
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of PtZn nanoalloy catalysts for propane dehydrogenation through interface tailoring via atomic layer deposition†
Piyush
Ingale
 a,
Kristian
Knemeyer
a,
Phil
Preikschas
a,
Mengyang
Ye
b,
Michael
Geske
a,
Raoul
Naumann d'Alnoncourt
*a,
Arne
Thomas
b and
Frank
Rosowski
ac
a,
Kristian
Knemeyer
a,
Phil
Preikschas
a,
Mengyang
Ye
b,
Michael
Geske
a,
Raoul
Naumann d'Alnoncourt
*a,
Arne
Thomas
b and
Frank
Rosowski
ac
aBasCat – UniCat BASF JointLab, Technische Universität Berlin, Berlin 10623, Germany. E-mail: r.naumann@bascat.tu-berlin.de
bFunctional Materials, Department of Chemistry, Technische Universität Berlin, Berlin 10623, Germany
cBASF SE, Process Research and Chemical Engineering, Heterogeneous catalysis, Ludwigshafen 67056, Germany
First published on 18th November 2020
Abstract
Supported Pt nanoparticles are widely used for the catalytic dehydrogenation of propane to propene. Monometallic Pt catalysts are subject to fast deactivation. A successful strategy for stabilization is alloying Pt with a second metal. In this study, we present a novel approach for the precise formation of bimetallic nanoparticles via tailoring of the interface between metal nanoparticles and the support. An ultra-thin functional layer of ZnO is deposited via atomic layer deposition on SiO2. The supported Pt nanoparticles undergo a phase transformation and form Pt1Zn1 alloy nanoparticles under reductive thermal treatment. The resulting Pt1Zn1 catalyst showed a high and stable selectivity to propene over 12 hours of time on stream. The activity of the Pt1Zn1 catalyst was 1.5 times higher than that of a catalyst of the same composition prepared by incipient wetness impregnation. The nanoalloy formation causes electronic and geometric modification of Pt which reduces side reactions and leads to a stable and active propane dehydrogenation catalyst.
Introduction
Propene is an important intermediate used in the production of polypropene, propene oxide, acrylic acid, etc.1 Propene is mainly produced as a by-product of steam cracking and fluid catalytic cracking.2 As propene is a secondary product in both processes, the demand for propene is often not met by its production as cracking processes are volatile to gasoline prices.3 To answer the rising demand for propene due to e.g. a growing middle class in developing countries, the on-purpose production of propene is gaining attention. Propane dehydrogenation (PDH) is a cost effective and efficient technology for propene production. The non-oxidative dehydrogenation of propane is an endothermic reaction limited by thermodynamic equilibrium. Therefore, high reaction temperatures (∼600 °C) are required to achieve economically feasible conversions. However, high reaction temperatures lead to several side reactions such as cracking, isomerization and deep dehydrogenation of products.4Several dehydrogenation processes are applied in industry. The most important are the Catofin process (CB&I Lummus), the Oleflex process (UOP Honeywell) and the STAR process (UHDE).5 The Catofin process is based on CrOx catalysts while the other processes are using bimetallic Pt–Sn catalysts.6 The use of chromium oxide-based catalysts is an environmental challenge due to chromium's toxicity. Therefore, noble metal-based catalysts are generally preferred. However, platinum-based catalysts are subject to fast deactivation due to sintering and coke formation.7,8 The addition of a second inactive metal to a noble metal is a widely used strategy in heterogeneous catalysis to improve catalytic performance. An example for the PDH reaction is the modification of Pt on alumina catalysts by adding Sn.9–11 The increased stability and selectivity of Pt–Sn catalysts is due to the inhibition of hydrogenolysis, cracking and coke formation.12 The dehydrogenation of light alkanes itself is a structure insensitive reaction where all Pt surface atoms are active. However, side reactions such as hydrogenolysis require larger Pt ensembles.6 The Pt ensemble size can be reduced by alloying with a second non-active metal via a geometric effect. Thus, many alloy phases have been studied in literature, e.g. Pt–Ga,12,13 Pt–Zn,14–16 Pt–Cu,17,18 Pt–Co,19 Pt–Mn,20 and Pt–Ti.21 Catalytic cracking is related to the Brønsted and Lewis acid sites of catalysts due to formation of carbocation intermediates.22
Zinc is a highly abundant, nontoxic and cheap raw material. Therefore, Zn-based materials for catalytic applications are of special interest.23 Several studies on Zn based materials for PDH are reported in literature.24,25 Single site Zn2+ species and ZnO nanoclusters were investigated for activation of C–H bonds.26–28 However, single site Zn2+ species and ZnO nanoclusters are unstable under the harsh conditions of PDH. Many strategies have been applied to form bimetallic Pt/Zn nanoparticles.14,24,25 However, most synthesis methods result in an inhomogeneous distribution of metals on the support. Atomic layer deposition (ALD) is a synthetic method developed in the 1970's by Tuomo Suntola in Finland.29 Nowadays, ALD is widely used in the semiconductor industry. The potential of ALD has been widely investigated for catalyst development in the last two decades.30 ALD can be used to stabilize nanoparticles via physical over-coating with an inert oxide layer,31,32 formation of metallic/core–shell nanoparticles,33 deposition of promoters on bulk catalysts,34 and coating of powders with functional materials.35
Here, we use ALD to tailor the interface between Pt nanoparticles and the SiO2 used as support. The surface of the SiO2 is modified by homogenous deposition of a very thin functional ZnO layer resulting in a high surface area support with ZnO termination. ALD of zinc oxide is the second most widely studied process after ALD of alumina.36 A detailed report of this process on powder substrates is given elsewhere, along with characterization results for the ALD modified materials.37Fig. 1 summarizes the design strategy for the final nanoalloy catalyst. After the ALD process, Pt nanoparticles are deposited on the ZnO/SiO2 support via incipient wetness impregnation. This strategy ensures a direct and intimate contact between all Pt nanoparticles and ZnO. The formed Pt/ZnOALD/SiO2 is a precursor for the final catalyst. Pt1Zn1 nanoalloy particles are formed in situ under activation treatment conditions in the reactor used for PDH.
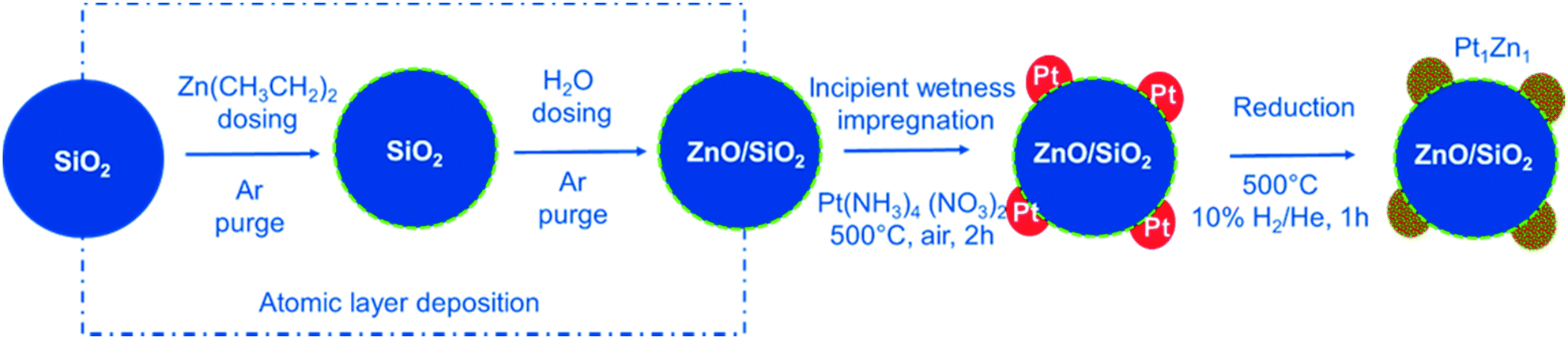 | ||
| Fig. 1 Synthesis approach used for the precise development of Pt1Zn1 nano-alloys via interphase tailoring using ALD. | ||
Experimental
Materials
Silica powder [SiO2, high-purity grade ≥99% (Davisil Grade 636), average pore size 60 Å, 35–60 mesh particle size, specific surface area 506 m2 g−1, Sigma-Aldrich, Germany] was used as ALD substrate and support for catalyst synthesis. Zinc oxide nanopowder [ZnO, nanopowder, <100 nm particle size, specific surface area 10–25 m2 g−1, Sigma-Aldrich, Germany] was used as support. Pt(NH3)4(NO3)2 [99.995% trace metal basis, Sigma-Aldrich, Germany] was used as metal salt for impregnation. Diethylzinc [Zn(C2H5)2, DEZn, elec. gr., 99.999% Zn, Strem Chemicals, Germany] and water (H2O, CHROMASOLV®, for HPLC, Riedel-de Haen) served as ALD precursors and were used without further purification. High purity N2, Ar, and He (99.999%) were used as carrier and purging gases.Atomic layer deposition of ZnO on SiO2
Atomic layer deposition experiments were conducted in a set-up designed and constructed in-house. A detailed description of the set-up is given elsewhere.38,39 High surface area SiO2 powder was filled into a tubular fixed bed reactor made of a quartz tube. The ALD process was carried out under a continuous total gas flow of 100 mL min−1 at atmospheric pressure and a deposition temperature of 80 °C. The diethylzinc (Zn(C2H5)2) saturator was heated to 50 °C while the water saturator was kept at room temperature. Both reactants were sequentially fed into the reactor in a flow of 100 mL min−1 carrier gas. The sequence used was (Zn(C2H5)2)/N2–Ar purge–H2O/He–Ar purge. The end of each half cycle times was determined by on-line mass spectrometry. Both reactants were dosed to the reactor until the mass traces for m/z = 93 (DEZn) and m/z = 18 (H2O) reached constant levels, respectively. The resulting material is denoted as ZnOALD/SiO2.Synthesis of Pt/SiO2, PtZnALD/SiO2, PtZnIWI/SiO2 and Pt/ZnO
Platinum nanoparticles supported on SiO2 and ZnOALD/SiO2 were prepared via incipient wetness impregnation. The ZnOALD/SiO2 was calcined in synthetic air at 500 °C for 2 h prior to the impregnation. The nominal metal loading of platinum was 1.0 wt% for both samples. An amount of 60 mg of tetraammineplatinum(II) nitrate (Pt(NH3)4(NO3)2) salt was dissolved in 2.2 mL and 1.8 mL HPLC grade water, respectively. The metal salt solution was added dropwise to 3 g of SiO2 (2.2 mL) and ZnOALD/SiO2 support (1.8 mL) and mixed thoroughly. The samples were left standing overnight at room temperature. The samples were then dried at 70 °C for 24 h. Finally, the samples were calcined at 500 °C for 2 h (heating rate: 10 K min−1) in a flow of 20% O2/N2 (500 mL min−1). The samples in the state after calcination are denoted as Pt/SiO2 and Pt/ZnOALD/SiO2.The reference Pt/ZnOIWI/SiO2 catalyst was synthesised by sequential incipient wetness impregnation of Zn and Pt. An amount of 1.36 g of Zn(NO3)2·6H2O was mixed with 2.2 mL HPLC grade water and stirred at 400 rpm for 30 min. The Zn metal salt solution was added dropwise to 3 g of SiO2. The sample was then dried at 70 °C for 24 h, followed by calcination at 500 °C for 2 h (heating rate: 10 K min−1) in a flow of 20% O2/N2 (500 mL min−1). The resulting material is denoted as ZnOIWI/SiO2. An amount of 65 mg of (Pt(NH3)4(NO3)2) salt was dissolved in 2.2 mL HPLC grade water added dropwise to the ZnOIWI/SiO2. The sample was dried again at 70 °C for 24 h followed by calcination at 500 °C for 2 h (heating rate: 10 K min−1, 500 mL min−1 of 20% O2/N2).
For a systematic comparison, Pt supported on ZnO without any SiO2 support was also prepared via incipient wetness impregnation. The nominal metal loading of platinum was kept at 1 wt%. An amount of 60 mg of tetraammineplatinum(II) nitrate (Pt(NH3)4(NO3)2) salt was dissolved in 0.2 mL HPLC grade water. The metal salt solution was added dropwise to 3 g of ZnO powder and mixed thoroughly. The sample was left standing overnight at a room temperature followed by drying in an oven at 70 °C for 24 h. The dried sample was calcined at 500 °C for 2 h (heating rate: 10 K min−1) in a flow of 20% O2/N2 (500 mL min−1). The resulting material is denoted as Pt/ZnO.
Prior to the catalytic test, all catalysts were activated in situ by a reduction in H2. The catalysts were reduced in 50 mL min−1 of 10% H2/He for 60 min at 600 °C with a heating rate of 10 K min−1. The activated catalysts are designated as PtZnALD/SiO2, PtZnIWI/SiO2, Pt/SiO2,ACT, and Pt/ZnOACT, respectively.
Characterization of the catalysts
For determination of specific surface areas, nitrogen physisorption was performed at 77 K using a Quadrasorb SI device manufactured by Quantachrome. Samples were degassed at 120 °C for 12 h prior to measurements. The surface area was determined by the Brunauer–Emmett–Teller (BET) method. The average pore size was calculated using non-local density functional theory (NLDF) method.Phase analysis was done by powder X-ray diffraction (XRD) measurements in Bragg–Brentano geometry on a D8 Advance II theta/theta diffractometer (Bruker AXS) using Ni-filtered Cu-Kα radiation and a position sensitive energy dispersive LynxEye silicon strip detector.
For microstructural analysis, scanning transmission electron microscopy (STEM) was conducted on a FEI Talos F200X microscope. The microscope was operated at an acceleration voltage of 200 kV. Elemental mappings were recorded with a Super-X system including four silicon drift energy-dispersive X-ray (EDX) detectors. All samples were prepared on holey carbon-coated copper grids (Plano GmbH, 400 mesh).
Chemical analysis was carried out by X-ray fluorescence spectroscopy (XRF) in a Bruker S4 Pioneer X-ray spectrometer. Samples were prepared by melting pellets with a ratio of 100 mg sample to 8.9 g of Li2B4O7.
The electronic state of Pt in the catalysts was investigated by X-ray photoelectron spectroscopy (XPS) measurements using a ThermoScientific K-Alpha+ X-ray photoelectron spectrometer. All samples were analysed using a micro-focused, monochromatic Al-Kα X-ray source (1486.68 eV; 400 μm spot size). The analyser had a pass energy of 200 eV (survey), and 50 eV (high-resolution spectra), respectively. To prevent any localized charge build-up during analysis the K-α+ charge compensation system was employed at all measurements. The samples were mounted on conductive carbon tape and the resulting spectra were analysed using the Avantage software (ThermoScientific). All peaks were calibrated by setting the binding energy (B.E.) for adventitious carbon peak C1s to 284.8 eV to compensate for the charging effect.
Catalytic experiments
The catalytic experiments were carried out at atmospheric pressure and 600 °C in set-up designed by Integrated Lab Solutions and equipped with a quartz tube as fixed bed reactor. The applied reactor had an inner diameter of 10 mm. The used catalyst amount resulted in a bed height of five mm. Catalyst amounts were fixed to 500 mg for all runs. In an activation step, all catalysts were first in situ reduced for 60 min at 600 °C in 50 mL min−1 of 10% H2/He. After purging with 50 mL min−1 He for 5 min, the gas flow was switched to 50 mL min−1 of 20 vol% C3H8/He. The resulting gas hourly space velocity (GHSV) was 6000 mL g−1 h−1. The effluent gas stream was analysed by an online gas chromatograph (Agilent 7890A) equipped with a flame ionization detector and a thermal conductivity detector. Propane conversion, propene selectivity and a deactivation factor were calculated according to eqn (1)–(3), respectively: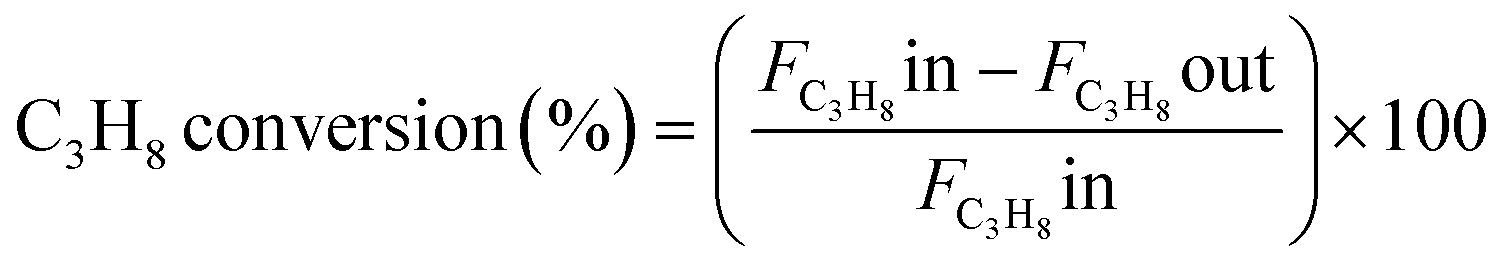 | (1) |
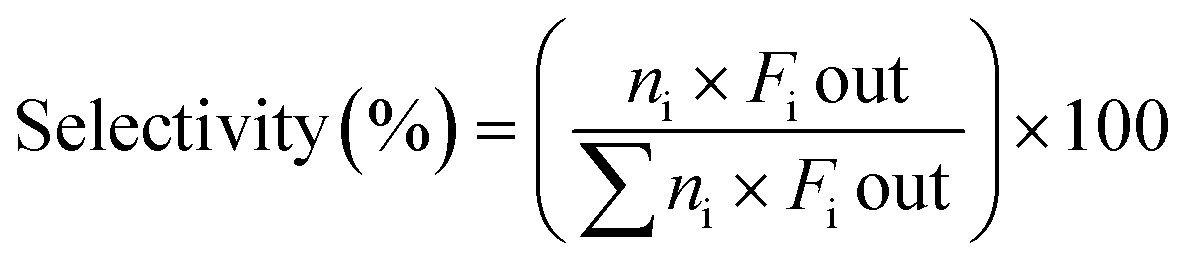 | (2) |
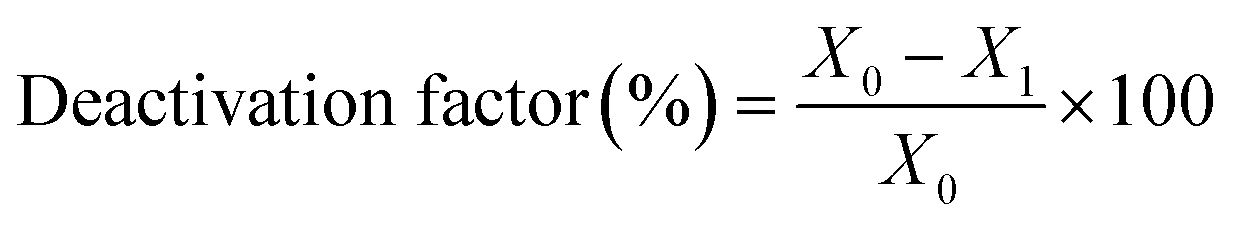 | (3) |
Results
The design strategy for the multistep synthesis applied in this study is summarized in Fig. 1. In the first step, the SiO2 support is modified by a thin layer of ZnO via ALD. The second step adds Pt nanoparticles via incipient wetness impregnation. This results in a direct contact between Pt and ZnO. The resulting Pt/ZnOALD/SiO2 material is then activated by reduction to yield well defined Pt–Zn nanoalloy particles. The ZnO ALD is carried out on a high surface area silica powder via an AB type ALD process of (Zn(C2H5)2)/H2O at 80 °C in a tubular fixed bed reactor. An in situ gravimetric study of the growth behaviour of ZnO on SiO2 is reported elsewhere.37 In the ZnO ALD process, diethyl zinc (Zn(C2H5)2) reacts with the surface hydroxyl groups forming surface chemisorbed Zn(C2H5) species while releasing C2H6 as by-product during the first half cycle, while in the second half cycle the surface hydroxyl groups are recreated by dosing H2O to form surface Zn(OH)x species with C2H6 as by-product. The reaction will continue until all of the surface groups are saturated.Characterization of synthesized catalysts
The elemental analysis (XRF) of ZnOALD/SiO2 after one ALD cycle gave a Zn content of 12.2 wt%. ZnOALD/SiO2 and the original SiO2 were used as support for Pt nanoparticles via incipient wetness impregnation. The physicochemical properties of the materials are reported in Table 1. The specific surface area and average pore volume decreased considerably after one ALD cycle of ZnO on SiO2 and the subsequent impregnation of Pt nanoparticles. A decrease in specific surface area and total pore volume can be rationalized by the coating of micro- and mesopores during the ALD process. Furthermore, the observed decrease in the specific surface area is partly due to the deposition of dense ZnO on low density SiO2, as the specific surface area is normalised to mass. The corresponding N2 adsorption isotherms are shown in Fig. S1.† The results for PtZnALD/SiO2 show a type IV isotherm with H1 hysteresis loop similar to the isotherm of the original SiO2 support, indicating the overall conformal coating with ZnO of the mesopores of SiO2 without loss of porosity due to pore blocking.| Material | Surface area (m2 g−1) | Pore size (nm) | Pore volume (mL g−1) | Pt content (wt%) | Zn content (wt%) |
|---|---|---|---|---|---|
| Specific surface area calculated by BET method, pore size from NLDFT method and Pt and Zn content calculated by XRF analysis. | |||||
| SiO2 | 505 | 4.9 | 0.75 | 0 | 0 |
| Pt/SiO2 | 465 | 4.6 | 0.71 | 1.15 | 0 |
| PtZnALD/SiO2 | 400 | 4.8 | 0.56 | 1.15 | 12.2 |
| PtZnIWI/SiO2 | 381 | 4.7 | 0.66 | 1.1 | 10 |
| Pt/ZnO | 13 | 2.8 | 0.04 | 1.2 | 78 |
The X-ray diffractograms of all materials in the state as synthesized and after activation are shown in Fig. 2. In the state before activation, no indications for Pt nanoparticles are observed in the diffractograms of Pt/SiO2 and Pt/ZnOIWI/SiO2. A very broad peak at 2θ ∼ 39.2°, corresponding to the (111) reflection of the face centered cubic (FCC) structure of Pt, can be found in the diffractogram of Pt/ZnOALD/SiO2. A peak at the same position, although much sharper, can be found in the diffractogram of Pt/ZnO. After the activation step a Pt (111) diffraction peak can be found in the diffractogram of Pt/SiO2,ACT. The peaks observed in the diffractograms of Pt/ZnOALD/SiO2 and Pt/ZnO are shifted to 2θ = 40.6° in the diffractograms of PtZnALD/SiO2 and Pt/ZnOACT. This shift can be rationalized by the formation of an alloy phase as literature reports a 2θ value of 40.6° for the (111) diffraction peak of the tetragonal Pt1Zn1 alloy (PDF-04-016-2848, spacing group P4/mmm). In contrast, the diffractogram of PtZnIWI/SiO2 shows no peaks indicating any Pt nanoparticles. The absence of such peaks can be rationalized by an average crystallite domain size for Pt below 5 nm, indicating either large polycrystalline nanoparticles, e.g. with stacking faults or twin faults, or highly dispersed, small nanoparticles.
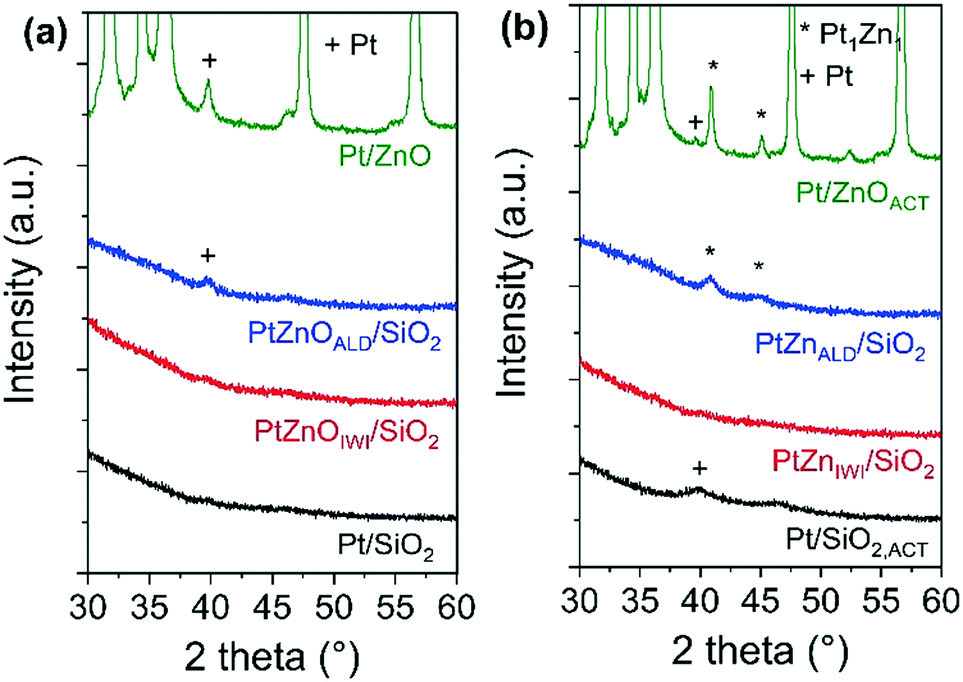 | ||
| Fig. 2 X-ray diffractograms of (a) as synthesized and (b) activated materials. | ||
The HAADF-STEM images and the corresponding EDX mappings of the catalysts in the state after synthesis and after activation are shown in Fig. S2† and 3, respectively. The STEM image of Pt/SiO2 (Fig. S2a†) shows highly dispersed Pt nanoparticles supported on SiO2. The average particle size of the Pt nanoparticles is about 5 nm, in agreement with the XRD results. A representative STEM image and corresponding EDX mappings for Pt/ZnOALD/SiO2 are shown in Fig. S2(b and d).† No indications for phase separation and formation of ZnO crystallites were found. The EDX mappings clearly show the well-dispersed distribution of Zn over the support indicating a homogenous coating of SiO2 with ZnO. The STEM-EDX analysis also confirms that all supported Pt nanoparticles are in direct and intimate contact with ZnO. The formation of PtZn nanoparticles during activation can be seen in a representative STEM image and the corresponding EDX mappings of PtZnALD/SiO2 shown in Fig. 3(a–c). The alloy formation can be deduced from the fact that in the EDX mappings Zn is enriched in the areas were Pt is concentrated. The average particle size of the supported PtZn nanoalloy particles is 8.6 nm. In contrast, the electron microscopy results for PtZnIWI/SiO2 cannot be summarized in a representative micrograph as the material is far too heterogeneous. The STEM images and corresponding EDX mappings shown in Fig. 3(d–i) indicate the presence of segregated ZnO nanoparticles with sizes up to 200 nm (Fig. 3i) as well as regions with finely dispersed ZnO agglomerates on SiO2 (Fig. 3f). The EDX mappings also show that Pt is not homogeneously distributed either. There are regions with very low Pt concentration (Fig. 3h) as well as regions with homogeneously dispersed Pt nanoparticles (Fig. 3e). It can be clearly seen that Zn is not enriched in the locations of Pt nanoparticles (Fig. 3e and f). The heterogeneous distribution of Pt and Zn can already be seen in STEM image and the corresponding EDX mappings of Pt/ZnOIWI/SiO2 in the state after synthesis shown in Fig. S2(e–g).† Therefore, it can be assumed that the segregation of ZnO observed in PtZnIWI/SiO2 is not due to the activation step but rather the way of ZnO deposition. The electron microscopy results regarding Pt/ZnO and Pt/ZnOACT are in full agreement with the corresponding XRD results. It can be clearly seen in Fig. S2(h–j)† that the IWI of Pt onto unsupported ZnO resulted in the formation of large Pt nanoparticles. The micrograph shows several Pt particles in an orientation without underlying ZnO, at the edge of a large ZnO particle. EDX mappings clearly show that these Pt nanoparticles contain no significant amount of Zn. In contrast, a similarly oriented Pt nanoparticle in Fig. 3(j–l) shows a high Zn content. Thus, the via XRD macroscopically observed phase transformation of Pt to Pt1Zn1 during activation can also be confirmed in microstructural analysis on the nm scale. The main difference to the other Pt containing catalysts is the size of the supported metal particles. The low specific surface area of the unsupported ZnO favours the formation of large Pt particles in the range above 10 nm. The differences between PtZnALD/SiO2 and PtZnIWI/SiO2 can be rationalized by the fact that ALD is based on chemisorption while IWI proceeds mainly via physisorption. Chemisorbed Zn species are more strongly bound to the SiO2 surface and therefore supposedly stay well dispersed during the calcination step prior to Pt impregnation. In contrast, physisorbed Zn species are more mobile on the SiO2 surface and thus may form easily ZnO aggregates and nanoparticles. The results obtained for Pt/ZnOACT support the idea that Pt which is in close contact to ZnO transforms into a PtZn upon activation as postulated in our design strategy.
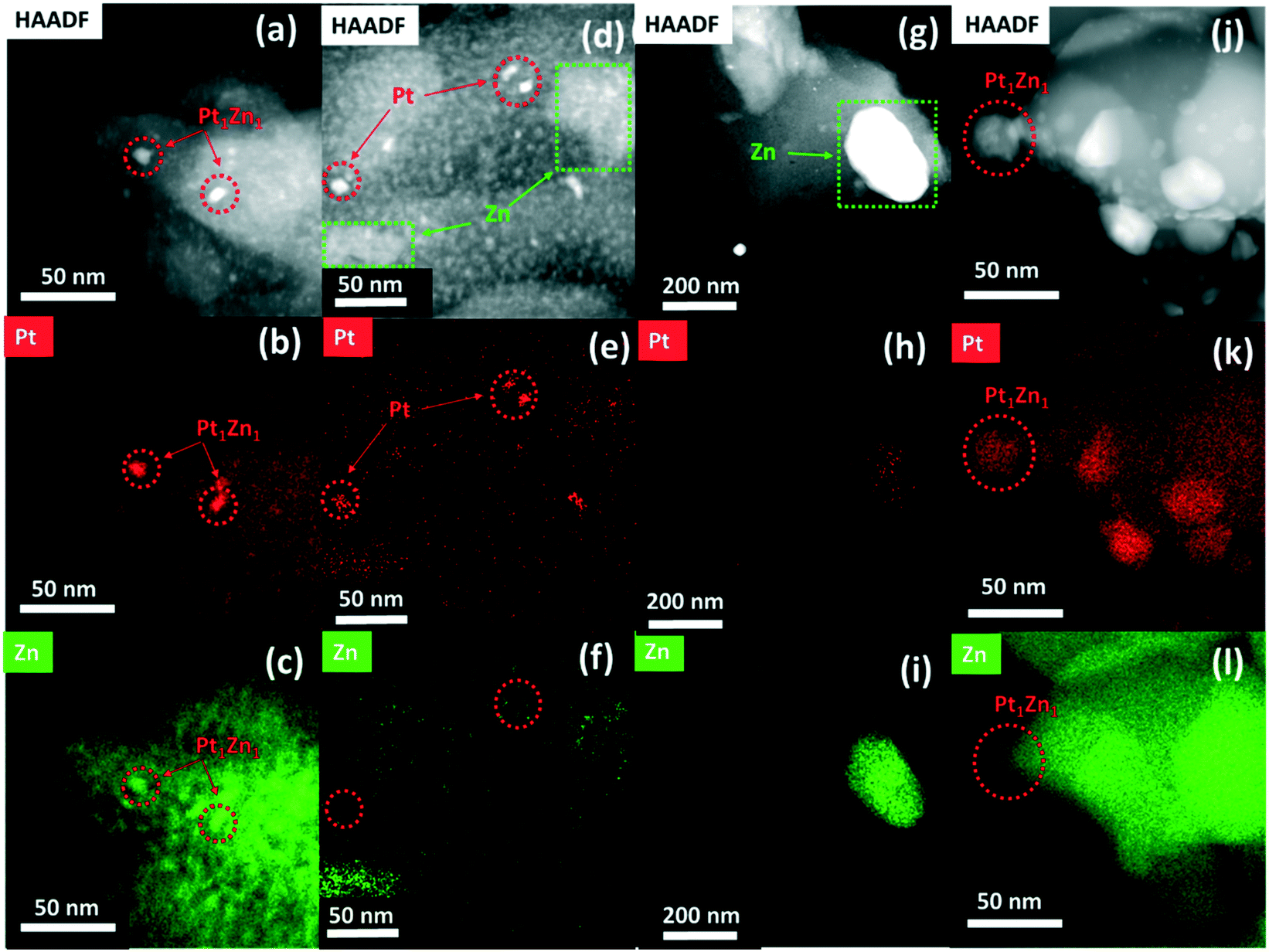 | ||
| Fig. 3 HAADF-STEM image and EDX maps of Pt and Zn in (a–c) PtZnALD/SiO2 showing homogenously dispersed Zn signal on SiO2 and Pt nanoparticles in direct contact with ALD deposited ZnO; (d–f) PtZnIWI/SiO2, indicating the inhomogeneous dispersion of Zn and Pt, where Zn is either in apart or in close proximity of Pt nanoparticles; (g–i) PtZnIWI/SiO2, where severe Zn aggregation can be visualised indicating poor dispersion of ZnO during impregnation; (j–l) Pt/ZnOACT, indicating larger Pt nanoparticles supported on bare ZnO support. | ||
XPS gives results in agreement with XRD and electron microscopy. The Pt 4f7/2 binding energy (B.E.) is different for Pt/SiO2,ACT and PtZnALD/SiO2 in the XPS spectra shown in Fig. 4 the Pt 4f7/2 B.E. for standalone Pt/SiO2,ACT is 71.2 eV indicating the presence of metallic Pt0. The Pt 4f7/2 peak for PtZnALD/SiO2 is at 71.7 eV. This positive shift of 0.5 eV in B.E. can be rationalized by the enhancement of the electrostatic field around Pt nuclei due to the vicinity of alloying Zn atoms. The alloying of Pt with Zn results in a reduction of the density of Pt 5d states around the Fermi level which leads to a shift of the Pt 5d band and the 4f core levels to higher binding energies.40,41
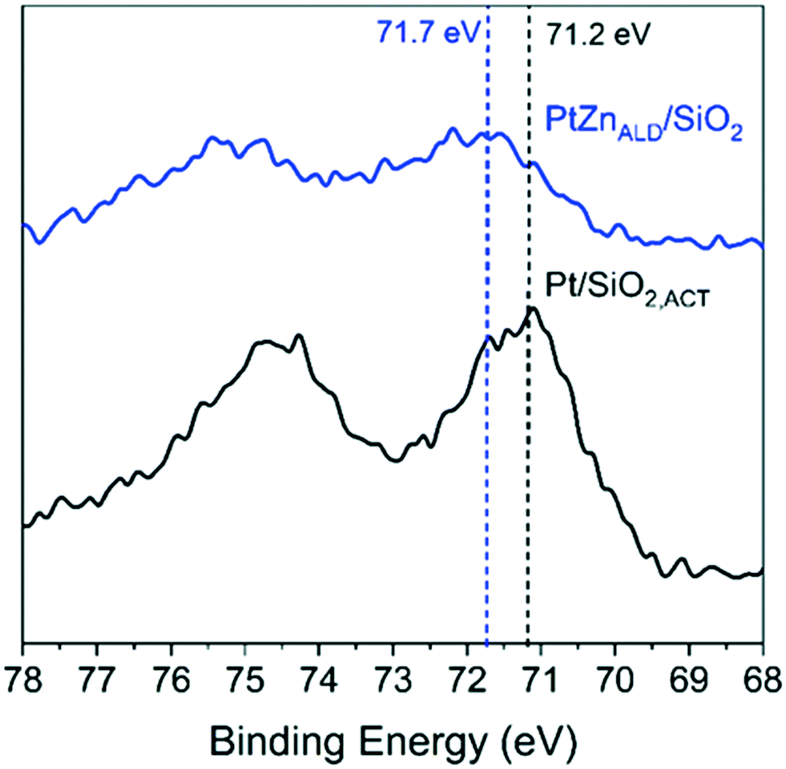 | ||
| Fig. 4 XPS analysis of Pt/SiO2 and PtZnALD/SiO2 after activation treatment in 10% H2/He at 600 °C for 1 h. | ||
Propane dehydrogenation
The propane dehydrogenation (PDH) experiments were conducted in a single tube quartz reactor at the reaction temperature of 600 °C. The catalytic performance of Pt/SiO2,ACT, PtZnALD/SiO2, PtZnIWI/SiO2 and Pt/ZnOACT are shown in Fig. 5 and Table 2. All four catalysts initially show a higher C3H8 conversion followed by deactivation within 0.5 h time on stream (TOS) and finally a stable performance for 12 h.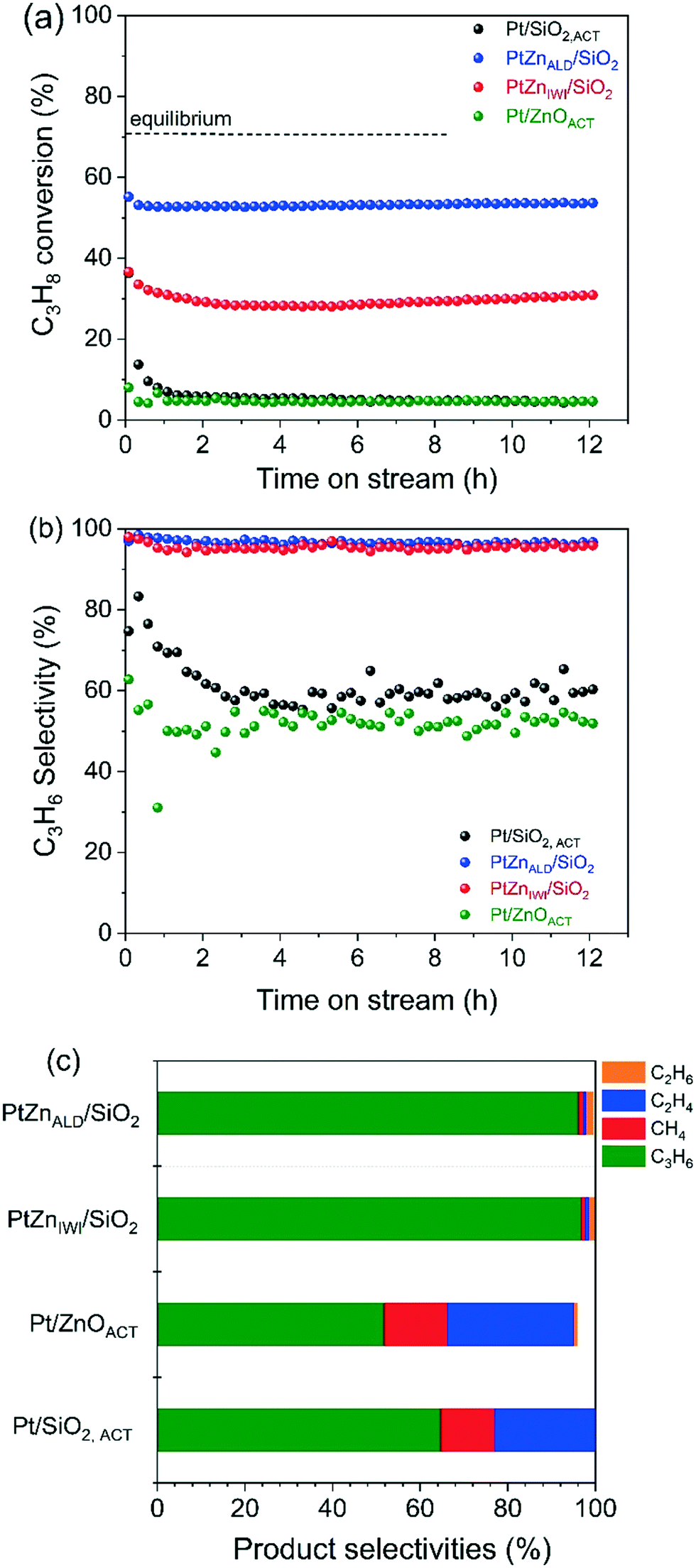 | ||
| Fig. 5 (a) C3H8 conversion, (b) C3H6 selectivity and (c) different product distributions at the end of 12 h time on stream; propane dehydrogenation was carried out at 600 °C under 20 vol% C3H8/He flow and 500 mg of catalyst. | ||
| Name of catalyst | C3H8 conversion X (%) | C3H6 selectivity S (%) | Deactivation factor D (%) | Carbon balance | ||
|---|---|---|---|---|---|---|
| X 0 | X 1 | S 0 | S 1 | |||
| a X 0 & S0: conversion of propane and selectivity towards propene at 5 min TOS. b X 1 & S1: conversion of propane and selectivity towards propene at 12 h TOS; carbon balance was calculated at end of 12 h TOS. | ||||||
| Pt/SiO2,ACT | 37 | 5 | 77 | 61 | 86 | 98 |
| PtZnALD/SiO2 | 53 | 49 | 97 | 97 | 7.5 | 99 |
| PtZnIWI/SiO2 | 40 | 31 | 97 | 96 | 22.5 | 99 |
| Pt/ZnOACT | 8 | 5 | 63 | 52 | 37.5 | 95 |
To compare the different catalysts regarding their stability, a deactivation factor (D) was calculated according to eqn (3). The performance of the reference Pt/SiO2,ACT catalyst is in general agreement with literature. An initial C3H8 conversion of 37% decreases within 0.5 h TOS giving a deactivation factor of 86% after 12 h time on stream. The C3H6 selectivity decreases over 12 h TOS from 77% to 61%. The second reference catalyst, Pt/ZnOACT, exhibits a much lower initial conversion of 8% and reaches a similar low performance after 12 h TOS. The lower initial performance is not surprising and can be clarified by the large size of the supported Pt nanoparticles and the resulting low dispersion of Pt.
Modification of the interface between Pt and SiO2 by deposition of ZnO before Pt is supported improves significantly the performance of the catalysts PtZnALD/SiO2 and PtZnIWI/SiO2. PtZnALD/SiO2 shows an initial C3H8 conversion of 53% and a low deactivation factor of 7.5% after 12 h TOS, with the complete deactivation occurring in the first 0.5 h. The C3H6 selectivity is at a high level 97% over the complete 12 h TOS. PtZnIWI/SiO2 shows an initial C3H8 conversion of 40% and a higher deactivation factor of 22.5%. The C3H6 selectivity of PtZnIWI/SiO2 is comparable to the selectivity shown by PtZnALD/SiO2 at a constant level of 96%.
The product distribution measured for all catalysts in the state after 12 h TOS is given in Fig. 5c. The deactivated state of Pt/SiO2,ACT and Pt/ZnOACT is very similar. The main product is propene with selectivities of 61% and 52%, respectively. Ethylene and methane are major byproducts, only minor amounts of ethane are found. One clear difference is the fact that the carbon balance for Pt/ZnOACT is not closed indicating ongoing formation of carbonaceous deposits. The byproduct distribution indicates hydrogenolysis of propene. Pt–Pt ensembles are known to perform deep dehydrogenation of alkanes followed by hydrogenolysis.42,43 On the other hand, Pt/ZnOACT shows the same product distribution. The results of a blank test using the quartz reactor filled with SiO2 are shown in Fig. S3.† The measured values for conversion of 3% and for propene selectivity of 53% can be attributed to homogeneous gas phase reaction. The values are in the same order of magnitude as measured for Pt/SiO2,ACT and Pt/ZnO after 12 h TOS, therefore the performance of these two catalysts after 12 h TOS cannot be reliably interpreted. The product distribution for PtZnALD/SiO2 and PtZnIWI/SiO2 is nearly identical, only small amounts of byproducts are found, in both cases methane, ethene and ethane. This can be taken as indication that the active sites are in both catalysts of the same nature. The difference in activity for the both catalysts can be rationalized by a different amount of active sites, with PtZnALD/SiO2 having more active sites than PtZnIWI/SiO2. An explanation could be that Pt1Zn1 nanoalloy particles are the active phase, while Pt nanoparticles are subject to fast deactivation. The macro- and microstructural analysis of the catalysts in the state as synthesized and after activation showed that the ZnO is more homogeneously distributed in the ALD modified samples, and that PtZnALD/SiO2 contained exclusively Pt1Zn1 nanoalloy particles while PtZnIWI/SiO2 contained a possibly some PtZn nanoalloy particles and mainly Pt nanoparticles. A hypothesis would be that the Pt nanoparticles in PtZnIWI/SiO2 are partly transformed into active Pt1Zn1 nanoalloy particles and partly deactivate in the form of Pt nanoparticles. In order to clarify whether ZnO supported on SiO2 has a positive contribution to the target reaction, ZnALD/SiO2 without Pt content was activated and tested. The results are shown in Fig. S4.† The C3H8 conversion is stable at a level of 8%, but the propene selectivity decreased within 4 h TOS from 98% to 65%. The activity is higher than that of pure SiO2, but the selectivity is poor, and thus the contribution of the exposed ZnO surface is in total not beneficial for the overall propene yield.
STEM micrographs and corresponding EDX mappings of spent catalysts are shown in Fig. 6. The elemental mapping of PtZnALD/SiO2 and Pt/ZnOACT shows that the PtZn nanoalloy particles observed in Fig. 3 can still be seen in Fig. 6(a–c) and (j–l), respectively. There is no hint for any phase segregation into Pt and ZnO. The results for the spent PtZnALD/SiO2 also show no hint for any aggregation or sintering of ZnO. The STEM images for PtZnIWI/SiO2 and the corresponding EDX mappings indicate clearly the presence of PtZn nanoalloy particles in Fig. 6(d–f) and Zn nanoparticles in Fig. 6(g–i). The results of the XRD analysis of all spent catalysts are shown in Fig. S5.† A peak at 2θ = 40.6° indicates the presence of Pt1Zn1 nanoalloy particles in PtZnALD/SiO2, PtZnIWI/SiO2, and Pt/ZnOACT, in agreement with the results from electron microscopy. To investigate whether the nanoalloy particles observed macroscopically by XRD and microscopically by STEM have the same composition, further STEM experiments were carried out investigating spent PtZnALD/SiO2. Many micrographs were analysed to find nanoalloy particles that are oriented at the edge of the supporting particles without any particles lying beneath or on top. The results are shown in Fig. S7.† Two suitable nanoparticles were chosen, and EDX analysis was performed. The results show that in both cases the mass fractions of Pt and Zn match very well the composition of the Pt1Zn1 nanoalloy particles observed by XRD. Therefore, it can be safely assumed that Pt1Zn1 nanoalloy particles are the active phase, and the under reaction conditions most stable alloy phase.
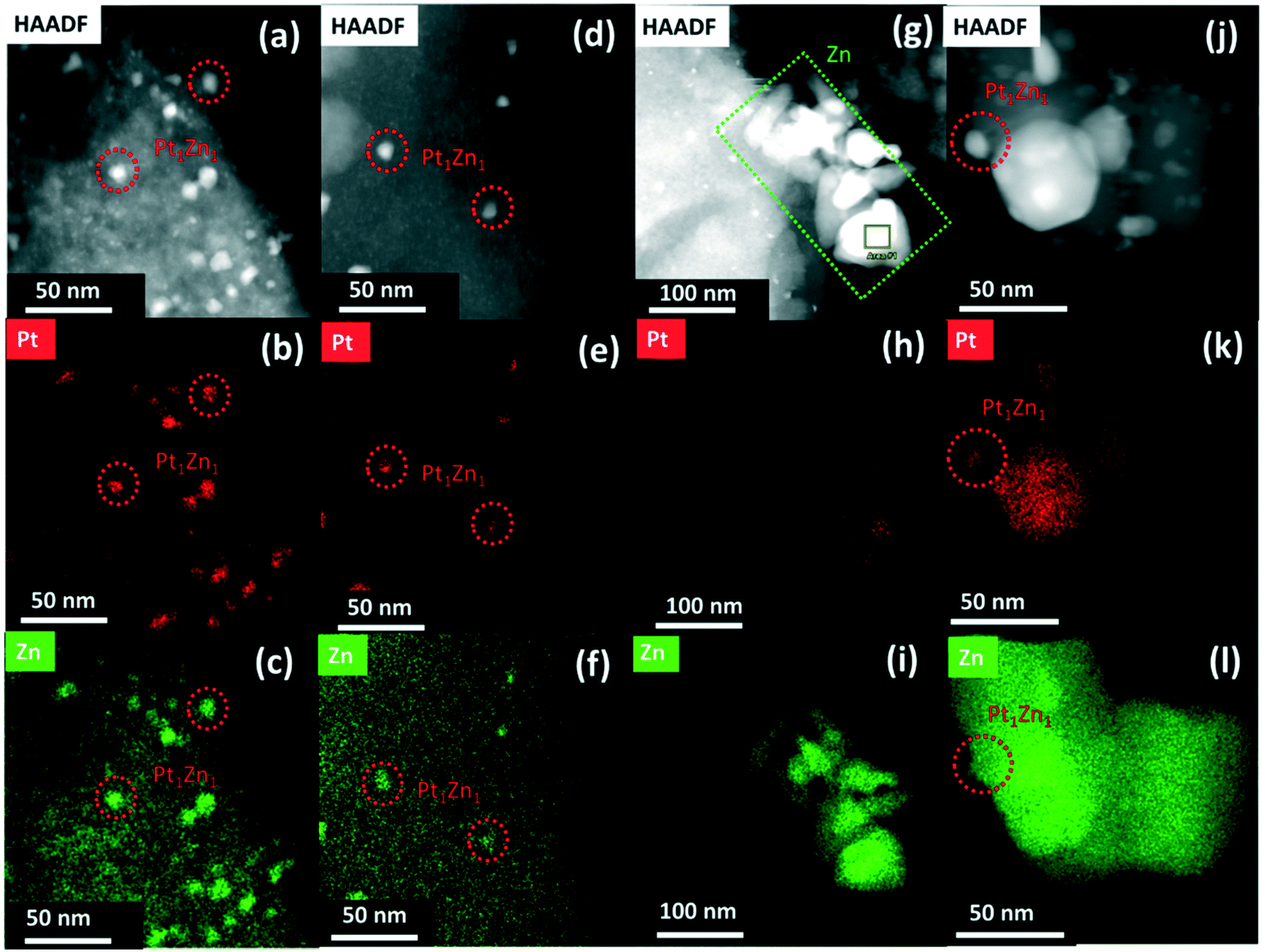 | ||
| Fig. 6 HAADF-STEM image and EDX maps of Pt and Zn in spent (a–c) PtZnALD/SiO2 showing formed Pt1Zn1 nanoalloys; (d–f) PtZnIWI/SiO2, indicating formed Pt1Zn1 nanoalloys as well as (g–i) severe aggregation of Zn away from Pt; (j–l) Pt/ZnOACT, indicating formed Pt1Zn1 alloy particles. | ||
Discussions
Previous studies reported that Zn2+ species are active in propane dehydrogenation.26,28,44–47 However, homogeneously coated ZnALD/SiO2 performed not very well and cannot be considered a relevant catalyst. Pt nanoparticles supported on SiO2 without ZnO exhibited a fast decrease of activity and selectivity. Bimetallic PtZn catalysts synthesized via ALD and IWI showed a high and stable activity and with high selectivity to C3H6. The promotional effect of Zn on Pt catalysts was clearly observed. The formation of Pt1Zn1 nanoalloy particles during activation or reaction was confirmed for all bimetallic catalysts. The alloy formation results in electronic modification of Pt but separation of larger Pt ensembles by Zn atoms may also play a role.15 The hydrogenolysis of propane to lower alkanes is favoured by larger Pt ensembles. Separation of Pt ensembles by Zn atoms via alloy formation is considered to be the main reason of the high propene selectivity of PtZn catalysts.48 The present study shows that the method used to deposit ZnO has a strong influence on the activity of a PtZn catalyst. It is crucial to bring all Pt nanoparticles in direct contact with ZnO to allow formation Pt1Zn1 nanoalloy particles under reducing conditions. ALD leads to a homogeneous dispersion of ZnO on SiO2, completely coating the whole surface of the SiO2 support with strongly chemisorbed ZnO species. In contrast, IWI yields more mobile and inhomogeneously dispersed ZnO particles on the SiO2 support. Only the homogeneously dispersed ZnO gave a direct contact between all Pt species and ZnO and thus ensured the complete formation of PtZn nanoalloy particles.V. J. Cybulskis et al.,15 studied the effect of promoting Pt with Zn for the catalytic dehydrogenation of ethane by in situ X-ray absorption and synchrotron X-ray diffraction. The study indicated that the addition of Zn to Pt nanoparticles lowers the energy level of occupied Pt 5d orbitals thereby weakening the bond between the Pt 5d orbital and adsorbates (reactant and product). Thus, the main dehydrogenation product will quickly desorb from surface of Pt, avoiding deep dehydrogenation and other side reactions. The geometric and electronic effect of promoting Pt with another element is also studied for Fe,48 Ce,49,50 Co,19 and Mn.20 In general, an increase in olefin selectivity is observed when Pt is combined with another element.
Many different strategies such as impregnation,14 ion exchange,41 and a single source precursor approach using bimetallic metalorganic precursors51 were investigated to form PtZn nanoalloy particles on different supports. Impregnation and ion exchange techniques typically lead to inhomogeneous deposition of Pt and Zn on the support. The single source precursor approach yields precisely formed PtZn nanoalloy particles.51 However, the single source precursor approach already requires immense efforts when a scale-up to the range of 10 to 25 g of catalyst is considered.
One previous study also used an ALD approach to synthesize a PtZn catalyst for butane dehydrogenation.52 However, the study focussed on the catalytic performance rather than the synthesis. Pt and Zn were both deposited via ALD, and it is not clear whether the observed effects are due to Zn or Pt ALD. The authors did not investigate the homogeneity of ZnO deposition in detail. The fact that Zn loading was varied by changing the Zn deposition temperature from 175 °C to 100 °C and the amount of substrate by a factor of ∼3 may be an indication for inhomogeneous deposition. The main reason for the homogeneity of via ALD prepared materials is the self-limitation of the reaction leading to a saturation of the surface. Generally, the independence of the loading from temperature and amount of dosed precursor is taken as proof that the conditions for ALD are fulfilled and the process operates within the ALD window.53–56 Furthermore, Libera et al.,57 reported that the deposition of Zn via ALD on SiO2 particles at temperatures above 150 °C yields metallic Zn and ZnO simultaneously. The presence of metallic Zn is reported as an indication of a chemical vapour deposition (CVD) process occurring in parallel. Please note that in general CVD leads to less homogeneously distributed deposition.58,59 The ALD process applied in this study for the deposition of ZnO on SiO2 particles was carefully developed and thoroughly investigated to ensure maximum homogeneity of the Zn distribution. Details and descriptions are given elsewhere.37 Therefore, to the best of our knowledge, this study is the first study that uses ALD of ZnO to synthesize in a controlled way a bimetallic PtZn nanoalloy catalyst for dehydrogenation of alkanes.
Conclusions
In summary, we applied a clear synthesis strategy to tailor the interface between Pt and SiO2 by deposition of a thin layer of ZnO. The direct contact between Pt and ZnO resulted in the complete transformation of all Pt nanoparticles into Pt1Zn1 nanoalloy particles under reducing conditions. The activated PtZnALD/SiO2 catalyst showed exceptional activity and stability in the reaction of propane dehydrogenation. Incorporation of Zn into Pt results in electronic as well as geometric modification of the active catalyst surface, which leads to a very high propene selectivity of 96%. Only one ALD cycle was needed to deposit a sufficient amount of ZnO for the boost in propane dehydrogenation performance. The present study established a novel yet easily scalable synthetic approach to design an interface between metal nanoparticles and the support with a functional layer in between. This synthesis strategy can be easily transferred to other catalyst systems where nanoalloy particles or strong metal support interactions (SMSI) play an important role.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the support by the Einstein Foundation Berlin (ESB) – Einstein Center of Catalysis (EC2). The work was conducted in the framework of the BasCat – UniCat BASF JointLab. The authors gratefully acknowledge the support by C. Eichenauer (AK Thomas, TU Berlin, N2 physisorption measurements), Dr. F. Girgsdies (FHI, Berlin, XRD measurements), Dr. B. Frank (BasCat, thermodynamic calculations), and Jan Meissner (BasCat, technical support).References
- J. C. Bricker, Top. Catal., 2012, 55, 1309–1314 CrossRef CAS
.
- B. V. Vora, Top. Catal., 2012, 55, 1297–1308 CrossRef CAS
- S. Kim, S. Jeong and E. Heo, Energy Sources, Part B, 2019, 14, 49–66 CrossRef CAS
- M. M. Bhasin, J. H. McCain, B. V. Vora, T. Imai and P. R. Pujadó, Appl. Catal., A, 2001, 221, 397–419 CrossRef CAS
-
D. E. Resasco and G. L. Haller, Catalysis, ed. J. Spivey and S. Agarwal, Royal Society of Chemistry, 2007, vol. 11, pp. 379–411 Search PubMed
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS
- H. Z. Wang, L. L. Sun, Z. J. Sui, Y. A. Zhu, G. H. Ye, D. Chen, X. G. Zhou and W. K. Yuan, Ind. Eng. Chem. Res., 2018, 57, 8647–8654 CrossRef CAS
- Q. Li, Z. Sui, X. Zhou, Y. Zhu, J. Zhou and D. Chen, Top. Catal., 2011, 54, 888–896 CrossRef CAS
- J. J. H. B. Sattler, A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2013, 15, 12095–12103 RSC
- H. N. Pham, J. J. H. B. Sattler, B. M. Weckhuysen and A. K. Datye, ACS Catal., 2016, 6, 2257–2264 CrossRef CAS
- S. Sahebdelfar, M. T. Ravanchi, F. Tahriri Zangeneh, S. Mehrazma and S. Rajabi, Chem. Eng. Res. Des., 2012, 90, 1090–1097 CrossRef CAS
- J. J. H. B. Sattler, I. D. Gonzalez-Jimenez, L. Luo, B. A. Stears, A. Malek, D. G. Barton, B. A. Kilos, M. P. Kaminsky, T. W. G. M. Verhoeven, E. J. Koers, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2014, 53, 9251–9256 CrossRef CAS
- K. Searles, K. W. Chan, J. A. Mendes Burak, D. Zemlyanov, O. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 11674–11679 CrossRef CAS
- C. Chen, M. Sun, Z. Hu, J. Ren, S. Zhang and Z. Y. Yuan, Catal. Sci. Technol., 2019, 9, 1979–1988 RSC
- V. J. Cybulskis, B. C. Bukowski, H. T. Tseng, J. R. Gallagher, Z. Wu, E. Wegener, A. J. Kropf, B. Ravel, F. H. Ribeiro, J. Greeley and J. T. Miller, ACS Catal., 2017, 7, 4173–4181 CrossRef CAS
- G. Liu, L. Zeng, Z. J. Zhao, H. Tian, T. Wu and J. Gong, ACS Catal., 2016, 6, 2158–2162 CrossRef CAS
- Z. Han, S. Li, F. Jiang, T. Wang, X. Ma and J. Gong, Nanoscale, 2014, 6, 10000–10008 RSC
- Z. Ma, Z. Wu and J. T. Miller, Catal., Struct. React., 2017, 3(1–2), 43–53 CrossRef CAS
- L. G. Cesar, C. Yang, Z. Lu, Y. Ren, G. Zhang and J. T. Miller, ACS Catal., 2019, 5231–5244 CrossRef CAS
- Z. Wu, B. C. Bukowski, Z. Li, C. Milligan, L. Zhou, T. Ma, Y. Wu, Y. Ren, F. H. Ribeiro, W. N. Delgass, J. Greeley, G. Zhang and J. T. Miller, J. Am. Chem. Soc., 2018, 140, 14870–14877 CrossRef
- F. Jiang, L. Zeng, S. Li, G. Liu, S. Wang and J. Gong, ACS Catal., 2015, 5, 438–447 CrossRef CAS
- N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS
- A. Kolodziejczak-Radzimska and T. Jesionowski, Materials, 2014, 7, 2833–2881 CrossRef CAS
- O. B. Belskaya, L. N. Stepanova, T. I. Gulyaeva, S. B. Erenburg, S. V. Trubina, K. Kvashnina, A. I. Nizovskii, A. V. Kalinkin, V. I. Zaikovskii, V. I. Bukhtiyarov and V. A. Likholobov, J. Catal., 2016, 341, 13–23 CrossRef CAS
- Z. Nawaz, T. Xiaoping and W. Fei, Korean J. Chem. Eng., 2009, 26, 1528–1532 CrossRef CAS
- C. Chen, Z. Hu, J. Ren, S. Zhang, Z. Wang and Z. Y. Yuan, ChemCatChem, 2019, 11, 868–877 CrossRef CAS
- T. Gong, L. Qin, J. Lu and H. Feng, Phys. Chem. Chem. Phys., 2016, 18, 601–614 RSC
- J. Camacho-Bunquin, P. Aich, M. Ferrandon, A. “Bean” Getsoian, U. Das, F. Dogan, L. A. Curtiss, J. T. Miller, C. L. Marshall, A. S. Hock and P. C. Stair, J. Catal., 2017, 345, 170–182 CrossRef CAS
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS
- B. J. Oneill, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef CAS
- J. Lu, B. Fu, M. C. Kung, G. Xiao, J. W. Elam, H. H. Kung and P. C. Stair, Science, 2012, 335, 1205–1208 CrossRef CAS
- P. Ingale, C. Guan, R. Kraehnert, R. Naumann d’Alnoncourt, A. Thomas and F. Rosowski, Catal. Today DOI:10.1016/j.cattod.2020.04.050
- K. Cao, Q. Zhu, B. Shan and R. Chen, Sci. Rep., 2014, 5, 8470 CrossRef
- V. E. Strempel, D. Löffler, J. Kröhnert, K. Skorupska, B. Johnson, R. Naumann d'Alnoncourt, M. Driess and F. Rosowski, J. Vac. Sci. Technol., A, 2016, 34, 01A135 CrossRef
- J. S. Jur and G. N. Parsons, ACS Appl. Mater. Interfaces, 2011, 3, 299–308 CrossRef CAS
- J. Lu, J. W. Elam and P. C. Stair, Surf. Sci. Rep., 2016, 71, 410–472 CrossRef CAS
- P. Ingale, K. Knemeyer, M. P. Hermida, R. Naumann d'Alnoncourt, A. Thomas and F. Rosowski, Nanomaterials, 2020, 10, 981 CrossRef CAS
- V. E. Strempel, R. Naumann D'Alnoncourt, M. Driess and F. Rosowski, Rev. Sci. Instrum., 2017, 88 Search PubMed
- V. E. Strempel, K. Knemeyer, R. Naumann
d’Alnoncourt, M. Driess and F. Rosowski, Nanomaterials, 2018, 8(6), 365 CrossRef
- J. A. Rodriguez and M. Kuhn, J. Chem. Phys., 1995, 102, 4279–4289 CrossRef CAS
- T. Iida, D. Zanchet, K. Ohara, T. Wakihara and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2018, 57, 6454–6458 CrossRef CAS
- J. H. Sinfelt, J. Phys. Chem., 1964, 68, 344–346 CrossRef CAS
- R. D. Cortright, R. M. Watwe and J. A. Dumesic, J. Mol. Catal. A: Chem., 2000, 163, 91–103 CrossRef CAS
- S. M. T. Almutairi, B. Mezari, P. C. M. M. Magusin, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2012, 2, 71–83 CrossRef CAS
- T. Gong, L. Qin, J. Lu and H. Feng, Phys. Chem. Chem. Phys., 2016, 18, 601–614 RSC
- S. M. T. Almutairi, B. Mezari, P. C. M. M. Magusin, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2012, 2, 71–83 CrossRef CAS
- N. M. Schweitzer, B. Hu, U. Das, H. Kim, J. Greeley, L. A. Curtiss, P. C. Stair, J. T. Miller and A. S. Hock, ACS Catal., 2014, 4, 1091–1098 CrossRef CAS
- E. C. Wegener, B. C. Bukowski, D. Yang, Z. Wu, A. J. Kropf, W. N. Delgass, J. Greeley, G. Zhang and J. T. Miller, ChemCatChem, 2020, 12, 1325–1333 CrossRef CAS
- Y. Qiu, X. Li, Y. Zhang, C. Xie, S. Zhou, R. Wang, S. Z. Luo, F. Jing and W. Chu, Ind. Eng. Chem. Res., 2019, 58, 10804–10818 CrossRef CAS
- W. Cai, R. Mu, S. Zha, G. Sun, S. Chen, Z. J. Zhao, H. Li, H. Tian, Y. Tang, F. Tao, L. Zeng and J. Gong, Sci. Adv., 2018, 4, eaar5418 CrossRef CAS
- L. Rochlitz, K. Searles, J. Alfke, D. Zemlyanov, O. V. Safonova and C. Copéret, Chem. Sci., 2020, 11, 1549–1555 RSC
- J. Camacho-Bunquin, M. S. Ferrandon, H. Sohn, A. J. Kropf, C. Yang, J. Wen, R. A. Hackler, C. Liu, G. Celik, C. L. Marshall, P. C. Stair and M. Delferro, ACS Catal., 2018, 20, 10058–10063 CrossRef
- A. W. Weimer, J. Nanopart. Res., 2019, 21, 9 CrossRef
- N. E. Richey, C. De Paula and S. F. Bent, J. Chem. Phys., 2020, 152, 40902 CrossRef CAS
- L. Cao and J. Lu, Cite this Catal. Sci. Technol., 2020, 10, 2695 RSC
- J. Lu, J. W. Elam and P. C. Stair, Surf. Sci. Rep., 2016, 71, 410–472 CrossRef CAS
- J. A. Libera, J. W. Elam and M. J. Pellin, Thin Solid Films, 2008, 516, 6158–6166 CrossRef CAS
- H. H. Sønsteby, A. Yanguas-Gil and J. W. Elam, J. Vac. Sci. Technol., A, 2020, 38, 020804 CrossRef
- G. L. Doll, B. A. Mensah, H. Mohseni and T. W. Scharf, J. Therm. Spray Technol., 2010, 19, 510–516 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01528h |
| This journal is © The Royal Society of Chemistry 2021 |