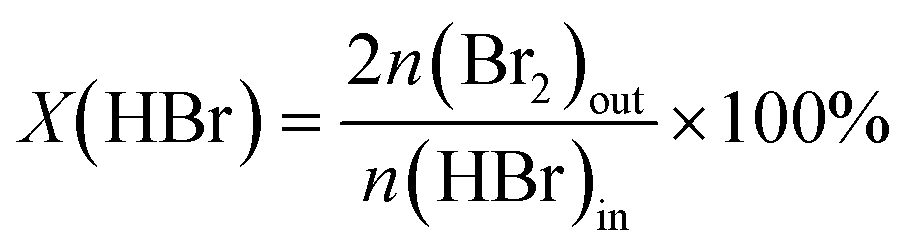Transformation of titanium carbide into mesoporous titania for catalysed HBr oxidation†
Received
20th April 2020
, Accepted 25th May 2020
First published on 1st June 2020
Abstract
Herein, we report TiC as precursor of a highly active and novel TiO2–TiC composite for the catalytic oxidation of HBr into Br2, which is a key technology for bromine looping in the halogen-mediated process for natural gas valorisation. In addition, we investigate the mechanism of TiO2 formation that is pivotal in deriving synthesis–structure–performance relationships. In-depth material characterisation, by means of X-ray diffraction, thermogravimetric analysis, N2 sorption, Raman, X-ray photoelectron, 47,49Ti and 13C nuclear magnetic resonance spectroscopies, as well as ex situ and in situ transmission electron microscopy reveal that the transformation of TiC proceeds via a combination of shrinking core and spot-oxidation mechanisms. This results in the formation of a TiO2–TiC composite, containing both anatase and rutile polymorphs, with enhanced mesoporosity, reaching up to a one order of magnitude higher surface area and pore volume compared to pristine TiC. Reactivity and kinetics studies are performed in HBr oxidation, showing that the catalyst activity follows a volcano behaviour, whose apex is found for the material calcined at 673 K. Its performance surpasses that of benchmark TiO2-P25 and is also found stable for 25 h on stream. Accordingly, the reactivity of the TiO2–TiC composite is rationalized based on the TiO2 generation and porosity formation, which are both functions of the oxidation temperature.
1. Introduction
The bromine-mediated functionalization of methane and of other alkanes has been demonstrated as an attractive technology for the valorisation of natural gas into chemicals and fuels at moderate temperatures (<833 K) and near-to-ambient pressures, which could overcome the challenges related to the capital- and energy-intensiveness of existing steam-based industrial processes.1–6 Nevertheless, the full recovery of by-product hydrogen bromide (HBr), which is formed in stoichiometric amounts during the alkane bromination and upgrading steps of the thus obtained bromoalkanes, constitutes a pivotal prerequisite for the potential commercialisation of this technology.1 The heterogeneously catalysed oxidation of HBr into molecular bromine (Br2) represents a highly attractive route to close the halogen loop, thanks to its low energy requirements, process simplicity, and continuous operation. However, catalysts typically suffer from deactivation due to extensive bromination of the sub-surface and bulk structure that occurs under stoichiometric feeds (O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HBr = 0.25), which are highly desirable to reduce reactor volume and to allow for the use of air as an O2 source.1,7–11 To date, titanium oxide (TiO2) represents the only earth-abundant catalytic material that was demonstrated stable under oxygen-lean conditions due to its ability to limit the bromination to its surface.8,9 The reactivity of this oxide originates from a self-doping mechanism, in which adsorbed HBr generates a Ti3+ defect that facilitates bromine evolution and enables the otherwise highly-endothermic O2 activation.10 Nevertheless, despite being fundamentally and practically relevant, the reactivity of self-synthesised TiO2-based catalysts has not been studied yet, since only commercial phases have been tackled so far.7–10 In this regard, we turned our attention to titanium carbide (TiC) as precursor of TiO2, since its controlled thermal oxidation enables to carefully tune the formation of the oxide phase. In addition, the generation of carbon dioxide (CO2) during the transformation of TiC into TiO2–TiC could lead to interesting structural alterations such as pore formation which often increases catalytic activity. This system has already been proven as a highly effective carrier in several applications, ranging from electrocatalysis to Fischer–Tropsch synthesis,12–16 although its use as an active phase has never been reported in thermal catalysis. Still, despite preliminary insights have been reported,16 there is a lack of understanding on the mechanism of oxidation of this material and on the concomitant evolution of its structural, morphological, and textural properties, which is indispensable to derive synthesis–structure–performance relationships that lead to optimal catalyst design criteria.
:HBr = 0.25), which are highly desirable to reduce reactor volume and to allow for the use of air as an O2 source.1,7–11 To date, titanium oxide (TiO2) represents the only earth-abundant catalytic material that was demonstrated stable under oxygen-lean conditions due to its ability to limit the bromination to its surface.8,9 The reactivity of this oxide originates from a self-doping mechanism, in which adsorbed HBr generates a Ti3+ defect that facilitates bromine evolution and enables the otherwise highly-endothermic O2 activation.10 Nevertheless, despite being fundamentally and practically relevant, the reactivity of self-synthesised TiO2-based catalysts has not been studied yet, since only commercial phases have been tackled so far.7–10 In this regard, we turned our attention to titanium carbide (TiC) as precursor of TiO2, since its controlled thermal oxidation enables to carefully tune the formation of the oxide phase. In addition, the generation of carbon dioxide (CO2) during the transformation of TiC into TiO2–TiC could lead to interesting structural alterations such as pore formation which often increases catalytic activity. This system has already been proven as a highly effective carrier in several applications, ranging from electrocatalysis to Fischer–Tropsch synthesis,12–16 although its use as an active phase has never been reported in thermal catalysis. Still, despite preliminary insights have been reported,16 there is a lack of understanding on the mechanism of oxidation of this material and on the concomitant evolution of its structural, morphological, and textural properties, which is indispensable to derive synthesis–structure–performance relationships that lead to optimal catalyst design criteria.
In this work, the transformation of TiC into TiO2–TiC composites was investigated and the resulting systems were assessed in HBr oxidation. An array of in situ and ex situ characterisation techniques were applied to follow the crystal structure, porous architecture, and morphological formations before, during, and after the transformation from carbide to oxide. The resulting material obtained from the intermediate temperature range was a mesoporous TiO2–TiC composite demonstrating superior activity and stability in the catalytic oxidation of HBr.
2. Experimental
Catalysts
Titanium carbide (TiC; Alfa Aesar, 99.8%) powder was calcined in flowing air (100 cm3 STP min−1) for 5 h at 598–1273 K with a heating rate of 5 K min−1. The resulting materials were denoted as TiC-T, where T indicates the calcination temperature in Kelvin. It is important to clarify that the catalyst nomenclature does not imply any phase composition, while it only represents the oxidation temperature used in the synthesis. Commercially available TiO2-anatase (Aldrich, 99.7%) and TiO2-rutile (Aldrich, 99.9%) as well as TiO2-P25 (Acros, 99.9%), which were utilized as reference for 47,49Ti nuclear magnetic resonance and as benchmark in HBr oxidation, respectively, were calcined in static air at 873 K for 5 h (5 K min−1) prior to their use. The solids were pressed and sieved to the desired average particle size (dp = 0.4–0.6 mm) prior to implementation in the catalytic tests.
Methods
X-ray diffraction (XRD) patterns were acquired in a PANalytical X'Pert PRO-MPD diffractometer with Bragg–Brentano geometry by applying Ni-filtered Cu-Kα radiation (λ = 1.54060 Å). The data were recorded in the 10–70° 2θ range with an angular step size of 0.033° and a counting time of 4.2 s per step. N2 sorption at 77 K was measured in a Micromeritics TriStar II analyser. Samples were evacuated at 423 K for 12 h prior to the measurement. The Brunauer–Emmett–Teller (BET) method was applied to calculate the total surface area, SBET. The pore volume, Vpore, was determined from the amount of N2 adsorbed at a relative pressure of p/p0 = 0.98. Raman spectroscopy was carried out on a WITec CRM200 confocal Raman system using a 532 nm laser with 20 mW power, a 100× objective lens with numerical aperture NA = 0.9 (Nikon Plan) as well as a fibre-coupled grating spectrometer (2400 lines per mm), giving a spectral sampling resolution of 0.7 cm−1. Thermogravimetric analysis (TGA) was performed using a Linseis TG-DSC analyser (STA PT 1600). The solids were heated to 1273 K at 5 K min−1 in a flowing (300 cm3 STP min−1) 20 vol% O2/Ar mixture. X-ray photoelectron spectroscopy (XPS) was measured in a Physical Electronics Quantum 2000 X-ray photoelectron spectrometer using monochromatic Al-Kα radiation, generated from an electron beam operated at 15 kV, and equipped with a hemispherical capacitor electron-energy analyser. The solids were mounted onto the sample holder by pressing the powders onto indium foil, and then analysed at the electron take-off angle of 45° and the pass energy of 46.95 eV. The spectrometer was calibrated for the Au 4f7/2 signal to be at 84.0 eV and used a minimum incremental energy step of 0.2 eV. To further compensate for charging of non-conductive samples, all spectra were referenced to the Ti 2p3/2 of TiO2 at 458.5 eV.17 The Ti 2p signal was fitted by mixed Gaussian–Lorentzian component profiles after Shirley background subtraction. The selected peak positions of the different titanium species are based on reported data and fixed with an error of ±0.2 eV. Depth profiles were recorded by employing alternating cycles of XPS analysis and sputtering with a focused 2 kV Ar+ beam rastered over an area of 4 mm2. The sputter yield was calibrated to be 10 nm min−1 on a 100 nm Ta2O5 reference film. The sputtering rate on the tested samples was estimated at 7.3 nm min−1 by adopting the reference value for TiO2, which is etched at 0.73 times the rate of Ta2O5.18 For non-sputtered samples, the TiO2 and TiC composition was determined by fitting the Ti 2p signal (Fig. S1 of the ESI†). For sputtered samples, this was not possible due to an argon beam-induced peak broadening, so the TiO2 and TiC composition was inferred by the relative atomic ratio of oxygen and carbon.1947,49Ti nuclear magnetic resonance (NMR) spectroscopy was performed on a Bruker Avance III HD 400 spectrometer equipped with a 9.4 T magnet at a resonance frequency of 22.5 MHz using a single channel static probehead with a 10 mm diameter coil. Data were acquired using a WURST-CPMG pulse sequence.20 A total of 64 echos were acquired with a delay of 0.95 ms between WURST-80 pulses (duration of 50 μs, 1 MHz frequency sweep) with a dwell time corresponding to a 1.25 MHz spectral width.21 A total of 36864 scans with a recycle delay of 0.1 s were co-added. The data were processed with MATLAB® scripts incorporating functions from the MatNMR toolbox.22 The individual echos were apodised with a cos2-function and summed before Fourier transform and plotting of the spectra in magnitude mode. The 13C NMR experiments were performed on a Bruker Avance NEO 400 spectrometer equipped with a 9.4 T magnet at a resonance frequency of 100.69 MHz. Samples were loaded into 4 mm ZrO2 rotors and measured in a 4 mm double resonance magic angle spinning probehead, using only a single channel tuned to the resonance frequency of 13C and without rotation. A standard static Hahn-echo pulse sequence was used with a 3.0 μs and 6.0 μs for the 90- and 180-degree pulse, respectively, and an echo delay of 200 μs, which allowed the acquisition of a full echo signal. The spectral width was chosen to be 1600 ppm centred at 300 ppm, the recycle delay was set to 1.0 s and a total of 16384 scans were accumulated. The magnetic field was calibrated using a 13C magic angle spinning NMR spectrum of adamantane at 38.52 ppm as an external standard. High-resolution transmission electron microscopy (HRTEM) and in situ TEM experiments were conducted on a double-corrected, JEM-ARM300F (Grand ARM, JEOL) microscope operated at 300 kV. For HRTEM, the samples were dispersed in ethanol and a few droplets were deposited onto perforated carbon foil supported on a copper grid. After evaporation of the ethanol, the grid was mounted on the single tilt holder of the microscope and treated for 5 min in an ion cleaner (EC-52000IC, JEOL) to minimize contamination before insertion into the microscope vacuum. In situ TEM experiments were performed inside the column of the Grand ARM where sample material was drop-deposited onto a MEMS-based heating chip, covered with a SiNx window chip and then mounted onto a DensSolutions climate holder. After insertion into the TEM, the in situ reactor was left exposed to the atmosphere. In order to avoid possible contamination during imaging, the sample was maintained at 473 K over night before starting the actual experiment. The transformation of particles was observed during heating from 590 to 720 K with a heating rate of 10 K min−1. All movies were recorded using a Gatan OneView camera.
Catalytic testing
The gas-phase HBr oxidation was performed at ambient pressure in a home-made continuous-flow fixed-bed reactor setup. HBr (Messer, 99.8%), O2 (PanGas, purity 5.0), and He (PanGas, purity 4.6) were supplied by Bronkhorst digital mass flow controllers. A quartz reactor (internal diameter, di = 8 mm) was charged with the TiC-T sample (catalyst weight, Wcat = 0.5 g, and particle size, dp = 0.4–0.6 mm) and heated to the desired temperature (T = 523–598 K) under He flow. Thereafter, the reaction mixture was fed with a total volumetric flow, FT, of 100 cm3 STP min−1 and desired feed composition of HBr:O2:He = 4.5:9:86.5 vol%, unless otherwise stated. Downstream lines and valves were heated at 393 K to prevent the condensation of products. Quantitative Br2 analysis was performed by purging the product stream through an impinging bottle containing a 0.1 M KI (Sigma-Aldrich, 99.5%) solution (Br2 + 3I− → 2Br− + I3−), followed by off-line iodometric titration (Mettler Toledo G20 Compact Titrator) of the formed triiodide with an aqueous 0.01 M Na2S2O3 (Alfa Aesar, Standard) solution (I3− + 2S2O32− → 3I− + S4O62−). The conversion of HBr, X(HBr), and the rate of Br2 production based on the catalyst mass, r, or its surface area, rS, were calculated using eqn (1), (2) and (3), respectively,| | 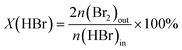 | (1) |
| |  | (2) |
| |  | (3) |
where n(HBr)in, n(Br2)out are the molar flow of HBr and Br2 at the reactor inlet and outlet, respectively, and SBET is the surface area of the catalyst. Evaluation of the dimensionless moduli based on the criteria of Carberry, Mears, and Weisz–Prater confirmed that all catalytic tests were performed in the absence of mass and heat transfer limitations.23,24
3. Results and discussion
Evolution of TiC upon oxidation
Structure.
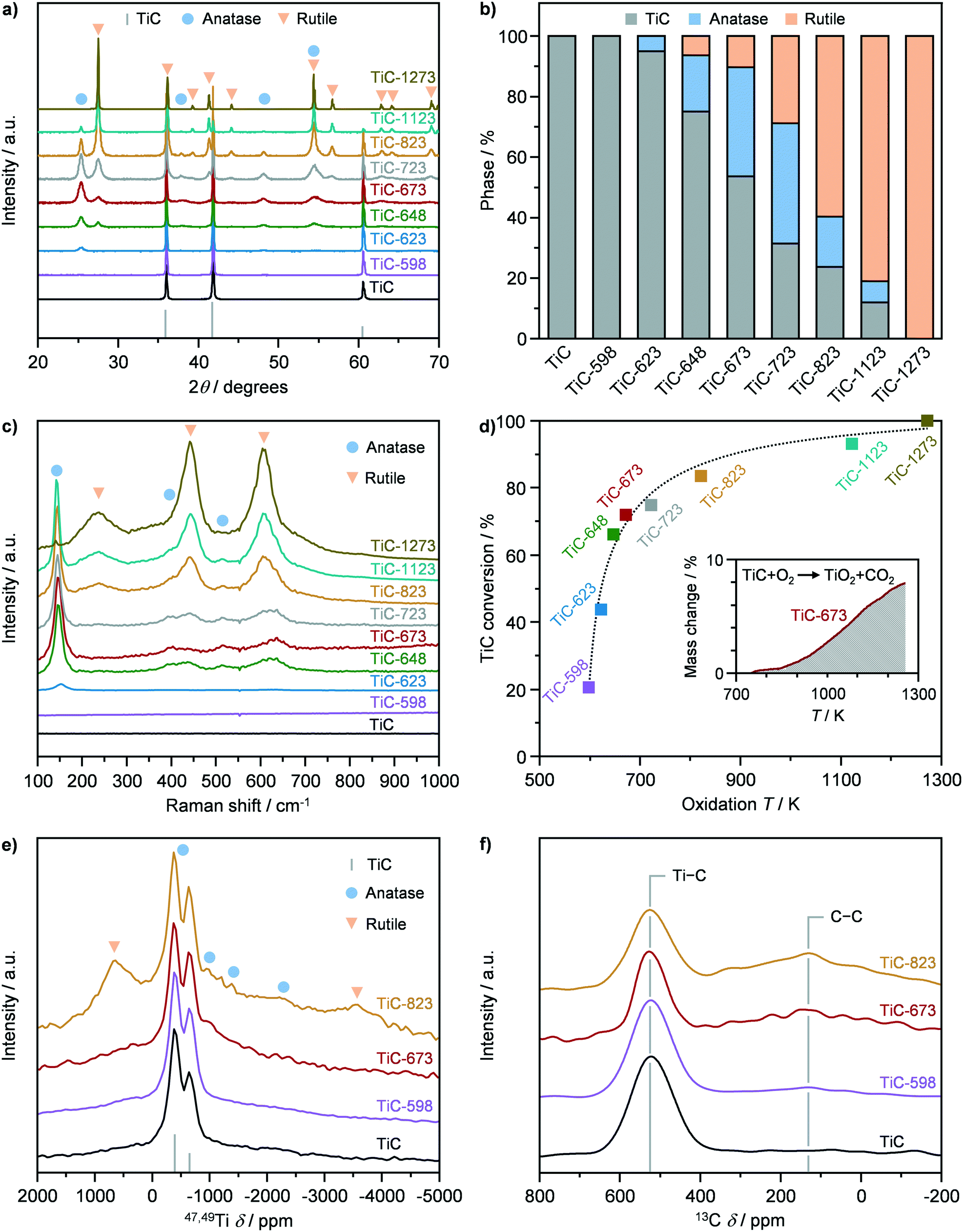
X-ray diffraction and Raman spectroscopy were adopted to assess the crystallographic properties of pristine TiC, prior to its use as a precursor for the synthesis of TiO2–TiC composites. The XRD pattern of TiC showed the presence of only cubic TiC, exhibiting the characteristic reflections (200), (111), and (220) (Fig. 1a and b). On the other hand, Raman spectroscopy showed no vibrational modes, indicating that TiC is Raman inactive (Fig. 1c), in agreement with literature observations.25 TiC was then used as a precursor to synthesize TiO2–TiC composites by thermal oxidation in flowing air. The resulting materials were denoted as TiC-T, where T indicates the oxidation temperature during synthesis, which was varied between 598 K and 1273 K. The structural properties of the thus obtained TiO2–TiC composites were characterised by X-ray diffraction and Raman spectroscopy. The onset of the oxidation occurred at 623 K, where the characteristic reflections and Raman band (145 cm−1) of TiO2-anatase were observed in the diffractograms and Raman spectrum of the TiC-623 sample (Fig. 1a–c),26 respectively. All samples that were oxidized at 648 K and above showed the formation of TiO2-rutile, as corroborated by its characteristic reflections and Raman bands (238 cm−1, 444 cm−1, and 609 cm−1) in the diffractograms and Raman spectra.26 Its portion steadily increased with oxidation temperature until it became the only phase present after oxidation at 1273 K (Fig. 1a and b). On the other hand, the formation of TiO2-anatase followed a volcano behaviour as a function of the oxidation temperature. This resembled the typical trend of a consecutive reaction, indicating that TiO2-rutile might form from the consecutive transformation of TiO2-anatase and not from the direct oxidation of TiC. Also, TiO2 polymorphs coexisted with unreacted TiC in the composite at oxidation temperatures ≤1173 K. This observation contrasts previous studies that have shown a step-like behaviour of the transformation of TiC into TiO2 at 623 K.15,16 However, the material characterisation in these works was mainly limited to XRD, precluding deeper insights on the oxidation mechanism. In order to further support these results, thermogravimetric analysis of the material was conducted. Fig. 1d shows the conversion of TiC as a function of the oxidation temperature, which was derived based on TGA profiles (Fig. S2†). Interestingly, TiC-598 and TiC-623 showed a TiC conversion of ca. 20% and 40%, respectively. This deviated from the results obtained by XRD and Raman, showing the complete or almost complete absence of TiO2, suggesting the presence of an amorphous TiO2 phase or of very small crystalline domains. At increased oxidation temperatures, the differences in the TiC conversion derived from XRD and from TGA analyses decreased, until disappearing in TiC-1273, indicating a general increase in the composite crystallinity.
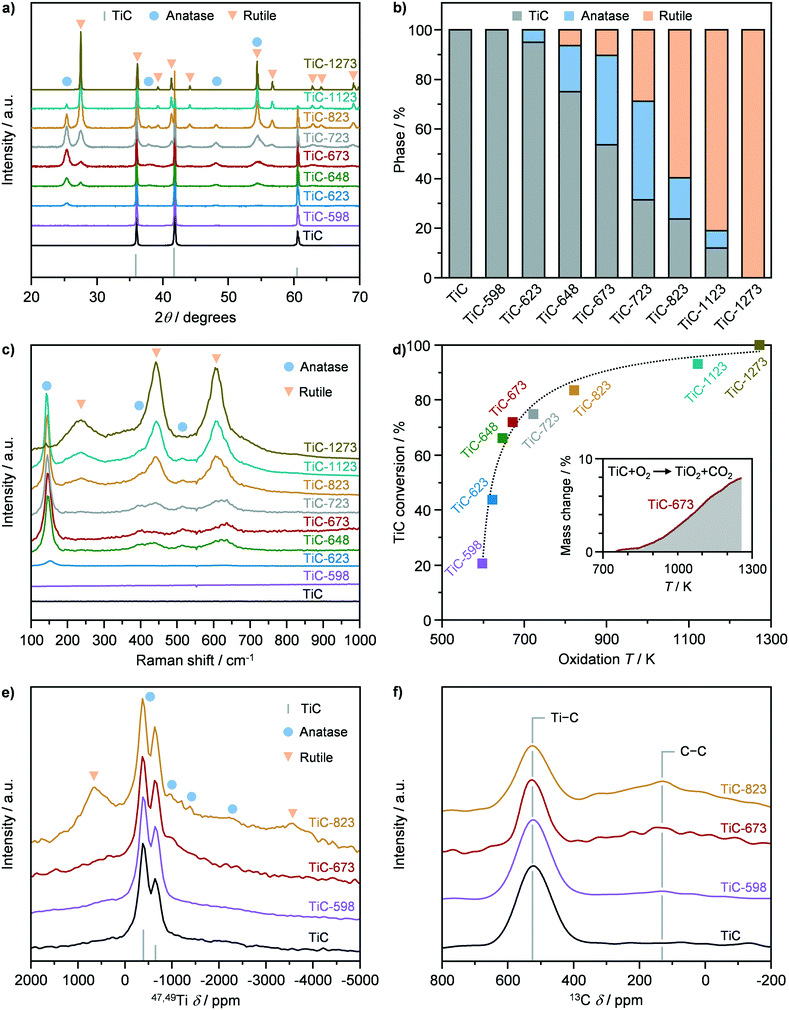 |
| | Fig. 1 a) X-ray diffraction (XRD) patterns, b) phase composition quantified from XRD, c) Raman spectra, d) conversion of TiC quantified from thermogravimetric analysis, as well as e) 47,49Ti and f) 13C nuclear magnetic resonance of TiC in fresh form and after thermal oxidation at different temperatures. | |
The Ti and C environment of the pristine TiC and of selected composites were assessed by 47,49Ti and 13C NMR (Fig. 1e and f). The samples were chosen based on their limited (ca. 20%; TiC-598), moderate (ca. 70%; TiC-673), and significant (ca. 85%; TiC-823) fraction of TiO2, according to TGA analysis. All materials showed two signals centred at −628 and at −362 ppm in the 47,49Ti NMR spectrum (Fig. 1e), which are the reported shifts for TiC,27 corroborating the presence of unreacted TiC in the composites oxidized at temperatures above 623 K. In addition, the 47,49Ti NMR spectrum of pristine TiC evidenced a broad, undifferentiated, underlying signal also known from literature.27 Similarly, 13C NMR evidenced the sole contribution of TiC by the appearance of a signal at 525 ppm. Analysis of the 47,49Ti NMR spectrum of the TiC-598 sample showed the absence of TiO2 bands in agreement with XRD and Raman, which evidenced the absence of crystalline oxide. TiC-673 displayed a relative increase of the broad underlying signal, which might be assigned to an increase in either unordered oxide or small domain of the anatase phase. The TiC-823 sample, on the other hand, clearly displayed the formation of a dominant amount of TiO2-rutile and, to a lesser extent, of anatase, in agreement with XRD and Raman results, as indicated by the presence of bands at −3554 and 703 ppm and at −2534, −1248, −761, and −495 ppm (Fig. 1e and S3†), respectively.28 Finally, 13C NMR spectra of the TiO2–TiC composites showed the presence of a signal at 130 ppm (Fig. 1f), which was associated with oxidized carbon residues or char.29
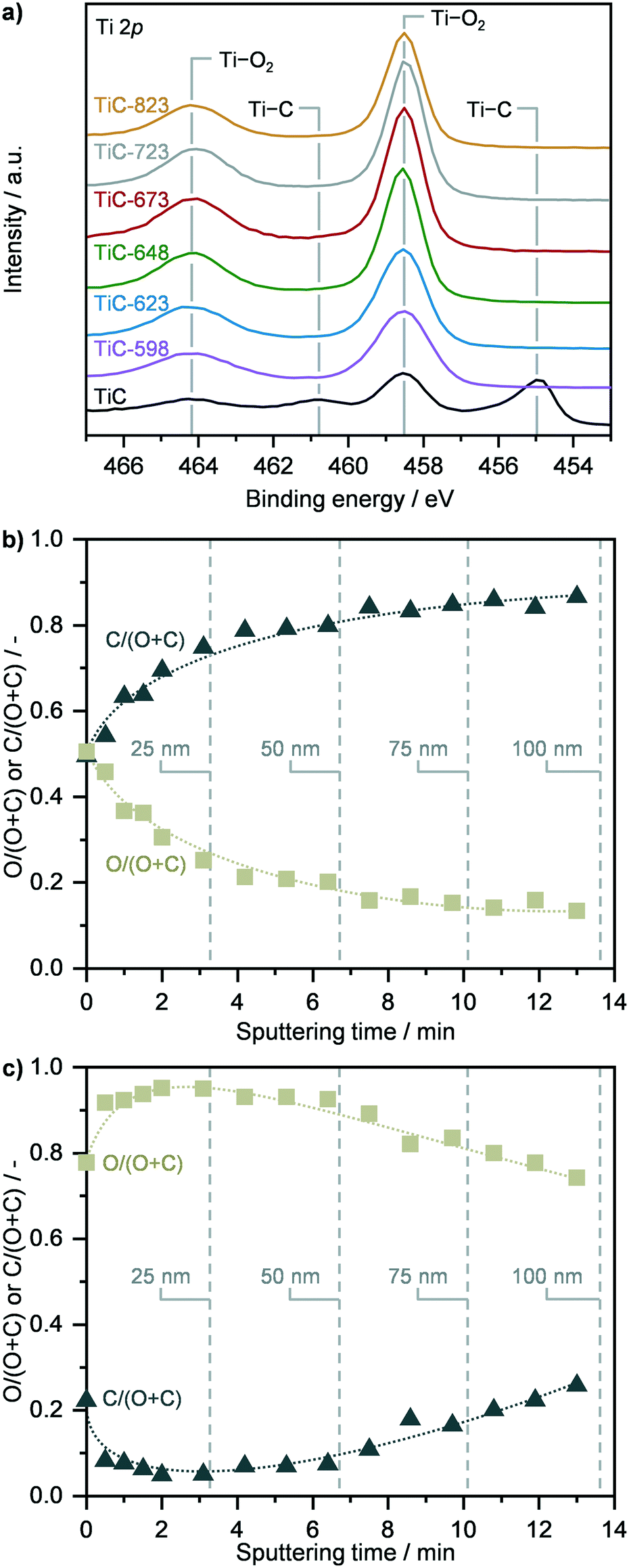
The surface properties of TiC and of TiO2–TiC composites were assessed by X-ray photoelectron spectroscopy (Fig. 2 and S4, Table S1†). Analysis of the Ti 2p and O 1s spectra as well as elemental composition revealed that the surface of pristine TiC was partially passivated (Fig. 2a and S4, Table S1†). The surface of the composite samples was found fully oxidized, and no signals associated to carbides were observed in the Ti 2p and C 1s spectra (Fig. 2a and S4†). Accordingly, the surface O:Ti atomic ratio increased from ca. 1.7 in pristine TiC to around 2.3 in the oxidised composites, which resembles the stoichiometric ratio of TiO2, and remained stable across all samples (Table S1†). Considering the continuous transformation of TiC as a function of the oxidation temperature as observed by XRD, TGA, Raman and NMR spectroscopies (vide supraFig. 1) together with the constant surface composition previously discussed, the increased formation of TiO2 might occur below the surface, suggesting that TiC transforms following a shrinking core mechanism, in agreement with previous reports.16 To further corroborate this hypothesis, XPS sputtering was conducted over TiC and over representative TiC-598 and TiC-673 samples (Fig. 2b, c and S5†). While only the immediate surface of parent TiC was found oxidized in accordance with previous observations, the analysis of TiC-598 showed a steadily decreasing atomic O:(O + C) ratio, indicating the presence of a TiC core (Fig. 2c). The deviation from the typical step-like function of a shrinking core model suggest that the surrounding layer could consist of a mixture of carbide and oxide. This indicates that a spatially-random spot-oxidation of TiC might occur at the same time, leading to the mixed carbide/oxide shell. A higher oxidation temperature of 673 K led to an atomic O:(O + C) ratio of ca. 1 across the 100 nm depth investigated (Fig. S5†), which suggests the presence of an oxide shell and points towards a shrinking core oxidation mechanism.
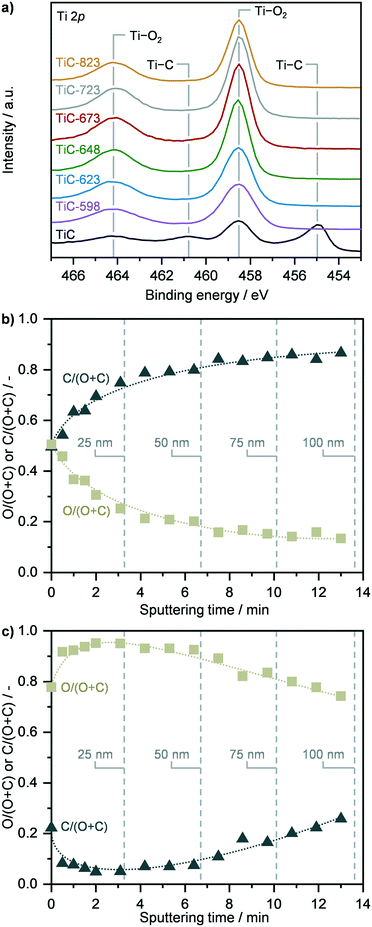 |
| | Fig. 2 a) Ti 2p X-ray photoelectron spectra (XPS) of TiC in fresh form and after thermal oxidation in air at different temperatures. Depth profiles of TiC b) in fresh form and c) after thermal oxidation at 598 K, showing the relative oxygen and carbon composition as a function of sputtering time, which is indicative of the content of titanium carbide and oxide in the TiC and TiC 598 samples. | |
Porosity.
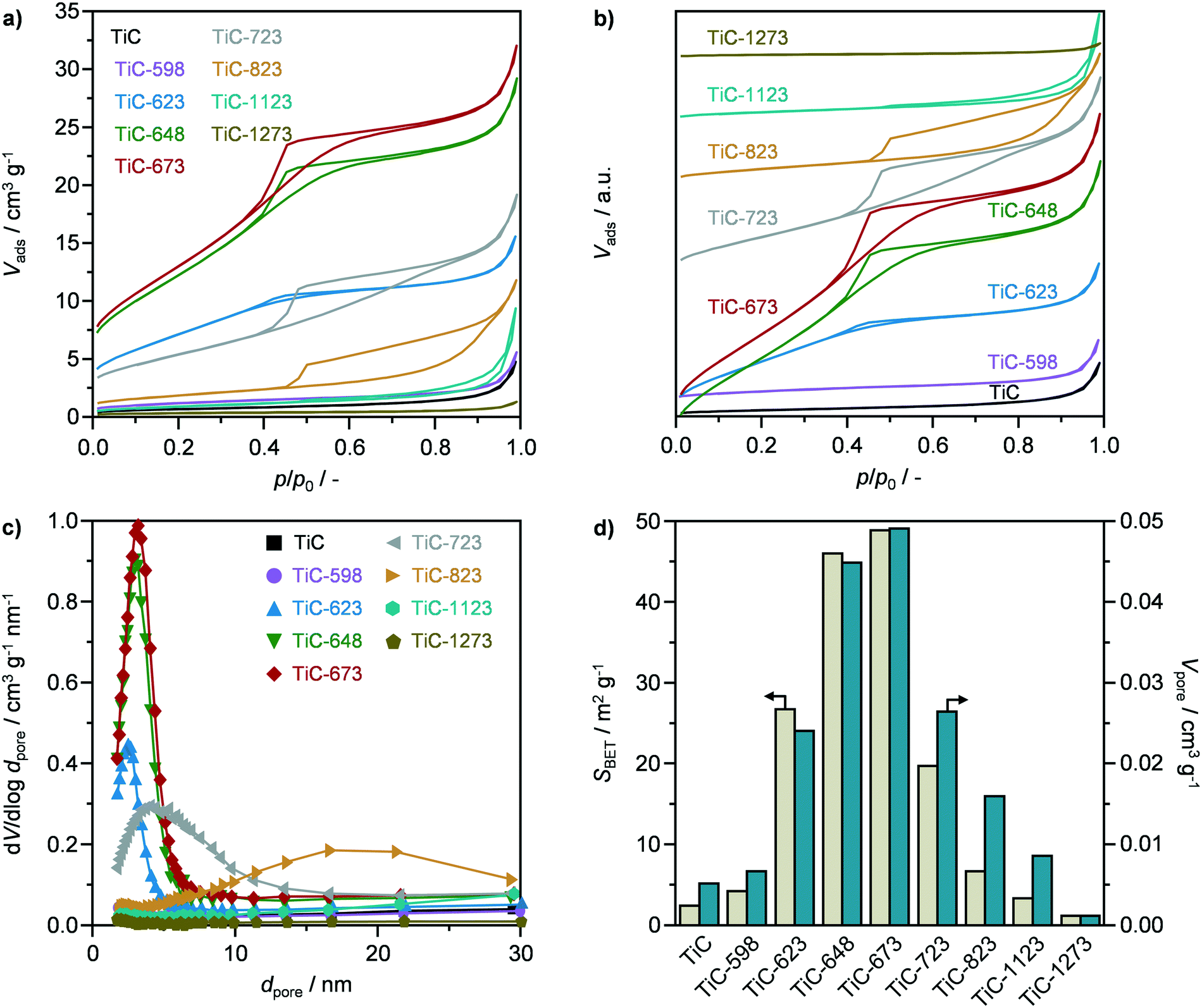
The textural properties of pristine TiC and of TiO2–TiC composites were assessed by N2 sorption in order to unravel the porosity evolution as a function of the oxidation temperature. The sorption isotherm of parent TiC, as shown in Fig. 3a and b, and the respective pore size distribution (Fig. 3c) evidenced the absence of porosity. Accordingly, the carbide showed very low values for the specific surface area (5 m2 g−1) and pore volume (0.005 cm3 g−1), as observed in Fig. 3d. While oxidation at 598 K did not significantly alter the textural properties, calcination at 623, 648, and 673 K resulted in the formation of mesopores with an average diameter of ca. 2.5, 3.0, and 3.2 nm, respectively, according to the Barrett–Joyner–Halenda method, as well as in the concomitant increase in the surface area and pore volume (Fig. 3). In particular, the latter reached a maximum of ca. 50 m2 g−1 and ca. 0.05 cm3 g−1 after oxidation at 648 K and especially at 673 K (Fig. 3d). The corresponding isotherms of TiC-648 and TiC-673 could be classified as type IV, and exhibited a hysteresis of type H2, which is typical of bottle-neck and/or occluded pores (Fig. 3a and b).30 Higher oxidation temperatures led to a reduction in porous properties, as evidenced by a sharp decline in the surface area and pore volume (Fig. 3). Interestingly, the samples calcined at 723 K and particularly at 823 K exhibited a change in hysteresis, which transformed into a H3 type, indicating the formation of slit-like pores (Fig. 3a and b).30 Accordingly, the average pore size increased to ca. 4.2 nm after oxidation at 723 K. On the other hand, the pores size distribution in the TiC-823 sample was centred around 18 nm (Fig. 3c), which is a typical artefact that can occur with the analysis of isotherms with H3 hysteresis.30 Even higher oxidation temperatures, i.e., 1123 and 1273 K, led to virtually the complete degradation of the porous properties, resembling those of pristine TiC. The observed volcano behaviour of the textural properties could be explained by the competition between evolution of CO2 during the calcination, promoting pore formation, and temperature-induced sintering. While the former is likely dominant below 673 K, the latter prevails at higher temperatures (≥723 K) leading to a non-porous material.
 |
| | Fig. 3 N2 sorption isotherms at 77 K with a) absolute values and b) offset values to display transformation, c) pore size distribution, and d) specific surface area and pore volume of TiC in fresh form and after thermal oxidation at different temperatures. | |
Morphology.
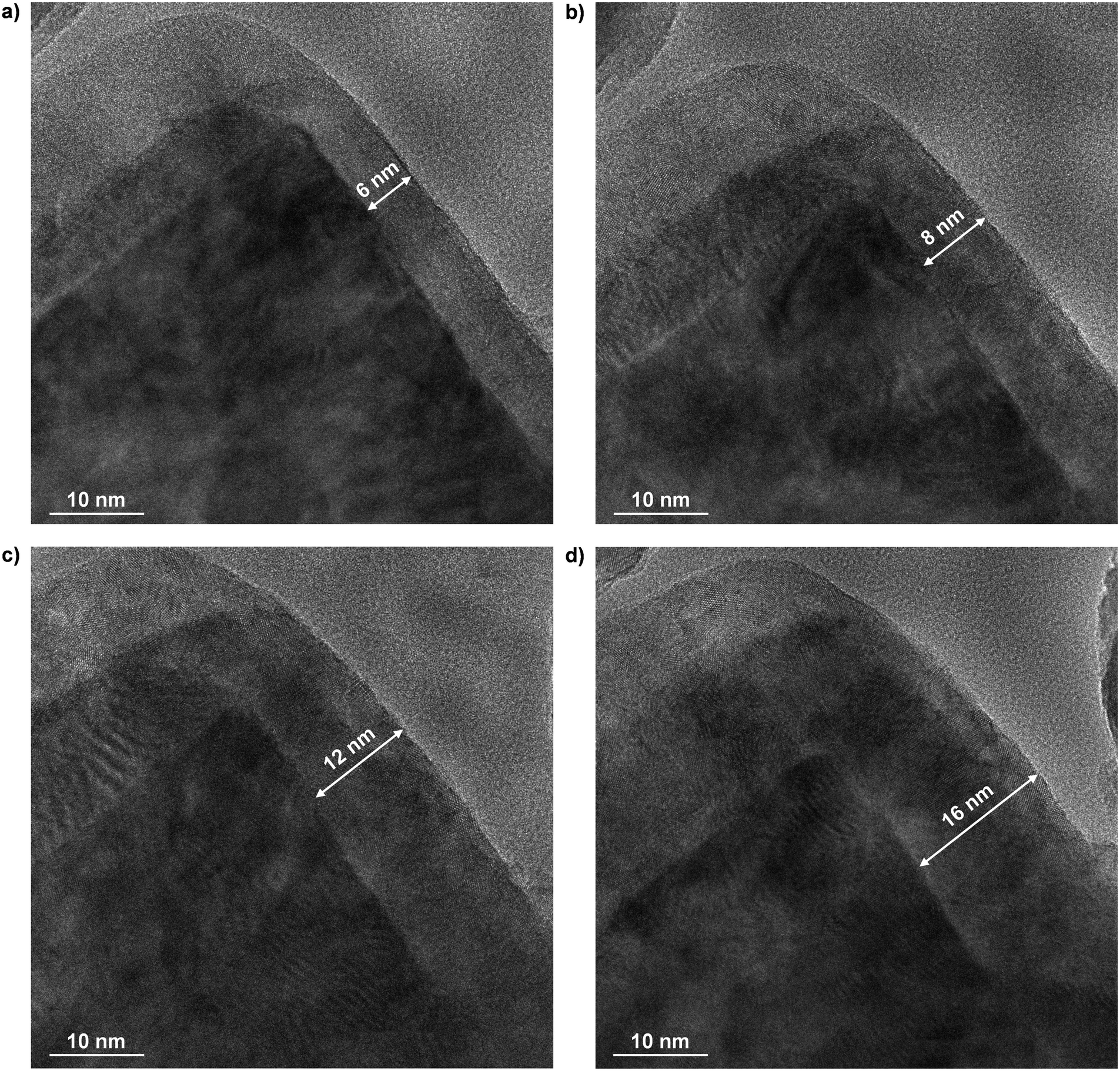
The morphological properties of bare TiC and of selected TiO2–TiC composites were investigated by high-resolution transmission electron microscopy. The samples were chosen based on the same rationale as discussed previously. As shown in Fig. 4a, parent TiC exhibited crystalline domains with a predominant TiC(200) orientation, in agreement with XRD results. In addition, an amorphous phase was also present, which could be attributed to surface passivation, in accordance to previous reports.31 Formation of islands of TiO2 could be observed across all TiO2–TiC composites investigated (Fig. 4b–d). In particular, both anatase and rutile polymorphs were detected, especially over TiC-673 and TiC-823, as seen from lattice distance analysis, revealing the characteristic interlayer distances corresponding to (101) of TiO2-anatase and (110) of TiO2-rutile planes (Fig. 4b–d). The relative anatase:rutile ratio decreased with increasing oxidation temperature, in agreement with XRD and Raman spectra. Interestingly, islands of crystalline TiO2-anatase were observed also over TiC-598, although it was not detected by neither Raman nor XRD. Nevertheless, these small islands were surrounded by amorphous TiO2 (Fig. 4b), which is still in line with TGA analysis. TEM images of all other samples can be found in Fig. S6.† Curiously, the micrographs showed the presence of islands of TiC in all composites investigated, indicating that both TiC and TiO2 phases are present over the surface of the composite even after oxidation at high temperatures. This suggests that formation of TiO2 occur following a spot-oxidation mechanism, which partially supports the results obtained by XPS. To shed light on this uncertainty, in situ TEM was conducted over TiC during a flowing-air oxidation process. As observed in Fig. 5, at ca. 590 K the sample exhibited a core–shell structure, with a ca. 6 nm thick shell, presumably of TiO2 considering the previously discussed XPS results. As the temperature rose, the shell increased in thickness reaching up to 16 nm at 720 K. This indicates that the material follows a shrinking core oxidation mechanism. Interestingly, the observations gathered from ex situ TEM could not be detected by in situ TEM. In addition to the halved magnification of the latter technique, this is likely due to enhanced charging of the sample at high temperatures, which is a common issue in in situ microscopy techniques32 that can limit resolution and prevent detailed analyses of lattice fringes. Still, based on the in-depth characterization conducted, it is plausible that a combination of shrinking core and spot-oxidation mechanisms occur, but at different scales. While the former drives the transformation of the bulk of the material, the latter pathway likely acts at the surface level, in agreement with ex situ TEM and XPS sputtering.
 |
| | Fig. 4 High-resolution transmission electron microscopy images of TiC a) in fresh form and after thermal oxidation at b) 598 K, c) 673 K, and d) 823 K. The coloured areas highlight the different phases identified as amorphous (green) and crystalline TiC (grey), TiO2-anatase (blue), and TiO2-rutile (orange). Insets at the corners of the panels highlight the interlattice distance that corresponds to a) TiC(200) (2.2 Å), b) TiO2 anatase(101) (3.5 Å), and c) TiO2 rutile(110) (3.2 Å). | |
 |
| | Fig. 5
In situ transmission electron microscopy images visualizing the thermal oxidation of TiC in air at a) 590 K, b) 620 K, c) 650 K, and d) 720 K with a heating rate of 10 K min−1. | |
Catalytic HBr oxidation
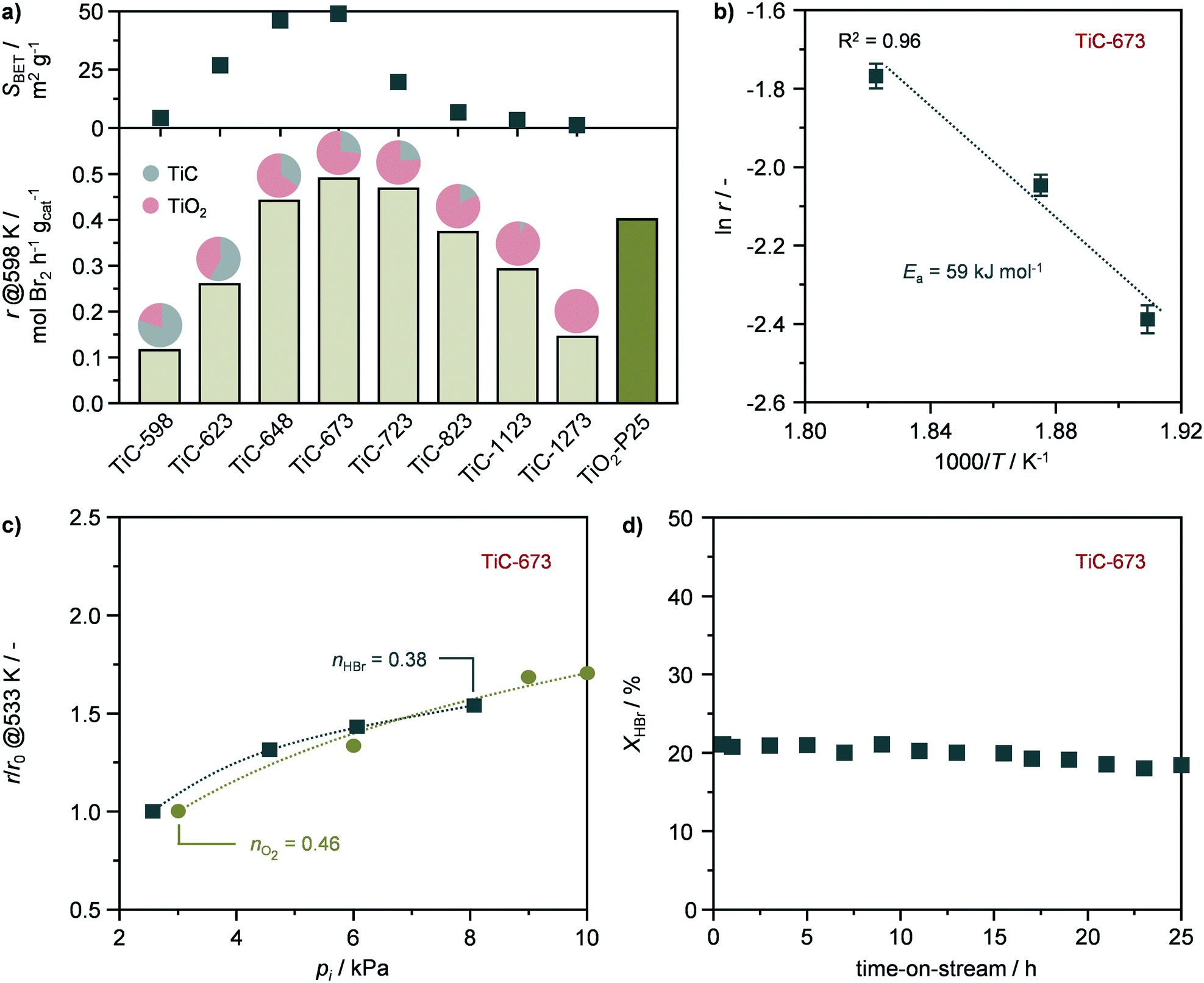
The TiO2–TiC composites were assessed in the catalytic oxidation of HBr to Br2. The reaction rates, expressed as the productivity of Br2 per mass of catalyst (eqn (2)) were measured at 598 K and at HBr conversion levels of ≤40% in the absence of mass and heat transfer constraints. To validate this assumption, the dimensionless moduli based on the criteria of Carberry, Mears, and Weisz–Prater were estimated and confirmed the lack of diffusion limitations and temperature gradients (ΔT ≤ 0.03 K) at inter and intraparticle levels under these conditions. These measured rates followed a volcano-like behaviour with the oxidation temperature (Fig. 6a), reaching a maximum of ca. 0.5 mol Br2 per h gcat over the TiC-673 sample, outperforming benchmark TiO2-P25. This system was chosen due to the presence of both anatase and rutile phases in its formulation in a comparable ratio (4:1) to that of the TiC-673 specimen. The activity of the TiO2–TiC composite originates from the presence of TiO2-anatase and TiO2-rutile, which, despite having intrinsically different reactivates, are known active phases in this reaction.8,10 These results represent the first application of TiO2–TiC composite as active phase in thermal catalysis.
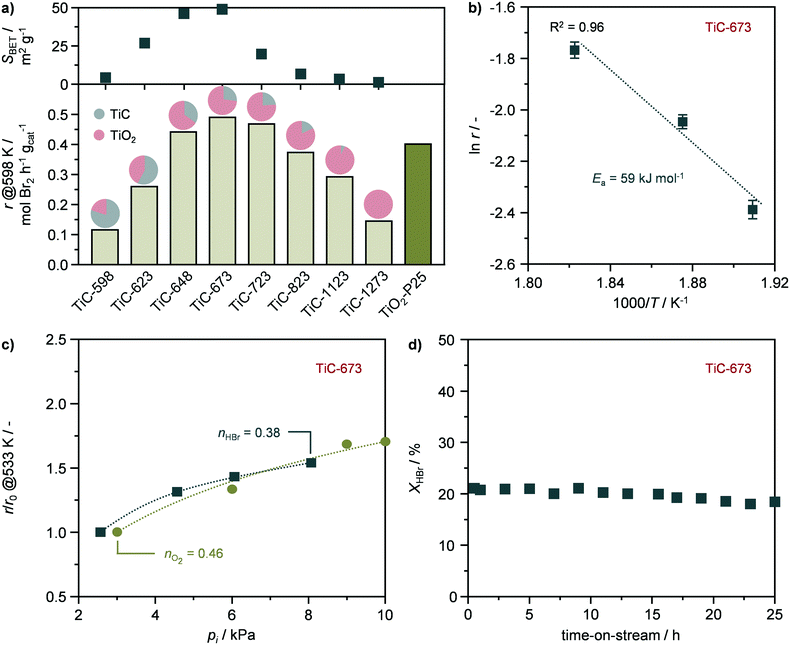 |
| | Fig. 6 a) Rate of Br2 production in the catalytic oxidation of HBr over the catalysts in a bar plot that includes the proportion of carbide and oxide phases as well as their corresponding surface areas for all catalysts. b) Arrhenius plot, c) HBr and O2 partial orders in HBr oxidation, and d) HBr conversion as a function of time-on-stream over TiC-673. Error bars are presented for each catalytic point in the Arrhenius analysis. Conditions: a, b and d) HBr:O2:He = 4.5:9:86.5; c) HBr:O2:He = 2.5–8:3–10:85.5–88.5; a–d) FT/Wcat = 200 cm3 STP min−1 gcat−1, T = 523–598 K, and P = 1 bar. | |
A kinetic study of the most active catalyst, TiC-673, was also carried out and performed under quasi differential conditions (≤15% conversion). The catalyst displayed an activation energy of 59 kJ mol−1 (Fig. 6b), which is in agreement with previous studies.8,10 In addition, the reaction orders with respect to HBr (nHBr) and oxygen (nO2) were estimated to 0.38 and 0.46 (Fig. 6c), respectively, which are in agreement with the values reported for TiO2-anatase and TiO2-rutile.8,10 This might indicate the interplay of both phases in the observed catalytic activity of the TiO2–TiC composite. Finally, the stability of representative TiC-673 was assessed by temperature cycle experiments and a long-term run. The catalytic activity of TiC-673 remained unaltered upon temperature cycling as well as after 25 h on stream (Fig. 6d and S7†). Characterisation by XRD showed that the material retained its crystallographic properties (Fig. S8†), indicating TiO2–TiC composite as a stable HBr oxidation catalyst.
Structure–performance relationships
The evolution of the structural, textural, and morphological properties upon the oxidation of TiC into TiO2–TiC composites was investigated by an array of techniques, providing key insights into the oxidation mechanism, and the resulting materials were found active in HBr oxidation due to the formation of TiO2 upon the transformation of TiC. Accordingly, the reactivity of the composites increased as the fraction of TiO2 in the material incremented until calcination at 673 K (Fig. 6a). However, despite the increased formation of TiO2, the reactivity of the samples steadily decreased at higher oxidation temperatures, until reaching comparable activity to TiC-598 that contained only ca. 20% of TiO2 (Fig. 6a). This volcano behaviour suggests a competition between two material properties that govern the catalytic activity of the composite. The transformation of TiC also led to the development of mesoporosity and to an increment of up to an order of magnitude in surface area and pore volume, especially after calcination at 673 K. Higher oxidation temperatures resulted in a gradual loss of porous properties until their virtually complete absence after calcination at 1273 K. Interestingly, the reactivity in HBr oxidation followed a very similar behaviour, with the most active catalyst being TiC-673 (Fig. 6a). This suggested a relation between catalytic activity and surface area, which have previously shown to be an important reactivity descriptor in hydrogen halide oxidation reactions.33,34 Given the similarity between the activity distribution and the porous properties as a function of the oxidation temperature, it is plausible to assume that the surface area of exposed TiO2 likely plays a key role in describing the reactivity of this composite. Accordingly, a positive trend was observed between the rate of Br2 production and the surface area of the composite (Fig. 7a), indicating that a high surface area is beneficial for high performance. The observed deviation from complete linearity seems to mainly arise from the low temperature calcined samples, as TiC-623 and especially TiC-598. This might be due to a series of factors such as (i) the minor content of TiO2 in these specimens, (ii) the fact that passivation of TiC at low temperature could result into a mixed TiCxOy having low activity in HBr oxidation, as well as (iii) structure-dependent effects, which highlights the complexity of this composite. By plotting the reaction rates expressed as the productivity of Br2 per surface area of catalyst (eqn (3)) as a function of the surface area (Fig. 7b), it could be observed an opposite trend in comparison to that shown in Fig. 7a. This suggests that a high calcination temperature is preferred to obtain a high specific activity, which is likely due to the considerably higher content of TiO2 in these composites, i.e., TiC-873, TiC-1123, TiC-1273. In addition, these materials have a considerably higher fraction of TiO2-rutile compared to best performing TiC-648, TiC-723, and particularly TiC-673, in which the oxide is mainly present as anatase polymorph (vide supraFig. 1). This is in line with recent studies that have shown the superior specific activity of the rutile phase compared to TiO2-anatase.10 Overall, these two trends indicate that both surface area and phase composition are key factors in determining the activity of these systems.
 |
| | Fig. 7 Rate of Br2 production based on the catalyst's a) mass and b) surface area in the catalytic oxidation of HBr over the systems as a function of the surface area of TiO2–TiC composite, SBET. c) Schematic representation of the evolution of the structural, porous, and morphological properties of the TiO2–TiC composite upon thermal oxidation of TiC in air and its synthesis–structure–performance relationships in HBr oxidation. The reactivity was rationalized based on the interplay between pore evolution and formation of anatase and rutile phases. Colour code: TiC: grey; TiO2-anatase: light blue; TiO2-rutile: orange. | |
In accordance to the previous analyses and hypotheses, it can be put forward that TiC transforms into TiO2–TiC composite through a combination of spot and shrinking core oxidation mechanisms. While the latter is dominant and concerns the bulk of the material, the former pathway is likely critical for the formation of pores as well as of islands of TiO2 over the surface of the TiO2–TiC composite, which are both important factors for the reactivity of this catalyst in HBr oxidation (Fig. 7c). Overall, these results indicate that the understanding of the TiC oxidation mechanism is crucial for deriving synthesis–structure–performance relationships to design optimal catalysts for HBr oxidation. This is critical in order to pave the way for the establishment of a bromine-mediate natural gas upgrading process, and opens new possibilities for the use of TiO2–TiC composites as highly active systems for TiO2-based catalytic applications.
Conclusions
In this study, we have demonstrated the first thermocatalytic application of TiO2–TiC composite in the oxidation of HBr into Br2. The system, which was synthesized via thermal oxidation of TiC in flowing air, has been extensively characterised in order to shed light on its oxidation mechanism and on the evolution of its structural, morphological, and textural properties, which was necessary to derive synthesis–structure–performance relationships. In-depth material characterisation, by means of X-ray diffraction, thermogravimetric analysis, N2 sorption, Raman, X-ray photoelectron, 47,49Ti and 13C nuclear magnetic resonance spectroscopies, as well as ex situ and in situ transmission electron microscopy revealed that TiC transforms via a combination of shrinking core and spot-oxidation mechanisms. This resulted in the formation of a TiO2–TiC composite, containing both anatase and rutile polymorphs, with enhanced mesoporosity, reaching up to an order of magnitude higher surface area and pore volume when calcined at 673 K as compared to pristine TiC. Reactivity and kinetic analyses were conducted in HBr oxidation, which showed that the catalyst activity followed a volcano behaviour. The best catalyst was obtained after thermal oxidation at 673 K, coinciding with the porosity apex, which outperformed benchmark TiO2-P25 and was demonstrated stable for 25 h on stream. Accordingly, the reactivity of the composite was rationalized based on the interplay between TiO2 formation and porosity generation, which can be tailored by tuning the oxidation temperature. These findings constitute a promising basis to design tuneable, high performance TiO2-based catalysts for HBr oxidation, paving the way for the establishment of a bromine-mediated process for efficient valorisation of natural gas.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by ETH Research Grant ETH-04 16-1. We thank the Scientific Center for Optical and Electron Microscopy and Prof. Ralph Spolenak of ETH Zurich for use of their facilities and Raman spectroscopy. Dr. Sharon Mitchell is thanked for the valuable discussions.
References
- R. Lin, A. P. Amrute and J. Pérez-Ramírez, Chem. Rev., 2017, 117, 4182–4247 CrossRef CAS PubMed.
- E. McFarland, Science, 2012, 338, 340 CrossRef CAS PubMed.
- V. Paunović, G. Zichittella, M. Moser, A. P. Amrute and J. Pérez-Ramírez, Nat. Chem., 2016, 8, 803–809 CrossRef PubMed.
- J. He, T. Xu, Z. Wang, Q. Zhang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2012, 51, 2438–2442 CrossRef CAS PubMed.
- G. Zichittella, S. Stähelin, F. M. Goedicke and J. Pérez-Ramírez, ACS Catal., 2019, 9, 5772–5782 CrossRef CAS.
- K. Ding, H. Metiu and G. D. Stucky, ACS Catal., 2013, 3, 474–477 CrossRef CAS.
- M. Moser, V. Paunović, Z. Guo, L. Szentmiklósi, M. G. Hevia, M. Higham, N. López, D. Teschner and J. Pérez-Ramírez, Chem. Sci., 2016, 7, 2996–3005 RSC.
- M. Moser, I. Czekaj, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2014, 53, 8628–8633 CrossRef CAS PubMed.
- V. Paunović, R. Lin, M. Scharfe, A. P. Amrute, S. Mitchell, R. Hauert and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2017, 56, 9791–9795 CrossRef PubMed.
- V. Paunović, M. Rellán-Piñeiro, N. López and J. Pérez-Ramírez, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.03.036.
- M. Moser, G. Vilé, S. Colussi, F. Krumeich, D. Teschner, L. Szentmiklósi, A. Trovarelli and J. Pérez-Ramírez, J. Catal., 2015, 331, 128–137 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- L. Calvillo, G. García, A. Paduano, O. Guillen-Villafuerte, C. Valero-Vidal, A. Vittadini, M. Bellini, A. Lavacchi, S. Agnoli, A. Martucci, J. Kunze-Liebhäuser, E. Pastor and G. Granozzi, ACS Appl. Mater. Interfaces, 2016, 8, 716–725 CrossRef CAS PubMed.
- Y. Liu, B. de Tymowski, F. Vigneron, I. Florea, O. Ersen, C. Meny, P. Nguyen, C. Pham, F. Luck and C. Pham-Huu, ACS Catal., 2013, 3, 393–404 CrossRef CAS.
- A. Ignaszak, C. Song, W. Zhu, J. Zhang, A. Bauer, R. Baker, V. Neburchilov, S. Ye and S. Campbell, Electrochim. Acta, 2012, 69, 397–405 CrossRef CAS.
- S. J. Stott, R. J. Mortimer, S. E. Dann, M. Oyama and F. Marken, Phys. Chem. Chem. Phys., 2006, 8, 5437–5443 RSC.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- D. R. Baer, M. H. Engelhard, A. S. Lea, P. Nachimuthu, T. C. Droubay, J. Kim, B. Lee, C. Mathews, R. L. Opila, L. V. Saraf, W. F. Stickle, R. M. Wallace and B. S. Wright, J. Vac. Sci. Technol., A, 2010, 28, 1060–1072 CrossRef CAS.
- S. Hashimoto and A. Tanaka, Surf. Interface Anal., 2002, 34, 262–265 CrossRef CAS.
- L. A. O'Dell and R. W. Schurko, Chem. Phys. Lett., 2008, 464, 97–102 CrossRef.
- E. Kupce and R. Freeman, J. Magn. Reson., Ser. A, 1995, 115, 273–276 CrossRef CAS.
- J. D. van Beek, J. Magn. Reson., 2007, 187, 19–26 CrossRef CAS PubMed.
-
J. J. Carberry, Physico-Chemical Aspects of Mass and Heat Transfer in Heterogeneous Catalysis, Catal. Sci. Technol, Springer-Verlag, Berlin, 1987, vol. 8, pp. 131–171 Search PubMed.
- D. Mears, J. Catal., 1971, 20, 127–131 CrossRef CAS.
- B. H. Lohse, A. Calka and D. Wexler, J. Appl. Phys., 2005, 97, 114912 CrossRef.
- O. Frank, M. Zukalova, B. Laskova, J. Kürti, J. Koltai and L. Kavan, Phys. Chem. Chem. Phys., 2012, 14, 14567–14572 RSC.
- K. J. D. MacKenzie, R. H. Meinhold, D. G. McGavin, J. A. Ripmeester and I. Moudrakovski, Solid State Nucl. Magn. Reson., 1995, 4, 193–201 CrossRef CAS PubMed.
- C. Gervais, M. E. Smith, A. Pottier, J. P. Jolivet and F. Babonneau, Chem. Mater., 2001, 13, 462–467 CrossRef CAS.
- X. Cao and K. Schmidt-Rohr, Environ. Sci. Technol. Lett., 2018, 5, 476–480 CrossRef CAS.
- J. C. Groen, L. A. A. Peffer and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2003, 60, 1–17 CrossRef CAS.
- O. Masahiro, K. Isao and M. Eizo, Bull. Chem. Soc. Jpn., 1986, 59, 689–695 CrossRef.
- J. Ditto, D. Krinsley and K. Langworthy, Scanning, 2012, 34, 279–283 CrossRef CAS PubMed.
- A. P. Amrute, C. Mondelli, M. Moser, G. Novell-Leruth, N. López, D. Rosenthal, R. Farra, M. E. Schuster, D. Teschner, T. Schmidt and J. Pérez-Ramírez, J. Catal., 2012, 286, 287–297 CrossRef CAS.
- C. Li, Y. Sun, I. Djerdj, P. Voepel, C.-C. Sack, T. Weller, R. Ellinghaus, J. Sann, Y. Guo, B. M. Smarsly and H. Over, ACS Catal., 2017, 7, 6453–6463 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Complementary characterisation and catalytic data. See DOI: 10.1039/d0cy00805b |
| ‡ Equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ali J.
Saadun
a,
Simon
Büchele
a,
Begoña
Puértolas
a,
René
Verel
a,
Ali J.
Saadun
a,
Simon
Büchele
a,
Begoña
Puértolas
a,
René
Verel