerant H2-evolving catalysts, demonstrates this bifunctionality.21 Here, we show that 1adt can react reversibly with O2 in the presence of reductants and previous electrochemical results from similar complexes also revealed the capacity for ORR chemistry albeit with limited stability during electrocatalysis. Further improvement of these diiron thiolate complexes are expected to enhance their stability during ORR and thus decrease oxygen sensitivity. Indeed, very recently, it was reported that the ORR and oxygen-tolerance of 1adt can be greatly improved via substitutions of the N-bridgehead atom, resulting in improving proton shuttling.22
Specifically with regards to [FeFe] hydrogenases, several O2 inactivated structures have been reported, many of which are considered to result in irreversible inactivation.23,24 For example, the oxygenation of sulfur at cysteine 169 (i.e. the formation of sulfenic acid) near the diiron center was observed after O2 exposure to [FeFe] hydrogenases from C. reinhardtii. However, Léger, Blumberger and co-workers have suggested that the reaction between O2 and [FeFe] hydrogenases is a multi-step process. The initial species formed upon exposure to O2 can be either reactivated, i.e. suggesting a reversible deoxygenation process, or further proceed to yield an irreversibly inactivated dead-end species.25,26 Here, we provide chemical precedence for such a direct oxygenation processes of the thiolate S atom via molecular oxygen, as well as the reversible deoxygenation process and degradation of oxygenated 1adt. Considering the structural similarities between 1adt and the H-cluster, this reactivity may also be relevant to [FeFe] hydrogenase.
Conclusions
In summary, this study reports on the oxygenation of two low valent diiron complexes, mimicking the [FeFe] hydrogenase cofactor, under reducing conditions. The preparation of O-1adt− and O-2pdt− provides a new venue for exploring the reactivity between molecular oxygen and H2-evolving catalysts. The results presented herein underscore the influence of the bridgehead nitrogen in the reaction towards O2, and the importance of protons for the reversibility of the process. Insights learned from studying these systems are expected to provide new guidelines for the design of oxygen-tolerant catalysts for H2 evolution and the engineering of [FeFe] hydrogenases. Detailed investigations of the oxygenation and deoxygenation mechanisms of these two complexes at low temperature conditions are currently ongoing.Experimental methods
Chemicals
Bis(cyclopentadienyl)cobalt(II), bis(pentamethylcyclopentadienyl)cobalt(II), potassium superoxide, hydrogen peroxide (30% w/w in water) and 18O2 (97%) were purchased from Sigma-Aldrich. Chloroacetic acid (>99.0%), trichloroacetic acid (> 99.0%) and 3-chloroperbenzoic acid (mCPBA) were also obtained from Sigma-Aldrich. The molecular oxygen (99.9%) from Air Liquide was used for this study. Bis(benzene)chromium was purchased from Alfa Aesar.Precursor to 1adt
The synthesis of the precursor to 1adt, Fe2(μ-S)2(CO)6 was adapted from literature procedures.27 A 500 mL three-necked round-bottom flask was equipped with a mechanical stirrer and a bubbler. To this was added iron(0) pentacarbonyl (6.3 mL, 47.9 mmol). Degassed methanol (32 mL) was added to the reaction vessel, followed by a freshly prepared and degassed 50 wt% aqueous solution of potassium hydroxide (20 mL). The reaction mixture was stirred for 30 minutes and then cooled to 0 °C. Sulfur (8.1663 g, 0.25 mol) was added to the reaction mixture over the course of 5 minutes. This mixture was stirred for one hour at 0 °C. Degassed water (60 mL) and degassed pentane (180 mL) was added to the reaction vessel. Ammonium chloride (21.8366 g, 0.41 mol) was added to the reaction mixture very slowly and in portions because the reaction produces lots of hydrogen sulfide gas. The reaction vessel was allowed to warm to room temperature, and stirred for 16 hours. After 16 hours, the reaction could be seen to form two layers, one black sticky aqueous phase, and a deep red pentane phase. The reaction mixture was passed through a 100 mL pad of Celite and washed with pentane until the black precipitate collected in the flask appeared slightly grey. The organic fraction was split into smaller volumes of 300 mL and washed with equal volumes of water. The organic phases were then dried over magnesium sulfate, filtered and pentane was evaporated under reduced pressure. The crude product was recombined and re-dissolved in a small volume of pentane before passing it through a silica column with pentane eluent. The solvent was removed under reduced pressure to produce a brown solid (Fe2(μ-s)2(CO)6, 2.7625 g, 8.03 mmol, 16% yield). FTIR (MeCN) 2002, 2041, and 2083 cm−1.Synthesis of 1adt and 2pdt
The synthesis of 1adt and 2pdt followed literature procedures.28,29Reactions of 1adt and 2pdt with molecular oxygen in the presence of reductants
All chemical solvent (mainly, dichloromethane) used in this study were obtained from solvent stills. Oxygen-free DCM was prepared by at least four freeze–pump–thaw cycles. Oxygen-free DI H2O or H2O2 were prepared by purging with N2 for an hour. The oxygen-saturated DCM was prepared by purging O2 for an hour and the concentration of saturated oxygen in DCM is estimated to be ca. 9 mM according to literature.30,31 The superoxide radicals were prepared by mixing potassium superoxide and 18-crown-6 ether (Sigma-Aldrich). All mixtures of 1adt or 2pdt with reductants (such as CoCp2, CoCp2*and Cr(Bz)2) in DCM were prepared inside an Ar-containing glovebox (0.5 ppm < O2 and 0.5 ppm < H2O). Molecular oxygen was either introduced from the oxygen-saturated DCM inside the glovebox or directly added excessive O2 into a sealed vial containing a mixture of 1adt or 2pdt and reductants outside the glove box. When reacting with superoxide, hydrogen peroxide and mCPBA, all experiments were conducted inside the glovebox. When samples were stored inside the glovebox, the sample vial was wrapped with tin foil to prevent photodissociation.Spectroscopic characterization
Nuclear magnet resonance measurements were recorded with a JEOL (400 YH magnet) Resonance 400 MHz. Infrared spectrum was collected with a Bruker IFS 66 v/S FT-IR spectrometer, in which the DLaTGS detector was employed with 2 cm−1 resolution. Sample solution was prepared inside a gas-tight IR cell with CaF2 or KBr as windows. For UV-visible spectrum, the Carry 5000 spectrometer was used and the sample solution was placed in a gas-tight UV-visible cuvette. The mass spectrum was collected in a negative mode with a Thermo Finnigan LCQ Deca XP Max LC/MS spectrometer through the direct injection mode. EPR spectra were recorded at X-band at 9.28 GHz, modulation frequency 100 kHz and modulation amplitude 5 Gauss on a Bruker ELEXYS E500 using an ER049X SuperX microwave bridge in a Bruker SHQ0601 cavity equipped with an Oxford Instruments continuous flow cryostat and an ITC 503 temperature controller (Oxford Instruments). A 1 mM CuSO4 and 10 eq. of EDTA dissolved in water was employed as a standard sample for spin counting.DFT calculation methods
The computational methods were followed as the previous reports.32,33 In brief, DFT calculations were conducted with the Gaussian 97 package using BP86 functionals with the Def2TZVP basis set32 or cc-pVDZ/cc-pVTZ/SDD basis set.33 Both basis sets were used to calculate similar molecular systems before. All geometry-optimized structures obtained in the gas phase were further optimized with the SMD solvation model for DCM solvent. The final optimized structure was used to simulate IR spectra. Compared to experimental IR spectra, both basis sets yielded the same trends for all simulated molecular complexes based on 1adt or 2pdt structure but the Def2TZVP basis set resulted in the less deviation of the absorption of carbonyl groups from 1adt and 2pdt. Here, only results from the method (BP86 functionals and Def2TZVP basis) are shown.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Swedish research Council (contract no. 621-2014-5670) and ERC (contract no. 714102, to G. B.). We acknowledge postdoc grants to V. W. and C. E. from the Wenner-Gren Foundations. We are very grateful to Jakob Thyr and Prof. Tomas Edvinsson, who made initial efforts on Raman spectroscopy and provided inspiration on IR measurements. We also thank Dr Patrícia Raleiras for conducting spin counting experiments and MSc. Robin Tyburski for supporting DFT calculations through his funding.References
- D. L. DuBois, Inorg. Chem., 2014, 53, 3935–3960 CrossRef CAS PubMed.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed.
- J. Y. Yang, R. M. Bullock, W. G. Dougherty, W. S. Kassel, B. Twamley, D. L. DuBois and M. R. DuBois, Dalton Trans., 2010, 39, 3001–3010 RSC.
- D. W. Wakerley, M. A. Gross and E. Reisner, Chem. Commun., 2014, 50, 15995–15998 RSC.
- S. Dey, A. Rana, S. G. Dey and A. Dey, ACS Catal., 2013, 3, 429–436 CrossRef CAS.
- S. Dey, A. Rana, D. Crouthers, B. Mondal, P. K. Das, M. Y. Darensbourg and A. Dey, J. Am. Chem. Soc., 2014, 136, 8847–8850 CrossRef CAS PubMed.
- D. S. Chong, I. P. Georgakaki, R. Mejia-Rodriguez, J. Samabria-Chinchilla, M. P. Soriaga and M. Y. Darensbourg, Dalton Trans., 2003, 4158–4163 RSC.
- S. J. Borg, T. Behrsing, S. P. Best, M. Razavet, X. M. Liu and C. J. Pickett, J. Am. Chem. Soc., 2004, 126, 16988–16999 CrossRef CAS.
- M. Bourrez, R. Steinmetz and F. Gloaguen, Inorg. Chem., 2014, 53, 10667–10673 CrossRef CAS.
- D. W. Mulder, E. S. Boyd, R. Sarma, R. K. Lange, J. A. Endrizzi, J. B. Broderick and J. W. Peters, Nature, 2010, 465, 248–251 CrossRef CAS.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592–6599 RSC.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS.
- S. Wang, S. Pullen, V. Weippert, T. Liu, S. Ott, R. Lomoth and L. Hammarström, Chem. – Eur. J., 2019, 25, 11135–11140 CrossRef CAS.
- A. Aster, S. Wang, M. Mirmohades, C. Esmieu, G. Berggren, L. Hammarström and R. Lomoth, Chem. Sci., 2019, 10, 5582–5588 RSC.
- I. A. de Carcer, A. DiPasquale, A. L. Rheingold and D. M. Heinekey, Inorg. Chem., 2006, 45, 8000–8002 CrossRef.
- T. B. Liu, B. Li, M. L. Singleton, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2009, 131, 8296–8307 CrossRef CAS PubMed.
- F. Eckert, I. Leito, I. Kaljurand, A. Kutt, A. Klamt and M. Diedenhofen, J. Comput. Chem., 2009, 30, 799–810 CrossRef CAS.
- P. Wulff, C. C. Day, F. Sargent and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6606–6611 CrossRef CAS PubMed.
- L. Lauterbach and O. Lenz, J. Am. Chem. Soc., 2013, 135, 17897–17905 CrossRef CAS PubMed.
- N. Kaeffer, A. Morozan and V. Artero, J. Phys. Chem. B, 2015, 119, 13707–13713 CrossRef CAS.
- M. E. Ahmed, S. Dey, M. Y. Darensbourg and A. Dey, J. Am. Chem. Soc., 2018, 140, 12457–12468 CrossRef CAS.
- S. T. Stripp, G. Goldet, C. Brandmayr, O. Sanganas, K. A. Vincent, M. Haumann, F. A. Armstrong and T. Happe, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17331–17336 CrossRef CAS.
- K. D. Swanson, M. W. Ratzloff, D. W. Mulder, J. H. Artz, S. Ghose, A. Hoffman, S. White, O. A. Zadvornyy, J. B. Broderick, B. Bothner, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2015, 137, 1809–1816 CrossRef CAS.
- C. Orain, L. Saujet, C. Gauquelin, P. Soucaille, I. Meynial-Salles, C. Baffert, V. Fourmond, H. Bottin and C. Leger, J. Am. Chem. Soc., 2015, 137, 12580–12587 CrossRef CAS PubMed.
- A. Kubas, C. Orain, D. De Sancho, L. Saujet, M. Sensi, C. Gauquelin, I. Meynial-Salles, P. Soucaille, H. Bottin, C. Baffert, V. Fourmond, R. B. Best, J. Blumberger and C. Leger, Nat. Chem., 2017, 9, 88–95 CrossRef CAS.
- J. L. Stanley, T. B. Rauchfuss and S. R. Wilson, Organometallics, 2007, 26, 1907–1911 CrossRef CAS.
- D. Seyferth, G. B. Womack, M. K. Gallagher, M. Cowie, B. W. Hames, J. P. Fackler and A. M. Mazany, Organometallics, 1987, 6, 283–294 CrossRef CAS.
- R. Zaffaroni, T. B. Rauchfuss, D. L. Gray, L. De Gioia and G. Zampella, J. Am. Chem. Soc., 2012, 134, 19260–19269 CrossRef CAS.
- K. Shirono, T. Morimatsu and F. Takemura, J. Chem. Eng. Data, 2008, 53, 1867–1871 CrossRef CAS.
- T. Sato, Y. Hamada, M. Sumikawa, S. Araki and H. Yamamoto, Ind. Eng. Chem. Res., 2014, 53, 19331–19337 CrossRef CAS.
- M. Mirmohades, S. Pullen, M. Stein, S. Maji, S. Ott, L. Hammarstrom and R. Lomoth, J. Am. Chem. Soc., 2014, 136, 17366–17369 CrossRef CAS PubMed.
- C. P. Liu, T. B. Liu and M. B. Hall, J. Chem. Theory Comput., 2015, 11, 205–214 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and further spectroscopic and mechanistic studies. See DOI: 10.1039/c9dt04618f |
| This journal is © The Royal Society of Chemistry 2020 |