DOI:
10.1039/C9DT04174E
(Paper)
Dalton Trans., 2020,
49, 1181-1190
Organometallic nucleoside analogues: effect of the metallocene metal atom on cancer cell line toxicity†
Received
27th October 2019
, Accepted 9th December 2019
First published on 16th December 2019
Abstract
A new chiral organometallic nucleoside analogue containing ruthenocene is reported, in which alkylthymine and alkylhydroxyl groups are attached in adjacent positions on one cyclopentadienyl ring. The synthetic procedures for this metallocene derivative and two control compounds are described, along with their characterisation by cyclic voltammetry and X-ray crystallography. Their biological activities in a human pancreatic cancer cell line (MIA-Pa-Ca-2) were significantly lower than those of three previously reported analogous ferrocene compounds, indicating that the choice of metallocene metal atom (Fe or Ru) plays a pivotal role in determining the anticancer properties of these nucleoside analogues, which in turn suggests a different mode of action from that of a conventional nucleoside analogue.
Introduction
A topical area within the field of metal-based anticancer drug research involves examining the effect of incorporating organometallic moieties into known organic drugs and related biological molecules.1,2 Ferrocene is a popular choice in this respect as a so-called bioisosteric group2 because of its stability and well-understood reactivity and electrochemistry. Its incorporation into the breast cancer drug tamoxifen to form the ferrocifen family of compounds3,4 has revealed potent activities in different cell lines compared to the parent compound. This suggests novel modes of action related to the redox properties of the ferrocene unit, which may help combat drug resistance in the clinic.
Nucleoside and nucleobase analogues are an important class of chemotherapeutic agents, with 5-fluorouracil (5-FU)5 and gemcitabine (Gem)6 being two examples of leading drugs on the market. This presents a similar opportunity to decorate and derivatise the components of DNA/RNA with organometallic groups to give a range of new biologically active and medicinally relevant compounds.7,8 As part of our work in this area, we previously reported the thymidine analogue 1-(S,Rp)-Fe in which the five-membered Cp ring of ferrocene replaced the five-membered sugar ring of the nucleoside (Fig. 1).9 This compound demonstrated excellent anticancer activities in a range of human cancer cell lines, with both the hydroxyl linker and the nucleobase moiety required for optimal cytotoxicity. A subsequent structure–activity relationship (SAR) study found a correlation between the IC50 values in cancer cells and the length of the hydroxyalkyl linker in these so-called ferronucleosides.10 In continuation of this line of enquiry, we next decided to consider the role played by the metal atom in the lead compound 1-(S,Rp)-Fe, the subject of this report. In metallocene-based drug discovery, changing the metal from iron to ruthenium is a worthwhile endeavour, given the stability of ruthenocene and its amenability to functionalisation. Furthermore, and of particular relevance from an SAR point of view, ruthenocenes have different redox properties to ferrocenes, having more positive oxidation potentials and less reversible electrochemistry.11 It follows that any difference in biological activity between the two metallocenes could indicate the role of redox processes in the mode of action. Indeed, work on ruthenocifen derivatives has indicated different anticancer activities to the ferrocifens, with their biological behaviour being more similar to the parent organic compound tamoxifen.3,12 However, despite these findings, other reports on the biological activities of ruthenocene compounds13 or organoruthenium nucleobase derivatives8a are relatively rare. Here we report the synthesis and anticancer properties of 1-(S,Rp)-Ru, the direct ruthenocene analogue of 1-(S,Rp)-Fe, and two related control compounds (Fig. 2). Our findings do indeed suggest an important role of the metal atom in controlling the anticancer activities of these metallocene-containing nucleoside analogues.
 |
| | Fig. 1 Structures of the nucleoside thymidine (left), a generic nucleoside analogue (middle), and a ferronucleoside drug candidate 1-(S,Rp)-Fe (right). | |
 |
| | Fig. 2 Structures of the main target compound 1-(S,Rp)-Ru (middle) and the two control compounds 2-(S) and 3 (left and right) respectively. | |
Results and discussion
Synthesis
We considered that the synthesis of 1-(S,Rp)-Ru and the two control compounds 2-(S) and 3 would allow a direct comparison with the three analogous ferrocene compounds previously reported9 and also enable similar synthetic routes to be followed. In the case of the main target 1-(S,Rp)-Ru, this meant building the compound up from the known acetoxy derivative 4-(R)14 (Scheme 1), which itself was prepared via a two-step route from acetylruthenocene (see ESI†). This was then treated with NHMe2 to give the ruthenocene version of Ugi's amine 5-(R),14 whose chiral purity was found to be greater than 98%, as evidenced by chiral HPLC (see ESI†). Its X-ray structure was determined for the first time from crystals grown from a solution of the racemate in DCM layered with hexane (see ESI†).‡ The next step was to introduce planar chirality through the diastereoselective synthesis of 6-(R,Sp)via treatment with n-BuLi in diethylether and then quenching with iodine in THF. This compound was then converted to the acetoxy derivative 7-(R,Sp) by heating at 50 °C for two hours in acetic anhydride. A short reaction time and a relatively low temperature were used to avoid the elimination of the amine group to give the alkene. The arm was then extended to three carbon atoms by reacting with freshly prepared 1-ethoxyvinyloxy trimethylsilane to give the ethyl ester product 8-(S,Sp). The ester was reduced to the corresponding alcohol 9-(S,Sp) using the mild reducing agent DIBAL-H, before being protected with the TBDPS group to give compound 10-(S,Sp). This compound was then formylated in dry ether in two steps in situ by reacting with n-BuLi in a lithium–halogen exchange reaction followed by addition of DMF to give compound 11-(S,Sp). A Wittig reaction of the aldehyde added another carbon atom to give the alkene 12-(S,Rp), which was then converted to the primary alcohol 13-(S,Rp) by hydroboration–oxidation with BH3·THF. Finally, a Mitsunobu coupling reaction of the alcohol 13-(S,Rp) with benzoyl-protected thymine gave the fully protected product, which was treated first with TBAF to remove the silyl group and then with ammonia in methanol to remove the benzoyl group, giving the target compound 1-(S,Rp)-Ru. HPLC analysis of this compound confirmed its formation in high chiral purity (>98%, see ESI†). Crystals suitable for X-ray crystallography were grown from an acetonitrile solution of the racemate at 0 °C (Fig. 3).‡ An internal O–H⋯O H bond is formed between the hydroxyl hydrogen atom and one carbonyl oxygen atom in the thymine base. Intramolecular H-bonding has previously been observed within other bioorganometallic compounds.8d,25
 |
| | Scheme 1 Synthesis route for the target compound 1-(S,Rp)-Ru from synthon 4-(R). | |
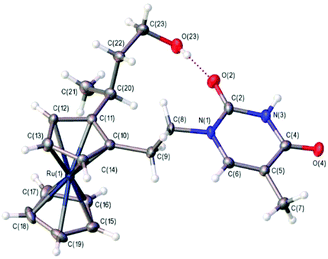 |
| | Fig. 3 Crystal structure of one of the two crystallographically-independent molecules of 1-(S,Rp)-Ru with ellipsoids drawn at the 50% probability level. The structure also contains two independent molecules of acetonitrile, which have been omitted for clarity. Intramolecular hydrogen bonding is shown using a dotted line. | |
The synthesis of control compounds 2-(S) and 3 also started from compound 4-(R) and proceeded through the routes depicted in Scheme 2. The chiral alcohol was obtained via a linker extension reaction using 1-ethoxyvinyloxy trimethylsilane, followed by reduction of the ester 14-(S) with LiAlH4. Achiral 3 was obtained in four steps, first involving elimination of the acetoxy group to give the vinyl ruthenocene 15. Crystals of this compound suitable for X-ray diffraction were successfully grown from a solution of DCM layered with hexane (see ESI†). A hydroboration–oxidation reaction then yielded the anti-Markovnikov product 16 containing the desired hydroxyethyl linker. The X-ray structure of this compound was also obtained from crystals grown using the same conditions (see ESI†). The route was completed using the same methodology described earlier via a Mitsunobu coupling reaction with the protected thymine base to give the protected product 17, which was then deprotected with ammonia to give the target compound 3. Crystals of the latter were grown by slow evaporation from a solution of ethyl acetate layered with hexane. The resulting X-ray structure, showing the correct bond connectivity, is depicted in Fig. 4.
 |
| | Scheme 2 Synthesis routes for control compounds 2-(S) and 3 from synthon 4-(R). | |
 |
| | Fig. 4 Crystal structure of 3 with ellipsoids drawn at the 50% probability level. | |
Electrochemistry
The electrochemistry of ruthenocene is more complicated than that of ferrocene. The 17-electron ruthenocenium cation is considerably more unstable and reactive than its ferrocene counterpart with Ru(VI) products formed from both the chemical15,16 and electrochemical11,16,17 oxidation of ruthenocene. The appearance of the cyclic voltammograms (CVs) of ruthenocene and those of its derivatives is highly dependent on the type of solvent and electrolyte.18–20 In non-coordinating electrolyte systems and in the presence of non-coordinating boron-containing electrolytes that do not form ion pairs, the cation has been reported to form two different dimers in a temperature-dependent ratio.20,21 In coordinating solvents and in the presence of more conventional electrolytes,18,19,22 the behaviour is different again. For example, a two-electron oxidation has been reported in acetonitrile using [NBu4][PF6] as a supporting electrolyte.18,23 with the cation binding to acetonitrile to form [MeCN-RuCp2]+, which then undergoes further oxidation to give [MeCN-RuCp2]2+ followed by a double reduction back to ruthenocene. Given these previous findings, it was decided to conduct CV experiments on the three target compounds 1-(S,Rp)-Ru, 2-(S) and 3 in acetonitrile in the presence of [NBu4][PF6] and compare them with 1-(S,Rp)-Fe under the same conditions. The experiments were performed in the presence of decamethylferrocene (dmfc) as an internal reference, as reported previously.10
All three ruthenocene compounds showed a similar EC (electrochemical-chemical) oxidation process at a positive potential value, with no return wave observed under the conditions used. Voltammograms of 1-(S,Rp)-Ru at different scan rates are displayed in Fig. 5, with those of 2-(S) and 3 presented in the ESI.† The Epa data for all three compounds are presented in Table 1, along with the corresponding value for the ferrocene analogue 1-(S,Rp)-Fe, which is considerably more negative (Epa = 455 mV, E1/2 = 424 mV (ref. 10)).
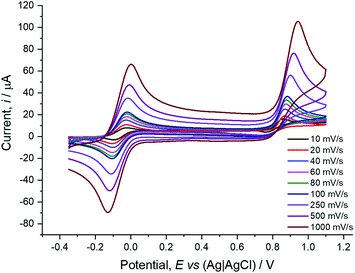 |
| | Fig. 5 Cyclic voltammograms of 1-(S,Rp)-Ru (1.0 mM) at various scan rates in dry acetonitrile with an electrolyte of [NBu4][PF6] (0.1 M) and dmfc (1.0 mM) as internal reference. | |
Table 1 Peak potential (Epa in mV) and IC50 values (in μM) in MIA-Pa-Ca-2 cancer cells of ferrocene compound 1-(S,Rp)-Fe and ruthenium compounds 1-(S,Rp)-Ru, 2(S) and 3
| Compound |
1-(S,Rp)-Fe
|
1-(S,Rp)-Ru
|
2-(S)
|
3
|
|
Mean values at scan rates 10–100 mV s−1versus the E1/2 of decamethylferrocene, at 1 mM concentration in dry acetonitrile containing 0.1 M TBAPF6, confidence limit = ±5 mV.
The mean from three independent biological experiments (n = 3), calculated using a variable slope 4 parameter model in Prism V8. Values in parentheses are the 95% CI. IC50 of cisplatin = 8.3 (5.1–17.0) μM.
|
|
E
pa
|
455 |
932 |
948 |
968 |
| IC50b |
9.3 (7.3–12.1) |
46.0 (37.7–56.6) |
>80 |
>80 |
This difference in value reflects the large difference in the relative stabilities between the oxidized and reduced forms of the two metallocenes, with the ferrocene derivative clearly being thermodynamically much easier to oxidise than its ruthenocene counterpart. The ferrocene derivative also shows reversible electrochemistry. The more positive value of Epa for compounds 2-(S) and 3 compared with 1-(S,Rp)-Ru can be explained by a greater inductive effect (+I) as the number of electron donating groups on the Cp ring increases, giving more stability to the charged ruthenocenium ion. The same trend is observed for the analogous ferrocene control compounds of 2-(S) and 3, which have Epa values of 497 mV and 540 mV respectively vs. dmfc (see ESI†).
Biological studies
The three ruthenocene compounds were next tested for cytotoxic activity in the pancreatic ductal adenocarcinoma cell line MIA-Pa-Ca-2 and compared with the ferrocene counterpart 1-(S,Rp)-Fe as well as with cisplatin. Assays were performed after 4 days incubation time using crystal violet staining. Cell viabilities, expressed as a percentage of a negative control, were plotted against concentration (μM) as shown in Fig. 6, with the resulting IC50 values presented in Table 1. As found previously for other cancer cell lines,9 the IC50 value for the ferrocene derivative 1-(S,Rp)-Fe is in the low micromolar range, with a value similar to that of cisplatin. However, the five-fold reduction in the toxicity of the ruthenocene analogue clearly shows that the identity of the metal ion has a significant impact on cytotoxicity.§ It is worth noting that the control compounds 2-(S) and 3 were even less toxic than 1-(S,Rp)-Ru, with IC50 values of >80 μM. This agrees with our previous findings on analogous and related ferrocene compounds,9,10 in that those metallocenes that are more electron rich, for example by having two groups attached to one cyclopentadienyl ring, are more cytotoxic. Indeed the previously published ferrocene analogues of 2-(S) and 3, which display more positive Epa values, are less toxic than 1-(S,Rp)-Fe.9 Overall the trend in the biological data supports the hypothesis that there is a significant relationship between the redox properties of the metallocene units in this series and cancer cell line toxicity.
 |
| | Fig. 6 Cytotoxicities of 1-(S,Rp)-Ru (green diamonds), 1-(S,Rp)-Fe (red triangles) and cisplatin (blue circles) towards MIA-Pa-Ca-2 pancreatic cancer cells determined from the crystal violet assay. Data are expressed as a percentage of the untreated control. Cells were treated for 4 days with each compound and the results represent the mean of three experiments ± SD (n = 3). Data were fitted to a variable slope 4 parameter model in Prism V8, R2 = 0.91 (cisplatin), 0.95 (1-(S,Rp)-Fe), 0.91 (1-(S,Rp)-Ru) respectively. | |
Conclusion
A ruthenocene-containing nucleoside analogue and two control compounds have been synthesised and fully characterized by a combination of spectroscopic, X-ray crystallography and electrochemical measurements. Their oxidation potentials were affected by the type and number of linker groups attached to the ruthenocene unit. All three compounds showed very low biological activities towards MIA-Pa-Ca-2 pancreatic ductal adenoma carcinoma cells, with IC50 values for the two mono-functionalised controls higher than that for the bis-functionalised target compound. The main finding of this study is the five-fold difference in cytotoxicity between 1-(S,Rp)-Ru and its ferrocene counterpart 1-(S,Rp)-Fe. Given their otherwise identical chemical structures and stereochemistries, this difference can confidently be attributed to the change in the metal atom from iron to ruthenium. While such a change would make little difference to a metallocene's size or lipophilicity, it clearly does affect its redox properties. The ferrocenes in this series demonstrate more reversible electrochemistry than their ruthenocene counterparts, with their oxidised forms accessible at significantly lower potentials. These differences in electrochemical behaviour signify an important role of the iron atom in determining the anticancer activity of the lead compound 1-(S,Rp)-Fe. This in turn suggests a mode of action different from that of a conventional nucleoside analogue, one that points more towards intra-cellular redox-triggered and ROS-mediated pathways leading to cell death. This line of enquiry is currently under investigation in our laboratory.
Experimental
Synthesis
(R)-1-α-N,N-Dimethylaminoethylruthenocene, 5-(R).
1-α-Acetoxyethylruthenocene 4-(R) (see ESI†) (0.1 g, 3.15 mmol) was dissolved in MeOH (3 mL). Dimethylamine (40% in H2O) was added and the resulting solution stirred for 24 h. The reaction was quenched with water, extracted with DCM, washed with water and then brine, and dried over MgSO4. The solvent was removed in vacuo and the crude product purified by flash column chromatography on silica gel using an eluent of 15% ether in hexane and 3% Et3N. The solvent was removed in vacuo to give the title compound as a pale-yellow solid (0.0263 g, 38%). δ H (400 MHz, CDCl3) 4.63–4.57 (m, 2H), 4.57–4.54 (m, 1H), 4.53 (s, 6H), 3.62–3.54 (m, 1H), 2.31 (s, 6H), 1.41 (d, J = 6.9 Hz, 3H). δ C (101 MHz, CDCl3) 72.4 (Cp), 71.1 (Cp), 70.2 (Cp), 70.1 (Cp), 69.5 (Cp), 59.6 (CH), 40.4 (CH3), 17.5 (α-CH3). HRMS (ES) (m/z) calcd for C14H20N102Ru 304.0639, found 304.0636. Vmax/cm−1 3200 (C–H), 2923 (CH2), 2851 (CH2), 2816 (CH2), 1453 (CH3), 1370 (CH3), 1100 (C–N), 806 (C–C), 720 (CH–Ar).
(R,Sp)-1-(α-N,N-Dimethylaminoethyl)-2-iodoruthenocene, 6-(R,Sp).
The Ugi amine 5-(R) (0.1 g, 0.33 mmol) was dissolved in Et2O (5 mL) at room temperature. n-BuLi (0.3 mL, 1.7 M, 2 eq.) was added and the mixture stirred overnight. The mixture was cooled to −78 °C, and iodine (0.23 g, 0.91 mmol), in THF (10 mL), was added over 10 min. The mixture was stirred at −78 °C for 90 min before being warmed to room temperature and then stirred for an additional 90 min. The reaction was then quenched at 0 °C with sodium thiosulfate (10 mL, 25% w/v). After dilution with Et2O (15 mL), the layers were separated, and the aqueous layer was washed further with Et2O (3 × 5 mL). The combined organic fractions were dried over MgSO4. The solvent was removed in vacuo before purification via flash column chromatography (10% Et2O and 4% TEA in hexane) to obtain the final oily product (0.07 g, 49%). δ H (400 MHz, CDCl3) 4.84 (dd, J = 2.2, 1.1 Hz, 1H), 4.57–4.52 (m, 2H), 4.51 (s, 5H), 3.43 (q, J = 6.8 Hz, 1H), 2.24 (s, 6H), 1.35 (d, J = 6.8 Hz, 3H). δ C (101 MHz, CDCl3) 94.9 (Cp), 77.1 (Cp), 73.7 (Cp), 71.0 (Cp), 68.7 (Cp), 57.7 (CH), 41.6 (CH3), 29.7 (ipso Cp), 18.7 (CH3). HRMS (ES) (m/z) calcd for C14H19N102Ru 429.9606, found 429.9604. Vmax/cm−1 3098 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H), 2963 (CH2), 2923 (CH2), 2851 (CH2), 1100 (C–H), 1453 (CH3), 1370 (CH3), 1261, 1156, 918 (CHCH), 806 (CH Ar). [α]20D = +27(±2) (c = 0.25 in CHCl3).
C–H), 2963 (CH2), 2923 (CH2), 2851 (CH2), 1100 (C–H), 1453 (CH3), 1370 (CH3), 1261, 1156, 918 (CHCH), 806 (CH Ar). [α]20D = +27(±2) (c = 0.25 in CHCl3).
(R,Sp)-1-(α-Acetoxyethyl)-2-iodoruthenocene, 7-(R,Sp).
Compound 6-(R,Sp) (1 g, 2.3 mmol) and acetic anhydride (8.3 mL, 81 mmol) were heated at 50 °C for 2 h. Acetic anhydride was removed under high vacuum (0.1 mmHg) and the residue purified by flash column chromatography (10% Et2O and 4%TEA in hexane) to obtain a light yellow oily product (0.9 g, 86%). δ H (400 MHz, CDCl3) 5.67 (q, J = 6.5 Hz, 1H), 4.87 (dd, J = 2.3, 1.1 Hz, 1H), 4.68 (d, J = 2.5 Hz, 1H), 4.55 (t, J = 2.4 Hz, 1H), 4.52 (s, 5H), 2.02 (s, 3H), 1.51 (d, J = 6.5 Hz, 3H). δ C (101 MHz, CDCl3) 170.3 (CO), 91.9 (ipso Cp), 78.2 (Cp), 73.8 (Cp), 71.7 (Cp), 69.4 (CH), 68.9 (Cp), 39.9 (ipso Cp), 21.2 (CH3), 19.4 (CH3). HRMS (ES) (m/z) calcd for C14H15O2Na102Ru127I 466.9058 found 466.9054. Vmax/cm−1 3098 (C–H), 2979 (CH2), 2929 (CH2), 2818 (CH2), 1727 (CO), 1449 (CH3), 1367 (CH3), 1229 (C–O), 1044, 1018, 806 (CHCH), 752 (CH–Ar). [α]20D = +25(±2) (c = 0.25 in CHCl3).
(S,Sp)-1-[α-Methyl(2-ethylpropanoate)]-2-iodoruthenocene, 8-(S,Sp).
Compound 7-(R,Sp) (1 g, 2.25 mmol) and 1-ethoxyvinyloxy trimethylsilane (0.9 g, 5.63 mmol) were dissolved in DCM (20 mL). The mixture was cooled to −78 °C, and BF3·OEt2 (0.3 mL, 2.7 mmol) was then added dropwise. The mixture was stirred for 15 min at −78 °C before being warmed to room temperature and quenched with saturated NaHCO3 (20 mL). The organic layer was separated, and the aqueous layer was washed further with DCM (20 mL). The combined organic fractions were dried over MgSO4. The solvent was removed in vacuo and the residue purified via flash column chromatography (10% Et2O in hexane) to obtain a yellow oily product (0.7 g, 66%). δ H (300 MHz, CDCl3) 4.80 (dd, J = 2.2, 1.2 Hz, 1H), 4.52 (s, 5H), 4.47–4.40 (m, 2H), 4.12 (q, J = 7.1 Hz, 2H), 2.97–2.81 (m, 1H), 2.60 (dd, J = 14.8, 4.2 Hz, 1H), 2.25 (dd, J = 14.8, 10.0 Hz, 1H), 1.29–120 (m, 6H, 2 × Me). δ C (101 MHz, CDCl3) 172.0 (CO), 98.6 (ipso Cp), 76.9 (Cp), 73.4 (Cp), 70.8 (Cp), 68.0 (Cp), 60.3 (CH2), 43.0 (CH2), 40.3 (ipso Cp), 30.6 (CH), 20.6 (CH3), 14.4 (CH3). HRMS (ES) (m/z) calcd for C16H19O2Na102Ru127I 494.9371, found 494.9373. Vmax/cm−1 3097 (C–H), 2973 (CH2), 2929 (CH2), 2823 (CH2), 1728 (CO), 1459 (CH3), 1369 (CH3), 1173, 1030 (C–O), 1100, 998 (CHCH), 805 (CH–Ar). [α]20D = −6(±2) (c = 0.25 in CHCl3).
(S,Sp)-1-[α-Methyl-(3-(hydroxyl)propyl)]-2-iodoruthenocene, 9-(S,Sp).
Compound 8-(S,Sp) (0.1586 g, 0.3365 mmol) was dissolved in Et2O (20 mL), and the solution was cooled to 0 °C. After stirring for 10 min, DIBAL-H (0.92 mL, 1 mmol) was slowly added at 0 °C. The mixture was stirred for 1 h at 0 °C before the reaction was quenched with saturated sodium potassium tartrate in water (10 mL). The aqueous layer was washed further with Et2O (15 mL). The combined organic fractions were dried over Na2SO4, the solvent was removed in vacuo, and the residue was purified via flash column chromatography (20% Et2O in hexane) to obtain a colorless solid product (0.119 g, 82%). δ H (400 MHz, CDCl3) 4.78 (dd, J = 2.2, 1.1 Hz, 1H), 4.50 (s, 5H), 4.44 (t, J = 2.3 Hz, 1H), 4.38 (dd, J = 2.4, 0.9 Hz, 1H), 3.64–3.56 (m, 2H), 2.55–2.45 (m, 1H), 2.03 (s, 1H, br s), 1.78–1.58 (m, 2H), 1.20 (d, J = 7.0 Hz, 3H). δ C (101 MHz, CDCl3) 100.5 (ipso Cp), 76.5 (Cp), 73.4 (Cp), 70.7 (Cp), 67.6 (Cp), 61.0 (CH2), 41.8 (CH2), 41.1 (ipso Cp), 29.8 (CH), 21.8 (CH3). HRMS (ES) (m/z) calcd for C14H17O23Na 102Ru127I 452.9265, found 452.9269. Vmax/cm−1 3287 br (OH), 3094 (C–H Fc), 2955 (CH2), 2924 (CH2), 2852 (CH2), 1524, 1458 (CH2), 1374 (CH3), 1054, 997 (C–O), 805 (CC). [α]20D = +10(±3) (c = 0.2 in CHCl3). m.p.: 98–100 °C.
(S,Sp)-1-[α-Methyl-(3-(tert-butyldiphenylsilyloxy)propyl)]-2-iodoferrocene, 10-(S,Sp).
Compound 9-(S,Sp) (0.2 g, 0.47 mmol) was dissolved in DCM (15 mL) at room temperature. TEA (0.098 mL, 0.699 mmol), tert-butyldiphenylsilyl chloride (0.18 mL, 0.699 mmol), and DMAP (catalytic amounts) were then added to the mixed solution. The reaction was then stirred overnight at room temperature before quenching with water (5 mL). The organic layer was separated, and the aqueous layer was washed with Et2O (2 × 10 mL). The combined organic fractions were dried over Na2SO4, the solvent was removed in vacuo, and the residue was purified via flash column chromatography (10% Et2O in hexane) to obtain a yellow oily product (0.3 g, 96%). δ H (400 MHz, CDCl3) 7.76–7.72 (m, 4H), 7.47–7.40 (m, 6H), 4.83 (dd, J = 2.2, 1.1 Hz, 1H), 4.56 (s, 5H), 4.48 (t, J = 2.3 Hz, 1H), 4.42–4.40 (m, 1H), 3.82–3.71 (m, 2H), 2.66–2.56 (m, 1H), 2.01–1.92 (m, 1H), 1.64–1.55 (m, 1H), 1.20 (d, J = 6.9 Hz, 3H), 1.12 (s, 9H). δ C (101 MHz, CDCl3) 135.7 (Ar), 134.1 (ipso Ar), 129.6 (Ar), 127.7 (Ar), 100.9 (ipso Cp), 76.7 (Cp), 73.3 (Cp), 70.6 (Cp), 67.7 (Cp), 62.4 (Cp), 52.3 (CH2), 41.5 (CH2), 40.8 (ipso Cp), 30.0 (CH), 27.0 (CH3), 20.9 (CH3), 19.3 (ipso t-Bu). HRMS (ES) (m/z) calcd for C30H35O23Na127I28Si102Ru 691.0446, found 691.0443. Vmax/cm−1 3070 (CH Fc), 2956 (CH2), 2928 (CH2), 2855(CH2), 1472 (CH2), 1387 (CH3), 1307, 1187, 1108, 821 (CH–Ar TBDPS), 701 (CC). [α]20D = +15(±2) (c = 0.25 in CHCl3).
(S,Sp)-1-[α-Methyl-(3-(tert-butyldiphenylsilyloxy)propyl)]-2-formylruthenocene, 11-(S,Sp).
Compound 10-(S,Sp) (0.4 g, 1.001 mmol) was dissolved in extra dry Et2O (30 mL) and de-oxygenated with argon for 15 min. The mixture was cooled to −78 °C, and fresh n-BuLi (0.2 mL, 2.0 mmol) was added dropwise. After 30 min, DMF (0.08 mL, 3.0 mmol) was added, and the mixture was stirred at −78 °C for another 30 min. The reaction was then warmed to room temperature and quenched with water (10 mL). The organic phase was separated, and the aqueous layer was washed with Et2O (2 × 10 mL). The combined organic fractions were dried over Na2SO4. The solvent was removed in vacuo and the residue purified via flash column chromatography (30% Et2O in hexane) to obtain a yellow oily product (0.3 g, 88%). δ H (400 MHz, CDCl3) 9.91 (s, 1H), 7.67–7.62 (m, 4H), 7.42–7.36 (m, 6H), 5.03 (dd, J = 2.2, 1.4 Hz, 1H), 4.74–4.71 (m, 2H), 4.61 (s, 5H), 3.65 (dt, J = 6.6, 1.4 Hz, 2H), 2.95 (q, J = 6.9 Hz, 1H), 1.79–1.61 (m, 2H), 1.15 (d, J = 6.9 Hz, 3H), 1.04 (s, 9H). δ C (101 MHz, CDCl3) 190.1 (CO), 135.57 (Ar), 1334.0 (ipso Ar), 133.8 (ipso Ar), 129.6 (Ar), 127.6 (Ar), 101.8 (ipso Cp), 82.4 (ipso Cp), 72.7 (Cp), 72.4 (Cp), 72.4 (Cp), 70.6 (Cp), 62.0 (CH2), 42.9 (CH2), 27.5 (CH), 26.9 (CH3), 20.9 (CH3), 19.2 (ipso t-Bu). HRMS (ES) (m/z) calcd for C31H36O2Na102Ru28Si 593.1432, found 593.1426. Vmax/cm−1 3096 (C–H Fc), 2958, 2931, 2856 (CH2), 1678 (CO), 1427 (CH2), 1388 (CH3), 1109, 998 (C–O), 820, 739 (CH Ar), 703 (CC). [α]20D = +7(±2) (c = 0.2 in CHCl3).
(S,Rp)-1-[α-Methyl-(3-(tert-butyldiphenylsilyloxy)propyl)]-2-vinylruthenocene, 12-(S,Rp).
Triphenylmethylphosphonium bromide (0.268 g, 0.708 mmol), potassium tert-butoxide (0.08 g, 0.78 mmol), and dibenzo-18-crown-6-ether (catalytic amount) were dissolved in dry THF (20 mL) under argon. The mixture was stirred for 30 min and then 11-(S,Sp) (0.269 g, 0.472 mmol) in dry THF (5 mL) was added to the mixture. The mixture was stirred overnight at room temperature, before quenching with water (5 mL) and extracting with Et2O (2 × 10 mL). The combined organic fractions were dried over Na2SO4, the solvent was removed in vacuo, and the residue was purified via flash column chromatography using hexane to obtain a yellow oily product (0.215 g, 80%). δ H (400 MHz, CDCl3) 7.66–7.62 (m, 4H), 7.42–7.34 (m, 6H), 6.48 (dd, J = 17.4, 10.8 Hz, 1H), 5.28 (dd, J = 17.4, 1.7 Hz, 1H), 4.89 (dd, J = 10.8, 1.7 Hz, 1H), 4.81 (t, J = 1.7 Hz, 1H), 4.45 (s, 5H), 4.43 (d, J = 3.9 Hz, 1H), 3.65–3.60 (m, 2H), 2.70–2.63 (m, 1H), 1.79–1.70 (m, 1H), 1.58–1.54 (m, 1H), 1.12 (d, J = 6.9 Hz, 3H), 1.04 (s, 9H, Me). δ C (101 MHz, CDCl3) 135.6 (Ar), 134.1 (ipso Ar), 132.7 (CH), 129.5 (Ar), 127.6 (Ar), 111.2 (CH2), 98.7 (ipso Cp), 86.7 (ipso Cp), 71.4 (Cp), 70.7 (Cp), 68.9 (Cp), 68.5 (Cp), 66.9 (Cp), 62.2 (CH2), 42.5 (CH2), 27.5 (CH), 26.9 (CH3), 20.6 (CH3), 19.2 (ipso t-Bu). HRMS (ES) (m/z) calcd for C32H39O102RuSi 569.1829, found 569.1814. Vmax/cm−1 3070 (C–H Fc), 2958 (CH2) 2928 (CH2), 2856 (CH2), 1627 (CC Ar), 1471 (CH2), 1388 (CH3), 1108, 1086 (C–O), 821, 806 (C–H Ar), 700 (vinyl/CC). [α]20D = +52(±3) (c = 0.2 in CHCl3).
(S,Rp)-1-[α-Methyl-(3-(tert-butyldiphenylsilyloxy)propyl)]-[2-(hydroxyl)ethyl] ruthenocene, 13-(S,Rp).
Compound 12-(S,Rp) (0.215 g, 0.379 mmol) was dissolved in dry THF (15 mL). BH3·THF (1 M, 1.1 mL, 1.084 mmol) was then added dropwise at room temperature and the mixture was stirred for 2 h. EtOH (1.4 mL), NaOH solution (3 M, 1.4 mL, 3.79 mol), and H2O2 (30 wt% in water, 0.73 mL, 8.338 mmol) were then added successively, and the mixture was stirred for 1 h at room temperature. The reaction mixture was added to DCM (20 mL), washed with brine (10 mL), and then dried over Na2SO4. The solvent was removed in vacuo and the residue purified via flash column chromatography (10% EtOAc in hexane) to obtain a yellow oily product (0.17 g, 77%). δ H (400 MHz, CDCl3) 7.68–7.62 (m, 4H), 7.43–7.34 (m, 6H), 4.52 (s, 5H), 4.51–4.50 (m, J = 1.0 Hz, 1H), 4.41 (dd, J = 2.2, 1.1 Hz, 1H), 4.37 (t, J = 2.3 Hz, 1H), 3.71–3.56 (m, 4H), 2.53–2.31 (m, 3H), 1.76–1.70 (m, 1H), 1.56–1.47 (m, 1H), 1.07 (d, J = 6.9 Hz, 3H), 1.05 (s, 9H). δ C (101 MHz, CDCl3) 135.6 (Cp), 134.0 (ipso Ar), 129.6 (CH Ar), 127.7 (Ar), 99.8 (ipso Cp), 87.4 (ipso Cp), 71.2 (Cp), 70.4 (Cp), 68.0 (Cp), 62.9 (CH2), 62.2 (CH2), 42.0 (CH2), 28.7 (CH2), 27.2 (CH), 26.9 (CH3) 21.4 (CH3), 19.2 (ipso t-Bu). HRMS (ES) (m/z) calcd for C32H40O2NaSi102Ru 609.1759, found 609.1739. Vmax/cm−1 3420 br (OH), 3071 (C–H Fc), 2955 (CH2), 2927 (CH2), 1589, 1470 (CH3), 1427 (CH2), 1388 (CH3), 1361, 1109, 997 (C–O), 822, 738 (C–H Ar), 702 (C–H). [α]20D = +45(±3) (c = 0.25 in CHCl3).
(S,Rp)-1-[α-Methyl-(3-(hydroxy)propyl)]-2-[(thyminyl)ethyl]-ruthenocene, 1-(S,Rp)-Ru.
Triphenylphosphine (160 mg, 0.6 mmol), N3-benzoylthymine (110 mg, 0.5 mmol), and 13-(S,Rp) (92 mg, 0.2 mmol) were dissolved in THF (10 mL) in a 100 mL Schlenk tube, and the mixture was stirred for 10 min at room temperature. The tube was then covered with foil, and DIAD (0.12 mL, 0.6 mmol) was added. The mixture was then heated to 65 °C for 2 h. The solvent was evaporated, and the resulting residue was dissolved in EtOAc (20 mL), washed with brine (10 mL) followed by water (5 mL), and dried over Na2SO4. The solvent was removed in vacuo and the residue purified via flash column chromatography (15% Et2O in hexane) to give the fully protected product. Deprotection was achieved by stirring the compound in TBAF (5 mL) for 2 h. The solvent was removed, and the residue was then redissolved in ammonia in methanol (7 N, 2 mL) and stirred at room temperature for 30 min. The solvent was evaporated, and the crude product was purified via flash column chromatography (5% MeOH in DCM) to obtain the product as a white solid (105 mg, 74%). δ H (400 MHz, CDCl3) 9.33 (s br, 1H), 7.04 (s, 1H), 4.50 (s, 5H), 4.45 (dd, J = 2.2, 1.2 Hz, 2H), 4.41 (t, J = 2.3 Hz, 1H), 3.86–3.77 (m, 1H), 3.74–3.62 (m, 3H), 2.68–2.54 (m, 3H), 2.40 (s br, 1H), 1.92 (s, 3H), 1.78–1.73 (m, 1H), 1.65–1.57 (m, 1H), 1.18 (d, J = 6.9 Hz, 3H). δ C (101 MHz, CDCl3) 164.2 (CO), 151.1 (CO), 140.4 (CH thymine), 111.0 (ipso thymine), 95.3 (ipso Cp), 80.7 (ipso Cp), 69.3 (Cp), 67.6 (Cp), 65.9 (Cp), 65.3 (Cp), 60.2 (CH2), 49.8 (CH2), 43.2 (CH2), 27.9 (CH2), 27.1 (CH), 19.2 (CH3 thymine), 12.3 (CH3). HRMS (ES) (m/z) calcd for C21H26N2O3NaFe 433.1191, found 433.1182. Vmax/cm−1 3462 br (OH), 3097 (C–H Fc), 2954 (CH2), 2926 (CH2), 1669 (CO), 1468, 1425 (CH3), 1425 (CH2), 1383 (CH3), 1353 (C–N) 1100, 1036, 997 (C–O), 805, 759 (C–H Ar), 690, 669 (C–H). [α]20D = +42(±3) (c = 0.1 in CH3CN).
(S)-1-[α-Methyl(2-ethylpropanoate)]ruthenocene, 14-(S).
1-Methoxyethylruthenocene 4-(R) (0.9 g, 2.84 mmol, 1.0 eq.) was dissolved in dry DCM and stirred at −78 °C for 5 min under an atmosphere of argon. 1-Ethoxyvinyloxy trimethylsilane (1 mL, 6.2 mmol, 2.2 eq.) was added and the mixture stirred for 5 min. BF3·Et2O (0.43 mL, 3.4 mmol, 1.2 eq.) was added dropwise and the solution stirred for 20 minutes at −78 °C. The solution was warmed to room temperature over 30 min and then stirred for another 30 min. The reaction was quenched with sat. NaHCO3, extracted with DCM and dried over MgSO4. The solvent was removed in vacuo and the crude product purified by flash column chromatography on silica gel using an eluent system of 15% Et2O in hexane. The solvent was removed in vacuo to give the title compound as a yellow oil (0.61 g, 63%). δ H (400 MHz, CDCl3) 4.52 (s, 5H), 4.48 (t, J = 1.7 Hz, 2H), 4.45–4.42 (m, 2H), 4.13 (q, J = 7.3 Hz, 2H), 2.86–2.75 (m, 1H), 2.52 (dd, J = 14.9, 5.4 Hz, 1H), 2.27 (dd, J = 14.9, 9.2 Hz, 1H), 1.25 (t, J = 7.2 Hz, 3H), 1.13 (d, J = 6.8 Hz, 3H). δ C (101 MHz, CDCl3) 172.76 (CO), 98.3 (ipso Cp), 70.5 (Cp), 70.1 (Cp), 69.5 (Cp), 68.9 (Cp), 60.3 (CH2), 43.7 (CH2), 29.6 (CH), 21.5 (CH3), 14.3 (CH3). HRMS (ES) (m/z) calcd for C16H19O2102Ru 346.0507, found 346.0508. Vmax/cm−1 3095 (C–H), 2964, 2929 (CH2), 1731 (CO), 1369 (CH3), 1030 (C–O), 817 (C–H). [α]20D = +10.5(±2) (c = 0.1 in CHCl3).
(S)-1-[α-Methyl-(3-(hydroxy)propyl)]ruthenocene, 2-(S).
Compound 14-(S) (0.48 g, 1.39 mmol, 1 eq.) was dissolved in dry THF and stirred at 0 °C for 5 min under an atmosphere of argon. LiAlH4 (0.11 g, 2.78 mmol, 2 eq.) was added slowly, and the solution stirred for two hours at RT. The reaction was quenched with a saturated solution of sodium potassium-tartrate (10 mL), extracted with diethylether and dried over MgSO4. The solvent was removed in vacuo and the crude product purified by flash column chromatography on silica gel using an eluent system of 15% Et2O in hexane. The solvent was removed in vacuo to give the title compound as a yellow oily product (0.35 g, 83%). δ H (400 MHz, CDCl3) 4.53 (s, 5H), 4.51–4.48 (m, 2H), 4.47–4.42 (m, 2H), 3.73–3.60 (m, 2H), 2.47–2.36 (m, 1H), 1.77–1.67 (m, 1H), 1.66–1.58 (m, 1H), 1.55 (s br, 1H), 1.12 (d, J = 6.9 Hz, 3H). δ C (101 MHz, CDCl3) 99.4 (ipso Cp), 70.5 (Cp), 70.1 (Cp), 69.4 (Cp), 69.2 (Cp), 68.6 (Cp), 61.3 (CH2), 41.4 (CH2), 29.3 (CH), 21.8 (CH3). HRMS (ES) (m/z) calcd for C14H18O102Ru 304.0401 found 304.0389. Vmax/cm−1 3328, 3092 (C–H), 2926 (CH2), 1372 (CH3), 1050, 1029 (C–O), 999, 815 (C–H). [α]20D = +13(±2) (c = 0.1 in CHCl3).
1-Vinylruthenocene, 15.
1-Methoxyethylruthenocene 4-(R) (0.23 g, 0.95 mmol, 1.0 eq.) and LiBr (0.8 g, 9.5 mmol, 10 eq.) were dissolved in DMF (7 mL) under an atmosphere of argon. The reaction was heated gradually to 80 °C and stirred for 30 min. The reaction was quenched with water (20 mL), extracted with DCM and dried over MgSO4. The solvent was removed in vacuo and the crude product purified by flash column chromatography on neutralized silica gel using 15% Et2O and 3% TEA in hexane. The solvent was removed in vacuo to give the title compound as a yellow solid (0.15 g, 61%). δ H (300 MHz, CDCl3) 6.33 (dd, J = 17.5, 10.7 Hz, 1H), 5.28 (dd, J = 17.5, 1.5 Hz, 1H), 4.89 (dd, J = 10.7, 1.5 Hz, 1H), 4.77 (t, J = 1.7 Hz, 1H), 4.56 (s, 5H), 4.51 (s, 3H). δ C (101 MHz, CDCl3) 133.8 (CH vinyl), 111.0 (CH2 vinyl), 87.6 (ipso Cp), 71.1 (Cp), 70.5 (Cp), 70.1 (Cp), 69.0 (Cp). HRMS (m/z) calcd for C12H12102Ru 257.9982, found 257.9979. Vmax/cm−1 3094 (C–H), 2958, 2915, 2848 (CH2), 1734, 1461, 1367 (CC), 805, 753 (C–H).
1-(β-Hydroxyethyl) ruthenocene, 16.
Compound 15 (0.23 g, 0.84 mmol, 1.0 eq.) was dissolved in dry THF (10 mL) under an atmosphere of argon. BH3·THF (1 M in THF) (1.3 mL, 1.25 mmol, 1.5 eq.) was added dropwise and the resulting solution was stirred for 2 h. EtOH (1.3 mL) was added dropwise followed by NaOH solution (3 M in H2O) (1.3 mL) and H2O2 (30 wt% in water, 1.3 mL) and the solution stirred for 2 h. The reaction was quenched with H2O, extracted with DCM and then dried over MgSO4. The solvent was removed in vacuo and the crude product purified by flash column chromatography on silica gel using an eluent of 20% EtOAc in hexane. The solvent was removed in vacuo to give the title compound as a yellow oil (0.15 g, 66%). δ H (400 MHz, CDCl3) 4.57–4.55 (m, 6H), 4.49 (t, J = 1.6 Hz,2H), 3.67 (t, J = 6.0 Hz, 2H), 2.66 (s br, 1H), 2.44 (t, J = 6.0 Hz, 3H). δ C (101 MHz, CDCl3) 88.5 (ipso-Cp), 71.2 (Cp), 70.8 (Cp), 69.8 (Cp), 62.9 (CH2), 30.8 (CH2). HRMS (m/z) calcd for C12H14ONa102Ru 298.9986, found 298.9988. Vmax/cm−1 3328, 3089 (C–H Fc), 2926, 2874 (CH2), 1037, 999 (C–O), 813 (C–H).
2-[(Thyminyl)ethyl]-ruthenocene, 3.
Triphenylphosphine (0.149 g, 0.567 mmol), N3-benzoylthymine (0.11 g, 0.49 mmol), and 16 (53 mg, 0.189 mmol) were dissolved in THF (10 mL) and the mixture was stirred for 10 min at room temperature. The flask was then covered with foil, and DIAD (0.11 mL, 0.57 mmol) was added. The mixture was heated to 65 °C for 2 h. The solvent was evaporated and the residue redissolved in EtOAc (15 mL), washed with brine (10 mL) and water (5 mL), and dried over Na2SO4. The solvent was removed in vacuo to obtain the protected product. Deprotection of the benzoyl group was achieved by treating the crude with ammonia solution (7 N in methanol, 5 mL) for 1 h. The solvent was then removed in vacuo and the residue purified via flash column chromatography (30% EtOAc in hexane) to give the product (43 mg, 59%). m.p.: 238–239 °C. δ H (400 MHz, CDCl3) 8.75 (s br, 1H), 6.84 (s, 1H), 4.54 (s, 5H), 4.47 (s, 4H), 3.76 (t, J = 7.4 Hz, 2H), 2.55 (t, J = 7.4 Hz, 2H), 1.88 (d, J = 1.1 Hz, 3H). δ C (101 MHz, CDCl3) 164.2 (CO), 150.6 (CO), 140.9 (CH–thymine), 110.2 (ipso thymine), 87.2 (ipso-Cp), 70.9 (Cp), 70.7 (Cp), 70.0 (Cp), 50.8 (CH2), 28.8 (CH2), 12.2 (CH3). HRMS (m/z) calcd for C17H18N2O2102Ru23Na 407.0309, found 407.0311. Vmax/cm−1 3210 (NH), 3089 (C–H Fc), 2954 (CH2), 2921 (CH2), 2850 (CH2), 1685, 1672 (CO), 1462, 1385 (CH3), 1353 (C–N), 803, 759 (CC).
Electrochemistry
Cyclic voltammetry measurements were performed using 1.0 mM solutions in dry and de-oxygenated MeCN containing tetrabutylammonium hexafluorophosphate (TBAPF6) electrolyte at a concentration of 0.1 M and 1.0 mM dfmc as internal reference. Data were measured with a BioAnalytical Systems Inc. (West Lafayette, IN) EC Epsilon potentiostat and with a C3 cell stand with a three-electrode unit. The cyclic voltammograms were recorded under argon at room temperature using a three-electrode cell. The electrodes were obtained from IJ Cambria (Llanelli, Wales). A platinum wire was used as a counter electrode (CE), a glassy carbon electrode with 3 mm diameter was used as a working electrode (WE) and an Ag|AgCl|3 M KCl electrode was used as a reference electrode (RE) and connected to the cell via a frit. Cleaning of all glassware was achieved by soaking overnight in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ammonia (35%) and hydrogen peroxide (30%), followed by multiple rinsing with ultrapure water (from a Millipore tandem Elix-A10 system, resistivity >18 MΩ cm, TOC < 5 ppb). The glassware was then left overnight in ultrapure water, then rinsed again and dried in an oven prior to use. The electrodes were cleaned as follows before their use: the RE was cleaned with dry acetonitrile and the CE was flame annealed. The WE was cleaned by polishing with aqueous slurries of successively finer grades of alumina (1.0 μm, 0.3 μm and 0.05 μm) and then rinsed with ultrapure water and MeCN, dried with a flow of argon, and then kept in an analyte solution.
:1 ammonia (35%) and hydrogen peroxide (30%), followed by multiple rinsing with ultrapure water (from a Millipore tandem Elix-A10 system, resistivity >18 MΩ cm, TOC < 5 ppb). The glassware was then left overnight in ultrapure water, then rinsed again and dried in an oven prior to use. The electrodes were cleaned as follows before their use: the RE was cleaned with dry acetonitrile and the CE was flame annealed. The WE was cleaned by polishing with aqueous slurries of successively finer grades of alumina (1.0 μm, 0.3 μm and 0.05 μm) and then rinsed with ultrapure water and MeCN, dried with a flow of argon, and then kept in an analyte solution.
X-ray crystallography
Crystal structure determination of 1-(S,Rp)-Ru.
C23H29N3O3Ru (M = 496.56 g mol−1): triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 12.1133(5) Å, b = 12.9420(7) Å, c = 15.0302(8) Å, α = 70.115(5)°, β = 84.171(4)°, γ = 84.068(4)°, V = 2198.4(2) Å3, Z = 4, T = 100.01(10) K, μ(CuKα) = 6.008 mm−1, Dcalc = 1.500 g cm−3, 15146 reflections measured (7.284° ≤ 2Θ ≤ 136.502°), 8029 unique reflections (Rint = 0.0314, Rsigma = 0.0440) which were used in all calculations. The final R1 was 0.0498 (I > 2σ(I)) and wR2 was 0.1326 (all data). The structure contains two crystallographically-independent molecules and also two independent molecules of acetonitrile. The hydrogen atoms bonded to N(3), O(23), N(103) and O(123) were located in the electron density map and their positions refined, with N(3)–H(3) subjected to a bond distance restraint. All remaining hydrogen atoms were fixed as riding models and the isotropic thermal parameters (Uiso) of all hydrogen atoms were based on the Ueq of the parent atom.
(no. 2), a = 12.1133(5) Å, b = 12.9420(7) Å, c = 15.0302(8) Å, α = 70.115(5)°, β = 84.171(4)°, γ = 84.068(4)°, V = 2198.4(2) Å3, Z = 4, T = 100.01(10) K, μ(CuKα) = 6.008 mm−1, Dcalc = 1.500 g cm−3, 15146 reflections measured (7.284° ≤ 2Θ ≤ 136.502°), 8029 unique reflections (Rint = 0.0314, Rsigma = 0.0440) which were used in all calculations. The final R1 was 0.0498 (I > 2σ(I)) and wR2 was 0.1326 (all data). The structure contains two crystallographically-independent molecules and also two independent molecules of acetonitrile. The hydrogen atoms bonded to N(3), O(23), N(103) and O(123) were located in the electron density map and their positions refined, with N(3)–H(3) subjected to a bond distance restraint. All remaining hydrogen atoms were fixed as riding models and the isotropic thermal parameters (Uiso) of all hydrogen atoms were based on the Ueq of the parent atom.
Crystal structure determination of 3.
C17H18N2O2Ru (M = 383.40 g mol−1): triclinic, space group P (no. 2), a = 5.9641(4) Å, b = 8.3166(7) Å, c = 15.9058(16) Å, α = 98.820(7)°, β = 95.605(6)°, γ = 104.953(6)°, V = 745.39(11) Å3, Z = 2, T = 100.01(10) K, μ(CuKα) = 8.584 mm−1, Dcalc = 1.708 g cm−3, 4176 reflections measured (11.206° ≤ 2Θ ≤ 136.494°), 4176 unique reflections (Rsigma = 0.0400) which were used in all calculations. The final R1 was 0.0369 (I > 2σ(I)) and wR2 was 0.0967 (all data). The crystal was a non-merohedral twin with the two domains related by 180° about the reciprocal direction [0 0 1] with the refined percentage ratio 59.4(1):40.6(1). The hydrogen atom bonded to N(3) was located in the electron density map and its position refined. All remaining hydrogen atoms were fixed as riding models and the isotropic thermal parameters (Uiso) of all hydrogen atoms were based on the Ueq of the parent atom.
Crystal structure determination of 5-(R).
C14H19NRu (M = 302.37 g mol−1): monoclinic, space group P21/c (no. 14), a = 7.6382(3) Å, b = 20.2298(9) Å, c = 8.0749(3) Å, β = 99.314(4)°, V = 1231.27(9) Å3, Z = 4, T = 100.01(10) K, μ(CuKα) = 10.053 mm−1, Dcalc = 1.631 g cm−3, 4460 reflections measured (8.742° ≤ 2Θ ≤ 138.252°), 2291 unique reflections (Rint = 0.0261, Rsigma = 0.0371) which were used in all calculations. The final R1 was 0.0280 (I > 2σ(I)) and wR2 was 0.0670 (all data).
Crystal structure determination of 15.
C12H12Ru (M = 257.29 g mol−1): monoclinic, space group P21/c (no. 14), a = 11.9888(12) Å, b = 5.6864(5) Å, c = 14.2237(16) Å, β = 100.876(9)°, V = 952.26(16) Å3, Z = 4, T = 100(2) K, μ(MoKα) = 1.590 mm−1, Dcalc = 1.795 g cm−3, 3039 reflections measured (5.834° ≤ 2Θ ≤ 55.172°), 3039 unique reflections (Rsigma = 0.0354) which were used in all calculations. The final R1 was 0.0732 (I > 2σ(I)) and wR2 was 0.2153 (all data). The crystal was a non-merohedral twin with the two domains related by 180° about the reciprocal direction [1 0 0] with the refined percentage ratio 54.2(3):45.8(3).
Crystal structure determination of 16.
C12H14ORu (M = 275.30 g mol−1): trigonal, space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (no. 147), a = 30.3009(5) Å, c = 5.97018(11) Å, V = 4747.12(18) Å3, Z = 18, T = 100.00(10) K, μ(CuKα) = 11.714 mm−1, Dcalc = 1.733 g cm−3, 21374 reflections measured (5.834° ≤ 2Θ ≤ 144.214°), 6213 unique reflections (Rint = 0.0387, Rsigma = 0.0342) which were used in all calculations. The final R1 was 0.0445 (I > 2σ(I)) and wR2 was 0.0953 (all data). The structure contains three crystallographically-independent molecules. The hydrogen atoms bonded to O(1), O(101) and O(201) were located in the electron density map and their positions and thermal parameters were freely refined.
(no. 147), a = 30.3009(5) Å, c = 5.97018(11) Å, V = 4747.12(18) Å3, Z = 18, T = 100.00(10) K, μ(CuKα) = 11.714 mm−1, Dcalc = 1.733 g cm−3, 21374 reflections measured (5.834° ≤ 2Θ ≤ 144.214°), 6213 unique reflections (Rint = 0.0387, Rsigma = 0.0342) which were used in all calculations. The final R1 was 0.0445 (I > 2σ(I)) and wR2 was 0.0953 (all data). The structure contains three crystallographically-independent molecules. The hydrogen atoms bonded to O(1), O(101) and O(201) were located in the electron density map and their positions and thermal parameters were freely refined.
CCDC 1953303–1953305, 1953307 and 1953308 contain the supplementary crystallographic data for this paper.†
Biological studies
MIAPaCa2 (85062806) pancreatic ductal adenoma cancer cells were purchased from the European Collection of Authenticated Cell Cultures. Cell culture media and supplements were purchased from Gibco (Thermo Scientific). All plasticware was purchased from Greiner Bio-One. Cells were maintained at 37 °C in a 5% CO2 humidified incubator and grown in T75 tissue culture flasks in DMEM supplemented with 10% (v/v) foetal bovine serum, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, and 2 mM L-glutamine. Cells were sub-cultured twice weekly before confluency using a standard trypsin-EDTA protocol. Cell cultures were confirmed to be free from Mycoplasma sp. contamination using the EZ-PCR mycoplasma detection kit according to the manufacturer's instructions.
Crystal violet assay.
Cells were sub-cultured in 96-well plates at a density of 6250 cells per well in 100 μL of complete DMEM and left overnight to allow the cells to attach. The next day culture media were removed and replaced with fresh media containing test compounds (0–80 μM) dissolved in DMEM with a final concentration of 0.5% v/v DMSO prepared from 50 mM stock solutions, except for cisplatin which was prepared as a 2 mM stock solution in phosphate buffered saline (PBS). All cultures were incubated for 72 h prior to commencement of the crystal violet assay as described below. The old media were removed, the cells were washed with 100 μL of PBS, and then 100 μL of 4% v/v paraformaldehyde was added. After 15 min this was removed and 100 μL of crystal violet solution (0.5% w/v in 10% v/v ethanol) was added and the plates were incubated for 20 min. Next the crystal violet solution was removed, and the plates were washed with PBS (4 × 100 μL) before being allowed to air dry for 20 min at room temperature. Finally, the samples were solubilized using 10% v/v acetic acid before measuring the absorbance at 590 nm using a well-plate reader.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
J. H. R. T. acknowledges support from the Engineering and Physical Sciences Research Council (EPSRC) for the award of a Leadership Fellowship (grant number EP/G007578/1) and the Leverhulme Trust for a research grant entitled “Expanding the range and versatility of ferrocene nucleic acids” (RPG-2013-100). The Centre for Chemical and Materials Analysis at the University of Birmingham is acknowledged for technical support. M. K. I. acknowledges the Ministry of Higher Education & Scientific Research in Iraq and The University of Kirkuk for funding and support. We thank the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data for 15.24
References
-
(a) A. Singh, I. Lumb, V. Mehra and V. Kumar, Dalton Trans., 2019, 48, 2840–2860 RSC;
(b) M. M. Santos, P. Bastos, I. Catela, K. Zalewska and L. C. Branco, Mini-Rev. Med. Chem., 2017, 17, 771–784 CrossRef CAS PubMed;
(c) G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- M. Patra and G. Gasser, Nat. Rev. Chem., 2017, 1, 0066 CrossRef CAS.
- G. Jaouen, A. Vessières and S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817 RSC.
- A. Vessieres, S. Top, W. Beck, E. Hillard and G. Jaouen, Dalton Trans., 2006, 529–541 RSC.
- K. Miura, M. Kinouchi, K. Ishida, W. Fujibuchi, T. Naitoh, H. Ogawa, T. Ando, N. Yazaki, K. Watanabe, S. Haneda, C. Shibata and I. Sasaki, Cancers, 2010, 2, 1717–1730 CrossRef CAS PubMed.
- S. Noble and K. L. Goa, Drugs, 1997, 54, 447–472 CrossRef CAS PubMed.
- For examples of metallocene derivatives of DNA/RNA nucleobases, see:
(a) M. Daniluk, W. Buchowicz, M. Koszytkowska-Stawińska, K. Jarząbek, K. N. Jarzembska, R. Kamiński, M. Piszcz, A. E. Laudy and S. Tyski, ChemistrySelect, 2019, 4, 11130–11135 CrossRef CAS;
(b) K. Kowalski, Coord. Chem. Rev., 2016, 317, 132–156 CrossRef CAS and references therein.
(c) P. James, J. Neudorfl, M. Eissmann, P. Jesse, A. Prokop and H. G. Schmalz, Org. Lett., 2006, 8, 2763–2766 CrossRef CAS PubMed;
(d) M. Hocek, P. Štěpnička, J. Ludvík, I. Císařová, I. Votruba, D. Řeha and P. Hobza, Chem. – Eur. J, 2004, 10, 2058–2066 CrossRef CAS PubMed.
- For recent examples of other organometallic derivatives of DNA/RNA nucleobases, see:
(a) P. R. Florindo, D. M. Pereira, P. M. Borralho, M. F. M. Piedade, M. C. Oliveira, A. M. Dias, C. M. P. Rodrigues and A. C. Fernandes, New J. Chem., 2019, 43, 1195–1201 RSC;
(b) M. I. P. S. Leitão, F. Herrera and A. Petronilho, ACS Omega, 2018, 3, 15653–15656 CrossRef PubMed;
(c) A. Collado, M. Gómez-Gallego and M. A. Sierra, Eur. J. Org. Chem., 2018, 1617–1623 CrossRef CAS;
(d) R. Kaczmarek, D. Korczynski, K. Krolewska-Golinska, K. A. Wheeler, F. A. Chavez, A. Mikus and R. Dembinski, ChemistryOpen, 2018, 7, 237–247 CrossRef CAS.
- H. V. Nguyen, A. Sallustrau, J. Balzarini, M. R. Bedford, J. C. Eden, N. Georgousi, N. J. Hodges, J. Kedge, Y. Mehellou, C. Tselepis and J. H. R. Tucker, J. Med. Chem., 2014, 57, 5817–5822 CrossRef CAS.
- J. L. Kedge, H. V. Nguyen, Z. Khan, L. Male, M. K. Ismail, H. V. Roberts, N. J. Hodges, S. L. Horswell, Y. Mehellou and J. H. R. Tucker, Eur. J. Inorg. Chem., 2017, 466–476 CrossRef CAS.
- S. Gubin, S. Smirnova, L. Denisovich and A. Lubovich, J. Organomet. Chem., 1971, 30, 243–255 CrossRef CAS.
-
(a) P. Pigeon, S. Top, A. Vessieres, M. Huche, E. A. Hillard, E. Salomon and G. Jaouen, J. Med. Chem., 2005, 48, 2814–2821 CrossRef CAS PubMed;
(b) H. Z. S. Lee, O. Buriez, E. Labbe, S. Top, P. Pigeon, G. Jaouen, C. Amatore and W. K. Leong, Organometallics, 2014, 33, 4940–4946 CrossRef CAS.
- For a recent example, see: S. Khanapure, M. Jagadale, P. Bansode, P. Choudhari and G. Rashinkar, J. Mol. Struct., 2018, 1173, 142–147 CrossRef CAS.
- T. Hayashi, A. Ohno, S.-J. Lu, Y. Matsumoto, E. Fukuyo and K. Yanagi, J. Am. Chem. Soc., 1994, 116, 4221–4226 CrossRef CAS.
- T. P. Smith, K. S. Kwan, H. Taube, A. Bino and S. Cohen, Inorg. Chem., 1984, 23, 1943–1945 CrossRef CAS.
- Y. S. Sohn, A. W. Schluete, D. N. Hendrick and H. B. Gray, Inorg. Chem., 1974, 13, 301–304 CrossRef CAS.
-
(a) R.-J. Gale and R. Job, Inorg. Chem., 1981, 20, 42–45 CrossRef CAS;
(b) T. Kuwana, D. E. Bublitz and G. Hoh, J. Am. Chem. Soc., 1960, 82, 5811–5817 CrossRef CAS.
- J. C. Swarts, A. Nafady, J. H. Roudebush, S. Trupia and W. E. Geiger, Inorg. Chem., 2009, 48, 2156–2165 CrossRef CAS PubMed.
- R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395–6401 CrossRef CAS.
- S. Trupia, A. Nafady and W. E. Geiger, Inorg. Chem., 2003, 42, 5480–5482 CrossRef CAS.
-
(a) K. C. Kemp, E. Fourie, J. Conradie and J. C. Swarts, Organometallics, 2008, 27, 353–362 CrossRef CAS;
(b) C. Amatore, M. Gareil and J. Savéant, J. Electroanal. Chem. Interfacial Electrochem., 1983, 147, 1–38 CrossRef CAS.
- S. Kukharenko, V. Strelets, A. Kudinov, A. Kreidlin, M. Peterleitner, L. Denisovich and M. Rybinskaya, J. Organomet. Chem., 1996, 519, 1–5 CrossRef CAS.
-
(a) M. Watanabe, M. Sato, A. Nagasawa, M. Kai, I. Motoyama and T. Takayama, Bull. Chem. Soc. Jpn., 1999, 72, 715–723 CrossRef CAS;
(b) K. Hashidzume, H. Tobita and H. Ogino, Organometallics, 1995, 14, 1187–1194 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- C. Biot, N. Chavain, F. Dubar, B. Pradines, X. Trivelli, J. Brocard, I. Forfar and D. Dive, J. Organomet. Chem., 2009, 694, 845–854 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1953303–1953305, 1953307 and 1953308. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/C9DT04174E |
| ‡ The X-ray structures of the chiral compounds were determined from crystals grown from solutions of racemic mixtures (see the ESI† for more details). |
| § The enantiomer 1-(R,Sp)-Ru, isolated from a racemic batch of the target compound (see ESI†), was found to be even less toxic, with an IC50 of >80 μM, the highest concentration investigated. These data suggest that stereochemistry also plays a significant role in determining the anticancer activity of these ferronucleosides. This aspect is currently being investigated further and will be included in a future report. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Katie A.
Armstrong
b,
Samantha L.
Hodder
b,
Sarah L.
Horswell
a,
Katie A.
Armstrong
b,
Samantha L.
Hodder
b,
Sarah L.
Horswell