
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzyme production of D-gluconic acid and glucose oxidase: successful tales of cascade reactions
Jakub F.
Kornecki†
a,
Diego
Carballares†
a,
Paulo W.
Tardioli
 b,
Rafael C.
Rodrigues
c,
Ángel
Berenguer-Murcia
d,
Andrés R.
Alcántara
e and
Roberto
Fernandez-Lafuente
*a
b,
Rafael C.
Rodrigues
c,
Ángel
Berenguer-Murcia
d,
Andrés R.
Alcántara
e and
Roberto
Fernandez-Lafuente
*a
aDepartamento de Biocatálisis, ICP-CSIC, Campus UAM-CSIC, 28049 Madrid, Spain. E-mail: rfl@icp.csic.es
bPostgraduate Program in Chemical Engineering (PPGEQ), Department of Chemical Engineering, Federal University of São Carlos, Rod. Washington Luís, km 235, 13565-905 São Carlos, SP, Brazil
cBiocatalysis and Enzyme Technology Lab, Institute of Food Science and Technology, Federal University of Rio Grande do Sul, Av. Bento Gonçalves, 9500, P.O. Box 15090, Porto Alegre, RS, Brazil
dDepartamento de Química Inorgánica e Instituto Universitario de Materiales, Universidad de Alicante, Alicante 03080, Spain
eDepartamento de Química en Ciencias Farmacéuticas, Facultad de Farmacia, Universidad Complutense de Madrid, Plaza de Ramón y Cajal, s/n, 28040-Madrid, Spain
First published on 14th July 2020
Abstract
This review mainly focuses on the use of glucose oxidase in the production of D-gluconic acid, which is a reactant of undoubtable interest in different industrial areas. The enzyme has been used in numerous instances as a model reaction to study the problems of oxygen supply in bioreactors. One of the main topics in this review is the problem of the generated side product, hydrogen peroxide, as it is an enzyme-inactivating reagent. Different ways to remove hydrogen peroxide have been used, such as metal catalysts and use of whole cells; however, the preferred method is the coupling glucose oxidase with catalase. The different possibilities of combining these enzymes have been discussed (use of free enzymes, independently immobilized enzymes or co-immobilized enzymes). Curiously, some studies propose the addition of hydrogen peroxide to this co-immobilized enzyme system to produce oxygen in situ. Other cascade reactions directed toward the production of gluconic acid from polymeric substrates will be presented; these will mainly involve the transformation of polysaccharides (amylases, cellulases, etc.) but will not be limited to those (e.g., gluconolactonase). In fact, glucose oxidase is perhaps one of most successful enzymes, and it is involved in a wide range of cascade reactions. Finally, other applications of the enzyme have been reviewed, always based on the production of D-gluconic acid, which produces a decrease in the pH, a decrease in the oxygen availability or the production of hydrogen peroxide; in many instances, cascade reactions are also utilized. Thus, this review presents many different cascade reactions and discusses the advantages/drawbacks of the use of co-immobilized enzymes.
1. D-Gluconic acid
1.1. Metabolic role
D-Gluconic acid, or (2R,3S,4R,5R)-2,3,4,5,6-pentahydroxyhexanoic acid (C6H12O7, also named dextronic acid), is the C1-oxidized form of D-glucose (or dextrose), where the aldehyde group has become oxidized to the corresponding carboxylic acid. It can also be obtained through the hydrolysis of D-gluconolactone. The gluconate shunt, shown in Fig. 1, illustrates the production of D-gluconic acid from D-glucose and the subsequent steps in its degradation, leading to shorter molecules.1 Metabolically, glucose, which is the main carbon source for most living organisms, after being phosphorylated to glucose 6-phosphate (G6P) in the first step of glycolysis, may follow two routes. It may either undergo glycolysis to provide energy as ATP (Embden–Meyerhof–Parnas pathway) or enter the pentose phosphate pathway (PPP) in order to furnish reducing equivalents (nicotinamide adenine dinucleotide phosphate [NADPH]) and pentose sugars. These compounds are precursors of nucleotide and serine/glycine/threonine biosynthesis.2 The derivation of glucose between these two metabolic alternatives, glycolysis and PPP, depends on the environmental situation (the nutrient concentration and presence or absence of stressors).1,3 In the gluconate shunt, glucose is oxidized by glucose dehydrogenase to furnish gluconate, the form in which D-gluconic acid is present at physiological pH. Subsequently, gluconate is phosphorylated by the action of gluconate kinase to produce 6-phosphogluconate, which is the second intermediate of the pentose phosphate pathway (Fig. 1, vide supra). This gluconate shunt is mainly found in plants, algae, cyanobacteria and some bacteria, which all use the Entner–Doudoroff pathway (Fig. 1, vide infra) to degrade glucose or gluconate; this generates 2-keto-3-deoxygluconate-6-phosphate, which is then cleaved to generate pyruvate and glyceraldehyde 3-phosphate.4 Glucose dehydrogenase and gluconate kinase activities are also present in mammals, fission yeast, and flies, none of which possess the key Entner–Doudoroff pathway enzymes.5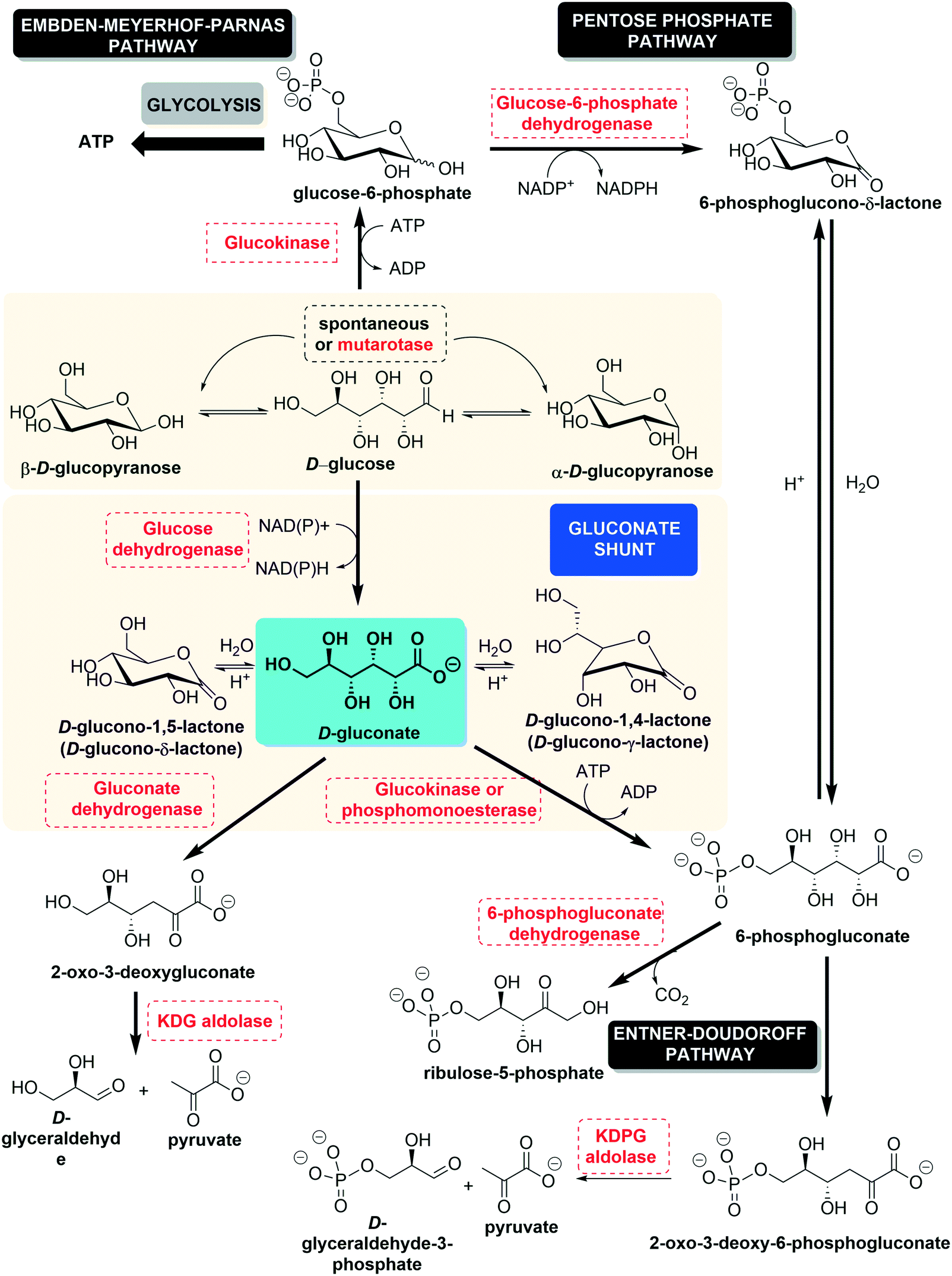 | ||
| Fig. 1 The gluconate shunt. | ||
1.2. Chemical properties
D-Gluconic acid is widely present in nature, especially in plants, fruits, rice, dairy products, vinegar, meat, wine (up to 0.25%) and honey (1%).6 Many fungi, yeasts and bacteria are able to produce D-gluconic acid.6,7 Historically, Hlasiwetz and Habermann discovered D-gluconic acid during the oxidation of glucose with chlorine in 1870, and it was first isolated as its barium or calcium salt; the history of the discovery and production of D-gluconic acid is well illustrated in articles from Ramachandran et al.6,8D-Gluconic acid is very soluble in water; the commercial 50% solution has a pH of 1.82 and a density of 1.23 g × cm−3 at 20 °C. On the other hand, the acid is only faintly soluble in ethanol and almost insoluble in nonpolar organic solvents.9 Free D-gluconic acid is difficult to prepare in crystalline form. The crystallization of the anhydrous substance (white and odorless crystalline power, specific rotation [α20D] − 6.7°, [α25D] − 5.4°)9 is possible below 30 °C, and a monohydrate (mp ca. 85 °C) has been reported to crystallize at 0 °C to 3 °C.9,10 By storing D-gluconic acid in a desiccant at room temperature or heating it above 50 °C, internal loss of water leads to the formation of lactones. This circumstance can be understood by looking at its chemical structure. D-Gluconic acid, being a poly-hydroxy acid, can undergo intramolecular cyclization to furnish either the D-glucono-δ-lactone ((3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-one) or the D-glucono-γ-lactone ((3R,4R,5R)-5-[(1R)-1,2-dihydroxyethyl]-3,4-dihydroxyoxolan-2-one) (Fig. 2). In aqueous media, an acid-catalyzed mechanism leads to an equilibrium where between 55% and 65% of the molecules exist as open D-gluconic acid because of its weak acidity (Ka = 1.99 × 10−4 at 25 °C, reported pKa from 3.62 to 3.7 at 25 °C).6,9,11 This conformational equilibrium (first order with respect to [H+])9 is reached very slowly, as the γ-lactone is produced around one hundred times more slowly than the δ-lactone.9,11
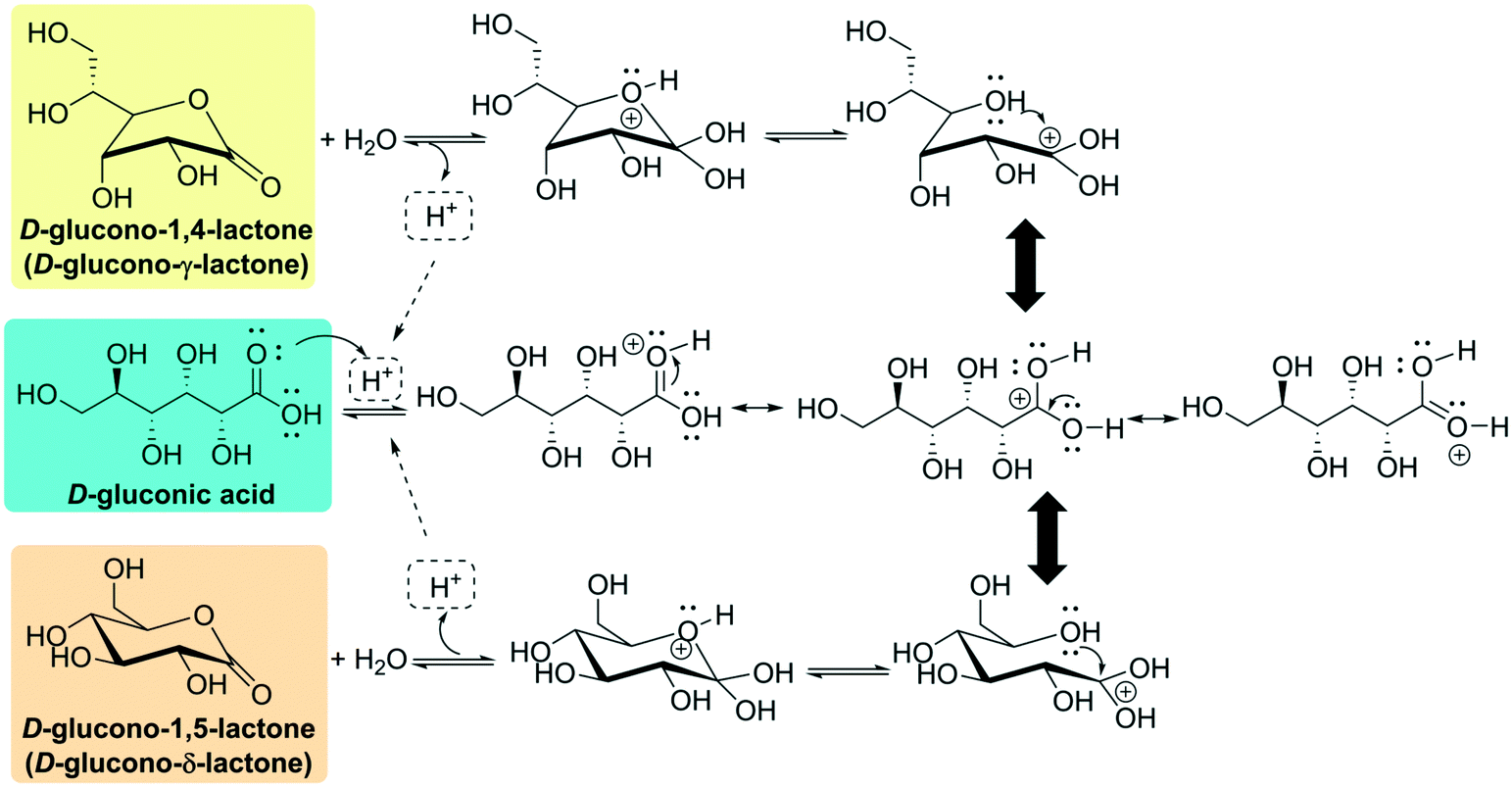 | ||
| Fig. 2 Acid-catalyzed cyclization of D-gluconic acid to D-glucono-δ-lactone or D-glucono-γ-lactone. | ||
Both lactones can be isolated in the solid state. δ-Lactone is a white, crystalline substance with a weakly sweet taste (melting point of 153 °C, [α20D] + 66.2 °C),9 while γ-lactone crystallizes as fine needles (melting point 134 °C to 136 °C, [α20D] + 67.8 °C).9 Because of the acid-catalyzed equilibrium, both the pH and the specific rotation of a lactone solution (up to 90 g of α-lactone can be dissolved in water at 20 °C, reaching 205 g at 50 °C, while only small amounts dissolve in organic solvents)9 decrease as hydrolysis proceeds.
By increasing the pH of the medium, upon addition of a base, the closed lactamic structures are quickly hydrolyzed9 to furnish gluconate, an anion which can effortlessly bind to di- or trivalent metallic cations to form very stable chemical salts11 (Fig. 3). Both the lactones and the calcium salts can be oxidized by reagents such as nitric acid or hydrogen peroxide under mild reaction conditions to furnish a mixture of 2- and 5-oxo (keto)-D-gluconate (2-KGA and 5-KGA). The former is the major product of anodic oxidations, treatment with sodium chlorate or biological fermentation. It is also possible to obtain 2,5-dioxo(keto)-D-gluconate (2,5-DKGA) at different percentages.9 Under extreme conditions (concentrated nitric acid or dinitrogen tetroxide), glucaric acid is produced.
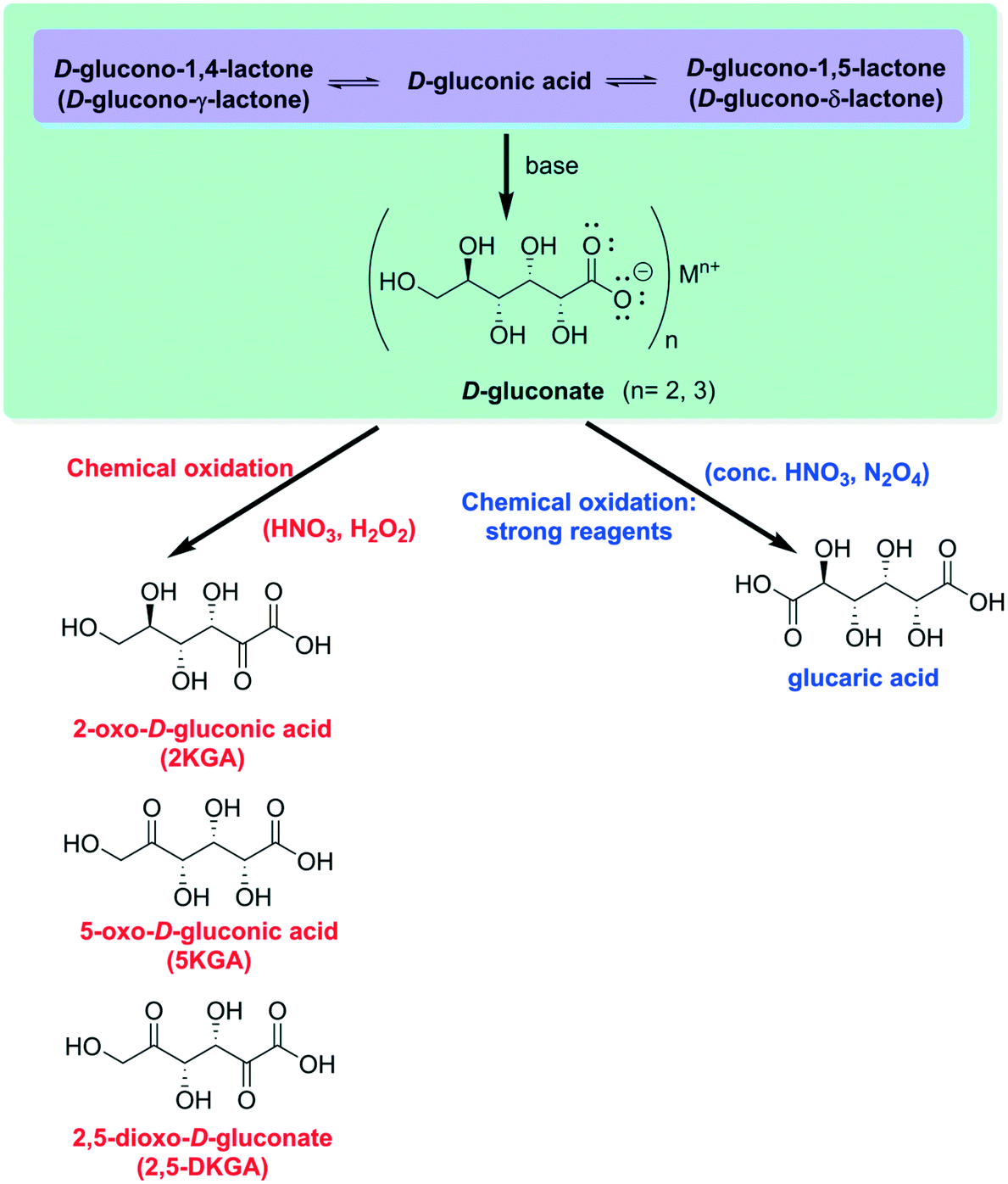 | ||
| Fig. 3 Formation of D-gluconate and oxidation products. | ||
1.3. Production
D-Gluconic acid and its derivatives are being increasingly used in different industries; their current production is estimated to be around 105 tons per year.7,11 The production costs vary from 1.20 US$ per kg for D-gluconic acid up to 8.50 US$ per kg for calcium gluconate and glucono-δ-lactone. The most broadly marketed derivative is sodium gluconate (more than 80% of the world's gluconic acid production).11 Most of the commercial methods for producing D-gluconic acid and its derivatives are based on the oxidation (chemical, electrolytical, catalytical or biochemical) of glucose or glucose-containing raw materials.6,7,11 Purely chemical oxidations (mainly using transition metals such as Pd, Pt or Au supported on titania, alumina or activated carbon)11 have the drawback of limited specificity and selectivity even when applying controlled and optimized reaction conditions; this leads to insufficient yields and the presence of several by-products. Thus, fermentation is the preponderant technique for the synthesis of D-gluconic acid. Many filamentous fungi and bacteria have been applied to obtain this product;6,11 currently, the key organisms employed are the fungi Aspergillus niger and the bacteria Gluconobacter oxydans. Current industrial processes are based on the original work of Blom et al. in 1952 (ref. 12) with some modifications introduced by Ziffer et al. in 1971,13 employing A. niger in a fed-batch process with periodical glucose feedings at 34 °C and using sodium hydroxide to maintain the fermentation pH value between 6.0 and 6.5. Using this procedure, glucose is converted to gluconic acid at a rate of 15 g L−1 h−1. Some other important patents related to D-gluconic acid production are summarized elsewhere.61.4. Uses
D-Gluconic acid, as a weak acid, can be used to dissolve combinations of polyvalent cations (oxides, hydroxides or carbonates) without attacking metallic or non-metallic surfaces, forming water-soluble complexes with the cations.9 Therefore, D-gluconic acid is extremely useful for removing calcareous and rust layers from copper, aluminum and other metal surfaces. These include beer and milk flakes on galvanized iron, oxide coatings from metal alloys or stainless steel, and the safe removal of paint and varnish without damaging the underlying surfaces.9 Conversely, sodium gluconate is present in many commercial cleansers due to its high stability to hydrolysis under extreme conditions (high temperature and pH values) as well as its sequestering properties for hardening agents in water. Although preferred for metal components, gluconate solutions are used in some food processing plants as cleaning agents, mainly for glassware, as it helps prevent scale formation.6 For example, the dairy industry uses solutions of D-gluconic acid derivatives to prevent the precipitation of calcium salts in their equipment and glass storage containers.11 Alkaline sodium gluconate solutions, when applied at 95 °C to 100 °C, are excellent agents for the effective removal of paint and varnish without affecting underlying surfaces. Gluconate has also been applied for the pretreatment of certain surfaces in metallurgy, replacing the highly toxic cyanide ion. Finally, the bakery industry uses δ-gluconolactone to diminish the absorption of fatty compounds.8D-Gluconic acid combined with magnesium salts is used in the textile industry as a stabilizer for peroxide bleach baths, while gluconate (sometimes mixed with polyphosphates) is employed for finishing natural cellulose fibers and desizing polyester or polyamide fabrics. Mixtures of gelatin and sodium gluconate are employed as sizing agents in the paper industry in order to obtain products displaying increased acid resistance.9
Sodium gluconate (0.02–0.2 wt%) is used as a highly effective agent for retarding the curing process of concrete. In fact, using this additive, a very homogeneous concrete is obtained, displaying high resistance to water, frost and cracking as well as better stability upon setting. Additionally, sodium gluconate improves the flow properties of wet concrete mixtures.8,9
However, the main applications of D-gluconic acid (E574) and its derivatives are as additives in the food industry. In 1986, δ-gluconolactone (E575) and sodium gluconate (E576) were granted GRAS (generally recognized as safe) status by the US FDA (Food and Drug Administration), authorizing their unlimited use as food ingredients. On the other hand, the United Nations agencies Food and Agriculture Organization (FAO) and World Health Organization (WHO) have also regulated the use of these food additives, including E577 (potassium gluconate), E578 (calcium gluconate), E579 (ferrous gluconate) and E580 (magnesium gluconate). For a detailed description of these additives, please see the review from Cañete-Rodríguez et al.11 Low concentrations of D-gluconic acid, ranging from 0.02% to 0.1%, promote sucrose inversion without triggering the resulting fructose to undergo any further reaction. Usually, trace elements are administered as gluconate salts because of their rapid absorption and broad tolerance. D-Gluconic acid improves the organoleptic properties of food products, conferring a fresh and less bitter taste. In fruit juices, it prevents clouding by binding some metals (calcium or iron, mainly) present at trace levels.11 These additives are extensively used in meat, dairy products and baked goods and as an ingredient of leavening agents for pre-leavened products. In sherbets, D-gluconic acid is used as a flavoring agent, while it can also be employed to reduce fat absorption in doughnuts or cones. Furthermore, δ-gluconolactone is present in tofu, yogurt, cottage cheese, bread, meat, confectionery and pickling foods.6 The lactone is generally preferred in cases where acidic conditions are required over a long period of time. Recently, the effects of D-gluconic acid and its derivatives as prebiotics, as well as their anti-oxidant properties, have been investigated, with promising results.11 Finally, it must be remarked that the pharmaceutical industry uses gluconate salts (Ca2+, Mg2+ and Fe2+) as supplements to treat hypocalcaemia, hypomagnesaemia and anemia, respectively.
2. Glucose oxidase: sources and mechanism of action
2.1. Mechanism of action
Glucose oxidase catalyzes the oxidation of β-D-glucose to D-glucono-δ-lactone producing hydrogen peroxide (H2O2) as a by-product, with molecular oxygen acting as an electron acceptor. The first product is non-enzymatically hydrolyzed to D-gluconic acid (Fig. 4).14,15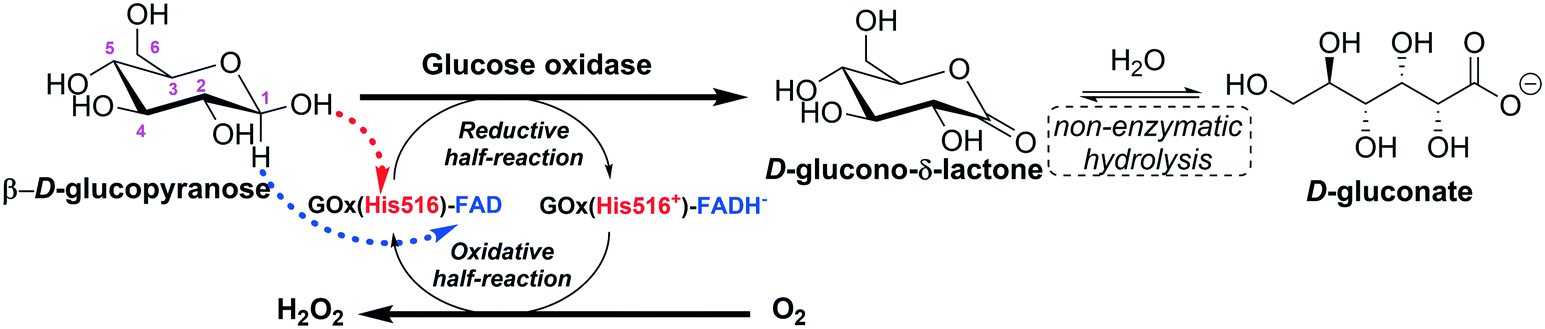 | ||
| Fig. 4 Representation of the action mechanism of glucose oxidase in the oxidation of glucose to D-glucono-δ-lactone (adapted from Leskovac et al.14 and Bankar et al.15). | ||
Glucose oxidase is highly specific for the β-anomer of D-glucopyranose (the best substrate), while the α-anomer is a very poor substrate (0.64% activity relative to β-D-glucose). Xylose and idose are very poor substrates (0.03% and 0.02% activities relative to β-D-glucose).14 In general, glucose oxidase exhibits at least a 20-fold higher affinity and a 5-fold higher turnover rate for β-D-glucopyranose than for any other monosaccharides. In the cases of D-mannose, D-galactose, and D-xylose, the specificity constants are 400, 1000, and 3000-fold lower than that of β-D-glucopyranose. The product of β-D-glucopyranose oxidation, D-glucono-δ-lactone, has very weak enzyme inhibitory effects.16
The catalytic action of glucose oxidase is dependent on the FAD cofactor, which acts as an initial electron acceptor (as a hydride) and undergoes reduction to FADH−. Then, FADH− is oxidized, transferring electrons (as a hydride) to molecular oxygen, which is reduced to hydrogen peroxide.14,16–18 The steady-state kinetics of the oxidation of β-D-glucopyranose by molecular oxygen obeys a ping pong bi–bi kinetic mechanism.14 For the reductive half-reaction, β-D-glucopyranose is oxidized to D-glucono-δ-lactone by transferring a proton from its C1-hydroxyl to a basic group on the enzyme (His516), and a direct hydride transfer occurs from its C1 position to the N5 position of the isoalloxazine ring of FAD.14 The oxidative half-reaction proceeds via one- or two-electron transfer mechanisms, depending on the type of the oxidizing substrate (oxygen, benzoquinones, naphthoquinones, ferrocene salts, organic metal complexes, etc.). In the case of oxygen, firstly, it diffuses into the active site very quickly (the diffusion of O2 to the active site is thermodynamically facilitated) and the reduced coenzyme (FADH−) is re-oxidized back to FAD by oxygen, which is reduced to hydrogen peroxide. This reaction proceeds via two single-electron transfer steps; a superoxide anion and a flavin semiquinone radical are the intermediates of the reaction.14
2.2. Glucose oxidase features and production
Glucose oxidase (β-D-glucose: oxygen 1-oxidoreductase; GOx, EC 1.1.3.4) is an oxidase with several large-scale technological applications, among them D-gluconic acid production.19–28A large number of microorganisms produce glucose oxidase, including plants, (red algae, citrus fruits), animals (e.g., insects), bacteria, and fungi. The main purpose of glucose oxidase in these sources is to generate hydrogen peroxide (a coproduct of the enzyme catalytic action), which acts as an anti-bacterial and anti-fungal agent.19,29–31 For example, glucose oxidase is found in honey, and the anti-bacterial activity is strongly dependent on the accumulation of hydrogen peroxide in the product.30,32
For commercial applications, glucose oxidase has been produced and purified from fungi, especially from Aspergillus and Penicillium species.19,33–45 The great success of these two species is mainly due to their metabolic versatility and their recognition as GRAS (generally recognized as safe).19,38 Among the Aspergillus species, remarkable attention has been given to A. niger over the last 60 years.46–65 Despite the great success of glucose oxidase production from Aspergillus and Penicillium species, efforts to find other glucose oxidase sources are being developed, including other glucose oxidase-producing fungi, e.g., Cladosporium neopsychrotolerans66 and Mucor circinelloides;67 other Aspergillus species, e.g., A. tubingensis,29A. terreus,68A. oryzae,69A. carbonarius,70 and A. nidulans;71 and some glucose oxidase-producing bacteria.72
The demand for glucose oxidase is increasing due to its growing applications in different industrial sectors. However, wild-type fungal strains producing glucose oxidase usually have low fermentation capacity, complicated purification processes and low enzyme efficiency. Additionally, impurities in glucose oxidase, including other enzymes (e.g., catalase, cellulase, and amylase), prevent its use in some applications15,19 and require full enzyme purification. In this way, genetic and non-genetic engineering tools have been applied to increase the productivity of glucose oxidase. Genetic engineering has stood out since 1990 as a tool to build excellent glucose oxidase expression systems; it permits the development of strains with high enzyme production.19,66,73–80 In addition, strategies of formulating the fermentation medium73,81 and computational tools82 have proven to be powerful tools to increase glucose oxidase productivity. As an example, Bankefa et al.73 reported a strategy of co-feeding complex carbon sources (e.g., yeast extract) with methanol, combining a secretory pathway engineering in a Pichia pastoris strain containing the gene encoding glucose oxidase from A. niger. Using this strategy, the volumetric extracellular activity of glucose oxidase in a 1 L fermenter was enhanced by around 3-fold compared to that reached when utilizing a recombinant P. pastoris strain containing only the gene encoding glucose oxidase. In another communication, Darvishi et al.82 reported the use of software to in silico analyze endogenous and exogenous signal peptides to secrete recombinant glucose oxidase in non-conventional Yarrowia lipolytica for further in vivo analysis. The recombinant Y. lipolytica strain produced up to 280 and 145 U L−1 of extracellular and intracellular glucose oxidase, respectively, from A. niger after 7 days in YPD medium. In other approaches, mutagenesis of A. niger by UV irradiation47 and chemical mutagens83 generated mutated strains that produced around two-fold and three-fold more glucose oxidase than their parental wild strains, respectively.
The structures of glucose oxidase from A. niger and P. amagasakiense are well-established. They were refined at 1.9 and 1.8 Å resolution, respectively.16 Both glucose oxidases are homodimers with 581 and 587 amino acid residues per monomer, respectively, one flavin adenine dinucleotide (FAD) per monomer non-covalently bound to the active site, and eight and seven potential N-glycosylation sites, respectively. The residues Asn89, Asn161, Asn355 and Asn388 are sites for attachment of N-acetylglucosamine moieties. The Asn89 site contains an extended carbohydrate structure consisting of a second N-acetylglucosamine and three mannose molecules. This carbohydrate moiety forms a bridge between the two molecules of the protein dimer. Salt bridges and hydrogen bonds further stabilize the protein dimers.16
In addition to FAD coenzyme, glucose oxidase enzymes from A. niger and P. amagasakiense have two histidines in the active site, His516 (pKa 6.9) and His559 (pKa > 8.0), which act as general bases and acids in the reductive and oxidative half-reactions of the oxidation of β-D-glucose to D-gluconic acid, as discussed above. His559 is strongly bound to Glu412 (pKa 3.4) by a hydrogen bond and to one water molecule in front of the flavin ring. These three residues of amino acids are essential for the catalysis; the protonation of these residues strongly influences the kinetic constants.14,16,19,84 Glycosylated glucose oxidases from A. niger and P. amagasakiense have a molecular mass of 150–160 kDa for the dimer, which is composed of two subunits of equal molecular mass. Both glucose oxidases are 10–16 wt% glycosylated with high-mannose type carbohydrates.14,16,18,75,84–87
Bankefa et al.73 reported that the SDS-PAGE analyses of glucose oxidase from A. niger extracellularly expressed by a recombinant P. pastoris strain showed a protein band of approximately 80 kDa, while its theoretical size was 63.5 kDa. This higher molecular mass of the enzyme subunit was attributed to enzyme glycosylation (around 21 wt%). Kalisz et al.84 reported that glycosylated and deglycosylated glucose oxidase from P. amagasakiense have molecular masses of 167 and 133 kDa (by PAGE analysis) for the dimer and 77 and 67 kDa for the monomers, respectively. On the basis of gas chromatography and SDS-PAGE analyses, for the glycosylated glucose oxidase, 13 wt% of the enzyme carbohydrates are high-mannose type.84
In general, fungal glucose oxidases have an isoelectric point in the range of pH 4.0–5.0,16 optimal activity in the pH range from 4.0 to 7.0,14,15,29,31,33,40,75,84 and optimal temperature in the range of 40–60 °C.15,29,33,40,75,84 Glucose oxidase from A. niger does not require metal ions to express its activity; however, it is inhibited by Ag+, Hg2+, Cu2+, Mg2+ and Ca2+.19 Very importantly in the context of this review, hydrogen peroxide is a strong deactivating agent of glucose oxidase.15,88
3. Glucose oxidase immobilization
Enzymes are potentially useful industrial biocatalysts because they operate under mild conditions with high activity, selectivity and specificity.89–93 However, due to their biological origin, they have some features that are far from ideal for industrial requirements.94 Current developments in all areas related to enzyme biocatalyst design (e.g., metagenomics,95–98 directed evolution,99–101 chemical modification,102 and immobilization103–108) can solve these problems. For example, several new strategies have been published, such as the design of plurizymes (enzymes bearing a natural and an artificially designed active center), the design of irreversible inhibitors directed to one of these active center bearing a metal catalytic center and the use of modified plurizymes (bearing a free active center while the other active center presented the inhibitor with a metal catalysts) in a cascade reaction.109Immobilization was the initial answer to the problem of enzyme solubility, which complicates the reuse of enzymes.106,110,111 Currently, with the decreasing price of enzymes, enzyme immobilization should address many other enzyme limitations, such as improving activity, stability, selectivity and specificity, decreasing inhibition104 or purifying the enzyme.103 This will increase the interest in immobilization and will enlarge the window of conditions in which the biocatalyst can be utilized.112,113 In this way, immobilization is not merely a tool to recover enzymes, but may also be a very powerful means to greatly improve enzyme performance.110
Immobilization may be also considered when using glucose oxidase. Although the enzyme is quite stable, the possibility of using it for longer times and many cycles may be very important for its industrial implementation. Therefore, proper glucose oxidase immobilization may be critical for the successful design of an industrial biocatalyst.110
Almost 1000 citations can be found using the keywords “glucose oxidase immob*” when searching the publication titles in Scopus (see Fig. 5). Obviously, a comprehensive assessment of this topic is beyond the objective of this review. Glucose oxidase used in biosensors has also been extensively reviewed;114–117 therefore, this topic will not be included in this new review.
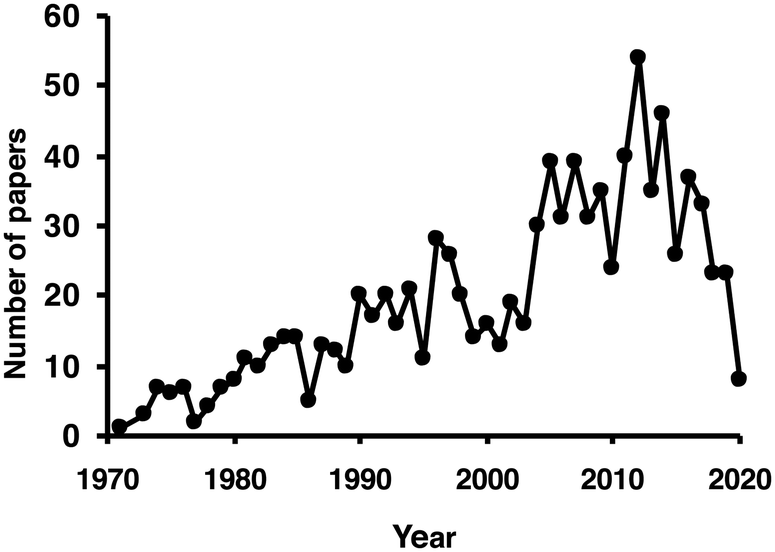 | ||
| Fig. 5 Number of papers per year including the term glucose oxidase and immob* in their titles. Source: Scopus (accessed 4/22/2020). | ||
Therefore, we have included only some of the more relevant immobilization papers, mainly those that offer new strategies or unexpected results. For example, a comparison between different supports to immobilize glucose oxidase has been reported and offered some unexpected results.118 Glucose oxidase was immobilized on glyoxyl agarose, epoxy Sepabeads and glutaraldehyde-activated supports. Although in general, glyoxyl-agarose supports provide higher stabilization of enzymes, in this instance, the highest stabilization was achieved using supports pre-activated with glutaraldehyde. The stability could be further improved if the enzyme was first ionically adsorbed on an aminated support and then treated with glutaraldehyde. This new biocatalyst was also more stable in the presence of ethanol or hydrogen peroxide.118
Glucose oxidase has been used as an example enzyme to develop multilayer biocatalysts. For example, layers of glucose oxidase immobilized on films of chitosan have been built for use in biosensors.119 This biocatalyst design was also used in the reduction of a gold salt to yield metallic gold nanoparticles.119 In another paper, nylon-based microfiltration membranes or hydrophobic polyvinylidene fluoride were used to build multilayers using in situ polymerization of acrylic acid to obtain a polyanion.120 An enzyme-based microcantilever sensor obtained by using layer-by-layer deposition of glucose oxidase was utilized in the design of a biosensor.121
3.1. Design of glucose-sensitive supports using immobilized glucose oxidase
This application of immobilized glucose oxidase is far from being applied in the production of D-gluconic acid, but it is interesting enough to comment on at least briefly. It has been known for a long time that some membranes have been identified as sensitive to changes in media. Some researchers have attempted to exploit these membrane features using glucose oxidase to change the medium conditions (usually decreasing the pH) in the presence of high concentrations of glucose, mainly to release insulin.122–125 For example, to achieve this goal, glucose oxidase has been immobilized on amine-containing polymers. When D-gluconic acid is produced by the action of glucose oxidase, it decreases the pH in the membrane, which then swells. This permits the release of drugs into the medium.125 A membrane was produced using poly(acrylic acid)-grafted porous cellulose film and glucose oxidase was immobilized on it to build an insulin delivery system that responds to glucose concentration levels.126 The system was based on conformational changes of the graft chains induced by changes in the medium pH as a consequence of changes in the ionic interactions in the matrix; the production of D-gluconic acid produced a decrease in the pH value, which led to a decrease in the electrostatic repulsion between the poly(acrylic acid) side chains. This transformed the graft chains into coil-like forms and opened the pores of the membrane, enabling the release of the drug.126Glucose-sensitive hydrogels can be used in a similar fashion; in some cases, they co-immobilize the enzyme with catalase to prevent hydrogen peroxide accumulation.127 In one interesting paper, β-cyclodextrin was grafted on a polyethyleneimine hydrogel (a gel that also swells at low pH values) and used to immobilize glucose oxidase.128 Other hydrogels have been described.129 To reinforce the immobilization, the enzyme was modified with 5 palmitic acid-N-hydroxysuccinimide ester molecules per mole of enzyme; therefore, the release of fluorescein isothiocyanate dextran from the support could increase as the concentration of glucose increased.128 Also, glucose-sensitive mesoporous silica nanoparticles have been designed for this goal.130,131 This includes an expanded-pore silica matrix coated with 1-propyl-1-H-benzimidazole groups and bearing insulin labeled with fluorescein isothiocyanate. Cyclodextrin-modified glucose oxidase is immobilized by interaction with the benzimidazole groups. When glucose is transformed into D-gluconic acid, de-threading of the benzimidazole–cyclodextrin-glucose oxidase complexes is induced, permitting cargo release.131 Multilayers of enzymes in this type of support have also been utilized for this goal.132 Nanogels and microgels,133,134 dextran nanoparticles,135 and metal organic framework nanoparticles136 have been also utilized for this purpose.
Co-immobilized enzymes have also been studied. The change of the membrane phase was studied in an interesting paper where two enzymes were coupled in a cascade reaction to accelerate the rate. Thus, gluconolactonase (an enzyme that hydrolyzes D-glucono-1,5-lactone) was co-immobilized with glucose oxidase.137 They studied the shrinking kinetics of the polyelectrolyte gels in the form of a cylinder composed of a copolymer of N-isopropylacrylamide and acrylic acid with low crosslinking. Using the co-immobilized glucose oxidase and gluconolactonase, the gel swelled rapidly, and the process was not governed by the reaction rate. Using only glucose oxidase, very slow shrinking was observed, and it was found that the rate was determined by the enzyme reaction. The more rapid shrinking using the co-immobilized enzyme was attributed to the rapid D-glucono-δ-lactone hydrolysis catalyzed by gluconolactonase after its production by glucose oxidase.137
4. The hydrogen peroxide challenge
As stated in point 2, hydrogen peroxide is the by-product of the reaction of glucose oxidase, and this by-product possesses some potential uses and dangers.138 Hydrogen peroxide is a simple and relatively mild oxidizing reagent; 47% of its weight is an oxidant, and it produces oxygen and water as its only by-products.139–141 Due to its electrochemical potential, hydrogen peroxide is suitable to be quantified in many of the applications of glucose oxidase in the biosensor area. Moreover, in some instances, glucose oxidase and glucose are utilized for the in situ production of this oxidant reagent as part of a cascade reaction involving other enzymes that use the in situ released hydrogen peroxide as a substrate (e.g., peroxidase, lipases).142,143However, the current review is mainly focused on D-gluconic acid production. In this reaction, hydrogen peroxide will be produced in an equimolecular ratio with D-gluconic acid; under industrially relevant concentrations of glucose, the enzyme will be exposed to moderate-to-high concentrations of hydrogen peroxide (e.g., over 100 mM). At these concentrations, hydrogen peroxide is a very efficient inactivating agent for enzymes, including glucose oxidase.118,144 The oxidation of proteins by hydrogen peroxide causes the conversion of some amino acid residues into carbonyl derivatives; chlorination, nitration or hydroxylation of aromatic amino acid moieties; hydroxylation of aliphatic groups; chlorination of primary amino groups; nitrosylation of sulfhydryl groups; sulfoxidation of methionine residues; etc.145–164 In this way, hydrogen peroxide is able to modify many of the amino acids of a protein (Arg, Cys, His, Lys, Met, Pro, Tyr, etc.).165–180 It can also promote the cleavage of some peptide bonds by either the diamide or α-amidation pathways; moreover, it can produce inter- or intramolecular protein crosslinks via different mechanisms.181–189 For a proper review of this topic, the reader can consult Stadtman and Levine's paper.144
In the case of D-gluconic acid production, the concentration of hydrogen peroxide must be reduced to prevent glucose oxidase inactivation, although in some cases this is not considered (see section 5). There are many possible routes to achieve this goal, e.g., destruction of hydrogen peroxide using metal catalysts (see section 6), use of whole cells as catalysts with their full metabolic machinery (section 7), and the use of catalase (section 8).
5. Oxidation of glucose catalyzed by glucose oxidase to produce D-gluconic acid
Although hydrogen peroxide is deleterious to the stability of glucose oxidase,138 there are some examples in the literature in which the reaction is studied in the absence of catalase or using another strategy to eliminate this dangerous compound. Usually, authors use the reaction to evaluate reactor configurations or mass transfer problems more than pursuing the production of D-gluconic acid as a real objective.For example, in well-established research, glucose oxidase was used to simulate the oxygen transfer rate in fermentation broths.190 If there is enough lactonase activity in the fungus, the D-gluconic acid production rate is proportional to the oxygen uptake rate. Glucose oxidase was immobilized on a carbon felt electrode with a catalytic current of 0.4 A and used to catalyze the oxidation of glucose employing benzoquinone to regenerate the electron acceptor.191 The coupling between electrochemical and enzymatic reactions was very efficient because of the proximity of the two reaction sites. Most importantly, by using benzoquinone instead of oxygen, the operational stability of the biocatalyst was increased 50 fold.191
D-Gluconic acid was also produced using glucose oxidase immobilized on a low-density polyethylene membrane grafted with 4-vinylpiridine (an anion-exchange membrane), affording relatively low yields.27 The immobilization of the enzyme on a membrane adjacent to the anion exchange membrane led to the selective removal of D-gluconic acid from the reaction medium and reduced enzyme inhibition by D-gluconic acid. The amount of D-gluconic acid increased 30 fold compared to the enzyme directly bound to the anion-exchange membrane. However, the concentration of glucose in this study was 1 g L−1, a very low concentration; this was very likely necessary because of the enzyme inactivation caused by accumulated hydrogen peroxide when using higher glucose concentrations.27 Another paper compared a Millipore stirred ultrafiltration cell with a Bioengineering Enzyme Membrane Reactor in the production of D-gluconic acid catalyzed by glucose oxidase.192 The maximum concentration of glucose was only 5.0 mM (also too low for practical applications), with a highest oxidation yield of 75% using 2.5 mM glucose. In other research, a falling-film micro-reactor was utilized in enzymatic glucose oxidation using free glucose oxidase.193 It was determined that the volumetric mass-transfer oxygen coefficient was the most critical parameter, and it was evaluated by employing the combined online determination of dissolved oxygen in all systems. The use of this bubble-free reactor afforded a 50% oxidation yield, while a conventional bubble column yielded only 27%.193 In other research, the inner wall of fused-silica capillaries was modified with different generations of polyamidoamine dendrimers by microwave irradiation and used to generate a capillary glucose oxidase micro-reactor.194 The oxidation efficiency increased with the number of dendrimer generations.
An ultra-scaled down membrane bioreactor based on a parameter scanning ultrafiltration device was utilized to optimize the production of D-gluconic acid using soluble glucose oxidase.195 The addition of sodium chloride reduced the yields, perhaps by reducing the concentration of available oxygen. Curiously, this paper showed that the addition of catalase had a negative effect on glucose conversion, which is difficult to explain considering that catalase should increase oxygen availability and also reduce enzyme inactivation by hydrogen peroxide.195 The catalase may have contained some contaminant enzyme that the authors could not identify.
However, in these studies using glucose oxidase, the industrial production of D-gluconic acid was not actually pursued.
6. Combinations of glucose oxidase to oxidize glucose and metal catalysts to eliminate hydrogen peroxide
Several metal catalysts very efficiently destroy hydrogen peroxide,196 although the reaction is not fully selective and can affect the enzyme.197,198 However, some examples of the combined use of glucose oxidase and metals can be found in the literature. For example, glucose oxidase was used as a model to compare the liquid–solid and the volumetric gas–liquid oxygen transfer coefficients using a normal bubble column, an internal loop airlift or an external loop airlift bubble column by the steady state method.199,200 In this study, glucose oxidase was trapped in a calcium alginate matrix in combination with fine palladium particles. These palladium particles were used to destroy the hydrogen peroxide in this study, and the external loop airlift bubble column offered the best results.7. Use of whole cell biocatalysts to produce D-gluconic acid from glucose
One solution to avoid the risks of glucose oxidase inactivation by hydrogen peroxide is the use of whole cells as biocatalysts, as the whole metabolic machinery may eliminate the produced hydrogen peroxide.201–204 However, this approach has some problems, such as risk of substrate contamination and side-reactions. For example, Aspergillus niger cells presenting catalase and glucose oxidase activities were permeabilized by treatment with organic solvents.205 Curiously, in this case, exogenous hydrogen peroxide was added to obtain oxygen after its destruction by catalase. The authors observed good stability of both enzymes in the cell. In other research, Gluconobacter oxydans suboxydans cells were described to accumulate D-gluconic acid in the medium in almost stoichiometric concentrations.206 These cells were entrapped in diverse alginate gels. The activity increased when the concentration of alginate and the size of the particles decreased and when the aeration increased (suggesting a diffusion limitation problem). When increasing the number of cells, the authors reported that the transport of oxygen became a rate-limiting parameter.206Aspergillus niger containing glucose oxidase was utilized in the oxidation of glucose employing oxygen-enriched air from 30% to 100% saturation at 1 bar, and a three-fold increase in the enzyme activity was observed.207 At the highest dissolved oxygen concentration, the activity of glucose oxidase increased more than three times, while the production rate of D-gluconic acid increased by a factor of two. It was found that the microorganism produced an endo- (main component) and an exo- (minority component) cellular glucose oxidase. Due to the oxygen transport limitations inside the mycelium, the activity of the endocellular glucose oxidase was reduced (reaching just 50–80%); however, it was still the main source of glucose oxidase activity.207 Later, the same research group compared the performance of Gluconobacter spp. and Aspergillus niger in D-gluconic acid production (using glucose oxidase and glucose dehydrogenase).208 The reactor was an airlift reactor (9.25 m) to generate a deep, wide gradient in the dissolved oxygen. The authors reported that the local dissolved oxygen concentration affected neither product formation nor cell growth, independently from the microorganism studied. This was explained by the short residence time for the cultures to pass through the area where the lowest dissolved oxygen concentration existed due to the single circulation loop. Aspergillus niger production of glucose oxidase increased with the local dissolved oxygen concentration. The authors claimed that a periodic high dissolved oxygen concentration was sufficient to induce enzyme synthesis, and this was obtained in tower-shaped reactors.208 Other researchers used Penicillium variabile P16, a strain presenting glucose oxidase and catalase activities, to study this reaction.209 The cells were immobilized using different protocols, and the best results in long-term experiments were obtained using polyurethane sponge. However, the highest mean volumetric productivity of D-gluconic acid was achieved using agar as a cell immobilizing matrix. Later, the same research group used this biocatalyst in a fluidized-bed reactor.209 Also, Aspergillus terreus was used to produce D-gluconic acid.210 It rapidly metabolized glucose to D-gluconic acid at pH 6.5 (with 70% conversion of D-glucose to D-gluconic acid); however, after using all the D-glucose in the reaction medium, the produced D-gluconic acid was employed as a carbon source. Later, glucose oxidase and a mutant enzyme (doubling the activity of the native enzyme at pH 5.5) were expressed in Aspergillus niger as C-terminal fusions with the Aga2 protein from Saccharomyces cerevisiae.211 This permitted expression of the enzymes on the surface of the yeast cells and their use for the production of D-gluconic acid.8. Use of catalases and glucose oxidase to produce D-gluconic acid: a pseudo-cascade reaction
Although metals can be used to destroy hydrogen peroxide, the most efficient way to reduce the hydrogen peroxide concentration in the glucose oxidase environment is the use of catalase in free, individually immobilized or co-immobilized (with glucose oxidase) forms (Fig. 6 and 7). The glucose–glucose oxidase-hydrogen peroxide-catalase system was studied in vitro in the field of “nonlinear chemical dynamics”.212 These reactions produce a natural basis of fluctuations in biological systems, belonging to the family of oscillatory enzymatic reactions.213,214 Catalases catalyze the decomposition of hydrogen peroxide in water and oxygen without risk of promoting some undesired side reactions. In fact, catalases produce oxygen that can be used again by glucose oxidase, even improving glucose oxidase activity.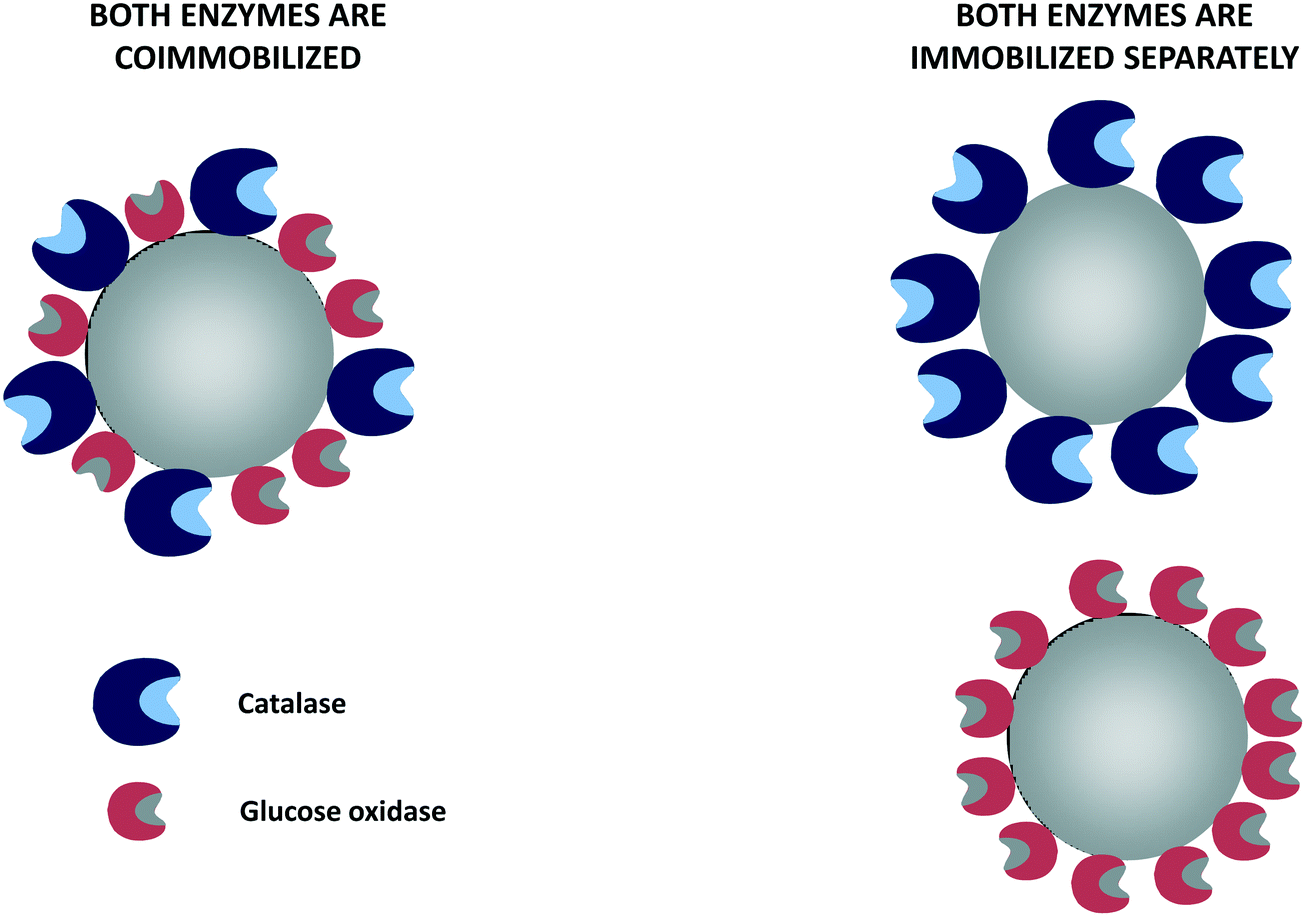 | ||
| Fig. 6 Glucose oxidase and catalase co-immobilized or immobilized separately using non-porous supports. | ||
 | ||
| Fig. 7 Glucose oxidase and catalase co-immobilized or immobilized separately using porous supports. | ||
A cascade reaction can be described as a reaction where the product of one enzyme is the substrate of another, and the final product is the target. The destruction of hydrogen peroxide to prevent glucose oxidase inactivation, even if it is not a real cascade reaction,212 can be considered as one because hydrogen peroxide is produced by glucose oxidase (enzyme 1), which also produces the target product (D-gluconic acid), while catalase (enzyme 2) destroys the by-product of enzyme 1 to yield only water and oxygen. In these cases, the researchers mainly used co-immobilized enzymes. As will be discussed below, glucose oxidase can participate in many cascade reactions.
8.1. Co-immobilized biocatalysts or individually immobilized enzymes?
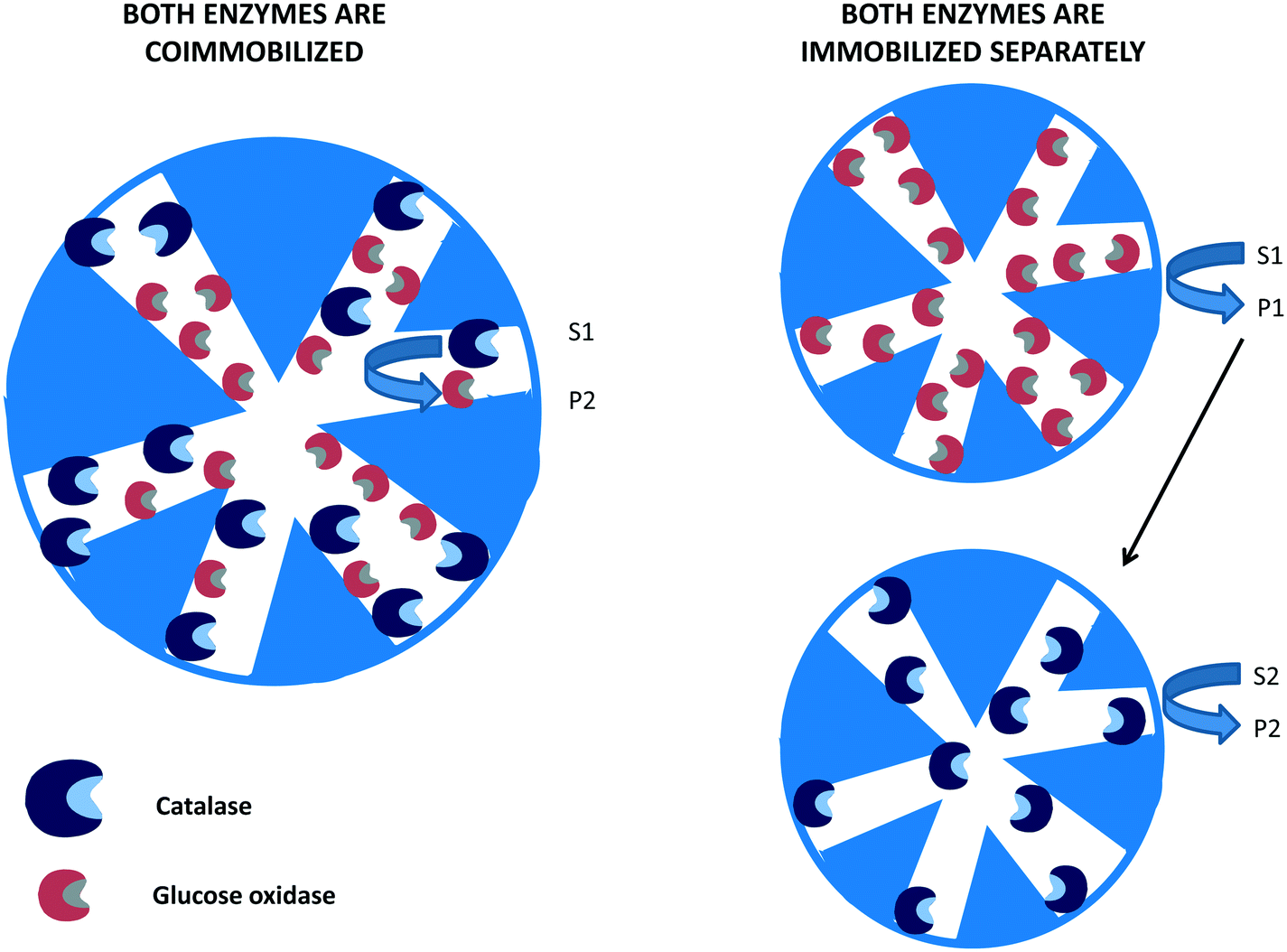
In many instances, if several enzymes are used in a cascade reaction, it is assumed that enzyme co-immobilization is the best alternative.215–221 However, co-immobilization, that is, the immobilization of two or more enzymes on the same particle, has advantages but also has some serious drawbacks that in many instances are not taken into account.222 Among the clearest advantages, we can note the shortening of the induction time for the second enzyme to act, which increases the initial rate of the global process105,106 (Fig. 8). This may be a key point if the intermediate product/products are unstable and can be destroyed if they are not rapidly modified.223–232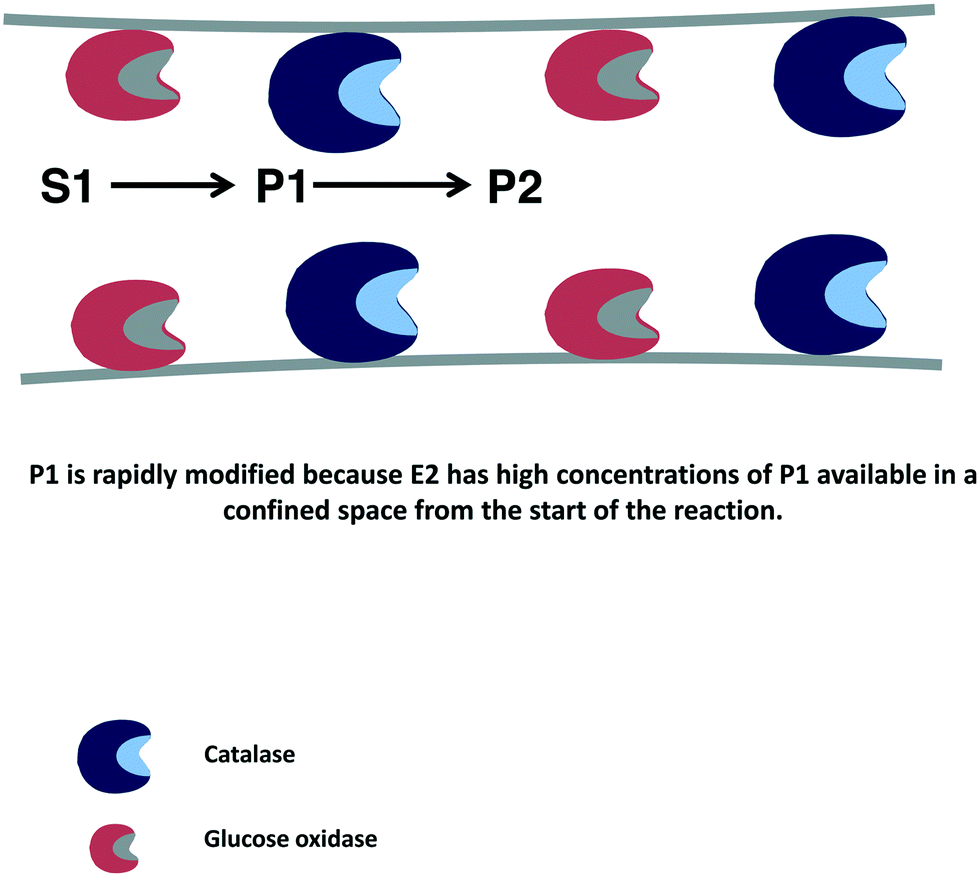 | ||
| Fig. 8 Reduction of time lapse in the destruction of hydrogen peroxide using catalase and glucose oxidase co-immobilized on porous supports. | ||
Co-immobilization may be also necessary if cofactors and enzymes are immobilized in porous supports, as the enzyme will only have access to the cofactors located in the same porous particles.233–236 On the negative side, it can be noted that in a co-immobilized biocatalyst, all enzymes should be immobilized using the same surface, and the stability of the least stable enzyme will determine the stability of the whole co-immobilized enzyme biocatalyst.106,237–239 Therefore, enzyme co-immobilization should be performed only after a careful evaluation of the pros and cons of individual immobilization versus enzyme co-immobilization.222
8.2. Examples of D-gluconic acid production using catalase and glucose oxidase
In a very old paper, a polarographic protocol to determine the activities of soluble catalase or catalase/glucose oxidase mixtures was designed based on the determination of hydrogen peroxide and oxygen.244 Later, the kinetics of the oxidation of glucose catalyzed by glucose oxidase in the presence of a large excess of catalase was studied both experimentally and theoretically.245 The mutarotation rate between the α and β forms was considered in these studies. The kinetics were adjusted to the Michaelis–Menten equation for glucose and oxygen, correlating the observed oxygen concentration profiles with those supplied by the model.245 In another paper, the authors utilized air-saturated solutions to study the kinetics of the oxygen consumption or the oxidation of glucose by glucose oxidase and the destruction of hydrogen peroxide to regenerate oxygen.246 The oxygen concentrations were determined by monitoring the phosphorescence decay of a Pd-phosphor in solution.
To analyze the adsorption rate of oxygen in a batch stirred tank reactor, the oxidation of glucose was studied using different loads of catalase and glucose oxidase.247 Pure oxygen was supplied to the D-glucose solution in the presence of active enzymes or using inactivated enzymes. In another paper, the oxidation of glucose to gluconic acid was performed in a membrane reactor.248 As catalysts, catalase and glucose oxidase were trapped between two semipermeable membranes of regenerated cellulose. The reaction mixture consisted of a glucose solution at pH 5.5 saturated with oxygen, which was continuously fed through one grooved section outside the membranes (liquid side). Oxygen was delivered at a pressure over 1 atm to the opposite grooved compartment (gas side). The studies were carried out under conditions where the oxygen transfer was a key process. It was concluded that the membrane permeation coefficient and oxygen pressure were the most important parameters influencing the reaction rate.248 In another publication, a commercial mixture of free glucose oxidase and catalase called hyderase was utilized to oxidize glucose.249 The kinetics studies agree with a Michaelis–Menten type reaction with two kinetic parameters, i.e., k1 denotes the reaction between the oxidized enzyme and glucose (the apparent activation energy was 49.6 kJ mol−1), and kc is related to a combination of the other reaction steps (the apparent activation energy was 26.7 kJ mol−1). The oxygen transfer enhancement in the oxidation of glucose by catalase/glucose oxidase was accomplished using an antifoam agent (KM-70), n-hexadecane or soybean oil as additives.250 Antifoam agents diminished the oxygen transfer rate; also, n-hexadecane or soybean oil (more significantly) decreased this parameter. This was correlated with the spreading coefficient of the oil on water. However, the combined use of antifoam agent and other additives increased the oxygen concentration. This was explained by the elimination of the antifoam-agent molecules away from the air–water interface. The enzyme kinetics remained unaltered by the addition of KM-70, n-hexadecane and/or soybean oil.250 A commercial pyranose 2-oxidase in combination with catalase was used to oxidize β-D-glucopyranose.251 The kinetic model of the reaction was ping-pong bi–bi type, and the authors also included the inactivation of the oxidase by hydrogen peroxide and the in situ hydrogen peroxide destruction catalyzed by catalase (with generalized Yano-Koya kinetics) in the model. Catalase reduced the oxidase inactivation; however, it also reduced the reaction rate.251
Some papers bear special interest, such as those where the authors modified free catalase and glucose oxidase to improve their performance while maintaining the enzymes in soluble form. For example, glutaraldehyde was utilized to produce soluble copolymerized catalase, glucose oxidase or catalase/glucose oxidase biocatalysts (when using glucose oxidase, bovine serum albumin was utilized as a feeder protein).252 These soluble aggregates exhibited better thermal stability than the untreated enzymes.
 | ||
| Fig. 9 Destruction of hydrogen peroxide by catalase in the oxidation of glucose catalyzed by glucose oxidase. | ||
The advantages of using co-immobilized catalase and glucose oxidases compared to glucose oxidase alone have been exemplified in many papers. For example, the enzymes were covalently immobilized in ultrafiltration membranes from acrylonitrile copolymer oxidized with hydrogen peroxide to obtain amide groups in the modified membranes.253 This system showed double the activity of glucose oxidase alone. Moreover, glucose oxidase without catalase was deactivated by hydrogen peroxide more rapidly than a co-immobilized biocatalyst. The enzymes were also immobilized onto an anion-exchange membrane reactor composed of low-density polyethylene modified with 4-vinylpiridine, then compared to co-immobilized catalase/glucose oxidase or glucose oxidase alone.27 The D-gluconic acid production was low using glucose oxidase alone. The amount of produced D-gluconic acid increased 30 fold if glucose oxidase was immobilized next to an anion-exchange membrane, as this permitted the selective removal of the D-gluconic acid and reduced the enzyme inhibition. Using the co-immobilized enzymes in this system, the oxidation yield reached a value of 95%.27
Moreover, in some publications, a real comparison between free, immobilized or co-immobilized enzymes is described. In an interesting paper, glucose oxidase and catalase, individually or co-immobilized in silanized nickel silica alumina activated with glutaraldehyde, were compared in the production of D-gluconic acid.254 The amount of product formed depended on the activity of both enzymes in the system. The oxidation efficiency increased as the ratio glucose oxidase/catalase decreased. Moreover, the efficiency increased when the activities of both enzymes increased even if the relationship between both enzymes was unaltered. The paper compared co-immobilized enzymes, individually immobilized enzymes and soluble enzymes. The results showed that the co-immobilized enzyme afforded the best results.254
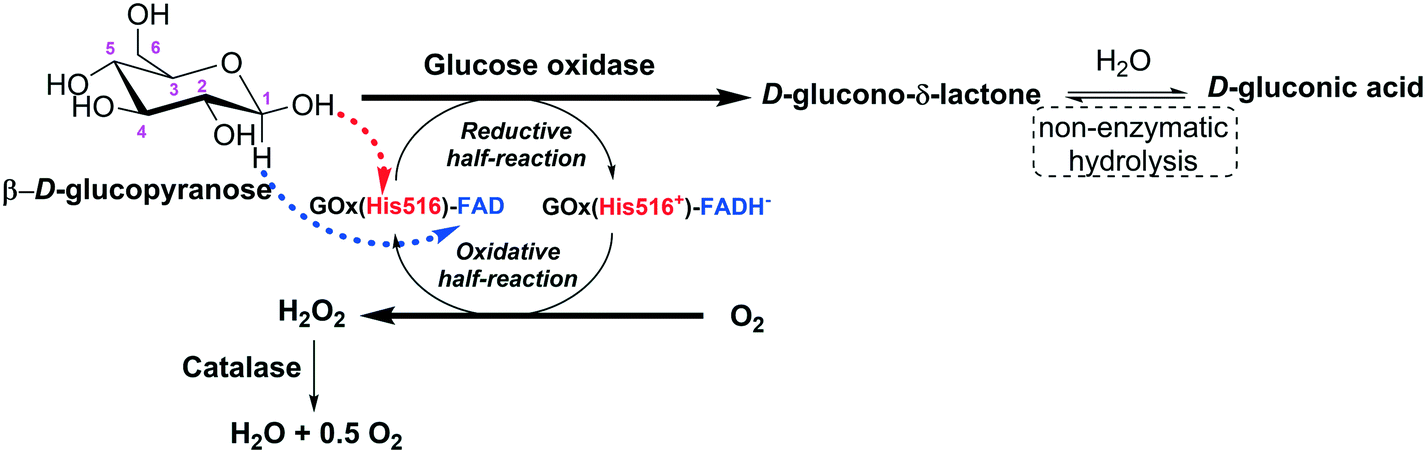
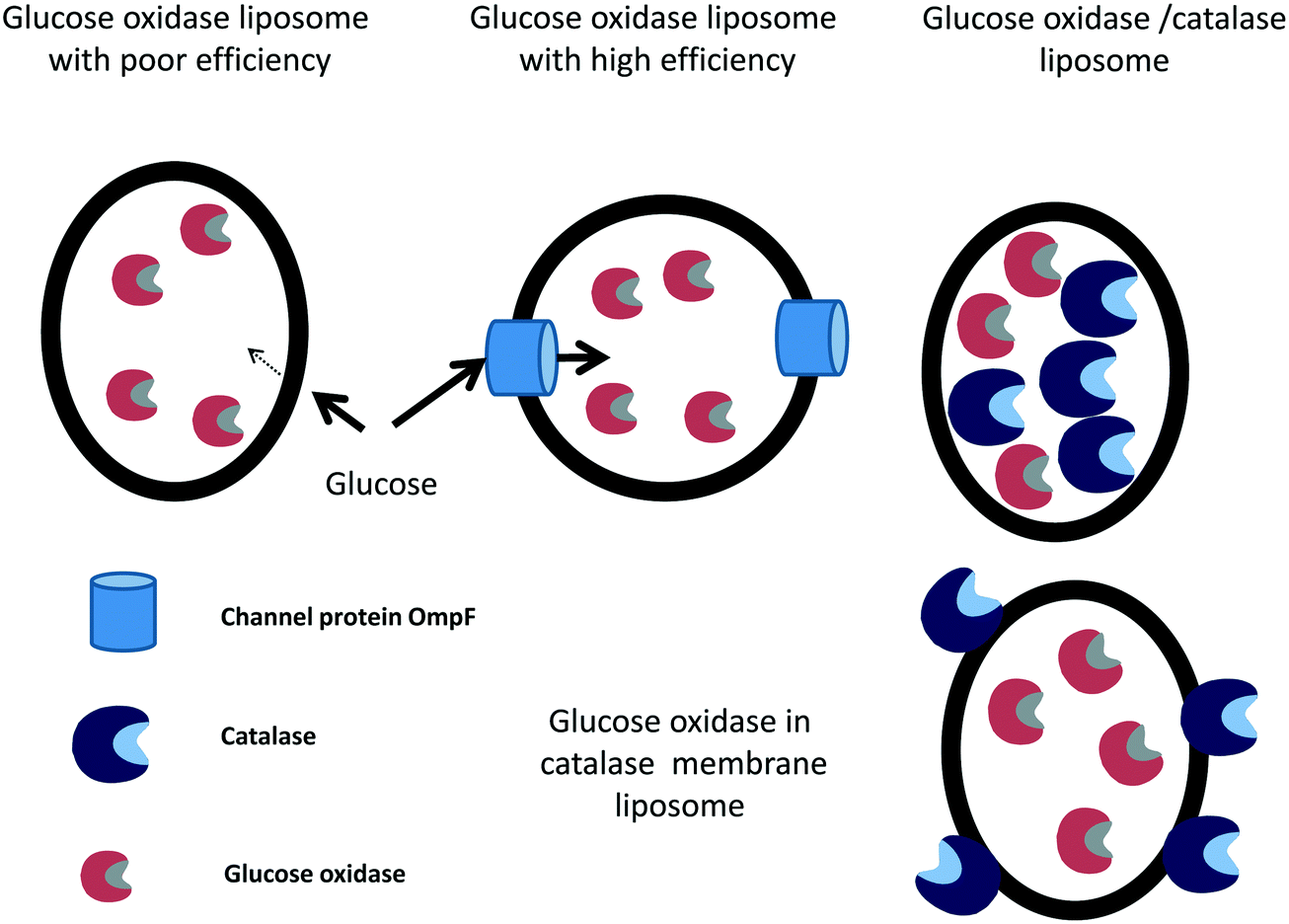
The efficiency of individually immobilized glucose oxidase-containing liposomes255 was improved by adding the channel protein OmpF from Escherichia coli to the liposome and co-immobilizing the glucose oxidase with catalase.256 OmpF increases the speed of entry of the glucose molecules into the liposome (Fig. 10). Both strategies increased the production of D-gluconic acid up to 17 times compared to the previous glucose oxidase, with no leakage of any of the enzymes. Then, the liposomes containing the enzymes and the transport protein were covalently immobilized in chitosan gel beads, improving the operational stability of the system.256 The same group later studied the stability of catalase immobilized in liposomes, without the membrane protein, adding hydrogen peroxide or co-immobilizing catalase and glucose oxidase and adding glucose and oxygen.257 A test tube and an external loop airlift bubble column were used as static liquid and circulating liquid flow systems, respectively. The immobilization of catalase inside the liposome reduced enzyme dissociation by increasing the catalase concentration, which increased the enzyme stability.258 Immobilized catalase was able to successively decompose 10 mM hydrogen peroxide, while the free enzyme lost activity with each hydrogen peroxide addition. This high stability of the liposome-catalase was explained by the diffusional limitation of the liposome towards hydrogen peroxide, which avoided exposure of the enzyme to the full concentration of this deleterious reagent.138 Using catalase and glucose oxidase and 10 mM of glucose in the static liquid in a test tube, the free catalase and immobilized catalase could be reused continuously because the low glucose oxidase activity (due to poor permeability of the liposome membrane to glucose) enabled the hydrogen peroxide to remain at low concentration. The D-gluconic acid production reaction rates were increased in the airlift. Much larger oxidation rates were observed because of an increase in the permeability of the liposome membrane to glucose in the gas–liquid two-phase flow. Using glucose oxidase immobilized in the liposomes and free catalase, the oxidation rate was lower than that using the co-immobilized system257 (Fig. 10). Later, catalase was conjugated to the membrane of the liposomes using 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(glutaryl), and this was utilized to trap different amounts of glucose oxidase259 (Fig. 10). After using the liposome for 75 h, scarce accumulation of hydrogen peroxide was appreciated. Utilizing liposome sub-lytic concentrations of cholate, the liposome permeability to glucose could be increased.259 The authors proposed the use of this catalase–liposome for other oxidase reactions. In a further research effort, these catalase–liposomes containing glucose oxidase were utilized in an external loop airlift bubble column to produce D-gluconic acid, and the results were compared with those achieved using a static liquid system.260 Using the latter, the activity was quite low because of the low permeability of the liposome membrane to the glucose molecules. In the airlift reactor, the reaction rates significantly increased because the reactor induced permeabilization of the liposomes. The structural stability of the liposomes in the airlift was dependent on its initial mean diameter: if it was 168.8 nm, the liposomes remained unaltered, while if it was 269.5 nm, the liposomes were partly disrupted and the catalase was deactivated.260
 | ||
| Fig. 10 Different liposomes developed for the oxidation of glucose. | ||
Another paper compares membrane containing glucose oxidase and catalase using independently immobilized and co-immobilized systems using a rotating disk electrode device.261 In this study, the intrinsic kinetic parameters and mass transfer (internal and external diffusion) of the different systems were analyzed. Enzyme immobilization decreases maximum enzyme intrinsic activity but does not affect Michaelis constants.261 In another paper, magnetic mesoporous bioactive glasses were modified with polyethylene glycol and were utilized to co-immobilize glucose oxidase and catalase using glutaraldehyde, giving better results than the individually immobilized enzymes.262 Another study shows the co-immobilization of previously chemically modified enzymes263 to enable the promotion of a self-assembling supramolecular structure. To achieve this goal, glucose oxidase and catalase were co-immobilized through the supramolecular recognition of β-cyclodextrin (attached to glucose oxidase) and adamantane (bound to catalase).264 This self-assembled multi-enzyme system was spontaneously formed immediately after mixing the two modified enzymes. The efficiency of the system improved compared to that of the free enzymes (activity by a 3.7 fold factor, kcat/KM by 7 times). This was related to the rapid destruction of hydrogen peroxide. The advantages of the co-immobilized biocatalyst increased as the glucose concentration increased.264
Curiously, one paper offers a result where enzyme co-immobilization was not the best solution. In that paper, the objective was to transform all glucose to D-gluconic acid; however, the aim was to facilitate the purification of xylose using free or immobilized catalase and glucose oxidase.265 In this paper, hydrogen peroxide was utilized as an oxygen source (without considering the negative effect of this reagent on the stability of the enzymes).138 To accomplish this, only immobilized glucose oxidase, co-immobilized glucose oxidase and catalase or soluble glucose oxidase and catalase were compared using a membrane bioreactor.265 The enzymes were immobilized on an ultrafiltration membrane. Under optimal conditions, the best productivity was obtained using the free enzymes because of the diffusion limitations found using co-immobilized enzymes and the poor performance of the individual immobilized catalyst of glucose oxidase.265
In other papers, the only objective was to develop strategies that permitted the co-immobilization of catalase and glucose oxidase without confirming the advantages/problems of this procedure.106,222 For example, glucose oxidase and catalase were co-immobilized via adsorption in controlled-pore titania particles.266 This yielded stable enzymes, and as expected, the authors found that catalase played a double role: it increased both the activity and stability of glucose oxidase. Other authors followed similar strategies and obtained similar results.267–269
Catalase and glucose oxidase were co-immobilized on a support of Ni-impregnated silica alumina, and the efficiency of the catalase as result of the activities of the two enzymes was assessed.270 The authors found that using a constant ratio of enzymes, the performance of the catalase increased when the global enzyme activities increased. The co-immobilized biocatalyst showed the maximum activity at pH 5.5, which corresponds to the optimal pH for glucose oxidase. This optimal pH was not observed using a system composed of individually immobilized enzymes or free enzymes.270 Catalase and glucose oxidase have been also immobilized in polyamide hollow fibers.271 The immobilization yield and final loading were improved if the fibers were coated with Cibacron Blue F3GA. Fluorescence spectrophotometry showed that the enzyme immobilization did not produce significant conformational changes.271 In other research, the enzymes were covalently co-immobilized onto magnesium silicate (florisil) by covalent coupling in the presence of glucose and oxygen (that is, the reaction produced D-gluconic acid and hydrogen peroxide).272 The activities of immobilized glucose oxidase and catalase reached a maximum upon the addition of glucose (15 and 20 mM glucose respectively). In another paper, this group analyzed the differences in the performance of co-immobilized biocatalysts that were sequentially and simultaneously co-immobilized at different enzyme activity ratios using the same support.273 The sequentially co-immobilized biocatalyst showed a higher efficiency than the simultaneously co-immobilized glucose oxidase–catalase. The individually immobilized glucose oxidase has 30% of the activity of the best co-immobilized biocatalyst. The improved activity and reusability of glucose oxidase of the co-immobilized biocatalyst was attributed to the protection afforded by the destruction of hydrogen peroxide by catalase and the generation of oxygen.273 The effect of the enzyme immobilization order was further studied. Glucose oxidase and catalase were sequentially co-immobilized in D301T resin to analyze the effects of the immobilization order and the amounts of enzymes in the final co-immobilized biocatalyst.274 The results were better when catalase was first immobilized (that is, immobilized in the external volume of the support).222 The treatment of the co-immobilized biocatalyst with glutaraldehyde, an efficient crosslinking reagent,275 improved the recovery activity and the operational stability of the co-immobilized biocatalyst (retaining over 85% of the initial activity after 10 reuses).274 However, in another paper, the results were completely different. This paper demonstrated that matching of the dimensions of the enzymes and the hierarchical pore sizes of the carriers activated with glutaraldehyde are critical to the success of immobilization processes.276 However, the simultaneous immobilization obtained in this case afforded better results than the sequential immobilization of the enzymes.
Catalase and glucose oxidase have been immobilized using non-porous glass beads coated with γ-aminopropyltriethoxysilane and polyethyleneimine activated with glutaraldehyde.277 Polyethyleneimine coating offered the best results. The concentrations of polyethyleneimine and glutaraldehyde, the glucose oxidase/catalase ratio, and the load of the beads were found to be relevant to the immobilization yields. Activation of carbon fiber by treatment with nitric acid or sulfuric acid afforded the best results. Using this support, the effects of the amounts of both enzymes, glutaraldehyde concentration and time of the glutaraldehyde treatment on the immobilization efficiency were studied. The optimal biocatalysts improved the storage and operational stabilities and enlarged the range of pH values and temperatures where the enzymes can be used. After 10 reaction cycles, more than 70% of the enzyme activity was recovered.278 In another study, the enzymes were also co-immobilized on carbon fiber and activated by following diverse support activation protocols.278 Recently, polyglycidyl methacrylate spheres were utilized to co-immobilize glucose oxidase and catalase.279 The support was activated with a combination of amination and plasma. Immobilization increases the optimal pH (from 5.5 to 7.5) and temperature (from 25 °C to 40 °C) so that 82% of the initial activity was maintained after three glucose oxidation batches; although this value was considered good by the authors, it is not high enough for industrial implementation of the biocatalyst.279
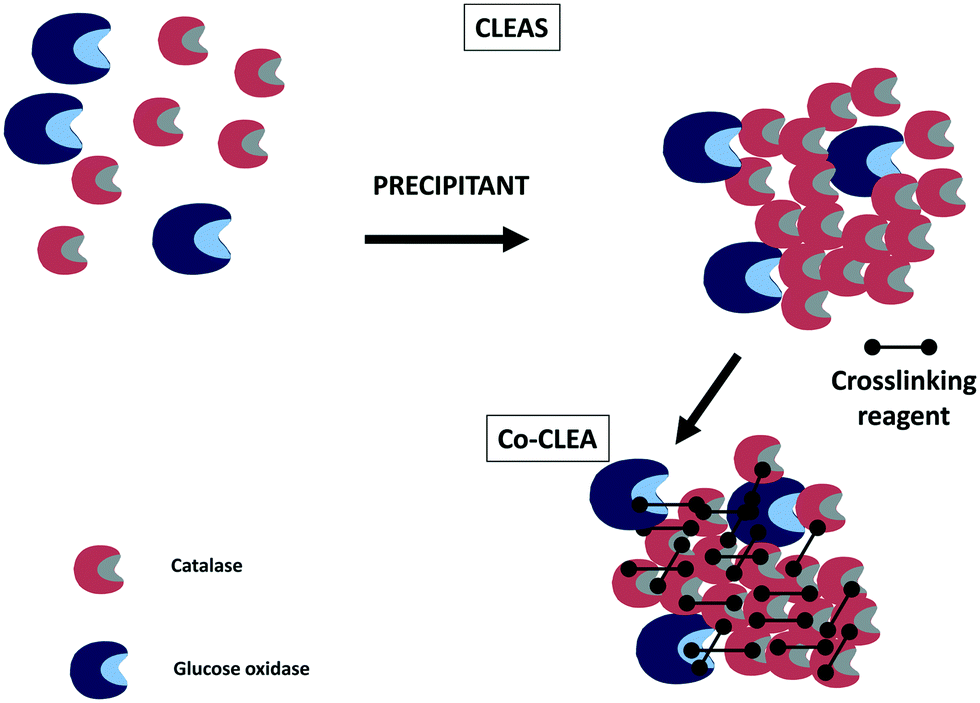
Crosslinked enzyme aggregates280–282 have been also utilized in some instances to co-immobilize catalase and glucose oxidase (Fig. 11). For example, the effects of the precipitant and crosslinking agents as well as the presence of a feeder protein were studied in the preparation of cross-linked aggregates of catalase and glucose oxidase.22 Differences in enzyme stabilities are a problem in enzyme co-immobilization;106,222 however, few papers demonstrate the stabilities of all the co-immobilized enzymes. This paper showed that the stabilities of both immobilized enzymes were very similar. Therefore, the difference in the stabilities of the co-immobilized enzymes was no longer a problem in this case. The biocatalyst enabled around 96% conversion of D-glucose into D-gluconic acid in a bubble column reactor.22
 | ||
| Fig. 11 Co-immobilization of glucose oxidase and catalase using crosslinking enzyme aggregate technology. | ||
Catalase and glucose oxidase have also been co-immobilized using the copolymer technique283 (Fig. 12). Catalase and glucose oxidase were crosslinked using genipin284 and subsequently employed for oxidizing glucose.20 Genipin-crosslinked glucose oxidase/catalase increased its activity 10-fold compared to the unmodified glucose oxidase/catalase mixture.
 | ||
| Fig. 12 Co-immobilization of glucose oxidase and catalase using copolymerization technology. | ||
In some instances, the co-immobilized catalase/glucose oxidase system is used to perform some kinetic studies, including substrate diffusional limitation problems, enzymes deactivation by hydrogen peroxide, etc. For example, the macro-kinetics of D-gluconic acid production was studied using co-immobilized glucose oxidase and catalase in continuous and batch reactions.285 The results showed that the effectiveness is improved using a co-immobilized biocatalyst. However, hydrogen peroxide caused the inactivation of both enzymes. This enzyme inactivation increases as the activity of glucose oxidase increases.285 Therefore, even when using co-immobilized enzymes, it is necessary to carefully design the biocatalyst to reduce the inactivation of both enzymes. Later, this research group performed a theoretical analysis of the interactions between the reaction and diffusion rates using this system.286 The efficiency of the glucose oxidase reaction depended on the concentrations of glucose and oxygen. They stated that external mass-transfer limitations may produce a change from glucose limitation to oxygen limitation. The predicted efficiency of the co-immobilized biocatalysts was higher than that for the uncoupled reaction; however, the main problem was enzyme deactivation. Similar results were found in another study, where a commercial cocktail containing glucose oxidase and catalase was co-immobilized on pumice and a titania carrier, then activated with a copolymer of phenylenediamine and glutaraldehyde.287 This co-immobilized biocatalyst was utilized in the production of D-gluconic acid in a differential-bed loop reactor. The kinetics of the reaction was first order regarding the oxygen concentration, suggesting severe external mass-transfer resistance in the range of the utilized flow rates. These results suggest that catalase is the first enzyme that becomes inactivated by hydrogen peroxide. Only by using a large excess of catalase can the inactivation of glucose oxidase be prevented.287
The problem of diffusional limitations of substrates was also studied using a co-immobilized biocatalyst in a flow micro-calorimetry study.288 To study the diffusivity of glucose, catalase and glucose oxidase were covalently co-immobilized in polyacrylonitrile membranes with different pore sizes and in a microfiltration polyamide membrane.289 As expected, the glucose diffusion increased when the pore size increased, and it also decreased when the enzyme membranes were inactivated.
In other research, using a continuous-flow stirred reactor coupled with a membrane and a continuous-flow reservoir containing hydrogen peroxide, evidence of periodic and aperiodic oscillations in an enzymatic system of glucose oxidase–catalase was obtained.290 This was accomplished by matching experimental data obtained under oscillatory instability with the stoichiometric restrictions of the mechanism formulated by considering the stability theory of the reaction networks, focusing on the catalase reaction. The model agreed with the experimentally achieved oscillatory dynamics, including apparently chaotic intermittent behavior.290
Another topic studied in this reaction system is enzyme inactivation; in many instances, it is promoted by hydrogen peroxide,138 while in others, it is caused by the dissociation of the monomers of the multimeric enzymes.258 This was the subject of a paper using a membrane containing both enzymes, using conditions where the inactivation was homogeneous throughout the membrane.291 The results pointed again towards hydrogen peroxide inactivation being the main reason for the decreased efficiency of the co-immobilized biocatalyst. In another paper, glucose oxidase and catalase were co-immobilized at optimal activity ratios on 2-amino-4-chloro-s-triazine-cellulose and acrolein/styrene copolymer activated with diaminohexane and glutaraldehyde and used in a recycling fluid-bed reactor to produce D-gluconic acid.292 The deactivation rates of the enzymes were first-order depending on the presence of hydrogen peroxide. Similar studies were performed using the enzyme co-immobilized in processed perlite, also using recycling fluid bed reactors.293 Other paper analyzed the inactivation of catalase and glucose oxidase co-immobilized in alginate beads in a continuous stirred tank reactor, proposing a general model of three-parameter irreversible deactivation.294
Thus, co-immobilization of catalase and glucose oxidase, if properly performed, is a successful example of the effects of enzyme co-immobilization on improving enzyme stability and activity because it is important to avoid any delay in the destruction of hydrogen peroxide.
9. Conversion of polysaccharides to D-gluconic acid
Glucose-based polysaccharides such as cellulose and starch have been used as substrates in cascade reactions using multienzymatic systems to obtain D-gluconic acid or its derivatives.9.1. Conversion of cellulose to D-gluconic acid
An interesting process involving an enzymatic cascade reaction is the conversion of cellulose to D-gluconic acid or its derivatives.295 The proposed enzymatic cascade reaction uses a combination of cellulases, β-glucosidase, glucose oxidase and catalase to produce D-gluconic acid from waste cellulosic material. This represents a five-enzyme cascade reaction because cellulase cocktails comprise endo and exoglucanases. In a first case, a DNA-guided approach was used to assemble the five-component enzyme co-immobilized biocatalyst for direct conversion of cellulose to D-gluconic acid and H2O2.296 The authors found that site-specific co-localization of β-glucosidase and glucose oxidase resulted in an improvement in peroxide production, highlighting the benefits of substrate channeling.The production of sodium gluconate from cellulose was also reported by a multi-enzymatic reaction.297,298 Firstly, a cellulase cocktail with high β-glucosidase activity was produced by a fed-batch process using Penicillium oxalicum I1-13 with simultaneous cellulose saccharification. Then, glucose oxidase and catalase were co-immobilized to produce sodium gluconate.297 Later, the same group tested the feasibility of a consolidated bioprocess for sodium gluconate production from cellulose. A recombinant strain named z19 was constructed from Penicillium oxalicum wild-type strain 114-2 for simultaneous expression of glucose oxidase and catalase from Aspergillus niger. The mutant maintained the cellulolytic ability of the wild-type strain and secreted certain amounts of glucose oxidase and catalase. Using cellulose (filter paper power) as the initial substrate, a total of 13.54 g L−1 sodium gluconate was obtained at the end of the fermentation.298
In a final example, cellulase and glucose oxidase were co-immobilized on graphene oxide to produce D-gluconic acid from carboxymethyl cellulose.299 The enzymes were immobilized by a step-by-step method. The graphene surface was activated with 2,4,6-trichlorotriazine followed by conjugation with cellulase (N-hydroxysuccinimide/1-ethyl-(3-dimethylaminopropyl) carbodiimide hydrochloride) and glutaraldehyde were used to activate the graphene residues and primary amino groups of cellulase, allowing glucose oxidase to be immobilized. The multi enzymatic biocatalyst could be reused for 7 cycles, and the conversion of cellulose to D-gluconic acid reached almost 64% in 2 h.299
9.2. Conversion of starch to D-gluconic acid
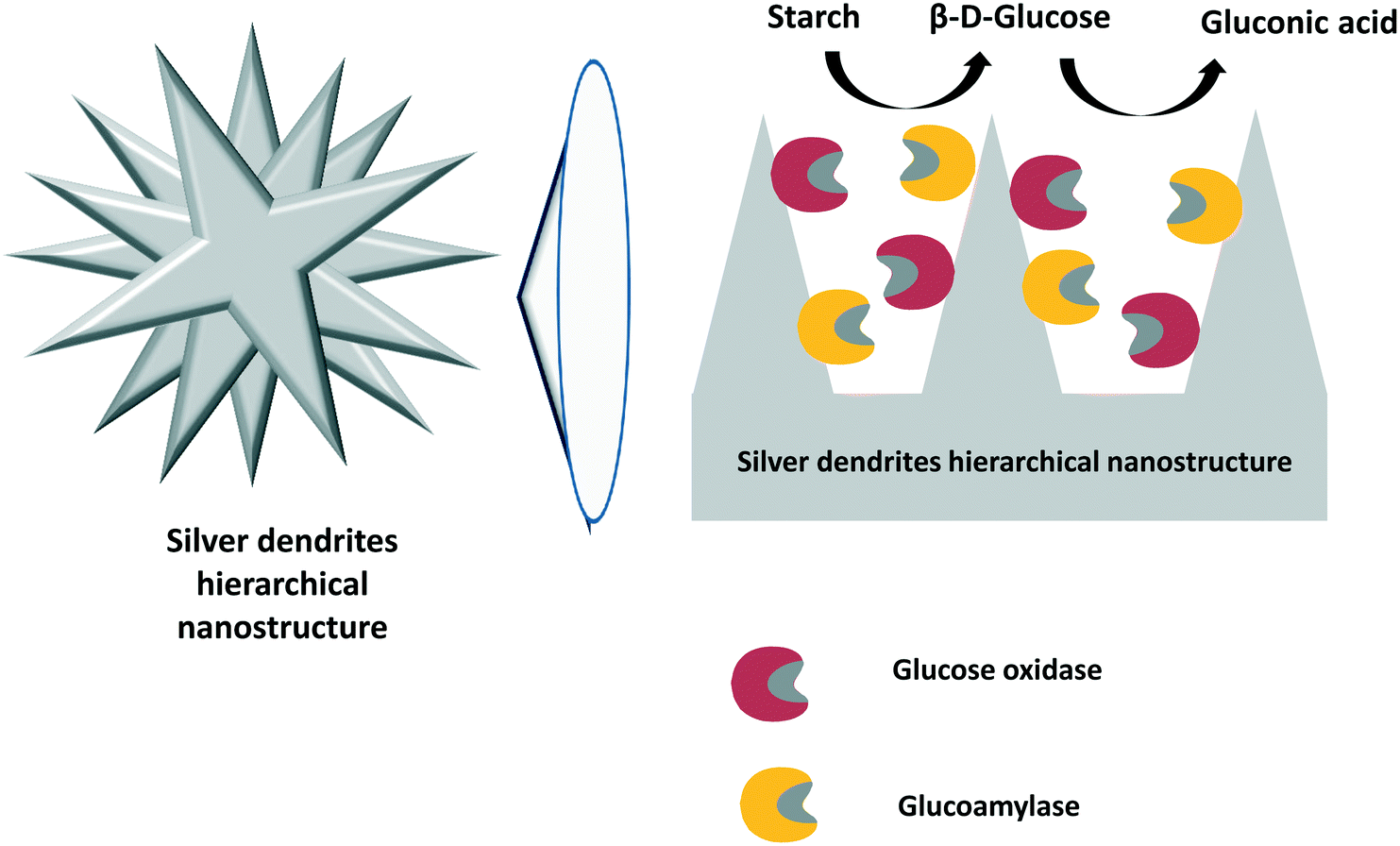
In a similar way, starch can be converted to D-gluconic acid by a cascade reaction involving its initial hydrolysis by a glucoamylase and further conversion of glucose to D-gluconic acid by glucose oxidase. Curiously, these examples do not use catalase to control the deleterious effects of hydrogen peroxide on the enzymes. In the first example, both glucoamylase and glucose oxidase were assembled in an ultrafilter by layer-by-layer adsorption using polyethyleneimine as a coupling poly-cation.300 Reaction products and unreacted starch were readily separated by filtration when the substrate solution was passed through these multi-enzyme films. Later, the same enzymes were co-immobilized by hydrophobic adsorption on chemically reduced graphene oxide.301 This multi-enzyme system was used as a biocatalyst in the starch-to-gluconic acid reaction in one pot, and it yielded 82% D-gluconic acid in 2 h.In an elegant example, glucoamylase and glucose oxidase were immobilized on silver dendrites with a hierarchical nanostructure302 (Fig. 13). The obtained biocatalyst was employed for the one-pot transformation of starch to D-gluconic acid, and the final product was obtained in high yield. Moreover, the co-immobilized biocatalyst exhibited high stability and reusability. The authors claimed that their results showed that the co-immobilization of enzymes on hierarchical structures could be extended to other biocatalytic cascade reactions.302
 | ||
| Fig. 13 Co-immobilization of glucose oxidase and glucoamylase using silver dendrite hierarchical nanostructures. | ||
9.3. Conversion of disaccharides to D-gluconic acid
Several disaccharides, such as sucrose, maltose and lactose, have been used for D-gluconic acid production by combining a disaccharide hydrolytic enzyme to release glucose molecules and glucose oxidase to convert glucose to D-gluconic acid. In some cases, the objective was the preparation of a biosensor to detect the respective disaccharide; in these cases, D-gluconic acid was not the final target. Biosensors were prepared to determine sucrose,303 maltose304 and lactose.305,306 The biosensors were assembled by co-immobilizing glucose oxidase with α-glucosidase, invertase or β-galactosidase for detection of maltose, sucrose and lactose, respectively. A special case is the assembly of an integrated nanodevice with bi-enzymatic cascade control for on-command cargo release. The nanocarrier is based on Au-mesoporous silica Janus nanoparticles capped at the mesoporous face with benzimidazole/β-cyclodextrin-glucose oxidase pH-sensitive gate-like ensembles and functionalized with invertase on the gold face. The rationale for this delivery mechanism is based on the invertase-mediated hydrolysis of sucrose to yield glucose, which is further transformed into D-gluconic acid by glucose oxidase, causing disruption of the pH-sensitive supramolecular gates at the Janus nanoparticles. This enzyme-powered device was successfully employed in the autonomous and on-demand delivery of doxorubicin in HeLa cancer treatment.307Some studies reported the use of maltose and sucrose as substrates for D-gluconic acid production in batches using catalase to destroy the hydrogen peroxide21,308,309 and using fed-batch,310 continuous311 and membrane-continuous309,310 processes. In general, all processes were satisfactory, reaching high yields of conversion of maltose and sucrose to D-gluconic acid.
10. Use of D-gluconic acid or glucose oxidase to alter the inhibition of other enzymes
Glucose oxidase has been used in other enzymatic reactions with the intention of removing glucose from the medium or to accumulate D-gluconic acid. This may alter the main enzyme inhibition mechanism (increasing or decreasing it depending on whether the inhibition is caused by D-gluconic acid). One example where the objective was to increase the imbibition of one enzyme is the production of cellobiose from cellulose. When the cellulase cocktail hydrolyzes cellulose, cellobiose is released synergistically by endo- and exoglucanases. The cellobiose is then hydrolyzed to glucose by β-glucosidase.312 Blocking β-glucosidase activity increased the accumulation of cellobiose. In this sense, D-gluconic acid inhibits β-glucosidase activity in that cellulase system. Thus, directly adding D-gluconic acid or adding glucose oxidase to produce in situD-gluconic acid by glucose oxidation resulted in accumulation of cellobiose.313,314 This indicates that both exo- and endoglucanases can act synergistically without the hydrolytic action of β-glucosidase in this cellulase cocktail during cellulose hydrolysis in the presence of D-gluconic acid as an inhibitor.Another interesting application of glucose oxidase, in this case to avoid enzyme inhibition, is in the synthesis of oligosaccharides from sucrose or lactose to produce fructooligosaccharides (FOS) or galactooligosaccharides (GOS). In the transglycosylation reaction, the galactosyl or the fructosyl moiety (from lactose or fructose, respectively) are transferred to another sugar molecule instead of water, resulting in oligosaccharide formation.315,316 Glucose is the by-product in both reactions and inhibits the transglycosylating activity. The reaction rate increases if glucose is removed; therefore, a higher yield of oligosaccharides can be obtained. Thus, the combination of glucose oxidase with β-fructofuranosidase or β-galactosidase has been reported to improve FOS and GOS yields by the removal of glucose from the reaction medium.317–320 The subsequent D-gluconic acid in the final product was easily removed by cationic ion exchange resin.
In another paper, trehalose was obtained from starch by a multi-enzymatic system. Initially, starch was hydrolyzed to maltose by amylases and pullulanase.321 The obtained high-maltose syrup was further converted into trehalose by a one-step enzymatic process using trehalose synthase as a biocatalyst.322 The final reaction product mixture contains glucose, trehalose and unreacted maltose. This mixture can be treated with glucoamylase and glucose oxidase to facilitate the separation of trehalose by sequential conversion of maltose to glucose and glucose to D-gluconic acid.323–325 After separation by ionic exchange, trehalose can be successfully purified, crystallized and identified with 92.6% purity and a recovery yield of 94.2%.325
11. Other applications of glucose oxidase
The glucose oxidase reaction, as an oxygen-depleting system, producer of D-gluconic acid or hydrogen peroxide, and pH-decreasing agent in combination (or not) with catalase, has many other applications in addition to the production of D-gluconic acid. We will end this review by summarizing some of these applications, excluding the field of biosensors, which has been the subject of many recent specific reviews.114–11711.1. Improvement of food quality
Glucose oxidase has found many applications in food quality improvement.326 The conversion of glucose to D-gluconic acid using glucose oxidase–catalase was used to prevent the deteriorative Maillard reaction.327 Albumen desugarization was used as a model reaction, and the treatment enabled reduction of the Maillard reaction by 40%.Glucose oxidase has been used to improve the organoleptic properties of many food products. For example, the treatment of white grape juice with glucose oxidase–catalase improved its color stability; moreover, the aroma and taste (non-flavonoid phenolic content) were enhanced.328 In another study, glucose oxidase/catalase, ascorbic acid and peroxidase systems were compared in the removal of oxygen to reduce the browning of Golden Delicious apple and Kaiser pear purées during storage.329 Glucose oxidase was the most effective treatment to reduce the dissolved oxygen content in the apple and pear purées and exhibited an ability to modulate the non-enzymatic browning during fruit processing and purée storage.
Another application of this enzyme in food quality management is in the stabilization of oil/water emulsions versus oxidation.330 Glucose oxidase/catalase was employed to stabilize salad dressing emulsions and measure peroxide values.331 The desired oxygen concentration decrease was improved at pH near neutrality using a glucose concentration of 0.5% and a temperature near 50 °C. The oxygen concentration also decreased with increasing amount of enzyme. In another study, the glucose oxidase/catalase system showed an antioxidant effect in mayonnaises containing different amounts of fish oil when stored at low temperature.332 The amount of enzyme required to slow the lipid oxidation increased when the lipid oxidation rate increased. This effect was achieved by the reduction of oxygen in the medium. Moreover, glucose oxidase/catalase was explored as an antioxidant in a food model consisting of an oil/water emulsion (a model of mayonnaises and other high-fat dressings) packed in an oxygen-permeable plastic bag.333 The results showed that the speed of lipid oxidation is inversely proportional to a term containing the glucose oxidase concentration. Later, the oxidative stability of a model salad dressing was studied by measuring the volatile compounds of sample bottles and peroxide values.334 Upon adding 1.0% glucose, glucose oxidase/catalase acted as an anti- or pro-oxidant depending on the enzyme concentrations. At moderate concentrations of enzymes, the volatile compounds and peroxide values decreased, that is, the biocatalyst behaved as an antioxidant. When using higher enzyme concentrations, the enzymes functioned as pro-oxidants.334 Obviously, the situation could change if the ratio of glucose oxidase to catalase were altered; however, the researchers did not alter it.
The enzyme was also used to improve the preservation of fish. Glucose oxidase and catalase were used in artificial seawater bearing 4% w/v glucose for on-board shrimp preservation.335 The system decreased browning by ∼80% and also inhibited the release of ammonia and total volatile nitrogen. Due to the increase in nitrogen compounds, it was necessary to replace the enzyme solution after 14 days.335
Preservation of eggs was also enhanced using glucose oxidase. Thus, glucose oxidase at different concentrations of glucose was evaluated as a bactericide agent in liquid whole egg.336 This treatment could fully eliminate Salmonella enteritidis, Micrococcus luteus and Bacillus cereus and presented bacteriostatic activity against Pseudomonas fluorescens. When the efficacy of this strategy was assayed on the natural contamination flora of liquid egg, it was found that this treatment prolonged the lag phase by 6 days. The authors explained this effect by the production of hydrogen peroxide.336 Other uses related to the egg industry involve improving the organoleptic properties of processed eggs. Catalase and glucose oxidase were co-immobilized on polyethylenimine-dressed cotton cloth by ion exchange and then treated with glutaraldehyde.337 The biocatalyst was utilized for the de-sugaring of egg mélange in a batch reactor to produce a spray-dried, sugar-free egg powder.
11.2. Reduction of sugar in juice to reduce the alcohol concentration after fermentation
Wine with reduced alcohol content may have healthy effects. To this end, without increasing the amount of remaining glucose, some authors have proposed to treat the grape juice prior to fermentation (Fig. 14). In this way, glucose oxidase was utilized to optimize the glucose content of grape juice to produce low-alcohol wines.338 The acidity of grape juice slowed the reaction and determined the conversion of glucose by glucose oxidase; however, after optimization, an 87% reduction in glucose was obtained (the D-gluconic acid concentration in the grape juice was 73 g l−1). Later, the same research group characterized the final properties of the wine obtained by reducing the glucose content (to around 20%).339 The ethanol concentrations of the produced wine ranged between 6.3% and 6.5%. Part of the formed D-gluconic acid precipitated, reducing the high D-gluconic acid concentration in the juice; the pH was fairly similar to that of the standard wine (with alcohol content over 13%), although the titrated acidity was higher, very likely due to the presence of more quinone and oxidizable phenolic compounds.339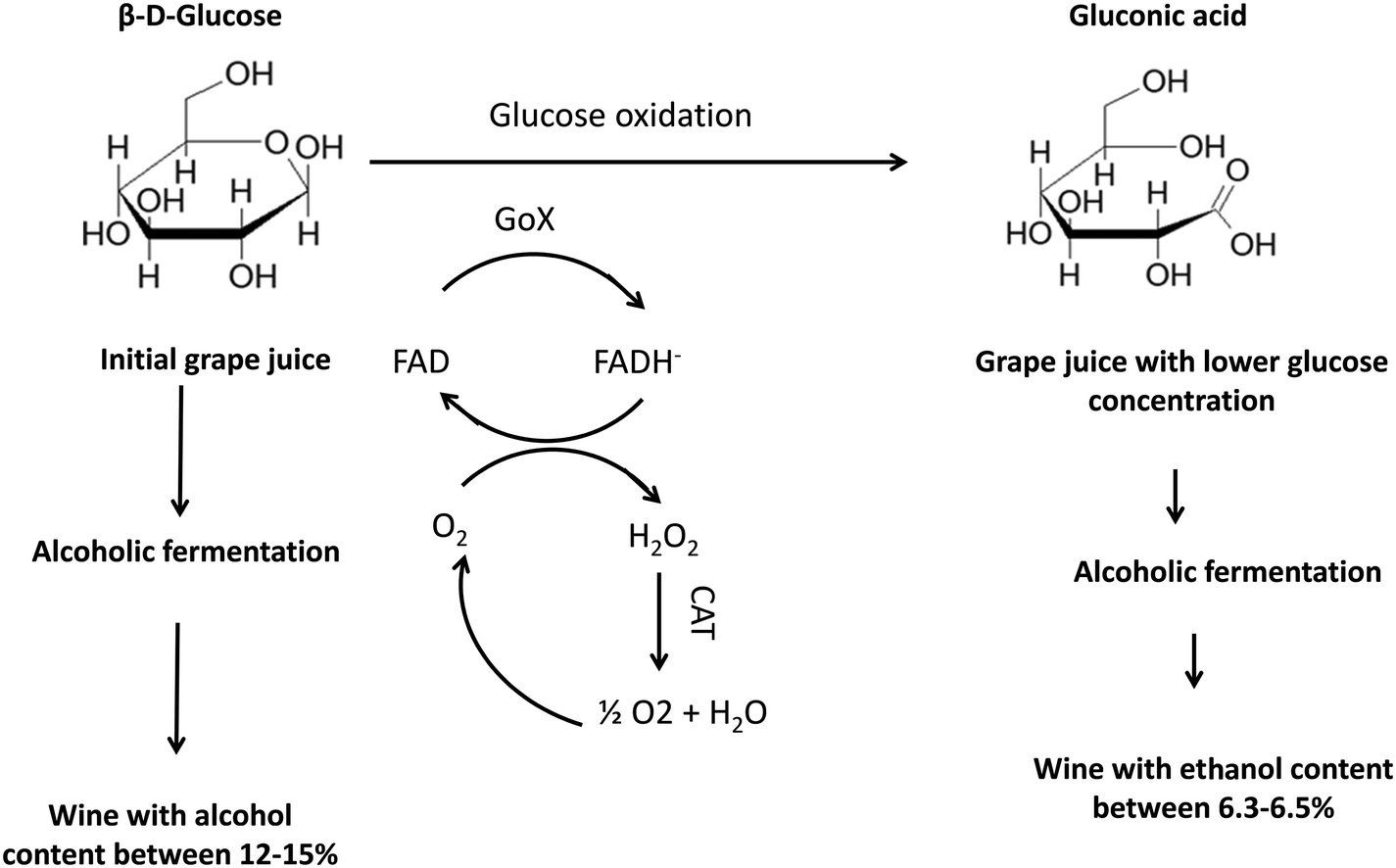 | ||
| Fig. 14 Reduction of glucose content in grape juice to reduce the final alcohol concentration after fermentation using a glucose oxidase–catalase biocatalyst. | ||
Later, the Aspergillus niger gene encoding glucose oxidase was cloned in Saccharomyces cerevisiae. This strategy, with glucose oxidase present during the fermentation, decreased the sugar content and, thus, the final alcohol concentration.340 This strategy also inhibited the growth of wine spoilage organisms (e.g., lactic acid or acetic acid bacteria). The results showed a reduction in ethanol content of 1.8–2.0%. A more recent paper discussed the problem of the increasing glucose levels in grapes due to global warming.341 In this instance, glucose oxidase was used in combination with catalase in its free form. Aeration influenced the wine, and the researchers obtained a 2% reduction in ethanol content.341
11.3. Use of glucose oxidase to facilitate monosaccharide purification
Glucose is presented in many monosaccharide mixtures and can complicate the purification of the most valuable sugars.342–345 The modification of glucose with glucose oxidase facilitates separation and the achievement of a higher added value product. For example, using a membrane system, the separation of glucose and xylose, which are very similar, is quite difficult.346 The full oxidation of glucose by glucose oxidase in the presence of catalase simplifies the separation by using the anionic nature of D-gluconic acid as a distinctive feature. In this way, xylose and D-gluconic acid can be easily separated.346 This was later utilized in biorefinery liquors obtained from hydrothermal pretreatment of wheat straw, corn stover and Miscanthus stalks.347 In another research effort, glucose oxidase and catalase were co-immobilized using a sol–gel protocol on silica inverse opals and utilized to oxidize glucose (with a yield of 98.97%) from commercial isomalto-oligosaccharide preparations.348 The operational stability requires improvement, as less than 80% of the enzyme activity remained after 6 reuses.11.4. Application of glucose oxidase in textile industries
Glucose oxidase has been used for the production of hydrogen peroxide required for bleaching cellulose fibers, e.g., using glucose during desizing.349 Also, the produced D-gluconic acid plays some role in this bleaching because of its outstanding chelating properties for metal ions. This process is milder from an environmental point of view than the common addition of hydrogen peroxide, and it has already been reviewed.35011.5. Glucose oxidase in fuel cells
The use of glucose oxidase in fuel cells was proposed a long time ago. In this contribution, it was suggested that because glucose is present in the bloodstream, glucose oxidase should be a good catalyst for supplying the electrical energy needed for the operation of devices implanted in the human body.351 Later, a glucose/O2 biofuel cell was built using a surface glucose oxidase monolayer as an anode, where the cathode presented a cytochrome c/cytochrome oxidase complex.352 Glucose oxidase oxidized glucose to D-gluconic acid, whereas the enzymes in the cathode reduced O2 to water. The appropriate genetic engineering of the proteins permitted a correct assembly on the electrodes, improving the power of the biofuel cell.352More recently, thiophene-3-acetic acid and the conducting copolymer 3-methylthiopene were used to modify a carbon paper electrode on which glucose oxidase was covalently immobilized.353 The modification of the paper increased the surface area, permitting higher enzyme loading and a high speed of electron transfer between the electrode and the electron mediator (p-benzoquinone). In another paper, carboxylated multi-walled carbon nanotubes were used to coat a polyester-supported screen-printed carbon paste electrode; then, glucose oxidase, 2,5-dihydroxybenzaldehyde, bovine serum albumin, and glutaraldehyde were used to crosslink the composite.354 This composite increased the electron-shuttling process and exhibited an electrocatalytic effect on D-gluconic acid, which enabled the removal of more electrons from a glucose molecule. The authors showed that this glucose biofuel cell coupled to a laccase/ABTS cathode generated electric power of 45 μW cm−2 in the presence of 1 M glucose at 37 °C.354 In another paper, glucose oxidase and laccase were suspended in a membraneless, single-chamber cell.355 Nickel mesh was the oxidative current collector and a carbon-based air electrode enclosed in an acrylic casing was the reductive current collector. The anolyte was glucose oxidase, 200 mM glucose and 3.8 mM FAD, while the catholyte contained laccase enzyme and 216 μM syringaldazine (Fig. 15). An open circuit voltage at pH 5.0 of around 960 mV was obtained, and the electrode could sustain a continuous discharge current of 30 μA for about 31.75 h.355 Later, glucose oxidase was entrapped in Nafion and multiwalled carbon nanotubes as an anode to build an EFC device that employed sulfonated polyether ketone as an electrolyte membrane.356 In another paper, glucose oxidase was immobilized on cross-linked carbon-chitosan, which functioned as a conductive enzymatic membrane for electrode applications in biofuel cells.357 The conductivity was increased by modifying the matrix with ferrocene derivatives. The stability of the enzyme was increased by glutaraldehyde treatment. This device produced a power output of up to 32.8 μW cm−2 depending on the redox mediator utilized. Another study demonstrated a cell chamber with oxygen in the cathode cell and glucose in the anode cell; a Nafion membrane was used as an ion transporter.358 The prototype cell chamber used NaCl in the cathode cell and glucose oxidase in the anodic chamber. Du Toit and collaborators immobilized glucose oxidase onto highly porous gold electrodes and assayed it as an anode in a glucose/O2 enzymatic biofuel cell, leading to a peak power density of 6 μW·cm−2 at a potential of 0.2 V.359 Glucose oxidase was immobilized on ZnO nanoparticles used to coat electrodes, using ferrocenyl π-stacked into a supramolecular architecture as a mediator.360 This provides a highly efficient architecture and delivers a catalytic current for glucose oxidation. This electrode was incorporated in a glucose/air Nafion-based biofuel cell under “air breathing” conditions at room temperature. A conventional platinum–carbon electrode was utilized as a cathode.360 The stability of glucose oxidase was increased by immobilization in Nafion, and it maintained its activity for 120 days.361 This biocatalyst was bound into a carbon cloth electrode and assembled in a EFC fed with glucose. Nafion permitted the process of electron transfer to the electrode. A micro-fluidic channel plate in which the glucose solution was driven into the fuel cell by capillary force was designed and fabricated using glucose oxidase.362 The fuel cell proposal comprised both end plates and a micro-fluidic flow channel plate. The results showed a lower output power than that obtained using an active pumping method; however, in the long term, a constant voltage suggested that the self-pumping fluid enzymatic fuel cell may function stably under a continuous fuel supply.362 Glucose oxidase was immobilized on a double-layered hydroxide of NiAl electrode and utilized to carry out oxidation of glucose present in blood for energy conversion to be employed as a self-feeding electrode in Lab-on-a-Chip devices.363
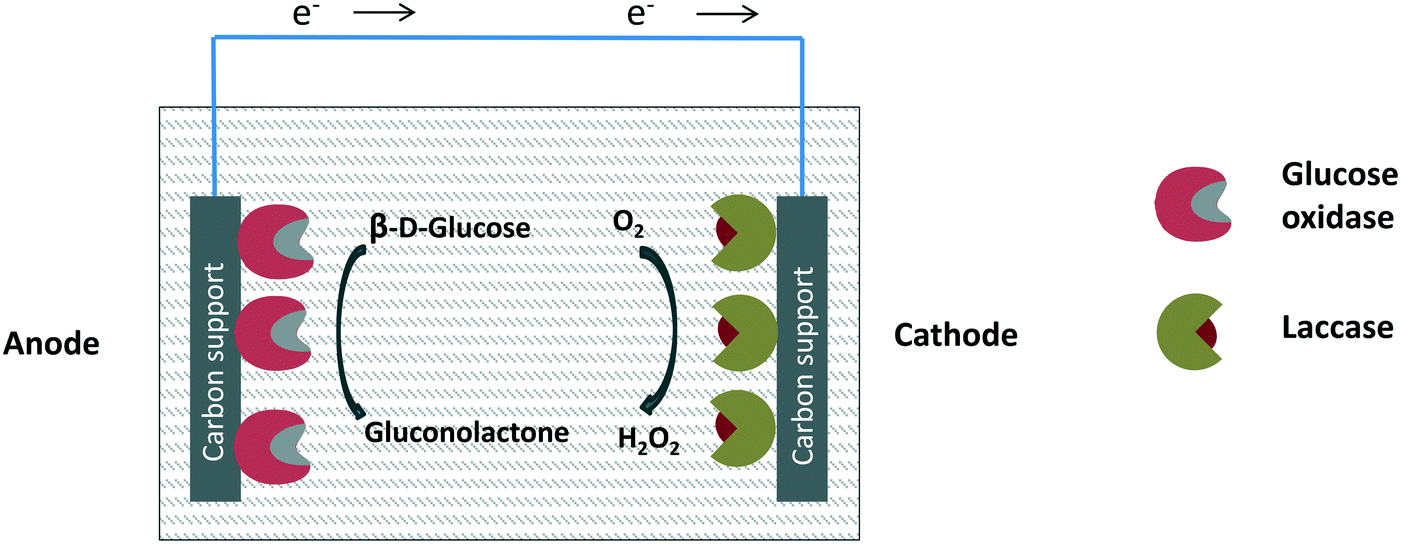 | ||
| Fig. 15 Co-immobilization of glucose oxidase and laccase using carbon nanotubes as an electrode for fuel cell design. | ||
Glucose oxidase does not always produce the best results. A paper suggested that if laccase is used in a cathode, the negative effect of the hydrogen peroxide on the laccase stability would make it advisable to change glucose oxidase to FAD-dependent glucose dehydrogenase (an O2-insensitive enzyme).364 The cathode was rapidly inactivated using glucose oxidase; meanwhile, its stability was much higher when using the new enzyme. The same group, in another communication, compared both enzymes in an enzymatic biological fuel cell using the cathode with direct electrocatalytic bilirubin oxidase using anthracene-modified multi-walled carbon nanotubes.365 A glucose oxidase cell produced in an open circuit at pH 6.5 using 200 mM glucose obtained higher maximum current densities and maximum power densities. In this case, the hydrogen peroxide does not affect the short term stability of the cathode; however, it has negative effects in the long term.365 Another research group compared the same enzymes again when using multi-walled carbon nanotubes to produce enzymatic electrodes.366 Glucose oxidase provided a higher current density at 0.12 V but a lower current density versus Ag/AgCl in synthetic medium; however, at physiologically relevant glucose concentrations (5 mM), flavin-dependent glucose dehydrogenase afforded higher current densities.
Another study compared the performance of glucose oxidase and aldose dehydrogenase as oxidizing agents in an anode, while ABTS and a fungal laccase were employed for reduction of oxygen at the cathode.367 Cells based on filter paper coated with conducting carbon inks, enzymes and mediators were used. The highest power production, although with low stability, was obtained using aldose dehydrogenase anodes mediated by an osmium complex.367
Glucose oxidase and catalase were co-immobilized on carbon nanotubes coated with polyethyleneimine,368 and their performance in glucose oxidation was analyzed369 (Fig. 16). Utilizing the current density peak derived from the FAD redox reaction, the authors determined the Michaelis–Menten constants, electron transfer rate, and sensitivity. The electrical performance using a membraneless glucose biofuel cell was determined; a maximum power density of 180.8 ± 22.3 μW cm−2 was produced, which is the highest value among MPDs obtained by adoption of catalysts.369
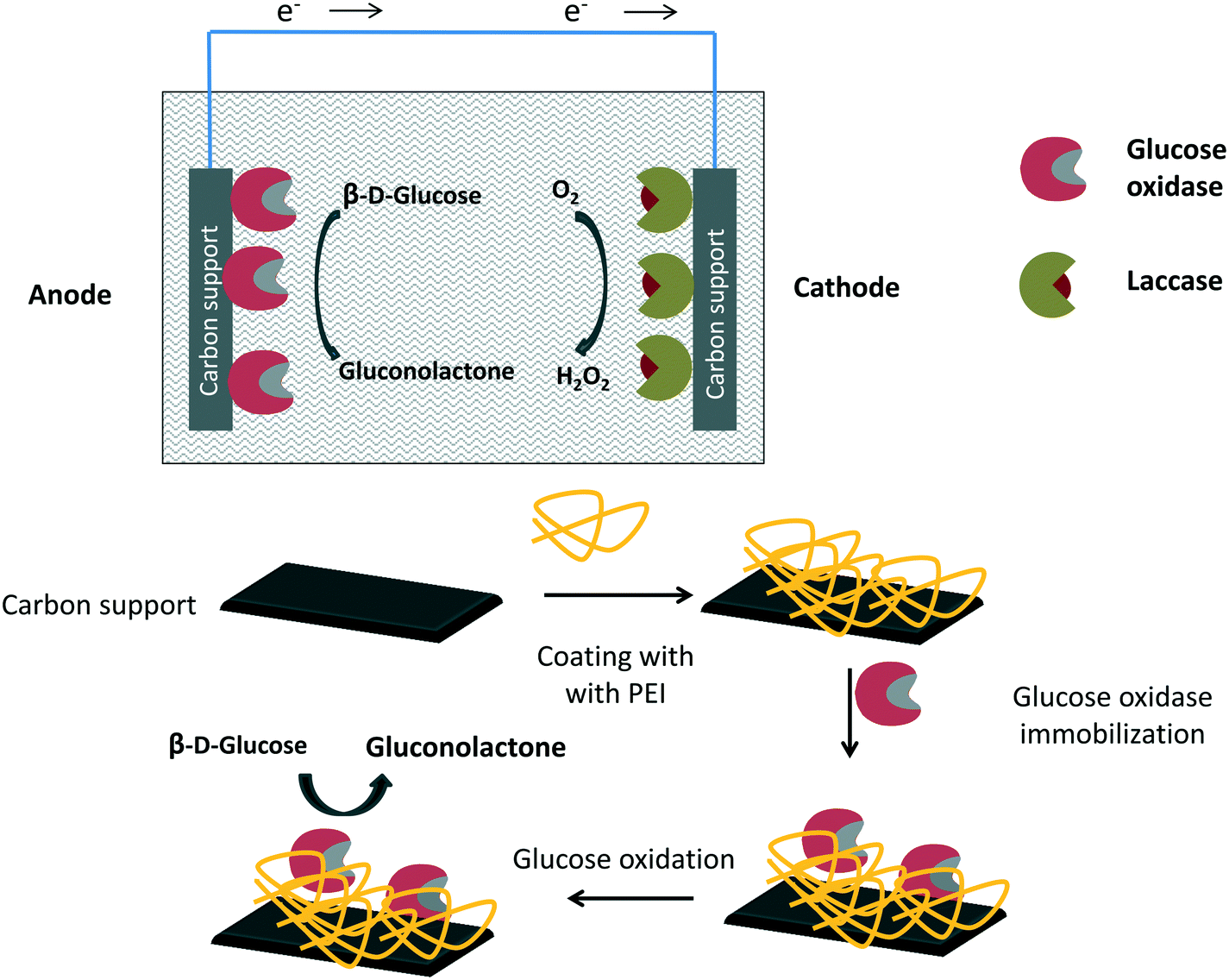 | ||
| Fig. 16 Co-immobilization of glucose oxidase and laccase using carbon coated with polyethylenimine as a support. | ||
11.6. Use of glucose oxidase and glucose as antimicrobial agents
Hydrogen peroxide has strong antibacterial effects; however, it may also produce some side-damage.370–372 Therefore, the production of hydrogen peroxide by glucose oxidase has been used in some papers to eliminate or reduce the infectivity of some pathogens, to reduce bacterial infections and to facilitate destruction of bacteria in a milder way than by the direct use of hydrogen peroxide. Glucose oxidase from the caterpillar Helicoverpa zea was used to decrease the infectivity of Pseudomonas aeruginosa and Serratia marcescens when supplemented with glucose.373 In another paper, an ultrathin two-dimensional metal–organic framework (two-dimensional 2Cu-TCPP(Fe)) nano-sheet with peroxidase-like activity was used to immobilize glucose oxidase.374 In this way, glucose could be converted to hydrogen peroxide and D-gluconic acid, decreasing the damaging side effects that directly result from high concentrations of hydrogen peroxide. Moreover, D-gluconic acid decreased the pH to 3.0–4.0, promoting the activation of the peroxidase-like activity of the metal–organic framework and producing a hydroxyl radical that presented antibacterial capacity.37411.7. Use of glucose oxidase and glucose as an antitumoral cell agent
Glucose oxidase has different applications in cancer research. It has been used in different methods for the detection of cancer biomarkers.375 Even more interestingly, various cancer healing approaches have been developed based on the use of this enzyme. This has been recently reviewed;375 as a short summary, the positive effects may be derived from the consumption of glucose (which produces cancer-starvation therapy); the reduction of oxygen (which increases tumor hypoxia, the so-called hypoxia-activated therapy); and the generation of D-gluconic acid, which produces medium acidification and can be used to release a drug that is contained in a pH-responsive particle, as discussed in section 3.1. for insulin release; the production of hydrogen peroxide induces oxidative stress in tumors and can produce toxic hydroxyl radicals that can kill tumor cells upon exposure to light irradiation or via the Fenton reaction.376 Glucose oxidase can also be combined with hypoxia-activated prodrugs, Fenton or photosensitizer reagents, or other enzymes to generate multi-modal synergistic cancer therapies based on cancer oxidation therapy, hypoxia-activated therapy, starvation therapy, photodynamic therapy, and/or photothermal therapy.375We will mention a few recent contributions in this regard. In the area of tumor biosensors, this technology is based on the fact that cancer cells undergo a transition from oxidative phosphorylation to anaerobic glycolytic metabolism, which provides proof of cancer progression. In an interesting study, nano-pipettes were developed to determine glucose concentrations in individual cells.377 These devices were used to covalently immobilize glucose oxidase, followed by measuring the change in impedance generated by the decrease in the medium pH value. The nanopipettes could measure this parameter at different times in the cells, as this change did not affect their viability.377
In the area of cancer therapy, one example of drug-controlled release using glucose oxidase oxidation of metabolic glucose is the use of anti-PD-1 as a target molecule.378,379 To achieve this goal, a microneedle was fabricated using hyaluronic acid and with pH-sensitive dextran nanoparticles that encapsulate anti-PD-1 and glucose oxidase.380 When glucose oxidase converts glucose into D-gluconic acid and the pH decreases, the dextran nanoparticles dissociated and released anti-PD-1 more effectively than from a microneedle without this pH-trigged release or employing the free antibody, using B16F10 mouse melanoma as a model system. In another paper, glucose oxidase was immobilized on a polymer nano-gel, and the biocatalyst was employed for synergistic cancer-starving and oxidation therapy.381 This immobilized biocatalyst showed high anti-melanoma efficacy without detectable systemic toxicity, whereas the free enzyme was nonfunctional and systemically toxic. In other research, glucose oxidase was trapped in hollow mesoporous organo-silica nanoparticles combined with L-Arg.382 Hydrogen peroxide at acidic pH produces NO from Arg. In this way, this system starves the tumor by eliminating glucose in the tumor. On the other hand, the decrease in the pH and the presence of hydrogen peroxide produced a remarkable H2O2–NO cooperative anticancer effect with minimal adverse effects.382,383
In another approximation, glucose oxidase was immobilized on stealth liposomes to reduce oxygen and glucose availability for tumor growth. Intravenous injection permitted the effective tumor retention of these liposomes, and this exhausts the glucose and the oxygen inside the tumor (Fig. 10). The release of cytotoxic hydrogen peroxide contributes to the anticancer effect of this strategy. If this system is combined with stealth liposomes loaded with banoxantrone dihydrochloride (a hypoxia-activated pro-anticancer drug),384–386 synergistic inhibition of tumor growth is accomplished.
In a final example, glucose oxidase was immobilized on Fe3O4 functionalized with polypyrrole to achieve photothermal-enhanced, tumor microenvironment-specific sequential nano-catalytic tumor therapy and diagnostic imaging-guided cancer therapy.387 When the glucose oxidase consumed intra-tumoral glucose, the hydrogen peroxide concentration increased, and Fe3O4 catalyzed the transformation of hydrogen peroxide into very toxic hydroxy radicals that promote cancer-cell death. The high photo-thermal-conversion efficiency of the poly-pyrrole increased the local tumor temperature, improving the metal catalytic activity. All these synergetic processes promoted great anticancer effects with minimal side effects.387
11.8. Unexpected uses of glucose oxidase
Some uses of glucose oxidase are very different from the usual applications in food, medicinal or analytical applications. However, all utilize the consumption of oxygen, the properties of D-gluconic acid or hydrogen peroxide or the decrease in the pH promoted during the oxidation of D-glucose to D-gluconic acid. For example, glucose oxidase and catalase were co-immobilized on nano-porous SiO2, and the biocatalyst was utilized for the removal of oxygen dissolved in water in order to avoid boiler corrosion.388 In another application, a glucose oxidase/catalase system using glucose as a substrate was employed to generate D-gluconic acid; this system was used to neutralize and precipitate hydrous tin oxide powders under mild conditions.389 A high concentration of the enzyme was required to obtain full precipitation. The hydrous tin oxide, which had a large surface area, was obtained when a high concentration of enzymes was used as a solution. The precipitates were converted to tin oxide via calcination at temperatures >400 °C.389One interesting use of glucose oxidase is its use in the decontamination of water using both D-gluconic acid and hydrogen peroxide features. The Fenton reaction is an oxidation technology utilized for the oxidation of pollutants.390–392 Some researchers propose the use of glucose and glucose oxidase (both in free and immobilized forms) to generate on-site D-gluconic acid, which acts as a chelating agent, and hydrogen peroxide, which acts as an oxidant.393 This enables the de-chlorination of 2,4,6-trichlorophenol under very mild conditions, first by fully transforming glucose in hydrogen peroxide and D-gluconic acid, then by performing the Fenton reaction.
12. Conclusion
The production of D-gluconic acid using glucose oxidase is a reaction that is receiving ongoing interest because of the many applications of this compound and the possibility of obtaining it from fully renewable sources. Moreover, this reaction is used as a model for many enzyme reaction design problems. It is a bi-substrate reaction (glucose and oxygen) and is a good model for oxygen diffusional limitation problems. Therefore, many studies use this reaction to analyze the possibilities of addressing these limitations. Even more interestingly from a basic point of view, due to its side product (hydrogen peroxide), this system is adequate for studying the effects of the inactivation of the enzyme by this deleterious agent in different reactor operation models. One of the logical evolutions in the development of this reaction is the design, perhaps using directed evolution, of glucose oxidases with high stability in high concentrations of hydrogen peroxide. The current solution to this problem is the destruction of this deleterious reagent; among the different solutions, the preferred one is the use of catalase. Therefore, this reaction is one of the most studied enzymatic cascade reactions. As in this case, the critical point is to avoid the exposition of glucose oxidase to high concentrations of hydrogen peroxide; enzyme co-immobilization has been the preferred solution. However, in many papers, this advantage has not been examined. It is granted that enzyme co-immobilization must be the best solution while ignoring some of the problems that this co-immobilization may cause. Although many methods of catalase/glucose oxidase co-immobilization have been developed, the stabilities of the two immobilized enzymes have been analyzed in only a few, and no research has attempted to solve the problems derived from the co-immobilization of two enzymes with very different stabilities.Glucose oxidase has also been co-immobilized with other enzymes, such as gluconolactonase; it has been utilized in many other enzymatic cascade reactions, in some instances involving even a half dozen enzymes, to achieve the one-pot transformation of polysaccharides to D-gluconic acid. In these reactions, the role of glucose oxidase/catalase has changed from simply providing D-gluconic acid to facilitating the purification of valuable saccharides by eliminating glucose and producing ionic compounds. Therefore, the production of D-gluconic acid using glucose oxidase is one of the successful examples involving a wide range of enzymatic cascade reactions, in some cases expanding the concept of combi-enzymes.394 With the current focus on bio-refineries as a solution to environmental problems (involving lignocellulosic materials), the necessity for the chemical industry to use renewable sources to substitute current petroleum chemistry, and the green chemistry requirements arising from societal demand, the number of processes, in many cases cascade processes, where glucose oxidase may play relevant roles should even increase in the immediate future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully recognize the financial support from Ministerio de Ciencia e Innovación-Spanish Government and FEDER funds (project number CTQ2017-86170-R, RTI2018-095291-B-I00, MAT2017-87579-R) and Generalitat Valenciana (PROMETEO/2018/076). DC thank to Ministerio de Ciencia e Innovacion-Spanish Government by a FPI. PWT thanks to the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001.References
- M. E. Corkins, S. Wilson, J.-C. Cocuron, A. P. Alonso and A. J. Bird, J. Biol. Chem., 2017, 292, 13823–13832 CrossRef CAS PubMed.
- B. C. Mulukutla, A. Yongky, T. Le, D. G. Mashek and W.-S. Hu, Trends Biotechnol., 2016, 34, 638–651 CrossRef CAS PubMed.
- E.-J. Kim, Y.-J. Cho, W.-H. Chung and J.-H. Roe, FEBS J., 2020, 287, 878–896 CrossRef CAS PubMed.
- X. Chen, K. Schreiber, J. Appel, A. Makowka, B. Fähnrich, M. Roettger, M. R. Hajirezaei, F. D. Sönnichsen, P. Schönheit, W. F. Martin and K. Gutekunst, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5441–5446 CrossRef CAS PubMed.
- N. Rohatgi, T. K. Nielsen, S. P. Bjørn, I. Axelsson, G. Paglia, B. G. Voldborg, B. O. Palsson and Ó. Rolfsson, PLoS One, 2014, 9, e98760 CrossRef PubMed.
- S. Ramachandran, S. Nair, C. Larroche and A. Pandey, in Current Developments in Biotechnology and Bioengineering, ed. A. Pandey, N. Negi and C. R. Soccol, Elsevier, Amsterdam, 2017, pp. 577–599 Search PubMed.
- P. Pal, R. Kumar and S. Banerjee, Chem. Eng. Process., 2016, 104, 160–171 CrossRef CAS.
- S. Ramachandran, P. Fontanille, A. Pandey and C. Larroche, Food Technol. Biotechnol., 2006, 44(2), 185–195 CAS.
- H. Hustede, H.-J. Haberstroh and E. Schinzig, in ULLMANN'S Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2000, pp. 37–44 Search PubMed.
- R. O. Marshall, E. R. Kooi and G. M. Moffett, Science, 1957, 125, 648–649 CrossRef CAS PubMed.
- A. M. Cañete-Rodríguez, I. M. Santos-Dueñas, J. E. Jiménez-Hornero, A. Ehrenreich, W. Liebl and I. García-García, Process Biochem., 2016, 51, 1891–1903 CrossRef.
- R. H. Blom, V. F. Pfeifer, A. J. Moyer, D. H. TraufRer, H. F. Conway, C. K. Crocker, R. E. Farison and D. V. Hannibal, Ind. Eng. Chem., 1952, 44, 435–440 CrossRef CAS.
- J. Ziffer, A. S. Gaffuey, S. Rothenberg and T. J. Cairney,British Pat., 1249347, 1971 Search PubMed.
- V. Leskovac, S. Trivić, G. Wohlfahrt, J. Kandrač and D. Peričin, Int. J. Biochem. Cell Biol., 2005, 37, 731–750 CrossRef CAS PubMed.
- S. B. Bankar, M. V. Bule, R. S. Singhal and L. Ananthanarayan, Biotechnol. Adv., 2009, 27, 489–501 CrossRef CAS PubMed.
- G. Wohlfahrt, S. Witt, J. Hendle, D. Schomburg, H. M. Kalisz and H. J. Hecht, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 969–977 CrossRef CAS PubMed.
- P.-R. Kommoju, Z. Chen, R. C. Bruckner, F. S. Mathews and M. S. Jorns, Biochemistry, 2011, 50, 5521–5534 CrossRef CAS PubMed.
- H. J. Hecht, H. M. Kalisz, J. Hendle, R. D. Schmid and D. Schomburg, J. Mol. Biol., 1993, 229, 153–172 CrossRef CAS PubMed.
- M. K. Dubey, A. Zehra, M. Aamir, M. Meena, L. Ahirwal, S. Singh, S. Shukla, R. S. Upadhyay, R. Bueno-Mari and V. K. Bajpai, Front Microbiol, 2017, 8, 1–22 Search PubMed.
- C. Cui, H. Chen, B. Chen and T. Tan, Appl. Biochem. Biotechnol., 2017, 181, 526–535 CrossRef CAS PubMed.
- A. C. O. Mafra, F. F. Furlan, A. C. Badino and P. W. Tardioli, Bioprocess Biosyst. Eng., 2015, 38, 671–680 CrossRef CAS PubMed.
- A. C. O. Mafra, L. G. Ulrich, J. F. Kornecki, R. Fernandez-Lafuente, P. W. Tardioli and M. P. A. Ribeiro, Catalysts, 2019, 9, 657 CrossRef CAS.
- X. Shen, M. Yang, C. Cui and H. Cao, Colloids Surf., A, 2019, 568, 411–418 CrossRef CAS.
- R. Jaquish, A. K. Reilly, B. P. Lawson, E. Golikova, A. M. Sulman, B. D. Stein, N. V. Lakina, O. P. Tkachenko, E. M. Sulman, V. G. Matveeva and L. M. Bronstein, Int. J. Biol. Macromol., 2018, 120, 896–905 CrossRef CAS PubMed.
- D. Pečar, D. Vasić-Rački and A. Vrsalović Presečki, Chem. Biochem. Eng. Q., 2018, 32, 511–522 CrossRef.
- E. J. Tomotani and M. Vitolo, Enzyme Microb. Technol., 2007, 40, 1020–1025 CrossRef CAS.
- T. Godjevargova, R. Dayal and S. Turmanova, Macromol. Biosci., 2004, 4, 950–956 CrossRef CAS PubMed.
- G. Richter and H. Heinecker, Starch/Staerke, 1979, 31, 418–422 CrossRef CAS.
- M. Kriaa, I. Hammami, M. Sahnoun, M. C. Azebou, M. A. Triki and R. Kammoun, Bioprocess Biosyst. Eng., 2015, 38, 2155–2166 CrossRef CAS PubMed.
- M. Bucekova, I. Valachova, L. Kohutova, E. Prochazka, J. Klaudiny and J. Majtan, Naturwissenschaften, 2014, 101, 661–670 CrossRef CAS PubMed.
- C. M. Wong, K. H. Wong and X. D. Chen, Appl. Microbiol. Biotechnol., 2008, 78, 927–938 CrossRef CAS PubMed.
- A. I. Schepartz and M. H. Subers, Biochim. Biophys. Acta, Spec. Sect. Enzymol. Subj., 1964, 85, 228–237 CAS.
- B. A. Hassan, M. A. Jebor and Z. M. Ali, Int. J. Pharm. Qual. Assur., 2018, 9, 273–281 Search PubMed.
- R. Mikhailova, A. G. Lobanok, Z. F. Shishko and M. I. Yasenko, Mikol. Fitopatol., 1998, 32, 60–61 Search PubMed.
- M. Petruccioli, M. Ceccarelli and F. Federici, World J. Microbiol. Biotechnol., 1993, 9, 77–79 CrossRef CAS PubMed.
- T. Nakamatsu, T. Akamatsu, R. Miyajima and I. Shiio, Agric. Biol. Chem., 1975, 39, 1803–1811 CAS.
- K. Jagathy, K. Kalpana and S. Rajeshkumar, Res. J. Pharm. Technol., 2017, 10, 1924 CrossRef.
- T. Konishi, T. Aoshima, F. Mizuhashi, S. S. H. Choi and A. Roberts, Regul. Toxicol. Pharmacol., 2013, 66, 13–23 CrossRef CAS PubMed.
- R. G. Bodade, C. N. Khobragade and S. Arfeen, Eng. Life Sci., 2010, 10, 35–39 CrossRef CAS.
- H. N. Bhatti and N. Saleem, Food Technol. Biotechnol., 2009, 47, 331–335 CAS.
- S. Sabir, H. N. Bhatti, M. A. Zia and M. A. Sheikh, Food Technol. Biotechnol., 2007, 45, 443–446 CAS.
- T. V. Semashko, R. V. Mikhailova and A. G. Lobanok, Microbiology, 2004, 73, 286–291 CAS.
- Z. I. Pavlovskaya, R. Mikhailova and I. V. Moroz, Mikol. Fitopatol., 2004, 38, 77–82 CAS.
- S. Manivannan and K. Kathiresan, Res. J. Microbiol., 2007, 2, 294–298 CrossRef CAS.
- M. Petruccioli, P. Piccioni and F. Federici, Enzyme Microb. Technol., 1997, 21, 458–462 CrossRef CAS.
- T. Tu, Y. Wang, H. Huang, Y. Wang, X. Jiang, Z. Wang, B. Yao and H. Luo, Food Chem., 2019, 281, 163–170 CrossRef CAS PubMed.
- I. Haq, A. Nawaz and A. Rehman, Pak. J. Bot., 2015, 47, 329–332 CAS.
- A. A. Khattab and W. A. Bazaraa, J. Ind. Microbiol. Biotechnol., 2005, 32, 289–294 CrossRef CAS PubMed.
- R. P. Kona, N. Qureshi and J. S. Pai, Bioresour. Technol., 2001, 78, 123–126 CrossRef CAS PubMed.
- A. Rothberg, J. Weegar, R. von Schalien, K. Fagervik, M. Rydström and K. Lind, Bioprocess Eng., 1999, 21, 307–312 CAS.
- T.-H. Li and T.-L. Chen, J. Ferment. Bioeng., 1994, 78, 298–303 CrossRef CAS.
- F. B. Cor Witteveen, P. van de Vondervoort, K. Swart and J. Visser, Appl. Microbiol. Biotechnol., 1990, 33, 683–686 CrossRef.
- J. Markwell, L. G. Frakes, E. C. Brott, J. Osterman and F. W. Wagner, Appl. Microbiol. Biotechnol., 1989, 30, 166–169 CrossRef CAS.
- Z. Fencl and M. Novák, Folia Microbiol., 1969, 14, 314–321 CrossRef CAS PubMed.
- S. Nakamura and Y. Ogura, J. Biochem., 1968, 63, 308–316 CrossRef CAS PubMed.
- J. Pásková and V. Munk, Folia Microbiol., 1963, 8, 215–220 CrossRef PubMed.
- V. Munk, J. Pásková and J. Hanus, Folia Microbiol., 1963, 8, 203–214 CrossRef CAS PubMed.
- G. Kovačević, M. Blažić, B. Draganić, R. Ostafe, M. Gavrović-Jankulović, R. Fischer and R. Prodanović, Mol. Biotechnol., 2014, 56, 305–311 CrossRef PubMed.
- M. A. Farid, E. A. Ghoneimy, M. A. El-Khawaga, A. Negm-Eldein and G. E. A. Awad, Ann. Microbiol., 2013, 63, 523–531 CrossRef CAS.
- R. Samreen, A. Z. Muhammad, A. S. Munir and I. Tehreema, Afr. J. Biotechnol., 2011, 10, 14522–14533 CrossRef.
- S. Khurshid, M. A. Kashmiri, Z. Quershi and W. Ahmad, Afr. J. Biotechnol., 2011, 10, 1674–1678 CAS.
- C. Yang, C. Li and T. Tan, Beijing Huagong Daxue Xuebao, Ziran Kexueban, 2011, 38, 97–100 CAS.
- S. B. Bankar, M. V. Bule, R. S. Singhal and L. Ananthanarayan, Food Bioprocess Technol., 2009, 2, 344–352 CrossRef CAS.
- M. H. Sarrafzadeh and A. R. Jafari, Pak. J. Biotechnol., 2008, 5, 11–20 Search PubMed.
- O. V. Singh, Appl. Biochem. Biotechnol., 2006, 135, 43–57 CrossRef CAS PubMed.
- J. Ge, X. Jiang, W. Liu, Y. Wang, H. Huang, Y. Bai, X. Su, B. Yao and H. Luo, Food Chem., 2020, 310, 125970 CrossRef PubMed.
- A. Bredenkamp, H. Velankar, W. H. van Zyl and J. F. Görgens, Yeast, 2010, 27, 849–860 CrossRef CAS PubMed.
- A. A. N. Gunny, D. Arbain and L. Sithamparam, Pak. J. Biol. Sci., 2013, 16, 960–964 CrossRef CAS PubMed.
- V. Silano, C. Bolognesi, L. Castle, K. Chipman, J.-P. Cravedi, P. Fowler, R. Franz, K. Grob, R. Gürtler, T. Husøy, S. Kärenlampi, W. Mennes, M. R. Milana, K. Pfaff, G. Riviere, J. Srinivasan, M. D. F. Tavares Poças, C. Tlustos, D. Wölfle, H. Zorn, A. Chesson, B. Glandorf, L. Herman, K.-D. Jany, F. Marcon, A. Penninks, A. Smith, H. van Loveren, D. Želježic, M. Andryszkiewicz, Y. Liu, A. Rossi and K.-H. Engel, EFSA J., 2018, 16(7), e05319 Search PubMed.
- L. Yang, M. Lübeck and P. S. Lübeck, AMB Express, 2014, 4, 54 CrossRef PubMed.
- R. Luque, M. Orejas, N. I. Perotti, D. Ramón and M. E. Lucca, J. Appl. Microbiol., 2004, 97, 332–337 CrossRef CAS PubMed.
- H. Xiao-Yue, M. Ri-Jian, X. De-Feng, Y. Ki-Ying, S. Li-Jun and W. Ya-Ling, Zhongguo Shengwu Zhipinxue Zazhi, 2018, 31, 1268–1273 Search PubMed.
- O. E. Bankefa, M. Wang, T. Zhu and Y. Li, Bioresour. Bioprocess., 2018, 5, 25 CrossRef.
- X. Liu, J. Zhang, J. Chen and G. Du, Shipin Yu Shengwu Jishu Xuebao, 2018, 37, 1233–1241 Search PubMed.
- F. Khadivi Derakshan, F. Darvishi, M. Dezfulian and C. Madzak, Mol. Biotechnol., 2017, 59, 307–314 CrossRef CAS PubMed.
- M. Zakhartsev and C. Momeu, J. Chromatogr., B, 2007, 858, 151–158 CrossRef CAS PubMed.
- K. Kojima, W. Tsugawa and K. Sode, Biochem. Biophys. Res. Commun., 2001, 282, 21–27 CrossRef CAS PubMed.
- A. Kapat, J.-K. Jung and Y.-H. Park, Biotechnol. Lett., 1998, 20, 319–323 CrossRef CAS.
- A. Kapat, J.-K. Jung and Y.-H. Park, Biotechnol. Lett., 1998, 20, 683–686 CrossRef CAS.
- K. R. Frederick, J. Tung, R. S. Emerick, F. R. Masiarz, S. H. Chamberlain, A. Vasavada, S. Rosenberg, S. Chakraborty, L. M. Schopter and V. Massey, J. Biol. Chem., 1990, 265(7), 3793–3802 CAS.
- Y. Shen, L. Gu, J. Zhang, J. Chen and G. Du, Shengwu Gongcheng Xuebao, 2013, 29, 927–936 CAS.
- F. Darvishi, A. Zarei and C. Madzak, World J. Microbiol. Biotechnol., 2018, 34, 128 CrossRef PubMed.
- I. Haq, A. Nawaz, H. Mukhtar, Z. Mansoor, M. Riaz, M. Ahmed and S. Ameer, Pak. J. Bot., 2014, 46, 1109–1114 Search PubMed.
- H. M. Kalisz, J. Hendle and R. D. Schmid, Appl. Microbiol. Biotechnol., 1997, 47, 502–507 CrossRef CAS PubMed.
- J. H. Pazur, K. Kleppe and A. Cepure, Arch. Biochem. Biophys., 1965, 111, 351–357 CrossRef CAS PubMed.
- J. J. O'Malley and J. L. Weaver, Biochemistry, 1972, 11, 3527–3532 CrossRef PubMed.
- H. Tsuge, O. Natsuaki and K. Ohashi, J. Biochem., 1975, 78, 835–843 CrossRef CAS PubMed.
- J. Bao, K. Furumoto, M. Yoshimoto, K. Fukunaga and K. Nakao, Biochem. Eng. J., 2003, 13, 69–72 CrossRef CAS.
- X. Huang, M. Cao and H. Zhao, Curr. Opin. Chem. Biol., 2020, 55, 161–170 CrossRef CAS PubMed.
- A. Fryszkowska and P. N. Devine, Curr. Opin. Chem. Biol., 2020, 55, 151–160 CrossRef CAS PubMed.
- J. M. Woodley, Curr. Opin. Green Sustain. Chem., 2020, 21, 22–26 CrossRef.
- D. J. Pollard and J. M. Woodley, Trends Biotechnol., 2007, 25, 66–73 CrossRef CAS PubMed.
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- H. E. Schoemaker, Science, 2003, 299, 1694–1697 CrossRef CAS PubMed.
- P. Lorenz, K. Liebeton, F. Niehaus and J. Eck, Curr. Opin. Biotechnol., 2002, 13, 572–577 CrossRef CAS PubMed.
- J. M. Almeida, R. C. Alnoch, E. M. Souza, D. A. Mitchell and N. Krieger, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140320 CrossRef PubMed.
- I. Castilla, D. Woods, F. Reen and F. O'Gara, Mar. Drugs, 2018, 16, 227 CrossRef PubMed.
- L. Fernández-Arrojo, M.-E. Guazzaroni, N. López-Cortés, A. Beloqui and M. Ferrer, Curr. Opin. Biotechnol., 2010, 21, 725–733 CrossRef PubMed.
- U. T. Bornscheuer and M. Pohl, Curr. Opin. Chem. Biol., 2001, 5, 137–143 CrossRef CAS PubMed.
- A. Currin, N. Swainston, P. J. Day and D. B. Kell, Chem. Soc. Rev., 2015, 44, 1172–1239 RSC.
- C. A. Denard, H. Ren and H. Zhao, Curr. Opin. Chem. Biol., 2015, 25, 55–64 CrossRef CAS PubMed.
- B. G. Davis, Curr. Opin. Biotechnol., 2003, 14, 379–386 CrossRef CAS PubMed.
- O. Barbosa, C. Ortiz, Á. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, Biotechnol. Adv., 2015, 33, 435–456 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- P. V. Iyer and L. Ananthanarayan, Process Biochem., 2008, 43, 1019–1032 CrossRef CAS.
- I. Eş, J. D. G. Vieira and A. C. Amaral, Appl. Microbiol. Biotechnol., 2015, 99, 2065–2082 CrossRef PubMed.
- S. Alonso, G. Santiago, I. Cea-Rama, L. Fernandez-Lopez, C. Coscolín, J. Modregger, A. K. Ressmann, M. Martínez-Martínez, H. Marrero, R. Bargiela, M. Pita, J. L. Gonzalez-Alfonso, M. L. Briand, D. Rojo, C. Barbas, F. J. Plou, P. N. Golyshin, P. Shahgaldian, J. Sanz-Aparicio, V. Guallar and M. Ferrer, Nat. Catal., 2020, 3, 319–328 CrossRef CAS.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- R. Di Cosimo, J. Mc Auliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- L. Dal Magro, J. F. Kornecki, M. P. Klein, R. C. Rodrigues and R. Fernandez-Lafuente, Int. J. Biol. Macromol., 2019, 138, 234–243 CrossRef CAS PubMed.
- L. Dal Magro, J. F. Kornecki, M. P. Klein, R. C. Rodrigues and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2020, 132, 109397 CrossRef CAS PubMed.
- J. M. Harris, C. Reyes and G. P. Lopez, J. Diabetes Sci. Technol., 2013, 7, 1030–1038 CrossRef PubMed.
- J. H. T. Luong, J. D. Glennon, A. Gedanken and S. K. Vashist, Microchim. Acta, 2017, 184, 369–388 CrossRef CAS.
- Q. Husain, Biointerface Res. Appl. Chem., 2018, 8, 3060–3074 CAS.
- L. V. Danyleyko, O. N. Schuvailo, V. M. Arkhypova, A. P. Soldatkin and S. V. Dzyadevych, Biopolym. Cell, 2003, 19, 76–80 Search PubMed.
- L. Betancor, F. López-Gallego, A. Hidalgo, N. Alonso-Morales, G. Dellamora-Ortiz, J. M. Guisán and R. Fernández-Lafuente, J. Biotechnol., 2006, 121, 284–289 CrossRef CAS PubMed.
- L. Caseli, D. S. dos Santos, M. Foschini, D. Gonçalves and O. N. Oliveira, Mater. Sci. Eng., C, 2007, 27, 1108–1110 CrossRef CAS.
- S. Datta, C. Cecil and D. Bhattacharyya, Ind. Eng. Chem. Res., 2008, 47, 4586–4597 CrossRef CAS PubMed.
- X. Yan, X. K. Xu and H.-F. Ji, Anal. Chem., 2005, 77, 6197–6204 CrossRef CAS PubMed.
- L.-Y. Chu, Y. Li, J.-H. Zhu, H.-D. Wang and Y.-J. Liang, J. Controlled Release, 2004, 97, 43–53 CrossRef CAS PubMed.
- K. Zhang and X. Y. Wu, J. Controlled Release, 2002, 80, 169–178 CrossRef CAS.
- T. Traitel, Y. Cohen and J. Kost, Biomaterials, 2000, 21, 1679–1687 CrossRef CAS PubMed.
- T. A. Horbett, J. Kost and B. D. Ratner, Swelling behavior of glucose sensitive membranes, in Polymers as Biomaterials, ed. S. W. Shalaby, A. S. Hoffman, B. D. Ratner and T. A. Horbett, Springer USA, Boston, MA, 1984, pp. 193–207 Search PubMed.
- Y. Ito, M. Casolaro, K. Kono and Y. Imanishi, J. Controlled Release, 1989, 10, 195–203 CrossRef CAS.
- R. Luo, H. Li and K. Y. Lam, Biomaterials, 2009, 30, 690–700 CrossRef CAS PubMed.
- X. Yang and J.-C. Kim, Int. J. Biol. Macromol., 2011, 48, 661–666 CrossRef CAS PubMed.
- S. Il Kang and Y. H. Bae, J. Controlled Release, 2003, 86, 115–121 CrossRef.
- M. Sönmez, D. Ficai, A. Ficai, L. Alexandrescu, M. Georgescu, R. Trusca, D. Gurau, M. A. Titu and E. Andronescu, Int. J. Pharm., 2018, 549, 179–200 CrossRef PubMed.
- M. Oroval, P. Díez, E. Aznar, C. Coll, M. D. Marcos, F. Sancenón, R. Villalonga and R. Martínez-Máñez, Chem. – Eur. J., 2017, 23, 1353–1360 CrossRef CAS PubMed.
- W. Zhao, H. Zhang, Q. He, Y. Li, J. Gu, L. Li, H. Li and J. Shi, Chem. Commun., 2011, 47, 9459–9461 RSC.
- C. Li, X. Liu, Y. Liu, F. Huang, G. Wu, Y. Liu, Z. Zhang, Y. Ding, J. Lv, R. Ma, Y. An and L. Shi, Nanoscale, 2019, 11, 9163–9175 RSC.
- Y. Wu, H. Hu, J. Hu and S. Liu, Macromol. Rapid Commun., 2012, 33, 1852–1860 CrossRef CAS PubMed.
- S. Jamwal, B. Ram, S. Ranote, R. Dharela and G. S. Chauhan, Int. J. Biol. Macromol., 2019, 123, 968–978 CrossRef CAS PubMed.
- W.-H. Chen, G.-F. Luo, M. Vázquez-González, R. Cazelles, Y. Sohn, R. Nechushtai, Y. Mandel and I. Willner, ACS Nano, 2018, 12, 7538–7545 CrossRef CAS PubMed.
- K. Ogawa, T. Nakajima-Kambe, T. Nakahara and E. Kokufuta, Biomacromolecules, 2002, 3, 625–631 CrossRef CAS PubMed.
- K. Hernandez, A. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, Curr. Org. Chem., 2012, 16, 2652–2672 CrossRef CAS.
- G. De Faveri, G. Ilyashenko and M. Watkinson, Chem. Soc. Rev., 2011, 40, 1722–1760 RSC.
- A. Podgoršek, M. Zupan and J. Iskra, Angew. Chem., Int. Ed., 2009, 48, 8424–8450 CrossRef PubMed.
- S. Schrader and E. V. Dehmlow, Org. Prep. Proced. Int., 2000, 32, 123–152 CrossRef CAS.
- D. M. Vriezema, P. M. L. Garcia, N. Sancho Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2007, 46, 7378–7382 CrossRef CAS PubMed.
- S. F. M. van Dongen, M. Nallani, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem. – Eur. J., 2009, 15, 1107–1114 CrossRef CAS PubMed.
- E. R. Stadtman and R. L. Levine, Amino Acids, 2003, 25, 207–218 CrossRef CAS PubMed.
- S. P. Wolff, A. Garner and R. T. Dean, Trends Biochem. Sci., 1986, 11, 27–31 CrossRef CAS.
- S. K. Grandhee and V. M. Monnier, J. Biol. Chem., 1991, 266, 11649–11653 CAS.
- T. G. Huggins, M. C. Wells-Knecht, N. A. Detorie, J. W. Baynes and S. R. Thorpe, J. Biol. Chem., 1993, 268, 12341–12347 CAS.
- J. W. Heinecke, W. Li, H. L. Daehnke and J. A. Goldstein, J. Biol. Chem., 1993, 268, 4069–4077 CAS.
- R. C. Armstrong and A. J. Swallow, Radiat. Res., 1969, 40, 563–579 CrossRef CAS PubMed.
- R. V. Winchester and K. R. Lynn, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1970, 17, 541–548 CrossRef CAS PubMed.
- K. Kikugawa, T. Kato and Y. Okamoto, Free Radical Biol. Med., 1994, 16, 373–382 CrossRef CAS PubMed.
- R. L. Levine, L. Mosoni, B. S. Berlett and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 15036–15040 CrossRef CAS PubMed.
- R. L. Levine, J. Moskovitz and E. R. Stadtman, IUBMB Life, 2000, 50, 301–307 CrossRef CAS PubMed.
- W. Vogt, Free Radical Biol. Med., 1995, 18, 93–105 CrossRef CAS PubMed.
- V. Y. Reddy, P. E. Desrochers, S. V. Pizzo, S. L. Gonias, J. A. Sahakian, R. L. Levine and S. J. Weise, J. Biol. Chem., 1994, 269, 4683–4691 CAS.
- M. A. Ciorba, S. H. Heinemann, H. Weissbach, N. Brot and T. Hoshi, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 9932–9937 CrossRef CAS PubMed.
- M. A. Smith, L. M. Sayre, V. E. Anderson, P. L. R. Harris, M. F. Beal, N. Kowall and G. Perry, J. Histochem. Cytochem., 1998, 46, 731–735 CrossRef CAS PubMed.
- C. E. Robinson, A. Keshavarzian, D. S. Pasco, T. O. Frommel, D. H. Winship and E. W. Holmes, Anal. Biochem., 1999, 266, 48–57 CrossRef CAS PubMed.
- Z. Maskos, J. D. Rush and W. H. Koppenol, Arch. Biochem. Biophys., 1992, 296, 521–529 CrossRef CAS PubMed.
- M. C. Wells-Knecht, T. G. Huggins, D. G. Dyer, S. R. Thorpe and J. W. Baynes, J. Biol. Chem., 1993, 268, 12348–12352 CAS.
- I. Balakrishnan and M. P. Reddy, J. Phys. Chem., 1970, 74, 850–855 CrossRef CAS.
- K. J. Davies, M. E. Delsignore and S. W. Lin, J. Biol. Chem., 1987, 262, 9902–9907 CAS.
- R. T. Dean, S. Gieseg and M. J. Davies, Trends Biochem. Sci., 1993, 18, 437–441 CrossRef CAS PubMed.
- S. Proteasome, C. Giulivisq, K. J. A. Daviesti, C. Giulivi and K. J. Davies, J. Biol. Chem., 1993, 268, 8752–8759 Search PubMed.
- C. Taggart, D. Cervantes-Laurean, G. Kim, N. G. McElvaney, N. Wehr, J. Moss and R. L. Levine, J. Biol. Chem., 2000, 275, 27258–27265 CAS.
- B. Nidetzky, Biochem. Soc. Trans., 2007, 35, 1588–1592 CrossRef CAS PubMed.
- E. R. Stadtman, B. S. Berlett and P. B. Chock, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 384–388 CrossRef CAS PubMed.
- J. M. Farber and R. L. Levine, J. Biol. Chem., 1986, 261, 4574–4578 CAS.
- I. Climent, L. Tsai and R. L. Levine, Anal. Biochem., 1989, 182, 226–232 CrossRef CAS PubMed.
- J. A. Sahakian, L. I. Szweda, B. Friguet, K. Kitani and R. L. Levine, Arch. Biochem. Biophys., 1995, 318, 411–417 CrossRef CAS PubMed.
- S. P. Fernandes, R. Dringen, A. Lawen and S. R. Robinson, Neurosci. Lett., 2011, 490, 27–30 CrossRef CAS PubMed.
- W. M. Garrison, Chem. Rev., 1987, 87, 381–398 CrossRef CAS.
- S. S. Ju, L. L. Lin, H. R. Chien and W. H. Hsu, FEMS Microbiol. Lett., 2000, 186, 215–219 CAS.
- C. Li, S. Sun, D. Park, H. O. Jeong, H. Y. Chung, X.-X. Liu and H.-M. Zhou, Int. J. Biol. Macromol., 2011, 49, 910–916 CrossRef CAS PubMed.
- H. Zhou, H. Singh, Z. D. Parsons, S. M. Lewis, S. Bhattacharya, D. R. Seiner, J. N. LaButti, T. J. Reilly, J. J. Tanner and K. S. Gates, J. Am. Chem. Soc., 2011, 133, 15803–15805 CrossRef CAS PubMed.
- J. A. Rosas-Rodríguez and E. M. Valenzuela-Soto, Chem.-Biol. Interact., 2011, 191, 159–164 CrossRef PubMed.
- A. Slavica, I. Dib and B. Nidetzky, Appl. Environ. Microbiol., 2005, 71, 8061–8068 CrossRef CAS PubMed.
- R. L. Levine, J. Biol. Chem., 1983, 258, 11828–11833 CAS.
- A. J. Rivett, J. E. Roseman, C. N. Oliver, R. L. Levine and E. R. Stadtman, Prog. Clin. Biol. Res., 1985, 180, 317–328 CAS.
- L. Fucci, C. N. Oliver, M. J. Coon and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 1521–1525 CrossRef CAS PubMed.
- H. Schuessler and K. Schilling, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1984, 45, 267–281 CrossRef CAS PubMed.
- K. Uchida, Y. Kato and S. Kawakishi, Biochem. Biophys. Res. Commun., 1990, 169, 265–271 CrossRef CAS.
- J. Q. Zhou and A. Gafni, J. Gerontol., 1991, 46, B217–B221 CrossRef CAS PubMed.
- A. E. Brodie and D. J. Reed, Arch. Biochem. Biophys., 1990, 276, 212–218 CrossRef CAS PubMed.
- R. Takahashi and S. Goto, Arch. Biochem. Biophys., 1990, 277, 228–233 CrossRef CAS PubMed.
- N. Verzijl, J. Degroot, E. Oldehinkel, R. A. Bank, S. R. Thorpe, J. W. Baynes, M. T. Bayliss, J. W. J. Bijlsma, F. P. J. G. Lafeber and J. M. Tekoppele, Biochem. J., 2000, 350, 381–387 CrossRef CAS PubMed.
- K. J. Wells-Knecht, D. V. Zyzak, J. E. Litchfield, S. R. Thorpe and J. W. Baynes, Biochemistry, 1995, 34, 3702–3709 CrossRef CAS PubMed.
- K. Uchida and E. R. Stadtman, J. Biol. Chem., 1993, 268, 6388–6393 CAS.
- B. Friguet, E. R. Stadtman and L. I. Szweda, J. Biol. Chem., 1994, 269, 21639–21643 CAS.
- D. P. H. Hsieh, R. S. Silver and R. I. Mateles, Biotechnol. Bioeng., 1969, 11, 1–18 CrossRef CAS PubMed.
- C. Bourdillon, R. Lortie and J. M. Laval, Biotechnol. Bioeng., 1988, 31, 553–558 CrossRef CAS PubMed.
- E. J. Tomotani, L. C. M. das Neves and M. Vitolo, Appl. Biochem. Biotechnol., 2005, 121, 149–162 CrossRef PubMed.
- S. Illner, C. Hofmann, P. Löb and U. Kragl, ChemCatChem, 2014, 6, 1748–1754 CrossRef CAS.
- S. Wang, P. Su, H. E and Y. Yang, Anal. Biochem., 2010, 405, 230–235 CrossRef CAS PubMed.
- J. Liu and Z. Cui, J. Membr. Sci., 2007, 302, 180–187 CrossRef CAS.
- I. Ullah, M. Sirajuddin, M. Khan, A. Shah and A. Badshah, J. Chem. Soc. Pak., 2013, 35, 775–782 Search PubMed.
- S. Otokesh, E. Kolvari, A. Amoozadeh and N. Koukabi, RSC Adv., 2015, 5, 53749–53756 RSC.
- J. Mlochowski and H. Wojtowicz-Mlochowska, Mini-Rev. Org. Chem., 2017, 13, 473–487 CrossRef.
- K. Nakao, T. Harada, K. Furumoto, A. Kiefner and M. Popovic, Can. J. Chem. Eng., 1999, 77, 816–825 CrossRef CAS.
- K. Nakao, A. Kiefner, K. Furumoto and T. Harada, Chem. Eng. Sci., 1997, 52, 4127–4133 CrossRef CAS.
- X. Zhang, N. Xu, J. Li, Z. Ma, L. Wei, Q. Liu and J. Liu, Enzyme Microb. Technol., 2020, 136, 109530 CrossRef CAS PubMed.
- J.-W. Song, J.-H. Seo, D.-K. Oh, U. T. Bornscheuer and J.-B. Park, Catal. Sci. Technol., 2020, 10, 46–64 RSC.
- W. A. Duetz, J. B. Van Beilen and B. Witholt, Curr. Opin. Biotechnol., 2001, 12, 419–425 CrossRef CAS PubMed.
- R. León, P. Fernandes, H. M. Pinheiro and J. M. S. Cabral, Enzyme Microb. Technol., 1998, 23, 483–500 CrossRef.
- W. Hartmeier and T. Döppner, Biotechnol. Lett., 1983, 5, 743–748 CrossRef CAS.
- J. Tramper, K. C. A. M. Luyben and W. J. J. van den Tweel, Eur. J. Appl. Microbiol. Biotechnol., 1983, 17, 13–18 CrossRef CAS.
- M. Traeger, G. N. Qazi, U. Onken and C. L. Chopra, J. Chem. Technol. Biotechnol., 1991, 50, 1–11 CrossRef CAS.
- M. Träger, G. N. Qazi, R. Buse and U. Onken, J. Ferment. Bioeng., 1992, 74, 274–281 CrossRef.
- M. Petruccioli, P. Piccioni, M. Fenice and F. Federici, Biotechnol. Lett., 1994, 16, 939–942 CrossRef CAS.
- C. Dowdells, R. L. Jones, M. Mattey, M. Benčina, M. Legiša and D. M. Mousdale, Lett. Appl. Microbiol., 2010, 51, 252–257 CrossRef CAS PubMed.
- M. Blazic, G. Kovacevic, O. Prodanovic, R. Ostafe, M. Gavrovic-Jankulovic, R. Fischer and R. Prodanovic, Protein Expression Purif., 2013, 89, 175–180 CrossRef CAS PubMed.
- M. Číp, L. Schreiberová and I. Schreiber, Russ. J. Phys. Chem. A, 2011, 85, 2322–2326 CrossRef.
- I. Schreiber, Y.-F. Hung and J. Ross, J. Phys. Chem., 1996, 100, 8556–8566 CrossRef CAS.
- S. Sasaki, T. Uchida, D. Nakano and T. Hideshima, Electroanalysis, 2004, 16, 1598–1602 CrossRef CAS.
- E. T. Hwang and S. Lee, ACS Catal., 2019, 9, 4402–4425 CrossRef CAS.
- O. Idan and H. Hess, Curr. Opin. Biotechnol., 2013, 24, 606–611 CrossRef CAS PubMed.
- F. Jia, B. Narasimhan and S. Mallapragada, Biotechnol. Bioeng., 2014, 111, 209–222 CrossRef CAS PubMed.
- F. Kazenwadel, M. Franzreb and B. E. Rapp, Anal. Methods, 2015, 7, 4030–4037 RSC.
- S. Ren, C. Li, X. Jiao, S. Jia, Y. Jiang, M. Bilal and J. Cui, Chem. Eng. J., 2019, 373, 1254–1278 CrossRef CAS.
- J. Wang and G. Zhang, Shengwu Gongcheng Xuebao, 2015, 31, 469–480 CAS.
- Y. Zhang and H. Hess, ACS Catal., 2017, 7, 6018–6027 CrossRef CAS.
- S. Arana-Peña, D. Carballares, R. Morellon-Sterling, Á. Berenguer-Murcia, A. R. Alcántara, R. C. Rodrigues and R. Fernandez-Lafuente, Biotechnol. Adv. DOI:10.1016/j.biotechadv.2020.107584 , in press.
- S. Butò, L. Pollegioni, L. D'Angiuro and M. S. Pilone, Biotechnol. Bioeng., 1994, 44, 1288–1294 CrossRef PubMed.
- A. Chmura, S. Rustler, M. Paravidino, F. Van Rantwijk, A. Stolz and R. A. Sheldon, Tetrahedron: Asymmetry, 2013, 24, 1225–1232 CrossRef CAS.
- R. Fernández-Lafuente, V. Rodriguez and J. M. Guisán, Enzyme Microb. Technol., 1998, 23, 28–33 CrossRef.
- A. Lopalco and V. J. Stella, J. Pharm. Sci., 2016, 105, 2879–2885 CrossRef CAS PubMed.
- F. Lopez-Gallego, L. Betencor, A. Hidalgo, C. Mateo, R. Fernandez-Lafuente and J. M. Guisan, Adv. Synth. Catal., 2005, 347, 1804–1810 CrossRef CAS.
- E.-M. Trost and L. Fischer, J. Mol. Catal. B: Enzym., 2002, 19–20, 189–195 CrossRef CAS.
- R. Upadhya, H. Nagajyothi and S. G. Bhat, Process Biochem., 1999, 35, 7–13 CrossRef CAS.
- G. Volpato, R. Rodrigues and R. Fernandez-Lafuente, Curr. Med. Chem., 2010, 17, 3855–3873 CrossRef CAS PubMed.
- A. H. Orrego, F. López-Gallego, A. Espaillat, F. Cava, J. M. Guisan and J. Rocha-Martin, ChemCatChem, 2018, 10, 3002–3011 CrossRef CAS.
- F. van Rantwijk, C. Mateo, A. Chmura, B. C. M. Fernandes and R. A. Sheldon, in Modern Biocatalysis, ed. W.-D. Fessner and T. Anthonsen, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009, pp. 261–272 Search PubMed.
- S. Velasco-Lozano, A. I. Benítez-Mateos and F. López-Gallego, Angew. Chem., Int. Ed., 2017, 56, 771–775 CrossRef CAS PubMed.
- S. Velasco-Lozano and F. López-Gallego, Biocatal. Biotransform., 2018, 36, 184–194 CrossRef CAS.
- A. I. Benítez-Mateos, M. L. Contente, S. Velasco-Lozano, F. Paradisi and F. López-Gallego, ACS Sustainable Chem. Eng., 2018, 6, 13151–13159 CrossRef.
- A. I. Benítez-Mateos, E. San Sebastian, N. Ríos-Lombardía, F. Morís, J. González-Sabín and F. López-Gallego, Chem. – Eur. J., 2017, 23, 16843–16852 CrossRef PubMed.
- S. Peirce, J. J. Virgen-Ortíz, V. G. Tacias-Pascacio, N. Rueda, R. Bartolome-Cabrero, L. Fernandez-Lopez, M. E. Russo, A. Marzocchella and R. Fernandez-Lafuente, RSC Adv., 2016, 6, 61707–61715 RSC.
- S. Arana-Peña, C. Mendez-Sanchez, N. S. Rios, C. Ortiz, L. R. B. Gonçalves and R. Fernandez-Lafuente, Int. J. Biol. Macromol., 2019, 131, 989–997 CrossRef PubMed.
- N. S. Rios, S. Arana-Peña, C. Mendez-Sanchez, C. Ortiz, L. R. B. Gonçalves and R. Fernandez-Lafuente, Catalysts, 2019, 9, 487 CrossRef CAS.
- L. Giorno and E. Drioli, Trends Biotechnol., 2000, 18, 339–349 CrossRef CAS PubMed.
- P. Jochems, Y. Satyawali, L. Diels and W. Dejonghe, Green Chem., 2011, 13, 1609–1623 RSC.
- G. T. Vladisavljević, Phys. Sci. Rev., 2019, 1, 1–35 Search PubMed.
- E. Nagy, Curr. Org. Chem., 2017, 21, 1713–1724 CAS.
- J. Pickova and O. Svabensky, J. Food Sci., 1964, 29, 400–406 CrossRef CAS.
- V. Linek, P. Beneš, J. Sinkule, O. Holeček and V. Malý, Biotechnol. Bioeng., 1980, 22, 2515–2527 CrossRef CAS.
- Z. Tao, R. A. Raffel, A.-K. Souid and J. Goodisman, Biophys. J., 2009, 96, 2977–2988 CrossRef CAS PubMed.
- S. Fukushima, A. Uyama and S. Katayama, J. Chem. Eng. Jpn., 1978, 11, 227–233 CrossRef CAS.
- M. Hashimoto, T. Tsukamoto, S. Morita and J. Okada, Chem. Pharm. Bull., 1983, 31, 1–11 CrossRef CAS.
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, J. Catal., 2004, 228, 282–287 CrossRef CAS.
- T.-H. Li, Y.-F. Su, C.-H. Hong and T.-L. Chen, J. Chem. Eng. Jpn., 1994, 27, 205–210 CrossRef CAS.
- G. Maria, M. D. Ene and I. Jipa, J. Mol. Catal. B: Enzym., 2012, 74, 209–218 CrossRef CAS.
- M. T. Atallah and H. O. Hultin, J. Food Sci., 1977, 42, 7–10 CrossRef CAS.
- T. Godjevargova, R. Dayal and I. Marinov, J. Appl. Polym. Sci., 2004, 91, 4057–4063 CrossRef CAS.
- J. C. Bouin, M. T. Atallah and H. O. Hultin, Biochim. Biophys. Acta, Enzymol., 1976, 438, 23–36 CrossRef CAS.
- S. Wang, M. Yoshimoto, K. Fukunaga and K. Nakao, Biotechnol. Bioeng., 2003, 83, 444–453 CrossRef CAS PubMed.
- M. Yoshimoto, S. Wang, K. Fukunaga, D. Fournier, P. Walde, R. Kuboi and K. Nakao, Biotechnol. Bioeng., 2005, 90, 231–238 CrossRef CAS PubMed.
- M. Yoshimoto, Y. Miyazaki, Y. Kudo, K. Fukunaga and K. Nakao, Biotechnol. Prog., 2006, 22, 704–709 CrossRef CAS PubMed.
- R. Fernandez-Lafuente, Enzyme Microb. Technol., 2009, 45, 405–418 CrossRef CAS.
- M. Yoshimoto, N. Takaki and M. Yamasaki, Colloids Surf., B, 2010, 79, 403–408 CrossRef CAS PubMed.
- M. Yoshimoto, M. Inoue and N. Takaki, J. Chem. Eng. Jpn., 2012, 45, 645–650 CrossRef CAS.
- P. H. S. Tse, J. K. Leypoldt and D. A. Gough, Biotechnol. Bioeng., 1987, 29, 696–704 CrossRef CAS PubMed.
- P.-W. Li, R.-R. Li, D.-L. Liu, D.-D. Min, Q. Yang and Q. Hao, Xiandai Shipin Keji, 2013, 29, 2180–2185+2130 CAS.
- N. Rueda, J. C. S. dos Santos, C. Ortiz, R. Torres, O. Barbosa, R. C. Rodrigues, Á. Berenguer-Murcia and R. Fernandez-Lafuente, Chem. Rec., 2016, 16, 1436–1455 CrossRef CAS PubMed.
- C. Cui, Y. Fang, B. Chen and T. Tan, Catal. Sci. Technol., 2019, 9, 477–482 RSC.
- S. T. Morthensen, A. S. Meyer, H. Jørgensen and M. Pinelo, Biochem. Eng. J., 2017, 117, 41–47 CrossRef CAS.
- R. A. Messing, Biotechnol. Bioeng., 1974, 16, 897–908 CrossRef CAS PubMed.
- P. E. Markey, P. F. Greenfield and J. R. Kittreli, Biotechnol. Bioeng., 1975, 17, 285–289 CrossRef CAS.
- P. F. Greenfield and R. L. Laurence, J. Food Sci., 1975, 40, 906–910 CrossRef CAS.
- J. C. Bouin, M. T. Atallah and H. O. Hultin, Methods Enzymol., 1976, 44, 478–488 CAS.
- J. C. Boujn, P. H. Dudgeon and H. O. Hultin, J. Food Sci., 1976, 41, 886–890 CrossRef.
- S. Akgöl, H. Yavuz, S. Şenel and A. Denizli, React. Funct. Polym., 2003, 55, 45–51 CrossRef.
- G. Ozyilmaz and S. S. Tukel, Appl. Biochem. Microbiol., 2007, 43, 29–35 CrossRef CAS.
- S. S. T. G. Ozyilmaz, J. Microbiol. Biotechnol., 2007, 17, 960–967 Search PubMed.
- T. T. F. Cui, Beijing Huagong Daxue Xuebao, Ziran Kexueban, 2012, 39, 63–67 Search PubMed.
- O. Barbosa, C. Ortiz, Á. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, RSC Adv., 2014, 4, 1583–1600 RSC.
- W. Zhuang, J. Huang, X. Liu, L. Ge, H. Niu, Z. Wang, J. Wu, P. Yang, Y. Chen and H. Ying, Food Chem., 2019, 275, 197–205 CrossRef CAS PubMed.
- S. B. Bankar, M. V. Bule, R. S. Singhal and L. A. Ananthanarayan, Int. J. Food Eng., 2011, 7, 8 Search PubMed.
- M. Hui, Z. Dong, Q. Tian, J. Huang and Y. Zi, Zhongguo Shipin Xuebao, 2018, 18, 134–140 Search PubMed.
- L. Liao, Y. Meng, R. Wang, B. Jia and P. Li, Front. Bioeng. Biotechnol., 2019, 7, 426 CrossRef PubMed.
- L. Cao, F. Van Rantwijk and R. A. Sheldon, Org. Lett., 2000, 2, 1361–1364 CrossRef CAS.
- R. Schoevaart, M. W. Wolbers, M. Golubovic, M. Ottens, A. P. G. Kieboom, F. van Rantwijk, L. A. M. van der Wielen and R. A. Sheldon, Biotechnol. Bioeng., 2004, 87, 754–762 CrossRef CAS PubMed.
- R. A. Sheldon, Appl. Microbiol. Biotechnol., 2011, 92, 467–477 CrossRef CAS PubMed.
- A. Pollak, H. Blumenfeld, M. Wax, R. L. Baughn and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 6324–6336 CrossRef CAS.
- V. G. Tacias-Pascacio, E. García-Parra, G. Vela-Gutiérrez, J. J. Virgen-Ortiz, Á. Berenguer-Murcia, A. R. Alcántara and R. Fernandez-Lafuente, Catalysts, 2019, 9, 1035 CrossRef CAS.
- K. Buchholz and B. Gödelmann, Biotechnol. Bioeng., 1978, 20, 1201–1220 CrossRef CAS.
- M. Reuss and K. Buchholz, Biotechnol. Bioeng., 1979, 21, 2061–2081 CrossRef CAS PubMed.
- J. E. Prenosil, Biotechnol. Bioeng., 1979, 21, 89–109 CrossRef CAS PubMed.
- R. K. Owusu and A. Finch, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1986, 872, 83–91 CrossRef CAS.
- R. Dayal and T. Godjevargova, Enzyme Microb. Technol., 2006, 39, 1313–1318 CrossRef CAS.
- F. Muzika, R. Jurašek, L. Schreiberová, V. Radojković and I. Schreiber, J. Phys. Chem. A, 2017, 121, 7518–7523 CrossRef CAS PubMed.
- P. H. S. Tse and D. A. Gough, Biotechnol. Bioeng., 1987, 29, 705–713 CrossRef CAS PubMed.
- L. Tarhaqn and A. Telefoncu, Appl. Biochem. Biotechnol., 1990, 26, 45–57 CrossRef.
- L. Tarhan and A. Telefoncu, Process Biochem., 1992, 27, 11–15 CrossRef CAS.
- L. E. Romero and D. Cantero, Prog. Biotechnol., 1998, 15, 107–112 CAS.
- A. V. Ruales-Salcedo, J. C. Higuita, J. Fontalvo and J. M. Woodley, Z. Naturforsch., C: J. Biosci., 2019, 74, 77–84 CAS.
- Q. Chen, S. Yu, N. Myung and W. Chen, J. Biotechnol., 2017, 263, 30–35 CrossRef CAS PubMed.
- X. Han, G. Liu, W. Song and Y. Qu, Bioresour. Technol., 2018, 248, 248–257 CrossRef CAS PubMed.
- X. Han, G. Liu, Y. Pan, W. Song and Y. Qu, Bioresour. Technol., 2018, 251, 407–410 CrossRef CAS PubMed.
- H. Zhang, S.-F. Hua and L. Zhang, J. Chem. Technol. Biotechnol., 2020, 95, 1116–1125 CAS.
- M. Onda, Y. Lvov, K. Ariga and T. Kunitake, J. Ferment. Bioeng., 1996, 82, 502–506 CrossRef CAS.
- F. Zhao, H. Li, Y. Jiang, X. Wang and X. Mu, Green Chem., 2014, 16, 2558–2565 RSC.
- S. Rezaei, A. Landarani-Isfahani, M. Moghadam, S. Tangestaninejad, V. Mirkhani and I. Mohammadpoor-Baltork, Chem. Eng. J., 2019, 356, 423–435 CrossRef CAS.
- H. Gülce, S. S. Çelebi, H. Özyörük and A. Yildiz, J. Electroanal. Chem., 1995, 397, 217–223 CrossRef.
- L. B. Marshall, G. D. Christian and A. Kumar, Analyst, 1977, 102, 424–428 RSC.
- F. Conzuelo, M. Gamella, S. Campuzano, M. A. Ruiz, A. J. Reviejo and J. M. Pingarrón, J. Agric. Food Chem., 2010, 58, 7141–7148 CrossRef CAS PubMed.
- W. Yang, P. Pang, X. Gao, Q. Cai, K. Zeng and C. A. Grimes, Sens. Lett., 2007, 5, 405–410 CrossRef CAS.
- S. Jimenez-Falcao, N. Joga, A. García-Fernández, A. Llopis Lorente, D. Torres, B. de Luis, F. Sancenón, P. Martínez-Ruiz, R. Martínez-Máñez and R. Villalonga, J. Mater. Chem. B, 2019, 7, 4669–4676 RSC.
- Y. K. Cho and J. E. Bailey, Biotechnol. Bioeng., 1977, 19, 185–198 CrossRef CAS.
- A. R. da Silva, E. J. Tomotani and M. Vitolo, Braz. J. Pharm. Sci., 2011, 47, 399–408 CrossRef.
- F. A. Taraboulsi, E. J. Tomotani and M. Vitolo, Appl. Biochem. Biotechnol., 2011, 165, 1708–1724 CrossRef CAS PubMed.
- T. T. H. Obata and J. Tanishita, Technol. Rep. Kansai Univ., 1983, 193–198 Search PubMed.
- B. Henrissat, T. T. Teeri and R. A. J. Warren, FEBS Lett., 1998, 425, 352–354 CrossRef CAS PubMed.
- M. Kim and D. F. Day, Appl. Biochem. Biotechnol., 2010, 162, 1379–1390 CrossRef CAS PubMed.
- D. F. Day, G. Dequeiroz, C. H. Chung and M. Kim, Int. Sugar J., 2008, 110, 7–11 CAS.
- A. Gosling, G. W. Stevens, A. R. Barber, S. E. Kentish and S. L. Gras, Food Chem., 2010, 121, 307–318 CrossRef CAS.
- M. B. Alvarado-Huallanco and F. Maugeri Filho, Catal. Sci. Technol., 2011, 1, 1043–1050 RSC.
- D. C. Sheu, P. J. Lio, S. T. Chen, C. T. Lin and K. J. Duan, Biotechnol. Lett., 2001, 23, 1499–1503 CrossRef CAS.
- K. Hoon Jung, J. Hwan Kim, Y. Joong Jeon and J. Heung Lee, Biotechnol. Lett., 1993, 15, 65–70 CrossRef.
- C. Fischer and T. Kleinschmidt, Enzyme Microb. Technol., 2019, 121, 45–50 CrossRef CAS PubMed.
- F. Rico-Rodríguez, J. C. Serrato, A. Montilla and M. Villamiel, Ultrason. Sonochem., 2018, 44, 177–183 CrossRef PubMed.
- S.-W. Chang, W.-H. Chang, M.-R. Lee, T.-J. Yang, N.-Y. Yu, C.-S. Chen and J.-F. Shaw, J. Agric. Food Chem., 2010, 58, 2908–2914 CrossRef CAS PubMed.
- T. Nishimoto, M. Nakano, T. Nakada, H. Chaen, S. Fukuda, T. Sugimoto, M. Kurimoto and Y. Tsujisaka, Biosci., Biotechnol., Biochem., 1996, 60, 640–644 CrossRef CAS PubMed.
- T.-T. Wu, S.-C. Lin and J.-F. Shaw, Biotechnol. Prog., 2013, 29, 83–90 CrossRef CAS PubMed.
- T.-T. Wu, C.-C. Ko, S.-W. Chang, S.-C. Lin and J.-F. Shaw, Process Biochem., 2015, 50, 928–934 CrossRef CAS.
- S. W. Chang, P. T. Liu, L. C. Hsu, C. S. Chen and J. F. Shaw, Food Chem., 2012, 134, 1745–1753 CrossRef CAS PubMed.
- L. S. Priputina, I. D. Obbarius and E. A. Nikol'skaia, Vopr. Pitan., 1971, 30, 39–42 CAS.
- D. Scott, J. Agric. Food Chem., 1953, 1, 727–730 CrossRef CAS.
- M. Castellari, L. Matricardi, G. Arfelli, G. Carpi and S. Galassi, Food Sci. Technol. Int., 2000, 6, 17–23 CrossRef CAS.
- G. P. Parpinello, F. Chinnici, A. Versari and C. Riponi, Lebensm.-Wiss. Technol., 2002, 35, 239–243 CrossRef CAS.
- B. Mistry and D. B. Min, J. Food Sci., 1992, 57, 196–199 CrossRef CAS.
- D. B. Min and B. S. Mistry, in Developments in Food Science, ed. G. Charalambous, Elsevier, Amsterdam, 1992, vol. 29, pp. 617–631 Search PubMed.
- A. Isaksen and J. Adler-Nissen, Lebensm.-Wiss. Technol., 1997, 30, 841–846 CrossRef CAS.
- A. Isaksen and J. Adler-Nissen, Lebensm.-Wiss. Technol., 1997, 30, 847–852 CrossRef CAS.
- S. Min, B. S. Mistry and H.-O. Lee, J. Food Sci., 2003, 68, 1272–1275 CrossRef CAS.
- C. A. Kantt, J. Bouzas, M. Dondero and J. A. Torres, J. Food Sci., 1993, 58, 104–107 CrossRef CAS.
- D. Dobbenie, M. Uyttendaele and J. Debevere, J. Food Prot., 1995, 58, 273–279 CrossRef CAS PubMed.
- K. Sankaran, S. S. Godbole and S. F. D'Souza, Enzyme Microb. Technol., 1989, 11, 617–619 CrossRef CAS.
- G. J. Pickering, D. A. Heatherbell and M. F. Barnes, Food Res. Int., 1998, 31, 685–692 CrossRef CAS.
- G. J. Pickering, D. A. Heatherbell and M. F. Barnes, Am. J. Enol. Vitic., 1999, 50, 291–298 CAS.
- D. F. Malherbe, M. Du Toit, R. R. Cordero Otero, P. Van Rensburg and I. S. Pretorius, Appl. Microbiol. Biotechnol., 2003, 61, 502–511 CrossRef CAS PubMed.
- J. Röcker, M. Schmitt, L. Pasch, K. Ebert and M. Grossmann, Food Chem., 2016, 210, 660–670 CrossRef PubMed.
- A. E. Gal, Anal. Biochem., 1968, 24, 452–461 CrossRef CAS PubMed.
- G. Karlsson, S. Winge and H. Sandberg, J. Chromatogr. A, 2005, 1092, 246–249 CrossRef CAS PubMed.
- K. H. Mah, H. W. Yussof, N. A. Jalanni, M. N. Abu Seman and N. Zainol, J. Teknol., 2014, 70, 93–98 Search PubMed.
- K. H. Mah, H. W. Yussof, M. N. Abu Seman and A. W. Mohammad, Sep. Purif. Technol., 2019, 209, 211–222 CrossRef CAS.
- S. T. Morthensen, J. Luo, A. S. Meyer, H. Jørgensen and M. Pinelo, J. Membr. Sci., 2015, 492, 107–115 CrossRef CAS.
- S. T. Morthensen, B. Zeuner, A. S. Meyer, H. Jørgensen and M. Pinelo, Sep. Purif. Technol., 2018, 194, 73–80 CrossRef CAS.
- B. Zhao, L. Zhou, L. Ma, Y. He, J. Gao, D. Li and Y. Jiang, Int. J. Biol. Macromol., 2018, 107, 2034–2043 CrossRef CAS PubMed.
- H. Schacht, W. Kesting and E. Schollmeye, Textilveredlung, 1995, 30, 237–243 CAS.
- N. Špička and P. Tavčer, Tekstilec, 2011, 54, 16–29 Search PubMed.
- L. B. Wingard Jr and C. C. Liu, Proc 8th Int Conf on Med & Bio Eng & 22nd Annu Conf on Eng in Med & Biol, Chicago, 1969, 1 Search PubMed.
- E. Katz, I. Willner and A. B. Kotlyar, J. Electroanal. Chem., 1999, 479, 64–68 CrossRef CAS.
- T. Kuwahara, H. Ohta, M. Kondo and M. Shimomura, Bioelectrochemistry, 2008, 74, 66–72 CrossRef CAS PubMed.
- C.-M. Yu, M.-J. Yen and L.-C. Chen, Biosens. Bioelectron., 2010, 25, 2515–2521 CrossRef CAS PubMed.
- N. Asrul, R. Othman, M. Z. A. Yahya, H. M. Salleh, F. Yusof and A. A. Ahmad, Adv. Mater. Res., 2012, 512–515, 1499–1502 CAS.
- B. Mecheri, A. D'Epifanio, A. Geracitano, P. Targon Campana and S. Licoccia, J. Appl. Electrochem., 2013, 43, 181–190 CrossRef CAS.
- C. E. La Rotta and E. R. González, J. Electrochem. Soc., 2013, 160, G37–G45 CrossRef CAS.
- S. Naidoo, Q. Naidoo, H. Blottnitz and G. Vaivars, IOP Conf. Ser.: Mater. Sci. Eng., 2013, 49, 012062 Search PubMed.
- H. du Toit and M. Di Lorenzo, Electrochim. Acta, 2014, 138, 86–92 CrossRef CAS.
- R. Haddad, J.-G. Mattei, J. Thery and A. Auger, Nanoscale, 2015, 7, 10641–10647 RSC.
- B. Mecheri, D. De Porcellinis, P. T. Campana, A. Rainer, M. Trombetta, A. Marletta, O. N. Oliveira and S. Licoccia, ACS Appl. Mater. Interfaces, 2015, 7, 28311–28318 CrossRef CAS PubMed.
- Y.-F. Tsai, C.-J. Shieh and H. Yang, Microsyst. Technol., 2017, 23, 3927–3935 CrossRef CAS.
- J. Galindo-De-La-Rosa, M. G. Araiza-Ramírez, A. Hernández-Torres, J. Ledesma-García and L. G. Arriaga, J. Phys.: Conf. Ser., 2019, 1407, 0–4 Search PubMed.
- R. D. Milton, F. Giroud, A. E. Thumser, S. D. Minteer and R. C. T. Slade, Phys. Chem. Chem. Phys., 2013, 15, 19371–19379 RSC.
- R. D. Milton, F. Giroud, A. E. Thumser, S. D. Minteer and R. C. T. Slade, Electrochim. Acta, 2014, 140, 59–64 CrossRef CAS.
- I. Osadebe and D. Leech, ChemElectroChem, 2014, 1, 1988–1993 CrossRef CAS.
- P. Jenkins, S. Tuurala, A. Vaari, M. Valkiainen, M. Smolander and D. Leech, Bioelectrochemistry, 2012, 87, 172–177 CrossRef CAS PubMed.
- J. J. Virgen-Ortíz, J. C. S. dos Santos, Á. Berenguer-Murcia, O. Barbosa, R. C. Rodrigues and R. Fernandez-Lafuente, J. Mater. Chem. B, 2017, 5, 7461–7490 RSC.
- M. Christwardana, Y. Chung and Y. Kwon, Nanoscale, 2017, 9, 1993–2002 RSC.
- C. Zhang, S. Zhang, W. Liu, T. Guo, R. Gu and J. Kong, Appl. Biochem. Biotechnol., 2019, 189, 822–833 CrossRef CAS PubMed.
- W.-N. Du and S.-T. Chen, Biocontrol Sci., 2019, 24, 97–101 CrossRef CAS PubMed.
- M. G. C. Baldry, J. Appl. Bacteriol., 1983, 54, 417–423 CrossRef CAS PubMed.
- R. O. Musser, H. S. Kwon, S. A. Williams, C. J. White, M. A. Romano, S. M. Holt, S. Bradbury, J. K. Brown and G. W. Felton, Arch. Insect Biochem. Physiol., 2005, 58, 138–144 CrossRef CAS PubMed.
- X. Liu, Z. Yan, Y. Zhang, Z. Liu, Y. Sun, J. Ren and X. Qu, ACS Nano, 2019, 13, 5222–5230 CrossRef CAS PubMed.
- L.-H. Fu, C. Qi, J. Lin and P. Huang, Chem. Soc. Rev., 2018, 47, 6454–6472 RSC.
- L.-H. Fu, C. Qi, Y.-R. Hu, J. Lin and P. Huang, Adv. Mater., 2019, 31, 1808325 CrossRef PubMed.
- R. A. S. Nascimento, R. E. Özel, W. H. Mak, M. Mulato, B. Singaram and N. Pourmand, Nano Lett., 2016, 16, 1194–1200 CrossRef CAS PubMed.
- B. Choi, J. S. Lee, S. J. Kim, D. Hong, J. B. Park and K.-Y. Lee, Cancer Lett., 2020, 478, 56–69 CrossRef CAS PubMed.
- M. Gao, T. Wang, L. Ji, S. Bai, L. Tian and H. Song, Front. Immunol., 2020, 11, 366 CrossRef PubMed.
- C. Wang, Y. Ye, G. M. Hochu, H. Sadeghifar and Z. Gu, Nano Lett., 2016, 16, 2334–2340 CrossRef CAS PubMed.
- W. Zhao, J. Hu and W. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 23528–23535 CrossRef CAS PubMed.
- W. Fan, N. Lu, P. Huang, Y. Liu, Z. Yang, S. Wang, G. Yu, Y. Liu, J. Hu, Q. He, J. Qu, T. Wang and X. Chen, Angew. Chem., Int. Ed., 2017, 56, 1229–1233 CrossRef CAS PubMed.
- R. Zhang, L. Feng, Z. Dong, L. Wang, C. Liang, J. Chen, Q. Ma, R. Zhang, Q. Chen, Y. Wang and Z. Liu, Biomaterials, 2018, 162, 123–131 CrossRef CAS PubMed.
- W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393–410 CrossRef CAS PubMed.
- S. R. McKeown, R. L. Cowen and K. J. Williams, Clin. Oncol., 2007, 19(6), 427–442 CrossRef CAS PubMed.
- K. M. Bailey, J. W. Wojtkowiak, A. I. Hashim and R. J. Gillies, Adv. Pharmacol., 2012, 65, 63–107 CAS.
- W. Feng, X. Han, R. Wang, X. Gao, P. Hu, W. Yue, Y. Chen and J. Shi, Adv. Mater., 2019, 31, 1805919 Search PubMed.
- F. Mahdizadeh and M. Eskandarian, J. Ind. Eng. Chem., 2014, 20, 2378–2383 CrossRef CAS.
- S. Kato, H. Unuma, T. Ota and M. Takahashi, J. Am. Ceram. Soc., 2000, 83, 986–988 CrossRef CAS.
- B. Jain, A. K. Singh, H. Kim, E. Lichtfouse and V. K. Sharma, Environ. Chem. Lett., 2018, 16, 947–967 CrossRef CAS.
- M. Kahoush, N. Behary, A. Cayla and V. Nierstrasz, Process Biochem., 2018, 64, 237–247 CrossRef CAS.
- P. V. Nidheesh, RSC Adv., 2015, 5, 40552–40577 RSC.
- D. K. Ahuja, L. G. Bachas and D. Bhattacharyya, Chemosphere, 2007, 66, 2193–2200 CrossRef CAS PubMed.
- S. Arana-Peña, D. Carballares, A. Berenguer-Murcia, A. R. Alcántara, R. C. Rodrigues and R. Fernandez-Lafuente, Catalysts, 2020, 10, 605 CrossRef.
Footnote |
| † Both authors have evenly contributed to this study. |
| This journal is © The Royal Society of Chemistry 2020 |