
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative dehydrogenation of propane on silica-supported vanadyl sites promoted with sodium metavanadate†
Manouchehr
Nadjafi
 a,
Agnieszka M.
Kierzkowska
a,
Paula M.
Abdala
a,
Rene
Verel
b,
Olga V.
Safonova
c,
Alexey
Fedorov
*a and
Christoph R.
Müller
*a
a,
Agnieszka M.
Kierzkowska
a,
Paula M.
Abdala
a,
Rene
Verel
b,
Olga V.
Safonova
c,
Alexey
Fedorov
*a and
Christoph R.
Müller
*a
aDepartment of Mechanical and Process Engineering, ETH Zürich, Leonhardstrasse 21, CH-8092 Zürich, Switzerland. E-mail: fedoroal@ethz.ch; muelchri@ethz.ch
bDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1-5, CH-8093 Zürich, Switzerland
cPaul Scherrer Institute, CH-5232 Villigen, Switzerland
First published on 7th September 2020
Abstract
The promotion of silica-supported vanadyl species [VO4]/SiO2 (1) by α-NaVO3 or β-NaVO3 enhances the specific rate of the propene formation in oxidative dehydrogenation of propane (ODP) by, respectively, 30 and 125% at 450 °C and ca. 1 V nm−2 nominal coverage. The increased rate of propene formation is offset only moderately by a decreased selectivity to propene, which declines by 10 and 15% relative to 1 (74%) in α-NaVO3/1 and β-NaVO3/1, at 5.8 and 8.2% propane conversion. The structural characterization of the promoted catalysts by Raman mapping, X-ray absorption near edge structure (XANES), transmission electron microscopy (TEM) and solid-state nuclear magnetic resonance (51V and 23Na MAS NMR) allowed for associating the higher specific activity of β-NaVO3/1 with a higher dispersion of vanadium sites on the silica support, while the agglomeration of these sites with the concomitant formation of a poorly dispersed Na1+xV3O8 phase is related to a decreased catalytic activity. Surprisingly, solid-state 51V NMR and Raman spectroscopies reveal that the α-NaVO3/1 and β-NaVO3/1 catalysts contain the metastable β-NaVO3 phase, explained by a more favorable interaction of Na1+xV3O8/SiO2, formed after calcination in both materials, with β-NaVO3 than with α-NaVO3.
Introduction
Propene, a fundamental building block in the production of bulk chemicals and polymers,1 is typically obtained as a byproduct from fluid catalytic cracking (FCC) and steam cracking of naphtha.2 However, the ongoing replacement of naphtha by shale gas3 decreases the propene production through this route, which occurs simultaneously with a growing demand for propene.3,4 Industrial, “on-purpose” propene production technologies via propane dehydrogenation (PDH) rely currently on CrOx/Al2O3 or Pt–Sn/Al2O3, both catalysts promoted with Na/K (Catofin and Oleflex processes, respectively).5 These technologies have drawbacks, including coking, high energy demand (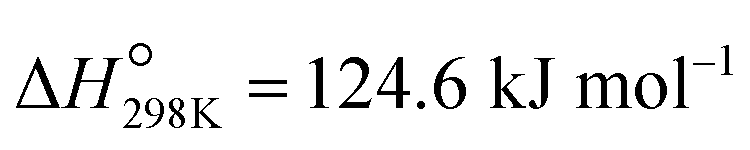 ), low conversions (at the thermodynamic equilibrium, 25% at 527 °C), high price of Pt, and toxicity of CrVI.2,5–7 An alternative to PDH is the oxidative dehydrogenation of propane (ODP) that exothermally converts propane and oxygen to propene and water (
), low conversions (at the thermodynamic equilibrium, 25% at 527 °C), high price of Pt, and toxicity of CrVI.2,5–7 An alternative to PDH is the oxidative dehydrogenation of propane (ODP) that exothermally converts propane and oxygen to propene and water (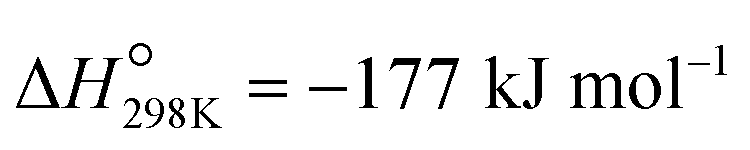 ) at lower temperatures (ca. 450 °C). Propane conversion is not limited by thermodynamics in ODP and coking is avoided due to the use of oxygen, thus providing usually a stable catalytic performance. Despite its potential, no ODP process has yet been industrialized, primarily because of the insufficient selectivity to propene at high propane conversions.6
) at lower temperatures (ca. 450 °C). Propane conversion is not limited by thermodynamics in ODP and coking is avoided due to the use of oxygen, thus providing usually a stable catalytic performance. Despite its potential, no ODP process has yet been industrialized, primarily because of the insufficient selectivity to propene at high propane conversions.6
At low vanadium loadings (such as those used in this work), dehydrated vanadia on oxide supports features mostly site-isolated, surface-grafted tripodal vanadium oxo sites, (–O)3V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, often denoted [VO4] sites.8–20 These species are among the best-performing ODP catalysts.2,6,8,21,22 Alkali dopants were reported to improve propene selectivity of supported VOx catalysts, which was, however, associated with a lower catalytic activity than in the undoped catalysts.23,24 It was argued that the basic alkali doping decreased the strong acidity of the undoped catalysts,23–25 in addition to weakening the VO bond.24,26 Furthermore, Na doping can improve the dispersion of VOx sites on a silica support by increasing the reactivity of surface OH groups of the silica support.26–28
O, often denoted [VO4] sites.8–20 These species are among the best-performing ODP catalysts.2,6,8,21,22 Alkali dopants were reported to improve propene selectivity of supported VOx catalysts, which was, however, associated with a lower catalytic activity than in the undoped catalysts.23,24 It was argued that the basic alkali doping decreased the strong acidity of the undoped catalysts,23–25 in addition to weakening the VO bond.24,26 Furthermore, Na doping can improve the dispersion of VOx sites on a silica support by increasing the reactivity of surface OH groups of the silica support.26–28
Recently, we have shown that a catalyst derived from silica supported sodium decavanadate (Na6V10O28) provides 65% selectivity to propene at 6% propane conversion at 450 °C.29 When heated under air to 600 °C, Na6V10O28 decomposes on the silica surface to the metastable β-NaVO3 phase along with a Na1+xV3O8 phase interacting with the silica support (Na1+xV3O8/SiO2). The formation of β-NaVO3 in the calcined Na6V10O28/SiO2 material under these conditions was surprising because the transformation of bulk and silica-supported β-NaVO3 to α-NaVO3 proceeds at notably lower temperatures than 600 °C and therefore an α-NaVO3 phase would have been expected.30,31 The presence of β-NaVO3 in the calcined Na6V10O28/SiO2 suggests that Na1+xV3O8/SiO2 plays a role in stabilizing β-NaVO3.29
With the objective of improving our understanding of the interaction between the NaVO3 phases and the vanadyl sites on the silica surface, we incipient wetness impregnated (IWI) an aqueous solution of either α-NaVO3 or β-NaVO3 onto a SiO2 support, followed by overnight drying at 100 °C and an IWI of an aqueous solution of NH4VO3. This procedure gave, after calcination, α-NaVO3/1 and β-NaVO3/1 materials with a similar nominal vanadium loading of ca. 1 V nm−2 and a Na/V ratio of ca. 0.6. We find that the phase of the NaVO3 promoter used for the impregnation influences the increase of the initial specific rate for propene formation of the reference catalyst 1, i.e. an increase by 30 and 125% is observed, respectively, for α-NaVO3/1 and β-NaVO3/1. Interestingly, solid state 51V NMR and Raman spectroscopy suggest that α-NaVO3/1 and β-NaVO3/1 contain the metastable β-NaVO3 phase. Yet the catalytic activity of α-NaVO3/1 and β-NaVO3/1 and their deactivation with time on stream (TOS) are different, with the rate of propene formation decreasing after 4 h by 12 and 21%, respectively. Agglomeration of Na and V species in the used catalyst was identified as the driving force for the deactivation. We explain the higher activity of β-NaVO3/1 compared to α-NaVO3/1 by the higher dispersion of β-NaVO3-Na1+xV3O8/SiO2 species in β-NaVO3/1 (Scheme 1).6,20,32
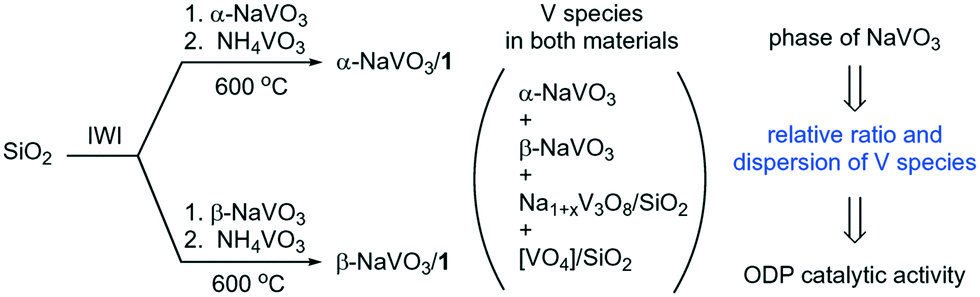 | ||
| Scheme 1 Silica-supported materials and vanadium species prepared in this work. (IWI stands for incipient wetness impregnation). | ||
Results and discussion
Incipient wetness impregnation of ammonium metavanadate6,8 was used to prepare [VO4]/SiO2 (1, 2.1 wt% V by ICP, Table S1†) containing ca. 1 V nm−2, which is below the monolayer coverage for SiO2 support.17 Vanadyl sites on silica have been characterized in details in previous reports.13,14,17,28,29,33–36 Since the nature of the supported VOx species changes with hydroxylation of the support,17,32,33 the materials discussed below were treated under synthetic air (500 °C, 1 h, 30 ml min−1) and stored in a glovebox (H2O and O2 < 0.5 ppm), indicated by the respective subscript notation, for instance 1(500-air). To prepare α-NaVO3/1 and β-NaVO3/1 materials, silica was impregnated first with aqueous solutions of α-NaVO3 or β-NaVO3 (0.6 V nm−2) followed by overnight drying at 100 °C and a subsequent IWI of NH4VO3 (0.4 V nm−2). Calcined α-NaVO3/1 and β-NaVO3/1 contained 1.9 and 2.0 wt% of vanadium and a molar ratio of Na/V of 0.63 and 0.62 (by ICP), respectively, corresponding to a nominal silica coverage of ca. 1 V nm−2. Although the nominal vanadium loading of the as impregnated α- and β-NaVO3 promoted catalysts were similar to that of the benchmark catalyst (1 V nm−2, ca. 2 wt%), the surface density of V calculated from the ICP-determined V loading and the specific surface area of the material (according to BET N2 physisorption measurements) was notably higher for the promoted catalysts compared to 1 (i.e. 1.6 and 2.0 V nm−2vs. 1.1 V nm−2, see Table S1,† entries 1–4). This is explained by a reduced surface area of the silica support due to the etching effect caused by Na-containing precursors.25 We have therefore optimized the loading of vanadium precursors in order to obtain a comparable surface density of V in the promoted catalysts and in 1. This was achieved for α- and β-NaVO3 promoted catalysts with a lower nominal vanadium loading (i.e. 0.7 V nm−2, denoted in a subscript), resulting in 1.2 and 1.0 V nm−2, respectively, after the calcination (Table S1,† entries 5–6). Lastly, note that the dissolution of β-NaVO3 in water gives dihydrate species, NaVO3·(2H2O) that transform, above ca. 34 °C, to β-NaVO3.31,37 The irreversible transformation of β-NaVO3 to α-NaVO3 was reported to occur at 403–405 °C.30The Raman spectrum of 1(500-air) features a characteristic sharp peak at 1039 cm−1 owing to the vanadium oxo stretching vibration29,33,38,39 that is significantly reduced in intensity in β-NaVO3/1(500-air) and α-NaVO3/1(500-air) (Fig. 1a and S1†). The latter materials contain also bands of β-NaVO3 and α-NaVO3 at 945 and 954 cm−1, respectively, but with diverging intensities. The characteristic Raman band of β-NaVO3 is minor and the band of α-NaVO3 is major in β-NaVO3/1(500-air). In α-NaVO3/1(500-air), the band of β-NaVO3 is more intense than the band of α-NaVO3, although both bands are less intense than in β-NaVO3/1(500-air). In addition, two broad peaks at 804 and 737 cm−1 that match the peak positions in the Na1+xV3O8/SiO2(500-air) reference are observed in β-NaVO3/1(500-air) and, to a larger extent, in α-NaVO3/1(500-air) (Fig. 1a).29 In this Na1+xV3O8/SiO2(500-air) material, the Na1+xV3O8 phase interacts with the SiO2 support, as evidenced by Raman, 51V and 23Na NMR data, although the exact nature of the formed sites is currently unclear.29 The Raman peaks at 804 and 737 cm−1 in α-NaVO3/1(500-air) are more intense in comparison to β-NaVO3/1(500-air), probably due to the lower dispersion and increased long-range order of the Na1+xV3O8 phase in α-NaVO3/1(500-air); besides, the β-NaVO3 peak has a lower intensity in β-NaVO3/1(500-air). Considering that α-NaVO3/SiO2(500-air) (i.e. the catalyst made by IWI of α-NaVO3 on silica at a nominal vanadium loading of 1 V nm−2) does not feature peaks at 804 and 737 cm−1,29 these bands must have been formed owing to an interaction between [VO4]/SiO2 and α-/β-NaVO3. These species may feature different degrees of dispersion and/or crystallinity, which leads to different intensities in Raman spectra (vide infra).33,40
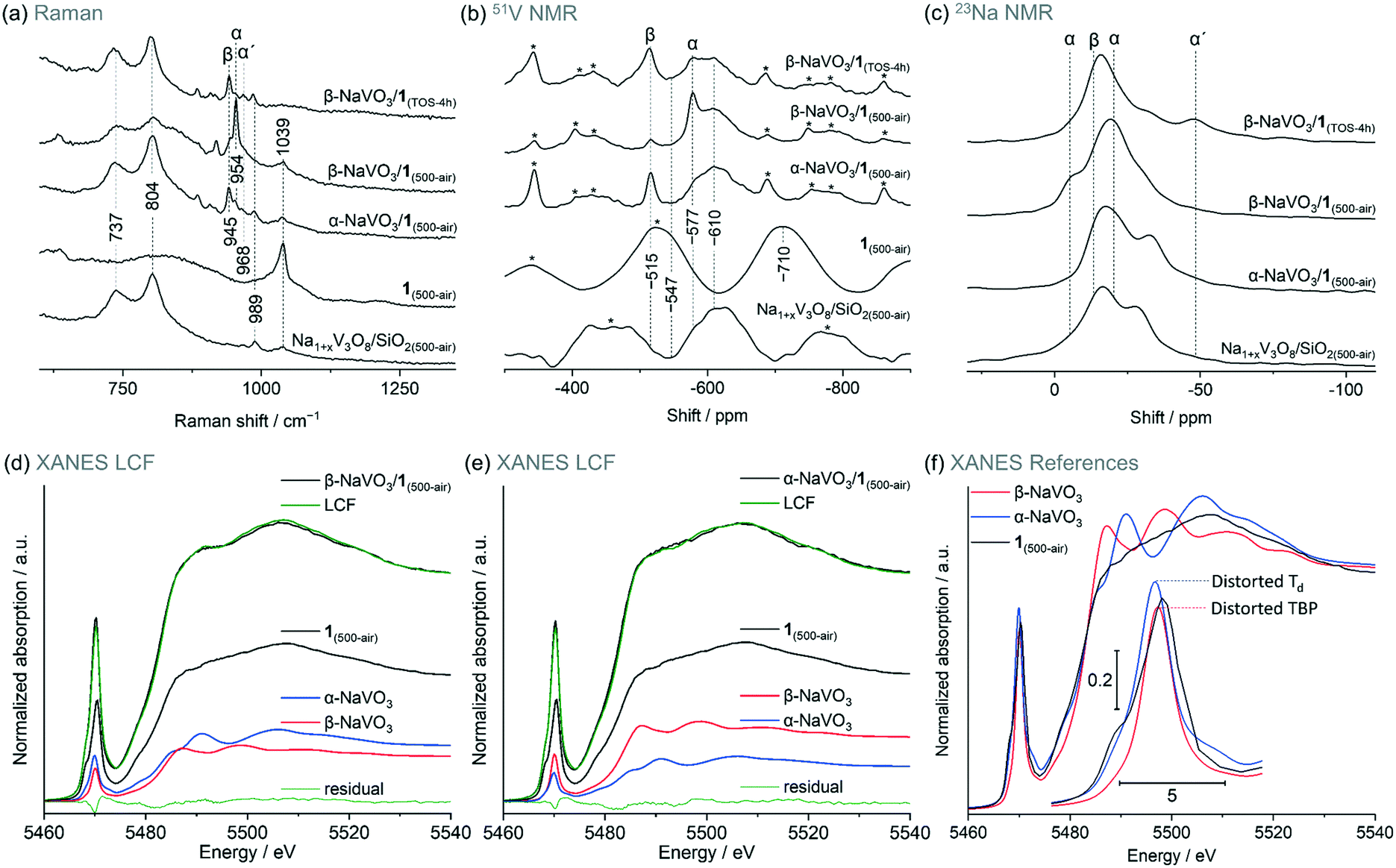 | ||
| Fig. 1 (a) Raman, (b) 51V and (c) 23Na MAS NMR spectra of the studied materials (see labels in the panels); (d) and (e) are linear combination fittings (LCF, see Table 1 for details) of the V K-edge XANES of α-NaVO3/1 and β-NaVO3/1 dehydroxylated at 500 °C as well as f) V K-edge XANES spectra of the reference materials. Subscript TOS in hours indicates a used catalyst that was cooled down to room temperature while flowing the ODP gas mixture and handled in pristine conditions. Side bands of the NMR spectra are marked by asterisks; a spinning rate of 15–18 kHz was used. Notations α, β, and α′ indicate α-NaVO3, β-NaVO3 and α′-NaV2O5 phases, respectively. Characterization of Na1+xV3O8/SiO2(500-air) was reported by us previously and is reproduced here for comparison purposes.29 | ||
It is conceivable that the melting of α-NaVO3 on the SiO2 surface upon calcination and its subsequent recrystallization during cooling yielded the metastable β-NaVO3 polymorph, owing to the more favorable interaction of this polymorph with the V-based, supported species. To test this hypothesis, we calcined α-NaVO3/1 to ca. 600 °C in situ in a Raman cell (Linkam CCR1000) under flow of synthetic air (30 ml min−1). By recording spectra from the various regions of the specimen heated to ca. 600 °C and then cooled down to room temperature, we observed an inhomogeneous distribution of vanadium species, which is possibly related to heat transfer gradients in the in situ Raman cell. Specifically, two distinct areas were found, viz. areas with peaks of α-NaVO3 and isolated vanadyl sites, as well as areas containing predominantly peaks of β-NaVO3 and Na1+xV3O8/SiO2 (beam spot size was ca. 1.6 μm, Fig. S2†). In a control experiment, calcination of α-NaVO3/1 in a muffle furnace at 600 °C for 4 h with the subsequent exposure to air gave a more homogeneous material that predominately features peaks of β-NaVO3 and Na1+xV3O8; only occasionally areas with α-NaVO3 and [VO4] sites are found (Fig. S3†). These experiments suggest that α-NaVO3 may react with [VO4]/SiO2 to give Na1+xV3O8/SiO2 and β-NaVO3. This mechanism for the formation of metastable β-NaVO3 does not necessarily require recrystallization of the molten NaVO3. Indeed, a differential scanning calorimetry (DSC) experiment of the calcination of α-NaVO3/1 reveals no clear features due to melting and recrystallization (Fig. S4†).
51V magic angle spinning (MAS) NMR spectra of β-NaVO3, α-NaVO3, and 1(500-air) give signals at −515, −577, and −710 ppm, respectively. In the 23Na NMR spectrum of β-NaVO3, one peak is observed ca. −13 ppm while two signals centered at −5 and −20 ppm are observed for α-NaVO3 (Fig. 1b and S5†).11,29,36,41 In line with Raman spectroscopy results, peaks due to β- and α-NaVO3 are observed in the 51V NMR spectra of α-NaVO3/1(500-air) and β-NaVO3/1(500-air); the signal from β-NaVO3 is more intense for α-NaVO3/1(500-air). A broad feature at ca. −610 ppm is observed for both promoted catalysts and Na1+xV3O8/SiO2(500-air). At least in part, this broad feature may be due to the vanadyl sites interacting with a Na+ cation, which induces a downfield shift by 100 ppm compared to that in 1(500-air) (Fig. 1b).26,28,29 Note that a broad shoulder at the same position of ca. −610 ppm is also observed for α-NaVO3/SiO2(500-air), and it is likely due to a partial decomposition of silica-supported α-NaVO3 to vanadyl sites interacting with the nearby sodium cations on surface siloxides ((–O)3VO⋯Na+, Fig. S6†). Consistent with our inferences from the Raman and 51V NMR data, 23Na MAS NMR of the promoted catalysts shows peaks that can be ascribed to α-, β-NaVO3, and Na1+xV3O8/SiO2 species (Fig. 1c). Interestingly, the feature due to β-NaVO3 centered at −13 ppm is more prominent in α-NaVO3/1(500-air) while two features due to α-NaVO3 (centered at −21 and −5 ppm) are more prominent in β-NaVO3/1(500-air) and are not noticeable in α-NaVO3/1(500-air). 23Na MAS NMR spectra of Na1+xV3O8/SiO2(500-air) and α-NaVO3/1(500-air) are similar, with a ca. 5 ppm upfield shift of peaks in the latter material. This indicates that Na1+xV3O8/SiO2 is a major phase in α-NaVO3/1(500-air) and that this material has nearly no Na atoms in the environment of α-NaVO3. Features of Na1+xV3O8/SiO2 are less prominent in β-NaVO3/1(500-air) (assessed by the peak at −28 ppm) and this is offset by more intense features of α- and β-NaVO3.
The intensity of the pre-edge peak in V K-edge XANES depends on the symmetry of the ligand sphere around the vanadium atom such that a more centro-symmetric environment gives lower pre-edge peak heights in the order: tetrahedral (Td) > distorted tetrahedral > square pyramidal (SP) > distorted octahedral (Oh) > octahedral (Oh, Fig. S7†).17,29,42 The spectra of 1(air-500) is consistent with V sites in a Td coordination.29 The V K-edge XANES spectra of Na1+xV3O8/SiO2 and 1 (exposed to air or dehydroxylated) are similar (Fig. S8 and S9†), indicating structural similarities of the V sites in those materials. To quantify the different V species present in the promoted materials we used linear combination fitting (LCF) of the V K-edge XANES spectra of the α- and β-NaVO3/1(500-air). In this analysis, we used the well-defined material 1(air-500) as one of the references, as well as α-NaVO3 and β-NaVO3. LCF analysis yielded a slightly higher fraction of α-NaVO3 than β-NaVO3 in β-NaVO3/1(500-air) (24 and 20%, respectively, Table 1, entry 1) and a moderately higher fraction of β-NaVO3 in α-NaVO3/1(500-air) in comparison to α-NaVO3 (28 and 16%, respectively, Table 1, entry 2, and Fig. 1d–f). These obtained phase percentages are consistent with the Raman and MAS NMR observations described above. Similar values for 1(500-air) were obtained for both promoted materials (56%).
| Entry | Material | β-NaVO3 | α-NaVO3 | 1 (500-air) |
|---|---|---|---|---|
| a Representing Na1+xV3O8/SiO2(500-air) (see Fig. S8 and S9†). | ||||
| 1 | β-NaVO3/1(500-air) | 20 | 24 | 56 |
| 2 | α-NaVO3/1(500-air) | 28 | 16 | 56 |
| 3 | β-NaVO3/1(TOS-4h) | 28 | 21 | 51 |
To investigate the dispersion of the α-NaVO3, β-NaVO3 and Na1+xV3O8/SiO2(500-air) phases in α-NaVO3/1(500-air) and β-NaVO3/1(500-air), Raman maps were collected. The freshly calcined materials were sealed in quartz capillaries under an inert atmosphere and Raman maps acquired from in total 225 points (15 × 15) separated by 4 μm (the laser spot size was ca. 1.6 μm). The intensities of the characteristic Raman peaks at 954, 945, 804 cm−1 (±2 cm−1) were used to map α-NaVO3, β-NaVO3, and Na1+xV3O8/SiO2, respectively (Fig. 2a and S10†). We observe that α-NaVO3 is less uniformly dispersed than β-NaVO3 in α-NaVO3/1(500-air) or β-NaVO3/1(500-air). In addition, Na1+xV3O8/SiO2 appears more abundant and less well dispersed in α-NaVO3/1(500-air) relative to β-NaVO3/1(500-air). Yet for both promoted materials, the distribution of the intensities of β-NaVO3 and Na1+xV3O8/SiO2 in the Raman maps is relatively similar, i.e. these phases are found similarly dispersed, which suggests an interaction between these two phases. Furthermore, EDX mapping of β-NaVO3/1 and α-NaVO3/1 (exposed to air during the sample transfer) shows a more uniform dispersion of Na and V in β-NaVO3/1 compared to α-NaVO3/1 that shows agglomerates of a Na/V rich phase (Fig. 2b).
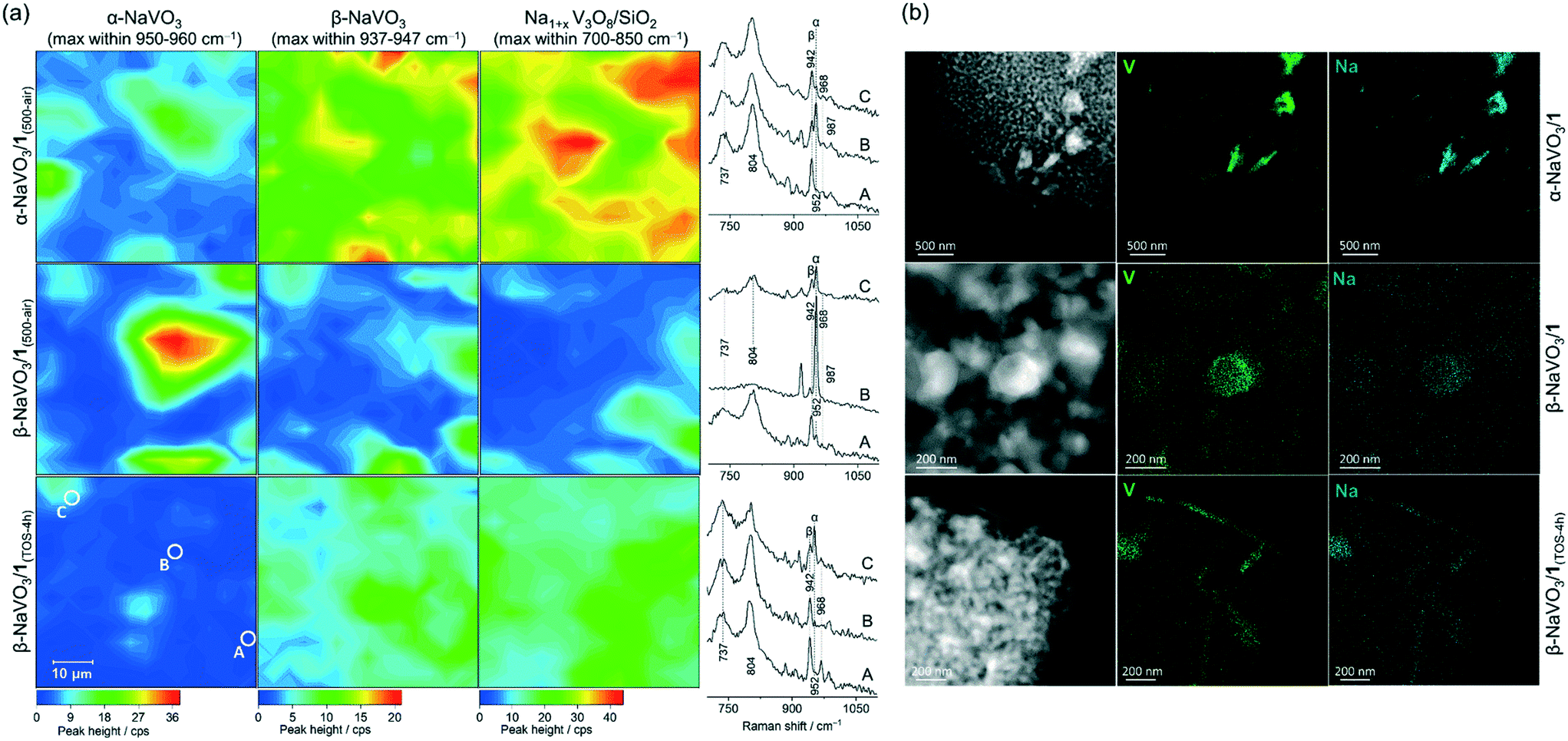 | ||
| Fig. 2 (a) Raman mapping of α-NaVO3/1(500-air), β-NaVO3/1(500-air) and β-NaVO3/1(TOS-4h) cooled down in an ODP atmosphere; the spectra shown correspond to the areas marked with A, B, and C (bottom left panel). (b) EDX mapping of α- and β-NaVO3/1 as well as β-NaVO3/1(TOS-4h) (exposed to air). | ||
The reducibility of supported vanadium-based catalysts for the oxidative dehydrogenation of propane and methanol (with vanadium loading below the monolayer coverage) was previously correlated with the turn over frequency (TOF) of those catalysts, such that higher reducibility is typically associated with higher activity.6,43,44 However, a counter example is crystalline V2O5 on Al2O3 promoted with molybdenum that showed higher conversions and selectivities in ODP with decreasing reducibility of vanadium, as assessed by the temperature corresponding to the maximum of H2 consumption (Tmax) in the H2 temperature-programmed reduction (TPR) experiment.45 This indicates that the activity and selectivity of the V-based catalysts for ODP does not depend solely on their reducibility6,43 but is influenced by other factors, for instance, the interaction with the support,20,43 dispersion of the active phase,29,46,47 V–O binding energy,45 and acid–base properties.48
Considering that α-NaVO3/1 and β-NaVO3/1 contain similar species, but show distinct catalytic activity (vide infra), we were interested to compare H2-TPR profiles of these two catalysts. A slightly lower Tmax was observed for the more active catalyst, β-NaVO3/1, as compared to α-NaVO3/1 (555 and 580 °C, respectively, Fig. S11†), while Tmax of the less active Na1+xV3O8/SiO2 and 1 catalysts were centered at 602 and 473 °C, consistent with the different nature of vanadium species in studied catalysts.
In summary, Raman spectroscopy, 51V and 23Na MAS NMR data as well as LCF of XANES spectra show that α- and β-NaVO3 as well as Na1+xV3O8 interacting with the silica support are present in α-NaVO3/1(500-air) and β-NaVO3/1(500-air) materials, albeit in different relative amounts. The dispersion of Na and V species is higher in β-NaVO3/1(500-air) compared to α-NaVO3/1(500-air) according to Raman and EDX mapping. By NMR and Raman spectroscopies, a higher fraction of Na1+xV3O8/SiO2 and β-NaVO3 is found in α-NaVO3/1(500-air) relative to β-NaVO3/1(500-air). The unexpected formation of the metastable β-NaVO3 polymorph from the thermodynamically stable α-NaVO3 is likely due to the stabilizing interaction between β-NaVO3 and Na1+xV3O8/SiO2 species, as compared to the respective interaction with α-NaVO3. This is supported by the fact that the calcination of β-NaVO3 on silica without vanadyl sites leads to α-NaVO3 (Fig. S12†).29 The materials Na1+xV3O8/SiO2 and α-NaVO3/SiO2(500-air) likely contain structurally similar (–O)3VO⋯Na+ sites, as suggested by the characteristic broad feature in the respective 51V MAS NMR spectra at −610 ppm (Fig. 1b and S6a†). However, Raman bands at 737 and 804 cm−1 due to V–O–V bonds, diagnostic for Na1+xV3O8/SiO2(500-air), are not observed for α-NaVO3/SiO2(500-air).17,29 Studies to refine our understanding of the nature of sites in Na1+xV3O8/SiO2 go beyond the scope of this work.
When the weight loadings of vanadium in the promoted catalysts were similar to that of the benchmark catalyst 1 (ca. 2 wt%, Na/V = 0.6), a higher initial specific activity was obtained for α- and β-NaVO3/1 catalysts (3.4 and 5.7 mmol C3H6 mol V−1 s−1) than for 1 (2.5 mmol C3H6 mol V−1 s−1), albeit the propene selectivities of α-NaVO3/1 and β-NaVO3/1 (64 and 59%) were lower than of 1 (74%, Fig. 3a). Yet α-NaVO3/1 and β-NaVO3/1 deactivate with TOS, after 4 h by 12 and 21%, respectively. However, the specific activity of α-NaVO3/1(0.7V) and β-NaVO3/1(0.7V), i.e. materials with a similar vanadium surface density to that of 1 (ca. 1.1 V nm−2 obtained at ca. Na/V ratio of 0.3, Table S1†) were 2.5 and 3.5 mmol C3H6 mol V−1 s−1, respectively, while a similar selectivity of 74% was observed for all three these catalysts. Noteworthy, α-NaVO3/1(0.7V) and β-NaVO3/1(0.7V) did not deactivate with TOS after 240 min (at ca. 2.9 and 4.0% conversion, respectively, Fig. S14†). These results demonstrate that the surface density of Na and V influence catalyst activity, selectivity and stability.
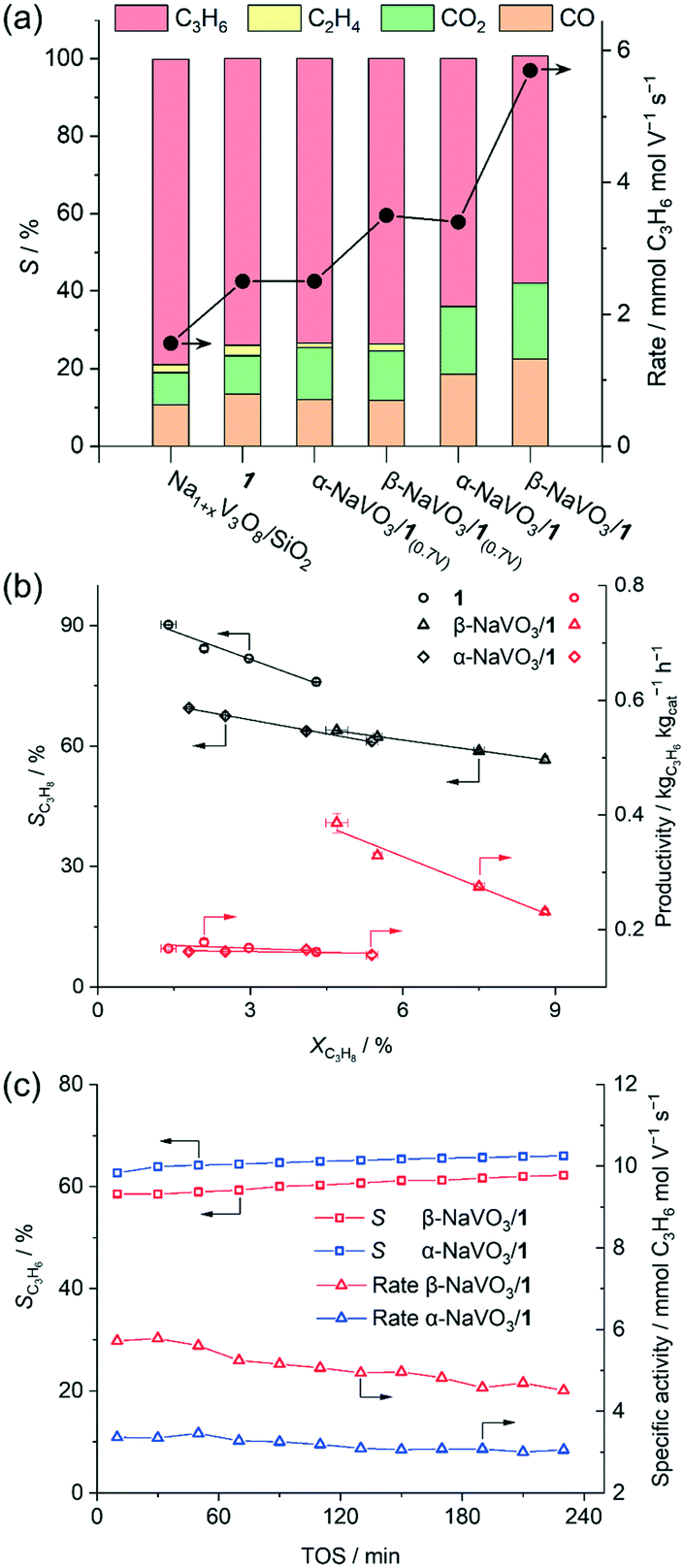 | ||
| Fig. 3 (a) Initial catalytic activity (TOS = 30 min), (b) propene selectivity and productivity vs. propane conversion for the studied catalysts (see Table S1†). WHSV was varied between 5.1–13.6 h−1 by changing the total feed flow (15.8–42 ml min−1). (c) Changes with TOS of β- and α-NaVO3/1 under ODP conditions (C3H8: air = 2: 5, total flow of 21 ml min−1, 450 °C). | ||
By increasing the contact time, the conversion increases, yet the selectivity of the benchmark catalyst (1) drops with a higher rate compared to both promoted catalysts (Fig. 3b). For a nominal V loading of 1 V nm−2, the benchmark catalyst 1 shows a higher selectivity to propene compared to the promoted catalysts at similar conversions that did not exceed 8% (Fig. 3b). Our promoted catalysts show higher propene selectivities at conversions exceeding 10%, i.e. 58, 51 and 41% at 13, 15 and 13% propane conversion for β-NaVO3/1, α-NaVO3/1 and 1, respectively (Fig. S13†). Overall, these values translate into higher initial productivities (within 30 min) for the promoted catalysts relative to 1.
Furthermore, the productivity of β-NaVO3/1 is ca. 2.4 times higher than that of 1 at similar propane conversion (0.39 vs. 0.16 kgC3H6 kgcat−1 h−1 at 4.7 vs. 4.3%, respectively, Fig. 3b). Interestingly, Na1+xV3O8/SiO2 showed the highest initial selectivity to propene among the studied catalysts, reaching 80% at a 2.2% propane conversion, i.e. slightly higher than the sodium-free benchmark catalyst 1 (77% at 3.6% propane conversion at 450 °C). In addition to propene, Na1+xV3O8/SiO2 and 1 produced up to ca. 3% C2H4 while α-NaVO3/1 and β-NaVO3/1 only gave propene and COx, with an initial propene selectivity of 64 and 59% at 5.8 and 8.2% conversions, respectively. At similar reaction conditions (WHSV = 6.8 h−1), the Na6V10O28/SiO2 catalyst reported by us previously29 showed 65% propene selectivity at 6% propane conversion.
Notably, while the ODP activities of catalysts 1 and Na1+xV3O8/SiO2 are stable (Fig. S15,† typical for V-based ODP catalysts),29 the catalytic activity of both promoted materials decreases with TOS (Fig. 3c). We observe a decline of the specific activity by 12 and 21% within 4 h TOS for α-NaVO3/1 and β-NaVO3/1, respectively. Comparison of the TEM images and EDX mapping of β-NaVO3/1 after 4 h TOS to the fresh catalyst reveals agglomeration of the Na and V species on the silica surface after the ODP reaction, which is a likely reason for deactivation (Fig. 2b). The Raman spectrum of the reacted β-NaVO3/1 (denoted β-NaVO3/1(TOS-4h); the material was handled without exposure to air) shows a tangible increase in the intensity of the peaks at 737 and 804 cm−1 compared to the fresh catalyst. This can indicate the formation of a less dispersed Na1+xV3O8 phase compared to the fresh catalyst and would be in line with the TEM analysis. The formation of three-dimensional V2O5 crystals has been reported to deactivate VOx-based ODP catalysts.6,20 Furthermore, the strongly reduced intensity of the 954 cm−1 peak of α-NaVO3 might be due to its reaction with [VO4] sites and transformation to Na1+xV3O8/SiO2 with TOS that decrease the number of active sites (possibly, (–O)3VO⋯Na+ sites). That being said, no notable change was observed in the characteristic peak of β-NaVO3 (945 cm−1, Fig. 1a). In addition, a low-intensity peak at 968 cm−1 due to α′-NaV2O5 is also observed in β-NaVO3/1(TOS-4h) (Fig. 1a). Note that we have previously shown that α′-NaV2O5 forms on the silica surface under inert conditions owing to the reaction between β-NaVO3 and Na1+xV3O8/SiO2; α′-NaV2O5 is poorly active for ODP.29 In agreement with the Raman data, 51V MAS NMR of β-NaVO3/1(TOS-4h) shows a decreased intensity of α-NaVO3 signatures and an increased intensity of β-NaVO3 that also broadens (Fig. 1b).49 MAS NMR data on 23Na nucleus shows a new peak centered at −48 ppm for β-NaVO3/1(TOS-4h), due to the formation of the α′-NaV2O5 phase. A decreased intensity of the α-NaVO3 phase (peaks at −21 and −5 ppm, Fig. 1c) is also observed.
We discussed above that Raman mapping of β-NaVO3/1(500-air) shows inhomogeneous distribution of α-NaVO3 in this material, while β-NaVO3 and Na1+xV3O8/SiO2 are more homogeneously dispersed compared to α-NaVO3 (Fig. 2a). After 4 h TOS, i.e. β-NaVO3/1(TOS-4h), Raman peaks of the α-NaVO3 phase have decreased notably, while peaks of β-NaVO3 and Na1+xV3O8/SiO2 phases have increased (Fig. 2a). As mentioned, this might be due to a reaction of the remaining vanadyl sites on silica with α-NaVO3 forming crystalline (three-dimensional) Na1+xV3O8 that is associated with a decreased catalytic activity. Raman maps in Fig. 2a also show that the increased intensity of signals related to Na1+xV3O8/SiO2 correlates with an increased intensity of the β-NaVO3 peak, which is consistent with the aforementioned hypothesis of an increased stabilization of the metastable β-NaVO3 by Na1+xV3O8/SiO2, in preference to the formation of α-NaVO3, which is consumed with TOS in β-NaVO3/1(500-air) (Fig. 2a).
Conclusions
[VO4]/SiO2 promoted with α- or β-polymorphs of NaVO3 shows an increase in the initial rate of propene formation for ODP by 30 and 125%, respectively, at similar vanadium loadings (ca. 1 V nm−2 nominal coverage and 2 wt%), albeit offset by a 10 and 15% decrease in propene selectivity. Both catalysts lose activity with TOS due to the agglomeration and formation of less well dispersed (and possibly crystalline) Na1+xV3O8. Calcination of vanadyl sites promoted by β-NaVO3 leads to the α-NaVO3 phase, while promotion of vanadyl sites with α-NaVO3 gives notable amounts of the β-NaVO3 phase, which is unexpected considering that β-NaVO3 (both bulk and silica-supported) transforms completely to α-NaVO3 already at ca. 400 °C. The stabilization of the metastable β-NaVO3 on [VO4]/SiO2 is associated with the formation of dispersed Na1+xV3O8 that interacts with the silica support. The interaction of [VO4]/SiO2 with β-NaVO3 seems to lead to Na1+xV3O8 with a higher dispersion on silica than when promoted by α-NaVO3, resulting in a higher catalytic activity for the β-NaVO3 promoted catalyst. We are currently exploring other ways (such as support effect) to maintain a high dispersion of Na1+xV3O8.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Scientific Centre for Optical and Electron Microscopy (ScopeM) of ETH Zürich for providing access to electron microscopes. The Paul Scherer Institute (PSI), SuperXAS beamline is acknowledged for providing access to the synchrotron X-ray facility.Notes and references
- J. S. Plotkin, The changing dynamics of olefin supply/demand, Catal. Today, 2005, 106, 10–14 CrossRef CAS.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Catalytic Dehydrogenation of Light Alkanes on Metals and Metal Oxides, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS.
- E. McFarland, Unconventional Chemistry for Unconventional Natural Gas, Science, 2012, 338, 340–342 CrossRef CAS.
- J. T. Grant, J. M. Venegas, W. P. McDermott and I. Hermans, Aerobic Oxidations of Light Alkanes over Solid Metal Oxide Catalysts, Chem. Rev., 2018, 118, 2769–2815 CrossRef CAS.
- M. M. Bhasin, J. H. McCain, B. V. Vora, T. Imai and P. R. Pujadó, Dehydrogenation and oxydehydrogenation of paraffins to olefins, Appl. Catal., A, 2001, 221, 397–419 CrossRef CAS.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, Critical Literature Review of the Kinetics for the Oxidative Dehydrogenation of Propane over Well-Defined Supported Vanadium Oxide Catalysts, ACS Catal., 2014, 4, 3357–3380 CrossRef CAS.
- Y. Wang, H. Su, Y. Gu, X. Song and J. Zhao, Carcinogenicity of chromium and chemoprevention: a brief update, OncoTargets Ther., 2017, 10, 4065–4079 CrossRef.
- R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Catalytic Applications of Vanadium: A Mechanistic Perspective, Chem. Rev., 2019, 119, 2128–2191 CrossRef CAS.
- H. Eckert, G. Deo, I. E. Wachs and A. M. Hirt, Solid state 51V NMR structural studies of vanadium(V) oxide catalysts supported on TiO2 (anatase) and TiO2 (rutile). The influence of surface impurities on the vanadium(V) coordination, Colloids Surf., 1990, 45, 347–359 CrossRef CAS.
- O. B. Lapina, V. M. Mastikhin, L. G. Simonova and Y. O. Bulgakova, Characterization of surface species of supported V2O5-Al2O3 catalysts by 51V NMR, J. Mol. Catal., 1991, 69, 61–73 CrossRef CAS.
- N. Das, H. Eckert, H. Hu, I. E. Wachs, J. F. Walzer and F. J. Feher, Bonding states of surface vanadium(V) oxide phases on silica: structural characterization by vanadium-51 NMR and Raman spectroscopy, J. Phys. Chem., 1993, 97, 8240–8243 CrossRef CAS.
- N. R. Jaegers, J.-K. Lai, Y. He, E. Walter, D. A. Dixon, M. Vasiliu, Y. Chen, C. Wang, M. Y. Hu, K. T. Mueller, I. E. Wachs, Y. Wang and J. Z. Hu, Mechanism by which Tungsten Oxide Promotes the Activity of Supported V2O5/TiO2 Catalysts for NOX Abatement: Structural Effects Revealed by 51V MAS NMR Spectroscopy, Angew. Chem., Int. Ed., 2019, 58, 12609–12616 CrossRef CAS.
- N. R. Jaegers, C. Wan, M. Y. Hu, M. Vasiliu, D. A. Dixon, E. Walter, I. E. Wachs, Y. Wang and J. Z. Hu, Investigation of Silica-Supported Vanadium Oxide Catalysts by High-Field 51V Magic-Angle Spinning NMR, J. Phys. Chem. C, 2017, 121, 6246–6254 CrossRef.
- T. Tanaka, H. Yamashita, R. Tsuchitani, T. Funabiki and S. Yoshida, X-ray absorption (EXAFS/XANES) study of supported vanadium oxide catalysts. Structure of surface vanadium oxide species on silica and γ-alumina at a low level of vanadium loading, J. Chem. Soc., Faraday Trans., 1988, 84, 2987–2999 RSC.
- E. L. Lee and I. E. Wachs, In Situ Raman Spectroscopy of SiO2-Supported Transition Metal Oxide Catalysts: An Isotopic 18O−16O Exchange Study, J. Phys. Chem. C, 2008, 112, 6487–6498 CrossRef CAS.
- S. T. Oyama, G. T. Went, K. B. Lewis, A. T. Bell and G. A. Somorjai, Oxygen chemisorption and laser Raman spectroscopy of unsupported and silica-supported vanadium oxide catalysts, J. Phys. Chem., 1989, 93, 6786–6790 CrossRef CAS.
- X. Gao, S. R. Bare, B. M. Weckhuysen and I. E. Wachs, In Situ Spectroscopic Investigation of Molecular Structures of Highly Dispersed Vanadium Oxide on Silica under Various Conditions, J. Phys. Chem. B, 1998, 102, 10842–10852 CrossRef.
- X. Gao, M. A. Bañares and I. E. Wachs, Ethane and n-Butane Oxidation over Supported Vanadium Oxide Catalysts: An in Situ UV–Visible Diffuse Reflectance Spectroscopic Investigation, J. Catal., 1999, 188, 325–331 CrossRef.
- X. Gao and I. E. Wachs, Investigation of Surface Structures of Supported Vanadium Oxide Catalysts by UV−vis−NIR Diffuse Reflectance Spectroscopy, J. Phys. Chem. B, 2000, 104, 1261–1268 CrossRef CAS.
- C. A. Carrero, C. J. Keturakis, A. Orrego, R. Schomacker and I. E. Wachs, Anomalous reactivity of supported V2O5 nanoparticles for propane oxidative dehydrogenation: influence of the vanadium oxide precursor, Dalton Trans., 2013, 42, 12644–12653 RSC.
- C. Carrero, M. Kauer, A. Dinse, T. Wolfram, N. Hamilton, A. Trunschke, R. Schlogl and R. Schomacker, High performance (VOx)n-(TiOx)m/SBA-15 catalysts for the oxidative dehydrogenation of propane, Catal. Sci. Technol., 2014, 4, 786–794 RSC.
- J. T. Grant, C. A. Carrero, F. Goeltl, J. Venegas, P. Mueller, S. P. Burt, S. E. Specht, W. P. McDermott, A. Chieregato and I. Hermans, Selective oxidative dehydrogenation of propane to propene using boron nitride catalysts, Science, 2016, 354, 1570–1573 CrossRef CAS.
- A. A. Lemonidou, L. Nalbandian and I. A. Vasalos, Oxidative dehydrogenation of propane over vanadium oxide based catalysts: Effect of support and alkali promoter, Catal. Today, 2000, 61, 333–341 CrossRef CAS.
- G. Garcia Cortez, J. L. G. Fierro and M. A. Bañares, Role of potassium on the structure and activity of alumina-supported vanadium oxide catalysts for propane oxidative dehydrogenation, Catal. Today, 2003, 78, 219–228 CrossRef CAS.
- S. Takenaka, T. Tanaka, T. Yamazaki, T. Funabiki and S. Yoshida, Structure of Active Species in Alkali-Ion-Modified Silica-Supported Vanadium Oxide, J. Phys. Chem. B, 1997, 101, 9035–9040 CrossRef CAS.
- J. T. Grant, A. M. Love, C. A. Carrero, F. Huang, J. Panger, R. Verel and I. Hermans, Improved supported metal oxides for the oxidative dehydrogenation of propane, Top. Catal., 2016, 59, 1545–1553 CrossRef CAS.
- S. Irusta, A. Marchi, E. Lombardo and E. Miré, Characterization of surface species on V/SiO2 and V, Na/SiO2 and their role in the partial oxidation of methane to formaldehyde, Catal. Lett., 1996, 40, 9–16 CrossRef CAS.
- J. T. Grant, C. A. Carrero, A. M. Love, R. Verel and I. Hermans, Enhanced Two-Dimensional Dispersion of Group V Metal Oxides on Silica, ACS Catal., 2015, 5, 5787–5793 CrossRef CAS.
- M. Nadjafi, P. M. Abdala, R. Verel, D. Hosseini, O. V. Safonova, A. Fedorov and C. R. Müller, Reducibility and Dispersion Influence the Activity in Silica-Supported Vanadium-Based Catalysts for the Oxidative Dehydrogenation of Propane: The Case of Sodium Decavanadate, ACS Catal., 2020, 10, 2314–2321 CrossRef CAS.
- I. Lukács and C. Strusievici, Über die Polymorphie von Natriummetavanadat, Z. Anorg. Allg. Chem., 1962, 315, 323–326 CrossRef.
- S. Seetharaman, H. L. Bhat and P. S. Narayanan, Raman spectroscopic studies on sodium metavanadate, J. Raman Spectrosc., 1983, 14, 401–405 CrossRef CAS.
- I. E. Wachs, Catalysis science of supported vanadium oxide catalysts, Dalton Trans., 2013, 42, 11762–11769 RSC.
- S. Xie, E. Iglesia and A. T. Bell, Effects of Hydration and Dehydration on the Structure of Silica-Supported Vanadia Species, Langmuir, 2000, 16, 7162–7167 CrossRef CAS.
- A. M. Love, C. A. Carrero, A. Chieregato, J. T. Grant, S. Conrad, R. Verel and I. Hermans, Elucidation of Anchoring and Restructuring Steps during Synthesis of Silica-Supported Vanadium Oxide Catalysts, Chem. Mater., 2016, 28, 5495–5504 CrossRef CAS.
- K. C. Szeto, B. Loges, N. Merle, N. Popoff, A. Quadrelli, H. Jia, E. Berrier, A. De Mallmann, L. Delevoye, R. M. Gauvin and M. Taoufik, Vanadium Oxo Organometallic Species Supported on Silica for the Selective Non-oxidative Dehydrogenation of Propane, Organometallics, 2013, 32, 6452–6460 CrossRef CAS.
- B. Schimmoeller, Y. Jiang, S. E. Pratsinis and A. Baiker, Structure of flame-made vanadia/silica and catalytic behavior in the oxidative dehydrogenation of propane, J. Catal., 2010, 274, 64–75 CrossRef CAS.
- K. Kato and E. Takayama, Das Entwasserungsverhalten des Natriummetavanadatdihydrats und die Kristallstruktur des β-Natriummetavanadats, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 102–105 CrossRef.
- M. D. Argyle, K. Chen, A. T. Bell and E. Iglesia, Effect of Catalyst Structure on Oxidative Dehydrogenation of Ethane and Propane on Alumina-Supported Vanadia, J. Catal., 2002, 208, 139–149 CrossRef CAS.
- G. T. Went, S. T. Oyama and A. T. Bell, Laser Raman spectroscopy of supported vanadium oxide catalysts, J. Phys. Chem., 1990, 94, 4240–4246 CrossRef CAS.
- P. M. A. Sherwood, Vibrational spectroscopy of solids, CUP Archive, 1972 Search PubMed.
- J. Skibsted, N. C. Nielsen, H. Bildsoe and H. J. Jakobsen, Magnitudes and relative orientation of vanadium-51 quadrupole coupling and anisotropic shielding tensors in metavanadates and potassium vanadium oxide (KV3O8) from vanadium-51 MAS NMR spectra. Sodium-23 quadrupole coupling parameters for α- and β-NaVO3, J. Am. Chem. Soc., 1993, 115, 7351–7362 CrossRef CAS.
- J. Wong, F. W. Lytle, R. P. Messmer and D. H. Maylotte, K-edge absorption spectra of selected vanadium compounds, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 5596–5610 CrossRef CAS.
- A. Dinse, B. Frank, C. Hess, D. Habel and R. Schomäcker, Oxidative dehydrogenation of propane over low-loaded vanadia catalysts: Impact of the support material on kinetics and selectivity, J. Mol. Catal. A: Chem., 2008, 289, 28–37 CrossRef CAS.
- G. Deo and I. E. Wachs, Reactivity of Supported Vanadium Oxide Catalysts: The Partial Oxidation of Methanol, J. Catal., 1994, 146, 323–334 CrossRef.
- S. Chen, L. Zeng, R. Mu, C. Xiong, Z.-J. Zhao, C. Zhao, C. Pei, L. Peng, J. Luo, L.-S. Fan and J. Gong, Modulating Lattice Oxygen in Dual-Functional Mo–V–O Mixed Oxides for Chemical Looping Oxidative Dehydrogenation, J. Am. Chem. Soc., 2019, 141, 18653–18657 CrossRef CAS.
- E. V. Kondratenko, N. Steinfeldt and M. Baerns, Transient and steady state investigation of selective and non-selective reaction pathways in the oxidative dehydrogenation of propane over supported vanadia catalysts, Phys. Chem. Chem. Phys., 2006, 8, 1624–1633 RSC.
- P. Gruene, T. Wolfram, K. Pelzer, R. Schlögl and A. Trunschke, Role of dispersion of vanadia on SBA-15 in the oxidative dehydrogenation of propane, Catal. Today, 2010, 157, 137–142 CrossRef CAS.
- I. Rossetti, G. F. Mancini, P. Ghigna, M. Scavini, M. Piumetti, B. Bonelli, F. Cavani and A. Comite, Spectroscopic Enlightening of the Local Structure Of VOX Active Sites in Catalysts for the ODH of Propane, J. Phys. Chem. C, 2012, 116, 22386–22398 CrossRef CAS.
- A. A. Zvyagin, Temperature dependence of the electron paramagnetic resonance linewidth in NaV2O5, Phys. Rev. B, 2001, 63, 172409 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, XRD, NMR, Raman, H2 temperature programmed reduction (TPR), XAS, DSC, and TEM data. See DOI: 10.1039/d0cy01234c |
| This journal is © The Royal Society of Chemistry 2020 |