
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-oxidative dehydrogenation of isobutane over supported vanadium oxide: nature of the active sites and coke formation†
Alberto
Rodriguez-Gomez
 a,
Abhishek Dutta
Chowdhury
a,
Mustafa
Caglayan
a,
Jeremy A.
Bau
a,
Edy
Abou-Hamad
b and
Jorge
Gascon
*a
a,
Abhishek Dutta
Chowdhury
a,
Mustafa
Caglayan
a,
Jeremy A.
Bau
a,
Edy
Abou-Hamad
b and
Jorge
Gascon
*a
aKAUST Catalysis Center (KCC), Advanced Catalytic Materials, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia. E-mail: jorge.gascon@kaust.edu.sa
bCore Labs, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia
First published on 1st July 2020
Abstract
We combine Raman spectroscopy, electron paramagnetic resonance (EPR), X-ray photoelectron spectroscopy (XPS), temperature-programmed reduction (TPR), X-ray diffraction (XRD), high-field 51V-solid-state magic angle spinning NMR spectroscopy (ssNMR), transmission electron microscopy (TEM) and N2-physisorption to unravel structure–activity relationships during the non-oxidative dehydrogenation of isobutane over a V-based catalyst. The use of SBA-15 as a support favours the formation of oligomeric tetrahedral VOx species along with a smaller amount of V2O5 clusters. EPR, 51V-ssNMR and XPS suggest the formation of mostly V4+ species under reaction conditions. Investigation of “coke” species by dynamic nuclear polarization surface enhanced solid-state NMR (DNP SENS) reveals the co-existence of aliphatic, olefinic/aromatic, acetal/alkoxy and carbonyl-based organic moieties in the post-reacted catalyst. Together with TPR and XRD results, we postulate that oxygenated coke species are the main components responsible for vanadium clustering, which results in the irreversible deactivation of the catalyst.
Introduction
The development of direct routes for the production of light olefins has gained interest over the last few decades. The main on-purpose route is the catalytic dehydrogenation of light alkanes, with platinum and chromium supported catalysts being the preferred option for industrial processes such as Catofin and Oleflex.1 In the search for catalysts based on abundant and low toxicity elements, vanadium has been identified as a suitable alternative to Pt and Cr.Vanadium-based catalysts have been widely characterized by different techniques in order to determine the structure and geometry of vanadia sites, highlighting EPR,2–9 solid-state NMR,5–7,9–15 Raman,4–6,8,11,13,16–23 FTIR4–6,12,13,19,23 and UV-vis spectroscopy.6–8,11–13,16–20 In spite of the interest, the actual nature of the active sites in vanadium-based dehydrogenation catalysts is still a matter of debate. The main reason is that vanadium displays, in most catalysts, a broad speciation under reaction conditions. In order to address this issue, a large number of techniques have already been applied in the literature to unravel the nature of the active sites in a large variety of V-based catalysts, including in situ DRIFTS,24–27 Raman,28–30 or UV-vis spectroscopy.7,8,31–33 It has been seen that both structural and chemical states of vanadium determine its catalytic performance. Isolated tetrahedral vanadium and two-dimensional V–O–V species directly bonded to the support have commonly been found in very active catalysts,32,34 which can be easily achieved in high surface area silica/alumina supports.5–8,11–13,16,17,19–23 In regard to the oxidation state of vanadium, it is also commonly accepted that V3+, V4+ and V5+ are all present during alkane dehydrogenation reactions, but only V3+ and V4+ are catalytically active.1,35,36 According to Kaichev et al., the oxidative dehydrogenation of propane requires a redox mechanism involving the participation of V3+, V4+ and V5+ species, whereas coordinatively unsaturated V3+ is the main catalytic site for the reaction under non-oxidative conditions.25 However, V3+ sites also promote cracking and isomerization.36 An in situ DRIFTS study over vanadium supported on γ-Al2O3 suggested that although isolated V3+ is the most active site for the non-oxidative dehydrogenation of propane, the formation of less active hydroxylated V4+Ox could prevent coke formation.26 Similarly, isolated oxo-vanadium species are also prone to reduction under reaction conditions, resulting in the formation of V–O–H acidic sites which promote the formation of aromatics and coke.31,37 Thus, oligomerization of VOx species might be beneficial by preventing secondary reactions, including coking.38
However, the literature is still lacking an in-depth structure–reactivity study, which could help us upgrade the process towards industrial maturity. To achieve this stage, it is also important to identify the nature of organic species trapped in the post-reacted material, which constitutes an extreme challenge due to the existence of non-diamagnetic metal centres within the catalyst. To overcome this limitation, in this work, we present a systematic in-depth study on the use of SBA-15 as a support for V species for the non-oxidative dehydrogenation of isobutane, including intensive characterization of V species prior to and after the reaction along with a thorough analysis of coke species and the deactivation mechanism. Temperature-programmed oxidation and UV-Raman spectroscopy have been shown to be useful to correlate different vanadium species with the formation of structured coke on the spent catalyst.26,32 On the other hand, advanced solid-state NMR spectroscopy has recently established itself as a compelling technique to elucidate the molecular structure(s) of coke species in heterogeneous catalysis, particularly involving porous catalytic materials.39–45 However, in the case of coke analysis/identification, the success of this approach typically depends on the natural abundance of the reactant feed during catalysis, since 13C possesses only 1.1% natural abundance.39–43,46–52 Ruling out the possibility of using highly expensive 13C-enriched reactants to address low sensitivity, we have recently demonstrated that the utilization of dynamic nuclear polarization surface-enhanced NMR spectroscopy (DNP SENS) is an ideal alternative.53 Prior to this work, DNP SENS was conventionally applied for the characterization of inorganic materials,54–62 including surface organometallic fragments/catalysts, metal–organic frameworks (MOFs), and amorphous aluminosilicates.63–67 Here, we further demonstrate the potential application of this advanced technique to elucidate the nature of coke deposits during the dehydrogenation of isobutane. To the best of our knowledge, this work represents a unique attempt to provide a thorough coke analysis by DNP SENS in heterogeneous catalysis (particularly on non-diamagnetic metal-containing materials).
Materials and methods
Catalyst preparation
The mesoporous silica support SBA-15 was synthesized according to a modification of the method previously described by Zhao et al.68,69 using a TEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P123:HCl:NaF:H2O (TEOS: Sigma-Aldrich, CAS: 78-10-4; HCl 37%: Alfa-Aesar, CAS: 7647-01-0; Pluronic P-123: Sigma-Aldrich, CAS: 9003-11-6; NaF: Sigma-Aldrich, CAS: 7681-49-4) molar ratio of 1:0.02:0.32:0.04:204. Typically, 7.6 g of P123 was dissolved in 300 mL of a HCl/water solution at pH = 1.5 in a glass bottle and stirred after total solution of the polymeric surfactant. After that, 17 mL of TEOS was added drop by drop to the solution and the mixture was kept under stirring at room temperature for 3 hours. Then, 140 mg of NaF was added, which is known to increase the crystallization rate and provide greater hydrothermal stability to the final product.70 The resulting solution was heated up to 45 °C and kept aging with stirring for 72 hours. Finally, the obtained white solid product was filtered, washed with 400 mL of deionized water and 200 mL of acetone, dried in an oven at 120 °C for 24 hours and calcined in air for 3 hours at 550 °C using a heating ramp of 1 °C min−1.
:P123:HCl:NaF:H2O (TEOS: Sigma-Aldrich, CAS: 78-10-4; HCl 37%: Alfa-Aesar, CAS: 7647-01-0; Pluronic P-123: Sigma-Aldrich, CAS: 9003-11-6; NaF: Sigma-Aldrich, CAS: 7681-49-4) molar ratio of 1:0.02:0.32:0.04:204. Typically, 7.6 g of P123 was dissolved in 300 mL of a HCl/water solution at pH = 1.5 in a glass bottle and stirred after total solution of the polymeric surfactant. After that, 17 mL of TEOS was added drop by drop to the solution and the mixture was kept under stirring at room temperature for 3 hours. Then, 140 mg of NaF was added, which is known to increase the crystallization rate and provide greater hydrothermal stability to the final product.70 The resulting solution was heated up to 45 °C and kept aging with stirring for 72 hours. Finally, the obtained white solid product was filtered, washed with 400 mL of deionized water and 200 mL of acetone, dried in an oven at 120 °C for 24 hours and calcined in air for 3 hours at 550 °C using a heating ramp of 1 °C min−1.
The supported vanadium catalyst was prepared by incipient wetness impregnation of vanadyl sulphate (VOSO4·5H2O: Sigma-Aldrich, CAS: 12439-96-2) for a metal loading of 10 wt% in the final product. Briefly, 2 g of as-synthesized SBA-15 was added directly to a solution containing 1.1 g of VOSO4·5H2O dissolved in 4.2 mL of water, previously calculated for the incipient wetness impregnation of the support. The mixture was homogenized by ultrasonic treatment for 1 hour, dried at 110 °C for 24 hours and finally calcined in air for 3 hours at 550 °C using a heating ramp of 1 °C min−1.
N2 adsorption
Nitrogen isotherms were obtained on TriStar II 3020 equipment (Micromeritics) at 77 K. The solids were pre-treated under vacuum at 100 °C for 12 hours prior to the experiment. Specific surface areas were estimated according to the BET method71 in the relative pressure range of 0.05–0.25. The pore size distribution was analysed by applying the NLDFT method.72 The total pore volume, VT, was estimated from a single point adsorption measurement at P/P0 = 0.94, for pore sizes below 35 nm, disregarding macropores.PXRD
Powder X-ray diffractograms were obtained with Bruker D8C equipment using an anode of Cu Kα (λ = 1.5418 Å) in a Bragg–Brentano configuration operating at 40 eV and 40 mA. Data acquisition was carried out in the 2θ range of 10–80° with an acquisition period of 3 seconds and a step size of 0.04°. The analysis and identification of peaks was carried out by Diffrac.Suite EVA software (Bruker).Temperature programmed reduction
TPR profiles were measured on an Altamira instrument, model AMI-200. After a pre-treatment in argon at 120 °C, the sample was cooled down to room temperature and then subjected to a treatment in 10% H2/Ar until 900 °C using a heating ramp of 10 °C min−1. The hydrogen consumption was monitored by using a thermal conductivity detector and the results analysed by AMI-Analysis v2.21 software.TEM imaging
Transmission electron microscopy was carried out using a Tecnai Twin microscope (FEI) operating at 120 kV in bright-field mode. The catalyst was dispersed in ethanol, dropped onto a copper grid coated with a lacey carbon film, and dried for 30 min.Inductively coupled plasma
Analysis by ICP-OES for V and S was carried out on a 5100 ICP-OES instrument using argon as the gas carrier. Digestion of the silica matrix was carried out with a solution containing nitric acid and HF at max 240 °C and max 35 bar on an UltraWAVE apparatus (Milestone).Raman
Measurements having a spectral resolution of ca. 1.1 cm−1 were collected using a Horiba Labram Aramis Raman spectrometer. The 473 nm laser line was applied via a Cobolt Blues laser. The maximum power of the laser line at the source was reported to be 50 mW. The spectrum of the spent catalyst was collected by using a leak-proof cell having a quartz window after the sample was placed in it under an Ar atmosphere. For all measurements, a 25% neutral density filter was applied.Solid-state NMR spectroscopy
The 51V MAS NMR spectra were recorded on a Bruker AVANCE III operating at 236.7 MHz (21.2 T) using a 3.2 mm MAS probe. The 51V MAS NMR spectrum was recorded using single pulse excitation with a small pulse angle (π/8). Three spinning rates (22, 20, and 18 kHz) were used to determine the isotropic chemical shift. The delay between the scans was set to 1 s and the number of scans ranged between 30000 and 100000. The 51V chemical shift was referenced with respect to neat VOCl3 (δ = 0 ppm).
Dynamic nuclear polarization NMR spectroscopy (DNP SENS)
Two different DNP agents (TEKPol and AMUPol) and three different solvent matrixes (TCE, glycine + water, and DMSO + water; DMSO: dimethylsulfoxide, TCE: tetrachloroethane) were used in this study. Data were acquired at the Core Labs of King Abdullah University of Science and Technology using a 263 GHz/400 MHz Avance III Bruker DNP solid-state NMR spectrometer (νL(13C) = 100.6 MHz) equipped with a 3.2 mm Bruker triple resonance low-temperature magic angle spinning (LTMAS) probe and the experiments were performed at ca. 100 K with 263 GHz gyrotron microwave irradiation. The sweep coil of the main magnetic field was set for the microwave irradiation occurring at the 1H positive enhancement maximum of the TEKPol or AMUPol biradical (see the ESI† for further details).XPS
X-ray photoelectron measurements were conducted on Axis Ultra DLD (Kratos) equipment under ultra-high vacuum conditions (10−9 mbar). Air sensitive samples were kept under an inert atmosphere in the catalytic reactor until introduced to the analysis chamber. The samples were excited by a monochromated Al Kα source (1486.6 eV) at 15 kV and 10 mA. The analysis spot size was set at 400 μm. The binding energy scale was calibrated by setting the C1s intensity maximum at 284.8 eV. High resolution V2p, O1s, Si2p and C1s spectra were recorded at 20 eV pass energy and a resolution of 0.1 eV. The results were processed and analysed in CasaXPS software, version 2.3.14dev5.EPR
Electron paramagnetic resonance spectroscopy was performed on an X-band continuous wave Bruker EMX PLUS spectrometer (BrukerBioSpin, Rheinstetten, Germany) equipped with a standard resonator for high sensitivity CW-EPR at a frequency of 9.43 GHz. Spectra were measured at 25 dB microwave attenuation, 1 G modulation amplitude, and 100 kHz modulation frequency.Catalytic tests
The catalytic performance of V/SBA-15 in the non-oxidative dehydrogenation of isobutane was measured at atmospheric pressure in a tubular quartz reactor (Ø = 8 mm) with 0.5 g of pelletized and sieved catalyst (150–250 μm) in fixed-bed mode. The catalyst was pre-treated at 600 °C in 10% H2/He using a total flow rate of 50 mL min−1 for 1 hour and then brought to the reaction temperature under helium flow. The reaction mixture consisted of 4 mL min−1 of 50% iC4H10/N2. In the reaction–regeneration cycle experiment, the catalyst was treated in air at 600 °C for 1 hour (heated up from 550 °C) after each reaction cycle of 4 hours using a flow rate of 50 mL min−1. After regeneration, the sample was cooled down until the reaction temperature in helium flow. The outgoing flow was analysed by gas chromatography conducted on a Varian 450-GC gas chromatograph equipped with a Molsieve 13X column and TCD for the analysis of nitrogen and a HP-Alumina/KCl column and FID for the analysis of hydrocarbons. Nitrogen was used as the internal standard. The isobutane conversion, selectivity to different products and carbon balance were calculated as follows:

| CxHy = hydrocarbon product x = number of carbons |

Results and discussion
Physico-chemical characterization of calcined V/SBA-15
The main physicochemical properties of the sample of interest in this study are summarized in Table 1. As shown, the high surface area of the mesoporous silica support SBA-15 decreased from 776 to 322 m2 g−1 after impregnation of the vanadyl precursor and calcination. The N2-adsorption/desorption isotherms of both samples show a hysteresis loop at P/P0 > 0.5 typically associated with ordered mesoporous materials (type IV, H1, Fig. 1a),68,73 as well as an important adsorption increase above 0.8 due to interparticle capillary condensation.74 The pore size distribution, as calculated from NLDFT, shows a unique pore width of 90 Å, even after vanadium impregnation. However, the pore volume decreases from 1.03 to 0.49 m2 g−1, presumably meaning that some of the pores in the pristine SBA-15 are not accessible after vanadium impregnation and calcination.| Catalyst | S BET/m2 g−1 | V T/cm3 g−1a | ICP/wt% | D Scherrer/nm | XPS V2p3/2 BE/eV | [Vn+]surface/at%b | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| V | V2O5 | V2O3 | V5+(I) | V5+(II) | V4+ | V3+ | V5+ | V4+ | V3+ | |||
| a V T is the total pore volume estimated from a single point measurement at P/P0 = 0.94 of the N2-adsorption isotherm. b Calculated from the deconvoluted V2p3/2 XPS peak. c V/SBA-15iC4,550°C: spent catalyst after dehydrogenation of isobutane. | ||||||||||||
| SBA-15 | 776 | 1.03 | — | — | — | — | — | — | — | — | — | |
| V/SBA-15 | 322 | 0.49 | 10.2 | 35 | n.d. | 518.0 | 516.8 | 515.7 | — | 80.7 | 19.3 | — |
| V/SBA-15iC4,550°C | 313 | 0.47 | 10.5 | n.d | 23 | 518.2 | 516.9 | 515.7 | 514.1 | 42.6 | 50.5 | 6.9 |
 | ||
| Fig. 1 N2-Adsorption/desorption isotherms and pore size distributions of mesoporous silica SBA-15 and both pristine and spent* V/SBA-15 (a), diffraction patterns of pristine, spent* and regenerated** V/SBA-15 (b), temperature programmed reduction profiles of bulky V2O5 (Sigma-Aldrich) and both pristine and regenerated** V/SBA-15 (c), and bright field TEM images of pristine V/SBA-15 (d). * indicates V/SBA-15iC4H10,550°C. ** indicates V/SBA-15regen.,600°C. | ||
Fig. 1b shows the PXRD pattern of the V/SBA-15 sample after calcination at 550 °C. The presence of orthorhombic V2O5 (JCPDS 001-0359) is clear from the diffractogram, with main peaks at 15.3, 20.2, 26.1 and 40.0° and an average crystallite size of around 32 nm, estimated by applying the Scherrer equation. Fig. 1c shows the temperature programmed reduction profile of the same catalyst. It is composed of a broad peak with two components in the temperature range of 450–650 °C. The first component, centred at ca. 565 °C, can be attributed to the presence of oligomeric tetrahedral vanadium species (O3V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).7,13,75 The second component, with the maximum at around 592 °C, is presumably associated with the reduction of bulk-like V2O5 clusters.7,13,75,76 As a comparison, the reduction profile of bulk V2O5 shows two sharp peaks at around 665 and 699 °C previously attributed to the consecutive reduction of V2O5 to V6O13 and V2O4, and also a broad peak at ca. 825 °C associated with the formation of V2O3.76 As expected, the mesoporous support has a clear effect on the dispersion of vanadium which results in an enhanced reducibility.
O).7,13,75 The second component, with the maximum at around 592 °C, is presumably associated with the reduction of bulk-like V2O5 clusters.7,13,75,76 As a comparison, the reduction profile of bulk V2O5 shows two sharp peaks at around 665 and 699 °C previously attributed to the consecutive reduction of V2O5 to V6O13 and V2O4, and also a broad peak at ca. 825 °C associated with the formation of V2O3.76 As expected, the mesoporous support has a clear effect on the dispersion of vanadium which results in an enhanced reducibility.
Transmission electron microscopy (Fig. 1d) reveals the characteristic features of SBA-15, with well-ordered pores of ca. 8.5 nm (in good agreement with N2 adsorption, vide supra) and a particle size of ca. 0.10–15 × 0.5–0.8 μm (more pictures in Fig. S1†). Vanadium seems to be well dispersed, with most of the visible clusters (dark spots) homogeneously distributed along the SBA-15 support and a minority of larger aggregates of ca. 20–30 nm necessarily located on the external surface. However, it is difficult to conclude anything about the arrangement of oligomeric species or small clusters since they are not distinguishable.
To understand more in detail the chemical nature of vanadium species in this system, a combination of different spectroscopic techniques, including Raman, XPS, EPR and 51V ssNMR, were used. The main drawback of 51V ssNMR spectroscopy is related to the quenching effect when paramagnetic species are present at a high concentration, including V4+, broadening the signal and thus making it more difficult to detect and assign chemical shifts.6 Contrarily, EPR in conventional magnetic fields is only sensitive to paramagnetic V4+ species, while V5+ and V3+ are invisible.77 Complementarily, Raman spectroscopy is able to detect different vanadium oxide environments; however, it is not sensitive enough for V4+ or V3+. On the other hand, X-ray photoelectron spectroscopy (XPS) can be used to determine and quantify the relative ratio of vanadium species in different chemical states but it is limited to the surface of the material. Hence, a complementary and multimodal spectroscopic approach is essential to gain a fundamental insight.
Fig. 2 shows the Raman spectra of calcined and spent V/SBA-15. The calcined sample displays the characteristic bands of V2O5 (281, 298, 406, 479, 522, 694, and 992 cm−1, see Fig. S2a and Table S1†) and an additional band at ca. 1035 cm−1 assigned to the VO stretching mode of tetrahedral vanadate species.12,20,27,78,79 Although the band observed around 1036 cm−1 is usually associated with the VO vibration of monomeric vanadate species, it would not be theoretically correct to exclude the possibility of the existence of polymeric vanadate species which may have VO vibration bands in close proximity. Thus, it can be suggested that the vanadium sites of the pristine catalyst mainly consist of V2O5 nanocrystals and tetrahedral vanadate species. Following the approach proposed by Xie et al.,80 who estimated that the scattering cross section of V2O5 particles is ca. 10 times larger than that of isolated mono/polyvanadates, a V2O5 to tetrahedral vanadate species ratio of 0.19 can be calculated for this sample.
 | ||
| Fig. 2 Raman spectra of calcined and spent V/SBA-15 catalysts (for clarity, the spectra are offset). | ||
Probing the 51V nucleus (diamagnetic, spin I = 7/2, 99.75% natural abundance and relatively short relaxation times) is a highly challenging affair from the perspective of solid-state NMR spectroscopy (particularly in high-field).13,81 In this case, our aim was to identify the number, oxidation state, and type of non-identical/equivalent V-sites. The range of 51V chemical shift is usually very broad (0 to −2000 ppm), as it is highly sensitive to all the parameters mentioned above.12 In the literature, for analogous samples (i.e., loaded V on mesoporous silica), two major chemical shifts at ∼500 and ∼600 ppm commonly appear, which are due to the presence of crystalline vanadium(V) oxide clusters in the internal (minor) and external (major) walls of mesoporous channels, respectively.13,81 Experimentally, it has been well-documented that V5+ species at the internal walls are not accessible (hence, catalytically irrelevant), as they are well-buried into the amorphous pore walls. In the present case, we do see only one peak at ∼613 ppm in our calcined catalyst (Fig. S3†), which means that all V5+ oxide clusters were exclusively loaded at the external wall, as expected from the preparation by incipient wetness impregnation followed by calcination. In this case, such VOx species are presumably in a distorted tetrahedral geometry with three ‘V–O–Si’ bonds bonded to the external wall of the silica framework along with one ‘VO’ double bond exposed to the pore surface.25,31,37,82–84
Additional information about the surface chemical state of vanadium in the calcined catalyst is obtained from the V2p XPS spectrum, illustrated in Fig. 3. The strong influence of the O1s signal over the V2p region makes background subtraction not trivial. Thus, we focus our peak analysis on the V2p3/2 region using a Tougaard approximation for background simulation. The deconvolution of the peak gives clearly at least three components, presumably attributed to V5+ (green) or V4+ (blue) since oxidation states below 4+ are not expected taking into account the nature of the vanadium precursor (VO(SO4)·5H2O) and the calcination treatment at 550 °C.85 The component centred at 518.1 eV could be attributed to V5+ species in close contact with the support.85–88 Wark et al. associated this high value of binding energy with the presence of V–O–Si bonds in vanadium-containing zeolites prepared by ion exchange.89 In the same way, Liu et al. also attributed the high binding energy to highly dispersed V5+ species interacting strongly with silica in a vanadium-containing dendritic mesoporous silica synthesized by a sol–gel method.30 Besides, the contributions at around 516.8 and 515.7 eV could be associated with V5+ (in the polymeric form) and V4+, respectively.90 An analysis by deconvolution of the V2p3/2 peak shows that the estimated atomic concentration of these species on the surface of the pristine catalyst is ca. 81 and 19%, respectively.
 | ||
| Fig. 3 XPS spectra of the calcined and spent V/SBA-15 samples in the O1s + V2p region (a) and detailed/deconvoluted V2p3/2 spectra (b). V5+, V4+ and V3+ components are marked in green, blue and orange, respectively. The red line corresponds to the envelope of the fitted peaks. | ||
Similar conclusions could be derived from the EPR spectra shown in Fig. 4, where low concentrations of isolated V4+ can be observed in the calcined catalyst. We arrived at this conclusion upon comparing the EPR signal of V4+ in the calcined and spent catalysts (vide infra) and from the hyperfine splitting features in the EPR spectrum of the calcined catalyst. In both cases, a relative isolation of individual V4+ atoms is apparent.91 Based on spin counting, V4+ accounts for 0.8% of the total vanadium content in the calcined sample.
 | ||
| Fig. 4 EPR spectra of the calcined and spent V/SBA-15. | ||
Catalytic performance in the non-oxidative dehydrogenation of isobutane
The activity of the catalyst in the non-oxidative dehydrogenation of isobutane was tested at 550 °C for 9 hours. Operation conditions were optimized in terms of temperature, WHSV and conversion to maximize the isobutylene yield at selectivities higher than 80% (see Fig. S4†). Fig. 5a shows the conversion of isobutane and selectivity to the main products, i.e. isobutylene, propylene, methane and C4= isomers. The initial conversion of isobutane exceeds the equilibrium value for this reaction (65% at 550 °C, PiC4H10 = 0.05 MPa),92 mainly due to coke formation, as indicated by the poor carbon balance in the gas phase. However, after stabilization, the selectivity to C4= increases to 94% and remains stable during the rest of the reaction, with a conversion and carbon balance in the gas phase of ca. 38 and 100%, respectively. The formation of isobutylene isomers (9%) indicates a certain catalytic activity of V/SBA-15 in isomerization. Blank experiments in the absence of a catalyst did not show any isomerization activity (see Fig. S5†). A slow catalyst deactivation can be observed from a moderate decline in isobutane conversion. Additional experiments performed after successive reaction–regeneration cycles demonstrate an irreversible loss of activity upon regeneration in air at 600 °C (Fig. 5b).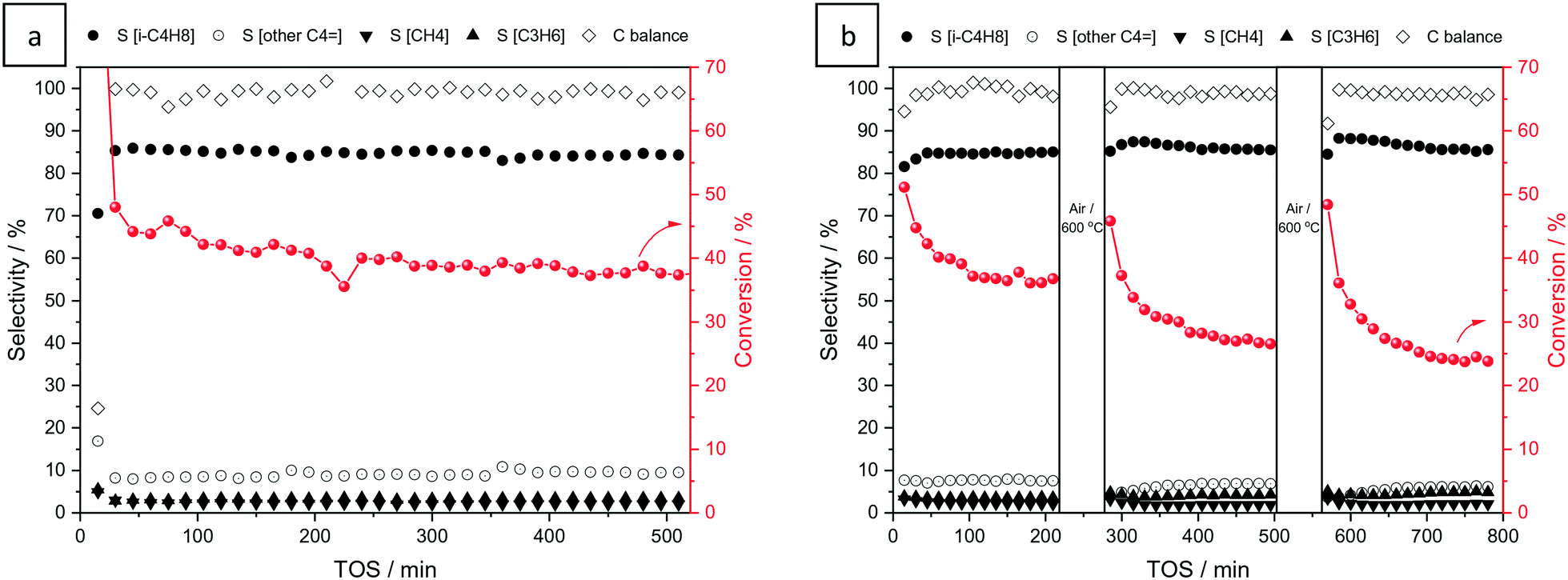 | ||
| Fig. 5 Catalytic performance of V/SBA-15 in the non-oxidative dehydrogenation of isobutane: conversion (right axis), carbon balance (left axis) and selectivity to different products (S[---], left axis) at 550 °C and WHSV = 240 mL min−1 gcat−1 for 520 minutes of experiment (a) and by applying cycles of reaction–regeneration* of 4–1 hours (b). *Regeneration conditions: 50 mL min−1 of air at 600 °C for 1 hour. | ||
Physicochemical characterization of spent V/SBA-15
In order to better understand the observed reversible and irreversible deactivation of the catalysts, we performed extensive characterization of the spent catalyst after 3 reaction cycles. The samples were kept under an inert atmosphere prior to analysis.PXRD (Fig. 1b) indicates the formation of bulk V2O3 (JCPDS 034-0187), with characteristic peaks at 24.3, 33.0, 36.2 and 53.9° and an average crystallite size of 23 nm. However, V2O3 is not Raman sensitive under vacuum/inert atmosphere, due to the small Raman cross-section of reduced vanadium oxide species, and is prone to oxidation to V2O5 under ambient conditions due to the laser-induced heating.93 For this reason, the spectrum of the spent sample was collected under an argon atmosphere. During this measurement, the quartz window of the cell unavoidably affected the spectrum. However, it does not mean that it is not possible to put any interpretation. First of all, the spent sample (Fig. 2) shows the appearance of carbon D and G bands (1346 and 1599 cm−1, respectively) as the fingerprint of coke formation. Next, if we omit the characteristic Raman modes (447, 487, 603, 816, 1060 and 1180 cm−1) of fused quartz,94,95 the peak around 1035 cm−1 may indicate the existence of 4-fold coordinated vanadium. It is well known in the literature that low vanadium loadings on SBA-15 only resulted in V5+ related Raman vibrations in the VO stretching mode.96,97 And it is also clear that none of the V2O5 Raman vibrations exist, even those not intersecting with fused quartz vibrations (281, 298, 694 and 992 cm−1). Furthermore, when we exposed the spent catalyst to air and collected another spectrum subsequently, we observed typical V2O5 Raman modes (Fig. S6†). This means that some reduced vanadium species are getting oxidized due to laser induced heating under air. This could explain the additional band observed by 51V NMR at 712 ppm (Fig. S3b,† in addition to the ∼617 ppm band), related to V5+ ions located at the external wall of the silica framework, where a difference of about 100 ppm indicates that both species reside in different chemical environments. Moreover, the poor signal-to-noise (S/N) ratio is mainly attributed to the strong presence of paramagnetic V4+ species as well as the shielding effect due to the deposited coke species. This is confirmed by the EPR spectrum shown in Fig. 4, which indicates a greatly increased proportion of V4+ in the catalyst after the reaction, 12.0% of V in the sample. Furthermore, while hyperfine splitting was still present, an underlying broad signal was observed in the spent catalyst, reflecting the accumulation of V4+ species at high local concentrations.98
In the same way, the V2p XPS signal of the spent catalyst (Fig. 3a) shows a general shift to lower binding energy due to the reduction of vanadium under reaction conditions. The deconvolution of the V2p3/2 peak (Fig. 3b) confirms a significant increase of the contribution at 515.7 eV (V4+, blue) as well as the presence of a small peak at 514.1 associated with V3+ (orange), estimated at 50.5 and 6.9 at%, respectively. Furthermore, V5+ (green) is still present at a surface concentration of 42.6 at%.
Summarizing, under reaction conditions, V/SBA-15 suffers a strong change. TPR, XRD and Raman demonstrate that vanadium is largely reduced; however ssNMR and XPS suggest that V5+ is still present to some extent along with V4+, which is present at a much higher concentration, as shown by EPR, and mostly localised on the surface as shown by XPS. The average oxidation state (AOS) of V in similar catalysts has been shown to be highly dependent on dispersion, with supports tending to stabilize V4+ species instead of V2O3.13,75 The observed formation of large clusters of V2O3 is most likely responsible for the irreversible deactivation of the catalyst.1 In spite of the initial excellent dispersion of the as-synthesized catalyst, it is obvious from the post-mortem characterization that sintering occurs under reaction conditions. In fact, the recovered catalyst after three reaction–regeneration cycles showed a PXRD pattern similar to the calcined V/SBA-15, but with sharper peaks of vanadium pentoxide (Fig. 1b), associated with larger crystallite sizes of ca. 45 nm (Scherrer). Additionally, the TPR analysis of the regenerated sample (Fig. 1c) shows differences with respect to the calcined V/SBA-15. Apart from a more complex profile, the two main components, previously associated with the reduction of highly dispersed tetrahedral vanadium species (565 °C) and bulk-like V2O5 clusters (592 °C), have been shifted one with respect to the other. The displacement of the second peak to higher temperature, with the maximum at 611 °C, indicates the formation of less reducible bulk-like species, in agreement with sintering of vanadium oxide.13,75,76 Furthermore, the roughness of the new profile is a clear signal of vanadium redistribution in different cluster types. To demonstrate that this effect is associated with the catalytic process, the fresh (calcined) catalyst was subjected to both reduction (H2, 600 °C) and reoxidation (air, 600 °C) treatments without being subjected to the catalytic process. The TPR profile of the reoxidized material showed no major changes compared to that of the fresh catalyst (Fig. S7†), which confirms that clustering is mainly associated with the catalytic process and successive regeneration.
Advanced characterization of deposited carbon species in the spent catalyst by MAS ssNMR
To get more insight into the deactivation mechanisms and derive structural information on coke species trapped in the mesoporous structure of SBA-15, advanced MAS ssNMR was performed on the spent catalyst. In a typical DNP experiment,63,99–113 the post-reacted catalytic material is initially impregnated with a solution of DNP agent (i.e., an exogenous nitroxide biradical) in a hydrogen-enriched solvent.63–65,99,106–112 As a result of microwave irradiation, the polarization of protons of the solvent matrix and neighbouring surface of the material is enhanced by the DNP agent, and next, transferred to the hetero-nuclei (i.e., carbons from trapped coke species) of the catalytic material through cross-polarization (CP) at cryogenic temperature. This phenomenon eventually leads to the enhancement of the signal from the residual coke species, even without any isotope-enrichment in the reactant feed.63–65,99,106,107 Following this philosophy, very recently, magic angle spinning DNP SENS was successfully implemented by us during the zeolite-catalysed methanol-to-hydrocarbon reaction (without the necessity for a 13C-enriched methanol feed) to illustrate the Brønsted–Lewis acid synergy in the formation of deactivating coke species.53It is well-documented that this DNP-based approach is highly sensitive to the environment, particularly on the nature of the nitroxide biradical and the solvent. It could be attributed to the fact that the success of this approach solely relies on the CP-like coherence magnetization transfer schemes to transfer the enhanced polarization to the dilute spins of the medium. Inspired by the seminal works from different ssNMR research groups in materials science,54–62 the efficacy of the solvent and DNP agent has been explored in this work for the characterization of coke species. Herein, we have explored two different biradicals (TEKPol and AMUPol) and three different solvent matrixes (TCE, glycine + water, and DMSO + water; DMSO: dimethylsulfoxide, TCE: tetrachloroethane) to illustrate their non-identical behaviours while elucidating the nature of coke species.
For DNP SENS measurements, samples were prepared using incipient wetness impregnation with a solution of 16 mM (i) TEKPol in TCE, AMUPol in (ii) glycine + water, and (iii) DMSO + water (see Fig. 6). There is a significant dissimilarity in the type of identified coke species in each case. At the beginning of our NMR study, we have performed conventional magic angle spinning 1D 1H–13C cross-polarization (CP) experiments (i.e., without microwave irradiation), where we could not detect any peak at reasonable intensity after a long acquisition time (see the bottom spectra in Fig. 6a, in black). This observation could be attributed to the lower natural abundance of residual coke species along with their lower hydrogen content (hence, less efficient CP). Overall, it also provides sufficient justification of how challenging is it to do coke analysis of metal-containing heterogeneous catalysts by means of ‘conventional’ solid-state NMR spectroscopy. Next, in the 1D 1H–13C cross-polarization DNP SENS of the spent catalytic material after impregnation with TEKPol in TCE (see the top spectra in Fig. 6a, in blue), the following two features were primarily observed: (i) 30–35 ppm saturated hydrocarbon moieties (i.e. aliphatic/paraffinic species) and (ii) 118–135 ppm unsaturated hydrocarbon moieties (i.e., olefinic/aromatics).40,48 Surprisingly, 1D 1H–13C cross-polarization DNP SENS of the spent catalytic material after impregnation with AMUPol in glycerol + water (Fig. 6b) demonstrates primarily the response from the aliphatic/paraffinic species, a peak centred around 33 ppm. To investigate the reason behind the absence of an aromatic/olefinic peak, in this case, we have performed 1D 1H–13C cross-polarization magic-angle spinning DNP SENS experiments at different CP contact times (ct) to study diffusion characteristics. Herein, we noticed that (albeit small) peaks due to the presence of unsaturated hydrocarbon moieties (i.e., olefinic/aromatics) only exist at shorter CP contact times (ct), while the intensity of the aliphatic/paraffinic peak is increasing proportionally to the increase of CP contact time (ct). It essentially means that saturated aliphatic/paraffinic coke molecules are preferentially located within the SBA-15 framework and that the aromatic coke species primarily reside on the surface. Another interesting feature is the relatively narrow line-width of aliphatic resonance in both samples (Fig. 6avs.Fig. 6b). This signifies that the aliphatic coke species are primarily mobile in nature, since the cross-polarization-based dipolar/through-space magnetization transfer schemes typically display such a narrower feature while probing mobile molecules, particularly within porous materials.
 | ||
| Fig. 6 1D 1H–13C cross-polarization (CP) magic-angle spinning (MAS) DNP SENS spectra of the post-reacted catalytic material (a) before (in black, without microwave irradiation) or after (in blue, ct = 0.5 ms, under microwave irradiation) the impregnation with TEKPol solution in 1,1,2,2-tetrachloroethane (TCE) (under identical measurement conditions), as well as with AMUPol solution (b) in water (D2O:H2O = 9:1) + glycerol (Gly) (under microwave irradiation), and (c) in water (D2O:H2O = 9:1) + dimethyl sulfoxide (DMSO) (under microwave irradiation) at different CP contact times (ct). See the ESI† for more details (* = spinning side-bands, 8 kHz MAS, recycle delay = 5 s, mW: microwave irradiation). | ||
This observation eventually led us to try another solvent combination (water + DMSO) with AMUPol as the DNP agent (Fig. 6c). Since the response from DMSO itself is overlapping with the aliphatic/paraffinic resonances, it is not possible to analyse aliphatic coke species in this case. However, to our surprise, we have detected two additional classes of organic moieties in this sample, which were absent previously. In this case, the 1D 1H–13C cross-polarization DNP SENS of the spent catalytic material showcases three following features: (i) 70–85 ppm active methylene/alkyl moieties (i.e., aliphatic species in the vicinity of electronegative atoms), (ii) 120–140 ppm unsaturated hydrocarbon moieties (i.e., olefinic/aromatics), and (iii) 165–170 ppm carbonyl moieties. However, contrary to Fig. 6b, this sample showcases the increase in intensity of all three resonances with the rise of CP contact time (ct), i.e., showing a uniform distribution of coke species. It should be worth mentioning that no oxygenate was co-fed during the reaction, and still, we have detected oxygen containing species (like acetals and carbonyls). The only oxygen sources could be reducible VO groups and V2O5 clusters on SBA-15. To derive more structural information on coke species, it is now necessary to perform 2D correlation spectroscopy.
Next, 2D 1H–13C correlation (cross-polarization HETero nuclear CORrelation spectroscopy (CP HETCOR)) solid-state NMR spectroscopy was performed to identify the nature of trapped organic species on all three samples (Fig. S8†). In the 2D 1H–13C CP HETCOR spectra of the TEKPol (in TCE) impregnated spent catalyst material (Fig. S8a†), we noticed that unsaturated hydrocarbons/coke species are primarily olefinic (116–133 ppm (13C) and 5–6.5 (1H) ppm) in nature. To our surprise, aliphatic peaks were not resolved properly in the HETCOR spectra, although they are clearly visible in the corresponding 1D spectra. However, the same aliphatic coke species could also be identified in the 2D 1H–13C CP HETCOR spectra of the AMUPol (in glycerol + water) impregnated spent catalyst (Fig. S8b†). In general, the correlations between 32–35 ppm (13C) and 0.5–1.5 (1H) indicates that aliphatics are the characteristically branched-paraffinic type of coke species. Again, no aromatic coke species were highlighted, which were detected solely in the 2D 1H–13C CP HETCOR spectra of the AMUPol (in DMSO + water) impregnated spent catalyst (Fig. S8c†). In this case, both olefinic (∼126 ppm (13C) and ∼5 (1H) ppm) and (poly)aromatic (128–138 ppm (13C) and 7.5–9 (1H) ppm) types of unsaturated hydrocarbons were independently recognized. In addition, two dependent correlations between 76.4 ppm (13C) and 1.32/3.99 ppm (1H) also confirm the existence of active methylene groups, either an acetal (–O–CH2–O) or alkoxy (V–O–CH2–R, R = H, alkyl) type of species. Although a carbonyl peak was detected (∼166 ppm) in the 1D 1H–13C CP spectrum of this sample, its correlation to any kind of proton is absent. It inevitably means that this is a carbonate species, possibly an ester-type carbonyl that is attached to ‘VO’ (V–O–(CO)–).
Altogether from DNP SENS, we have identified three different types of coke species: primarily, (i) olefinic and (ii) paraffinic species, along with (iii) (poly)aromatics in lower quantity. Moreover, we have identified two different active intermediates in the deactivated catalyst, i.e. (i) acetal/alkoxy and (ii) metal-carbonate species, presumably associated with partially reduced vanadyl groups in the vicinity of hydrocarbons, which might play a crucial role in catalysis, but also in catalyst deactivation, since the formation of oxygenated coke species could promote the aggregation of vanadium observed during the regeneration treatment in air. Finally, the D (1334 cm−1) and G (1598 cm−1) bands observed in the spectrum of the spent sample (Fig. 2), linked to polycrystalline-disordered graphitic and ordered graphitic coke,114 respectively, suggest that the coke on the external surface contains mainly sp2 hybridized C atoms, which is confirmed from the strong asymmetry of the C1s XPS peak, centred at 284.8 eV (see Fig. S9a†), commonly associated with sp2 carbon species.115,116 As a result, this indicates the heterogeneity of coke formation during isobutane dehydrogenation and supports the information described in the analysis of DNP SENS measurements.
Conclusions
In this work we analysed the physicochemical state of vanadium prior to and after the non-oxidative dehydrogenation of isobutane, highlighting the formation of both V4+ and V3+ species at the expense of V5+ along the reaction. It seems that the high dispersion of vanadium along with the mesoporous structure of SBA-15 promotes the formation of V4+ during the reaction while larger clusters of V2O5 are reduced to V2O3, located necessarily outside of the ordered pore structure. The combination of results from different characterization techniques allow us to conclude that V4+ is the main vanadium species generated during the catalytic process with V3+ present to a minor extent. The high activity of the pristine sample, with a stable conversion and selectivity to C4= of 38 and 94%, is impaired after catalyst regeneration. In this sense, the existence of reaction intermediates such as methylene groups, acetal/alkoxy, and carbonate species that may be responsible for the irreversible catalyst deactivation after several reaction–regeneration cycles has been observed, which promotes the sintering of vanadium in large clusters.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by King Abdullah University of Science and Technology (KAUST). We also thank Sandra Ramirez Cherbuy for TOC art.References
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- A. Brückner, Chem. Soc. Rev., 2010, 39, 4673–4684 RSC.
- A. Brückner, Appl. Catal., A, 2000, 200, 287–297 CrossRef.
- S. S. R. Putluru, L. Schill, A. Godiksen, R. Poreddy, S. Mossin, A. D. Jensen and R. Fehrmann, Appl. Catal., B, 2016, 183, 282–290 CrossRef CAS.
- G. C. Bond and S. F. Tahir, Appl. Catal., 1991, 71, 1–31 CrossRef CAS.
- B. M. Weckhuysen and D. E. Keller, Catal. Today, 2003, 78, 25–46 CrossRef CAS.
- H. Berndt, A. Martin, A. Brückner, E. Schreier, D. Müller, H. Kosslick, G. U. Wolf and B. Lücke, J. Catal., 2000, 191, 384–400 CrossRef CAS.
- O. Ovsitser, M. Cherian, A. Brückner and E. V. Kondratenko, J. Catal., 2009, 265, 8–18 CrossRef CAS.
- A. S. L. Thankamony, S. Knoche, S. Bothe, A. Drochner, A. P. Jagtap, S. T. Sigurdsson, H. Vogel, B. J. M. Etzold, T. Gutmann and G. Buntkowsky, J. Phys. Chem. C, 2017, 121, 20857–20864 CrossRef CAS.
- T. Gutmann, A. Schweitzer, M. Wächtler, H. Breitzke, A. Buchholz, W. Plass and G. Buntkowsky, Z. Phys. Chem., 2008, 222, 1389 CrossRef CAS.
- P. Bai, Z. Ma, T. Li, Y. Tian, Z. Zhang, Z. Zhong, W. Xing, P. Wu, X. Liu and Z. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 25979–25990 CrossRef CAS PubMed.
- M. de Oliveira, D. Seeburg, J. Weiss, S. Wohlrab, G. Buntkowsky, U. Bentrup and T. Gutmann, Catal. Sci. Technol., 2019, 9, 6180–6190 RSC.
- Y.-M. Liu, Y. Cao, N. Yi, W.-L. Feng, W.-L. Dai, S.-R. Yan, H.-Y. He and K.-N. Fan, J. Catal., 2004, 224, 417–428 CrossRef CAS.
- A. Fenn, M. Wächtler, T. Gutmann, H. Breitzke, A. Buchholz, I. Lippold, W. Plass and G. Buntkowsky, Solid State Nucl. Magn. Reson., 2009, 36, 192–201 CrossRef CAS PubMed.
- A. Schweitzer, T. Gutmann, M. Wächtler, H. Breitzke, A. Buchholz, W. Plass and G. Buntkowsky, Solid State Nucl. Magn. Reson., 2008, 34, 52–67 CrossRef CAS PubMed.
- X.-L. Xue, W.-Z. Lang, X. Yan and Y.-J. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 15408–15423 CrossRef CAS PubMed.
- R. Chlosta, G. Tzolova-Müller, R. Schlögl and C. Hess, Catal. Sci. Technol., 2011, 1, 1175–1181 RSC.
- C. Hess, M. H. Looi, S. B. A. Hamid and R. Schlögl, Chem. Commun., 2006, 451–453 RSC.
- X. T. Gao, S. R. Bare, B. M. Weckhuysen and I. E. Wachs, J. Phys. Chem. B, 1998, 102, 10842–10852 CrossRef CAS.
- C. Hess, J. D. Hoefelmeyer and T. D. Tilley, J. Phys. Chem. B, 2004, 108, 9703–9709 CrossRef CAS.
- C. Hess, G. Tzolova-Müller and R. Herbert, J. Phys. Chem. C, 2007, 111, 9471–9479 CrossRef CAS.
- P. S. Waleska and C. Hess, J. Phys. Chem. C, 2016, 120, 18510–18519 CrossRef CAS.
- C. Hess, U. Wild and R. Schlögl, Microporous Mesoporous Mater., 2006, 95, 339–349 CrossRef CAS.
- G. Liu, Z.-J. Zhao, T. Wu, L. Zeng and J. Gong, ACS Catal., 2016, 6, 5207–5214 CrossRef CAS.
- V. V. Kaichev, Y. A. Chesalov, A. A. Saraev and A. M. Tsapina, J. Phys. Chem. C, 2019, 123, 19668–19680 CrossRef CAS.
- Z.-J. Zhao, T. Wu, C. Xiong, G. Sun, R. Mu, L. Zeng and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 6791–6795 CrossRef CAS PubMed.
- C. Hess, ChemPhysChem, 2009, 10, 319–326 CrossRef CAS PubMed.
- Q. Liu, J. Li, Z. Zhao, M. Gao, L. Kong, J. Liu and Y. Wei, Catal. Sci. Technol., 2016, 6, 5927–5941 RSC.
- Z. Wu and P. C. Stair, J. Catal., 2006, 237, 220–229 CrossRef CAS.
- Q. Liu, Z. Yang, M. Luo, Z. Zhao, J. Wang, Z. Xie and L. Guo, Microporous Mesoporous Mater., 2019, 282, 133–145 CrossRef CAS.
- I. Takahara, M. Saito, M. Inaba and K. Murata, Catal. Lett., 2005, 102, 201–205 CrossRef CAS.
- S. D. Jackson and S. Rugmini, J. Catal., 2007, 251, 59–68 CrossRef CAS.
- X. T. Gao, J. M. Jehng and I. E. Wachs, J. Catal., 2002, 209, 43–50 CrossRef CAS.
- Z. Wu, H.-S. Kim, P. C. Stair, S. Rugmini and S. D. Jackson, J. Phys. Chem. B, 2005, 109, 2793–2800 CrossRef CAS PubMed.
- M. E. Harlin, V. M. Niemi and A. O. I. Krause, J. Catal., 2000, 195, 67–78 CrossRef CAS.
- U. Rodemerck, M. Stoyanova, E. V. Kondratenko and D. Linke, J. Catal., 2017, 352, 256–263 CrossRef CAS.
- R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Chem. Rev., 2019, 119, 2128–2191 CrossRef CAS PubMed.
- U. Rodemerck, S. Sokolov, M. Stoyanova, U. Bentrup, D. Linke and E. V. Kondratenko, J. Catal., 2016, 338, 174–183 CrossRef CAS.
- D. C. Apperley, R. K. Harris and P. Hodgkinson, Solid-state NMR: basic principles & practice, Momentum Press, 2012 Search PubMed.
- A. D. Chowdhury, K. Houben, G. T. Whiting, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 15840–15845 CrossRef CAS PubMed.
- A. D. Chowdhury, A. L. Paioni, K. Houben, G. T. Whiting, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2018, 57, 8095–8099 CrossRef CAS PubMed.
- A. D. Chowdhury, K. Houben, G. T. Whiting, S.-H. Chung, M. Baldus and B. M. Weckhuysen, Nat. Catal., 2018, 1, 23–31 CrossRef CAS.
- Z. Ristanović, A. D. Chowdhury, R. Y. Brogaard, K. Houben, M. Baldus, J. Hofkens, M. B. J. Roeffaers and B. M. Weckhuysen, J. Am. Chem. Soc., 2018, 140, 14195–14205 CrossRef PubMed.
- C. Wang, M. Hu, Y. Chu, X. Zhou, Q. Wang, G. Qi, S. Li, J. Xu and F. Deng, Angew. Chem., Int. Ed., 2020, 59, 7198–7202 CrossRef CAS PubMed.
- R. H. Meinhold and D. M. Bibby, Zeolites, 1990, 10, 121–130 CrossRef CAS.
- D. Xiao, S. Xu, X. Han, X. Bao, Z. Liu and F. Blanc, Chem. Sci., 2017, 8, 8309–8314 RSC.
- M. Zhang, S. Xu, J. Li, Y. Wei, Y. Gong, Y. Chu, A. Zheng, J. Wang, W. Zhang, X. Wu, F. Deng and Z. Liu, J. Catal., 2016, 335, 47–57 CrossRef CAS.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, Chem. Soc. Rev., 2015, 44, 7155–7176 RSC.
- W. Wang and M. Hunger, Acc. Chem. Res., 2008, 41, 895–904 CrossRef CAS PubMed.
- X. Wu, S. Xu, W. Zhang, J. Huang, J. Li, B. Yu, Y. Wei and Z. Liu, Angew. Chem., Int. Ed., 2017, 56, 9039–9043 CrossRef CAS PubMed.
- D. Xiao, S. Xu, N. J. Brownbill, S. Paul, L.-H. Chen, S. Pawsey, F. Aussenac, B.-L. Su, X. Han, X. Bao, Z. Liu and F. Blanc, Chem. Sci., 2018, 9, 8184–8193 RSC.
- A. Dutta Chowdhury, I. Yarulina, E. Abou-Hamad, A. Gurinov and J. Gascon, Chem. Sci., 2019, 10, 8946–8954 RSC.
- V. Klimavicius, S. Neumann, S. Kunz, T. Gutmann and G. Buntkowsky, Catal. Sci. Technol., 2019, 9, 3743–3752 RSC.
- A. S. Lilly Thankamony, J. J. Wittmann, M. Kaushik and B. Corzilius, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 102–103, 120–195 CrossRef CAS PubMed.
- V. Aladin and B. Corzilius, Solid State Nucl. Magn. Reson., 2019, 99, 27–35 CrossRef CAS PubMed.
- T. Wolf, S. Kumar, H. Singh, T. Chakrabarty, F. Aussenac, A. I. Frenkel, D. T. Major and M. Leskes, J. Am. Chem. Soc., 2019, 141, 451–462 CrossRef CAS PubMed.
- A. Harchol, G. Reuveni, V. Ri, B. Thomas, R. Carmieli, R. H. Herber, C. Kim and M. Leskes, J. Phys. Chem. C, 2020, 124, 7082–7090 CrossRef CAS PubMed.
- A. Svirinovsky-Arbeli, D. Rosenberg, D. Krotkov, R. Damari, K. Kundu, A. Feintuch, L. Houben, S. Fleischer and M. Leskes, Solid State Nucl. Magn. Reson., 2019, 99, 7–14 CrossRef CAS PubMed.
- D. J. Kubicki, G. Casano, M. Schwarzwälder, S. Abel, C. Sauvée, K. Ganesan, M. Yulikov, A. J. Rossini, G. Jeschke, C. Copéret, A. Lesage, P. Tordo, O. Ouari and L. Emsley, Chem. Sci., 2016, 7, 550–558 RSC.
- M.-A. Geiger, A. P. Jagtap, M. Kaushik, H. Sun, D. Stöppler, S. T. Sigurdsson, B. Corzilius and H. Oschkinat, Chem. – Eur. J., 2018, 24, 13485–13494 CrossRef CAS PubMed.
- A. G. M. Rankin, J. Trébosc, F. Pourpoint, J.-P. Amoureux and O. Lafon, Solid State Nucl. Magn. Reson., 2019, 101, 116–143 CrossRef CAS PubMed.
- E. Pump, J. Viger-Gravel, E. Abou-Hamad, M. K. Samantaray, B. Hamzaoui, A. Gurinov, D. H. Anjum, D. Gajan, A. Lesage, A. Bendjeriou-Sedjerari, L. Emsley and J.-M. Basset, Chem. Sci., 2017, 8, 284–290 RSC.
- W. R. Gunther, V. K. Michaelis, M. A. Caporini, R. G. Griffin and Y. Román-Leshkov, J. Am. Chem. Soc., 2014, 136, 6219–6222 CrossRef CAS PubMed.
- M. Valla, A. J. Rossini, M. Caillot, C. Chizallet, P. Raybaud, M. Digne, A. Chaumonnot, A. Lesage, L. Emsley, J. A. van Bokhoven and C. Copéret, J. Am. Chem. Soc., 2015, 137, 10710–10719 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, J. Canivet, S. Aguado, O. Ouari, P. Tordo, M. Rosay, W. E. Maas, C. Coperet, D. Farrusseng, L. Emsley and A. Lesage, Angew. Chem., Int. Ed., 2012, 51, 123–127 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, F. Hegner, M. Schwarzwälder, D. Gajan, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2012, 134, 16899–16908 CrossRef CAS PubMed.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS PubMed.
- A. Rodriguez–Gomez, R. Pereniguez and A. Caballero, J. Phys. Chem. B, 2018, 122, 500–510 CrossRef PubMed.
- T. Jiang, H. Tao, J. Ren, X. Liu, Y. Wang and G. Lu, Microporous Mesoporous Mater., 2011, 142, 341–346 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- P. I. Ravikovitch and A. V. Neimark, J. Phys. Chem. B, 2001, 105, 6817–6823 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- J. Rouquerol, F. Rouquerol, P. Llewellyn, G. Maurin and K. S. W. Sing, Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, Elsevier Science, 2013 Search PubMed.
- B. Solsona, T. Blasco, J. M. López Nieto, M. L. Peña, F. Rey and A. Vidal-Moya, J. Catal., 2001, 203, 443–452 CrossRef CAS.
- K. V. R. Chary, G. Kishan, K. Ramesh, C. P. Kumar and G. Vidyasagar, Langmuir, 2003, 19, 4548–4554 CrossRef CAS.
- J. Krzystek, A. Ozarowski, J. Telser and D. C. Crans, Coord. Chem. Rev., 2015, 301–302, 123–133 CrossRef CAS.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, ACS Catal., 2014, 4, 3357–3380 CrossRef CAS.
- G. Mitran, R. Ahmed, E. Iro, S. Hajimirzaee, S. Hodgson, A. Urdă, M. Olea and I.-C. Marcu, Catal. Today, 2018, 306, 260–267 CrossRef CAS.
- S. Xie, E. Iglesia and A. T. Bell, J. Phys. Chem. B, 2001, 105, 5144–5152 CrossRef CAS.
- D. Santhanaraj, C. Suresh, A. Selvamani and K. Shanthi, New J. Chem., 2019, 43, 11554–11563 RSC.
- J. Le Bars, A. Auroux, M. Forissier and J. C. Vedrine, J. Catal., 1996, 162, 250–259 CrossRef CAS.
- K. C. Szeto, B. Loges, N. Merle, N. Popoff, A. Quadrelli, H. Jia, E. Berrier, A. De Mallmann, L. Delevoye, R. M. Gauvin and M. Taoufik, Organometallics, 2013, 32, 6452–6460 CrossRef CAS.
- O. B. Lapina, A. A. Shubin, D. F. Khabibulin, V. V. Terskikh, P. R. Bodart and J. P. Amoureux, Catal. Today, 2003, 78, 91–104 CrossRef CAS.
- N. S. Youssef, A. N. Mahdy and M. F. Abadir, Thermochim. Acta, 1990, 157, 155–161 CrossRef CAS.
- T. P. Moser and G. L. Schrader, J. Catal., 1987, 104, 99–108 CrossRef CAS.
- S. Albonetti, F. Cavani, F. Trifirò, P. Venturoli, G. Calestani, M. López Granados and J. L. G. Fierro, J. Catal., 1996, 160, 52–64 CrossRef CAS.
- I.-S. Park, S. Y. Choi, J. S. Ha and J. Khim, Chem. Phys. Lett., 2007, 444, 161–166 CrossRef CAS.
- M. Wark, M. Koch, A. Brückner and W. Grünert, J. Chem. Soc., Faraday Trans., 1998, 94, 2033–2041 RSC.
- G. Sawatzky and E. Antonides, J. Phys., Colloq., 1976, 37, C4-117–C4-123 CrossRef.
- M. Chiesa, V. Meynen, S. Van Doorslaer, P. Cool and E. F. Vansant, J. Am. Chem. Soc., 2006, 128, 8955–8963 CrossRef CAS PubMed.
- B. C. Gates and H. Knoezinger, Advances in Catalysis, Elsevier Science, 2001 Search PubMed.
- G. Xu, X. Wang, X. Chen and L. Jiao, RSC Adv., 2015, 5, 17782–17785 RSC.
- D. Tuschel, Spectroscopy, 2016, 31, 14–23 Search PubMed.
- C. Li, W. Zheng, Q. Zhu, J. Chen, B. Y. Wang and X. Ju, Nucl. Instrum. Methods Phys. Res., Sect. B, 2016, 384, 23–29 CrossRef CAS.
- C. A. Carrero, C. J. Keturakis, A. Orrego, R. Schomäcker and I. E. Wachs, Dalton Trans., 2013, 42, 12644–12653 RSC.
- N. Das, H. Eckert, H. Hu, I. E. Wachs, J. F. Walzer and F. J. Feher, J. Phys. Chem., 1993, 97, 8240–8243 CrossRef CAS.
- M. Baltes, K. Cassiers, P. Van Der Voort, B. M. Weckhuysen, R. A. Schoonheydt and E. F. Vansant, J. Catal., 2001, 197, 160–171 CrossRef CAS.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed.
- F. Blanc, Investigation of Catalytic Surfaces with Surface-Enhanced Solid-State NMR Spectroscopy, Wiley-VCH Verlag GmbH & Co. KGaA, 2017, pp. 1003–1028 Search PubMed.
- N. J. Brownbill, R. S. Sprick, B. Bonillo, S. Pawsey, F. Aussenac, A. J. Fielding, A. I. Cooper and F. Blanc, Macromolecules, 2018, 51, 3088–3096 CrossRef CAS.
- A. L. Paioni, M. A. M. Renault and M. Baldus, eMagRes, 2018, 7, 51–61 CAS.
- A. Jantschke, E. Koers, D. Mance, M. Weingarth, E. Brunner and M. Baldus, Angew. Chem., Int. Ed., 2015, 54, 15069–15073 CrossRef CAS PubMed.
- E. A. W. van der Cruijsen, E. J. Koers, C. Sauvée, R. E. Hulse, M. Weingarth, O. Ouari, E. Perozo, P. Tordo and M. Baldus, Chem. – Eur. J., 2015, 21, 12971–12977 CrossRef CAS PubMed.
- T. Kobayashi, F. A. Perras, I. I. Slowing, A. D. Sadow and M. Pruski, ACS Catal., 2015, 5, 7055–7062 CrossRef CAS.
- B. Plainchont, P. Berruyer, J.-N. Dumez, S. Jannin and P. Giraudeau, Anal. Chem., 2018, 90, 3639–3650 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, A. Lesage, C. Copéret and L. Emsley, Acc. Chem. Res., 2013, 46, 1942–1951 CrossRef CAS PubMed.
- D. Mance, J. van der Zwan, M. E. Z. Velthoen, F. Meirer, B. M. Weckhuysen, M. Baldus and E. T. C. Vogt, Chem. Commun., 2017, 53, 3933–3936 RSC.
- E. Pump, A. Bendjeriou-Sedjerari, J. Viger-Gravel, D. Gajan, B. Scotto, M. K. Samantaray, E. Abou-Hamad, A. Gurinov, W. Almaksoud, Z. Cao, A. Lesage, L. Cavallo, L. Emsley and J.-M. Basset, Chem. Sci., 2018, 9, 4866–4872 RSC.
- J. C. Mohandas, E. Abou-Hamad, E. Callens, M. K. Samantaray, D. Gajan, A. Gurinov, T. Ma, S. Ould-Chikh, A. S. Hoffman, B. C. Gates and J.-M. Basset, Chem. Sci., 2017, 8, 5650–5661 RSC.
- A. Lesage, M. Lelli, D. Gajan, M. A. Caporini, V. Vitzthum, P. Miéville, J. Alauzun, A. Roussey, C. Thieuleux, A. Mehdi, G. Bodenhausen, C. Coperet and L. Emsley, J. Am. Chem. Soc., 2010, 132, 15459–15461 CrossRef CAS PubMed.
- F. A. Perras, J. D. Padmos, R. L. Johnson, L.-L. Wang, T. J. Schwartz, T. Kobayashi, J. H. Horton, J. A. Dumesic, B. H. Shanks, D. D. Johnson and M. Pruski, J. Am. Chem. Soc., 2017, 139, 2702–2709 CrossRef CAS PubMed.
- D. Xiao, X. Han, X. Bao, G. Hou and F. Blanc, RSC Adv., 2019, 9, 12415–12418 RSC.
- A. Zawadzki, J. D. A. Bellido, A. F. Lucrédio and E. M. Assaf, Fuel Process. Technol., 2014, 128, 432–440 CrossRef CAS.
- N. P. Blanchard, R. A. Hatton and S. R. P. Silva, Chem. Phys. Lett., 2007, 434, 92–95 CrossRef CAS.
- A. Kovtun, D. Jones, S. Dell'Elce, E. Treossi, A. Liscio and V. Palermo, Carbon, 2019, 143, 268–275 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01174f |
| This journal is © The Royal Society of Chemistry 2020 |