
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of the stability of NiFe-(oxy)hydroxide anodes in alkaline water electrolysis under industrially relevant conditions†
Marco
Etzi Coller Pascuzzi
 ,
Alex J. W.
Man
,
Andrey
Goryachev‡
,
Jan P.
Hofmann§
* and
Emiel J. M.
Hensen
*
,
Alex J. W.
Man
,
Andrey
Goryachev‡
,
Jan P.
Hofmann§
* and
Emiel J. M.
Hensen
*
Laboratory of Inorganic Materials and Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: j.p.hofmann@tue.nl; e.j.m.hensen@tue.nl
First published on 17th July 2020
Abstract
NiFe-(Oxy)hydroxide is one of the most active electrocatalysts for the oxygen evolution reaction (OER) in alkaline conditions. Herein we investigated the stability of NiFe-(oxy)hydroxide anodes at high current densities (100 mA cm−2) at different temperatures (25, 75 °C) and base concentrations (1, 5, 10 M KOH). While polarization led to minor structural and compositional changes under standard conditions (25 °C, 1 M KOH), the anodes were severely impacted at higher temperature (75 °C) and base concentrations (5, 10 M KOH). Overall leaching and preferential leaching of Fe (resulting in a lower Fe/Ni ratio) led to decreased OER performance and increased charge transfer resistance for the samples tested at industrially relevant conditions. A dramatic loss in the catalytic activity occurred for the sample polarized at 75 °C in 10 M KOH: besides extensive leaching, a transformation of Ni(OH)2 into NiO was noted in this case. For pure NiOxHy, incorporation of Fe impurities from the electrolyte during polarization at 75 °C in 5 M KOH led to an improvement in the catalytic activity and charge-transfer properties, approaching the performance of NiFeOxHy.
1. Introduction
Electrochemical water splitting is a promising technology to convert excess electricity produced by intermittent renewable energy sources into a chemical fuel (hydrogen).1 The anodic oxygen evolution reaction (OER), being a counter reaction to the H2 evolution, constitutes a major bottleneck due to its sluggish kinetics.2 Electrocatalysts are therefore needed to overcome kinetic limitations and to increase the overall efficiency of electrochemical water splitting devices. An ideal OER catalyst should combine high catalytic activity and stability with earth-abundancy and low price. First-row transition metal oxides and hydroxides offer interesting opportunities to enhance the rate of the OER, especially in the alkaline regime, without the use of expensive and scarce metals such as Ir or Ru.3–6Fe-Doped Ni oxides, and especially (oxy)hydroxides,7 are the most active among noble-metal-free OER catalysts.8–11 Some have reported that the alkaline OER activity of such anodes can be higher than those based on IrO2 and RuO2.12–16 Fe impurities were found to enhance the OER activity of Ni-oxide already in 1987.17 Subsequently, most efforts were directed towards understanding the role of Fe and the synthesis of highly active and porous anodes to maximize the activity.18 Some reports mention that Fe enhances the activity of Ni sites by altering their redox properties,19,20 while others propose that Fe atoms are the active sites for the OER.21–24 Recent findings have pointed to the formation of mixed phase in which bridging Ni–O–Fe motifs are responsible for the enhanced catalytic activity.25 Although the exact role of Fe is still debated, various synthetic strategies were developed to maximize the catalytic performance.18
Despite the efforts made to enhance the activity and to understand the role of Fe in NiFe-(oxy)hydroxide, the stability aspect of this material has not been extensively studied yet. Stability tests are usually conducted under standard conditions (T = 25 °C, [KOH] = 1 mol L−1), whereas the industrial alkaline water electrolysis requires much higher temperatures and base concentrations (T = 70–80 °C, [KOH] = 25–30 wt%).26 These harsh conditions may cause structural changes in the electrodes and their premature degradation. These aspects are usually not captured in electrocatalyst evaluation protocols under standard conditions. However, performing experiments under such harsh conditions becomes challenging at the lab scale.
Recently, Andronescu et al. reported that NiFe layered double hydroxide (LDH) is sensitive to the applied anodic treatment: operation in 5 M KOH at 60 °C transforms the LDH into a mixture of NiO and NiFe2O4.27 The same authors also studied the structural changes of a NiFe-LDH electrode upon immersion in 7.5 M KOH at 80 °C.28 Even without electrochemical polarization, the activity decreased considerably due to chemical degradation. We can thus expect that electrochemical polarization in harsh conditions could considerably affect anode stability.
Given the relevance of NiFe-(oxy)hydroxide for the alkaline OER, we investigated herein the stability of this material under electrochemical treatments conducted in non-standard, industrially relevant temperature and basicity regimes. The catalysts were comprehensively characterized by a combination of X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), Raman spectroscopy, X-ray diffraction (XRD), and cyclic voltammetry (CV) to gain understanding into the structural, morphological, compositional, and activity change induced by anodic polarization under non-standard conditions.
2. Experimental
2.1 Anode preparation
NiFe-(Oxy)hydroxide films were prepared on Au-coated Si substrates (Sigma-Aldrich, 100 nm) via electrodeposition, in accordance with previous literature.19,29 The substrates were cleaned with absolute ethanol and then rinsed with copious amounts of Milli-Q water prior to the deposition to remove contaminants.The solution for electrodeposition was prepared by dissolving 5 mM nickel(II) sulfate hexahydrate (≥99.0%, Merck) and 0.5 mM iron(II) sulfate heptahydrate (≥99.0%, Sigma Aldrich) in Milli-Q water (18.2 MΩ cm). Before dissolving the metal salts, the water was purged by N2 bubbling for 30 min to prevent the oxidation of Fe2+ into Fe3+.
Each deposition was carried out from the fresh solution (20 mL) by applying a constant cathodic current density of 50 μA cm−2 for 1125 s.19 The geometric areas of the samples (in the range of 1.0–1.8 cm2) were evaluated from digital photographs using ImageJ software.30 The reproducibility of the preparation method was validated by XPS analysis of the fresh samples, which showed maximal deviations in the atomic Ni and Fe percentage of ±3%. An iron-free Ni-(oxy)hydroxide anode (NiOxHy) was prepared using the same synthetic procedure but without the addition of iron salts to the electrodeposition bath. The as-prepared anodes were subjected to the stability tests described in the following section or tested for activity evaluation.
2.2 Electrochemical characterization
Electrochemical measurements were performed using an Autolab PGSTAT30N potentiostat (Metrohm Autolab B.V.). The stability tests were conducted in a two-electrode Teflon cell under vigorous stirring, using a Pt foil (area 5 cm2) as counter electrode. A total of 6 NiFeOxHy anodes were used for the stability evaluation in different conditions. Each sample was subjected to a chronopotentiometry at 100 mA cm−2 for 1 h at different temperatures (25 °C or 75 °C) and base concentrations (1, 5, or 10 M KOH).Activity evaluation of fresh and used samples was performed in 1 M KOH (KOH, Alfa Aesar, ACS reagent ≥85.0%) at room temperature. Milli-Q water (18.2 MΩ cm) was used in all the dilution and cleaning steps. The tests were conducted in a standard three-electrode configuration. A platinum wire and an XR440 red rod electrode (Ag/AgCl without KCl, Radiometer Analytical, E = +0.48 VSHE) were used as counter and reference electrodes, respectively. Cyclic voltammetry (CV) was conducted with a scan rate of 50 mV s−1. Five cycles were applied in order to obtain stable voltammograms. The fifth cycle is reported in the graphs shown throughout the manuscript. Electrochemical impedance spectroscopy (EIS) measurements were conducted at E = +1.54 VRHE in a frequency range from 10−1 to 103 Hz with an AC amplitude of 10 mV. Step chronoamperometry was conducted by increasing the potential in 20 mV steps and applying a hold time of 30 s: the current densities measured at the end of each step were plotted versus the iR-corrected values of overpotential to construct Tafel plots. Tafel slopes were evaluated by linear fits of the experimental data with an error of ±1 mV dec−1.
2.3 Materials characterization
XPS measurements were performed on a K-alpha XPS spectrometer (Thermo Scientific) equipped with a monochromatic Al Kα (1486.6 eV) X-ray source. The spot size was 400 μm and the pass energy was set at 200 eV and 50 eV for survey and high-resolution spectra, respectively. A flood gun was used for charge compensation. Binding energy calibration of the spectra was applied by setting the C 1s binding energy of the adventitious sp3 carbon component at 284.8 eV. Ni 2p and Fe 3p regions were used to determine the metal content at the surface of the electrodes. All the spectra were processed using CasaXPS software version 2.3.22 (Casa Software Ltd.).Scanning electron micrographs (SEM) were acquired on a FEI Quanta 3D FEG microscope at an accelerating voltage of 5 kV. Energy dispersive X-ray (EDX) spectroscopy was performed on a Phenom Pro-X microscope with an accelerating voltage of 10 kV.
Raman spectra were acquired on a Witec Alpha 300R Raman microscope with λexc = 532 nm using a Zeiss 50×, NA = 0.55 objective. Spectra were acquired at a power of 10 mW, with a collection time of 10 s, averaging 10 accumulations for each spectrum.
X-ray diffraction (XRD) patterns were acquired using a Bruker D2 Phaser diffractometer equipped with a Cu Kα radiation source, using a step size of 0.02° and acquisition time of 1 s per step.
Inductively coupled plasma optical emission spectrometry (ICP-OES) measurements of catalytic films and used electrolytes were conducted on a SPECTROBLUE spectrometer equipped with an axial plasma source (Ar). The films were dissolved by immersing the coated substrates overnight in 1 mL of concentrated nitric acid (HNO3 65%, Sigma) under standard conditions and sealing with parafilm to prevent evaporation. Then, the solutions were diluted 20 times to reach a HNO3 concentration of ∼3% to prevent damage of the instrument. Four independent samples were measured using this protocol.
For ICP-OES analysis of used electrolytes, as KOH concentrations exceeding 0.1 M could damage the instrument, the solutions were diluted to this concentration prior to the analysis. Quantities below 5 μg L−1 could not be detected because of the sensitivity limit of the instrument.
3. Results and discussion
NiFe-(Oxy)hydroxide films (NiFeOxHy) were deposited via cathodic electrodeposition from an aqueous solution of Ni(II) and Fe(II) sulfates. The deposition method is expected to form predominantly a Ni(OH)2 phase, with Fe3+ ions substituting Ni2+. Elemental analysis of four independent samples measured by ICP-OES revealed total metal loadings of 13.5 (±2.6) μg cm−2, with Ni and Fe contents of 73 at% and 27 at% (±5 at%), respectively. Surface analysis of the films was conducted by means of XPS.Ni 2p, Fe 2p, and O 1s XP spectra are reported in Fig. 1. The Ni 2p spectrum reveals a major contribution from Ni2+ species in a Ni(OH)2 environment with the Ni 2p3/2 and Ni 2p1/2 contributions located at binding energies (BE) of 855.5 eV and 873.0 eV, respectively.14,15,31,32 Satellite peaks at higher binding energies than the main photoemission lines are an additional fingerprint of Ni2+ species and originate from the interaction of unpaired core electrons with unpaired electrons in the valence band.32,33 A small contribution of Ni3+ species (BE(Ni 2p3/2) = 857.3 eV) indicates a minor presence of NiOOH in the fresh films.15 The fraction of Ni3+ was in the range of 9–20% for the fresh samples. The Fe 2p spectrum shows two main peaks located at 711.7 eV (Fe 2p3/2) and 725.4 eV (Fe 2p1/2) and broad satellite features identified as shake-up peaks. These peaks are a clear sign of the presence of Fe3+ in the fresh samples.31,34,35 It is however difficult to identify and quantify the presence of different Fe phases (oxides, hydroxides) from the Fe 2p spectrum because of the multiple overlapping components in a narrow binding energy range.33 The O 1s spectrum was fitted with three components located at binding energies of 529.4 eV, 531.0 eV, and 533.1 eV, which can be ascribed to lattice oxygen, oxygen in hydroxides, and adsorbed water, respectively.32,36
 | ||
| Fig. 1 a) Ni 2p, b) Fe 2p, and c) O 1s XPS spectra of as-prepared NiFeOxHy films. Black points represent the experimental data, red lines the fit. | ||
The Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe ratio of the as-prepared samples was evaluated by XPS and EDX in order to determine the surface and bulk composition of the films, respectively. Both techniques revealed similar Ni:Fe ratios (74:26 by XPS vs. 70:30 by EDX), which is an indication of the absence of surface enrichment in any of the metals. The results are also in agreement with the ICP-OES elemental analysis of the films (Ni:Fe ratio of 73:27). The composition evaluated by EDX can be considered as the bulk composition because the probing depth of EDX is higher than the thickness of the films. This assumption is confirmed by the strong signal arising from the underlying Au-coated Si substrate (Fig. S1†). The Fe content at the surface (26% as determined by XPS) of our samples is in the range reported to be optimum for the OER (15–50%).8,17,19,29 XRD revealed no other peaks than those originating from the Au-coated Si substrate (Fig. S2†). The electrodes were then subjected to an anodic electrochemical treatment at different temperatures and KOH concentrations (Fig. S3†). The impact of these treatments on the electrodes will be discussed in the following.
:Fe ratio of the as-prepared samples was evaluated by XPS and EDX in order to determine the surface and bulk composition of the films, respectively. Both techniques revealed similar Ni:Fe ratios (74:26 by XPS vs. 70:30 by EDX), which is an indication of the absence of surface enrichment in any of the metals. The results are also in agreement with the ICP-OES elemental analysis of the films (Ni:Fe ratio of 73:27). The composition evaluated by EDX can be considered as the bulk composition because the probing depth of EDX is higher than the thickness of the films. This assumption is confirmed by the strong signal arising from the underlying Au-coated Si substrate (Fig. S1†). The Fe content at the surface (26% as determined by XPS) of our samples is in the range reported to be optimum for the OER (15–50%).8,17,19,29 XRD revealed no other peaks than those originating from the Au-coated Si substrate (Fig. S2†). The electrodes were then subjected to an anodic electrochemical treatment at different temperatures and KOH concentrations (Fig. S3†). The impact of these treatments on the electrodes will be discussed in the following.
As anodic polarization can lead to dissolution of the active metals, we analyzed the compositional changes of the anodes after the electrochemical treatment in terms of Ni:Fe ratio at the surface and in the bulk using XPS and EDX. XPS analysis (Fig. 2a) shows the changes in the surface composition of the anodes upon electrochemical treatment under different conditions. After treatment at 25 °C, a very small increase in the Ni:Fe ratio at the surface is noted by XPS. The influence of the base concentration is very small. Similar treatment at 75 °C led to more substantial changes in the surface composition, which became larger with increased basicity. The sample anodically polarized in 10 M KOH at 75 °C shows a very high Ni content (97%). The results of EDX analysis (Fig. 2b) are in line with the XPS results. While the samples tested at 25 °C show a slightly higher Ni:Fe ratio as compared to fresh samples, the changes are more substantial for the samples tested at 75 °C. These changes are also larger at higher KOH concentration, especially for the sample tested at 75 °C in 10 M KOH. The XPS and EDX results show preferential Fe leaching due to the electrochemical treatment. There is no large difference between these compositional changes at the surface and in the bulk, suggesting that the changes are uniform. This is likely because the films are very thin and composed of small particles. The overall changes before and after anodic testing are very minor in 1 M KOH, regardless of the temperature, showing that at low base concentration the electrodes are stable. Preferential Fe leaching is substantial for samples tested at 75 °C in 5 M KOH and 10 M KOH. As the films are very thin, it was not possible to quantitatively determine absolute leaching ratios by elemental analysis and, in this way, to establish how much Ni was leached. Fe leaching is most likely due to the applied electrochemical treatment rather than chemical dissolution, as no significant changes in the Ni:Fe ratio were observed by XPS after 1 hour of electrode immersion in 10 M KOH at 75 °C (data reported in Fig. S4†).
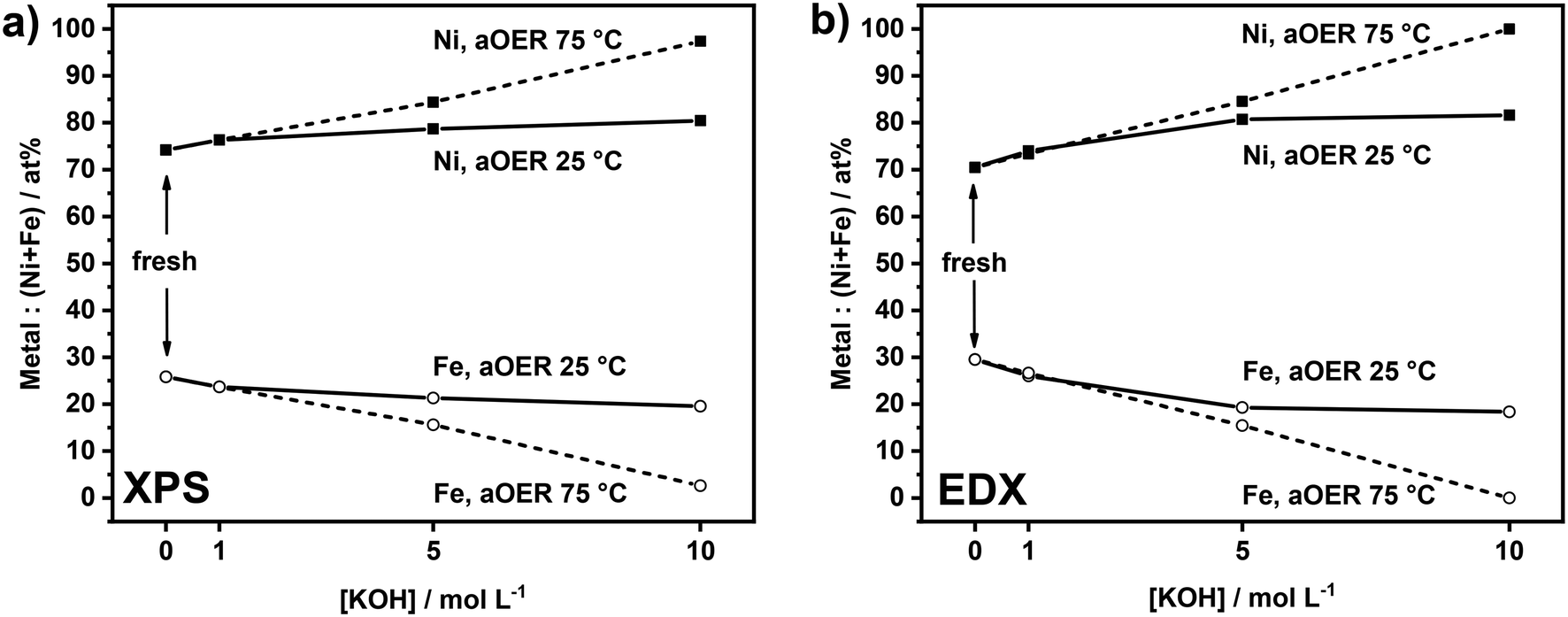 | ||
| Fig. 2 a) XPS and b) EDX analysis of NiFeOxHy films before and after OER treatment (aOER) conducted at j = 100 mA cm−2 for 1 h at different temperatures and KOH concentrations. | ||
The Ni 2p, Fe 2p, and O 1s XPS spectra of the used samples reported in Fig. S5, S6, and S7,† respectively, do not show significant changes as compared to those of fresh samples, except for the sample polarized in 10 M KOH at 75 °C. For this latter sample, the Ni 2p3/2 region contains an additional component at lower binding energy (BE = 853.9 eV) corresponding to NiO.32,37 Its appearance goes together with an increasing signal of the lattice O component observed in the O 1s XPS spectrum. Notably, the Fe signal in this sample was very weak (Ni:Fe ratio = 97:3). We can infer from this that part of Ni-hydroxide was converted into Ni-oxide during electrochemical treatment at the most severe conditions (10 M KOH, 75 °C). This finding is in keeping with a previous study where NiFe-LDH was found to transform into a mixture of NiO and Ni2FeO4 after prolonged electrolysis (100 h) in 5 M KOH at 60 °C.27 The transformation of Ni-(oxy)hydroxide into Ni-oxide could negatively affect the anode performance because NiO has a lower OER activity than Ni(OH)2.38
Moreover, after electrochemical treatment at 75 °C in 10 M KOH, the loss of active phase led to the exposure and corrosion of the underlying Au coating on the substrate. The XPS survey spectrum (Fig. S8†) showed an increase in the intensity of the Au peaks and the appearance of Si peaks arising from the substrate after polarization at the harshest conditions.
Changes in film morphology upon electrochemical treatment were investigated by SEM (Fig. 3). The micrograph of the bare substrate is reported in Fig. S9† as reference. The as-deposited sample exhibits a platelet-like morphology, characteristic of NiFe-(oxy)hydroxide films,39 with some thicker agglomerates on the electrode surface. There were no substantial changes upon anodic polarization of the samples in 1 M KOH irrespective of the temperature. This is in line with the small chemical changes evident from XPS and EDX analyses under these conditions. Testing in 5 M KOH did not lead to morphological changes at 25 °C, although the number of platelets was lower after anodic polarization at 75 °C. Most of the platelets disappeared after anodic polarization at 10 M KOH. At 25 °C, the platelets were changed into small and compact flakes, while the sample obtained after testing at 75 °C only contained fine round grains on a flat surface. The absence of platelet-like features points toward a structural instability of the layered NiFeOxHy phase under electrochemical conditions relevant to the OER. The severe changes are consistent with loss of material during the stability tests at high temperatures and KOH concentrations.
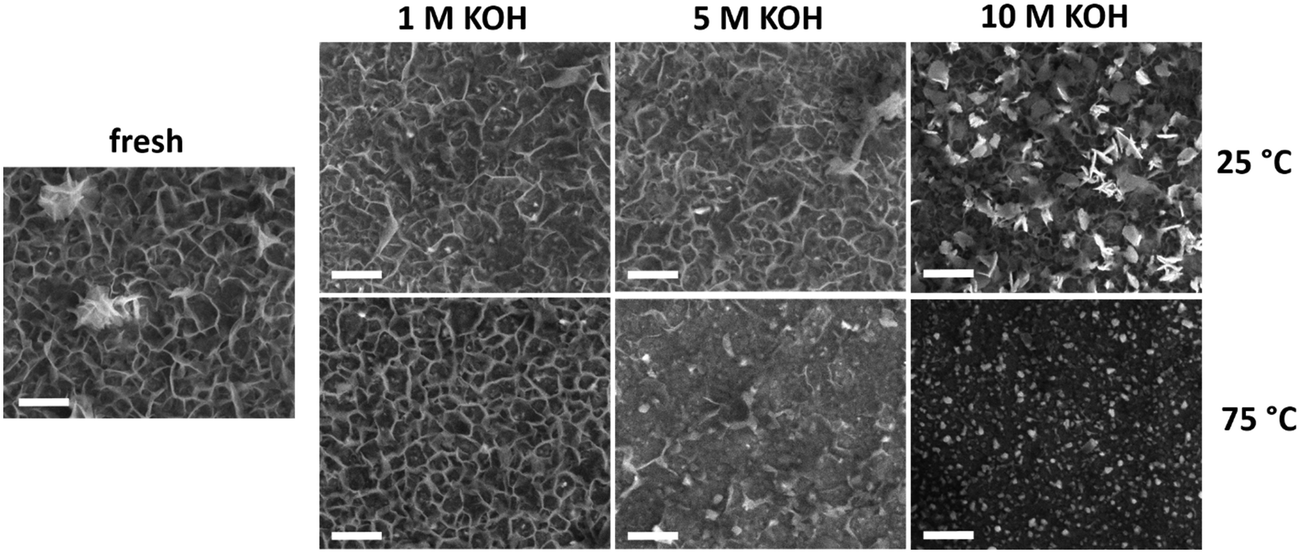 | ||
| Fig. 3 SEM images of fresh and used NiFeOxHy films after the stability tests conducted at different temperatures and electrolyte concentrations (scale bars 500 nm). | ||
The structure and phase composition of the NiFeOxHy films were also investigated by Raman spectroscopy (Fig. 4). The Raman spectra of NiFeOxHy exhibit three characteristic bands at 458 cm−1, 540 cm−1, and 675 cm−1. The band at 458 cm−1 is ascribed to α-Ni(OH)2 and β-Ni(OH)2 phases, which have Ni–O bands at similar frequencies (α-Ni(OH)2: 460–465 cm−1, β-Ni(OH)2: 445–449 cm−1).19,21,40,41 The band at 540 cm−1 is ascribed to Ni–O vibrations in defective or highly disordered Ni(OH)2 with a low degree of crystallinity.19,40,41 The position of this band was found to be dependent on the amount of incorporated Fe in the Ni(OH)2 films, shifting toward higher wavenumber at higher Fe incorporations.19 The feature at 675 cm−1, due to the presence of Fe, can be attributed to Fe3O4, γ-Fe2O3, and γ-FeOOH, which all exhibit a band at nearly similar frequencies.42–44 The spectrum of NiFeOxHy after activity evaluation (CV) exhibits similar but more intense features compared to fresh NiFeOxHy, which can be attributed to enhanced Raman scattering on a roughened surface. The spectra of NiFeOxHy samples after anodic polarization in 1 M and 5 M KOH show some changes compared to the spectrum of NiFeOxHy. The lower intensity of the band at 458 cm−1 indicates a loss of Ni(OH)2, while the shift of the band from 540 cm−1 to 530 cm−1 implies a lower substitution degree of Fe in the Ni(OH)2 structure. These changes are in line with the preferential leaching of Fe as discussed above. To confirm this hypothesis, we compared the position of this band with a NiOxHy measured as a reference. We used the Raman spectrum of a NiOxHy after the activity test (cyclic voltammetry) for this purpose because we could not identify any Raman feature in the spectrum of fresh sample, possibly because of the low roughness of the as-deposited material. NiOxHy showed the presence of the feature at 515 cm−1, confirming that the position of this band is sensitive to the amount of Fe. The negative shift occurred for NiFeOxHy after the stability tests is, therefore, an indication of lower Fe incorporation as a result of the electrochemical treatment. The lower amount of Fe in the tested samples is also reflected in the lower intensity of the band at 675 cm−1, related to Fe oxide/oxyhydroxide. For the sample tested in 10 M KOH, we can observe differences depending on the temperature used in the electrochemical test. In the spectrum of the sample tested at 25 °C, in addition to the previous changes, we can also distinguish a broad band appearing at ∼580 cm−1, potentially due to the oxidation of the underlying Au.45 This can be due to a lower amount of catalytic phase resulting in parts of the substrate left uncovered. This band appears as a very broad feature grouped with the Ni–O band in the Raman spectrum of the sample tested at 75 °C in 10 M KOH, resulting in a very weak and large band extending from 500 cm−1 to 600 cm−1. The decrease in band intensity suggests that a massive film leaching occurred during the electrochemical treatment at the harshest condition (75 °C and 10 M KOH).
 | ||
| Fig. 4 Raman spectra of fresh NiOxHy, NiOxHy after CV, fresh NiFeOxHy, NiFeOxHy after CV, and NiFeOxHy after anodic polarization at j = 100 mA cm−2 at a) 25 °C and b) 75 °C. | ||
The OER activity and redox properties of the samples before and after the electrochemical polarization were evaluated by cyclic voltammetry (Fig. 5). The voltammogram of fresh NiFeOxHy anode contains a redox peak associated with Ni2+ → Ni3+ oxidation, followed by an increase of the current density ascribed to oxygen evolution at higher potentials. The split of the redox peak can be associated with the presence of α-Ni(OH)2 and β-Ni(OH)2 phases in the fresh films, which respectively oxidize to γ-NiOOH and β-NiOOH. The conversion of α-Ni(OH)2 to γ-NiOOH occurs at a lower potential than the conversion of β-Ni(OH)2 to β-NiOOH.21,39,46 The cyclic voltammograms of NiFeOxHy samples previously subjected to anodic polarization at 25 °C are reported in Fig. 5a. The catalytic activity of the used anodes at 25 °C is lower than that of the fresh NiFeOxHy films, regardless of the different KOH concentration used in the stability test. We can also observe a decrease in the redox peak areas compared to the fresh anode, which is most likely due to dissolution of part of the active phase in the electrode. For samples previously tested at 75 °C (Fig. 5b), the decrease in the catalytic activity is more pronounced than for samples tested at 25 °C. This is consistent with the more pronounced Fe leaching in the samples tested under more severe conditions. The KOH concentration in the stability test also affects the catalytic activity, a higher base concentration resulting in a lower OER activity, especially after treatment in 10 M KOH. Similar to the samples tested at 25 °C, the redox peak area of the samples tested at 75 °C is lower, which is also due to film degradation. In fact, for the most severely tested sample, the redox peak is hardly visible in the voltammogram.
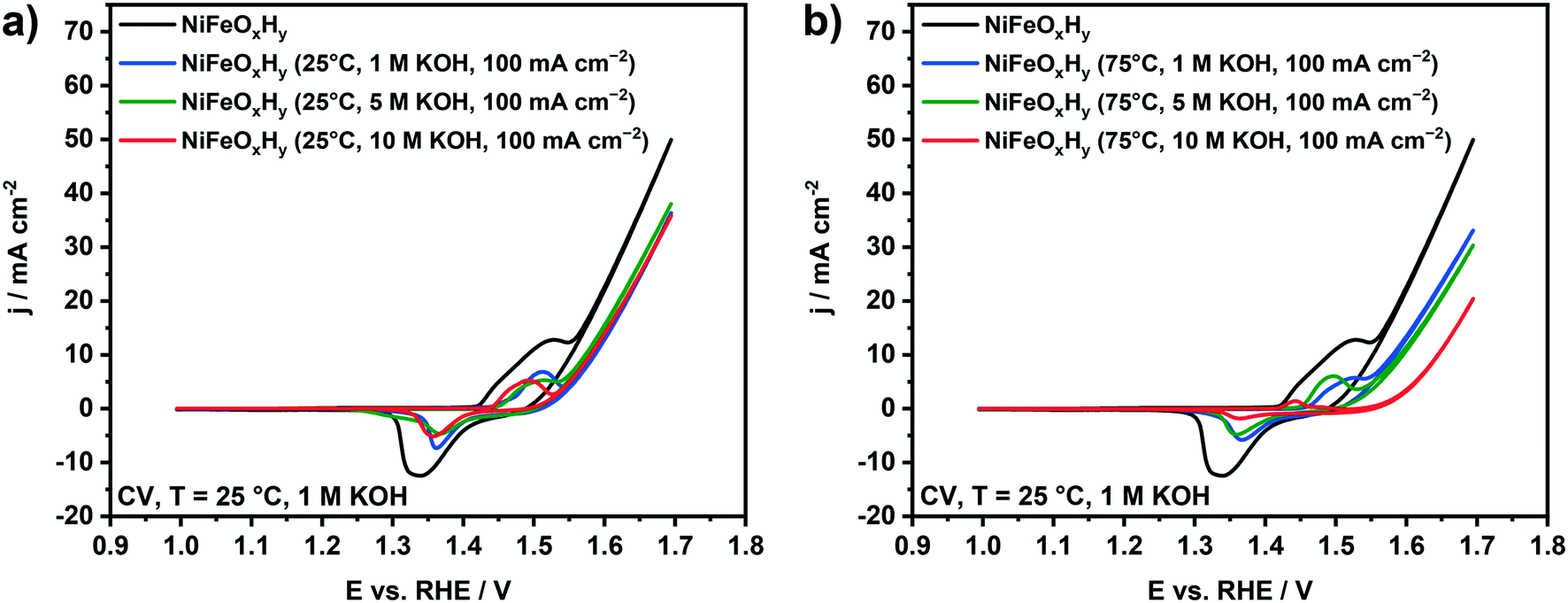 | ||
| Fig. 5 Cyclic voltammetry (CV) recorded at 25 °C in 1 M KOH at a scan rate of 50 mV s−1 of NiFeOxHy before and after anodic polarization at j = 100 mA cm−2 in alkaline conditions at a) 25 °C and b) 75 °C. | ||
The important result is that anodic polarization at elevated temperature and increased base concentration leads to loss of the active metals with a preferential removal of Fe, negatively affecting the OER performance of NiFeOxHy anodes. Under the most severe testing conditions (75 °C, 10 M KOH), the conversion of Ni-(oxy)hydroxide to Ni-oxide was also observed, which will also contribute to the lowered activity.
We next compared the stability of NiFeOxHy with that of NiOxHy after 1 hour of anodic polarization at 75 °C in 5 M KOH, similar to the conditions applied in commercial alkaline electrolyzers (Fig. S10†). The effect on the redox properties and electrocatalytic activity is shown in Fig. 6a. In accordance with previous findings, the redox peak ascribed to the transformation Ni2+/Ni3+ in the voltammogram of NiOxHy is cathodically shifted and the catalytic activity is significantly lower as compared to NiFeOxHy.17,19,47 After anodic polarization at 75 °C in 5 M KOH, differently than in the case of NiFeOxHy, the activity of NiOxHy increased. The electrochemical test conducted at high temperature and molar concentration was thus beneficial for the performance of NiOxHy.
 | ||
| Fig. 6 a) Cyclic voltammetry (CV) recorded in 1 M KOH with a scan rate of 50 mV s−1 of NiOxHy and NiFeOxHy before and after anodic polarization at 75 °C in 5 M KOH, j = 100 mA cm−2; b) experimental (points) and fitted (lines) Nyquist plots recorded at 25 °C in 1 M KOH at E = +1.54 VRHE; c) atomic Fe percentage out of the total (Ni + Fe) content measured by XPS before and after anodic polarization at 75 °C in 5 M KOH, j = 100 mA cm−2. | ||
The electrocatalytic behavior of the anodes was also evaluated by Tafel analysis (Fig. S11†). The Tafel slopes of fresh NiFeOxHy and NiOxHy (Table 1) clearly show the beneficial effect of Fe on the kinetics of OER on Ni(OH)2: the Tafel slopes for the Fe-doped sample (37 mV dec−1) are lower than for pure NiOxHy (48 mV dec−1). The Tafel slopes are in agreement with previous values available in literature.19,48 Values around 40 mV dec−1 indicate that a second-electron transfer is rate-determining in the OER reaction pathway.48,49 After an electrochemical treatment conducted at 75 °C in 5 M KOH, NiFeOxHy maintained the same Tafel slope (37 mV dec−1), indicating that the reaction mechanism and the rate-determining step were unchanged by the applied electrochemical treatment. On the other hand, the Tafel slope of NiOxHy decreased after anodic polarization at 75 °C in 5 M KOH (to 39 mV dec−1), exhibiting a slope comparable to that of NiFeOxHy (37 mV dec−1). This finding indicates improved kinetics for NiOxHy after anodic polarization.
| Sample ID | Tafel slope, mV dec−1 | R ct, Ω cm2 | C dl, mF cm−2 |
|---|---|---|---|
| NiFeOxHy | 37 | 3.14 | 7.05 |
| NiFeOxHy (75 °C, 5 M KOH, j = 100 mA cm−2) | 37 | 10.29 | 3.20 |
| NiOxHy | 48 | 61.2 | 20.8 |
| NiOxHy (75 °C, 5 M KOH, j = 100 mA cm−2) | 39 | 26.8 | 12.0 |
To further investigate the electrochemical properties of the samples, electrochemical impedance spectroscopy was applied (Fig. 6b). Each point in the Nyquist plot represents the real (Z′) and imaginary (Z′′) part of the impedance determined at different frequencies (decreasing from left to right) of a sinusoidal electrochemical perturbation of the voltage.50 The Nyquist plots of the fresh and tested samples show typical semicircles attributed to the charge-transfer resistance (Rct) at the electrode surface.13,51,52 The real value of impedance at low Z′ (i.e., at high frequencies) corresponds to the resistance of the electrolyte and the circuit (Rs). The spectra were fitted with an Rs(RctCdl) circuit, to account for the series resistance, the charge-transfer resistance, and the double-layer capacitance of the samples.50,53 The results are summarized in Table 1.
Analysis of the values of Rct shows that the charge-transfer resistance of the Fe-doped sample increased by a factor of three after anodic polarization, while for pure NiOxHy the values of Rct were significantly lower after polarization. While the electrochemical treatment detrimentally affected the charge-transfer properties of NiFeOxHy, improved charge transfer was found for NiOxHy.
We also evaluated the double-layer capacitances (Cdl) of the samples, which are proportional to the electrochemical surface area of the samples.53–56 The values of Cdl of NiOxHy were higher than for NiFeOxHy before the electrochemical test; after anodic polarization, the values for both samples were roughly halved, indicating that a loss of surface area occurred due to applied electrochemical treatment. The lower electrochemical surface areas could be due to lower catalyst loading after the electrochemical treatment because of leaching. SEM images of NiOxHy after anodic polarization at 75 °C in 5 M KOH reveal a loss of active phase compared to fresh sample (Fig. S12†), similar to what we previously observed for NiFeOxHy.
NiOxHy is known to incorporate Fe impurities present in KOH, which results in an activity enhancement.21,39,57 We thus evaluated the surface composition on fresh and used NiOxHy by means of XPS analysis. The results reported in Fig. 6c conducted on NiOxHy show that after anodic polarization at 75 °C in 5 M KOH the NiOxHy sample incorporated a small amount of Fe during the electrochemical treatment, responsible for the increase in catalytic activity compared to fresh NiOxHy. These results indicate that differences in Fe content and electrocatalytic properties of NiOxHy and NiFeOxHy become small after electrochemical polarization at 75 °C in 5 M KOH, because of the similar final low Fe content.
4. Conclusions
We investigated the stability of thin NiFe-(oxy)hydroxide films upon alkaline water oxidation at a current density of 100 mA cm−2 at different temperatures and KOH concentrations, which is close to conditions prevalent in industrial alkaline water electrolyzers. Depending on the temperature and KOH concentration, the anodes were affected in terms of structure and performance. While anodic polarization at 25 °C did not significantly impact the anode, a higher temperature (75 °C) caused faster degradation of the anodes, especially in 5 or 10 M KOH. XPS, EDX, and Raman characterization pointed to preferential leaching of Fe over Ni, especially when high temperatures and KOH concentrations were applied. The platelet-like morphology characteristic of NiFeOxHy disappeared during polarization at the harshest conditions, suggesting a loss of active phase, which was confirmed by a decreased electrochemical surface area. These changes also impacted the catalytic activity of the samples, which was decreased after electrochemical testing at high temperature and base concentrations. For the sample tested under the harshest conditions (75 °C, 10 M KOH), a conversion from Ni(OH)2 to NiO was observed, resulting in further deterioration of the catalytic properties of this sample. On the other hand, pure NiOxHy easily incorporates Fe from the KOH electrolyte impurities during anodic polarization at 75 °C in 5 M KOH, which leads to an increase of its catalytic activity despite the loss of surface area during the electrochemical test. The Fe content, electrocatalytic activity, and Tafel slope of NiOxHy were almost similar to those of NiFeOxHy after electrochemical treatment at industrially relevant conditions. These results highlight the importance of conducting stability tests at more realistic conditions to evidence degradation mechanisms of the electrodes during industrial operations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Adelheid Elemans-Mehring for ICP-OES measurements. M. E. C. P. acknowledges funding by a Graduate School program from the Netherlands Organization for Scientific Research (NWO). A. G. and E. J. M. H. acknowledge funding from an NWO Vici grant.References
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- M. Schalenbach, A. R. Zeradjanin, O. Kasian, S. Cherevko and K. J. J. Mayrhofer, Int. J. Electrochem. Sci., 2018, 13, 1173–1226 CrossRef CAS.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 RSC.
- F. Lyu, Q. Wang, S. M. Choi and Y. Yin, Small, 2019, 15, 1–17 Search PubMed.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737–3742 CrossRef CAS PubMed.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- J. Landon, E. Demeter, I. Nilay, C. Keturakis, I. E. Wachs, R. Vasic, A. I. Frenkel and J. R. Kitchin, ACS Catal., 2012, 2, 1793–1801 CrossRef CAS.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- D. A. Corrigan and R. M. Bendert, J. Electrochem. Soc., 1989, 136, 723–728 CrossRef CAS.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- T. Zhou, Z. Cao, P. Zhang, H. Ma, Z. Gao, H. Wang, Y. Lu, J. He and Y. Zhao, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
- K. He, Z. Cao, R. Liu, Y. Miao, H. Ma and Y. Ding, Nano Res., 2016, 9, 1856–1865 CrossRef CAS.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- D. A. Corrigan, J. Electrochem. Soc., 1987, 134, 377–384 CrossRef CAS.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 2174 Search PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486–1491 CrossRef CAS PubMed.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- M. B. Stevens, C. D. M. Trang, L. J. Enman, J. Deng and S. W. Boettcher, J. Am. Chem. Soc., 2017, 139, 11361–11364 CrossRef CAS PubMed.
- H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2016, 138, 313–318 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- M. Görlin, P. Chernev, P. Paciok, C. W. Tai, J. Ferreira de Araújo, T. Reier, M. Heggen, R. Dunin-Borkowski, P. Strasser and H. Dau, Chem. Commun., 2019, 55, 818–821 RSC.
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- C. Andronescu, S. Barwe, E. Ventosa, J. Masa, E. Vasile, B. Konkena, S. Möller and W. Schuhmann, Angew. Chem., Int. Ed., 2017, 56, 11258–11262 CrossRef CAS PubMed.
- C. Andronescu, S. Seisel, P. Wilde, S. Barwe, J. Masa, Y. T. Chen, E. Ventosa and W. Schuhmann, Chem. – Eur. J., 2018, 24, 13773–13777 CrossRef CAS PubMed.
- S. Klaus, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 18303–18316 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- B. M. Hunter, W. Hieringer, J. R. Winkler, H. B. Gray and A. M. Müller, Energy Environ. Sci., 2016, 9, 1734–1743 RSC.
- N. S. Mcintyre and M. G. Cook, Anal. Chem., 1975, 47, 2208–2213 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- C. Roy, B. Sebok, S. B. Scott, E. M. Fiordaliso, J. E. Sørensen, A. Bodin, D. B. Trimarco, C. D. Damsgaard, P. C. K. Vesborg, O. Hansen, I. E. L. Stephens, J. Kibsgaard and I. Chorkendorff, Nat. Catal., 2018, 1, 820–829 CrossRef CAS.
- M. Etzi Coller Pascuzzi, E. Selinger, A. Sacco, M. Castellino, P. Rivolo, S. Hernández, G. Lopinski, I. Tamblyn, R. Nasi, S. Esposito, M. Manzoli, B. Bonelli and M. Armandi, Electrochim. Acta, 2018, 284, 294–302 CrossRef CAS.
- T. Sun, L. Xu, Y. Yan, A. A. Zakhidov, R. H. Baughman and J. Chen, ACS Catal., 2016, 6, 1446–1450 CrossRef CAS.
- I. G. Casella, M. R. Guascito and M. G. Sannazzaro, J. Electroanal. Chem., 1999, 462, 202–210 CrossRef CAS.
- L. A. Stern and X. Hu, Faraday Discuss., 2014, 176, 363–379 RSC.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- R. Kostecki and F. McLarnon, J. Electrochem. Soc., 1997, 144, 485–493 CrossRef CAS.
- J. Desilvestro, D. A. Corrigan and M. J. Weaver, J. Electrochem. Soc., 1988, 135, 885–892 CrossRef CAS.
- D. Thierry, D. Persson, C. Leygraf, N. Boucherit and A. Hugot-le Goff, Corros. Sci., 1991, 32, 273–284 CrossRef CAS.
- L. Slavov, M. V. Abrashev, T. Merodiiska, C. Gelev, R. E. Vandenberghe, I. Markova-Deneva and I. Nedkov, J. Magn. Magn. Mater., 2010, 322, 1904–1911 CrossRef CAS.
- D. L. A. de Faria, S. Venâncio Silva and M. T. de Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef CAS.
- B. S. Yeo, S. L. Klaus, P. N. Ross, R. A. Mathies and A. T. Bell, ChemPhysChem, 2010, 11, 1854–1857 CAS.
- M. E. G. Lyons and M. P. Brandon, Int. J. Electrochem. Sci., 2008, 3, 1386–1424 CAS.
- X. Li, F. C. Walsh and D. Pletcher, Phys. Chem. Chem. Phys., 2011, 13, 1162–1167 RSC.
- F. J. Pérez-Alonso, C. Adán, S. Rojas, M. A. Peña and J. L. G. Fierro, Int. J. Hydrogen Energy, 2014, 39, 5204–5212 CrossRef.
- E. Guerrini, M. Piozzini, A. Castelli, A. Colombo and S. Trasatti, J. Solid State Electrochem., 2008, 12, 363–373 CrossRef CAS.
- A. R. C. Bredar, A. L. Chown, A. R. Burton and B. H. Farnum, ACS Appl. Energy Mater., 2020, 3, 66–98 CrossRef CAS.
- M. Merrill and R. Dougherty, J. Phys. Chem. C, 2008, 112, 3655–3666 CrossRef CAS.
- B. Weng, F. Xu, C. Wang, W. Meng, C. R. Grice and Y. Yan, Energy Environ. Sci., 2017, 10, 121–128 RSC.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- M. Etzi Coller Pascuzzi, A. Goryachev, J. P. Hofmann and E. J. M. Hensen, Appl. Catal., B, 2020, 261, 118225 CrossRef CAS.
- F. Zhou, A. Izgorodin, R. K. Hocking, V. Armel, L. Spiccia and D. R. MacFarlane, ChemSusChem, 2013, 6, 643–651 CrossRef CAS PubMed.
- A. Goryachev, M. Etzi Coller Pascuzzi, F. Carlà, T. Weber, H. Over, E. J. M. Hensen and J. P. Hofmann, Electrochim. Acta, 2020, 336, 135713 CrossRef CAS.
- A. M. Smith, L. Trotochaud, M. S. Burke and S. W. Boettcher, Chem. Commun., 2015, 51, 5261–5263 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01179g |
| ‡ Present address: KAUST Catalysis Center, 4700 King Abdullah University of Science and Technology, Thuwal 23955-6900, Kingdom of Saudi Arabia. |
| § Present address: Surface Science Laboratory, Department of Materials and Earth Sciences, Technical University of Darmstadt, Otto-Berndt-Strasse 3, 64287 Darmstadt, Germany. |
| This journal is © The Royal Society of Chemistry 2020 |