
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of small promoter amounts on coke structure in dry reforming of methane over Ni/ZrO2†
Robert
Franz
ab,
Tobias
Kühlewind
b,
Genrikh
Shterk
c,
Edy
Abou-Hamad
 d,
Alexander
Parastaev
e,
Evgeny
Uslamin
a,
Emiel J. M.
Hensen
e,
Freek
Kapteijn
b,
Jorge
Gascon
c and
Evgeny A.
Pidko
*a
d,
Alexander
Parastaev
e,
Evgeny
Uslamin
a,
Emiel J. M.
Hensen
e,
Freek
Kapteijn
b,
Jorge
Gascon
c and
Evgeny A.
Pidko
*a
aInorganic Systems Engineering, Department of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: e.a.pidko@tudelft.nl
bCatalysis Engineering, Department of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands
cAdvanced Catalytic Materials, KAUST Catalysis Center, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia
dCore Labs, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia
eLaboratory of Inorganic Materials and Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands
First published on 22nd May 2020
Abstract
Coke deposition is one of the main challenges in the commercialisation of dry reforming of methane over supported Ni catalysts. Besides the coke quantity, the structure of the deposits is also essential for the catalyst lifetime. Accordingly, in this study, we analysed the effect of Na, K, and Cs promoters on both these variables over Ni/ZrO2 catalysts. Besides blocking the most active coke-forming sites already at low loading, the promoting effect of the alkali metals is also contributed to by their coke gasification activity. To evaluate the additional impact of the latter, the behaviour of alkali-doped catalysts was compared to that for Mn-doped catalysts, exclusively featuring the site-blocking promotion mechanism. While the conversion is barely affected by the type of promoter, it has a profound effect on the amount and the composition of carbon deposits formed during the reaction. Promoting with K or Mn reduces the coke content to a similar degree but with less carbon fibres observed in the case of K. The promotion by Cs and Na results in the lowest coke content. The superior performance of Cs and Na-doped Ni/ZrO2 catalysts is attributed to the enhanced coke gasification via carbonate species on top of the site blocking effects.
Introduction
Current prognoses for worldwide energy consumption predict an increase in global CO2 emissions for all except the most optimistic scenarios. At the same time, the demand for various chemical products, especially plastics, will keep increasing and thereby also the consumption of fossil fuels.1 Recycling CO2 as a raw material for the chemical industry would lead to a reduction of CO2 emissions and at the same time help to meet growing customer demands. The so-called dry reforming of methane (1), in which carbon dioxide and methane react to synthesis gas at elevated temperatures, is a potential route to achieve this.| CH4 + CO2 ⇌ 2CO + 2H2 ΔH298K = 247 kJ mol−1 | (1) |
The extensive experience of the (petro)chemical industry with the related process of methane steam reforming is a considerable advantage of dry reforming. However, one of the significant drawbacks of this process over steam reforming is the more extensive carbon formation, causing rapid deactivation of the catalyst during operation.2 The main coking reactions are the Boudouard reaction (2) and methane decomposition (3):
| 2CO ⇌ CO2 + C ΔH298K = −171 kJ mol−1 | (2) |
| CH4 ⇌ 2H2 + C ΔH298K = 75 kJ mol−1 | (3) |
The comparison of thermodynamic equilibrium constants for the target dry reforming and the undesirable side-reactions shown in Fig. 1 highlight the fundamental challenges of this process, especially when aiming for large-scale industrial application. Ideally, the reaction temperature should be low to reduce energy consumption and thus costs. High-pressure operation would also be economically preferable to a compression of the more voluminous syngas for use in syntheses at elevated pressure (e.g., Fischer–Tropsch (FT)4 or methanol5 syntheses).6 At low temperature and high pressure, the side-reactions producing coke are thermodynamically most favourable, however (Fig. 1). According to thermodynamics, high temperature and low-pressure operation is desired, if coke formation is to be minimised. A compromise of high temperature and pressure would lead to additional issues. Under such conditions, gas-phase reactions start becoming relevant, which also lead to coke formation.7 Coke formation appears, therefore, inevitable under practical conditions, which implies that a successful dry reforming catalyst should be highly resistant to coking.8
 | ||
| Fig. 1 Equilibrium constants of the dry reforming and the two main coking reactions as a function of temperature at 1 bar and 20 bar (insert); calculated using ASPEN V8.8, amended from.3 | ||
Both support and active phase need to be optimised to develop a coke-resistant catalyst. Precious metals such as Ru exhibit excellent performance but are scarce and expensive, shifting the research focus to alternative catalyst formulations based on earth-abundant 3d transition metals.9 Amongst these, nickel is the most studied element due to its wide availability and high catalytic activity in dry reforming.10–12 The downside of nickel is its considerable susceptibility to coking. Depending on the reaction temperature, Ni catalysts can promote the formation of various types of coke, such as surface graphite, graphene as well as carbon fibres.13–15 The latter represents the most critical challenge as the growth of carbon fibres in the course of the catalytic process breaks catalyst particles and results in reactor plugging.9
A popular approach to improve the coke resistance of Ni-based dry reforming catalysts is to deactivate the highly reactive sites on the surface by promoter addition. This approach has been earlier investigated in detail for various support–promoter combinations.16–20 A representative example is the Sulphur PAssivated ReforminG (SPARG) process developed by Haldor Topsøe, in which the feed contains a controlled amount of H2S poison.21 The H2S chemisorbs on the Ni catalyst, deactivating the most active sites, which are also responsible for most of the coke formation. As a result, the catalyst operates with reduced activity but in a much more stable fashion over extended periods of time.22
Single-crystal studies of methane decomposition on Ni have shown that a similar effect is achieved with Au and K.23,24 Step sites on Ni clusters are the most active sites for coke formation and all promoters mentioned above preferentially occupy these sites.25 Site blocking with alkaline and earth-alkaline oxides has already been investigated.18–20,26,27 Other additives such as Mn or Sn are also reported to reduce coking by blocking the Ni surface.28,29 For the earth-alkaline metals and manganese a caveat must be added. Besides blocking the Ni surface, these promoters are also assumed to increase the CO2 affinity of the catalyst, additionally reducing the coking. Although the site-sensitivity of the Boudouard reaction has been studied less extensively, the literature points to similar trends as for methane decomposition for this process with a higher reactivity of the step-edge sites.30 Furthermore, it has been proposed that promoter ions may also affect the electronic structure of the neighbouring Ni centres, reducing their activity towards dissociative methane chemisorption.23 At the same time, alkali addition can enhance Ni sintering in select cases, meaning that a delicate balance must be struck during synthesis.31–34
However, a detailed quantitative analysis of the effect on the coke structure of several different promoters operating through a similar mechanism has not been reported until now. Even small decreases in coking activity can delay the exchange of the catalyst bed or result in a milder regeneration procedure, extending the catalyst lifetime further. Consequently, the alkali elements Na, K and Cs were selected as the first batch of promoters. Potassium has been one of the prime examples of this site-blocking behaviour. Other alkali metals should thus represent a rather moderate change of promoter characteristics, allowing to probe the electronic effects mentioned previously.23 Amongst metal oxides, MnOX has been reported as a promoter in dry reforming literature.29,35 As mentioned above, at high loadings of manganese, an increased CO2-affinity of the catalyst is noticeable. For low loadings, the physical blocking of Ni sites dominates. This apparent similarity in function made MnOX an interesting comparison. MnOX is typically reported to have little to no activity in the gasification of carbon.36,37 In contrast, alkali metals are well-known for their activity in carbon gasification.38,39 Thus, manganese as the fourth promoter should also clarify, if this reaction contributes to the reduced coke content. All samples were supported on ZrO2 since it is less prone to forming mixed phases with Ni or the promoters, as is the case for Al2O3 or SiO2 (e.g.ref. 31 and 40). To avoid possible interference of additional species, pure ZrO2 without any stabilising additives was used. Additives such as CaO would be necessary to stabilise cubic ZrO2,41 which has been reported to be the superior ZrO2 phase for use as catalyst support.42 In contrast to other supports, ZrO2-based catalysts also tend to form more coke, which is desirable in the analysis of the impact of the promoters on the coke structure.43,44
Experimental
Chemicals
Ethylenediaminetetraacetic acid (EDTA, Thermo Fisher 99%), NH3 solution (VWR, 25%), ZrO2 (Alfa Aesar, 51 m2 g−1), Ni(NO3)2·6H2O (Merck, analysis quality), KNO3 (Acros, 99%+), NaNO3 (Riedel-De Haen, 99.5%), CsNO3 (Alfa Aesar 99.5%), Mn(NO3)2·4H2O (Thermo Fisher, analysis quality).All materials were used without further modification except for NH3 (aq), which was diluted with demineralised water in a volumetric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 before usage, and ZrO2. ZrO2 extrudates were ground to a fine powder before impregnation.
:1 before usage, and ZrO2. ZrO2 extrudates were ground to a fine powder before impregnation.
Catalyst synthesis
All catalysts were synthesised via incipient wetness impregnation on commercial ZrO2 support. A solution of the required concentration was prepared in the following fashion: in a first step EDTA was dissolved in a solution of 12.5% NH3. Afterwards, the required amount of Ni(NO3)2 and either KNO3, NaNO3, CsNO3 or Mn(NO3)2 were added to the solution. After the dissolution of all components, the required amount of liquid (0.4 mL g−1) was impregnated onto the dry ZrO2 powder. This was followed by thorough mixing, drying for 5 h at 80 °C and then calcination at 700 °C for 5 h (heating rate of 10 °C min−1). For all samples, the loading of Ni was set at 0.02 gNi gSupport−1. The amount of the promoter was calculated to achieve molar ratios promoter/Ni = 1/10 or 1/5. In all syntheses, a ratio of EDTA/Ni = 1 was maintained. An overview of the different catalysts is given in Table 1.| Promoter metal | Ratio promoter/Ni | Abbreviation |
|---|---|---|
| — | — | REF |
| K | 1/10 | 1K |
| K | 2/10 | 2K |
| Cs | 1/10 | 1Cs |
| Cs | 2/10 | 2Cs |
| Na | 1/10 | 1Na |
| Mn | 1/10 | 1Mn |
Temperature programmed reduction (TPR)
Temperature programmed reduction was carried out in a fixed-bed reactor system. 100 mg of freshly prepared sample was pelletised, crushed and sieved to a particle size of 212–355 μm and filled into a quartz reactor (I.D. 6 mm). The quartz reactor was placed into a furnace, and a flow of 10% H2/Ar (30 mL min−1) started. The furnace was then heated from room temperature to 900 °C with a rate of 5 °C min−1. At the outlet, the hydrogen signal was monitored with a thermal conductivity detector (TCD). Peak deconvolution of the TPR data was carried by the superposition of three Gaussian functions for each profile.H2-Chemisorption
Chemisorption was measured on a Micromeritics ASAP 2020C. Approx. 130 mg of sample was loaded into the setup and reduced at 650 °C for 2 h in 20% H2 in N2, thus mimicking the reduction conditions of the reactivity tests. Afterwards, the sample was cooled down to 30 °C, at which H2 chemisorption was measured with a static-volumetric method.Electron microscopy
ADF-STEM analysis and EELS elemental mapping of the samples were carried out with an FEI Titan G2 80–300 kV electron microscope operated at 300 kV. For the TEM analysis of the coked samples, a Jeol JEM 1400 plus TEM was used. The high-resolution images were obtained on a JEM3200-FSC.13C-MAS solid state NMR
For the 1D 13C MAS NMR, a known amount of sample was filled into zirconia rotors and recorded on a Bruker AVANCE III spectrometer. The system operated at resonance frequencies of 600 MHz (1H frequency), and a conventional double resonance 3.2 mm CPMAS probe was used. The spinning frequency was set to 10–15 kHz. NMR chemical shifts are reported with respect to TMS as the external reference. Spectra were recorded by a spin-echo pulse sequence (pulse length 3.4 μs) with four-phase alternation synchronised with the spinning rate for the MAS experiments to delete all background signals from the probe. The interscan delay was set to 15 s to allow the complete relaxation, and 5000–30000 scans were performed. An apodisation function (exponential) corresponding to a line broadening of 80 Hz was applied prior to the Fourier transformation.
Raman spectroscopy
Raman spectra were obtained with a Renishaw Via Reflex confocal spectrometer using a 532 nm laser excitation. The laser power was set to 100% and the sample was irradiated for 10 s with 6 accumulations. The laser has a maximum power of 30 mW.XRD
XRD measurements were carried out in a Bruker D8 Advance Diffractometer with monochromatic Co Kα radiation (λ = 0.179026 nm) at room temperature.Reactivity tests
The system used for catalytic testing has been described in detail in previous publications.45 In short, it consists of a parallel fixed-bed reactor system with six reactors (quartz tubes with 4 mm I.D.). The quartz tubes are inserted into steel tubes mounted within the furnace to provide for better heat conduction. For each experiment the reactors were filled in the following manner (top to bottom): quartz wool plug, a 15 cm SiC bed (212–425 μm), a thin quartz layer, the catalytic bed, a thin quartz wool layer, a 10 cm SiC bed, a quartz wool plug. For the standard catalytic experiments, the catalyst bed consisted of 30 mg of catalyst (212–355 μm) mixed with 70 mg of SiC (300–355 μm). These two components were mixed as described in the literature to ensure proper distribution of the sample.46 For a detailed coke content analysis, catalytic runs were carried out with a catalytic bed of 75 mg of undiluted catalyst (212–355 μm), to allow for a full recovery of coked catalyst without SiC contamination. In all experiments, the correct placement of the catalytic bed within the isothermal zone of the reactor was ensured.The catalytic setup allowed for premixing the reaction mixture in a separate mixing section upstream of the reactors. The custom mixture was then fed to each reactor individually through separate mass flow controllers (MFCs). Activity testing was carried out as follows. To reduce the catalysts, the reactors were heated under a flow of 40 mL min−1 (20% H2 in N2) to 650 °C with a rate of 10 °C min−1, followed by an isothermal period of 2 h. Next, the feed was switched to the reaction mixture. For the catalytic activity tests, the feed consisted of 20% CO2, 20% CH4 and 60% N2. For the separate coking runs 50% CO2 and 50% CH4 were used. In both cases, the flow of gas per reactor was 80 mL min−1, keeping the reactant to catalyst ratio identical between the two sets of experiments (32 LCH4 g−1 h−1). All runs were carried out for a total of 12 hours. The coke content was determined with the aid of a TGA-MS (Mettler Toledo TGA/DSC1 connected to a Pfeiffer Vacuum OmniSTAR) by quantifying the CO2 signal while heating the sample using synthetic air as an oxidant.
Product analysis was carried out by a GC equipped with both an FID and a TCD. The TCD was used for the analysis of all gases (columns: 0.3 m Hayesep Q 80–100 mesh with back-flush, 25 m × 0.53 mm Porabond Q, and 15 m × 0.53 mm molsieve 5A with bypass option) with N2 as the internal standard. The FID signal provided a quality check for the TCD signal.
Results and discussion
Catalyst characterisation
In STEM analysis of the synthesised catalysts Ni particles smaller than 20 nm were detected in all samples, with a major fraction smaller than 10 nm, again for all samples. The low loading of Ni and the low number of detected particles (14 for REF and 1Na, around 30 for all other samples) did not allow for a proper statistical analysis, especially when the possible variation of the PSD on the Ni/ZrO2 system is considered. For example, Charisiou et al. report a PSD from 20 nm to 80 nm for an 8 wt% Ni/ZrO2 catalyst characterised via STEM-HAADF.47 The different particles observed in STEM have irregular shapes, similar to previously published literature.48 Previous studies revealed that the nature of the support and the synthesis method have a strong impact on the observed Ni particle shapes.49,50 Another primary characterisation tool for the Ni-PSD of Ni/ZrO2 catalysts is H2 chemisorption. While this method allows estimating an averaged diameter, the exact ZrO2 morphology can also strongly impact the average Ni size.51 In contrast to frequent literature reports of successful H2 chemisorption, no H2 uptake at 30 °C was observed. The support can partially cover metal particles after prolonged reduction at high temperatures, which significantly reduces H2 uptake.52–54 Steib et al. have observed ZrOX clusters on Ni particles in a Ni/ZrO2 system.55 Therefore, the chemisorption results can be seen as proof of a partial coverage of the Ni particles by the support. The low loading of Ni prevented its detection in the XRD measurements and thus the determination of the Ni particle size via line broadening. XRD established that in all cases ZrO2 is present in the monoclinic phase.Temperature-programmed reduction (TPR) has been used in the past to compare the particle size of supported Ni catalysts. Deconvolution of the TPR profiles leads to three peaks in all cases with the maxima at 350 °C, 456 °C and 510 °C for pure Ni/ZrO2 (Fig. 2 and Table S1†). When considering the positions of peaks 1 and 3, no general statement can be made. The peak positions are shifted to higher or lower temperatures depending on the promoter with no visible systematic effect. Only the position of the intermediate peak 2 is shifted to lower temperatures for all promoted catalysts 1Mn must be mentioned separately, since here the positions of peaks 2 and 3 shift more substantially than for the catalysts promoted with alkali metals. A more interesting effect can be noted when analysing the relative peak area of the three reduction peaks. In short, all promoters increase the area of the highest temperature peak at the expense of the two others. Except for 1Mn, this increase comes mainly at the cost of the lowest temperature peak. Increasing the loading of Cs or K once again increases the lowest temperature peak, as seen in Fig. 3.
 | ||
| Fig. 2 TPR curves with peak deconvolution for K-promoted and Na-promoted (a, left) and Cs-promoted and Mn-promoted (b, right) catalysts. | ||
 | ||
| Fig. 3 Relative distribution of peak areas observed in TPR. | ||
Previous studies indicate that in TPR analysis of supported Ni catalysts, smaller NiO clusters are reduced at higher temperatures. In contrast, large NiO particles on the support can be considered bulk NiO.56–58 This rule cannot be applied universally, however. The available literature on Mn-promoted Co and Ni catalysts shows that shifts in reduction temperature are not necessarily caused by changes in Ni particle size but can also be affected by the formation of solid solutions during synthesis.59,60 The promoter itself can also decrease the reducibility of the Ni active phase. Mixed oxides of alkali metals and nickel have mainly been reported for Ni3+.61 However, we found no indication of Ni3+ in our samples, and this is not expected after calcination at 700 °C.
Instead, the TPR curves are in good agreement with the results of Peters et al. for pure Ni/ZrO2.58 We propose that a correlation between reducibility and particle size is also appropriate for alkali-promoted Ni/ZrO2. Thus, we attribute here peak 1 to bulk NiO and peaks 2 and 3 to dispersed Ni species with different degrees of interaction with the support.58
To summarise, our TPR data show, that the addition of alkali promoters reduces the amount of “bulk” nickel (peak 1). All alkali promoters increase the highest temperature peak also at the expense of the intermediate peak. However, an increase in alkali loading again increases the percentage of “bulk” nickel. It depends on the promoter in question, which of the two “dispersed nickel” peaks is affected by this readjustment. This concept of an optimal promoter loading for nickel distribution mirrors the TEM analyses of Park et al.62 When adding various alkali metals to Ni/SiO2, they also noticed a smaller average Ni particle size for low promoter loadings and a growth in particle size with increased promoter loading.
Catalytic activity
The catalytic runs were carried out with very low loadings of the active metal under conditions at which coking is thermodynamically favourable. This allowed us to observe a significant deactivation for all catalysts during the 12 h activity test. The results of the catalytic runs are summarised in Fig. 4, in which the methane conversion is shown for all samples. For the CO2 conversion, we refer to the ESI.† For all catalysts, the increase in the promoter loading gave rise to a decreased conversion. Interestingly, at identical loadings, the catalysts with different promoters show similar levels of conversion (Fig. 4). These data suggest that regardless of the choice of the promoter, a similar number of active sites in the catalyst was blocked by promoter addition. | ||
| Fig. 4 Methane conversion over promoted and non-promoted Ni/ZrO2 as a function of time-on-stream at 650 °C, 30 mg sample, 80 mL min−1 (20% CH4, 20% CO2 in N2). | ||
Following this line of thought, samples with higher promoter loadings should be more resistant to coking and deactivate less. Yet, in all cases, the conversion decreases considerably within 12 h TOS and after 6–8 h the measured values differ only marginally between the samples. Catalyst deactivation in dry reforming is frequently caused by coke formation, but sintering can also play a significant role. For example, the weak interactions between ZrO2 and Ni have been proposed to accelerate Ni sintering, and thus catalyst deactivation during dry reforming.43 Pronounced sintering would thus be a credible explanation for the observed behaviour.
A strong impact of sintering may obscure differences between the different promoters. The different promoters do appear to block a similar amount of active sites. The question that remains is, if the effect on the catalyst stability is the same or if differences exist between the promoters. For example, one promoter may be more effective at blocking the most coke forming sites. To elucidate these potential differences, the coke contents on the spent catalysts was analysed and quantified.
Coke characterisation
The carbon content of spent samples was analysed with TGA-MS. The data are summarised in Fig. 5. In short, the non-promoted REF contains 53 mgC g−1cat, and the coke loading of all promoted samples was substantially lower. In this group, samples 1Mn and 1K contain the most coke with 9 and 10 mgC g−1cat, respectively. Using caesium and sodium as promoters instead reduces the coke content by around 50% to 3 or 5 mgC g−1cat. Increasing the loading of potassium reduces the coke content further (3 mgC g−1cat for 2K). 2Cs is on a similar level with 4 mgC g−1cat. As mentioned previously, the increased promoter loadings of 2Cs and 2K also reduce the initial methane conversion substantially. The coke contents in REF sample is already rather low compared to the values commonly reported for different Ni-based catalyst systems.18,63 We attribute this to a low Ni loading and the short runtime of 12 h TOS used in the current study. Experiments at higher Ni loadings and longer reaction times have indicated, that ZrO2-based systems commonly form more coke than those based on Ce1−XZrXO2.44 Supports such as MgO or SiO2 have also been reported to lead to less carbon formation.43 | ||
| Fig. 5 Initial methane conversion of the samples plotted over the carbon content after 12 hours of coking. | ||
Published literature shows that increasing the loading of the alkali metal promoter reduces both the conversion and the amount of coke deposits.18 For the K-promoted samples, this correlation holds. Increasing the K/Ni ratio from 1/10 (1K) to 2/10 (2K) further reduces the coke, albeit only by small overall amounts when comparing the drop in coke content from REF to 1K. This is in line with the previously proposed concept of site blocking by the promoters, which implies that the promoter quickly blocks the most active coke forming sites.23,25 The remaining coke does not form on step-edge sites but Ni ensembles of minimal size.21,64 Larger quantities of the promoter are then necessary to cover all such ensembles sufficiently to prevent further coke formation. For Cs the situation is slightly different as a Cs/Ni ratio of 1/10 (1Cs) is already enough to reduce the carbon content to 3 mgC g−1cat. In our work, we were not able to obtain a catalyst that coked less.
To summarise, in our work caesium and sodium were more effective at reducing the overall coke content than manganese and potassium at low promoter loadings. At higher promoter loadings, the low total coke content prevents any differentiation between the samples. Literature has already hinted at Cs being more effective at reducing the carbon formation than K.20 Horiuchi et al. compared the relative effectiveness of sodium and potassium in suppressing coke formation. Still, their data do not establish a clear trend.26 Alkali metals are well-known coke gasification catalysts (e.g.ref. 65 and 66) and typically outperform manganese oxides in this respect.36,67 Thus, the comparable coke content of 1Mn and 1K implies the site-blocking mechanism dominating in these cases. The superior performance of 1Cs and 1Na may well be due to an increased gasification activity.
A different effectivity at reducing the coke content could result in different coke structures for the individual promoters. Besides the presence of carbon fibres (Fig. S1†), TEM analysis revealed carbon agglomerates of initially unclear structure. HRTEM analysis indicated that these agglomerates are composed at least partially of overlapping and intertwining fibres (Fig. S2†). Additionally, at high resolution, non-hollow coke structures could be identified on the catalyst surface (Fig. 6 and S3†). This structure also appears ordered when contrasted with published TEM images of amorphous carbon deposits.42 Lastly, it must be mentioned that the most carbon fibres were observed, when analysing samples REF and 1Mn.
 | ||
| Fig. 6 HRTEM images of non-hollow carbon (left – sample REF) and carbon filaments (right – sample 1K) after 12 h coking treatment. | ||
Raman spectroscopy is a potential tool to probe the nature of the carbon species in dry reforming catalysts.68 In particular the graphitic coke species are probed, since Raman spectra of carbons are typically dominated by the characteristic bands of sp2 species.69Fig. 7 shows the spectra for the pure Ni/ZrO2 and all samples with a 1/10 promoter ratio. The only exception is 1Cs for which no Raman spectrum of the carbon could be obtained.
 | ||
| Fig. 7 Raman spectra for coked catalysts. | ||
Sp2 carbons typically display two different Raman bands: the D band (approx. 1360 cm−1) and G band (approx. 1560 cm−1). The latter has been correlated to the tangential vibrations of C–C bonds, while the D-band indicates the defect density within the graphitic structure.68 In Fig. 7 all samples exhibit these two bands. To further differentiate between the samples, the relative intensity of the D- and G-bands can be calculated for the different samples. A high value of the ID/IG is a sign of graphitic carbon with high defect density or of nanocrystalline graphite.69,70 Carbon nanotubes are highly structured graphitic species and should thus lead to a more pronounced G-band relative to the D-band.70
The ID/IG values for all samples are in the range of 1.45–1.55, whereas Stroud et al. reported values between 0.8 and 2.0.68 However, they also reported different ratios for measurements carried out on different regions of the same catalyst. These observations can be explained by considering the contribution of carbon fibres with their low defect density. During the TEM analysis described above a distinct clustering of carbon fibres was observed. Stroud et al. did not report coke quantification, but the catalysts investigated contained roughly five times more Ni than the samples in this work and were tested for more extended periods of time.68 This ought to lead to significantly higher coke content and thus more clusters of carbon fibres. It is possible that the different ratios of ID/IG reported previously were caused by analysing areas of coke containing vastly different percentages of carbon fibres. Fewer clusters of fibres make their contribution to the measured Raman spectra unlikely in this work. The calculated ID/IG values of approx. 1.5 are too low for only the presence of nanocrystalline graphite.69,71 Instead they are indicative of the existence of amorphous carbon on all samples in addition to nanocrystalline graphite.
To check this hypothesis, the pure Ni/ZrO2 and all samples with a promoter/Ni ratio of 1/10 were analysed with 13C-NMR to investigate the character of the carbon species further. The presence of magnetic nickel on the catalysts required high spinning frequencies and short relaxation times to obtain information on the carbon without interference. At the same time, the low carbon content required measurement periods of approx. 24 h per sample. As can be seen in Fig. 8, the spectra of 1Cs, 1K and 1Na on the one hand and REF and 1Mn, on the other hand, are quite similar.
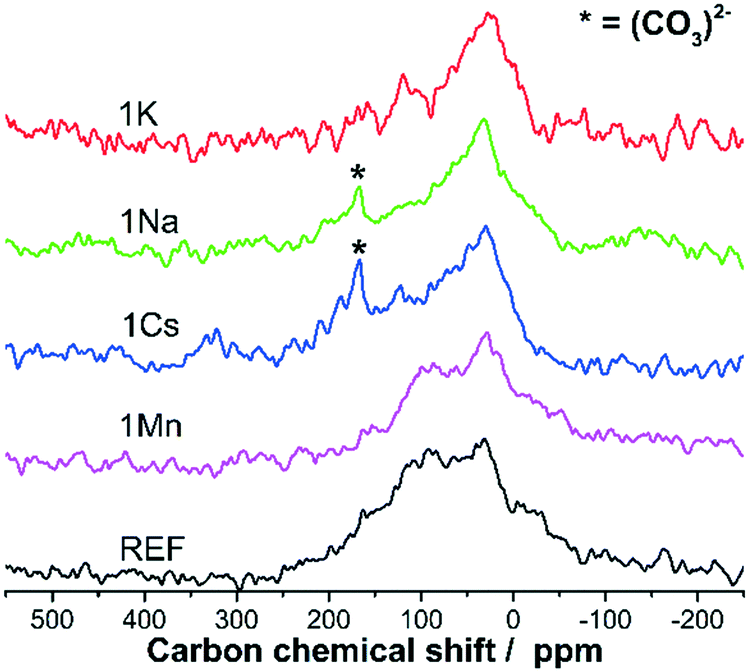 | ||
| Fig. 8 13C-NMR spectra of the non-promoted (REF) Ni/ZrO2 sample and all catalysts with a promoter loading of 1/10. | ||
All samples clearly show a peak in the range of 0–50 ppm, typically associated with alkanes and sp3-hybridized carbons. Therefore, this evidences the presence of amorphous carbon with its contributions to both the sp2 and sp3-range of the NMR spectrum.72,73 Pure amorphous carbon would consist of two peaks around 30 ppm and 120 ppm. Instead, REF and 1Mn contain a significant peak around 100 ppm. For 1K the main peak decreases slowly with increasing chemical shift, and a smaller peak is visible at approx. 120 ppm. For 1Cs and 1Na, instead of a distinct peak at 120 ppm, a shoulder can still be identified in this range. Additionally, these two samples also display a comparatively sharp peak at 167 ppm.
The contribution between 50–100 ppm may be due to several reasons. Firstly, hydrogen-poor amorphous carbon has been observed to display clear peaks or shoulders in the sp3-peak at around 70 ppm.73 The less hydrogen is present in the amorphous carbon, the more apparent this peak at 70 ppm becomes. Secondly, the existence of carbon fibres was proven by TEM for almost all samples. These fibres are typically compared to carbon nanotubes. Pristine nanotubes display a chemical shift of 100–130 ppm, depending on their diameter.74 At the same time, the encapsulation of hydrocarbons within carbon nanotubes typically reduces the chemical shift of these molecules in 13C-NMR.74,75 Sp2-hydrocarbons (typically 120–160 ppm) encapsulated in the fibres could then contribute in the range of 80–100 ppm.
Thus, we consider the carbon deposits to contain amorphous carbon with a noticeable sp3-contribution. For REF and 1Mn the NMR spectra further show the presence of significant amounts of carbon fibres. For the alkali-promoted samples, the small peak or shoulder at 120 ppm is more in keeping with a small sp2-contribution in amorphous carbon. The comparison of the different spectra shows that while all promoters reduce overall carbon levels, alkali metals are more effective at preventing the formation of carbon fibres. This is in line with TEM analysis, where carbon fibres were easier to detect for REF and 1Mn than for the alkali-promoted samples.
The NMR results for all alkali-promoted samples are mostly comparable, especially since at such low coke contents contaminations could quickly influence the signal. The only exception is the peak mentioned above at approx. 167 ppm for 1Cs and 1Na, which is absent for 1K. The peak is attributed to carbonate species present in the spent catalyst in the form of either alkali carbonate formation or as Zr(CO3)2. A clear identification was not possible since the chemical shift of the different carbonates differ only slightly.76–79 However, the coke contents of all three samples are in the same order of magnitude. If the peak were due to Zr(CO2)3, it should also be visible in the spectrum of 1K. This is a strong indication of the presence of Na2CO3 and Cs2CO3.
The detection of carbonates is in line with the previous proposal of enhanced coke gasification for the samples 1Cs and 1Na. Carbonate formation is commonly considered an important step in the (earth) alkali-catalysed gasification of carbon with CO2.39,80,81 The accepted order of catalytic activity in coal gasification of the alkali metals used in this work is Cs > K > Na.81 However, this relative activity is affected by the dispersion of the alkali metals on the carbon, since Na is known to agglomerate on carbon surfaces.39 At the same time, the CO2 absorption capacity of Na2ZrO3 has been reported previously.82,83 For Na-promoted Ni/ZrO2 a coke gasification cycle including Na2ZrO3 and Na2CO3 has already been proposed for dry reforming of methane.27 Thus, whereas Cs itself is a very active gasification catalyst, the activity of Na may be increased through interaction with the support.
Summarising, the coke quantification shows that all promoters reduce the coke content significantly compared to the non-promoted REF Ni/ZrO2 catalyst. However, 1Na and 1Cs only contain approx. 50% of the carbon deposited on 1Mn and 1K. Raman spectroscopy did not reveal any differences in the structure of the non-fibrous carbon on the different samples. At the same time TEM and 13C-NMR show a higher propensity of 1Mn and REF to form carbon fibres. The relative amount of the various carbon species and thus, the overall carbon composition appears to be a function of the promoter. We did not observe any effect of the promoters on the structure of the individual carbon species. Additionally, NMR showed significant contributions of sp3-hybridized carbon in all samples, which is assumed to be amorphous carbon. Operation at temperatures such as 650 °C and higher is expected to produce coke with high levels of graphitization.14,84 This is such an accepted assumption that in reviews on methane reforming, the words graphite and coke have been used interchangeably.13 The presence of sp3 signals shows, that, at least at 650 °C, full graphitisation cannot yet be assumed.
Furthermore, the NMR spectra of 1Na and 1Cs show the presence of carbonates indicating the presence of Cs2CO3 and Na2CO3 and thus carbon gasification activity for the two samples with the lowest coke content. The results in this publication show that the same promoted catalyst can achieve site blocking and carbon gasification. However, the overall catalytic effectiveness is not directly correlated to the results of classical coal gasification. It appears that the superior interaction of Na and ZrO2 compared to Na and coal cause 1Na to outperform 1K, despite K being reported to be the superior coal gasification catalyst.
Conclusion
A series of Ni/ZrO2 catalysts with different promoter metals and promoter ratios was synthesised. TPR studies led to the observation of different effects of the metal promotors on Ni reducibility, which indicate smaller Ni particles when adding small promoter amounts. STEM analysis reveals that for all samples the majority of the Ni particles is smaller than 10 nm before the reaction. Despite varying effects on the reducibility of Ni, the different promoters lead to almost identical conversion in the dry reforming of methane at 650 °C. A difference only becomes noticeable in the analysis of coked samples.While all promoters strongly reduce the coke content compared to the pristine Ni/ZrO2, the degree of coke reduction depends on the promoter. 13C-NMR analysis shows the presence of sp3-hybridized carbon in all samples, despite literature frequently assuming full graphitisation at these temperatures. In combination with TEM analysis, it could be established that alkali metals are more effective at suppressing fibre growth than manganese. At the same time, samples promoted with sodium or caesium coked less than a sample coked with potassium. These two samples are also the only ones, for which carbonate species could be detected in NMR. Therefore, it is proposed that the lower coke content is caused by coke gasification. Comparison with literature indicates that the higher than expected activity of the Na-promoted sample can be attributed to superior interactions between Na and the ZrO2 support compared to Na on carbon in typical coal gasification.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the CatC1Chem project of NWO, BASF, SABIC and Sasol is gratefully acknowledged.Notes and references
- BP Energy Outlook 2019 edition, https://www.bp.com/content/dam/bp/business-sites/en/global/corporate/pdfs/energy-economics/energy-outlook/bp-energy-outlook-2019.pdf, (accessed Apr. 17, 2020).
- T. L. Roussière, PhD, Karlsruhe Institute of Technology, 2013 Search PubMed.
- J. Zhang, H. Wang and A. K. Dalai, J. Catal., 2007, 249, 300–310 CrossRef CAS.
- I. K. van Ravenhorst, C. Vogt, H. Oosterbeek, K. W. Bossers, J. G. Moya-Cancino, A. P. van Bavel, A. M. J. van der Eerden, D. Vine, F. M. F. de Groot, F. Meirer and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2018, 57, 11957–11962 CrossRef CAS PubMed.
- R. Gaikwad, A. Bansode and A. Urakawa, J. Catal., 2016, 343, 127–132 CrossRef CAS.
- L. Tillmann, J. Schulwitz, A. van Veen and M. Muhler, Catal. Lett., 2018, 148, 2256–2262 CrossRef CAS.
- L. C. S. Kahle, T. Roussière, L. Maier, K. Herrera Delgado, G. Wasserschaff, S. A. Schunk and O. Deutschmann, Ind. Eng. Chem. Res., 2013, 52, 11920–11930 CrossRef CAS.
- J. A. Moulijn, A. E. van Diepen and F. Kapteijn, Appl. Catal., A, 2001, 212, 3–16 CrossRef CAS.
- N. A. K. Aramouni, J. G. Touma, B. A. Tarboush, J. Zeaiter and M. N. Ahmad, Renewable Sustainable Energy Rev., 2018, 82, 2570–2585 CrossRef CAS.
- H. Düdder, K. Kähler, B. Krause, K. Mette, S. Kühl, M. Behrens, V. Scherer and M. Muhler, Catal. Sci. Technol., 2014, 4, 3317–3328 RSC.
- B. AlSabban, L. Falivene, S. M. Kozlov, A. Aguilar-Tapia, S. Ould-Chikh, J.-L. Hazemann, L. Cavallo, J.-M. Basset and K. Takanabe, Appl. Catal., B, 2017, 213, 177–189 CrossRef CAS.
- S. A. Theofanidis, V. V. Galvita, H. Poelman and G. B. Marin, ACS Catal., 2015, 5, 3028–3039 CrossRef CAS.
- J. R. Rostrup-Nielsen, J. Sehested and J. K. Nørskov, in Adv. Catal., Academic Press, 2002, vol. 47, pp. 65–139 Search PubMed.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- R. T. K. Baker, P. S. Harris, J. Henderson and R. B. Thomas, Carbon, 1975, 13, 17–22 CrossRef CAS.
- I. Chen and F. L. Chen, Ind. Eng. Chem. Res., 1990, 29, 534–539 CrossRef CAS.
- F. Frusteri, F. Arena, G. Calogero, T. Torre and A. Parmaliana, Catal. Commun., 2001, 2, 49–56 CrossRef CAS.
- J. Juan-Juan, M. C. Román-Martínez and M. J. Illán-Gómez, Appl. Catal., A, 2006, 301, 9–15 CrossRef CAS.
- T. Osaki and T. Mori, J. Catal., 2001, 204, 89–97 CrossRef CAS.
- C. Ping, H. Zhao-Yin and Z. Xiao-Ming, Chin. J. Chem., 2005, 23, 847–851 CrossRef.
- J. R. Rostrup-Nielsen, J. Catal., 1984, 85, 31–43 CrossRef CAS.
- S. Arora and R. Prasad, RSC Adv., 2016, 6, 108668–108688 RSC.
- H. S. Bengaard, I. Alstrup, I. Chorkendorff, S. Ullmann, J. R. Rostrup-Nielsen and J. K. Nørskov, J. Catal., 1999, 187, 238–244 CrossRef CAS.
- F. Besenbacher, I. Chorkendorff, B. S. Clausen, B. Hammer, A. M. Molenbroek, J. K. Nørskov and I. Stensgaard, Science, 1998, 279, 1913–1915 CrossRef CAS PubMed.
- H. S. Bengaard, J. K. Nørskov, J. Sehested, B. S. Clausen, L. P. Nielsen, A. M. Molenbroek and J. R. Rostrup-Nielsen, J. Catal., 2002, 209, 365–384 CrossRef CAS.
- T. Horiuchi, K. Sakuma, T. Fukui, Y. Kubo, T. Osaki and T. Mori, Appl. Catal., A, 1996, 144, 111–120 CrossRef CAS.
- M. Németh, D. Srankó, J. Károlyi, F. Somodi, Z. Schay, G. Sáfrán, I. Sajó and A. Horváth, Catal. Sci. Technol., 2017, 7, 5386–5401 RSC.
- Z. Hou, O. Yokota, T. Tanaka and T. Yashima, Appl. Surf. Sci., 2004, 233, 58–68 CrossRef CAS.
- S.-H. Seok, S. H. Han and J. S. Lee, Appl. Catal., A, 2001, 215, 31–38 CrossRef CAS.
- D. W. Flaherty, W.-Y. Yu, Z. D. Pozun, G. Henkelman and C. B. Mullins, J. Catal., 2011, 282, 278–288 CrossRef CAS.
- A. Díaz, D. R. Acosta, J. A. Odriozola and M. Montes, J. Phys. Chem. B, 1997, 101, 1782–1790 CrossRef.
- W. D. Mross, Catal. Rev.: Sci. Eng., 1983, 25, 591–637 CrossRef CAS.
- A. Parmaliana, F. Arena, F. Frusteri, N. Mondello and N. Giordano, in Stud. Surf. Sci. Catal., ed. C. H. Bartholomew and J. B. Butt, Elsevier, 1991, vol. 68, pp. 489–492 Search PubMed.
- A. Ehrmaier, L. Löbbert, M. Sanchez-Sanchez, R. Bermejo-Deval and J. Lercher, ChemCatChem, 2020 DOI:10.1002/cctc.202000349.
- P. Littlewood, X. Xie, M. Bernicke, A. Thomas and R. Schomäcker, Catal. Today, 2015, 242, 111–118 CrossRef CAS.
- M. Keller, H. Leion and T. Mattisson, Energy Technol., 2013, 1, 273–282 CrossRef CAS.
- D. W. McKee, Carbon, 1974, 12, 453–464 CrossRef CAS.
- J. A. Moulijn and F. Kapteijn, in Carbon and Coal Gasification: Science and Technology, ed. J. L. Figueiredo and J. A. Moulijn, Springer, Netherlands, Dordrecht, 1986, pp. 181–195, DOI:10.1007/978-94-009-4382-7_6.
- J. A. Moulijn and F. Kapteijn, Carbon, 1995, 33, 1155–1165 CrossRef CAS.
- J. L. Rogers, M. C. Mangarella, A. D. D'Amico, J. R. Gallagher, M. R. Dutzer, E. Stavitski, J. T. Miller and C. Sievers, ACS Catal., 2016, 6, 5873–5886 CrossRef CAS.
- R. H. Nielsen and G. Wilfing, in Ullmann's Encyclopedia of Industrial Chemistry, 2010, DOI:10.1002/14356007.a28_543.pub2.
- C. Wang, N. Sun, N. Zhao, W. Wei, J. Zhang, T. Zhao, Y. Sun, C. Sun, H. Liu and C. E. Snape, ChemCatChem, 2014, 6, 640–648 CrossRef CAS.
- R.-J. Zhang, G.-F. Xia, M.-F. Li, Y. Wu, H. Nie and D.-D. Li, J. Fuel Chem. Technol., 2015, 43, 1359–1365 CrossRef CAS.
- A. Wolfbeisser, O. Sophiphun, J. Bernardi, J. Wittayakun, K. Föttinger and G. Rupprechter, Catal. Today, 2016, 277, 234–245 CrossRef CAS.
- C. Nederlof, V. Zarubina, I. Melián-Cabrera, H. J. Heeres, F. Kapteijn and M. Makkee, Catal. Sci. Technol., 2013, 3, 519–526 RSC.
- R. J. Berger, J. Pérez-Ramírez, F. Kapteijn and J. A. Moulijn, Chem. Eng. Sci., 2002, 57, 4921–4932 CrossRef CAS.
- N. D. Charisiou, S. L. Douvartzides, G. I. Siakavelas, L. Tzounis, V. Sebastian, V. Stolojan, S. J. Hinder, M. A. Baker, K. Polychronopoulou and M. A. Goula, Catalysts, 2019, 9, 676 CrossRef CAS.
- P. M. Mortensen, D. Gardini, H. W. P. de Carvalho, C. D. Damsgaard, J.-D. Grunwaldt, P. A. Jensen, J. B. Wagner and A. D. Jensen, Catal. Sci. Technol., 2014, 4, 3672–3686 RSC.
- W. L. Vrijburg, E. Moioli, W. Chen, M. Zhang, B. J. P. Terlingen, B. Zijlstra, I. A. W. Filot, A. Züttel, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2019, 9, 7823–7839 CrossRef CAS.
- W. L. Vrijburg, J. W. A. van Helden, A. Parastaev, E. Groeneveld, E. A. Pidko and E. J. M. Hensen, Catal. Sci. Technol., 2019, 9, 5001–5010 RSC.
- R. K. Singha, A. Shukla, A. Yadav, S. Adak, Z. Iqbal, N. Siddiqui and R. Bal, Appl. Energy, 2016, 178, 110–125 CrossRef CAS.
- E. Taglauer, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, 2008, pp. 1014–1029, DOI:10.1002/9783527610044.hetcat0051.
- G. Marcelin and J. E. Lester, J. Catal., 1985, 93, 270–278 CrossRef CAS.
- D. M. Stockwell, A. Bertucco, G. W. Coulston and C. O. Bennett, J. Catal., 1988, 113, 317–324 CrossRef CAS.
- M. Steib, Y. Lou, A. Jentys and J. A. Lercher, ChemCatChem, 2017, 9, 3809–3813 CrossRef CAS.
- C. Li and Y.-W. Chen, Thermochim. Acta, 1995, 256, 457–465 CrossRef CAS.
- B. Mile, D. Stirling, M. A. Zammitt, A. Lovell and M. Webb, J. Mol. Catal., 1990, 62, 179–198 CrossRef CAS.
- A. Peters, F. Nouroozi, D. Richter, M. Lutecki and R. Gläser, ChemCatChem, 2011, 3, 598–606 CrossRef CAS.
- F. Morales, D. Grandjean, A. Mens, F. M. F. de Groot and B. M. Weckhuysen, J. Phys. Chem. B, 2006, 110, 8626–8639 CrossRef CAS PubMed.
- W. L. Vrijburg, E. Moioli, W. Chen, M. Zhang, B. J. P. Terlingen, B. Zijlstra, I. A. W. Filot, A. Züttel, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2019, 7823–7839, DOI:10.1021/acscatal.9b01968.
- L. D. Dyer, B. S. Borie and G. P. Smith, J. Am. Chem. Soc., 1954, 76, 1499–1503 CrossRef CAS.
- C. Park and M. A. Keane, J. Colloid Interface Sci., 2002, 250, 37–48 CrossRef CAS PubMed.
- H.-S. Roh, H. S. Potdar, K.-W. Jun, J.-W. Kim and Y.-S. Oh, Appl. Catal., A, 2004, 276, 231–239 CrossRef CAS.
- N. T. Andersen, F. Topsøe, I. Alstrup and J. R. Rostrup-Nielsen, J. Catal., 1987, 104, 454–465 CrossRef CAS.
- F. Kapteijn, O. Peer and J. A. Moulijn, Fuel, 1986, 65, 1371–1376 CrossRef CAS.
- D. W. McKee, Fuel, 1983, 62, 170–175 CrossRef CAS.
- M. Arjmand, H. Leion, T. Mattisson and A. Lyngfelt, Appl. Energy, 2014, 113, 1883–1894 CrossRef CAS.
- T. Stroud, T. J. Smith, E. Le Saché, J. L. Santos, M. A. Centeno, H. Arellano-Garcia, J. A. Odriozola and T. R. Reina, Appl. Catal., B, 2018, 224, 125–135 CrossRef CAS.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus, A. Jorio, A. G. Souza Filho and R. Saito, Carbon, 2002, 40, 2043–2061 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- R. H. Jarman, G. J. Ray, R. W. Standley and G. W. Zajac, Appl. Phys. Lett., 1986, 49, 1065–1067 CrossRef CAS.
- J. Braddock-Wilking, S. H. Lin and B. J. Feldman, Solid State Commun., 2001, 119, 19–21 CrossRef CAS.
- E. Abou-Hamad, M. R. Babaa, M. Bouhrara, Y. Kim, Y. Saih, S. Dennler, F. Mauri, J. M. Basset, C. Goze-Bac and T. Wågberg, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 165417 CrossRef.
- N. A. Besley and A. Noble, J. Chem. Phys., 2008, 128, 101102 CrossRef.
- F. Takasaki, K. Fujiwara, Y. Nakajima, T. Nishikawa, H. Masu, M. Imanari, Y. Hidaka and N. Ogawa, Dalton Trans., 2015, 44, 645–652 RSC.
- National Center for Biotechnology Information. PubChem Database. Sodium carbonate, CID=10340, https://pubchem.ncbi.nlm.nih.gov/compound/Sodium-carbonate, (accessed Feb. 5, 2020).
- National Center for Biotechnology Information. PubChem Database. Cesium carbonate, CID=10796, https://pubchem.ncbi.nlm.nih.gov/compound/Cesium-carbonate, (accessed Feb. 5, 2020).
- National Center for Biotechnology Information. PubChem Database. Potassium carbonate, CID=11430, https://pubchem.ncbi.nlm.nih.gov/compound/Potassium-carbonate, (accessed Feb. 5, 2020).
- D. Cazorla-Amorós, A. Linares-Solano, F. H. M. Dekker and F. Kapteijn, Carbon, 1995, 33, 1147–1154 CrossRef.
- S. G. Chen and R. T. Yang, Energy Fuels, 1997, 11, 421–427 CrossRef CAS.
- H. G. Jo, H. J. Yoon, C. H. Lee and K. B. Lee, Ind. Eng. Chem. Res., 2016, 55, 3833–3839 CrossRef CAS.
- G. G. Santillan-Reyes and H. Pfeiffer, Int. J. Greenhouse Gas Control, 2011, 5, 1624–1629 CrossRef CAS.
- J. M. Ginsburg, J. Piña, T. El Solh and H. I. de Lasa, Ind. Eng. Chem. Res., 2005, 44, 4846–4854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy00817f |
| This journal is © The Royal Society of Chemistry 2020 |