
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insights into the selective oxidation of 5-(hydroxymethyl)furfural over silver-based catalysts†
Oliver R.
Schade
ab,
Abhijeet
Gaur
 ab,
Anna
Zimina
ab,
Erisa
Saraçi
ab and
Jan-Dierk
Grunwaldt
*ab
ab,
Anna
Zimina
ab,
Erisa
Saraçi
ab and
Jan-Dierk
Grunwaldt
*ab
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), 76131 Karlsruhe, Germany. E-mail: grunwaldt@kit.edu
bInstitute of Catalysis Research and Technology (IKFT), Karlsruhe Institute of Technology (KIT), 76344 Eggenstein-Leopoldshafen, Germany
First published on 2nd July 2020
Abstract
Silver-catalyzed oxidation of 5-(hydroxymethyl)furfural (HMF) to 5-hydroxymethyl-2-furancarboxylic acid (HFCA) was investigated using Ag/ZrO2 and Ag/TiO2 catalysts. The reaction proceeded very selectively without formation of the dicarboxylic acid in the presence of air and NaOH as a base. In situ X-ray absorption spectroscopy (XAS) performed systematically under varying reaction conditions in a specially designed cell evidenced that reduced silver particles are the catalytically active species in this reaction. Although an incomplete reduction of Ag/ZrO2 and Ag/TiO2 was observed after the catalyst preparation even after reduction in hydrogen, silver was reduced to the metallic state as soon as HMF was introduced to the reaction mixture at room temperature and stayed reduced throughout the reaction under conditions optimized for high HFCA yield. The degree of silver reduction and product formation differed for varying reaction conditions, indicating that reduced silver particles, a homogeneous base and oxygen are needed in order to achieve high HFCA yield. Based on the catalytic and spectroscopic experiments, a detailed reaction mechanism is proposed involving a dehydrogenation pathway of an intermediately formed geminal diol in basic aqueous solution.
Introduction
In view of the depletion of fossil resources, the sustainable synthesis of chemicals from renewable raw materials such as biomass is of great importance and has received much attention in recent years.1–3 Motivated by current chemical value chains, the production of bio-derived platform molecules is considered a promising approach for transitioning the chemical industry towards renewable feedstock. Among others, 5-(hydroxymethyl)furfural (HMF) is a very versatile platform molecule that can be produced from hexose-containing biomass, including their polymers, for example lignocellulose.4–6 As a platform molecule, HMF can be used for the synthesis of a great variety of products that can be used e.g. as fuel additives or for the production of fine chemicals.5 The production of monomers is regarded specifically interesting, since most monomers are based on fossil resources in a constantly growing market.7,8 In this context, the selective oxidation of HMF can yield possible monomers. One of the most important oxidation products that can serve this purpose is 2,5-furandicarboxylic acid (FDCA), which is formed by selective oxidation of both functional groups of HMF. FDCA was listed as one of 12 important chemicals that can be produced from biomass on an industrial scale by the US Department of Energy,9 since its structural similarity to terephthalic acid might allow it to replace current fossil-derived monomers in future green plastics. Many studies focus on the synthesis of FDCA and the routes range from stoichiometric chemical transformations10 to bio-,11,12 electro-13–15 or heterogeneous metal catalysts. In the latter case, mostly noble metals like Pd,16–18 Pt,19–21 Au22–25 or alloys thereof26–28 show high activity.Another interesting product obtained from selective oxidation of the aldehyde moiety of HMF is 5-hydroxymethyl-2-furancarboxylic acid (HFCA) with attractive applications in the polymer industry29–31 and pharmacy sector as HFCA shows antitumor activity and can be used for the synthesis of an interleukin inhibitor.32,33 Nevertheless, only a limited number of studies focus on the targeted synthesis of HFCA by applying stoichiometric34 or biocatalytic oxidation,35–37 and the Cannizzaro reaction.38 However, these approaches have some drawbacks like the generation of waste or difficult process management. In addition, HFCA yield is naturally limited to 50% in the Cannizzaro reaction. Therefore, advantages like easy recovery, recyclability and possibly more sustainable reaction conditions make the use of heterogeneous catalysts more desirable. Recently, it has been reported that HFCA can be produced from HMF in 87% yield within 3 h over montmorillonite K-10 clay in toluene under optimal conditions of 100 °C and oxygen flow.39 In another study, HFCA was synthesized over a heterogeneous ruthenium catalyst in p-chlorotoluene with a maximum yield of 73% after 12 h at 130 °C and oxygen flow.40 For a more sustainable process, we recently introduced highly active silver-based catalysts for HMF oxidation in aqueous medium.41 With a Ag/ZrO2 catalyst prepared by deposition–precipitation, HFCA was produced in up to quantitative yield under mild reaction conditions of 50 °C in the presence of one equivalent of NaOH within 1 h. A further modification of the catalyst was reported by An et al.42 using poly(vinylpyrrolidone) as a capping agent for the silver particles. The high selectivity of silver catalysts towards HFCA suggests that these do not convert the alcohol moiety of HMF, although silver catalysts have been reported to be active in the oxidation of a variety of alcohols.43,44 Beier et al.43 speculated that silver oxygen species, which are generated upon heating of silver catalysts at different temperatures, are the catalytically active species in alcohol oxidation in organic solvents, similar to silver-catalyzed reactions in the gas phase.45–49
Although the oxidation of HMF in general has been studied extensively, the number of studies focusing on the mechanism by characterizing the working catalysts is limited.50 Davis et al.51 studied the oxidation of HMF over Au- and Pt-based catalysts using isotope labelling, which revealed the role of both base and oxygen in the catalytic cycle. They speculate that the base forms a geminal diol, which is then oxidized on the catalyst surface. The electrons originating from the oxidation process are then removed by oxygen, which thus has an indirect role in the mechanism. However, the precise role and structure of the metal remains unclear due to a lack of spectroscopic studies under reaction conditions. Consequently, the role of silver and its active chemical state in the liquid phase oxidation of HMF has not yet been clarified. Since catalysts show dynamic behavior under reaction conditions,52 mechanistic studies under reaction conditions (i.e. in situ) have to be conducted.53,54 Among the possible approaches that enable in situ or real-time characterization, synchrotron-based X-ray absorption spectroscopy (XAS) has evolved as a well-established technique for understanding the structural changes that accompany activation or deactivation of catalysts. XAS spectra allow to monitor the structure of even amorphous species and clusters in solution and can be divided into the X-ray absorption near-edge structure (XANES) region, which is sensitive to the oxidation state and coordination geometry of the absorbing atoms and extended X-ray absorption fine structure (EXAFS), which can be used to extract structural information (interatomic distances, coordination numbers and type of backscattering atoms).55 The great advantage of high-energy X-rays is the high penetration depth allowing the experiments to be conducted under realistic reaction conditions, i.e. high temperature, pressure, both in gas56,57 and even in liquid phase.58–60
In this study, we applied in situ XAS to characterize the chemical state and atomic arrangement of silver in Ag/TiO2 and Ag/ZrO2 prepared by deposition–precipitation under working conditions for the oxidation of HMF. For this, we used a specially designed cell, which can probe the catalyst both in liquid and solid phase to investigate the silver-based catalysts during the reaction at 50 °C and 10 bar pressure, supported by comprehensive catalytic tests and ex situ characterization.
Experimental
Materials
All chemicals were of analytical grade and have been used without further purification: HMF, FDCA, TiO2 P25 (Sigma-Aldrich), HFCA, NaOH, 5-formyl-2-furoic acid, 2,5-diformylfurane (TCI Chemicals), ZrO2 (Alfa Aesar) and synthetic air (Air Liquide).Catalyst preparation
A Ag/ZrO2 catalyst was prepared by deposition–precipitation with NaOH similar to the description in an earlier study.41 In brief, a suspension of ZrO2 (89 m2 g−1) and AgNO3 was brought to a pH of 9 with 0.05 M NaOH. After heating to 80 °C and keeping it at this temperature for 2 h, the suspension was stirred at room temperature overnight.Ag/TiO2 was prepared using a modified deposition–precipitation method. In this case, a suspension of TiO2 (52 m2 g−1) was brought to pH 12 using 0.5 M NaOH and an aqueous solution of AgNO3 was added dropwise at room temperature. In both cases, the resulting catalyst was filtered, washed and dried at 110 °C prior to calcination (350 °C, 2 h) and subsequent reduction (350 °C, 2 h, 3 L min−1, 10% H2 in N2, denoted as pre-reduction).
Characterization methods
X-Ray diffraction (XRD) measurements of the powder catalysts were performed on rotating sample holders using a PANalytical X'Pert Pro instrument (Cu Kα, 1.54060 Å, 2θ from 20° to 80°, step size 0.017°, 0.53 s acquisition time).The specific surface area was determined by N2 physisorption using the Brunauer–Emmett–Teller (BET) method. Prior to the measurements, the catalysts were heated to 300 °C for 2 h under reduced pressure and the measurements were performed on a Rubotherm BELSORP-mini II instrument.
Metal loading of solid catalysts and the concentration of ions in solution were determined using X-ray fluorescence (XRF, Bruker S4 Pioneer) and inductively coupled plasma optical emission spectrometry (ICP-OES, Agilent 720/725-ES), respectively.
Catalytic tests
Home-built stainless steel autoclaves were charged with 5 mL of a 0.2 M aqueous solution of HMF, a desired amount of a 2.5 M solution of NaOH and the resulting solution was diluted to 10 mL with deionized water in a typical reaction. After adjusting the air pressure, the magnetically stirred autoclaves were heated using heating sleeves and the starting point of the reaction was set after reaching the adjusted temperature for the first time. The reactors were cooled in an ice bath after the reactions, depressurized and the catalysts were recovered by decantation. In a final step, the solutions were filtered and diluted for HPLC analysis (cf. ESI,† Fig. S1). Conversion, yield and selectivity were calculated based on concentrations obtained from HPLC before and after the catalytic reactions. The measurements were performed on a Hitachi Primaide instrument at 50 °C and 50 bar (Bio-Rad Aminex HPX-87H column, solvent 5 mM H2SO4).In situ X-ray absorption spectroscopy and data analysis
In situ XAS measurements were performed in a specially designed batch reactor cell (Fig. 1) allowing measurements under realistic reactions conditions, comparable to laboratory experiments. The cell was optimized based on a previously reported one61 but was adjusted to the experimental conditions (temperature up to 100 °C, pressure up to 50 bar). It allows probing the liquid phase as well as the solid catalyst to study a possible contribution of homogeneous catalysis and structural changes of the catalyst, respectively.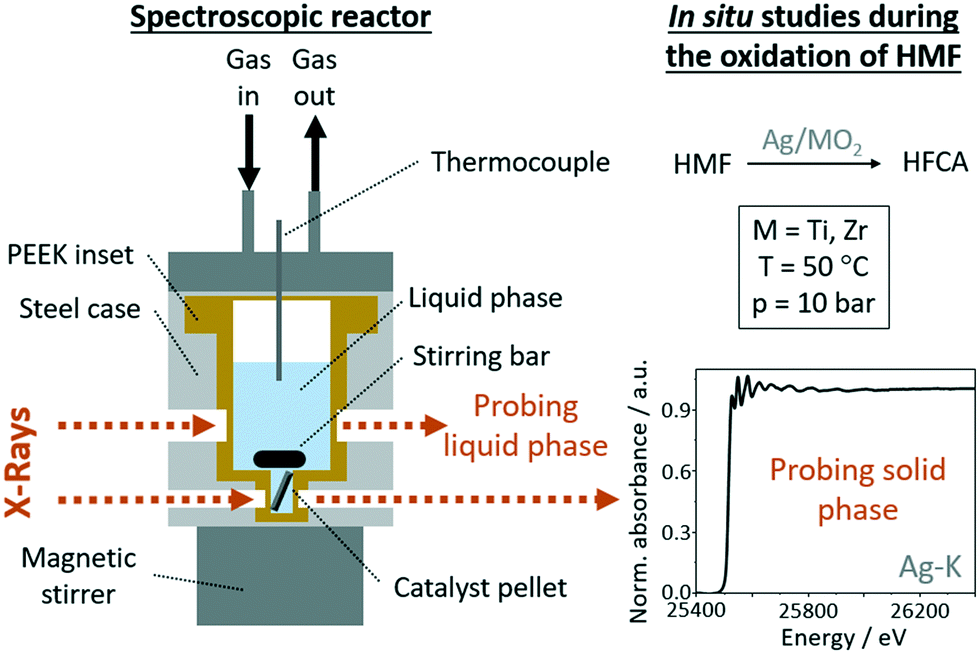 | ||
| Fig. 1 Schematic illustration of the spectroscopic batch reactor cell that was used for in situ spectroscopic experiments.61 | ||
The reaction solution is located in an exchangeable polyether ether ketone (PEEK) inset with thin windows transparent for high energy X-rays inside the steel body (Fig. 1) of the cell, which is heated with four heating cartridges and the reaction temperature and pressure are recorded continuously. The cell has two pairs of windows, which allow probing the liquid phase to study a possible contribution of homogeneous catalysts and probing the solid catalyst to gain insight into the state of the catalyst during the catalytic reaction. In a typical experiment, a catalyst pellet (10 mm diameter) was placed in the cell, then an aqueous solution of HMF was added together with an appropriate amount of a 2.5 M NaOH solution to give a total volume of 4 mL. Then the reactor was sealed and the desired pressure was set. Samples from the solution were taken before and after the reaction and diluted for HPLC analysis. After the reactions, the cell was allowed to cool to room temperature before starting with another experiment. All spectroscopic experiments were conducted at the CAT-ACT beamline at the KIT synchrotron in Karlsruhe, Germany (2.5 GeV, 100–150 mA ring current).62 A double crystal monochromator (DCM) with a pair of Si(311) crystals was used and higher harmonics were rejected by a Pt coated Si mirror in front of the DCM. The spectra were recorded at the Ag K edge (25.514 keV) in transmission mode using ionization chambers (Ohyo Koken Kogyo Co. Ltd., Japan). The beam size was about 1 × 1 mm2. The energy calibration is made on Ag metal foil by assigning the energy of the absorption edge to the tabulated value. XAS spectra of the solid catalyst were recorded in transmission mode at room temperature, during heating and under reaction conditions at 50 °C. A measurement time of 30 min per scan resulted in the corresponding time resolution. In addition to in situ studies, XAS spectra of the catalyst pellets after different stages of preparation were recorded. AgO, Ag2O, Ag2CO3 in form of pellets and a Ag foil were used as references.
EXAFS data analysis was performed using the IFEFFIT/Demeter package (version 0.9.25).63 With the Athena package, the raw data normalized to the intensity of the incident beam were processed by subtracting a smooth background and by normalizing the edge step to 1. μ(E) data were transformed to EXAFS χ(k) data, which were Fourier transformed into R-space (k-weighting of 2). Artemis package was employed for the fitting of the EXAFS data by generating theoretical models from available crystallographic data of reference compounds, i.e., Ag2CO3, AgO and Ag metal. The models were fitted to the experimental data to determine energy shifts (ΔE0) and structural parameters, including changes in the path length (ΔR), passive electron reduction factor (S02), coordination number (CN) and relative mean-square displacement of the atoms (Debye–Waller factor, σ2). For the analysis of the EXAFS data, the input parameter Rbkg, was set to 1.0 Å. For reduced samples, Fourier transformation was performed in the k-range of 2.7–10.9 Å−1. Theoretically modelled data were fitted to the experimental data in R-space using kw = 2 and in a range of 1.0–3.8 Å. For calcined samples, a k-range of 2.7–10.9 Å−1 and R range of 1.0–3.6 Å were used. In addition, linear combination fitting (LCF) to reference spectra has been performed to quantify the degree of oxidation of Ag.
Please note that the catalytic tests and in situ spectroscopic measurements have been performed at elevated pressure and should thus only be conducted by experienced personnel.
Results and discussion
Catalyst characterization
Two types of catalysts were prepared via deposition–precipitation, Ag/ZrO2 and Ag/TiO2. The Ag/ZrO2 catalyst was similar to the one reported in ref. 41 and was chosen as a benchmark because it was reported as the most active catalyst in HMF oxidation, hence it was also used to study the reaction mechanism. Ag/ZrO2 had a metal loading of 1 wt% based on XRF, which corresponds to a deposition efficiency of 50%. The specific surface area of this catalyst was 89 m2 g−1, the same as the pure support material. No changes in the XRD patterns and the absence of characteristic Ag reflections in the XRD show that the support was stable in the preparation process and hints at the presence of small metal particles (Fig. S2†).Yet the downside of ZrO2 for in situ XAS studies is its high absorbance of X-rays, which may compromise the data quality. Therefore, in order to increase the data quality for the Ag species and to complement the mechanistic studies, TiO2 was chosen as a second support material despite lower activity.41 The metal loading of this catalyst was increased to achieve comparable activity with the ZrO2-supported counterpart, which required a higher pH of precipitation.64 The metal loading of Ag/TiO2 was 4% based on XRF corresponding to a quantitative deposition of the used metal precursor due to the higher pH. No change in the specific surface area of the used TiO2 was observed (52 m2 g−1), indicating that the deposited metal does not block the pores of the support material and that its structure was stable at high pH during preparation. XRD measurements confirmed the preservation of the TiO2 structure in form of a mixture of rutile and anatase (Fig. S2†) before and after preparation of the catalyst. Besides these phases, two small and broad reflections at 32.3° and 46.4° in the XRD patterns of the calcined and pre-reduced Ag/TiO2 can be attributed to oxidized Ag species, indicating the presence of larger oxidized noble metal particles. Fig. 2a and b shows the XANES spectra of the catalysts after different stages of preparation. After precipitation and subsequent calcination at 350 °C, the catalysts are mainly in an oxidized state, as in previous studies.43 Comparison with different reference spectra shows that the calcined catalysts have edge features and an edge shift similar to that of Ag2CO3, which probably formed by reaction with atmospheric CO2.65 For Ag/TiO2, this is in line with the XRD results, which had reflections of oxidized Ag species in the XRD patterns. After pre-reduction in hydrogen at 350 °C, a decrease in the white line intensity (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 520 eV) and emerging features similar to that of Ag metal indicate a partial reduction, i.e. 50% for Ag/ZrO2 and 34% for Ag/TiO2, based on LCF (Fig. 2 and S4†). Note that the pellets have not been prepared under inert atmosphere, which might have led to a re-oxidation of the freshly reduced catalyst. In addition, EXAFS analysis of the calcined Ag/TiO2 catalyst reveals that the Ag–O (2.30 Å) shell has a coordination number of 2.8 ± 0.4 and the Ag–Ag (2.85 Å) shell a coordination number of 2.8 ± 0.5 indicating a Ag2CO3 structure (Table 1, entry 1). In the pre-reduced catalyst, the coordination number of Ag–O (2.27 Å) slightly decreases to 2.4 ± 0.7 while that of the Ag–Ag (2.86 Å) shell increases to 5.0 ± 0.5 (Table 1, entry 2). Thus, even in the pre-reduced catalyst, the Ag–O shells are present, possibly due to re-oxidation during pellet preparation in air.
520 eV) and emerging features similar to that of Ag metal indicate a partial reduction, i.e. 50% for Ag/ZrO2 and 34% for Ag/TiO2, based on LCF (Fig. 2 and S4†). Note that the pellets have not been prepared under inert atmosphere, which might have led to a re-oxidation of the freshly reduced catalyst. In addition, EXAFS analysis of the calcined Ag/TiO2 catalyst reveals that the Ag–O (2.30 Å) shell has a coordination number of 2.8 ± 0.4 and the Ag–Ag (2.85 Å) shell a coordination number of 2.8 ± 0.5 indicating a Ag2CO3 structure (Table 1, entry 1). In the pre-reduced catalyst, the coordination number of Ag–O (2.27 Å) slightly decreases to 2.4 ± 0.7 while that of the Ag–Ag (2.86 Å) shell increases to 5.0 ± 0.5 (Table 1, entry 2). Thus, even in the pre-reduced catalyst, the Ag–O shells are present, possibly due to re-oxidation during pellet preparation in air.
 | ||
| Fig. 2 XANES spectra at the Ag K-edge of the (a) Ag/ZrO2 and (b) Ag/TiO2 catalysts after different stages of preparation i.e. calcination in air and pre-reduction in hydrogen, (c and d) influence of NaOH addition and (e and f) reaction temperature on the product distribution in HMF oxidation over both catalysts. Arrows in (a) indicate the assignment of the peaks to the corresponding reference spectra for Ag/ZrO2. Reaction conditions of catalytic tests: 10 bar air, 5 h reaction time, 1 mmol HMF in 10 mL total volume, (c) 50 °C, HMF:NaOH 1:0/1/4, HMF:Ag 200:1, (d) 50 °C, HMF:NaOH 1:0/1/4, HMF:Ag 125:1, (e) HMF:NaOH 1:4, HMF:Ag 200:1, (f) HMF:NaOH 1:1, HMF:Ag 125:1. | ||
:NaOH:Ag 1:0:6, 107 min
| Entry | Catalyst/reaction | Ag–O | Ag–Ag | χ ν 2 | E 0/eV | ||||
|---|---|---|---|---|---|---|---|---|---|
| R/Å | CN | σ 2/Å2 | R/Å | CN | σ 2/Å2 | ||||
| 1 | Ag/TiO2 calcined | 2.30 | 2.8 ± 0.4 | 0.0153 ± 0.0028 | 2.85 | 2.8 ± 0.5 | 0.0154 ± 0.0019 | 29 | 3.2 ± 0.8 |
| 2 | Ag/TiO2 pre-reduced | 2.30 | 2.4 ± 0.7 | 0.0146 ± 0.0049 | 2.85 | 5.0 ± 0.5 | 0.0122 ± 0.0008 | 54 | 0.6 ± 0.2 |
| 3 | After addition of HMF at rt | — | — | — | 2.87 | 4.8 ± 1.3 | 0.0083 ± 0.0024 | 6 | 1.8 ± 0.4 |
| 4 | T = 80 min at 50 °C | — | — | — | 2.86 | 7.3 ± 1.6 | 0.0108 ± 0.0021 | 7 | 1.4 ± 0.2 |
Catalytic activity – influence of added base, temperature and time
The catalytic activity of Ag/ZrO2 and Ag/TiO2 in HMF oxidation was tested in home-built batch reactors. For a first screening, the oxidation was carried out in the presence of four equivalents of NaOH, in relation to HMF, at 50 °C, which has previously been proven to favor HFCA synthesis.22,41,66 Quantitative HMF conversion was obtained using Ag/ZrO2, which led to the production of HFCA in 98% yield. HMF conversion was 95% over Ag/TiO2 yielding 23% HFCA (24% selectivity), emphasizing the need to use a higher Ag-loading on this support. Because of the different metal loadings of both catalysts, the amount of Ag added to the solution differed slightly. The normalized yield of the catalysts to the amount of metal and reaction time was calculated to better compare the catalysts and resulted in productivity rates of 39 molHFCA h−1 molAg−1 for Ag/ZrO2 and 6 molHFCA h−1 molAg−1 for Ag/TiO2. As HMF conversion proceeds very rapidly, the calculation of initial rates, i.e. TOFs, is not possible as they require low conversions. Hence, the productivity gives the most comparable information. The high productivity of the ZrO2 supported Ag indicates its superior activity, which might be attributed to the lower oxygen storage capacity of ZrO2.67 In general, the support material greatly affects HMF oxidation also by oxygen vacancies68 or Brønsted acidity.24It is well-known that HMF is unstable in alkaline aqueous solution.13 Therefore, the influence of added NaOH on the oxidation of HMF over both catalysts was studied. While, as expected,41,42 there was no activity in absence of added base, the addition of one equivalent of NaOH gave already 94% HMF conversion and 92% HFCA yield over Ag/TiO2 and 100% HMF conversion and 94% HFCA yield over Ag/ZrO2 (Fig. 2c and d). Increasing the amount of base led to a deterioration of the HFCA yield over Ag/TiO2, due to the rapid decomposition of HMF in highly alkaline aqueous solution. The higher activity of Ag/ZrO2 is further underlined by an almost constant HFCA yield at four equivalents of NaOH. Even under these more challenging conditions in terms of HMF stability, HFCA was produced in high yield indicating rapid aldehyde oxidation before degradation of HMF.
The catalytic performance as function of the reaction temperature is shown in Fig. 2e and f. For Ag/TiO2, both the conversion of HMF and the HFCA yield passed through a maximum at 50 °C, where also the productivity of the catalyst towards HFCA reached its highest value of 23 molHFCA molAg−1 h−1. Over pure TiO2, HFCA was formed in 1% yield at 41% HMF conversion under identical conditions. The higher production of HFCA at a rather mild temperature is well in line with literature, especially for Ag-based catalysts.22,41,42,66 Further increase to 75 °C led to a decreasing carbon balance at almost the same conversion, which can be attributed to limited thermal stability of HMF in basic aqueous solution. The same trend was observed for Ag/ZrO2 with the only difference that the HMF conversion was always 100%. The FDCA yield was negligible and similar to blank experiments. No HFCA was formed in the presence of pure ZrO2, hence blank experiments evidence the high catalytic activity of the Ag species. To investigate a possible contribution of side-reactions to the HFCA yield, HMF was stirred at 50 °C in the presence of 4 equivalents of NaOH for 5 h at 10 bar air pressure. In this reaction, HFCA was produced in 30% yield at 96% HMF conversion. Generally, side-reactions like the Cannizzaro reaction can lead to HFCA formation at high base concentration, but the presence of the pure support materials prevents product formation. Therefore, HFCA originates solely from the Ag-catalyzed reaction in the catalytic experiments. In general, HMF oxidation requires highly active catalysts in order to convert HMF before its thermal degradation,69 which makes the possible use of low temperatures for this reaction even more relevant and adds more sustainability value to the process.70 In addition, the reaction time was reduced to 1 h under these optimized conditions for Ag/TiO2, which afforded 93% selectivity towards HFCA at 78% HMF conversion resulting in a productivity of 92 molHFCA molAg−1 h−1 (not shown). Hence, the selective oxidation of HMF over Ag/TiO2 proceeds very selectively, but longer reaction times are required to obtain high HFCA yields. The higher activity of Ag/ZrO2 is also reflected in 83% HFCA formation with a productivity of 166 molHFCA molAg−1 h−1 upon reduction of the reaction time to 1 h. In conclusion, ZrO2 is the more suitable support material in terms of catalytic activity, but Ag is also active when supported on TiO2, which enables in situ XAS studies at a later stage. In addition, we observed that the catalytic activity of Ag/ZrO2 was dependent on the state of the catalyst (Fig. 3). The freshly prepared and pre-reduced catalyst gave the highest HFCA yield of 98%, whereas the calcined catalyst was less active giving 73% of HFCA. Storing the fresh and pre-reduced catalyst in air for four weeks led to both a change in the color of the solid and a decrease in catalytic activity, giving HFCA in 84% yield. No changes in terms of catalytic activity were observed upon storage of the catalyst for up to one year. Hence, the performance approaches that of the calcined catalyst, which indicates that reduced Ag particles provide the catalytically active species in this reaction. Also here, FDCA was not formed in yields higher than in blank reactions. Based on these findings, one can speculate that the reactivity of silver depends on its structure e.g. the oxidation state, therefore identifying the active species of the Ag-based catalysts in HMF oxidation is crucial.
 | ||
| Fig. 3 Dependence of the catalytic activity on the state of Ag/ZrO2 catalyst. Reaction conditions: 50 °C, 10 bar air, 5 h reaction time, 1 mmol HMF in 10 mL H2O, HMF:NaOH 1:4, HMF:Ag 200:1. | ||
Mechanistic studies using in situ XAS
In order to gain first insight into the catalytically active species of Ag/ZrO2, the calcined, pre-reduced and stored catalysts were characterized using XAS. The XANES spectra of these ex situ samples show a variation in oxidation state of the catalysts tested above (Fig. 3, spectra given in Fig. S3†). Comparing the XANES spectrum of the stored catalyst at the Ag K-edge with the spectra of Ag/ZrO2 after different stages of preparation and reference compounds, the freshly reduced and more active catalyst was more reduced than the one stored in air whose lower activity can thus be attributed to a re-oxidation (Fig. S3†). Although these results indicate that reduced Ag is catalytically more active, such studies cannot be used to fully clarify the reaction mechanism, since the catalysts change their structure during air exposure and are rather dynamic systems.52:Ag ratio was decreased to enhance product formation.
For both catalysts, reduction of the pre-reduced and partially oxidized samples was identified by the XANES spectra at the Ag K-edge immediately after addition of the reaction mixture at room temperature. In case of Ag/ZrO2 (Fig. 4a), increasing the temperature to 50 °C and during the course of the reaction (230 min), the characteristic spectroscopic features of the Ag metal phase in the near edge region augmented, indicating a further reduction or an increase in crystallinity/particle size. The degree of reduction of Ag/ZrO2 increased from 50% to 92% based on LCF and HFCA was produced in 72% yield after 4 h. To verify the suitability of the used batch reactor cell with a catalyst pellet, a blank reaction under the same reaction conditions was performed using the pure ZrO2 support. In this reaction, 34% of HMF was converted and no product was formed. Hence, HFCA originates from the Ag-catalyzed process and the contribution of side-reactions like the Cannizzaro reaction is negligible.
 | ||
| Fig. 4
In situ XANES spectra at the Ag K-edge of pre-reduced (a) Ag/ZrO2, (b) Ag/TiO2 and (c) FT EXAFS spectra (k2-weighted) of Ag/TiO2 during the oxidation of HMF. The insets in (a) and (b) zoom into the increasing characteristic XANES features with time on stream. Reaction conditions: 50 °C, 10 bar air, HMF:NaOH 1:1, HMF:Ag 20:1, 4 h reaction time. | ||
Similarly, the in situ XANES spectra of the Ag/TiO2 catalyst under the same reaction conditions (Fig. 4b) show a significant reduction of the silver particles from 34% to 86% upon addition of HMF at room temperature. By increasing the temperature to 50 °C, the spectroscopic features of metallic Ag phase slightly enhanced and the degree of reduction increased to 92% based on LCF. This can also be observed in the Fourier-transformed (FT) EXAFS spectra (Fig. 4c, k2-weighted, not corrected for phase shift) from which the coordination number of Ag–Ag (2.86 Å) was derived to be 10.5 ± 0.8 at room temperature and 11.4 ± 0.9 at 50 °C, indicating an increase in crystallinity of Ag particles (Table S6†). As both catalysts reach the same degree of reduction of 92%, a possible influence of the support material or different particle sizes71 on the reduction are unlikely. This in situ experiment gave HFCA in 28% yield (37% selectivity) compared to no HFCA formation at 21% HMF conversion in a blank experiment using pure TiO2, further underlining the suitability of the used batch reactor cell. The lower HFCA yields compared to studies in the lab-scale reactor can be attributed to mass transport limitations in the batch reactor cell, where a catalyst pellet was used instead of a powder.72,73 However, the catalysts were still active, which was verified by blank experiments under identical conditions. Also, the use of a catalyst pellet instead of a powder was beneficial for the temporal resolution and the signal-to-noise ratio of the spectroscopic experiments (see Fig. S8†). The slow diffusion of the reaction solution into the catalyst pellet may also be the reason for some remaining oxidized Ag in the inner part of the pellet. Despite mass transfer effects, it seems justified to derive metallic Ag as catalytically active, since HMF cannot reach the inner part of the pellet, thus the reaction proceeds on the metallic outer area. Additionally, the liquid phase was monitored continuously during the reaction by XANES. The absence of a significant edge jump at the Ag K-edge in the solution and a constant one of the solid catalyst unravelled that the catalyst did not dissolve and thus did not participate as a homogeneous catalyst in contrast, for example, to Pd-catalyzed Heck reactions.60,74 This was also confirmed by the absence of Ag in the solution after the reaction by ICP-OES. These results suggest that reduced silver particles provide the catalytically active sites acting as heterogeneous catalyst.
In addition, spectra of the pre-reduced catalyst were recorded in pure water at both room temperature (rt) and at 50° showing no changes in the state of Ag (Fig. S6†). Therefore, changes in the oxidation state of Ag can solely be attributed to interaction with the reaction mixture. To investigate whether also catalysts in a more oxidized state were reduced by the reaction mixture, the calcined Ag/ZrO2 catalyst was studied under the optimized reaction conditions of 50 °C in the presence of one equivalent of NaOH at 10 bar air pressure. This also led to a reduction of Ag (cf. ESI† and Fig. S8). To substantiate these conclusions, we extended our spectroscopic studies to further selected reaction conditions. As the oxidation of HMF over Ag-based catalysts only proceeds in the presence of a homogeneous base and oxygen, we investigated in a next step their influence on the catalyst structure by omitting one after the other (details, cf. Tables S1 and S2†).
 | ||
| Fig. 5
In situ XANES spectra at the Ag K-edge of pre-reduced (a) Ag/ZrO2 and (b) Ag/TiO2 and (c) k2-weighted FT EXAFS spectra of Ag/TiO2 in HMF solution without NaOH. The insets in (a) and (b) zoom into the increasing characteristic XANES features with time on stream during the oxidation of HMF. Spectra of calcined Ag/TiO2 are shown for comparison. Reaction conditions: 50 °C, 10 bar air, HMF:NaOH 1:0, HMF:Ag 25:1 (a), HMF:NaOH 1:0, HMF:Ag 6:1 (b), 107 min. | ||
In the base-free reaction over pre-reduced Ag/TiO2, reduction was observed after addition of the aqueous HMF solution at room temperature. However, as in the case of Ag/ZrO2, the degree of reduction was lower in the absence of NaOH based on an edge shift towards Ag foil and the features in the XANES spectra (inset in Fig. 5b, LCF Tables S1 and S2†) compared to the reaction with NaOH. By increasing the temperature to 50 °C, the EXAFS-oscillations and the contributions in the FT EXAFS spectra (Ag–Ag scattering peak in Fig. 5c) typical for the metallic phase were enhanced. As the regions of Ag–O and Ag–Ag backscattering overlap, this makes it hard to resolve, but is possible with EXAFS analysis. The EXAFS fitting shows that for Ag/TiO2 after different stages of preparation (Fig. 2b and 5c and Table 1), the Ag–O shells are present even in the pre-reduced catalyst. As stated above (catalyst characterization), pre-reduction leads to a slightly lower CN of Ag–O (2.27 Å) with an increase in the CN of Ag–Ag (2.86 Å), so Ag–O shells are present even in the pre-reduced catalyst.
On introducing the reaction mixture at room temperature, the CN of the Ag–Ag (2.86 Å) shell increases to 4.8 ± 1.4 and no measurable CN for Ag–O can be observed indicating Ag mostly in metallic phase (Table 1 entry 3). Increasing the temperature to 50 °C leads to a further increase in the CN of Ag–Ag (2.86 Å) to 7.3 ± 1.6 indicating growth of Ag particles, which is still smaller as compared to that observed in reaction with base. The increase in particle size can be observed for Ag/ZrO2 in the XRD pattern by an emerging reflection (Fig. S5†). Derivation of CNs from EXAFS analysis was not possible due to the strong absorption of the ZrO2 support. TEM analysis is not sufficiently informative due to low contrast between Ag and ZrO241 as well as re-oxidation of the spent catalyst in air. Also in this reaction, 7% of Ag leached into the solution based on ICP-OES.
The Ag leaching into the solution during the reaction without the base occurs probably via oxidative dissolution, which depends on the pH.64,75,76 Both the presence of a base and with that an increased pH and the presence of HMF reduce or completely prevent the dissolution of Ag41 as no Ag was detected in the reaction solutions of the above reactions. Leaching may also be influenced by the degree of reduction. To study this assumption, the base-free reaction was also performed over the calcined, i.e. more oxidized, Ag/TiO2 catalyst. No HMF conversion in this reaction in combination with more leaching of Ag (14%, cf. ESI,† Fig. S9) further underlined this assumption. Therefore, the presence of base does not only enhance the reaction rate, but also prevents the loss of the active metal via leaching. Considering that no HMF was converted in this reaction and the absence of an absorption edge in the liquid phase during presence of base with no detection of Ag by ICP-OES further confirms that the catalyst in fact acts as a heterogeneous catalyst.
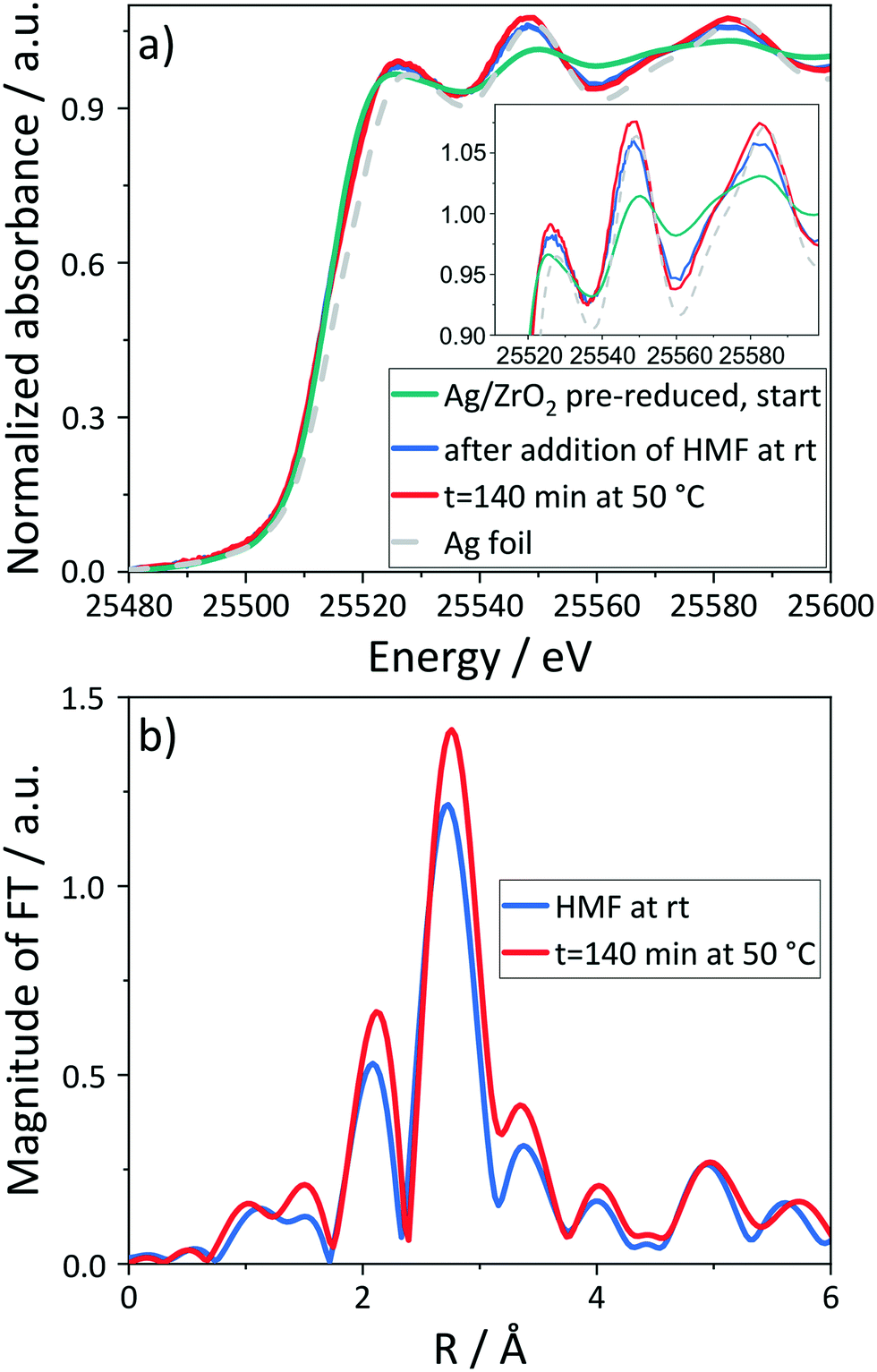 | ||
| Fig. 6
In situ Ag K-edge XANES (a) and FT EXAFS spectra (b) of the pre-reduced Ag/ZrO2 catalyst in a basic HMF solution under nitrogen. The inset in (a) shows a zoom into the increasing characteristic spectroscopic features in the near the edge range with time on stream during the oxidation of HMF. Reaction conditions: 50 °C, 10 bar N2, HMF:NaOH 1:1, HMF:Ag 24:1, 193 min. | ||
As Ag/ZrO2 and Ag/TiO2 were in reduced state throughout the reaction under optimized conditions, the oxidation reaction does not involve any silver–oxygen species, in contrast, to some previously reported Ag-catalyzed oxidation reactions, which were not performed in aqueous medium.43,45–49 In general, calcination of silver-based catalysts at higher temperatures leads to both a reduction and the generation of different silver oxygen species.43–45 Temperatures below 330 °C result in surface adsorbed oxygen (Oα), bulk dissolved oxygen (Oβ) is formed at higher temperatures before strongly bound surface species (Oγ) are formed at 630 °C.45,47 In our experiments, the calcination was performed at 350 °C after precipitation, thus no reduction of silver and formation of oxygen species took place in our catalysts (Fig. 2). Subsequent pre-reduction of both catalysts in hydrogen led to a higher activity in HMF oxidation compared to the oxidized i.e. calcined catalyst.41 In addition, the slight loss of catalytic activity of Ag/ZrO2 upon storage of the catalyst in air can be attributed to a re-oxidation of the reduced catalyst (Fig. S3†) as indicated by re-emerging features of oxidized silver species (oxidation degree of 89% based on LCF). The absence of Ag–O species, that were active in alcohol oxidation in organic solvents in the above-mentioned studies, may be a reason for the selectivity towards HFCA rather than FDCA in water.
Omitting NaOH in the reaction solution led to the lowest degree of reduction during the in situ experiments (Fig. S10†). Therefore, the presence of a homogeneous base exhibits a key role in the reaction as it facilitates the reduction to metallic Ag (e.g. due to formation of hydrogen on the surface during dehydrogenation, see below) and therefore activation of the catalyst. In addition, no leaching of Ag occurs in the presence of NaOH. Its presence probably leads to the formation of a diol by a nucleophilic attack of a hydroxide ion on the aldehyde moiety of HMF.51 This diol is subsequently converted to the carboxylic acid by the catalyst, probably by dehydrogenation, which is also the dominant mechanism e.g. in the oxidation of alcohols over Pd-based catalysts.59,78,79 The remaining hydrogen on the catalyst surface then reduces the Ag catalyst, before it is removed by oxygen, as evidenced isotope labelling.51,80 Since the diol is formed less in the absence of base, this leads to a lower degree of reduction and no significant product formation.
In addition, reduction of Ag was observed in the absence of air. However, the degree of reduction was lower as compared to the optimized conditions in the presence of both base and oxygen (Fig. S10†). Under these conditions, more diol is formed which might lead to the deposition of some hydrogen on the catalyst surface and with that a more pronounced reduction of Ag. Due to the indirect role of O2, this hydrogen cannot be removed without oxygen, which also explains the lower HFCA yields in these reactions.
Based on these conclusions drawn from the spectroscopic experiments combined with catalytic data, a reaction mechanism can be proposed in which the reaction is heterogeneously catalyzed on reduced silver particles and the key step in the catalytic cycle consists of the dehydrogenation of an intermediately formed diol from HMF in aqueous solution (Fig. 7).
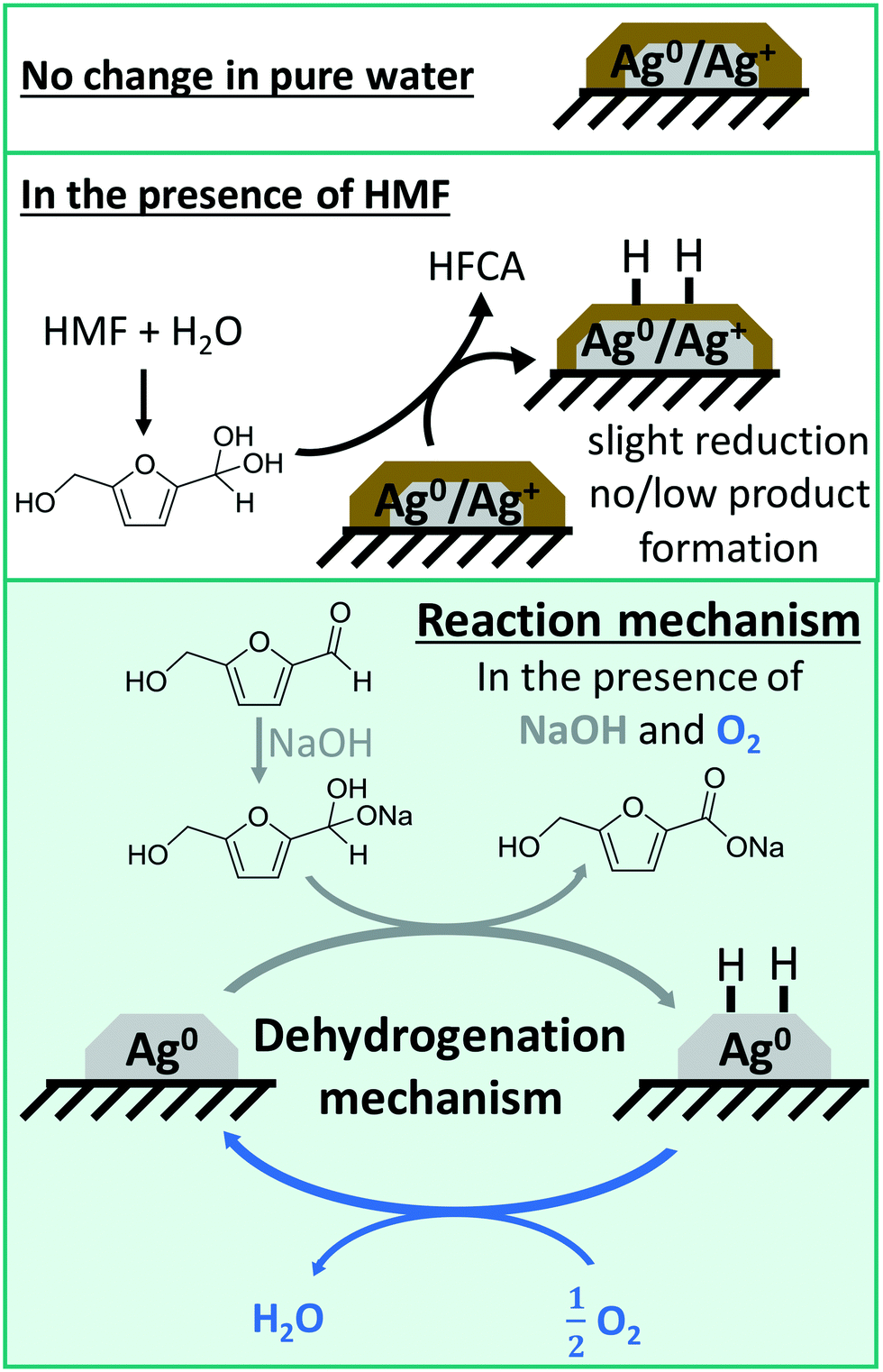 | ||
| Fig. 7 Schematic structure of the silver catalysts under various reaction conditions and schematic reaction mechanism of the heterogeneously catalyzed oxidation of HMF over silver-based catalysts in the presence of base and oxygen. | ||
In a first step, the geminal diol is formed through a nucleophilic attack of a hydroxide ion to the aldehyde in solution, which required at least one equivalent of NaOH. In the presence of NaOH, the diol is then dehydrogenated on the solid catalyst first leading to a reduction of Ag and then to adsorbed H-species on the surface that need to be removed. Since the diol can be formed in sufficient quantities in the presence of NaOH, the degree of reduction increases and some HFCA is produced. In the early stage of the reaction, i.e. after the addition of HMF at room temperature, this hydrogen from the dehydrogenation reaction reduced the silver particles and forms the active catalyst. During the course of the reaction, the Ag surface is covered by hydrogen, which is then removed by oxygen,51 similar to mechanisms reported for alcohol oxidation on Pd-based catalysts.59,78,79
In the absence of air, the hydrogen remains on the Ag surface, therefore, HFCA is formed to a less extent. In addition, because the used calcination temperature is too low to create silver oxygen species44,45 and the catalysts stay in reduced state throughout the reaction, silver–oxygen species are not involved in the catalytic cycle under the selected reaction conditions. This may be the reason for the observed selectivity to HFCA, since Ag–O species are active in alcohol oxidation in organic solvents.43 Another reason may be a high energy barrier of C–H activation, which differs for different metals.80 Here, the catalytic cycle is closed by oxygen, which removes the surface bound hydrogen via formation of water and releases the active sites of the Ag catalyst. Therefore, the reaction proceeds on the Ag particles and the support material is not involved in the catalytic cycle, which is reflected in high activities and productivity rates of both the TiO2- and ZrO2-supported catalyst. These first in situ studies on HMF oxidation under relevant conditions can be used as a basis for further studies, for example the influence of different HMF:NaOH or HMF:Ag ratios on the catalyst structure to gain even deeper insight into the reaction mechanism. As metallic Ag was proven to provide catalytically active sites, further studies like DFT calculations may shed more light on the distinct selectivity towards HFCA.
Conclusions
In this study, the silver-catalyzed oxidation of HMF was systematically studied by catalytic data under varying reaction conditions combined with in situ XAS. A fast reduction of both pre-reduced and oxidized Ag/ZrO2 and Ag/TiO2 to metallic silver evidenced reduced silver particles to be the catalytically active species. The degree of reduction was dependent on the reaction conditions, which therefore allowed drawing conclusions about the molecular processes on the catalyst surface. Based on these results, a dehydrogenation reaction mechanism is proposed. The formation of a geminal diol in solution is a crucial step, since the reaction proceeds via dehydrogenation of this diol on reduced Ag particles. The dehydrogenation leads to a reduction of the Ag-based catalysts and oxygen is indirectly involved in the catalytic cycle by removing hydrogen and releasing active sites of the catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded and supported by KIT as well as the recently accepted FNR-project (KEFIP, FKZ: 22010718). We acknowledge the KIT light source for provision of beamtime at the CAT-ACT beamline and the Institute of Beam Physics and Technology (IBPT) for the operation of the storage ring, the Karlsruhe Research Accelerator (KARA). We are grateful to Dr. Tim Prüßmann (IKFT/KIT) for providing assistance and technical support during the beamtime as well as Matthias Stehle, Marc-André Serrer, Kai Kalz and Weiss Naim (KIT) for beamtime support. The authors also thank Angela Deutsch (ITCP/KIT) for N2 physisorption measurements and Armin Lautenbach (IKFT/KIT) for ICP-OES analyses.Notes and references
- D. Esposito and M. Antonietti, Chem. Soc. Rev., 2015, 44, 5821–5835 RSC.
- B. J. Nikolau, M. A. D. N. Perera, L. Brachova and B. Shanks, Plant J., 2008, 54, 536–545 CrossRef CAS PubMed.
- E. Henrich, N. Dahmen, E. Dinjus and J. Sauer, Chem. Ing. Tech., 2015, 87, 1667–1685 CrossRef CAS.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- D. Steinbach, A. Kruse and J. Sauer, Biomass Convers. Biorefin., 2017, 7, 247–274 CrossRef CAS.
- M. Rose and R. Palkovits, Macromol. Rapid Commun., 2011, 32, 1299–1311 CrossRef CAS PubMed.
- A. Llevot, P. K. Dannecker, M. von Czapiewski, L. C. Over, Z. Soeyler and M. A. Meier, Chem. – Eur. J., 2016, 22, 11510–11521 CrossRef CAS PubMed.
- T. Werpy and G. Petersen, Natural Renewable Energy Laboratory, US Department of Energy, 2004 Search PubMed.
- T. Miura, H. Kakinuma, T. Kawano and H. Matsuhisa, US Pat., US020070232815A1, 2007 Search PubMed.
- M. Krystof, M. Pérez-Sánchez and P. Domínguez de María, ChemSusChem, 2013, 6, 826–830 CrossRef CAS PubMed.
- W. P. Dijkman, D. E. Groothuis and M. W. Fraaije, Angew. Chem., Int. Ed., 2014, 53, 6515–6518 CrossRef CAS PubMed.
- K. R. Vuyyuru and P. Strasser, Catal. Today, 2012, 195, 144–154 CrossRef CAS.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhöfer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS PubMed.
- B. J. Taitt, D.-H. Nam and K.-S. Choi, ACS Catal., 2018, 9, 660–670 CrossRef.
- B. Siyo, M. Schneider, M.-M. Pohl, P. Langer and N. Steinfeldt, Catal. Lett., 2014, 144, 498–506 CrossRef CAS.
- B. Liu, Y. Ren and Z. Zhang, Green Chem., 2015, 17, 1610–1617 RSC.
- C. Chen, X. Li, L. Wang, T. Liang, L. Wang, Y. Zhang and J. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 11300–11306 CrossRef CAS.
- S. E. Davis, L. R. Houk, E. C. Tamargo, A. K. Datye and R. J. Davis, Catal. Today, 2011, 160, 55–60 CrossRef CAS.
- H. A. Rass, N. Essayem and M. Besson, Green Chem., 2013, 15, 2240–2251 RSC.
- W. Niu, D. Wang, G. Yang, J. Sun, M. Wu, Y. Yoneyama and N. Tsubaki, Bull. Chem. Soc. Jpn., 2014, 87, 1124–1129 CrossRef CAS.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS PubMed.
- Y. Y. Gorbanev, S. K. Klitgaard, J. M. Woodley, C. H. Christensen and A. Riisager, ChemSusChem, 2009, 2, 672–675 CrossRef CAS PubMed.
- C. Megías-Sayago, K. Chakarova, A. Penkova, A. Lolli, S. Ivanova, S. Albonetti, F. Cavani and J. A. Odriozola, ACS Catal., 2018, 8, 11154–11164 CrossRef.
- C. Ferraz, M. Zielinski, M. Pietrowski, S. Heyte, F. Dumeignil, L. M. Rossi and R. Wojcieszak, ACS Sustainable Chem. Eng., 2018, 6, 16332–16340 CrossRef CAS.
- A. Villa, M. Schiavoni, S. Campisi, G. M. Veith and L. Prati, ChemSusChem, 2013, 6, 609–612 CrossRef CAS PubMed.
- C. A. Antonyraj, N. T. T. Huynh, S.-K. Park, S. Shin, Y. J. Kim, S. Kim, K.-Y. Lee and J. K. Cho, Appl. Catal., A, 2017, 547, 230–236 CrossRef CAS.
- S. Campisi, S. Capelli, D. Motta, F. Trujillo, T. Davies, L. Prati, N. Dimitratos and A. Villa, C, 2018, 4, 48 Search PubMed.
- H. Hirai, J. Macromol. Sci., Chem., 1984, 21, 1165–1179 CrossRef.
- M. Ventura, A. Dibenedetto and M. Aresta, Inorg. Chim. Acta, 2018, 470, 11–21 CrossRef CAS.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- A. C. Braisted, J. D. Oslob, W. L. Delano, J. Hyde, R. S. McDowell, N. Waal, C. Yu, M. R. Arkin and B. C. Raimundo, J. Am. Chem. Soc., 2003, 125, 3714–3715 CrossRef CAS PubMed.
- M. Munekata and G. Tamura, Agric. Biol. Chem., 1981, 45, 2149–2150 CAS.
- T. Reichstein, Helv. Chim. Acta, 1926, 9, 1066–1068 CrossRef CAS.
- K. Mitsukura, Y. Sato, T. Yoshida and T. Nagasawa, Biotechnol. Lett., 2004, 26, 1643–1648 CrossRef CAS PubMed.
- Y.-Z. Qin, Y.-M. Li, M.-H. Zong, H. Wu and N. Li, Green Chem., 2015, 17, 3718–3722 RSC.
- M. Sayed, S.-H. Pyo, N. Rehnberg and R. Hatti-Kaul, ACS Sustainable Chem. Eng., 2019, 7, 4406–4413 CrossRef CAS.
- E.-S. Kang, D. W. Chae, B. Kim and Y. G. Kim, J. Ind. Eng. Chem., 2012, 18, 174–177 CrossRef CAS.
- Z. Zhang, B. Liu, K. Lv, J. Sun and K. Deng, Green Chem., 2014, 16, 2762–2770 RSC.
- F. Wang and Z. Zhang, J. Taiwan Inst. Chem. Eng., 2017, 70, 1–6 CrossRef CAS.
- O. R. Schade, K. F. Kalz, D. Neukum, W. Kleist and J.-D. Grunwaldt, Green Chem., 2018, 20, 3530–3541 RSC.
- J. An, G. Sun and H. Xia, ACS Sustainable Chem. Eng., 2019, 7, 6696–6706 CrossRef CAS.
- M. J. Beier, T. W. Hansen and J.-D. Grunwaldt, J. Catal., 2009, 266, 320–330 CrossRef CAS.
- E. Skrzynska, S. Zaid, A. Addad, J. S. Girardon, M. Capron and F. Dumeignil, Catal. Sci. Technol., 2016, 6, 3182–3196 RSC.
- X. Bao, M. Muhler, B. Pettinger, R. Schlögl and G. Ertl, Catal. Lett., 1993, 22, 215–225 CrossRef CAS.
- H. Schubert, U. Tegtmeyer, D. Herein, X. Bao, M. Muhler and R. Schlögl, Catal. Lett., 1995, 33, 305–319 CrossRef CAS.
- V. I. Bukhtiyarov, M. Hävecker, V. V. Kaichev, A. Knop-Gericke, R. W. Mayer and R. Schlögl, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 235422 CrossRef.
- Z. Qu, M. Cheng, W. Huang and X. Bao, J. Catal., 2005, 229, 446–458 CrossRef CAS.
- Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer and P. C. Redfern, Science, 2010, 328, 224–228 CrossRef CAS PubMed.
- L. Ardemani, G. Cibin, A. J. Dent, M. A. Isaacs, G. Kyriakou, A. F. Lee, C. M. A. Parlett, S. A. Parry and K. Wilson, Chem. Sci., 2015, 6, 4940–4945 RSC.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147 RSC.
- H. Topsøe, J. Catal., 2003, 216, 155–164 CrossRef.
- M. G. O'Brien, A. M. Beale, S. D. Jacques, M. Di Michiel and B. M. Weckhuysen, Appl. Catal., A, 2011, 391, 468–476 CrossRef.
- J.-D. Grunwaldt and A. Baiker, Phys. Chem. Chem. Phys., 2005, 7, 3526–3539 RSC.
- A. Gaur, B. D. Shrivastava and H. Nigam, Proc. Indian Natl. Sci. Acad., 2013, 79, 921–966 Search PubMed.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed.
- A. M. Gänzler, M. Casapu, A. Boubnov, O. Müller, S. Conrad, H. Lichtenberg, R. Frahm and J.-D. Grunwaldt, J. Catal., 2015, 328, 216–224 CrossRef.
- P. J. Ellis, I. J. Fairlamb, S. F. Hackett, K. Wilson and A. F. Lee, Angew. Chem., 2010, 122, 1864–1868 CrossRef.
- C. M. Parlett, C. V. Gaskell, J. N. Naughton, M. A. Newton, K. Wilson and A. F. Lee, Catal. Today, 2013, 205, 76–85 CrossRef CAS.
- S. Reimann, J. Stötzel, R. Frahm, W. Kleist, J.-D. Grunwaldt and A. Baiker, J. Am. Chem. Soc., 2011, 133, 3921–3930 CrossRef CAS PubMed.
- J.-D. Grunwaldt, M. Ramin, M. Rohr, A. Michailovski, G. R. Patzke and A. Baiker, Rev. Sci. Instrum., 2005, 76, 054104 CrossRef.
- A. Zimina, K. Dardenne, M. Denecke, D. Doronkin, E. Huttel, H. Lichtenberg, S. Mangold, T. Pruessmann, J. Rothe, T. Spangenberg, R. Steininger, T. Vitova, H. Geckeis and J.-D. Grunwaldt, Rev. Sci. Instrum., 2017, 88, 113113 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- B. Molleman and T. Hiemstra, Environ. Sci.: Nano, 2017, 4, 1314–1327 RSC.
- G. I. Waterhouse, G. A. Bowmaker and J. B. Metson, Phys. Chem. Chem. Phys., 2001, 3, 3838–3845 RSC.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824–827 RSC.
- R. Sahu and P. L. Dhepe, React. Kinet., Mech. Catal., 2014, 112, 173–187 CrossRef CAS.
- Q. Li, H. Wang, Z. Tian, Y. Weng, C. Wang, J. Ma, C. Zhu, W. Li, Q. Liu and L. Ma, Catal. Sci. Technol., 2019, 9, 1570–1580 RSC.
- T. Pasini, M. Piccinini, M. Blosi, R. Bonelli, S. Albonetti, N. Dimitratos, J. A. Lopez-Sanchez, M. Sankar, Q. He, C. J. Kiely, G. J. Hutchings and F. Cavani, Green Chem., 2011, 13, 2091–2099 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- V. Schwartz, D. R. Mullins, W. Yan, B. Chen, S. Dai and S. H. Overbury, J. Phys. Chem. B, 2004, 108, 15782–15790 CrossRef CAS.
- J.-D. Grunwaldt, M. Caravati, S. Hannemann and A. Baiker, Phys. Chem. Chem. Phys., 2004, 6, 3037–3047 RSC.
- J.-D. Grunwaldt and A. Baiker, AIP Conf. Proc., 2007, 882, 577–581 CrossRef CAS.
- K. Köhler, W. Kleist and S. S. Pröckl, Inorg. Chem., 2007, 46, 1876–1883 CrossRef PubMed.
- G. A. Sotiriou, A. Meyer, J. T. Knijnenburg, S. Panke and S. E. Pratsinis, Langmuir, 2012, 28, 15929–15936 CrossRef CAS PubMed.
- B. Molleman and T. Hiemstra, Langmuir, 2015, 31, 13361–13372 CrossRef CAS PubMed.
- C. Keresszegi, T. Mallat and A. Baiker, New J. Chem., 2001, 25, 1163–1167 RSC.
- J.-D. Grunwaldt, M. Caravati and A. Baiker, J. Phys. Chem. B, 2006, 110, 25586–25589 CrossRef CAS PubMed.
- J.-D. Grunwaldt, C. Keresszegi, T. Mallat and A. Baiker, J. Catal., 2003, 213, 291–295 CrossRef CAS.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further catalyst characterization and in situ studies. See DOI: 10.1039/d0cy00878h |
| This journal is © The Royal Society of Chemistry 2020 |